
A novel, environment-friendly method to prepare pyranones from furfural alcohols via photocatalytic O2 oxidation in an aqueous phase†
Bei
Zhou
 a,
Yun-Feng
Tao
a,
Yu-Juan
He
a,
Lan-Xiang
Liu
a,
Zu-Hui
Chang
a,
Xiang-Hong
Li
a,
Tong
Lin
*b and
Guan-Ben
Du
*a
a,
Yun-Feng
Tao
a,
Yu-Juan
He
a,
Lan-Xiang
Liu
a,
Zu-Hui
Chang
a,
Xiang-Hong
Li
a,
Tong
Lin
*b and
Guan-Ben
Du
*a
aInternational Joint-Research Center for Bio-Materials, Ministry of Science and Technology, Key Laboratory of Forest Resources Conservation and Utilization in the Southwest Mountains of China, Ministry of Education, Yunnan Province Key Laboratory of Wood Adhesives and Glued Products, School of Materials and Chemical Engineering (and College of Life Science), Southwest Forestry University, Kunming 650224, China. E-mail: guanben@swfu.edu.cn
bState Key Laboratory of Separation Membranes and Membrane Processes, School of Textile Science and Engineering, Tiangong University, Tianjin 300387, China. E-mail: tong.lin@tiangong.edu.cn
First published on 13th October 2022
Abstract
This study developed a novel photocatalytic method to synthesize pyranones from furfural alcohols. By using 0.2 mol% equivalent of tris-Ir(ppy)3 as a photocatalyst under visible light and O2 as the reaction atmosphere, furfural alcohols were rapidly oxidized and hydrolyzed in the aqueous reaction solution to produce diol intermediates, which further underwent hydrolysis, furan ring-opening and rearrangement to form the final product pyranones. The entire reaction was carried out at room temperature with a yield as high as 86% and a conversion efficiency of 90%. This method is general and suitable for furfural alcohols with different substituted groups on the 5- and α-OH sites. We used in situ IR and real-time NMR to examine the reaction kinetics, GC-MS and isotope-labeling experiments to track the source of atom transfer in the reaction, and EPR to identify the singlet oxygen. The results showed that the reaction followed a first-order rate equation, and O2 oxidation was the rate-determining step. The transferred H atom came from the H2O solvent, the O atom from the O2 atmosphere, and the singlet oxygen from the O2 photocatalysis. The UV-vis and cyclic voltammetry results indicated that this photocatalysis reaction involved a single electron transformation process. We also used DFT calculation to simulate the reaction routes and showed that the tris-Ir(ppy)3 photocatalysis O2 oxidation in the aqueous phase was the critical step of endoperoxide oxidation, which differs from oxygen onium ion oxidation in the classic Achmatowicz rearrangement reaction. Our method can carry out a 10.0 gram-scale reaction with a 78% yield. It avoids stoichiometric consumption of toxic or corrosive oxidative reagents in the Achmatowicz rearrangement reaction. It may provide a novel, efficient, and “green” alternative to the Achmatowicz rearrangement reaction.
Introduction
Pyranone (6-hydroxy-2H-pyran-3(6H)-one) and its derivatives are critical precursors in monosaccharide synthesis,1O-glycosylation,2 [5 + 2] cycloaddition,3 Kishi reduction,4 Ferrier allylation,5 metallizations,6 redox isomerization,7 and Tsuji–Trost arylation.8 These synthetic building blocks address the challenge of bio-active structural complex molecule synthesis,9 including indole diterpenoids shearinines10 and paspalicines,11alstofolinine,12uprolide,13norhalichondrin,14ipomoeassin,15 and glycosides.2 Furthermore, pyranones are a vital intermediate in synthesizing medicines, such as aspergillides16 and damone pheromones.17The Achmatowicz rearrangement (AR) reaction is the major method widely used to prepare pyranones from furfural alcohols (FALs), developed in 1971.1,18 It is also the only method for large-scale preparation of pyranones. The AR reaction typically uses stoichiometric bromine (Br2) to oxidize the furan-ring into furan-bromonium ions and sequentially ring-opens and rearranges the furan in an acid (e.g., H2SO4) to form the pyran-ring. It causes pollution issues due to using toxic Br2. Although bromine surrogates, e.g., N-bromosuccinimide,19meta-chloroperbenzoic acids,20 and other oxidants, such as potassium peroxomonosulfate,21 can be used to replace Br2, they add to the cost. Recently, enzymatic aerobic ring rearrangement,22 Fenton chemistry (using FeBr2 or CeBr3 combined with H2O2),23tert-butyl hydroperoxides combined with VO(OiPr)3 catalysis,24 and [Mn]-H2O2 catalysis25 have been reported to improve the AR reaction. However, they are still not ideal because they use stoichiometric and excessive oxidants. Other methods have been explored besides the AR reaction, such as the multistep manipulations of sugars.26 These methods were seldom used for pyranone preparation because the harsh reaction conditions and complicated reactions usually lead to a very low yield. Finding a novel, effective method to prepare pyranones without consuming toxic and corrosive oxidants is still challenging.
Oxidation through singlet oxygen (1O2) is very common in nature27 and plays an important role in synthesizing natural products and drugs.28–33 A few attempts were reported to convert FAL into pyranone using a singlet-oxygen photocatalytic oxidation method. However, trioxolanes were always formed in the reaction,34 and their removal required stoichiometric triphenylphosphine (PPh3)35 or dimethyl sulfide (DMSO).36 Recently, Gilmore's group reported a photocatalysis method to prepare pyranone from FAL using [Ru(bpy)Cl2·6H2O] as the catalyst to generate singlet oxygen persulfate radicals.37 It used stoichiometric sodium persulfate (Na2S2O8) for stepwise furan-ring oxidation. However, a simple and green singlet oxygen source for effective furan-to-pyranone conversion has yet to be reported in the research literature.
This study demonstrates a novel photocatalytic synthesis method to convert FALs into pyranones. Using 0.2 mol% equivalent of tris-Ir(ppy)3 as a photocatalyst under visible light irradiation and atmospheric O2 as an oxidative agent, we showed that FALs could be quickly oxidized and hydrolyzed into pyranone products (Fig. 1). The reaction was undertaken at room temperature with mild reaction conditions, reaching a good yield of 86% and a high conversion efficiency (90%). This method was general and suitable for FALs with different substituted functional groups on the 5- and α-OH sites (most from biomass).
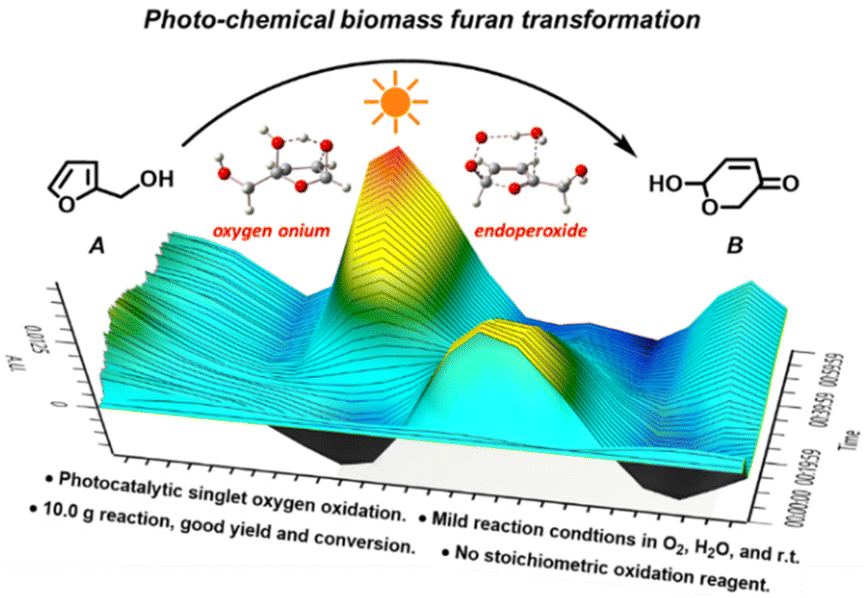 | ||
| Fig. 1 Photocatalytic furfural alcohol (A)-to-pyranone (B) conversion. | ||
We used in situ FTIR and real-time NMR to examine the reaction kinetics, GC-MS and isotope-labeling experiments to track the source of atom transfer in the reaction, and EPR to identify the singlet oxygen. The results showed that the reaction followed a first-order rate equation, and O2 oxidation was the rate-determining step. The transferred H atom came from the H2O solvent, the O atom from the O2 atmosphere, and the singlet oxygen from the O2 photocatalysis. The UV-vis and cyclic voltammetry experiments indicated that this photocatalysis reaction involved a single electron transformation process. We also simulated the reaction routes using a density functional theory (DFT) method. We showed that the tris-Ir(ppy)3 photocatalysis O2 oxidation in the aqueous phase was the critical step of endoperoxide oxidation, which differs from oxygen onium ion oxidation in the classical Achmatowicz rearrangement reaction. Our method can carry out a 10.0 gram-scale reaction with a 78% yield. It avoids stoichiometric consumption of oxidative reagents in the classic Achmatowicz reaction.
Results and discussion
Basic photocatalytic synthesis reaction
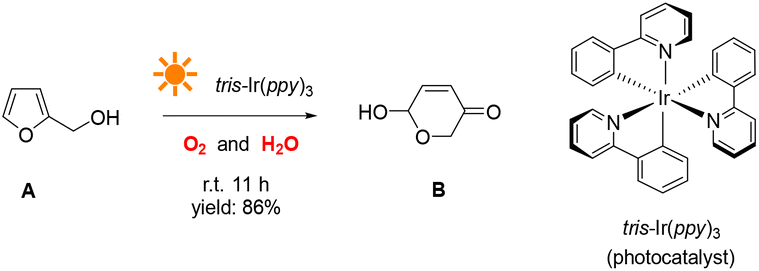
In a typical photocatalytic reaction, the aqueous solution of FAL (A, 0.5 gram-scale) and tris-(2-phenylpyridine)-iridium (tris-Ir(ppy)3) was irradiated with visible light (Fig. 1). The reaction was carried out under an oxygen atmosphere at ambient temperature. (Tris-Ir(ppy)3) fine powder sublimate was used to ensure good dispersion in the reaction mixture. After 11 h, pyranone (B) was isolated by column chromatography (with neutral alumina, Al2O3). The yield was 86%, and the conversion efficiency was 90% (see the details in the ESI†).We conducted this reaction under different reaction conditions to examine the effect of the reaction parameters on the reaction yield. The experiment details and results are listed in Table 1. No pyranone B was obtained without tris-Ir(ppy)3 (Table 1, entry 2). When the reaction was performed without photo-irradiation, no pyranone B was obtained either, even at an increased temperature (80 °C) for 24 h (Table 1, entry 3). LEDs with different spectral bands, e.g., 365–370 nm, 395–400 nm, 410–415 nm, 460–465 nm, 520–525 nm, 590–595 nm, and 620–625 nm, and white light (18 W for all LEDs), were used for the reaction (see more details in the ESI†). The reaction irradiated with a shorter wavelength of light consumed the reactant but resulted in less targeted products, hence a lower yield but larger conversion efficiency (Fig. 2). The reaction under white light LED irradiation achieved the best yield (Table 1, entry 4). Therefore, 18 W light LED was chosen as the light source in further experiments.
