
New insights into urethane alcoholysis enable chemical full recycling of blended fabric waste†
Wen-Hao
Xu
,
Lin
Chen
 *,
Shun
Zhang
,
Rong-Cheng
Du
,
Xuehui
Liu
,
Shimei
Xu
and
Yu-Zhong
Wang
*
*,
Shun
Zhang
,
Rong-Cheng
Du
,
Xuehui
Liu
,
Shimei
Xu
and
Yu-Zhong
Wang
*
Collaborative Innovation Center for Eco-Friendly and Fire-Safety Polymeric Materials (MoE), State Key Laboratory of Polymer Materials Engineering, National Engineering Laboratory of Eco-Friendly Polymeric Materials (Sichuan), College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: linchen410@scu.edu.cn; yzwang@scu.edu.cn
First published on 25th November 2022
Abstract
Accumulation and mishandling of post-consumer polymers have created environmental concerns worldwide. Chemical recycling represents a promising strategy by converting waste polymers into valuable monomers, fuels or chemicals. However, due to multi-component features and similar chemical activities, the chemical recycling of major polymer components often comes at the expense of minor polymer components. Herein, starting from ubiquitous textiles, the effect of spandex on the chemical recycling of polyester is studied qualitatively and quantitatively. Although the spandex content in blended fabrics is low (6%), it has great impact on polyester recycled monomers (recovery rate decreased by 32.4% and chromaticity no longer meets standards). Urethane alcoholysis of spandex is the main depolymerization reaction. Importantly, we reveal for the first time that the amide-esterification reaction occurs during urethane alcoholysis, whereas the reported literature proposes that only ester-interchange reaction occurs. The new insights into urethane alcoholysis not only break the traditional inherent understanding and enrich urethane-based chemistry, but also enable chemical-full-recycling of both major-polyester and minor-spandex.
Introduction
The continuous development of polymer science and the diversity of polymeric materials have brought great progress to modern society. The annual global production of synthetic polymers also exceeds 350 million tons.1–4 However, just like every coin has two sides, the chemical inertness of most polymers makes their recycling and degradation in the natural environment difficult.1,5–7 Most post-consumer polymers are incinerated, landfilled or directly discarded in the environment, resulting in undesired ecological problems and waste of resources.8–11 Therefore, how to efficiently reuse and recycle waste polymers to achieve “waste to wealth” is beginning to be more valued and chosen.Chemical recycling at the molecular level enables a polymer circular economy by converting waste polymers into virgin monomers, fuels or valuable chemicals.12–20 Although there are extensive research studies on chemical recycling of different polymers, most of them focus on single-component primary resins or consumer products. Taking polyethylene terephthalate (PET) as an example, which presents the largest yield of a polycondensation polymer, most research studies focus on the chemical recycling of polyester bottles (pure PET).2,13,21–26 However, bottle consumption is only a small fraction of total PET production. More than 70%27–29 of PET is used to generate fibers and to prepare various textiles by blending with other fibers, such as spandex, nylon, cotton and so on. At present, the global fiber output has exceeded 100 million tons,30,31 of which polyester fiber (namely PET fiber or terylene) accounts for more than 50%.27 As per capita fiber consumption continues to increase, a large amount of waste terylene textiles is generated every year. It is because of the single component of PET bottles that its recycling rate in many countries has exceeded 80%,2,29,32,33 but the recycling rate of polyester textiles is less than 20%29,34 due to their multi-component feature.
The deeper reason is that many non-polyester fibers have similar chemical reactivity to polyester, and the co-depolymerization of non-polyester fibers occurs during the chemical recycling of polyester. The chemical recycling of polyester mainly includes glycolysis, hydrolysis and methanolysis.23,35–38 Among them, glycolysis has been widely utilized on a commercial scale, and the detailed process in actual industry is divided into two steps.35,39 As shown in Fig. 1, the first step (glycolysis) is the nucleophilic substitution26 between the ester bond of polyester and ethylene glycol (EG) to depolymerize into bis(2-hydroxyethyl) terephthalate (BHET). The second step (methanol transesterification) is to perform transesterification of the obtained BHET with methanol to prepare dimethyl terephthalate (DMT) and EG. Since it is easy to purify DMT by sublimation, high-purity DMT that meets the polymerization standard is obtained. Based on whether co-depolymerization occurs, non-polyester fibers can be divided into two categories. One type is fibers that do not undergo co-depolymerization, such as polypropylene fiber and polyacrylonitrile fiber. The main chain of these fibers is formed by chemically inert C–C bonds, which can be removed from the depolymerized solution of polyester by filtration. Another type is fibers that may undergo co-depolymerization, such as spandex40 and nylon.41 These fibers have chemically active groups such as urethane, amide and urea, which are prone to nucleophilic substitution42 with alcohol, ammonia, and water. In the actual industry, only the depolymerization and monomer recovery of polyester fibers, the major component in blended fabrics, are often involved. However, the minor co-depolymerizable non-polyester fibers are not only structurally destroyed in this process, but also directly wasted and sacrificed. Since most commodities are composed of a variety of different polymer components, establishing the depolymerization chemistry and quantitative interplays of each component becomes the key to realizing their chemical-full-recycling and circular polymer economy.
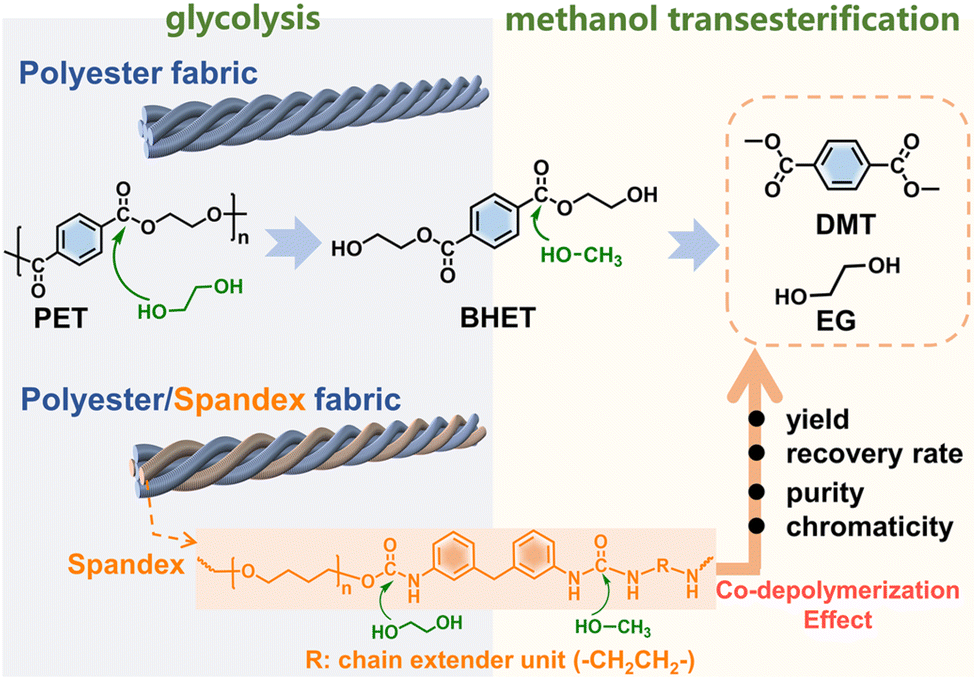 | ||
| Fig. 1 Diagram showing the two-step depolymerization process of glycolysis and methanol transesterification. | ||
Herein, starting from widely used textiles in life, the effect of spandex co-depolymerization on polyester chemical recycling has been studied for the first time. As shown in Fig. 1, spandex is a polyurethane–polyurea copolymer (a kind of polyurethane material), which is widely used in blended fabrics for its high elastic recovery and extensibility.43,44 The structural changes of spandex in the two-step depolymerization process of polyester are systematically studied, and the influence of spandex on polyester recovered monomers is qualitatively and quantitatively analyzed. The urethane group is the main linkage of the spandex macromolecular structural unit. Different from the reported literature proposing that only ester-interchange reaction occurs during urethane alcoholysis,45–48 we reveal for the first time that the amide-moiety of the urethane group can undergo esterification by experiments and density functional theory calculations. The new insights into urethane alcoholysis not only provide a totally new idea for the chemical-full-recycling of major polyester and minor spandex, but also enable “waste to wealth” by recycling high value-added chemicals.
Experimental section
Materials
Polyester–spandex blended fabric (94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 blend ratio, named P&S) and pure polyester fabric were purchased from Jingdong. Potassium carbonate (K2CO3) was supplied by Kelong Chemical Industrial Reagent Co., Ltd (Sichuan, China). Ethylene glycol (EG) and methanol were provided by Tianjin Zhiyuan Chemical Reagent Co., Ltd (Tianjin, China). Chromatographically pure tetrahydrofuran (THF) is obtained from Kemeiou Chemical Reagent Co., Ltd (Tianjin, China). Deuterated chloroform, deuterated dimethyl sulfoxide and deuterated N,N-dimethylformamide (DMF) were supplied by Chengdu Tenglong Microwave Technology Co. (Chengdu, China). 4,4-Diaminodiphenylmethane (MDA) was purchased from Energy Chemical Co., Ltd (Shanghai, China).
