
Photocatalytic syngas production from bio-derived glycerol and water on AuIn-decorated GaN nanowires supported by Si wafer†
Zhouzhou
Wang‡§
 ab,
Bowen
Sheng‡
c,
Yiqing
Chen‡
d,
Sharif Md.
Sadaf
e,
Jinglin
Li
a,
Jiajia
Yang
c,
Jun
Song
*d,
Lin
Yao
f,
Ying
Yu
*b,
Lei
Zhu
a,
Xinqiang
Wang
*cgh,
Zhen
Huang
a and
Baowen
Zhou
*a
ab,
Bowen
Sheng‡
c,
Yiqing
Chen‡
d,
Sharif Md.
Sadaf
e,
Jinglin
Li
a,
Jiajia
Yang
c,
Jun
Song
*d,
Lin
Yao
f,
Ying
Yu
*b,
Lei
Zhu
a,
Xinqiang
Wang
*cgh,
Zhen
Huang
a and
Baowen
Zhou
*a
aKey Laboratory for Power Machinery and Engineering of Ministry of Education, School of Mechanical Engineering, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, China. E-mail: zhoubw@sjtu.edu.cn
bInstitute of Nanoscience and Nanotechnology, College of Physical Science and Technology, Central China Normal University, Wuhan 430079, China. E-mail: yuying01@mail.ccnu.edu.cn
cState Key Laboratory of Artificial Microstructure and Mesoscopic Physics, School of Physics, Nano-Optoelectronics Frontier Center of Ministry of Education (NFC-MOE), Peking University, Beijing 10087, China. E-mail: wangshi@pku.edu.cn
dDepartment of Mining and Materials Engineering, McGill University, 3610 University Street, Montreal, QC H3A0C9, Canada. E-mail: jun.song2@mcgill.ca
eCentre Energie, Matériaux et Télécommunications, Institut National de la Recherche Scientifique (INRS)-Université du Québec, 1650 Boulevard Lionel-Boulet, Varennes, Quebec J3X1S2, Canada
fChina-UK Low Carbon College, Shanghai Jiao Tong University, 3 Yinlian Road, Shanghai 201306, China
gPeking University Yangtze Delta Institute of Optoelectronics, Nantong, Jiangsu 226010, China
hCollaborative Innovation Center of Quantum Matter, School of Physics, Peking University, Beijing 100871, China
First published on 5th December 2022
Abstract
Green syngas production from Earth-abundant and renewable resources is a viable means to achieve carbon neutrality. However, this critical path to realize net-zero carbon dioxide emissions has remained extremely challenging. In this work, by coupling GaN nanowires (NWs) with dual gold–indium nanoparticles (NPs) onto wafer-scale silicon, a novel and well-developed photocatalytic architecture of AuIn NPs/GaN NWs/Si has been assembled for the first time to produce syngas with bio-derived glycerol, water, and concentrated light as the only inputs. By correlative experimental and theoretical investigations, the AuIn/GaN interface is found to be synergistically favorable for dehydrogenating the edge functional group of –CH2OH of the glycerol skeleton, followed by the cleavage of the inert C(sp3)–C(sp3) bond toward an ethylene glycol-based intermediate with a greatly reduced reaction energy barrier in glycerol solution. Owing to the exceptional optoelectronic and catalytic properties, syngas yields a benchmarking activity of 149.3 mmol g−1 h−1 under mild conditions without an extra energy input other than light. The H2/CO ratio can be broadly tailored from 19.3 to 0.9, thus favoring various downstream products. Water plays a vital role in the highly efficient formation of syngas by providing active species e.g., hydroxyl radicals through photocatalysis. Owing to the widespread distribution of biomass, water, and sunlight, and the industrially available semiconductor platform, this work promises to efficiently and selectively produce next-generation distributed syngas.
Introduction
Syngas, a mixture of CO and H2, is considered as the building block of modern civilization by providing countless chemicals and fuels via the Fischer–Tropsch process.1–4 Unfortunately, over the last century, syngas has been primarily produced from fossil fuels, resulting in tremendous carbon dioxide emissions, energy crisis, and severe environmental issues.5,6 Moreover, the conventional syngas production from fossil fuels in the chemical industry has suffered from harsh reaction conditions e.g., high temperature, elevated pressure, and extensive thermal input.7–9 For sustainability, carbon-neutral production of syngas using renewable resources under mild conditions is a significant yet challenging thrust. As opposed to fossil fuel reforming, biomass, water, and sunlight can provide unlimited and widespread-distribution resources for the sustainable production of syngas.7,10 The construction of a rational photocatalytic architecture by coupling an appropriate light-harvesting platform with an efficient catalyst is at the core of this topic.11–15For carbon-neutral syngas generation from biomass, water, and sunlight, a couple of photocatalytic architectures have been actually pioneered using the conventional semiconductors e.g., TiO2, WO3, and CdS.11–13 There has been, however, limited success in achieving high-efficiency syngas production with tunable H2/CO ratios,16 which is, in principle, attributed to the following causes, first, it is extremely challenging to generate suitable charge carriers without compromising light harvesting using conventional semiconductors;17,18 second, severe electron–hole recombination in the bulk and/or surface of the semiconductors;19,20 third, there is lack of a rational catalytic architecture for effectively and selectively activating a variety of chemical bonds of biomass e.g., C–C, C–H, and –OH.21,22 A rational photocatalytic architecture for addressing the key issues above is urgently demanded.