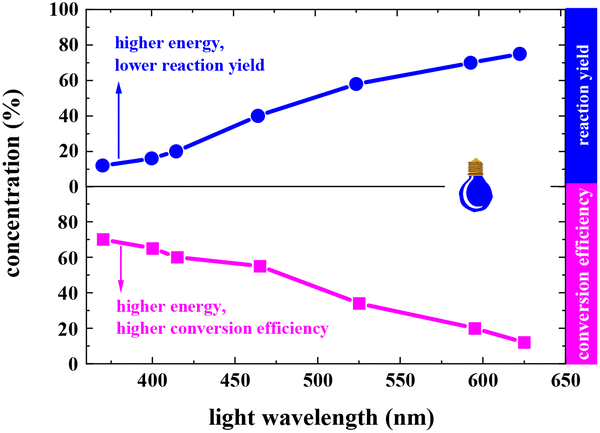 | ||
| Fig. 2 Effect of light wavelength on the reaction yield and conversion efficiency. | ||
|
|
||
|---|---|---|
| Entries | Variations of the standard reaction conditions | Yield (%) |
| a Standard reaction conditions: FAL A (0.5 g, 10.2 mmol), tris-Ir(ppy)3 (7.3 mg, 0.2mol%), and H2O (10.0 mL) were added to a two-necked quartz flask. The reaction was carried out under the irradiation of visible white LED (18 W) at room temperature under an O2 balloon atmosphere for 11 h. b Isolated yield by column chromatography with neutral alumina was used, and the yield was calculated after recycling the FAL reactant. “—” means no target pyranone was observed. | ||
| 1 | None | 86 |
| 2 | No tris-Ir(ppy)3 | — |
| 3 | No light irradiation, but 80 °C for 24 h | — |
| 4 | Light wavelength, 365 nm, 395 nm, 410 nm, 460 nm, 520 nm, 590 nm, 620 nm | 12–76 |
| 5 | Without H2O | — |
| 6 | CH3OH or C2H5OH to replace H2O | 32–36 |
| 7 | CH2Cl2, THF, dioxane, toluene, acetone, DMF or DMSO as a solvent | — |
| 8 | AcOH or NaOH (0.5 equiv.) addition | — |
| 9 | Ambient atmosphere | 22 |
| 10 | Ar atmosphere | — |
| 11 | Reaction time, 48 h | 38 |
| 12 | Ru(bpy)3Cl2 as photocatalyst | 50 |
| 13 | 4CzIPN as photocatalyst | 10 |
| 14 | Rose Bengal as photocatalyst | 10 |
| 15 | 0.1, 0.01 and 0.001 mol% tris-Ir(ppy)3 | 50, 25, 5 |
| 16 | 1.0 to 5.0 mol L−1 concentration A | 76, 82 |
No targeted pyranone B was obtained without H2O. In this case, 62% FAL A was consumed to form furan aldehydes, ketones, and acids (Table 1, entry 5, the products were analyzed by gas chromatography-mass spectrometry, GC-MS, see the details in the ESI†). The yield decreased when other protonic solvents, such as CH3OH and C2H5OH, were used as a solvent to replace H2O (Table 1, entry 6). When non-protonic solvents, such as CH2Cl2, THF, dioxane, toluene, acetone, DMF, and DMSO, were used, the reactant was consumed; however, no targeted pyranone B was obtained (Table 1, entry 7). Additionally, when 0.5 equivalent-mol of Lewis acid (e.g., AcOH) or Lewis base (e.g., NaOH) was added to the aqueous reaction solution, the majority of reactants vanished quickly, and the desired product was not obtained (Table 1, entry 8).
The atmospheric conditions impacted the reaction greatly. When the reaction was conducted under an ambient atmosphere, the yield was only 22% (Table 1, entry 9), much lower than under an oxygen atmosphere. When the reaction time was extended from 11 h to 24 h and 48 h, the yield increased from 22% to 33% and 46%, and the conversion efficiency was increased from 35% to 67% and 84%. It suggests that a longer reaction time would slightly increase the yield. However, when the reaction occurred under an argon atmosphere, no conversion occurred, with large amounts of reactant A recycled (Table 1, entry 10, the products were analyzed by GC-MS, see the details in the ESI†).
The effect of reaction time on the reaction yield was examined. Overoxidation occurred when increasing the reaction time from 11 h to 48 h. In this case, some products and reactants decomposed. The yield was reduced to 38%, with very little reactant A recycled (Table 1, entry 11).
The above results suggest that tris-Ir(ppy)3, visible light, water, and oxygen are essential for this reaction. Since tris-Ir(ppy)3 is a visible-light photocatalyst, which involves a typical single electron transfer oxidation as reported in the literature,38 we tested other three photocatalysts, tris-(2,2′-bipyridine) ruthenium(II) dichloride (Ru(bpy)3Cl2), 2,4,5,6-tetrakis-(carbazol-9-yl)-1,3-dicyano-benzene (4CzIPN) and Rose Bengal. Ru(bpy)3Cl2 has a metal–polypyridyl complex structure similar to tris-Ir(ppy)3. 4CzIPN39 is a non-metal photocatalyst with a redox potential close to that of tris-Ir(ppy)3, and it has a smaller energy gap between its singlet and triplet excited states. Rose Bengal is a general and widely used quinone-type natural photosensitizer.
Under the same conditions, the reaction catalyzed by Ru(bpy)3Cl2 showed a lower yield (50%, Table 1, entry 12). When 4CzIPN was used, the reactant was largely consumed, but only a small amount of pyranone B was obtained (yield: 10%, Table 1, entry 13). A similar result was found on Rose Bengal (Table 1, entry 14). These results indicated that a special photo-redox energy gap is key for efficiently converting FAL into pyranone.
We also tested the influence of the photocatalyst amount on the reaction yield and turnover frequency (TOF) (see the TOF calculation details in the ESI†). The yield decreased to 50% when the photocatalyst content was reduced from 0.2 mol% to 0.1 mol% (Table 1, entry 15), and 0.01 mol% photocatalyst led to a larger decrease in the yield (25%). The reaction showed a good yield when the FAL concentration was in the range of 1.0–5.0 mol (L−1) (Table 1, entry 16). Additionally, this tris-Ir(ppy)3 mediated visible-light catalysis presented a high TOF of 2750 h−1, significantly higher than the previously reported transformation for FALs (80–100 TOF).40
The above results were obtained from the reaction using FAL A. We also investigated the reaction versatility using a series of FALs with different functional groups, as depicted in Fig. 3 (see more details in the ESI†). The FALs had substituted groups on the hydroxymethyl-site (1R) and 5-site of furan ring (2R), such as methyl- (B2, B3, and B4, yield: 82, 84, and 80%), iso-propyl- (B5, 82%), cyclohexyl- (B6, 80%), spiro- (B7, 78%), phenyl- (B8, 81%), hydroxyl- (B9, and B10, yield: 62, and 60%), TBS: ((tert-butyldimethylsilyl)oxy)methyl- (B11, and B13, yield: 82, and 78%), benzyloxy- (B12, B14, and B15, yield: 81, 85 and 83%), acetyl- (B16, 76%), benzoyl- (B12, B14, and B15, yield: 82, 80 and 80%) showed a good yield and conversion efficiency. However, pyranones were not obtained when halogen (B20 and B21) or nitro (B22) group at the 5-site of furan reactant.
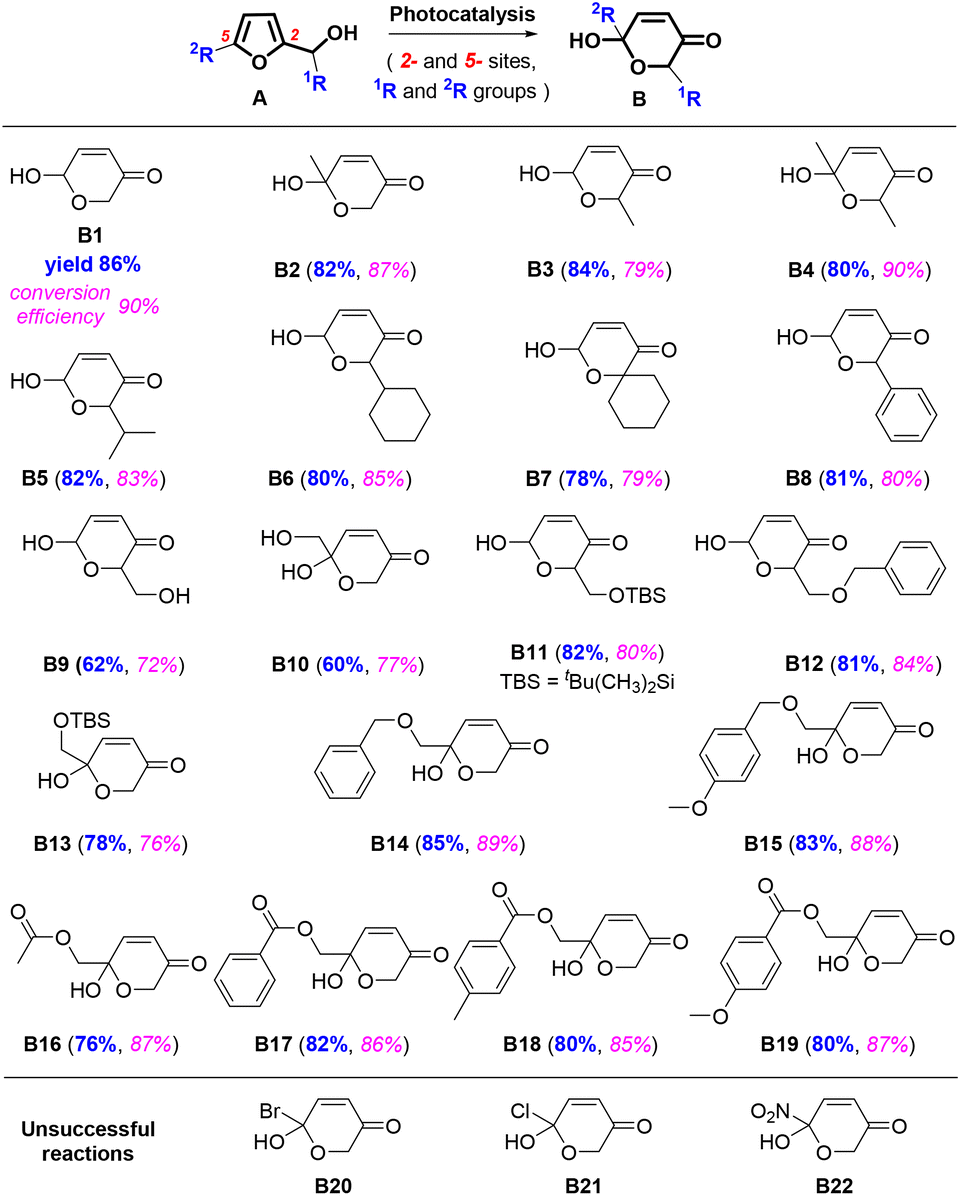 | ||
| Fig. 3 Chemical structures of the targeted products, including the yield, and conversion efficiency. | ||
Our FAL-to-pyranone conversion approach differs significantly from the classic AR reaction1 in that no stoichiometric oxidative reagent is used for furan-ring oxidation, and no acid additive is added for the rearrangement into pyranone. Oxidative chemicals, such as bromine radicals, peroxides, and metal–oxygen chelates, must be used in the AR reactions. A significant advantage of our reaction is that it takes place in the aqueous phase under an O2 atmosphere without toxic oxidants, which is environmentally friendly.
Our method is superior to Gilmore's method37 in many aspects. We use tris-Ir(ppy)3 as a photocatalyst. Under visible light irradiation, the tris-Ir(ppy)3 photoinduced the generation of singlet oxygen from atmospheric O2. Then, the singlet oxygen oxidized FAL into pyranone under mild conditions (temperature 25 °C). It achieved a yield as high as 86%. In comparison, Gilmore's method uses [Ru(bpy)3]2+ as a photocatalyst but has to be combined with sodium persulfate (Na2S2O8), which is corrosive. Under photo-redox reaction conditions, the [Ru(bpy)3]2+ and Na2S2O8 generated persulfate radicals, and the persulfate radicals oxidized FAL into pyranone. Gilmore's method usually has a yield of around ∼80%. Although our method and Gilmore's method both used [metal]-polybipyridine-type photocatalysts and under visible light irradiation, two different types of radicals were generated for the oxidative transformation of FAL. Additionally, our method is simpler and milder, does not involve corrosive oxidative reagents, and has a higher yield, which is superior and more environmentally friendly.