:6 blend ratio, named P&S) and pure polyester fabric were purchased from Jingdong. Potassium carbonate (K2CO3) was supplied by Kelong Chemical Industrial Reagent Co., Ltd (Sichuan, China). Ethylene glycol (EG) and methanol were provided by Tianjin Zhiyuan Chemical Reagent Co., Ltd (Tianjin, China). Chromatographically pure tetrahydrofuran (THF) is obtained from Kemeiou Chemical Reagent Co., Ltd (Tianjin, China). Deuterated chloroform, deuterated dimethyl sulfoxide and deuterated N,N-dimethylformamide (DMF) were supplied by Chengdu Tenglong Microwave Technology Co. (Chengdu, China). 4,4-Diaminodiphenylmethane (MDA) was purchased from Energy Chemical Co., Ltd (Shanghai, China).
Depolymerization process of different fabrics
The first step is glycolysis by ethylene glycol. In this process, K2CO3 (typical Lewis base) was chosen as the catalyst for glycolysis.36,49,50 Taking polyester fabric as an example, polyester fabric (5 g), K2CO3 (0.1 g) and EG (7.5 g) were added into a 50 mL round bottom flask. The mixture was stirred at 200 °C for 3 h to obtain a homogeneous solution (the main component is BHET). Pure spandex is separated from the polyester–spandex blended fabrics using DMF solvent. The P&S blend fabric was dissolved in DMF solvent at 75 °C for 3 h. The dissolved part is spandex, and the insoluble part is polyester. The spandex solution was obtained by filtration. The spandex solution was evaporated under reduced pressure to remove the solvent DMF, and the obtained solid was dried at 80 °C for 12 hours, which was the separated spandex. The glycolysis processes of the P&S fabric and spandex were exactly the same as that of the above polyester fabric.The second step is a transesterification process with methanol. Before performing methanol transesterification, the obtained above reaction solution was first distilled at 170 °C by vacuum distillation (10 kPa) to remove excess ethylene glycol. The separated ethylene glycol was collected and named EG1. 20 g methanol and 0.0075 g K2CO3 were added to the remaining reaction solution, and the reaction was carried out at 65 °C for 3 h. When the reaction was over, a large amount of solid was precipitated from the reaction solution. This solid was filtered to give crude DMT. Crude DMT was sublimed under reduced pressure (10 kPa) at 170 °C using a decompression S735060-sublimator to obtain purified DMT. The methanol transesterification process for the above P&S and spandex was exactly the same.
The yield and recovery rate of DMT were calculated using eqn (1) and (2), respectively.
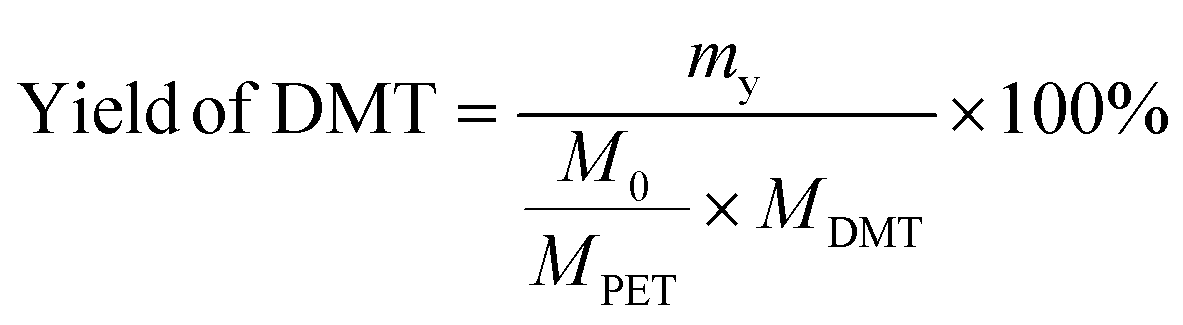 | (1) |
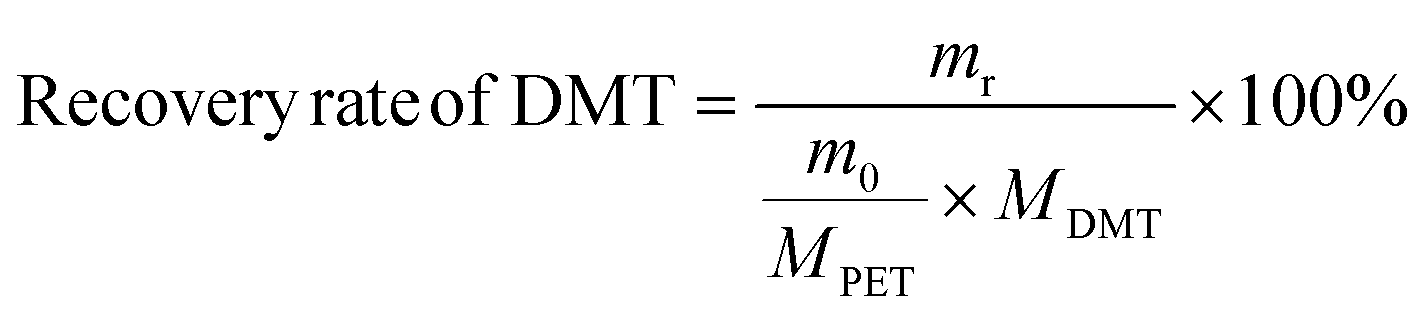 | (2) |
In order to explore the effect of spandex on recycled ethylene glycol in the depolymerization experiment, the filtrate was evaporated at 50 °C through a rotary evaporator to remove all methanol, and then vacuum distillation carried out at 170 °C to steam out the remaining EG and recorded as EG2. EG1 and EG2 were collected together, that is, the recovered monomer ethylene glycol.
Alcoholysis simulation experiments of phenylurethane
Phenylurethane (1.65 g, 0.01 mol), ethylene glycol (1.28 g, 0.02 mol), and potassium carbonate (0.01 g) were reacted at 100 °C, 150 °C and 200 °C for 1 h. The reaction product was analyzed by GC-MS and 1H NMR. When potassium hydroxide or dibutyltin dilaurate is used as a catalyst, the reaction steps and conditions are the same.Computational details
All calculations were performed using the Gaussian 16 suite of programs. Geometry optimisations were performed using the B3LYP density functional and the 6-311+G(d,p) basis set. Frequency optimisations at the same level of theory confirmed that the optimised structures were minima (zero imaginary frequencies) or transition states (one imaginary frequency) and then used to evaluate the ZPVE and the thermal vibrational corrections at T = 298 K. In order to include the solvent effects, all calculations using the SMD model have been performed, with the dielectric constant of ethylene glycol (e = 37.7).Characterization
Morphological characterization of fabrics at different depolymerization times was performed using a scanning electron microscope (SEM, JSM-7500F, 15 kV). Fourier Transform Infrared Spectroscopy (FT-IR) of the samples was performed using a Nicolet 6700 spectrometer with the wavenumber range from 400 cm−1 to 4000 cm−1. NMR testing in this work was performed on a Bruker 400 MHz Avance III spectrometer. Analysis of components in the two-step reaction solutions was conducted on a Finnigan TSQ high performance liquid chromatography mass spectrometer (LC-MS). The purity of recovered ethylene glycol was measured on a gas chromatography-mass spectrometer (GCMS-QP2010 Plus). The molecular weight of spandex was measured on a gel permeation chromatograph (HLC-8320 GPC). The samples were dissolved in DMF/LiBr solution with a concentration of 10 mg mL−1, injection volume = 10 μL, flow rate = 0.6 ml min−1, and run time = 22 min. The chromaticity of ethylene glycol was measured on a spectrophotometer (YS6010). The organic elemental analysis of the two-part alcoholysis product was performed on a Thermo Scientific elemental analyzer (Flashsmart).Results and discussion
Structural change of spandex in the chemical recycling process of polyester