Group III-nitrides (GaN and its related alloys), a mature semiconductor material system in the photonic and electronic industry, is a promising platform for photocatalysis because of its unique structural, optical, and electronic properties.23 In recent years, a series of pioneering breakthroughs have been made using III-nitride-based photocatalytic architectures for H2O splitting and CO2 fixation.24–27 It is worth noting that through state-of-the-art plasma-assisted molecular beam epitaxy technology, 1-dimensional III-nitride NWs with a nearly defect-free structure, high optical absorption, and efficient charge carrier separation/migration are facilely grown at the atomic level. More importantly, due to their high surface area and unique surface properties, epitaxial III-nitrides exhibit great potential for adsorbing and activating multifunctional groups of biomass in conjunction with a rational cocatalyst, which is distinct from the conventional semiconductors.28
In addition to light absorbing materials, a cocatalyst also plays a pivotal role in photocatalysis. Indium (In) is acknowledged to be an efficient material for oxidation reactions owing to its unique properties.29–32 However, this monofunctional metal is limited by non-tunable interaction with reactant molecules. Coupling with a secondary component can offer a flexible window for tailoring the catalytic properties. In this work, gold (Au), a known noble metal for CO evolution,33–36 is tentatively employed for integrating with indium to catalyze bio-derived glycerol reforming. Experimentally, using highly controllable molecular beam epitaxy together with straightforward photodeposition, dual gold–indium nanoparticles are anchored onto GaN nanowires supported by wafer-level silicon (Fig. S1†). By density functional theory (DFT) calculations, it has been revealed that the AuIn cluster, in synergy with GaN, is favored for activating and dehydrogenating the edge functional group of –CH2OH of the glycerol skeleton. Subsequently, the inert C(sp3)–C(sp3) bond is broken to produce an ethylene glycol-based intermediate with a greatly reduced reaction energy barrier in glycerol solution. Moreover, such a nanoarchitecture of AuIn NPs/GaN NWs/Si provides a multifunctional configuration for decoupling light absorption, energetic charge carrier separation, and cocatalyst loading. Under a concentrated light illumination of 4 W cm−2, an exceptional syngas activity of 149.3 mmol g−1 h−1 is achieved with a broad range of H2/CO ratios varying from 19.3 to 0.9, which is highly favorable for various downstream products. Herein, water is crucial for improving syngas production by offering active species e.g., hydroxyl radicals through photocatalysis. Benefitting from the widespread distribution of biomass, water, and concentrated light on the Earth and the industrial availability of the building blocks of the photocatalytic architecture, this work provides a promising approach for distributed production of green syngas. To our knowledge, this is the first attempt on using such a novel and well-developed III-nitride/Si-based photocatalytic architecture for syngas production from biomass, water, and concentrated light.
Results and discussion
Adsorption/activation behavior of glycerol over a AuIn/GaN interface