Previously, as shown in Table 2, other methods were reported to achieve a FAL-to-pyranone conversion, such as anodic oxidation/rearrangement, photochemical oxidation with singlet oxygen, and the enzyme method.22 Anodic oxidation/rearrangement usually causes environmental pollution and low transformation efficiency. Photochemical oxidation with singlet oxygen always needs different reduction methods of trioxolanes. The enzyme methods are only suitable for a limited number of FALs, and the reaction must be carried out at a very low reactant concentration. Our approach is superior to these methods. It is suitable for a broad spectrum of FALs. The reaction condition is mild and easy to control.
| Year | Work | Reagents | Solvents | Reaction conditions | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| a m-Chloroperbenzoic acid (m-CPBA), N-bromosuccinimide (NBS), tert-butyl hydroperoxide (TBHP), p-toluenesulfonic acid (p-TsOH), glucose oxidase (Gox), room temperature (rt). | ||||||
| 1969 | Cavill | Br2 | MeOH, H2O | (1) Na2CO3, −60 °C, 30 min (2) HCl, rta | 25 | 41 |
| 1971 | Achmatowicz | Br2 | MeOH, H2O | (1) Gaseous NH3, −60 °C, 30 min (2) H2SO4, rt | 50 | 1 |
| 1982 | Georgiadis | m-CPBAa | CHCl3 | (1) 10 °C, 3 h (2) KI (20%), Na2S2O3 (30%), NaHCO3 | 70 | 20 |
| 1992 | Georgiadis | NBSa | THF/H2O 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
(1) 10 °C, 3 h (2) KI (20%), Na2S2O3 (30%), NaHCO3 | 70 | 26 |
| 1991 | Lee | Photocatalysis: Rose Bengal, O2 | THF, acetone, or MeOH | 150 W visible light lamp, −78 °C (or 0 °C), 3 h | 50 | 36 |
| 2012 | VassilIkogiannakis | Photosensitizer: spirulina, O2 | H2O | (1) A xenon 300 W visible light lamp, 5 °C, 2 h (2) p-TsOHa | 50 | 34 |
| 2014 | Deska | Enzymatic: chloroperoxidase, Goxa | H2O | D-Glucose, citrate buffer, L-Methionine, r.t. 3 h | 68 | 42 |
| 2015 | Blackmond | VO(OiPr)3, TBHPa | DCM | (1) DCM, 2 h (2) PPh3 (or Me2S), <30 °C | 63 | 24 |
| 2016 | Tong | KBr/Oxone | THF/H2O (4:l) |
NaHCO3, 0 °C, 30 min | 80 | 21 |
| 2017 | Gilmore | Photocatalysis: Ru(bpy)3Cl2, Na2S2O8 | H2O/DMSO/MeCN | White light (red/green/blue LEDs), rt, 1 h | 80 | 37 |
| 2021 | Tong | CeBr3 (or FeBr2), H2O2, KBr | THF/H2O | (1) rt, 8 h, (2) Na2S2O3 | 80 | 43 |
| 2021 | Yu | Mn(ClO4)2 + 4,4′-diamino-2,2′-bipyridine as [Mn]-catalyst, H2O2 | CH3CN (or acetone) | (1) rt, 1 h (2) Na2S2O3 | 80 | 44 |
| 2022 | Our work | Photocatalysis: tris-Ir(ppy)3, O2 | H2O | rt, 11 h | 80 | |
Kinetic insight into this photocatalysis
To gain an insight into chemical changes during this photocatalysis, we measured the FTIR spectra in real-time during the reaction, as shown in Fig. 4 (see the ESI† for more details). An obvious decrease of a broad vibration peak at ∼2950 cm−1 was observed under irradiation of white light, resulting from the consumption of primary alcohol (R–CH2–OH, R = furan ring). This peak vanished after 9 h of reaction. Meanwhile, the peak at 2925 cm−1 showed an increasing and decreasing trend, indicating the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond migration in the furan ring, presumably from the O2 oxidation of furfural and formation of a furan-[O] intermediate (Fig. 4A). An obvious peak increase at 1490 cm−1 after 4 h of reaction indicated the occurrence of pyranone CH2 characteristics. The peak at 1600 cm−1 was assigned to the carbonyl group (CO) of pyranone (Fig. 4B). The greatly increased peaks at 750 cm−1 and 730 cm−1 correspond to the sp2 C–H bonds of the pyranone carbonyl conjugated CC bonds (Fig. 4C). A newly formed and gradually vanishing vibration peak at 1075 cm−1, originating from the sp2 C–H bond (CC bond), suggested that a furan-[O] intermediate had been formed and then consumed during the reaction (Fig. 4D).
C bond migration in the furan ring, presumably from the O2 oxidation of furfural and formation of a furan-[O] intermediate (Fig. 4A). An obvious peak increase at 1490 cm−1 after 4 h of reaction indicated the occurrence of pyranone CH2 characteristics. The peak at 1600 cm−1 was assigned to the carbonyl group (CO) of pyranone (Fig. 4B). The greatly increased peaks at 750 cm−1 and 730 cm−1 correspond to the sp2 C–H bonds of the pyranone carbonyl conjugated CC bonds (Fig. 4C). A newly formed and gradually vanishing vibration peak at 1075 cm−1, originating from the sp2 C–H bond (CC bond), suggested that a furan-[O] intermediate had been formed and then consumed during the reaction (Fig. 4D).
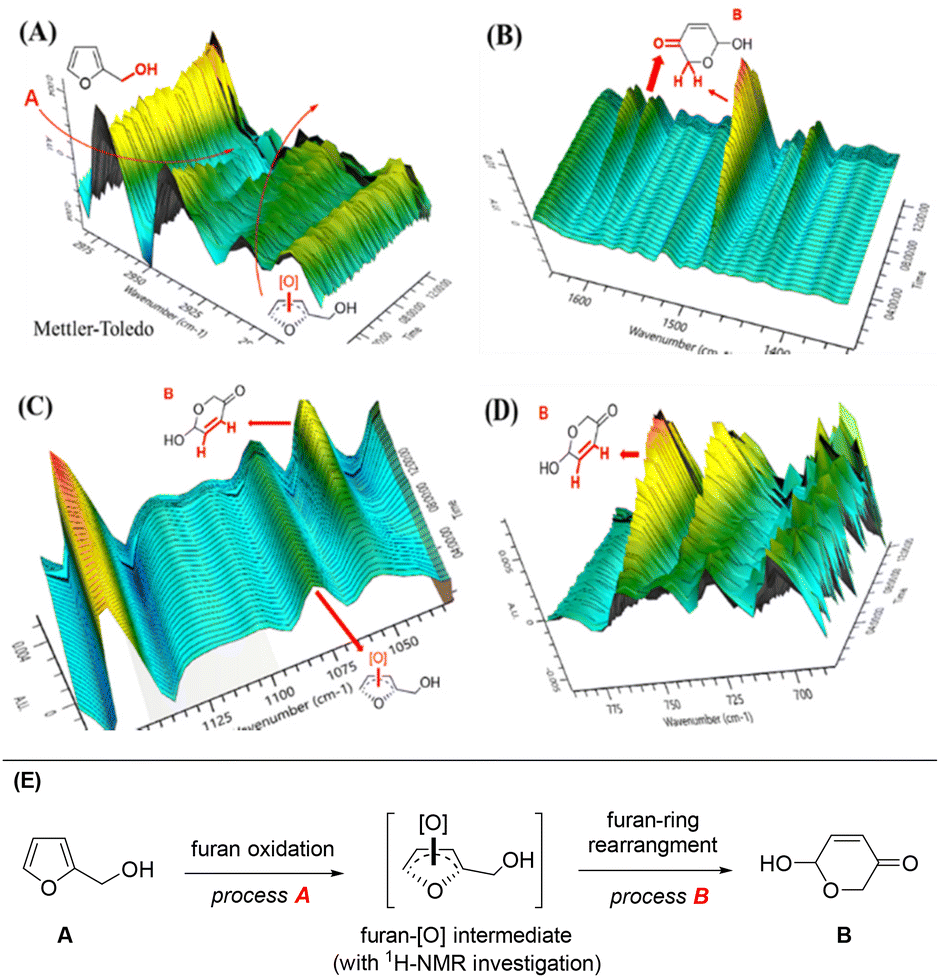 | ||
| Fig. 4 (A)–(D) In situ FTIR spectra during the reaction with waterfall plot map, and (E) a furan-[O] intermediate in the process during FAL transformation into pyranone. | ||
We also used 1H-NMR to examine the furan-[O] intermediate. After 8 h of reaction, a peak with the chemical shift of 7.5–8.0 was detected, explaining the CC bond migration in the furan rings (see the 1H-NMR spectra in the ESI†), confirming the formation of the furan-[O] intermediate. These results suggest that FAL (A) might pass through the furan-[O] intermediate to form pyranone (B), as described in Fig. 4E.
We next estimated the reaction kinetics based on the in situ FTIR results (see more details in the ESI†). Based on the specific IR vibration peaks, the concentrations of FAL, pyranone, and furan-[O] intermediate as a function of reaction time are obtained and are shown in Fig. 5. During the reaction, 10 mL of H2O was added to a reaction vessel, followed by adding 0.5 g (5.0 mmol, 0.5 mol L−1) of FAL and a catalytic amount of tris-Ir(ppy)3 (7.3 mg, 0.2mol%) to the reaction system. The tris-Ir(ppy)3 photocatalyst was not well dissolved at the beginning of the reaction. It was found that the reaction rate went through an accelerating period at the beginning of 1 h. After that, the reaction was approximately linear and remained quite constant, with the reactant decreasing and product increasing. We noted that upon the visible light irradiation at around 1 h, the reaction solution temperature rose from 39 °C to 44 °C. Therefore, a suitable warm-up period was allowed for initiating the photocatalysis. During the warm-up period, the furan-[O] intermediate was also quickly formed and consumed gradually until the end of the reaction. This result verifies that the FAL-to-pyranone conversion passes through the furan-[O] intermediate.
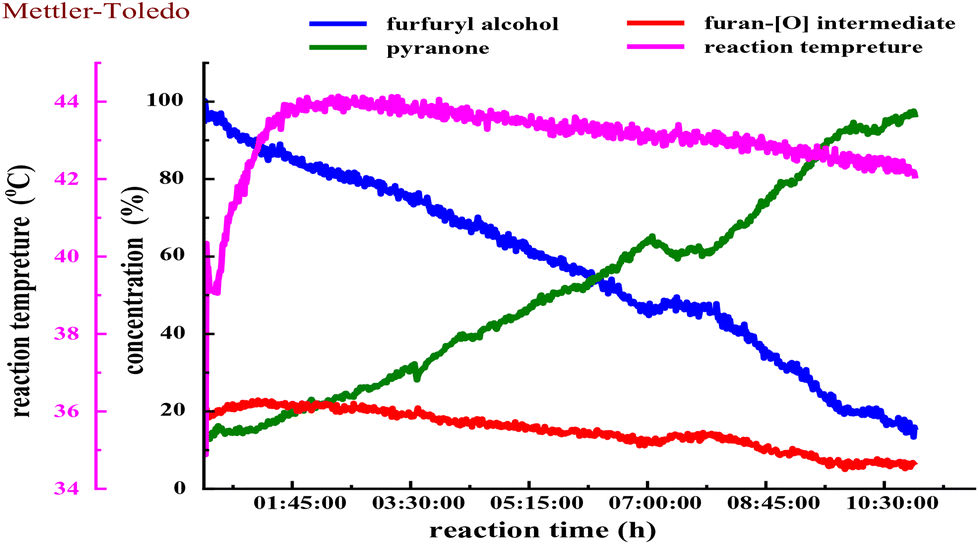 | ||
| Fig. 5 The reaction temperature and change of FAL, pyranone, and furan-[O] intermediate concentrations during the reaction process. The starting [FAL] = 0.5 mol L−1 and temperature = 25 °C sealed by an O2 balloon. ν-OHFAL = 2950 cm−1, ν-OH, CO, CCpyranone = 1500, 750 cm−1, and ν-OH, CCfuran-[O] intermediate = 2925, 1075 cm−1 were used to calculate the FAL, pyranone, and furan-[O] intermediate concentrations. | ||
We also performed the reaction by heating the solution to 50 °C or 80 °C (in an oil bath) for about 30 min before starting the photo-irradiation. However, the reaction showed low selectivity producing more side products. It suggests that the photocatalytic warm-up is an essential period for producing the furan-[O] intermediate and differs from the direct heating process. When the photocatalysis was kept at 0 °C (cooled in an ice bath) for 6 h, 85% of FAL was left with a low conversion efficiency, and only 11% yield of pyranone product was detected by GC analysis. When the reaction solution was heated to 50 °C (in an oil bath) for about 1 h to help the tris-Ir(ppy)3 photocatalyst disperse in the aqueous reaction solution and then cooled to room temperature before the photo-irradiation, the reaction also showed low selectivity producing more side products.