To investigate whether spandex would undergo co-depolymerization during the two-step chemical recycling of polyester fabrics, its structural changes and products were analyzed qualitatively and quantitatively. The first step (glycolysis) occurs at 200 °C for 3 h under K2CO3 (commonly used catalyst for polyester). As shown in Fig. 2a, the spandex formed a homogeneous solution and many sticky solids after glycolysis. The main structure of sticky solids is that of polytetrahydrofuran diols, which is derived from the depolymerization of spandex (Fig. 2b). For unreacted spandex, peak a is the urethane group. Peak b (connecting a benzene ring) and peak c (connecting an aliphatic chain extender) are hydrogens of urea. Peak d (7.0–7.6 ppm) is the benzene ring. Peak e and peak f are the methylene in polytetrahydrofuran units. Peak g is the methylene between two benzene rings. Peak R is the methylene of the chain extender. 4.48 ppm (–OH) is the terminal hydroxyl group of polytetrahydrofuran. For the sticky solid, there is no urea (peak b and c), and only a little urethane (peak a) and diphenylmethane unit (peak d) remain. This is due to EG attacking the carbonyl of the urethane and urea bonds, resulting in the scission of the spandex macromolecular chain at the urethane and urea sites. Therefore (ESI Table S1†), the molecular weight of the sticky solid (0.30 × 104 g mol−1) is significantly lower than that of the unreacted spandex (3.66 × 104 g mol−1). The formed solution of spandex during the glycolysis process was studied by LC-MS and GC-MS (Fig. 2c, Fig. S1–S4 and Table S2†). The main products are diphenylmethane-containing molecules (peaks a–c), polytetrahydrofuran diols with relatively low molecular weights (peak d, repeat unit number n is 4–13, 306–954 g mol−1) and 2-imidazolidinone (peak e).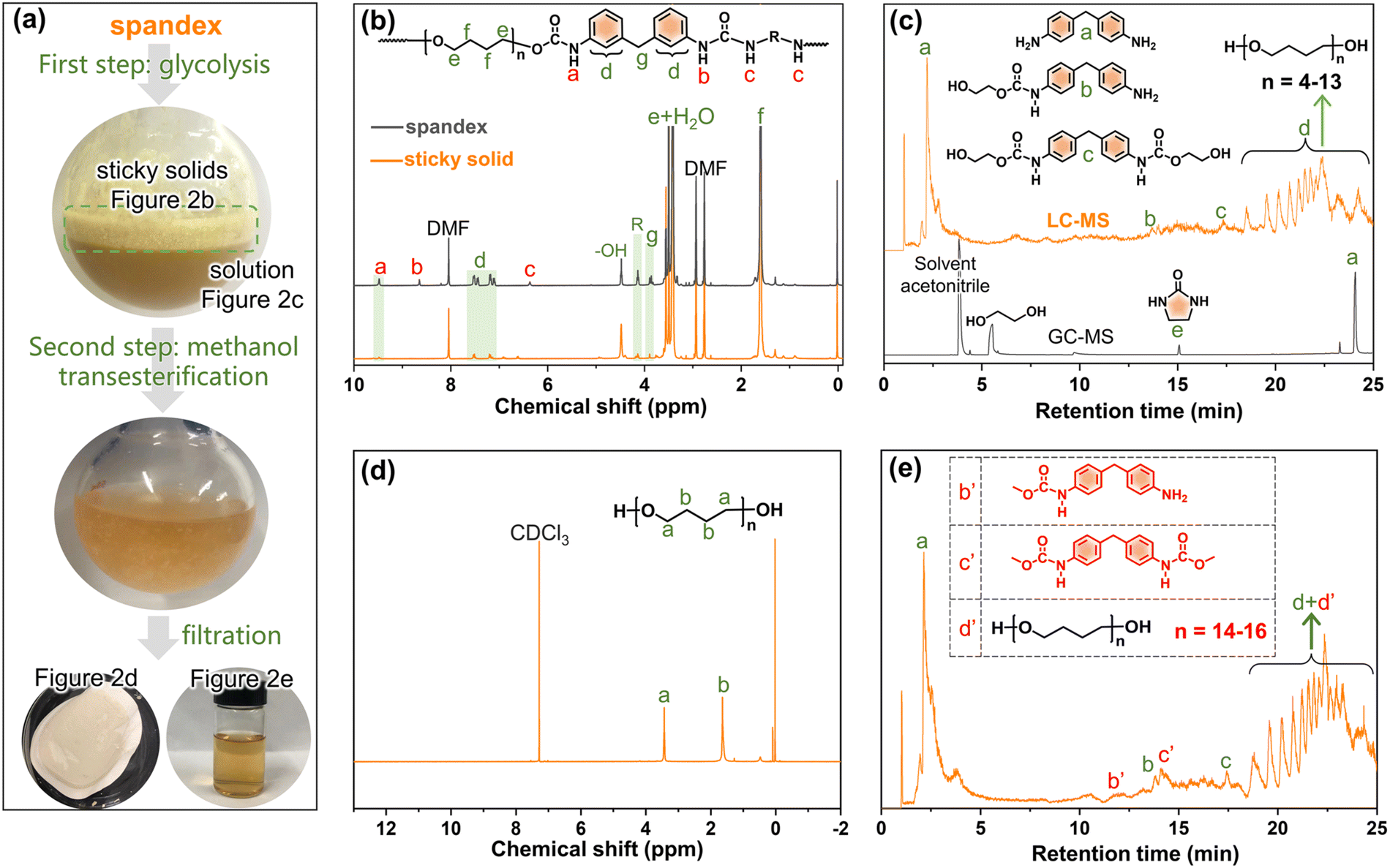 | ||
| Fig. 2 Structural change of spandex during polyester chemical recycling. (a) Photographs of spandex after glycolysis and methanol transesterification; (b) 1H NMR (in dimethyl formamide, DMF-d6) of unreacted spandex and the formed sticky solid; (c) LC-MS and GC-MS chromatograms of the formed solution from spandex after the glycolysis process; (d) 1H NMR (in CDCl3) of the formed solid and (e) LC-MS chromatogram of the formed solution from spandex after methanol transesterification. | ||
In the second step (methanol transesterification), the above solution and sticky solids are reacted with methanol at 65 °C for 3 h. These sticky solids are further transformed into solid powders (Fig. 2a), which are polytetrahydrofuran (Fig. 2d). This indicates that the residual small number of urethane bonds in the sticky solids are completely reacted, thereby removing the diphenylmethane units (dissolved in solution) and producing polytetrahydrofuran. The formed solution in this process is also studied by LC-MS (Fig. 2e, Fig. S5–S7 and Table S3†). Except for the same products as in Fig. 2c (peaks a, b, c, and d), new terminal-methyl-containing diphenylmethane molecules (peaks b′ and c′) and polytetrahydrofuran diols (peak d′, n is 14–16, 1026–1170 g mol−1) are generated by the reaction with methanol. The above results prove that in the two-step chemical recycling of polyester fabrics, spandex indeed undergoes co-depolymerization reactions, resulting in by-products: 2-imidazolidinone, diphenylmethane-containing molecules and polytetrahydrofuran diols.
Based on the above qualitative analysis, the relevant quantitative results are as follows (detailed calculations in Fig. S8†). During glycolysis, the formed sticky solids account for 65.0% (3.25 g) of the initial spandex mass (5.0 g). In solution, 4,4′-methylenedianiline (peak a in Fig. 2c), 2-imidazolidinone and polytetrahydrofuran diols account for 10.0% (0.50 g), 3.0% (0.15 g) and 14.2% (0.71 g) of the initial spandex mass, respectively. The sum of these substances accounts for 92.2% of the initial spandex mass. After methanol transesterification, the remaining insoluble solid powders only account for 4.4% (0.22 g) of the initial spandex mass, which indicates that much more low molecular weight polytetrahydrofuran diols enter the reaction solution.
Influence of spandex on polyester recovered monomers
Under the same two-step depolymerization, a comparative analysis of the polyester fabric and polyester–spandex blended fabric (polyester and spandex mass ratio is 94:6, named P&S) is carried out to qualitatively and quantitatively analyze the effect of spandex on polyester final recovered monomers (DMT and EG).
In the first glycolysis step, the polyester fabric reacts with EG to depolymerize into BHET (Fig. 1). The changes in the microscopic morphology of different fabrics are investigated by SEM (ESI Fig. S9†). As the glycolysis progresses, the surface of fabrics is gradually damaged and the fiber weaving structure begins to break down. After glycolysis (Fig. S10†), the polyester fabric forms homogeneous solution, while the P&S fabric forms solution and sticky solids (from spandex). The formed solutions are characterized (Fig. 3a). Peaks a, b and c, and d represent the benzene ring, methylene and hydroxyl of BHET, respectively. Peaks f and e are the –OH and –CH2 of EG, respectively. From the LC-MS (ESI Fig. S11 and Table S4†) of the solution for the polyester fabric, besides the main product BHET (∼86%), there are also oligomers (BHET dimer and BHET trimer). Therefore, peaks a′ and d′ in Fig. 3a represent the benzene and hydroxyl groups in the BHET oligomer, respectively. The two peaks at 7.0–7.5 ppm (benzene of diphenylmethane-containing molecules) and 1.51 ppm (methylene of polytetrahydrofuran) in the solution of P&S are attributed to the small molecules formed from spandex co-depolymerization.