The adsorption/activation of the reactant over the catalytic interface is the initial step of chemical reactions and plays a pivotal role in the performance. Glycerol adsorption characteristics were thereby studied by DFT calculations.37,38 First, four surface models, i.e., AuIn, GaN, In4/GaN, and In3Au/GaN with the optimized configurations were built (Fig. S2†). The Gibbs free energy of glycerol adsorption (ΔGad) on AuIn is discovered to have a minimal value of 0.03 eV under solvent effects, indicating a laborious interaction between AuIn and glycerol without GaN (Fig. 1a). However, pristine GaN illustrates a moderate ΔGad of −0.89 eV (Fig. S3a†). Meanwhile, covalent-like bonding between O atoms and Ga atoms is observed. Notably, the integration of metal clusters with GaN did not change the adsorption of glycerol a lot as a result of the solvation effects, as seen from ΔGad values of −0.90 and −0.78 eV for In4/GaN and In3Au/GaN, respectively (Table S1, Fig. 1b and Fig. S3b†). The results above reveal that GaN plays a pivotal role in glycerol adsorption/activation. The adsorption configurations were further elucidated and analyzed via charge density difference plots (see Fig. 1c and d), where similar electron charge density redistributions around the adsorption region were found. The electron transfer from the surface to the adsorbed glycerol was further studied by Bader charge analysis.39 It is highly interesting to find that glycerol attracted 0.011e and 0.035e respectively from the substrate of GaN and In3Au/GaN. This finding provides electronic evidence that In3Au and GaN are both critical for glycerol activation, which is beneficial for the subsequent reactions.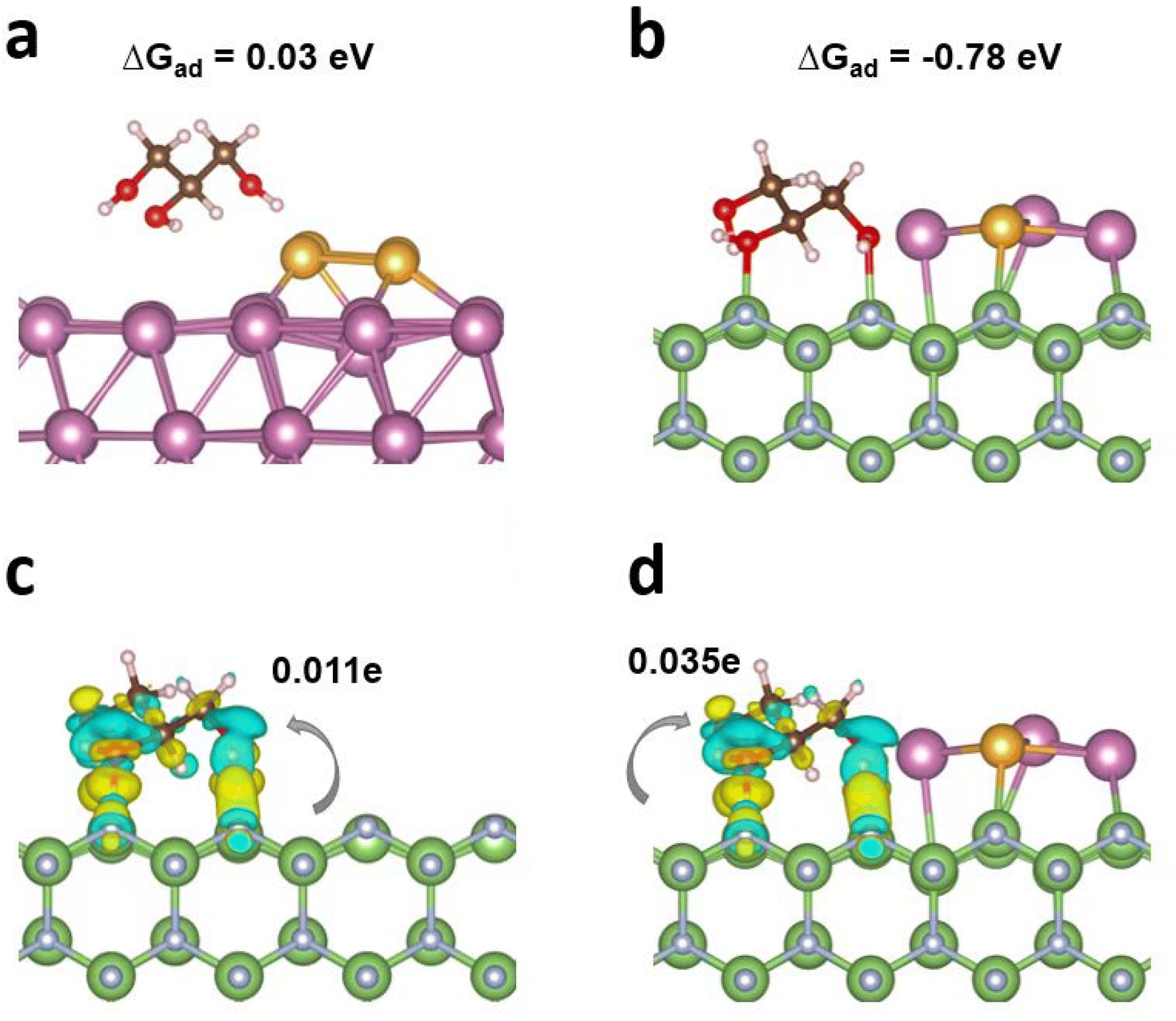 | ||
| Fig. 1 Adsorption behavior of glycerol under solvent effects. Side views of the optimized configurations of (a) glycerol adsorbed on Au/In, (b) In3Au/GaN, and charge density difference plots for glycerol adsorbed on (c) GaN and (d) In3Au/GaN. Ga, green; N, silver; In, purple; Au, gold; C, brown; H, pink; and O, red. Regions of yellow and cyan indicate the gain and loss of electronic charge respectively, with an isosurface of 0.003 e Å−1. | ||
Characterization of the assembled photocatalytic architectures
Inspired by the theoretical investigation above, a range of photocatalytic architectures were technically assembled by molecular beam epitaxy of GaN nanowires (NWs) on wafer-level silicon, followed by photo-deposition of various cocatalysts e.g., Au, In, and AuIn (Please see Experimental section in Supporting Information). The architectures were correlatively characterized by various microscopy and spectroscopy techniques. As shown in Fig. S4 and S5,† GaN NWs are vertically aligned onto a silicon wafer, showing an average length of 900 nm and diameters varying from 50 to 100 nm. Such an integrated design enables efficient light harvesting due to the narrow bandgap of the Si substrate and the low reflectance of GaN NWs (Fig. S6†). As characterized by photoluminescence spectroscopy, in this architecture, GaN NWs could be excited by photons < 365 nm to produce energetic charge carriers for biomass/water reforming (Fig. S7†). Moreover, the defined morphology of 1-dimensional NWs is beneficial for spatially decoupling photon absorption with charge separation and active site dispersion, thus benefiting the overall performance.37 