The reaction rate was estimated based on the in situ FTIR spectra (see more details in the ESI†). The FAL concentration, [FAL], change with the reaction time was recorded. Under the standard conditions (as listed in Table 1), the FAL reaction rate followed the first-order reaction of eqn (1)
| ln([FAL]/[FAL]0) = −kt + R | (1) |
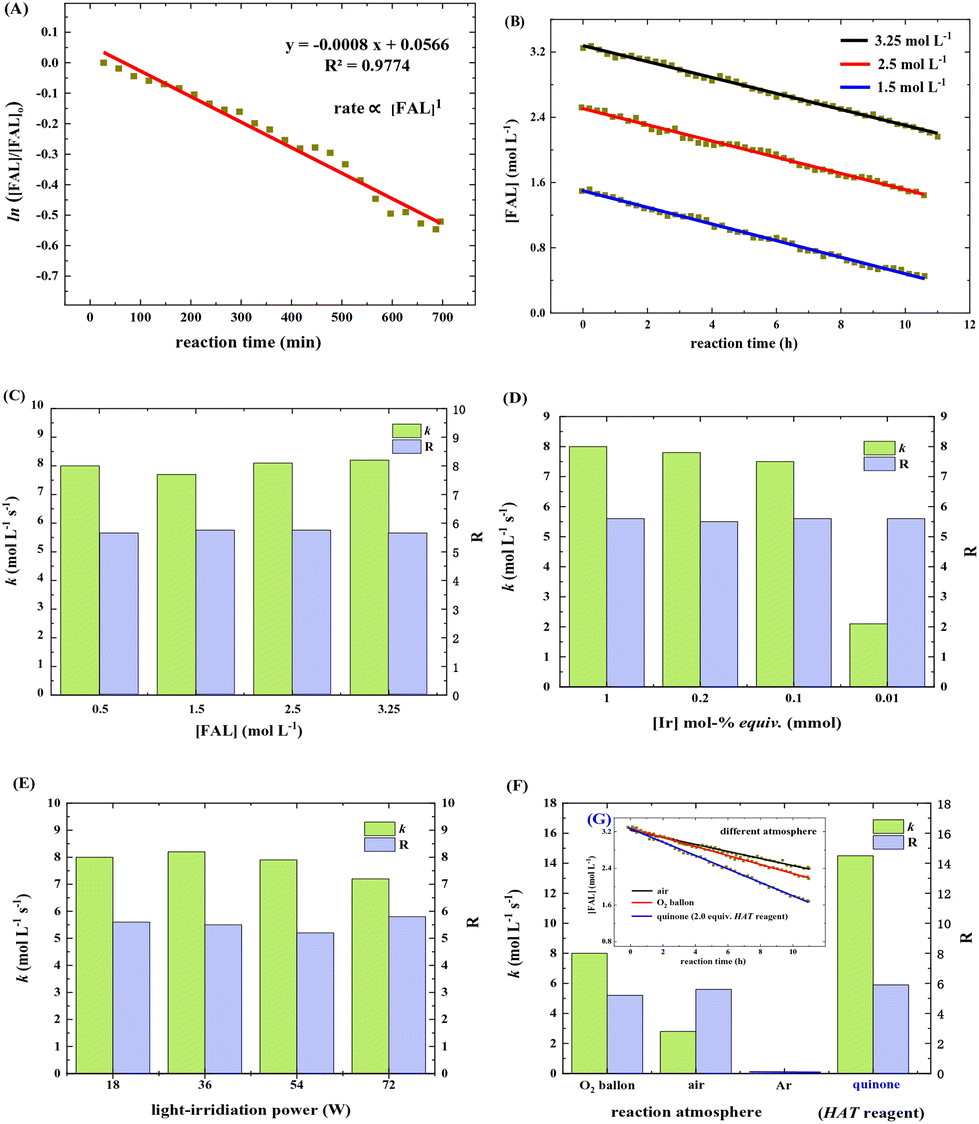 | ||
| Fig. 6 (A) ln([FAL]/[FAL]0) change with reaction time. (B) reaction progress with different [FAL]. (C) k and R of the reaction with a different initial [FAL]. (D) k and R of the reaction with different tris-Ir(ppy)3 amounts. (E) Influence of white light power on k and R. (F) influence of reaction atmosphere on k and R. (Ginset in F) reaction progress with different atmosphere and quinone (2.0 equiv.). | ||
It was also noted that the k and R values remained almost the same when the initial [FAL] varied in the range of 1.5–3.25 mol L−1 (Fig. 6C). Changing the tris-Ir(ppy)3 amount in the range of 1.0–0.001 mol% (based on FAL) led to k slowing down from 8 to 2.1 mol L−1 S−1. The equivalent of 0.1 mol% was the minimum amount to achieve a reaction yield of above 23% (reaction time 6 h). A small amount would sharply decrease the reaction speed (Fig. 6D, see more results in the ESI†). In addition, when FAL concentration was 0.5 mol L−1, the power of white light showed little impact on k and R (Fig. 6E).
When the reaction was carried out in different O2 atmosphere environments, which means different O2 concentrations in the reaction solution, the reaction exhibited a first-order dependence on [FAL]. As shown in Fig. 6F, pure O2 and air balloons were used separately as oxygen sources. Suppose the oxygen diffusion rate from the gas to the liquid is the same. The O2 concentration in the reaction solution was only 21% when using the air balloon compared to the pure O2 balloon. In this case, the k and R values for the air-involving reaction were 2.8 mol L−1 S−1 and 5.2, whereas the values for the pure O2 involving reaction were 8 mol L−1 S−1 and 5.6 (Fig. 6F and G).
We speculated that oxygen is activated in the form of singlet oxygen by tris-Ir(ppy)3 photocatalyst, which might be the rate-determining step. So we used quinone (2.0 equiv.) as an oxidation and hydrogen atom transformation (HAT) catalyst to perform the kinetic study. The k and R values were 14.5 mol L−1 S−1 and 5.9 (Fig. 6F and G), suggesting that the reaction rate has almost doubled in the photocatalysis reaction under an O2 balloon atmosphere.
Reaction mechanism
We further conducted a series of experiments to probe the reaction mechanism, such as light ON/OFF control, radical inhibition, electron paramagnetic resonance (EPR), isotope-labeling, ultraviolet-visible (UV-vis), fluorescence luminescence (FL), Stern–Volmer plot, cyclic voltammetry (CV), and quantum yield measurements.When conducted in the presence of 2,2,6,6-tetramethyl-1-oxylpiperidine (TEMPO), a stable oxyl-radical, the photocatalytic reaction was seriously inhibited (Fig. 7, see more details in the ESI†). This suggests that pyridyl radical generation is involved, presumably originating from tris-Ir(ppy)3. No reaction occurred when the reaction used other radical initiators such as tbutylated hydroxytoluene (BHT) and tri-nbutyltin hydride (nBu3SnH). They indicate that photocatalytic transformation is a radical reaction process.
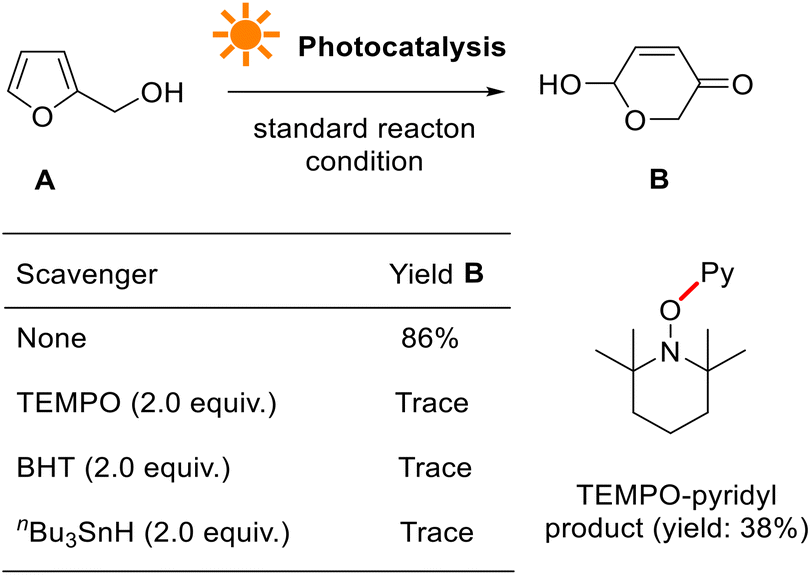 | ||
| Fig. 7 Radical inhibition experiments. | ||
To verify the oxygen radical in the photocatalytic oxidation reaction, we conducted EPR experiments using 2,2,6,6-tetramethyl-piperidine (TEMP) to capture oxygen radicals in the photocatalysis process. The EPR experiments were conducted using the hyperfine splitting constant aN = 16 G. Fig. 8 shows the EPR spectra (see the ESI† for details). The bar represents 3000 relative units, from 0 min to 30 min of photo-irradiation. The EPR response increased with exposure time, and the characteristic EPR signals of a stable nitroxide radical (TEMPO, g = 2.0056, aN = 15.96 G, aHβ = 14.82 G) were observed when TEMP was added. The EPR results suggest the photosensitized generation of 1O2 in the reaction.45
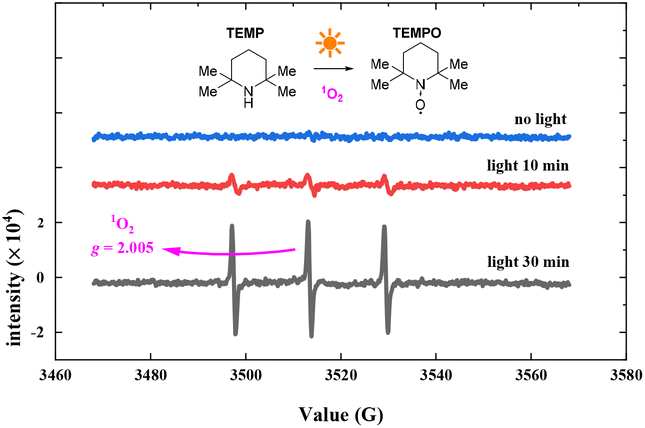 | ||
| Fig. 8 Radical intermediate identification by radical capture EPR experiments. | ||
The light ON/OFF control experiment was conducted, and the yield of the pyranone product was measured at different reaction times using gas chromatography (GC). As shown in Fig. 9, the product generation indicated that the yield was blocked immediately when the light was turned off (see more details in the ESI†). However, the reaction resumed when the light was turned on, indicating that constant light irradiation is essential for this transformation.
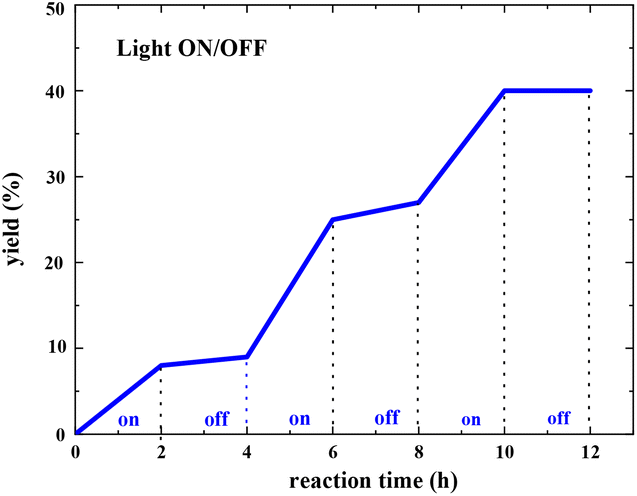 | ||
| Fig. 9 Light ON/OFF experiments for radical propagation test (the yield of pyranone product was measured by GC analysis). | ||
The 1O2 photocatalysis quantum yield over a reaction time of 2 h was measured under the irradiation of 18 W LED light (light intensity of 9.3 mW cm−2), and the average photocatalysis reaction photons were compared with the absorbed photons of photo irradiation (see the calculation details in the ESI†), resulting in a quantum yield of 11%. These results suggest that the transformation proceeds through a photo-redox catalytic radical initiation pathway rather than the previous radical chain propagation.46
The H and O atom transfers during the reaction were traced using D2O and H218O as the solvents. Fig. 10 shows the reaction results. When H218O was used, no 18O pyranone product was detected by GC-MS, indicating that the extra O atom in the pyranone product did not come from the solvent H2O and thus could most likely come from the O2 atmosphere (see more details in the ESI†). When D2O was used as the solvent, a deuterated hydroxyl group was formed on the D substituted pyranone product, suggesting that the solvent H2O provides hydrogen atoms in the transformation process.