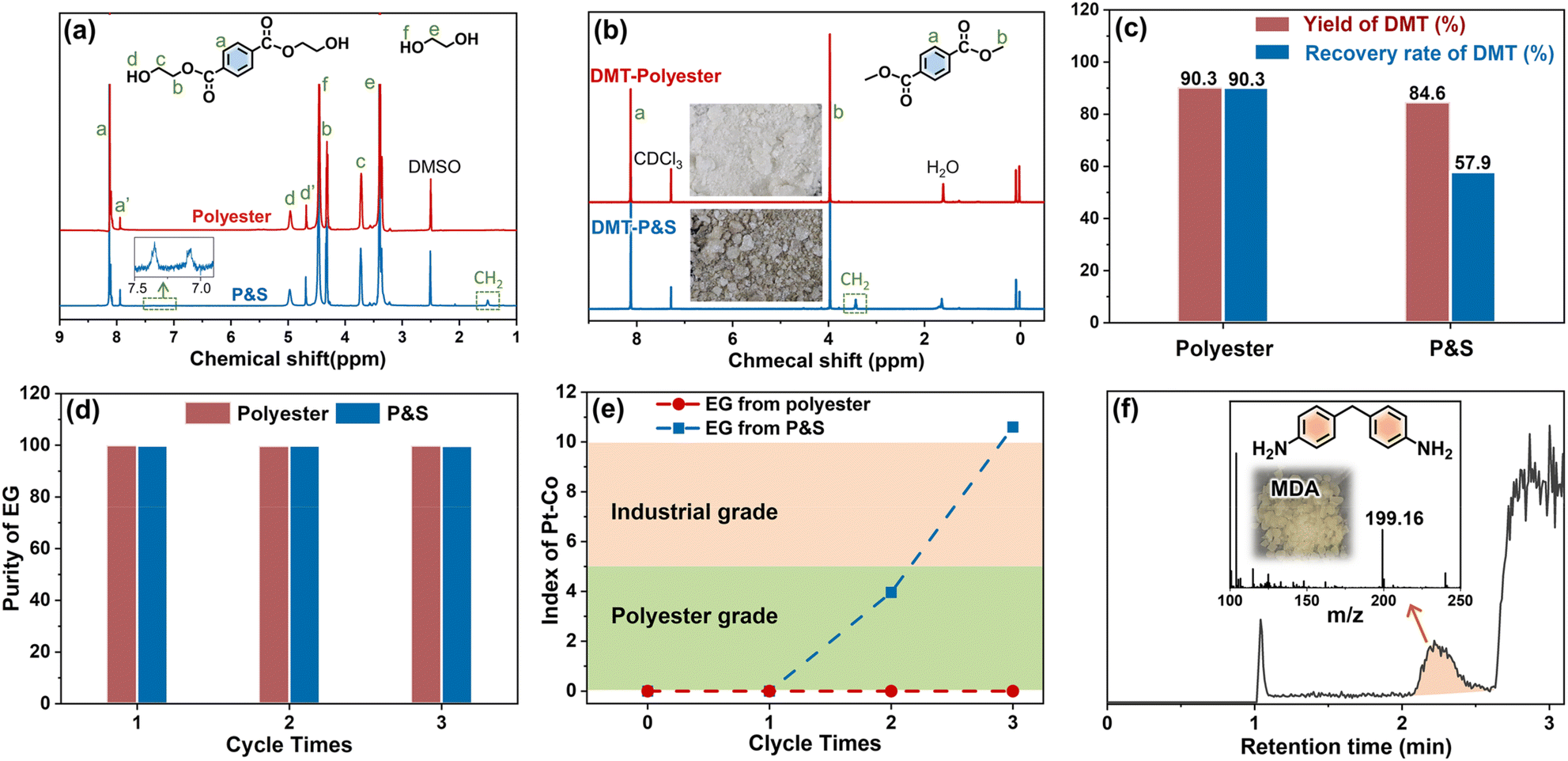 | ||
| Fig. 3 Influence of spandex on polyester recovered monomers. (a) 1H NMR (in DMSO-d6) of the obtained solutions from the polyester fabric and P&S fabric after glycolysis; (b) 1H NMR (in CDCl3) of crude DMT from the polyester fabric and P&S fabric (inset: photographs of crude DMT); (c) yield and recovery rate of DMT from the polyester fabric and P&S fabric; (d) purity of recovered EG at different cycle times; (e) index of Pt–Co for recovered EG at different cycle times; (f) LC-MS chromatogram of recovered EG from P&S (inset: mass spectrum of the marked peak and digital photograph of purchased 4,4′-methylenedianiline). | ||
In the second methanol transesterification step, the hydroxyl of methanol nucleophilically attacks the carbonyl carbon of BHET and BHET oligomers, and the substitution reaction occurs to generate monomers DMT and EG (Fig. 1). When the reaction ends, a large amount of solid is precipitated from the reaction solution, which is filtered to give crude DMT. Compared with DMT obtained from polyester (Fig. 3b), there is an impurity peak at 3.44 ppm (the methylene of polytetrahydrofuran from spandex) for the DMT obtained from P&S. From the inset photographs, the crude DMT obtained from the P&S fabric is obviously yellowish, while the crude DMT obtained from the polyester fabric appears as white crystals. In the FT-IR spectra (ESI Fig. S12†), there are obvious stretching vibrations (3325 cm−1) and deformation vibrations (1567 cm−1) of N–H from spandex impurities in the crude DMT obtained from P&S. The nitrogen content of crude DMT obtained from P&S is 0.22% (ESI Table S5†). That is, the co-depolymerization products of spandex can significantly affect the purity and color of crude DMT.
The yield of DMT from polyester and P&S fabrics is calculated by HPLC (ESI Fig. S13†) according to eqn (1), which represents the percentage of DMT content actually produced by polyester depolymerization to the theoretical content. In Fig. 3c, the DMT yield of the polyester fabric is 90.3%, which is higher than that of the P&S fabric (84.6%). This is due to that spandex depolymerization byproducts containing amino and hydroxyl groups, and these functional groups can have side reactions with the ester group of DMT to decrease its yield. Sublimation is commonly used in industry to purify crude DMT. As shown in eqn (2) and Fig. 3c, the DMT recovery rate is defined as the percentage of the mass of DMT collected after sublimation to the mass of DMT theoretically formed from polyester. The DMT recovery rate of polyester is 90.3%, which is much higher than that of the P&S fabric (57.9%). The crude DMT of the polyester fabric has minimal residue in the sublimator after sublimation, while the crude DMT of P&S still has a large amount of yellow solid left in the sublimator after sublimation (ESI Fig. S14†). The –OH and –NH containing viscous substances produced by the spandex co-depolymerization can adhere to a large amount of DMT during sublimation, resulting in a significant decrease in the recovery rate of DMT.
In addition, we study the co-depolymerization effect of spandex on the recycled EG. The two-step depolymerization of glycolysis and methanol transesterification is defined as one cycle. In Fig. 3d, the purity of the recovered EG from polyester and P&S has no obvious decrease after three cycles, and the purity is above 99% (calculated by GC-MS, ESI Fig. S15–S17†), but there is a significant difference in chromaticity (Fig. 3e). The chromaticity requirement for different purposes is different. It is stipulated that a Pt–Co value ≤ 5 is polyester grade, and a Pt–Co value ≤ 10 is industrial grade. The Pt–Co value of the EG recovered from the polyester fabric has no obvious change after three cycles, and it is still polyester grade. However, the chromaticity of the EG recovered from P&S became worse after three cycles, increasing from 0 to 10.6, and it no longer meets the industrial or polyester grade standard. In the LC-MS of EG recovered from P&S (Fig. 3f), the yellow 4,4′-methylenedianiline coming from spandex is detected, which can evaporate along with EG and reduce the EG chromaticity. Since EG is an excess feedstock in the first step of glycolysis, it is difficult to calculate the yield and recovery rate of recovered EG.
In conclusion, in the chemical recycling of polyester, spandex undergoes co-depolymerization reaction, and the main products are 2-imidazolidinone, diphenylmethane-containing molecules and polytetrahydrofuran oligomers. Although the content of spandex in blended fabrics is relatively low (6%), it has a great impact on polyester recycled monomers. The yield and recovery rate of recycled DMT reduce by 5.7% and 32.4%, respectively. At the same time, the chromaticity of recycled EG no longer meets the requirements of industrial or polyester grade. Because of this remarkable effect, the recycling rate of multi-component textiles is still less than 20%.
New insights into urethane alcoholysis
Before understanding the co-depolymerization mechanism, the synthetic process of spandex is briefly presented.43 As shown in Fig. 4a, the polyether diol and diisocyanate (molar ratio: 1:2, forming a urethane group) were reacted to prepare the prepolymer, and then the prepolymer was reacted with the same amount of diamine chain extender (forming a urea group) to obtain high molecular weight spandex. Therefore, in the chemical structure of spandex, the diisocyanate unit (diphenylmethane-containing) is mostly connected to the polyether unit by a urethane group at one end and the diamine chain extender unit by a urea group at the other end. Based on literature reports45–47,51,52 (Fig. 4b), when alcoholysis occurs under the action of an acid/base catalyst, the urethane group is cleaved from the C–O bond to generate new urethane and new alcohol, namely ester-interchange or transcarbamoylation reaction. The amide moiety of the urethane exhibits low reactivity due to the delocalization of the nitrogen lone pairs,53,54 and is therefore considered unchanged after alcoholysis. During alcoholysis, urea groups can cleave from two C–N bonds with different chemical environments to generate new amino groups and new urethane groups. Thus, in theory (Fig. 4c), the alcoholysis products of spandex should be mono-amino-containing and dual-urethane-containing diphenylmethane molecules (peaks b and c in Fig. 4c, which are also detected in Fig. 2c and e). However, in the experimental results (Fig. 2c and e), the content of 4,4′-methylenedianiline (peak a in Fig. 4c) is the largest (accounting for 10.0% of the initial spandex mass), which is inconsistent with the theoretical expectation. Based on the chemical structure of spandex, the possible reason for the high 4,4′-methylenedianiline content is that the amide part of urethane participates in the alcoholysis reaction, and thus the C–N bond is cleaved to form another amino group.