Scanning transmission electron microscopy (STEM) characterization and elemental mapping images illustrate that the dual AuIn nanoparticles (NPs) are seamlessly anchored on the surface of GaN NWs, exhibiting a solid solution feature with a size distribution ranging from 5 to 16 nm (Fig. 2b–g and Fig. S8†). Meanwhile, the high angle annular dark field (HAADF) image in Fig. 2c shows that the planar distance between the two adjacent plane is 0.26 nm, which is associated with the (002) plane of GaN.40 It is in agreement with the typical peak of the X-ray diffraction patterns appearing at a 2θ of 34.2 degree (Fig. S9†).40 Impressively, the nearly defect-free structure of GaN NWs with orderly stacked atoms is highly favorable for charge carrier migration.41 The oxidation states of various elements were analyzed by X-ray photoelectron spectroscopy (XPS) (Fig. S10†). As shown in Fig. 2h, when In was introduced, the Au 4f peak shifted to a higher binding energy. It is opposite to the slight shift of the In 3d peak to a lower binding energy upon incorporation of Au (Fig. 2i). These observations indicate that the outer electrons of Au are attracted to In, resulting in an electron-rich outer layer of In, which is conducive to the activation of *C in glycerol and promotes the reaction. The XPS spectra of N 1s shown in Fig. 2j and Fig. S11a† are nearly identical, which means that GaN NWs exist stably. It is noted that the introduction of In caused the Ga 3d peak to shift to a lower binding energy because the electronegativity of Ga is greater than that of In, leading to the redistribution of electrons from In to Ga (Fig. 2k and Fig. S11b†). It is well matched with the DFT investigations, in which glycerol is favorably adsorbed on the Ga atom of the GaN surface. Overall, the microscopy and spectroscopy investigations above indicate the success in constructing a hybrid architecture by coupling AuIn with GaN NWs/Si, which promises outstanding structural and optoelectronic properties for biomass/water reforming.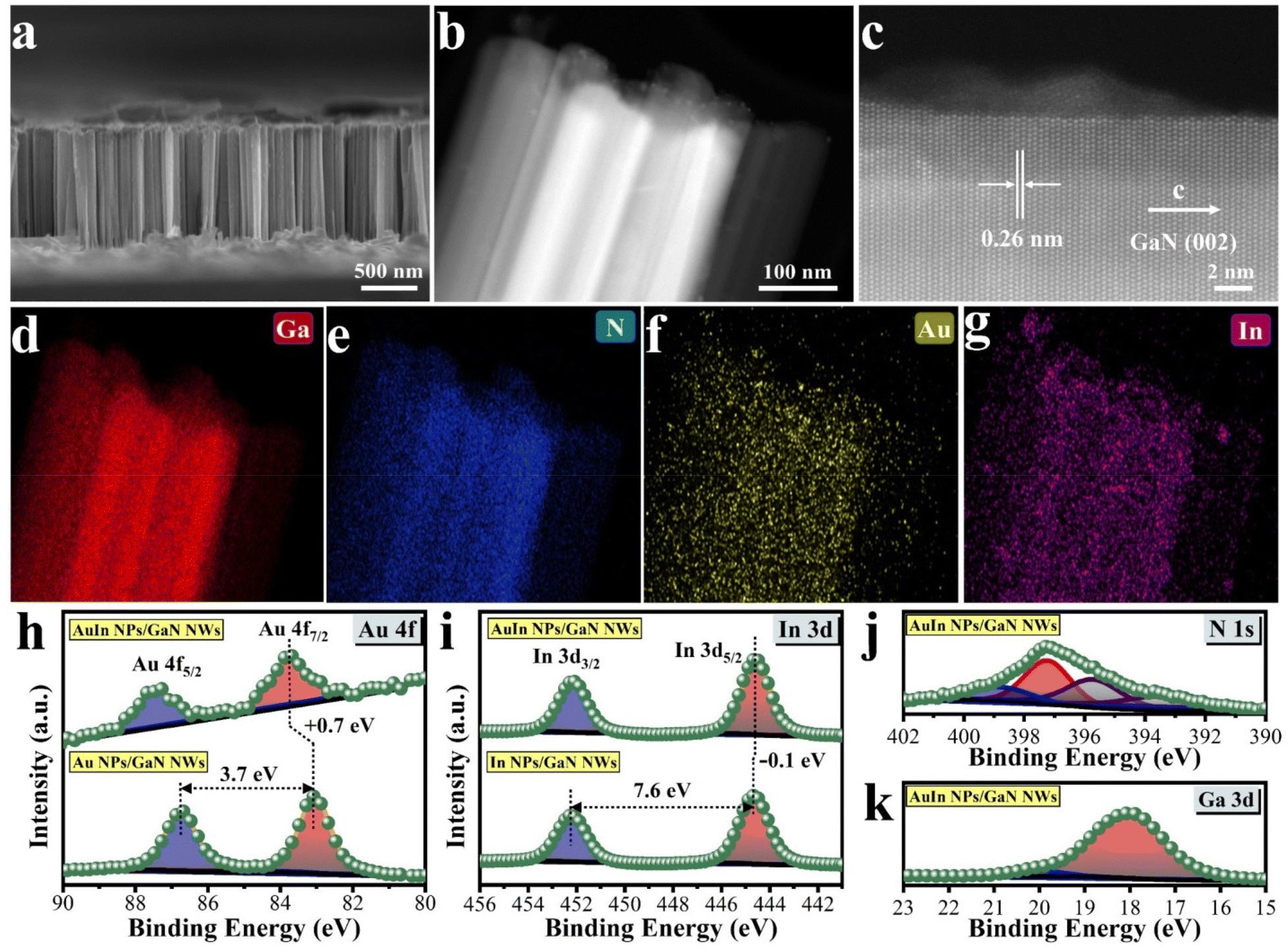 | ||
| Fig. 2 Characterization of the assembled photocatalytic architectures. (a) Front-view SEM image of AuIn NPs/GaN NWs/Si. HAADF-STEM images of AuIn NPs/GaN NWs at (b) low and (c) high resolutions. (d–g) The elemental mapping images of AuIn NPs/GaN NWs vertically aligned onto the planar silicon. The XPS measurements of (h) Au 4f for AuIn NPs/GaN NWs and Au NPs/GaN NWs, (i) In 3d for AuIn NPs/GaN NWs and In NPs/GaN NWs, (j) N 1s and (k) Ga 3d for AuIn NPs/GaN NWs. | ||
Bio-derived production of syngas
The performance of various architectures was evaluated in a quartz chamber without extra inputs except glycerol, light, and water (Fig. S12†). Illustrated in Fig. 3a, among the architectures evaluated, AuIn NPs/GaN NWs/Si shows a maximum activity, reaching 149.3 mmol g−1 h−1 with a high H2/CO ratio of 14.7. Under the same conditions, AuIn/Si is nearly not active for syngas generation because Si cannot generate energetic charge carriers to drive the reaction although it has a narrow bandgap for absorbing a large part of the incident light. In stark contrast, AuIn NPs/GaN/sapphire demonstrates a moderate syngas activity of 25 mmol g−1 h−1. These discoveries suggest that the rational design of AuIn NPs/GaN NWs/Si is critical for superior activity, which is associated