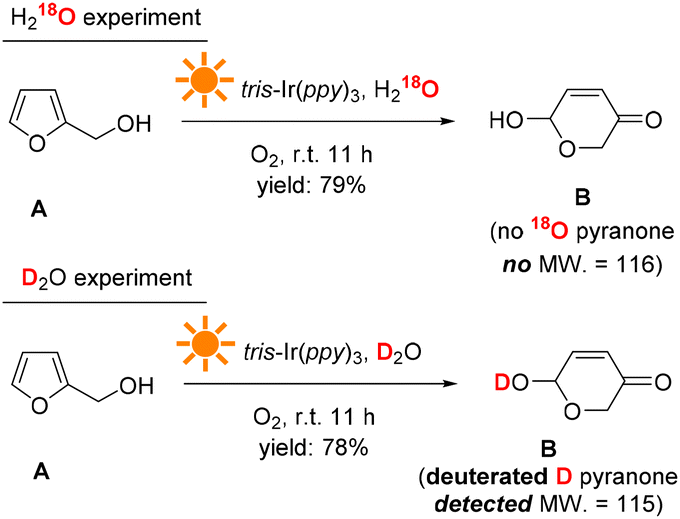 | ||
| Fig. 10 Isotope-labeling experiments. | ||
We further analyzed the reaction products from the photocatalysis reaction without an O2 atmosphere or H2O (see more details in the ESI†). As shown in Fig. 11, when the photocatalysis experiment was performed without H2O, the majority of the reactant furfural alcohol was left in the reaction mixture, and about 5mol% byproducts were detected, including aldehyde (C), acid (D), and furan-one (E and F). However, under the argon atmosphere, most of the reactant was left unchanged. These results indicate that O2 plays an important role in furan oxidation and initiates the entire reaction.
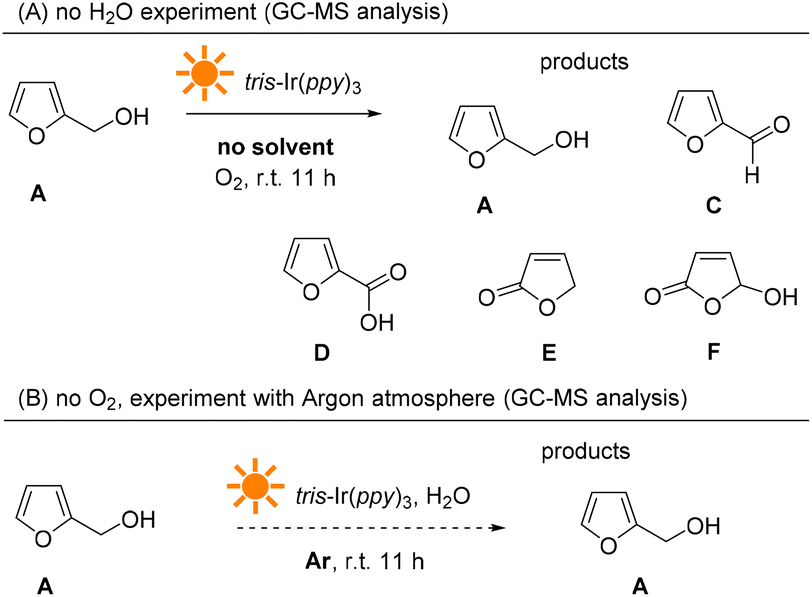 | ||
| Fig. 11 GC-MS analysis of the photocatalysis experiments without H2O or O2. | ||
Fig. 12 shows the optical absorption and fluorescence spectra of the reaction mixture (see more details in the ESI†). The absorption peak from FAL at 276 nm diminished with the reaction time, whereas a new peak at 268 nm appeared and increased gradually during the reaction from 0.5 h to 11 h (Fig. 12A). The optical absorption band from 276 to 268 nm was assigned to the n → π* transition of the furan-ring C–O bond breaking and the pyranone CO bond formation, similar to the literature.24 However, when the reaction was performed without O2, the 276 nm absorption peak gradually vanished over 11 h of visible light irradiation. No absorption appeared at around 268 nm (Fig. 12B). These results indicate a conjugated aromatic structure change in the FAL furan ring-opening rearrangement process and verify the key role of O2 in the photocatalysis reaction. Fig. 12C shows the fluorescence spectra of the reaction solution (excitation wavelength 270 nm). The single-emission peak appeared at around 290 nm, originating from the reaction intermediate of the furan-[O] intermediate. The peak reduced gradually with the reaction, indicating that the aromatic structure diminishes the FAL reactant. On the other side, emission spectroscopy in the reaction without O2 remained almost unchanged.
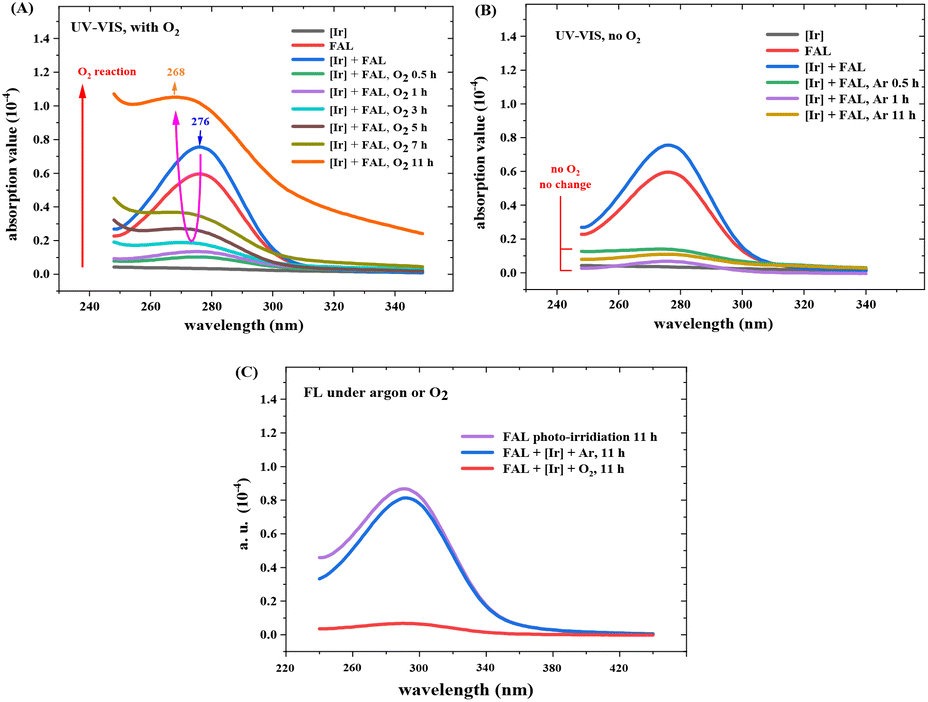 | ||
| Fig. 12 UV-vis spectra of FAL photocatalysis reaction mixture (A) under an O2 reaction atmosphere. (B) Under an argon atmosphere. (C) Fluorescence spectra of the reaction mixture in the presence and absence of O2. | ||
We also performed the Stern–Volmer analysis based on the steady-state luminescence data. The Stern–Volmer plot was collected from aqueous solutions consisting of FAL, H2O, O2, and FAL + O2 mixture (see the details in the ESI†). The UV-vis and fluorescence spectra of tris-Ir(ppy)3 were recorded. Tris-Ir(ppy)3 showed an optical absorption maximum at 200 nm,46 and under the excitation wavelength of 270 nm, it showed an emission peak at λem = 290 nm. Different [FAL] were used to monitor the emission intensities (see more details in the ESI†). Fig. 13 shows the Stern–Volmer plot spectroscopy. The electron transfer in the quenching process was analyzed using eqn (2):47
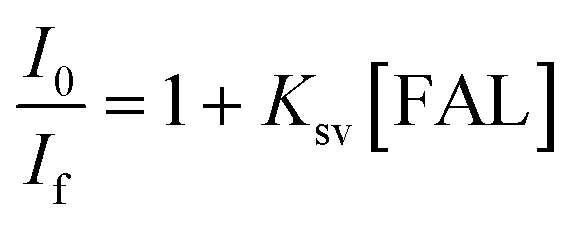 | (2) |
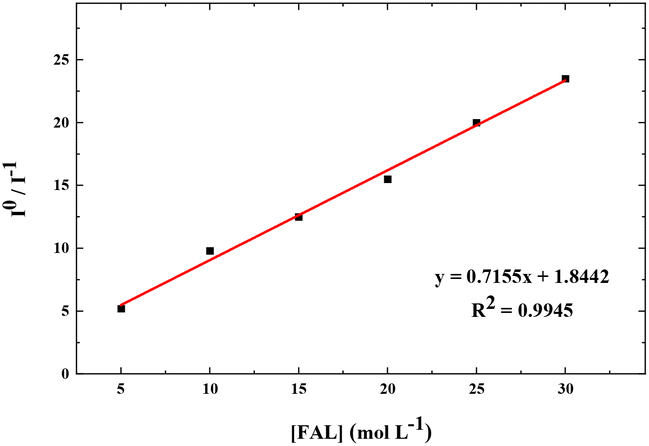 | ||
| Fig. 13 Stern–Volmer plot for FAL + [Ir] (10 μM) + O2 in an aqueous solution. The solid line is a linear least-square fit of the data. Ksv from the slope is 7.155 × 109 M−1 s−1. | ||
The redox current of FAL in different photocatalysis conditions was measured using a cyclic voltammetry (CV) method (see more results in the ESI†). FAL presents a low redox current of 8.1 × 10−7 A in an aqueous solution, as shown in the ESI.† When photo-irradiation and O2 atmosphere were added, the redox current increased from 0.7 × 10−6 A to 1.2 × 10−6 A (Fig. 14A), and after the addition of photocatalyst tris-Ir(ppy)3, the photocatalysis redox current increased to 3.2 × 10−6 A. This photocatalysis of FAL also presented good reversibility under standard reaction conditions (Fig. 14B).
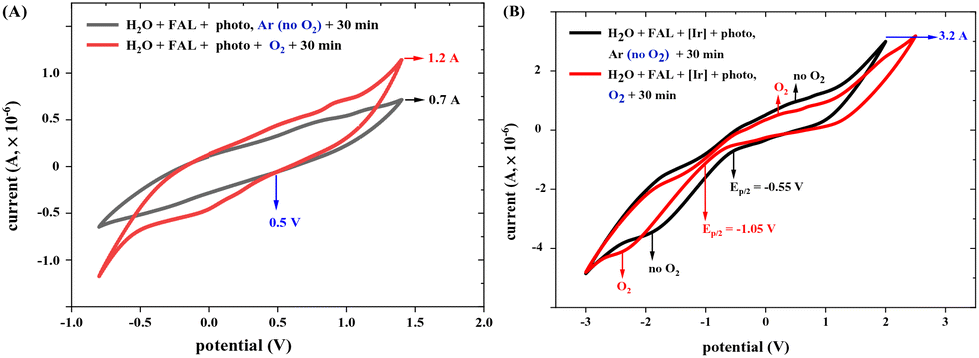 | ||
| Fig. 14 (A) CV curves of FAL photocatalysis under standard reaction conditions without tris-Ir(ppy)3. During the testing, solvent (H2O, 40 mL) containing FAL (80.0 mg, 0.02 mol L−1, 1 chemical equivalent) was used under an O2 or Ar atmosphere. A constant current of 0.01 mA. The scan rate was 0.10 V s−1, ranging from −0.8 V to 1.40 V. (B) CV curves of FAL photocatalysis with tris-Ir(ppy)3. Solvent (H2O, 40 mL) containing FAL (80.0 mg, 0.02 mol L−1, 1 chemical equiv.), (tris-Ir(ppy)3) (10.0 mg, 3.8 × 10−4 mol L−1, 3 × 10−3 chemical equiv.) under an O2 or Ar atmosphere was used. A constant current of 0.01 mA. The scan rate was 0.10 V s−1, ranging from −3 V to 2.5 V. | ||
Adding O2 reduced the FAL oxidation potential from −0.55 to −1.05 V (Ep/2) (Fig. 14B). Therefore, the excited-state reduction of the optimal photocatalyst (E1/2[Ir*III/II] = −0.60 V vs. SCE and E1/2[IrIV/*III] = −0.43 V vs. SCE)48 is sufficient enough to oxidize the FAL reactant (Ep/2 = −1.05 V). However, when the reaction was performed without tris-Ir(ppy)3, the redox potential of FAL remained unchanged at 0.5 V, which indicated that O2 is not suitable for FAL oxidation.