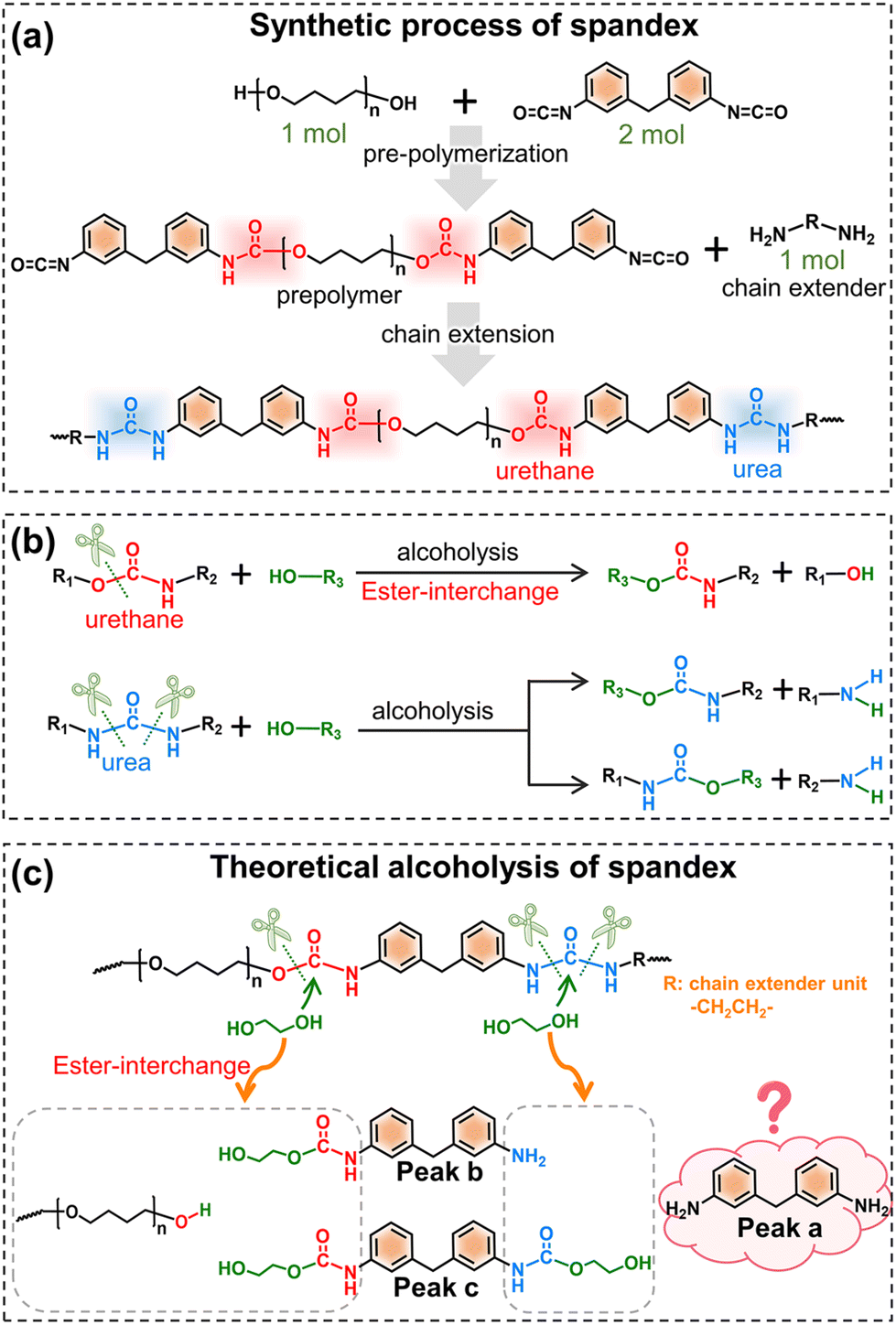 | ||
| Fig. 4 Synthetic and theoretical alcoholysis of spandex. (a) Synthetic process of spandex; (b) reported alcoholysis reaction of urethane and urea groups; and (c) theoretical alcoholysis of spandex. | ||
To eliminate the interference of the urea bond and study whether the amide part of urethane participates in the alcoholysis reaction, a simulation experiment of small molecule phenylurethane is designed. As shown in Fig. 5, phenylurethane (1.65 g, 0.01 mol, peak 2) is subjected to alcoholysis reaction with ethylene glycol (1.28 g, 0.02 mol, peak 1) at 100 °C, 150 °C and 200 °C for 1 h under the same catalyst (K2CO3, 0.01 g). The alcoholysis products are analyzed by GC-MS (Fig. 5 and ESI Fig. S18† for the mass spectrum) and 1H NMR (ESI Fig. S19–S27†). In Fig. 5(a), peak 2′ (isocyanatobenzene) is generated due to the decomposition of phenylurethane during GC-MS testing (ESI Fig. S28†). As the reaction temperature increased, the peak area of the reactant ethylene glycol did not change much because of its excess, but the reactant phenylurethane gradually decreased. The main reaction products are aniline (peak 3), 2-hydroxyethyl phenylcarbamate (peak 4), 3-phenyl-2-oxazolidinone (peak 5), ethylene carbonate (peak 6) and ethanol (peak 7). At 100 °C, only a small amount of phenylurethane participates in the reaction, and the products aniline (peak 3) and 2-hydroxyethyl phenylcarbamate (peak 4) are formed. At 150 °C, more phenylurethane is converted to aniline and 2-hydroxyethyl phenylcarbamate, and there is a marked increase of the 3-phenyl-2-oxazolidinone (peak 5) product, which is derived from the cyclo-elimination55 of 2-hydroxyethyl phenylcarbamate. At 200 °C, almost all the raw material phenylurethane participates in the reaction, and large amounts of aniline and 3-phenyl-2-oxazolidinone are generated. Meanwhile, a very small amount of 2-hydroxyethyl phenylcarbamate is observed because it is completely converted to the 3-phenyl-2-oxazolidinone product. In addition, the NMR results (ESI Fig. S19–S27†) also confirmed the presence of these products.
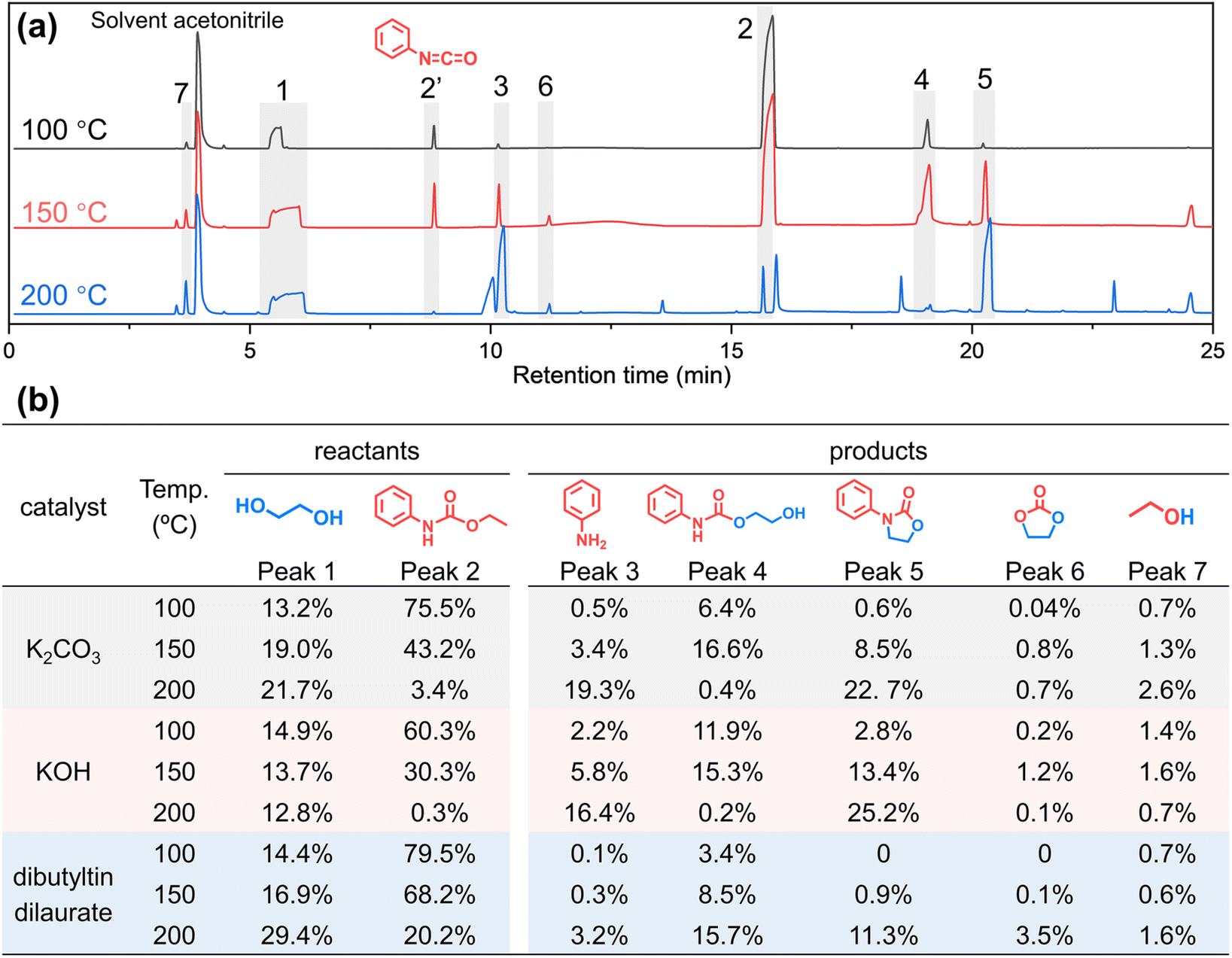 | ||
| Fig. 5 Alcoholysis experiments of small molecule phenylurethane. (a) GC-MS chromatograms of formed solutions after the reaction of phenylurethane with ethylene glycol (under a K2CO3 catalyst) at different temperatures for 1 h; (b) peak area percentages of substances detected by GC-MS after the reaction of phenylurethane with ethylene glycol (under different catalysts) at different temperatures for 1 h. | ||
To further prove the generality of the above results, the same experiments were carried out with two other common catalysts (KOH and dibutyltin dilaurate) for the alcoholysis reaction of the urethane group. As shown in Fig. 5b and ESI Fig. S29,† under different catalysts, the structures of the main products and their variation with reaction temperature are basically the same. The above results prove that when the urethane group undergoes alcoholysis, not only a new urethane (2-hydroxyethyl phenylcarbamate, peak 4) but also a new amino group (aniline, peak 3) is indeed formed. That is to say, not only the ester-moiety of urethane participates in the alcoholysis reaction to generate a new urethane group by ester-interchange, but also the amide-moiety of the urethane group participates in the alcoholysis reaction to generate a new amino moiety by amide-esterification.