with the integration of high optical absorption, appropriate energetic charge, and effective glycerol activation. The influence of co-catalysts was further studied (Fig. 3b and Fig. S13†). It is found that without a co-catalyst, both H2 and CO were slightly yielded over bare GaN NWs onto Si with evolution rates of 2.7 mmol g−1 h−1 and 1.1 mmol g−1 h−1, respectively, owing to the absence of active sites. The performance was clearly enhanced by decorating with co-catalysts. In particular, the performance of AuIn is remarkably superior to that of the single component of either gold (11.8 mmol g−1 h−1) or indium (24.2 mmol g−1 h−1), validating the synergy between Au and In for promoting the reaction. It is worth mentioning that by varying the Au/In ratios within AuIn, H2/CO ratios can be broadly tailored from 2.7 to 19.3 with varied syngas activity between 11.8 and 149.3 mmol g−1 h−1. As suggested by the DFT calculations, the varied performance is probably attributed to the tunable interaction between the cocatalyst with reactants/intermediates. Under the optimized conditions, Au5In40/GaN NWs/Si exhibits a superior syngas activity of 149.3 mmol g−1 h−1 with a H2/CO ratio of 14.7. As an essential metric of the catalytic activity, the turnover frequency (TOF) shown in Fig. 3c was calculated based on the loaded amount of the cocatalysts listed in Table S2.† It is observed that at a suitable AuIn loading of 0.053 μmol cm−2, the TOF of syngas is as high as 285 mol CO + H2 per mol AuIn per hour, which is higher than that of either Au (197 h−1) and In (36 h−1). However, when it is overloaded, the TOF is lowered because of the underutilized metal sites.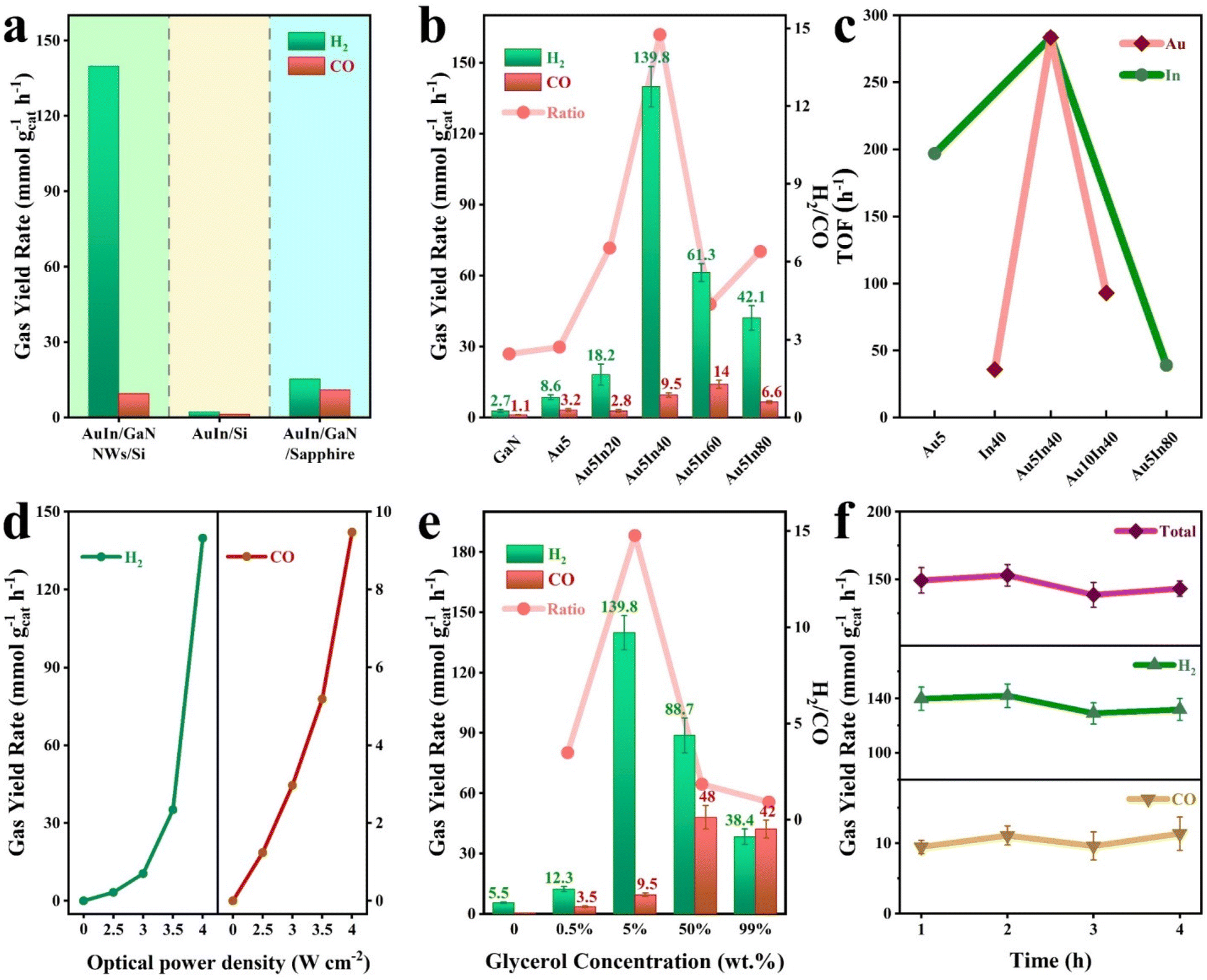 | ||
| Fig. 3 Performance of the syngas production from bio-derivative glycerol and water. (a) The performance of AuIn NPs deposited on GaN NWs/Si, Si, and GaN/sapphire. (b) The influence of the cocatalyst on the performance. (c) The effect of varied Au and In precursors′ concentration on the TOF of syngas. (d) The performance under different illumination intensities. (e) The influence of glycerol concentration on the performance. (f) Stability testing. | ||
The impacts of optical power density and glycerol concentration on the performance were also examined. As shown in Fig. 3d, without any extra energy input, both CO and H2 evolution rates increased considerably with the increasing optical power density from 2.5 to 4 W cm−2. Meanwhile, the H2/CO ratio increased from 2.8 to 14.7 (Fig. S14†). Herein, at a low optical power density, the low abundance of photoexcited charge carriers led to limited activity. Moreover, the activity of CO/H2 showed a notable reduction upon visible-infrared light illumination when other experimental conditions were not varied (Fig. S15†), suggesting that the photoexcitation of GaN is critical for driving the reaction. As expected, the reaction did not happen in the dark (Fig. 3d). Collectively, these findings indicate that the reaction proceeds via photocatalysis, and validate the viability of using concentrated light for bio-derived production of syngas from biomass and water.