The above results indicated that the photo-irradiation induced single-electron transfer (SET) process of photosensitizers Ir*III/II and IrIV/*III occurred with the photocatalyst tris-Ir(ppy)3. However, the SET process cannot directly lead to FAL oxidation. In contrast, the SET process introduced the generation of singlet oxygen radicals after the tris-Ir(ppy)3 photocatalysis of O2. The singlet oxygen initiated the FAL oxidation.
The electron transfer number (n-value) was calculated based on the CV curves (see more details in the ESI†). As shown in Fig. 15A, the maximum oxidation current ipa is approximately equal to the maximum reduction current ipc (pa means oxidation process, pc means reduction process). Thus, ipa1 = 2.85 × 10−6 mA, ipc1 = 2.81 × 10−6 mA, and ipa1/ipc1 = 1.02 ≈ 1. It indicates a reversible redox reaction of FAL. The relationship between the maximum oxidation potential Epa and current ipa can be calculated according to the Nernst equation in eqn (3).49
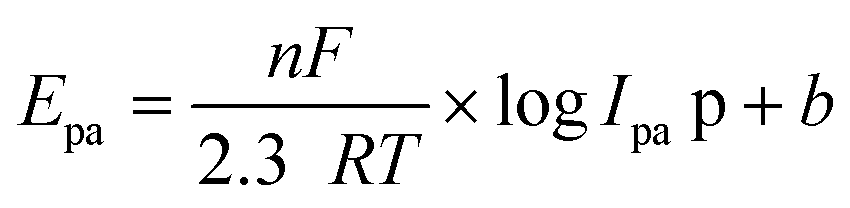 | (3) |
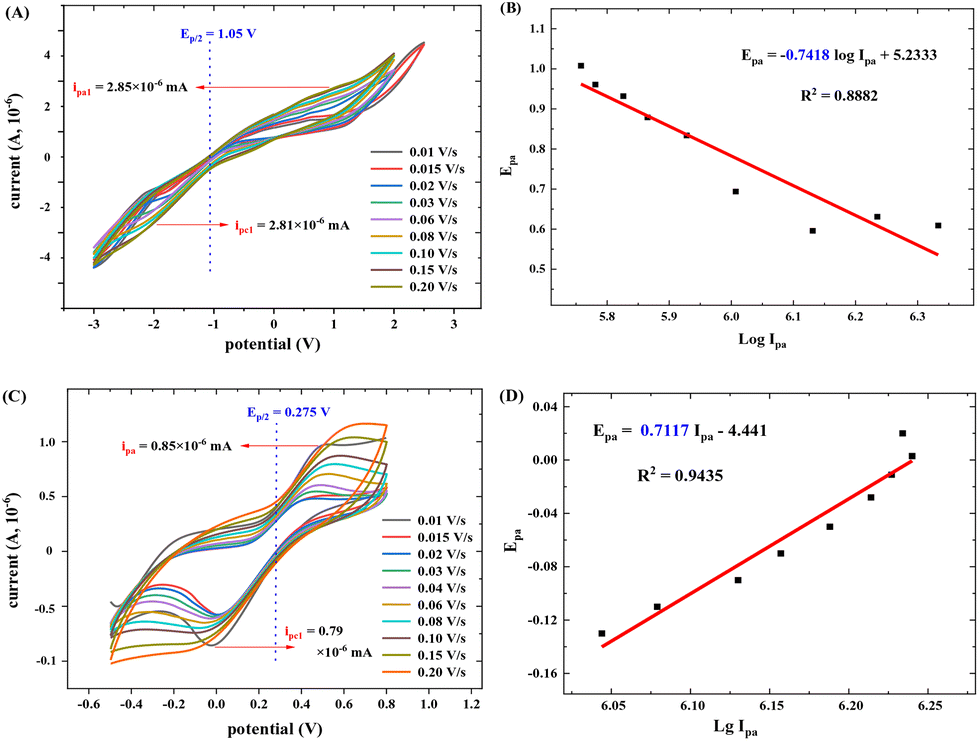 | ||
| Fig. 15 CV experiments with different scan rate experiments, (A) CV experiments of FAL, using H2O (40 mL) as a solvent in standard conditions, FAL (A, 80.0 mg) at a scan rate of 0.010 V s−1, from−3.0 V to 3.0 V; (B) Epa1 and Ipa1 linear relationship of FAL. (C) CV experiments of ferrocene, using H2O (40 mL) as a solvent, ferrocene (74.4 mg) at a scan rate of 0.010 V s−1, from −0.5 V to 0.9 V. (D) Epa and Ipa linear relationship of ferrocene. | ||
Here, F is Faraday's constant, R is the universal gas constant, n is the number of electrons, T is the temperature, and b is the standard potential of analyte in the system at equilibrium (including FAL). Fig. 15B shows Epa1 ∼ logIpa1 of the FAL and the linear fitting result. Because the number of electron transformed in the electrocatalytic redox process of Ferrocene/Ferrocene* is one, and Ferrocene/Ferrocene* is set as the standard reference object. As a comparison, we measured the CVs of ferrocene at different scan rates under the same electrocatalysis conditions as that of FAL (Fig. 15C and D). Results indicated that both oxidation potential of FAL (Epa1) and ferrocene (Epa) have linear relationships with logIpa, and the comparative number is n = 0.7117ferrocene/0.7418FAL = 0.96 ≈ 1, which indicated a one-electron transfer during the redox process of FAL (n-value = 1).50 Therefore, the photocatalytic O2 oxidation of FAL involves a single-electron transfer process (SET).
DFT simulation of reaction routes
In order to explore the reaction route, we performed a density functional theory (DFT) calculation and analyzed the potential energy surface (PES) and electro-static potentials (ESP) (see more details in the ESI†).51,52Fig. 16A shows the oxygen onium ion oxidation process. Singlet oxygen was generated under tris-Ir(ppy)3 photocatalysis of the O2 atmosphere. This singlet oxygen directly oxidized the reactant FAL 1 to form epoxidized furan 2 (the furan-[O] intermediate), which H2O hydrolyzed. Then the water hydrolyzed furan 3 passed through a hydrogen atom transformation (HAT) process of TS1 (bond distance a, b, c, and d of the optimized structure furan C4–H2O–O+˙ for 3 and TS1 were demonstrated in PES, as shown in Fig. 16B), with the first energy barrier of ΔGsol = 22.93 kcal mol−1. Next, the hydrolyzed furan underwent a ring-opening process through TS2, involving a second energy barrier of ΔGsol = 26.64 kcal mol−1. Finally, the rearrangement reached the final pyranone product 9, preferred through a lower H2O-induced transition state of TS4 (ΔGsol = 30.90 kcal mol−1) than a higher energy barrier of TS3 (ΔGsol = 45.66 kcal mol−1).
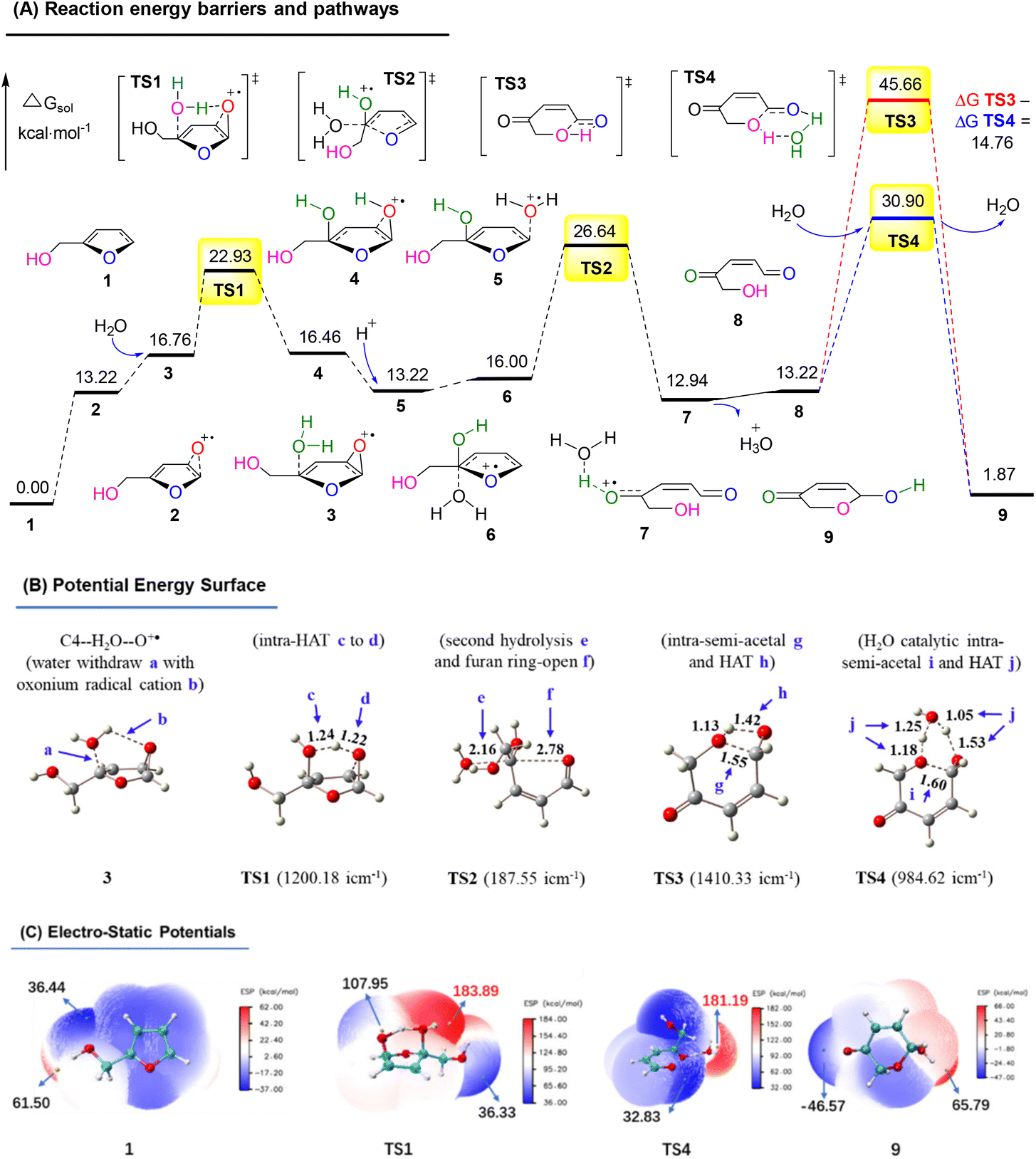 | ||
| Fig. 16 (A) DFT-calculated free energy surface and energy barriers. (B) Optimized structures of PES (distances in Å). (C) ESP mapped molecular vdW surface in kcal mol−1. | ||
Fig. 16C shows the ESP on the vdW (van der Waals) surface. The singlet oxygen-induced oxygen onium ion unit TS1 exhibited a nucleophilic site to abstract hydrogen atoms for the HAT. It led to the furan-ring opening with an ESP of 183.89 kcal mol−1. TS4 also showed a high reactivity in water-catalytic intra-semi-acetal HAT transformation, with an ESP of 181.19 kcal mol−1.