Based on the experimental results, the alcoholysis mechanism of phenylurethane with ethylene glycol under the action of the potassium carbonate catalyst is proposed (Fig. 6a), and the energy change is calculated by density functional theory (Fig. 6b, DFT computational details in the ESI†). The K+ cation (Lewis acid) protonates the carbonyl oxygen (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the urethane group, making it more electrophilic and therefore more susceptible to nucleophilic attack by ethylene glycol. At the same time, the anion CO32− pulls the hydrogen of the hydroxyl group to its side, so the oxygen in ethylene glycol has a higher electron density and it is easy for it to attack the CO of the urethane group. Therefore, the glycolysis reaction involves two transition states: nucleophilic attack of the hydroxyl group (TS1), followed by the elimination of the leaving group (TS2-a and TS2-b). The transition state 1 (TS1) is the nucleophilic attack of one of the hydroxyl groups of ethylene glycol on the carbonyl oxygen of the urethane group, thereby forming a tetrahedral intermediate. For the transition state TS2-a, the protonation of the hydroxyethyl moiety of the intermediate results in the elimination of a molecule of ethanol to obtain a stable complex product-a. For the transition state TS2-b, the protonation of the aromatic amine moiety of the intermediate results in the elimination of a molecule of aniline to obtain a stable complex product-b. The calculated results show that the energetic barrier for TS1, TS2-a and TS2-b is 28.38 kcal mol−1, 4.22 kcal mol−1 and 1.22 kcal mol−1, respectively. Thus, TS1 (nucleophilic attack of the hydroxyl group) is the rate-determining step of the reaction, and the following C–O cleavage (TS2-a, namely ester-interchange) and C–N cleavage (TS2-b, namely amide-esterification) can occur simultaneously. Since complex product-a has a lower potential energy than product-b, the ester-interchange occurs preferentially. Based on the above experimental and calculation results, we discovered and confirmed the amide-esterification during urethane alcoholysis for the first time.
O) of the urethane group, making it more electrophilic and therefore more susceptible to nucleophilic attack by ethylene glycol. At the same time, the anion CO32− pulls the hydrogen of the hydroxyl group to its side, so the oxygen in ethylene glycol has a higher electron density and it is easy for it to attack the CO of the urethane group. Therefore, the glycolysis reaction involves two transition states: nucleophilic attack of the hydroxyl group (TS1), followed by the elimination of the leaving group (TS2-a and TS2-b). The transition state 1 (TS1) is the nucleophilic attack of one of the hydroxyl groups of ethylene glycol on the carbonyl oxygen of the urethane group, thereby forming a tetrahedral intermediate. For the transition state TS2-a, the protonation of the hydroxyethyl moiety of the intermediate results in the elimination of a molecule of ethanol to obtain a stable complex product-a. For the transition state TS2-b, the protonation of the aromatic amine moiety of the intermediate results in the elimination of a molecule of aniline to obtain a stable complex product-b. The calculated results show that the energetic barrier for TS1, TS2-a and TS2-b is 28.38 kcal mol−1, 4.22 kcal mol−1 and 1.22 kcal mol−1, respectively. Thus, TS1 (nucleophilic attack of the hydroxyl group) is the rate-determining step of the reaction, and the following C–O cleavage (TS2-a, namely ester-interchange) and C–N cleavage (TS2-b, namely amide-esterification) can occur simultaneously. Since complex product-a has a lower potential energy than product-b, the ester-interchange occurs preferentially. Based on the above experimental and calculation results, we discovered and confirmed the amide-esterification during urethane alcoholysis for the first time.
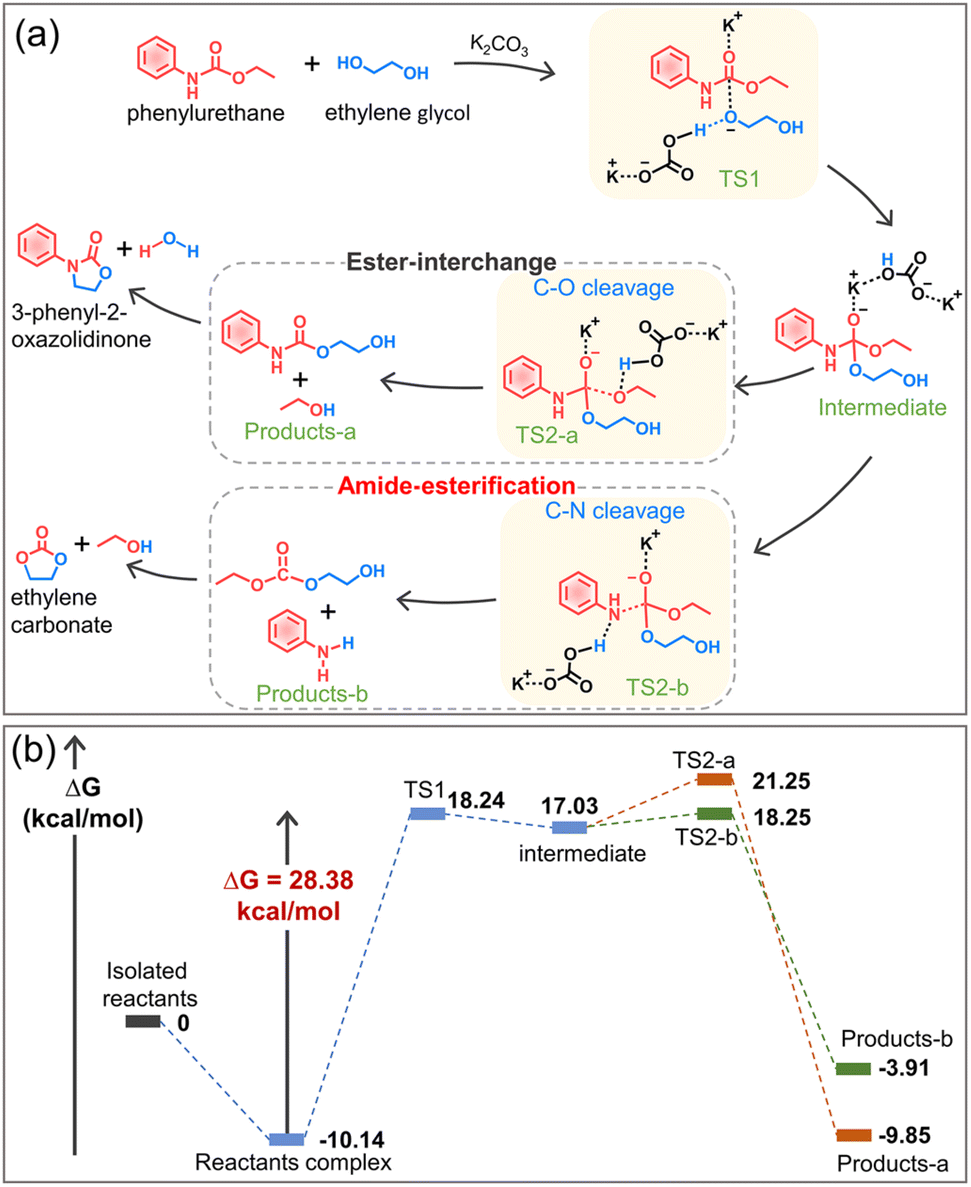 | ||
| Fig. 6 New insights into urethane alcoholysis. (a) Proposed mechanism for the alcoholysis reaction of phenylurethane with ethylene glycol; (b) computed reaction pathways for the glycolysis of phenylurethane. | ||
Interestingly, the subsequent intramolecular elimination reactions of ethyl (2-hydroxyethyl) carbonate and 2-hydroxyethyl phenylcarbamate yield ethylene carbonate26 and 3-phenyl-2-oxazolidinone (Fig. S27†), respectively. In particular, the generation of 3-phenyl-2-oxazolidinone also provides a new and facile route for its preparation, which is extensively used in pharmaceuticals for its bioactivity.56–58 In general, 3-aryl-2-oxazolidinones are prepared from toxic isocyanates or phosgene by the reaction of aryl halide with oxazolidin-2-one, CO2 with amino alcohols, or aryl iodide with amino alcohol carbamate.59–62 Most of these routes rely on toxic or expensive catalysts. This work provides a novel one-step synthesis for this important and expensive 3-aryl-2-oxazolidinone from cheap starting materials and catalysts.