As illustrated in Fig. 3e, the evolution rate of CO + H2 and the H2/CO ratio were found to be highly related to the glycerol concentration. In pure glycerol, CO and H2 were simultaneously produced at a total rate of 80.4 mmol g−1 h−1. In this case, CO evolution is dominant with a H2/CO ratio of 0.9. Upon incorporation of water, the overall activity could be significantly promoted to 149.3 mmol g−1 h−1. In particular, hydrogen evolution was greatly improved with greatly reduced CO rates when the reaction was conducted in a dilute glycerol aqueous solution of 5%. It is probably due to the highly reactive species of hydroxyl radicals yielded from water dissociation, as characterized by in situ electron paramagnetic resonance spectroscopy (EPR) (Fig. S16†). It was further verified by a control experiment. For example, the activity was clearly inhibited by introducing the hydroxyl radical scavenger of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) (Fig. S17†). It is worth mentioning that even in the absence of glycerol, hydrogen could be produced from water splitting because of the outstanding properties of the AuIn NPs/GaN NWs/Si architecture. Thereby, both glycerol and H2O can contribute to the formation of H2, and CO primarily originating from glycerol. This process has also been demonstrated by experiments with hydrogen isotopes. When the reaction was conducted in a mixture of water (the volume ratio of D2O/H2O is 1/1) and glycerol (5%, weight ratio), the atomic ratio of D/H in the formed hydrogen was estimated to be 0.41, suggesting that H2 originated from both water splitting and glycerol reforming (Fig. S18†). Additionally, the time evolution of syngas was plotted to determine the stability, and over the four-recycling test, the production of syngas did not vary a lot compared to the initial value, suggesting the appreciable stability of the architecture (Fig. 3f and Fig. S19†).
Glycerol reforming process
Considering that glycerol reforming is an extremely complex process involving multiple steps, a series of feedstocks were examined to figure out the first key step (Fig. 4a). Specifically, when 1,3-dihydroxyacetone was used as the starting feedstock, the activity of CO was as high as 872.8 mmol g−1 h−1, which significantly outperformed the CO activity of 9.5 mmol g−1 h−1 from glycerol under the same conditions. Meanwhile, 1,3-dihydroxyacetone showed a significantly low H2 activity of 2.5 mmol g−1 h−1 compared to glycerol. Next, when CH2OHCOOH derived from the C–C cleavage of 1,3-dihydroxyacetone was tested, a relatively high CO activity of 20.9 mmol g−1 h−1 was obtained. With a similar trend to 1,3-dihydroxyacetone, the H2 activity of CH2OHCOOH was much lower than that of glycerol. In contrast, when glycol was used as the feedstock, the evolution rates of H2 and CO were determined to be 330.2 and 74.7 mmol g−1 h−1, respectively, showing a matched trend with glycerol. These results exclude the possibility that syngas is predominantly produced via forming 1,3-dihydroxyacetone. AuIn NPs/GaN NWs prefer to activate the edge functional group of –CH2OH of the glycerol skeleton, followed by the C–C cleavage toward an ethylene glycol-based intermediate. Notably, a comparable amount of CO2 is produced when the reaction is carried out under an air atmosphere, which is in sharp contrast to the result under an argon atmosphere. This finding indicates that Ar is critical to prevent further oxidation of the products, thus facilitating high-efficiency formation of syngas (Fig. S20†). Furthermore, gas chromatography mass spectrometry (GC-MS) was used to detect the liquid products after the reaction. As illustrated in Fig. S21,† the m/z values of 27, 29, 31, 32, 33, 42, 43, 44, and 62 are well matched with the typical signals of ethylene glycol, being consistent with the discussion above. As characterized by GC and GC-MS, it was found that during the process, glycerol could be mainly converted into ethylene glycerol except for the mixture of CO and H2 without evidently forming CO2.