Similarly, we calculated the endoperoxide-furan process, as shown in Fig. 17A. In this case, two types of oxygen with singlet (O12) and triplet (O32) might become involved in the FAL oxidation, with an energy gap of 37.10 kcal mol−1 (doublet and quartet oxygen were excluded in this O2 photocatalysis). Reactant FAL 1 was oxidized into key intermediate TS1153 (the furan-[O] intermediate, bond distance k 1.85 Å of the optimized structure furan C1–O–O was demonstrated in PES, as depicted in Fig. 17B). After an intramolecular HAT process of TS12 and the following hydrolysis processes, furan ring-open product 27 was afforded. To reach the final pyranone product 28, specific two molecules of water catalysis for TS14 were preferred than TS13, with a gap of ΔGsol = 56.85 kcal mol−1. The endoperoxide-furan intermediate TS11 possessed a moderate nucleophilic activity of 12.8 kcal mol−1, and TS14 had a −30.25 kcal mol−1 of high reactivity in the specific two moles of water catalytic intra-semi-acetal transformation process (as ESP showed in Fig. 17C).
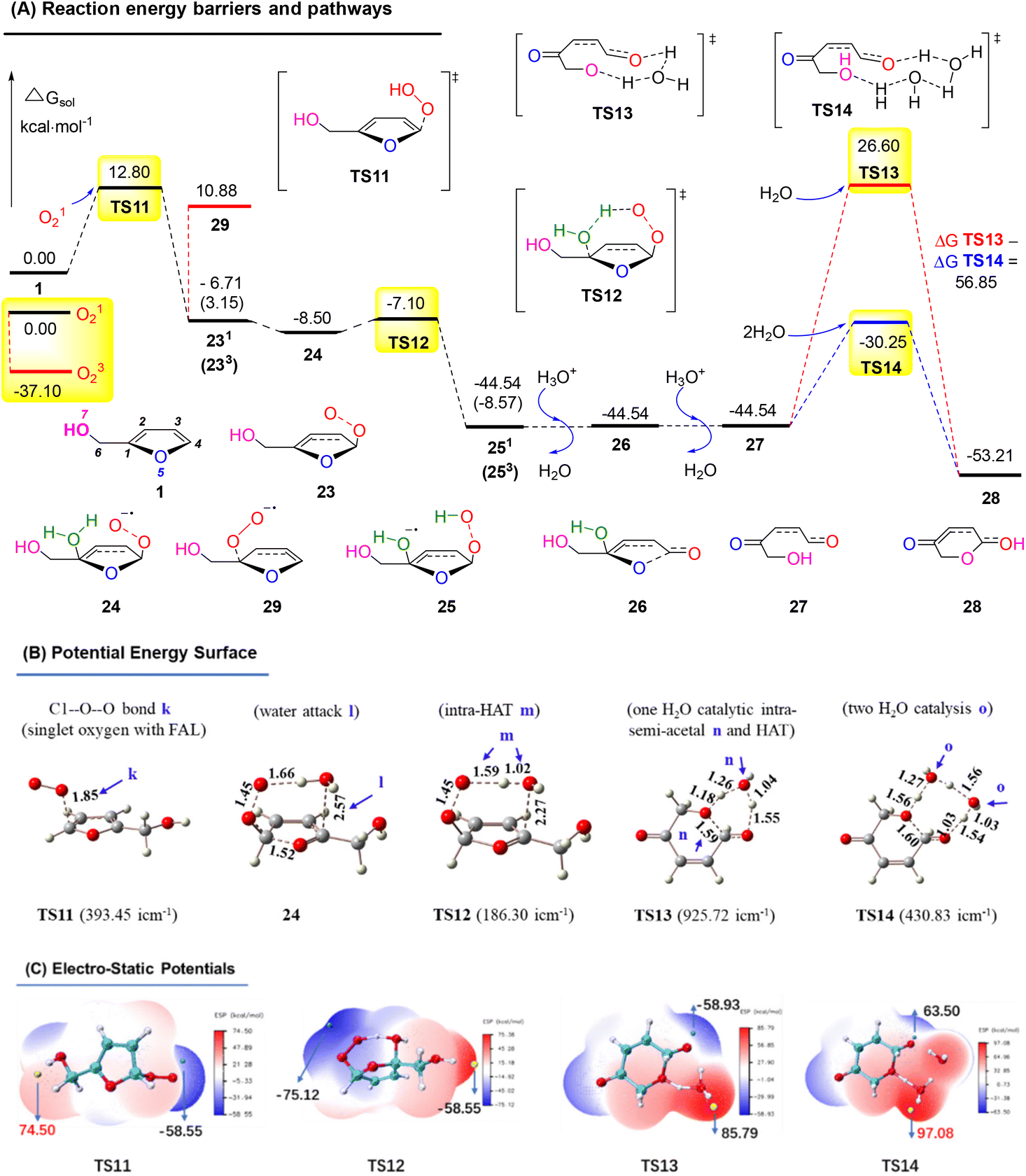 | ||
| Fig. 17 (A) DFT-calculated free energy surface and energy barriers. (B) Optimized structures of PES (distances in Å). (C) ESP mapped molecular vdW surface in kcal mol−1. | ||
Based on the above results, the oxygen onium ion oxidation process needs to overcome three energy barriers of 9.71, 13.42, and 17.7 kcal mol−1; however, the endoperoxide-furan process was easier with the first and primary energy barrier, 12.8 kcal mol−1, to initiate the entire reaction. Therefore, the FAL-to-pyranone transfer was more likely to happen through an endoperoxide-furan reaction pathway.
In the conventional AR reaction, FAL was first oxidized from the 4,5-sites CC bond and formed a tricyclic onium ion following the oxidation process (Fig. 16).1 We developed a novel endoperoxide-furan process oxidized by photocatalytic singlet oxygen from the O2 atmosphere, as shown in Fig. 17. Additionally, we found new water-assisted FAL rearrangement into pyranone and successfully avoided previous work of singlet oxygen reduction by stoichiometric PPh3 or DMSO.
A plausible mechanism for this photocatalysis FAL-to-pyranone conversion was proposed, as shown in Fig. 18. The tris-Ir(ppy)3 under visible light irradiation catalyze O2, which is supplied from the O2 atmosphere, into singlet oxygen. FAL was first oxidized to afford a key furan-[O] intermediate of endoperoxide 23. This singlet oxygen involved in the HAT initiates hydrolyzation. The endoperoxide radical 24 produces the diol intermediate 25. Further hydrolysis of this diol intermediate induces a single electron release and an open ring process of the furan ring to 27. Finally, a water-catalytic semi-acetal process from TS14 to pyranone product 9 is achieved.
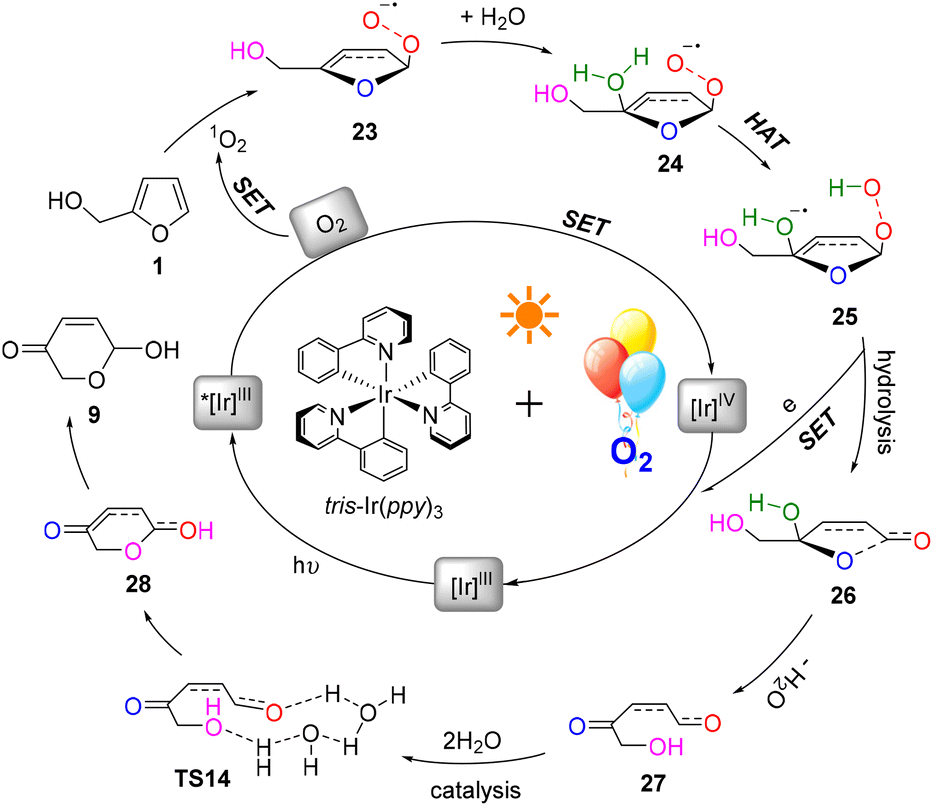 | ||
| Fig. 18 The proposed FAL-to-pyranone conversion photocatalytic oxidation mechanism. | ||
10.0 gram-scale reaction
Our photocatalysis reaction is scalable. We performed a 10.0-gram (102.0 mmol) reaction. By adding only 0.05 mol% tris-Ir(ppy)3 (36.5 mg, 0.051 mmol, 0.0005 equiv.), 7.735 g (67.3 mmol) of pyranone product B was obtained with a yield of 79% and conversion efficiency of 85% (see more details in the ESI†). In the 10.0 gram-scale reaction, a white photo-irradiation light source of 4 × 18 = 72 W was used. The reaction time was 48 h. The photocatalyst tris-Ir(ppy)3 had a turnover frequency (TOF) of 2750 h−1. The TOF is significantly higher than the previous transformation for furans (average about 80–100 TOF).40 It indicates a high transformation specificity and efficiency. Additionally, this photocatalysis is easy to operate.Conclusions
We have demonstrated a novel photocatalytic singlet oxygen oxidation for transforming biomass furfural alcohols into pyranones for the first time. The visible light catalyst tris-Ir(ppy)3 induced the formation of singlet oxygen from the O2 atmosphere. This method uses H2O as the solvent and hydrogen atom source to assist the furan-ring opening and rearrangement. Kinetic studies show that this photocatalysis is a first-order reaction, and the singlet oxygen oxidation of the furan ring is the rate-determining step. Our DFT calculations reveal that the singlet oxygen oxidation happens following an endoperoxide-furan reaction pathway rather than the conventional oxygen onium ion oxidation pathway. This method can generally tolerate different substituted groups on furfural alcohol molecules and is scalable to perform at least a 10.0-gram reaction with a good reaction yield and conversion efficiency.Experimental section
Basic photocatalysis for FAL transformation into pyranone
In an oven-dried quartz flask (25 mL) equipped with a stirring bar, furfural alcohol (500 mg, 5.0 mmol, 1.0 equiv.), tris-Ir(ppy)3 (7.3 mg, 0.1 mmol, 0.002 equiv.), and H2O (10.0 mL) were combined and added. The reaction was conducted under an oxygen atmosphere using an O2 balloon. The reaction mixture was stirred and illuminated by two 18 W visible LED lights (a total of 36 W) under room temperature for 11 h and cooled by a fan. For TLC, UV light was the visualizing agent, the reaction was monitored, and an ethanolic solution of phosphomolybdic acid, cerium sulfate, and heat was the developing agent. When the reactant material no longer decreased, the solution was extracted with ethyl acetate (EtOAc, 3 × 10 mL) and H2O (3 × 10 mL). The combined organic layer was dried with Na2SO4 and filtered. The solvent was removed with a rotary evaporator. The pure product was obtained by flash chromatography on neutral alumina (Al2O3, Merck KGaA, 70–230 mesh, and pH = 6.8–7.8) using EtOAc and petroleum ether (5
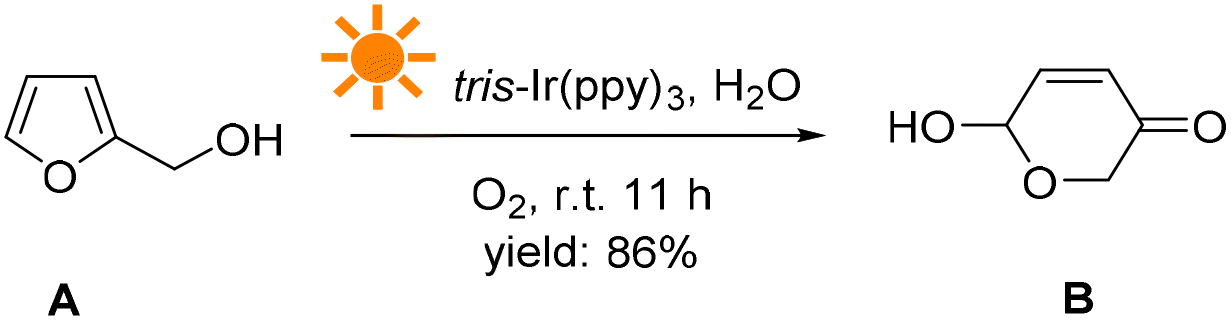 :1, vol/vol) as the eluent solvents. The pyranone product B was obtained in a yield of 421.4 mg, 86%, with a conversion efficiency of 90%.