Spandex co-depolymerization mechanism and chemical full recycling of blended fabrics
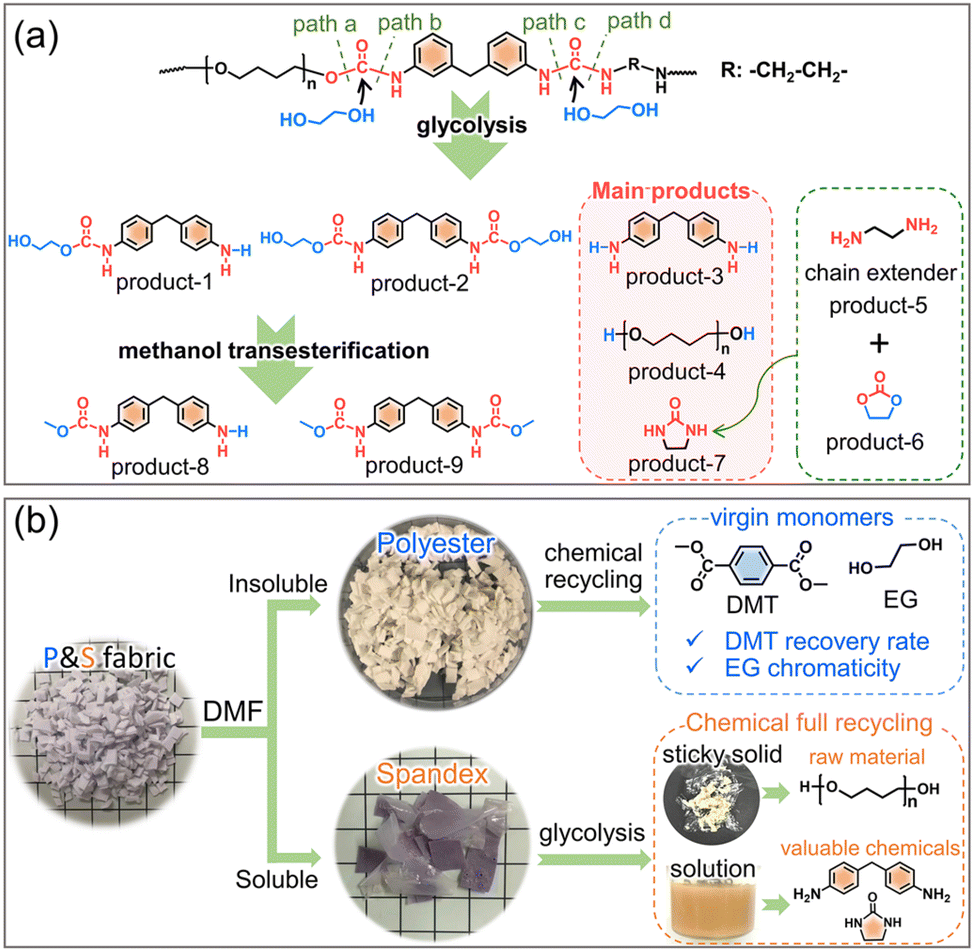
Based on the above results, the co-depolymerization mechanism of spandex is proposed in Fig. 7a. During glycolysis, ethylene glycol attacks the carbonyl carbon of the urethane and urea groups to form tetrahedral intermediates. The intermediate formed by the urethane group can be cleaved from the C–O bond (path a, ester-interchange) or from the C–N bond (path b, amide-esterification). The intermediates formed by the urea group also have two ways of breaking bonds (path c and path d). For the diphenylmethane-containing unit, product-1 results from path a + c or path b + d, and product-2 and product-3 come from path a + d and path b + c, respectively. For the polytetrahydrofuran unit, the C–O cleavage (path a) of urethane bonds on both sides generates polytetrahydrofuran diols (product-4). For the chain extender unit (marked as R), the C–N cleavage (path d) of urea bonds on two sides generates ethylenediamine (product-5). During glycolysis, the urea group generates a new urethane group regardless of the cleavage of any C–N bond (Fig. 4b). Thus, as the reaction progress, the remaining carbonyl (CO) of the urethane group is first transformed into ethylene carbonate (products 6). Subsequently, the carbonylation63,64 reaction of ethylenediamine with ethylene carbonate generates 2-imidazolidinone (products 7), which also promotes the amide-esterification reaction during urethane alcoholysis by consuming ethylene carbonate. During methanol transesterification, product-1 and product-2 are transformed to product-8 and product-9, respectively. Although these co-depolymerization products of spandex in turn have adverse effects on the recovered monomers of polyester, some of them are valuable chemicals. Among them, 4,4′-methylenedianiline (product-3), as a high value-added chemical raw material, is widely used in dyes, curing agents, adhesives, polyamides, rubber and so on. High-performance 2-imidazolidinone is also widely used in formaldehyde scavengers, plant growth regulators, antibiotic intermediates, etc.
 | ||
| Fig. 7 Spandex co-depolymerization mechanism and chemical-full-recycling of the blended fabric. (a) Proposed co-depolymerization mechanism of spandex during glycolysis and methanol transesterification and (b) chemical-full-recycling of major polyester and minor spandex in the blended fabric. | ||
Last but not least, the new findings of amide-esterification during urethane alcoholysis enable the chemical-full-recycling of major polyester and minor spandex in blended fabrics instead of sacrificing spandex (Fig. 7b). Considering that spandex has significant effects on polyester chemical recycling, and there are high value-added molecules in the alcoholysis products of spandex. Thus, before the waste polyester textile enters the chemical recycling process, spandex can be removed by solvent (DMF) pretreatment. On the one hand, the purified polyester can greatly contribute to its chemical-full-recycling by increasing the DMT recovery rate and unchanging EG chromaticity to obtain virgin monomers as much as possible. On the other hand, the separated spandex can also achieve its chemical-full-recycling by collecting the remaining sticky solids (raw material polytetrahydrofuran) and recycling abundant valuable chemicals (4,4′-methylenedianiline and 2-imidazolidinone) in solution.
Spandex is an important polyurethane material. This work also provides directional and theoretical guidance for the chemical-full-recycling of other post-consumer polyurethanes. Although due to the difference of the raw material diisocyanate, oligomer diol and chain extender, the structure of commercial polyurethane is very diverse. But the urethane group is the main linkage of the polyurethane structural unit. Thus, urethane alcoholysis has important industrial applications in polyurethane chemistry, especially for chemical recycling.48,65,66 Polyurethane is one of the most used polymers with annual global production exceeding 20 million tons,52,65 and undoubtedly a large amount of post-consumer polyurethane is generated every year. Alcoholysis, especially glycolysis, is by far the most widely used chemical recycling method for polyurethane to recycle polyols (namely, sticky solids after alcoholysis), while other substances (solution part) are wasted. The new findings of amide-esterification can not only well explain the still unclear urethane alcoholysis mechanism of polyurethane, but also provide totally new insights into the chemical-full-recycling of polyurethane materials by turning the wasted solution into wealth.
Conclusions
This work provides an in-depth study of the spandex co-depolymerization effect on polyester chemical recycling and presents new insights into the urethane alcoholysis. The urethane and urea groups of spandex are the depolymerization sites, leading to 4,4′-methylenedianiline, 2-imidazolidinone and polytetrahydrofuran oligomers as main products. Although spandex accounts for only 6% in a polyester–spandex fabric, its co-depolymerization products greatly reduce the recovery rate of DMT by 32.4% and markedly change the chromaticity of EG. Importantly, we reveal for the first time that amide-esterification can take place besides the reported only ester-interchange during urethane alcoholysis, which is applicable to both phenylurethane small molecule and spandex macromolecule. The amide-esterification reaction can well explain the still unclear alcoholysis mechanism of spandex, and also provide a theoretical basis and novel synthesis for useful chemical structures (such as carbonate and amine). By further utilizing this amide-esterification reaction during urethane alcoholysis, the chemical full recycling of the polyester–spandex blended fabric is enabled by using this spandex waste as a “cheaper and greener” alternative feedstock for the synthesis of high value-added chemicals (4,4′-methylenedianiline and 2-imidazolidinone). These interesting findings not only break the traditional inherent understanding of urethane alcoholysis and enrich urethane-based chemistry, but also underpin the sustainable development of polymer materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Key Research and Development Program of China (2020YFC1910304), the Fundamental Research Funds for the Central Universities (2020SCUNL205 and 2021SCU12149) and Sichuan University Postdoctoral Interdisciplinary Innovation Fund.Notes and references
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y.-H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef PubMed.