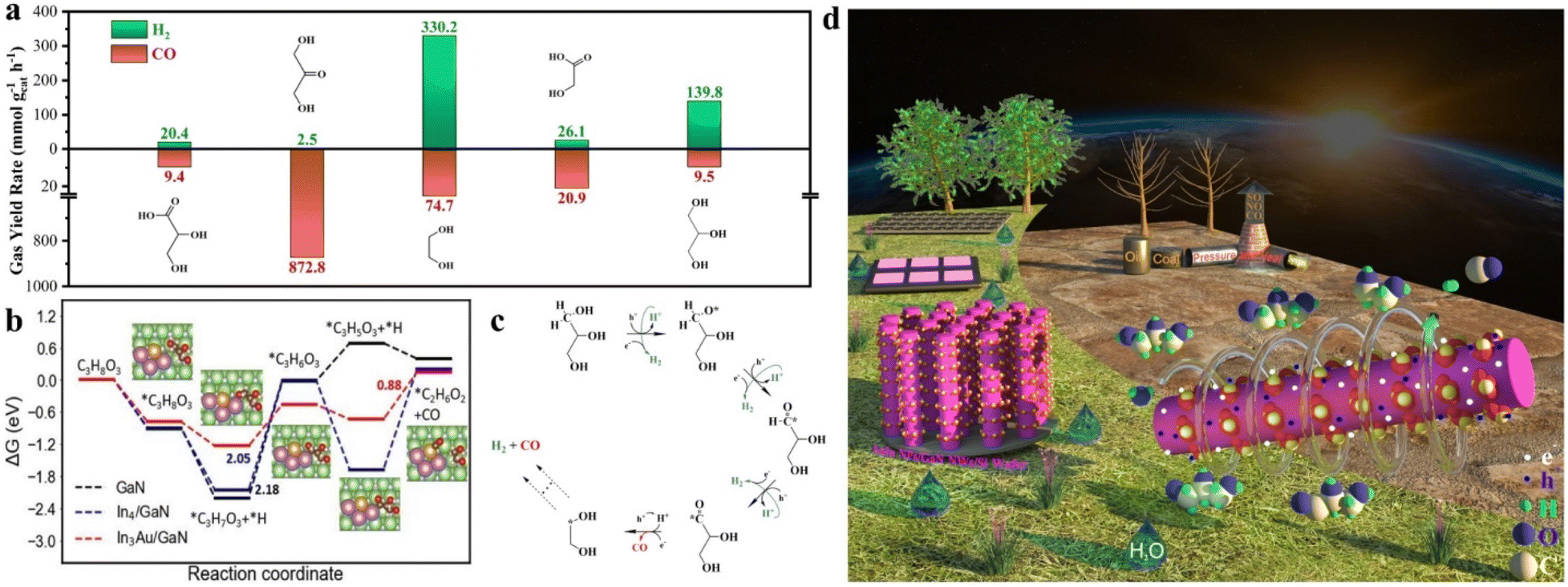 | ||
| Fig. 4 Glycerol reforming process. (a) The performance of several possible C2 and C3 intermediates in the reaction. (b) Calculated free-energy diagram for glycerol reforming toward glycol under solvent effects on GaN, In4/GaN and In3Au/GaN, respectively. The values in the figures indicate the energy barriers for the potential-determining step of the reaction. Ga, green; N, silver; In, purple; Au, gold; C, brown; H, pink; and O, red. (c) Photocatalytic glycerol reforming. (d) Schematic illustration of green syngas production from glycerol, water, and sunlight over AuIn NPs/GaN NWs/Si. | ||
DFT calculations were further performed to gain fundamental insight into the glycerol reforming process under solvent effects at the molecular level (Please see Theoretical calculations in Supporting Information). Fig. 4b demonstrates the free-energy diagram of the lowest-energy pathways of glycerol to ethylene glycol on GaN, In4/GaN and In3Au/GaN. For the case of GaN, the potential-determining step (PDS) is *C3H7O3 → *C3H6O3, with a free-energy change of 2.18 eV. For glycerol on In4/GaN, the PDS remains the same, but with a slightly reduced free-energy change of 2.05 eV. By contrast, the decoration of In3Au reduces the binding of *C3H6O3 and *H intermediates, and thus switches the PDS to *C2H6O2 formation. It also lowers the energy barrier of *C2H6O2 formation with an appreciably low free energy change of 0.88 eV. Therefore, it is expected that the efficiency of glycerol reforming on In3Au/GaN would be highest. The above analysis agrees well with the experimental results.
Based on the experimental and theoretical investigations above, the glycerol reforming process for syngas production is schematically described in Fig. 4c and d. In the photocatalytic cycle, glycerol is first adsorbed on the AuIn/GaN interface. Upon light illumination, the architecture of AuIn NPs/GaN NWs/Si is photoexcited to produce energetic electrons and holes with suitable redox potentials. The edge functional group of –CH2OH of the adsorbed glycerol is subsequently dehydrogenated by photogenerated holes with simultaneously releasing protons. The released protons are facilely reduced toward H2 by the photogenerated electrons. Afterwards, the C–C bond of the glycerol skeleton is broken toward glycol with a greatly reduced energy barrier, with the concurrent formation of CO. Glycol can be further converted into syngas with superior activity. In the presence of water, hydrogen evolution will be significantly enhanced due to the formation of highly active species of hydroxyl radicals. Benefitting from the widespread distribution of biomass, water, and sunlight, this work presents a promising avenue for distributed green syngas production, showing a sustainable alternative to the traditional route built on fossil fuels.