:1, vol/vol) as the eluent solvents. The pyranone product B was obtained in a yield of 421.4 mg, 86%, with a conversion efficiency of 90%.
DFT calculations
Gaussian program 16 was employed for DFT calculations, and /M06-2X/def2-SVP level was used for geometry optimization and frequency calculations. The 6-311 + G(2d,p) basis sets were employed in the calculations of triplet-singlet energy gaps. The model substrate's reaction profile calculations used 6-31G(d,p) basis sets. Optimizations were conducted without constraint using the implicit solvation model (SMD)19 in dichloromethane (ε = 8.93). Frequency analyses (at 298.15 K and 1 atm) were carried out to confirm that each structure is a minimum (no imaginary frequency) or a transition state (only one imaginary frequency). The search for minimal energy crossing points was conducted using a modified version of Harvey's code20 (sobMECP21) interfaced with Gaussian 16. The Gibbs free energies in water (ΔG) were discussed throughout this article unless otherwise specified. The single-point calculations were at M06-2X/def2-TZVP level. The SMD implicit solvent model accounted for the water solvation effect. The 3D images of the calculated structures and the orbital diagrams were prepared using CYLView22 or VMD.Characterization of pyranone products
Data availability
The authors declare that all other data supporting the findings of this study are available within the article and the ESI files,† and also are available from the corresponding author upon reasonable request.Author contributions
Conceptualization, project administration, and funding acquisition have been conducted by Guan-Ben Du, Tong Lin, and Bei Zhou. Yun-Feng Tao, Yu-Juan He, Lan-Xiang Liu, Zu-Hui Chang, and Xiang-Hong Li worked on the investigation and methodology. Writing – review and editing were conducted by Bei Zhou and Tong Lin.Conflicts of interest
The authors declare no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Funding support from the Program for Leading Talents (No. 2017HA013), Department of Science and Technology of Yunnan Province, the Programme of Introducing Talents of Discipline to Universities (the 111 Project, No. D21027), the National Natural Science Foundation of China (No. 32260363 and No. 31860188), and the Yunnan Fundamental Research Project (No. 202001AT070148) is acknowledged. The authors thank Key Laboratory of Forest Biotechnology in Yunnan Province (College of Life Science, Southwest Forestry University), the Shiyanjia Lab (https://www.shiyanjia.com) for the GC-MS analysis, and Hangzhou Yanqu Information Technology Co., Ltd for the computational work. Dr Teng Xu from Mettler-Toledo Co. Ltd (Shanghai, China) is acknowledged for the in situ IR (React IR 15™) experiments.References
- O. Achmatowicz, P. Bukowski, B. Szechner, Z. Zwierzchowska and A. Zamojski, Tetrahedron, 1971, 27, 1973–1996 CrossRef CAS.
- R. S. Babu and G. A. O'Doherty, J. Am. Chem. Soc., 2003, 125, 12406–12407 CrossRef CAS PubMed.
- C. G. Zhao, D. A. Glazier, D. S. Yang, D. Yin, I. A. Guzei, M. M. Aristov, P. Liu and W. P. Tang, Angew. Chem., Int. Ed., 2019, 58, 887–891 CrossRef CAS PubMed.
- A. K. Ghosh, H. M. Simpson and A. M. Veitschegger, Org. Biomol. Chem., 2018, 16, 5979–5986 RSC.
- D. S. Kim, F. G. Fang, H. W. Choi and H. Fang, Org. Lett., 2018, 20, 4295–4297 CrossRef CAS.
- M. A. Marquez-Cadena, J. Y. Ren, W. K. Ye, P. Y. Qian and R. B. Tong, Org. Lett., 2019, 21, 9704–9708 CrossRef CAS PubMed.
- H. Y. Wang, K. Yang, S. R. Bennett, S. R. Guo and W. P. Tang, Angew. Chem., Int. Ed., 2015, 54, 8756–8759 CrossRef CAS PubMed.
- G. D. Zhao, D. P. Canterbury, A. P. Taylor, X. Y. Cheng, P. Mikochik, S. W. Bagley and R. B. Tong, Org. Lett., 2020, 22, 458–463 CrossRef CAS.
- A. K. Ghosh and M. Brindisi, RSC Adv., 2016, 6, 111564–111598 RSC.
- N. Hauser, M. A. Imhof, S. S. Eichenberger, T. Kundig and E. M. Carreira, Angew. Chem., Int. Ed., 2022, 61, e202112838 CrossRef CAS PubMed.
- L. D. Guo, Z. J. Xu and R. B. Tong, Angew. Chem., Int. Ed., 2022, 61, e202115384 CAS.
- L. Zhang, Y. Zhang, W. T. Li and X. B. Qi, Angew. Chem., Int. Ed., 2019, 58, 10397–10397 CrossRef CAS PubMed.
- L. Y. Zhu, Y. Liu, R. Z. Ma and R. B. Tong, Angew. Chem., Int. Ed., 2015, 54, 627–632 CAS.
- K. L. Jackson, J. A. Henderson, H. Motoyoshi and A. J. Phillips, Angew. Chem., Int. Ed., 2009, 48, 2346–2350 CrossRef CAS PubMed.
- A. Furstner and T. Nagano, J. Am. Chem. Soc., 2007, 129, 1906–1907 CrossRef.
- S. Pabbaraja, N. Gantasala, S. Ydhyam, H. K. Namballa, S. Gundeboina, M. R. Lambu, S. Meena and D. Datta, Tetrahedron Lett., 2018, 59, 2570–2576 CrossRef CAS.
- H. B. Guo, J. J. La Clair, E. P. Masler, G. A. O'Doherty and Y. L. Xing, Tetrahedron, 2016, 72, 2280–2286 CrossRef CAS.
- R. Bielski and G. Grynkiewicz, Tetrahedron, 2021, 85, 132058 CrossRef CAS.
- E. A. Couladouros and M. P. Georgiadis, J. Org. Chem., 1986, 51, 2725–2727 CrossRef CAS.
- M. P. Georgiadis, E. A. Couladouros, M. G. Polissiou, S. E. Filippakis, D. Mentzafos and A. Terzis, J. Org. Chem., 1982, 47, 3054–3058 CrossRef CAS.
- Z. L. Li and R. B. Tong, J. Org. Chem., 2016, 81, 4847–4855 CrossRef CAS.
- D. Thiel, D. Doknic and J. Deska, Nat. Commun., 2014, 5, 5278 CrossRef CAS PubMed.
- G. D. Zhao, L. X. Liang, E. Y. Wang and R. B. Tong, ACS Catal., 2021, 11, 3740–3748 CrossRef CAS.
- Y. N. Ji, T. Benkovics, G. L. Beutner, C. Sfouggatakis, M. D. Eastgate and D. G. Blackmond, J. Org. Chem., 2015, 80, 1696–1702 CrossRef CAS PubMed.
- Q. Z. Xing, Z. Hao, J. Hou, G. Q. Li, Z. W. Gao, J. Gou, C. Q. Li and B. X. Yu, J. Org. Chem., 2021, 86, 9563–9586 CrossRef CAS PubMed.
- M. P. Georgiadis, E. A. Couladouros and A. K. Delitheos, J. Pharm. Sci., 1992, 81, 1126–1131 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS.
- Y. F. Lv, P. L. Bao, H. L. Yue, J. S. Li and W. Wei, Green Chem., 2019, 21, 6051–6055 RSC.
- L. Y. Xie, T. G. Fang, J. X. Tan, B. Zhang, Z. Cao, L. H. Yang and W. M. He, Green Chem., 2019, 21, 3858–3863 RSC.
- X. X. Meng, Q. Q. Kang, J. Y. Zhang, Q. Li, W. T. Wei and W. M. He, Green Chem., 2020, 22, 1388–1392 RSC.
- N. Meng, Y. F. Lv, Q. S. Liu, R. S. Liu, X. H. Zhao and W. Wei, Chin. Chem. Lett., 2021, 32, 258–262 CrossRef CAS.
- Z. W. Wang, Q. S. Liu, R. S. Liu, Z. Y. Ji, Y. Li, X. H. Zhao and W. Wei, Chin. Chem. Lett., 2022, 33, 1479–1482 CrossRef CAS.
- D. Noutsias, I. Alexopoulou, T. Montagnon and G. Vassilikogiannakis, Green Chem., 2012, 14, 601–604 RSC.
- Y. H. Kuo and K. S. Shih, Heterocycles, 1990, 31, 1941–1949 CrossRef CAS.
- G. C. M. Lee, E. T. Syage, D. A. Harcourt, J. M. Holmes and M. E. Garst, J. Org. Chem., 1991, 56, 7007–7014 CrossRef CAS.
- M. B. Plutschack, P. H. Seeberger and K. Gilmore, Org. Lett., 2017, 19, 30–33 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–235 CrossRef CAS.
- S. H. Dong, M. Z. Chen, J. R. Zhang, J. Z. Chen and Y. S. Xu, Green Energy Environ., 2021, 6, 715–724 CrossRef.
- G. W. K. Cavill, D. G. Laing and P. J. Williams, Aust. J. Chem., 1969, 22, 2145–2160 CrossRef CAS.
- D. Thiel, D. Doknic and J. Deska, Nat. Commun., 2014, 5, 5278 CrossRef CAS.
- G. D. Zhao, L. X. Liang, E. Wang and R. B. Tong, ACS Catal., 2021, 11, 3740–3748 CrossRef CAS.
- Q. Z. Xing, Z. Hao, J. Hou, G. Q. Li, Z. W. Gao, J. Gou, C. Q. Li and B. X. Yu, J. Org. Chem., 2021, 86, 9563–9586 CrossRef CAS PubMed.
- X. Liang, Z. F. Guo, H. X. Wei, X. Liu, H. Lv and H. Z. Xing, Chem. Commun., 2018, 54, 13002–13005 RSC.
- A. H. Hu, J. J. Guo, H. Pan, H. M. Tang, Z. B. Gao and Z. W. Zuo, J. Am. Chem. Soc., 2018, 140, 1612–1616 CrossRef CAS PubMed.
- M. Zhu, C. Zheng, X. Zhang and S. L. You, J. Am. Chem. Soc., 2019, 141, 2636–2644 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- S. Tang, S. Y. Wang, Y. C. Liu, H. J. Cong and A. W. Lei, Angew. Chem., Int. Ed., 2018, 57, 4737–4741 CrossRef CAS PubMed.
- B. Zhou, Z. C. Liu, W. W. Qu, R. Yang, X. R. Lin, S. J. Yan and J. Lin, Green Chem., 2014, 16, 4359–4370 RSC.
- B. Zhou, Y. J. He, Y. F. Tao, L. X. Liu, M. Hu, Z. H. Chang, H. Lei, J. Lin, T. Lin and G. B. Du, Green Chem., 2022, 24, 2859–2870 RSC.
- D. Q. Hu, Y. L. Zhou and X. F. Jiang, Natl. Sci. Rev., 2022, 9, nwab156 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03344e |
| This journal is © The Royal Society of Chemistry 2023 |