- E. Barnard, J. J. Rubio Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- M. R. Martinez and K. Matyjaszewski, CCS Chem., 2022, 4, 2176–2211 CrossRef.
- X. Zhang, Y. Sun, C. Zhang and X. Zhang, CCS Chem., 2022 DOI:10.31635/ccschem.022.202202072.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef.
- X. Liu, S. Xu, F. Zhang, X.-L. Wang and Y.-Z. Wang, Acta Polym. Sin., 2022, 53, 1005–1022 Search PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS.
- C. Ostle, R. C. Thompson, D. Broughton, L. Gregory, M. Wootton and D. G. Johns, Nat. Commun., 2019, 10, 1622 CrossRef PubMed.
- M. Bergmann, M. B. Tekman and L. Gutow, Nature, 2017, 544, 297 CrossRef CAS.
- S. Lievens, T. Slegers, M. A. Mees, W. Thielemans, G. Poma, A. Covaci and M. Van Der Borght, Environ. Pollut., 2022, 307, 119511 CrossRef PubMed.
- W. An, X.-L. Wang, X. Liu, G. Wu, S. Xu and Y.-Z. Wang, Green Chem., 2022, 24, 701–712 RSC.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van Der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef PubMed.
- Z. Huang, M. Shanmugam, Z. Liu, A. Brookfield, E. L. Bennett, R. Guan, D. E. V. Herrera, J. A. Lopez-Sanchez, A. G. Slater, E. J. L. McInnes, X. Qi and J. Xiao, J. Am. Chem. Soc., 2022, 144, 6532–6542 CrossRef PubMed.
- S. T. Nguyen, E. A. McLoughlin, J. H. Cox, B. P. Fors and R. R. Knowles, J. Am. Chem. Soc., 2021, 143, 12268–12277 CrossRef PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef PubMed.
- H. Chen, K. Wan, Y. Zhang and Y. Wang, ChemSusChem, 2021, 14, 4123–4136 CrossRef PubMed.
- A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef PubMed.
- H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, J. Am. Chem. Soc., 2022, 144, 4678–4684 CrossRef PubMed.
- D. D. Pham and J. Cho, Green Chem., 2021, 23, 511–525 RSC.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M.-L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS.
- J. J. R. Arias and W. Thielemans, Green Chem., 2021, 23, 9945–9956 RSC.
- V. Sinha, M. R. Patel and J. V. Patel, J. Polym. Environ., 2010, 18, 8–25 CrossRef CAS.
- A. B. Raheem, Z. Z. Noor, A. Hassan, M. K. Abd Hamid, S. A. Samsudin and A. H. Sabeen, J. Cleaner Prod., 2019, 225, 1052–1064 CrossRef CAS.
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruiperez, J. L. Hedrick, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2021, 60, 6710–6717 CrossRef CAS PubMed.
- Z. Guo, E. Adolfsson and P. L. Tam, Waste Manage., 2021, 126, 559–566 CrossRef CAS.
- Y. Liu, Q. Zhong, P. Xu, H. Huang, F. Yang, M. Cao, L. He, Q. Zhang and J. Chen, Matter, 2022, 5, 1305–1317 CrossRef CAS.
- J. Chu, Y. Cai, C. Li, X. Wang, Q. Liu and M. He, Waste Manage., 2021, 124, 273–282 CrossRef CAS PubMed.
- C. Stone, F. M. Windsor, M. Munday and I. Durance, Sci. Total Environ., 2020, 718, 134689 CrossRef.
- G. Suaria, A. Achtypi, V. Perold, J. R. Lee, A. Pierucci, T. G. Bornman, S. Aliani and P. G. Ryan, Sci. Adv., 2020, 6, eaay8493 CrossRef PubMed.
- M. Haupt, E. Waser, J. C. Wurmli and S. Hellweg, Waste Manage., 2018, 77, 220–224 CrossRef PubMed.
- S. Honma and J.-L. Hu, J. Cleaner Prod., 2021, 284, 125274 CrossRef.
- S. Jiang, Z. Xia, A. Farooq, M. Zhang, M. Li and L. Liu, Cellulose, 2021, 28, 3235–3248 CrossRef.
- P. S. Ho and S. H. Kim, Fash. Text., 2014, 1, 1–17 CrossRef.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- S. Zhang, W. Xu, R. Du, X. Zhou, X. Liu, S. Xu and Y.-Z. Wang, Green Chem., 2022, 24, 3284–3292 RSC.
- Y. Li, M. Wang, X. Liu, C. Hu, D. Xiao and D. Ma, Angew. Chem., Int. Ed., 2022, 61, e202117205 Search PubMed.
- K. Ishihara, K. Ishida, M. Miyamoto, M. Nakashima, K. Sato and H. Hasegawa, US6706843B1, 2004.
- Y. Jiang, A. Zheng, Y. Guan, D. Wei, X. Xu and W. Gong, Chem. Eng. J., 2021, 404, 125152 CrossRef.
- A. Ballistreri, D. Garozzo, M. Giuffrida and G. Montaudo, Macromolecules, 1987, 20, 2991–2997 CrossRef CAS.
- M. L. Bender, Chem. Rev., 1960, 60, 53–113 CrossRef.
- J. U. Otaigbes and A. Madbouly, Handbook of Textile Fibre Structure, 2009, vol. 1, pp. 325–351 Search PubMed.
- A. Quye, Polym. Degrad. Stab., 2014, 107, 210–218 CrossRef.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef.
- B. Jousseaume, C. Laporte, T. Toupance and J. M. Bernard, Tetrahedron Lett., 2002, 43, 6305–6307 CrossRef.
- D. K. Chattopadhyay and D. C. Webster, Prog. Polym. Sci., 2009, 34, 1068–1133 CrossRef.
- K. M. Zia, H. N. Bhatti and I. A. Bhatti, React. Funct. Polym., 2007, 67, 675–692 CrossRef.
- H. T. Kim, M. H. Ryu, Y. J. Jung, S. Lim, H. M. Song, J. Park, S. Y. Hwang, H.-S. Lee, Y. J. Yeon, B. H. Sung, U. T. Bornscheuer, S. J. Park, J. C. Joo and D. X. Oh, ChemSusChem, 2021, 14, 4251–4259 CrossRef.
- R. Lopez-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutierrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022–1028 CrossRef.
- K. Kanaya and S. Takahashi, J. Appl. Polym. Sci., 1994, 51, 675–682 CrossRef.
- T. Vanbergen, I. Verlent, J. De Geeter, B. Haelterman, L. Claes and D. De Vos, ChemSusChem, 2020, 13, 3835–3843 CrossRef.
- B. Jousseaume, C. Laporte, T. Toupance and J. M. Bernard, Tetrahedron Lett., 2003, 44, 5983–5985 CrossRef.
- T. Deguchi, H.-L. Xin, H. Morimoto and T. Ohshima, ACS Catal., 2017, 7, 3157–3161 CrossRef.
- P. ten Holte, L. Thijs and B. Zwanenburg, Org. Lett., 2001, 3, 1093–1095 CrossRef.
- B. Mallesham, B. M. Rajesh, P. R. Reddy, D. Srinivas and S. Trehan, Org. Lett., 2003, 5, 963–965 CrossRef PubMed.
- Z. Luo, B. Wang, Y. Liu, G. Gao and F. Xia, Phys. Chem. Chem. Phys., 2016, 18, 27951–27957 RSC.
- M. R. Barbachyn and C. W. Ford, Angew. Chem., Int. Ed., 2003, 42, 2010–2023 CrossRef.
- T. Baronsky, C. Beattie, R. W. Harrington, R. Irfan, M. North, J. G. Osende and C. Young, ACS Catal., 2013, 3, 790–797 CrossRef.
- R. L. Paddock, D. Adhikari, R. L. Lord, M.-H. Baik and S. T. Nguyen, Chem. Commun., 2014, 50, 15187–15190 RSC.
- W. Mahy, P. K. Plucinski and C. G. Frost, Org. Lett., 2014, 16, 5020–5023 CrossRef PubMed.
- C. J. Dinsmore and S. P. Mercer, Org. Lett., 2004, 6, 2885–2888 CrossRef.
- A. K. Qaroush, A. W. Alsayyed, A. F. Eftaiha, F. M. Al-Qaisi and B. A. Salameh, ChemistrySelect, 2022, 7, e202200478 CrossRef CAS.
- A. H. Tamboli, H. A. Bandal and H. Kim, Chem. Eng. J., 2016, 306, 826–831 CrossRef CAS.
- L. Zhao and V. Semetey, ACS Omega, 2021, 6, 4175–4183 CrossRef CAS.
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03663k |
| This journal is © The Royal Society of Chemistry 2023 |