Conclusion
In summary, a novel photocatalytic architecture of AuIn/GaN NWs/Si has been explored using well-developed building blocks. Both experimental and theoretical studies have revealed that GaN functions in synergy with AuIn for adsorbing/activating the glycerol molecule. As a consequence, upon photoexcitation, the monolithic architecture prefers to activate and dehydrogenate the edge functional group of –CH2OH in the glycerol skeleton, followed by the cleavage of the inert C(sp3)–C(sp3) bond to form an ethylene glycol-based intermediate with a greatly reduced reaction energy barrier in glycerol solution. Together with the distinguished optical and electronic properties of the monolithically integrated design, the architecture has demonstrated a superior syngas activity of 149.3 mmol g−1 h−1. Tailoring of a broad range of 19.3 to 0.9 H2/CO ratios can be enabled by engineering the architecture and modulating the experimental conditions, which is highly favorable for producing various downstream products. This work presents a promising avenue for distributed green syngas production with the only inputs of glycerol, water, and sunlight.Author contributions
B. Z. conceived the idea. Z. W. conducted the experiments. B. S., J. Y. and X. W. oversee the growth of the material. Y. C. and J. S. performed the theoretical calculations. S. S., J. L., L. Y., Y. Y., L. Z. and Z. H. participated in the project discussion and result analyses. Z. W., Y. C., J. S. and B. Z. wrote the paper with the contributions from all co-authors. Z. H. and B. Z. led the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from Shanghai Jiao Tong University, the National Natural Science Foundation of China (No. 22109095) and the Shanghai Pilot Program for Basic Research -Shanghai Jiao Tong University (21TQ1400211). B. S., J. Y. and X. W. are thankful for the financial support from the Beijing Outstanding Young Scientist Program (No. BJJWZYJH0120191000103), the Beijing Natural Science Foundation (No. Z200004), and the National Natural Science Foundation of China (No. 61734001). Y. C. and J. S. acknowledge the financial support from the Natural Science and Engineering Research Council of Canada (NSERC) Discovery grant (RGPIN-2017-05187) and the computing resource by Compute Canada.References
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi, Y. Dai, L. Gu, J. Hu, S. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84–87 CrossRef CAS.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS.
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS.
- M. E. Dry, Catal. Today, 2002, 71, 227–241 CrossRef CAS.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Fuel, 2010, 89, 1763–1783 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- J. Jin, L. Wang, M. Fu, X. Li and Y. Lu, Front. Energy, 2020, 14, 71–80 CrossRef.
- S. C. Tsang, J. B. Claridge and M. L. H. Green, Catal. Today, 1995, 23, 3–15 CrossRef CAS.
- R. R. Soares, D. A. Simonetti and J. A. Dumesic, Angew. Chem., Int. Ed., 2006, 45, 3982–3985 CrossRef CAS PubMed.
- M. Wang, M. Liu, J. Lu and F. Wang, Nat. Commun., 2020, 11, 1083 CrossRef CAS PubMed.
- A. Tanaka, K. Hashimoto and H. Kominami, J. Am. Chem. Soc., 2014, 136, 586–589 CrossRef CAS PubMed.
- Z. Zhang, M. Wang, H. Zhou and F. Wang, J. Am. Chem. Soc., 2021, 143, 6533–6541 CrossRef CAS.
- S. Fujita, H. Kawamori, D. Honda, H. Yoshida and M. Arai, Appl. Catal., B, 2016, 181, 818–824 CrossRef CAS.
- R. Su, R. Tiruvalam, A. J. Logsdail, Q. He, C. A. Downing, M. T. Jensen, N. Dimitratos, L. Kesavan, P. P. Wells, R. Bechstein, H. H. Jensen, S. Wendt, C. R. A. Catlow, C. J. Kiely, G. J. Hutchings and F. Besenbacher, ACS Nano, 2014, 8, 3490–3497 CrossRef CAS.
- R. S. Tan, T. A. Tuan Abdullah, A. Johari and K. Md Isa, Front. Energy, 2020, 14, 545–569 CrossRef.
- A. Wadsworth, Z. Hamid, J. Kosco, N. Gasparini and I. McCulloch, Adv. Mater., 2020, 32, 2001763 CrossRef CAS.
- B. Y. Zheng, H. Zhao, A. Manjavacas, M. McClain, P. Nordlander and N. J. Halas, Nat. Commun., 2015, 6, 7797 CrossRef PubMed.
- F. Ambrosio, J. Wiktor, F. D. Angelis and A. Pasquarello, Energy Environ. Sci., 2018, 11, 101–105 RSC.
- J. Zaumseil, C. Groves, J. M. Winfield, N. C. Greenham and H. Sirringhaus, Adv. Funct. Mater., 2008, 18, 3630–3637 CrossRef CAS.
- S. P. Morcillo, Angew. Chem., Int. Ed., 2019, 58, 14044–14054 CrossRef CAS.
- X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed.
- M. G. Kibria and Z. Mi, J. Mater. Chem. A, 2016, 4, 2801–2820 RSC.
- F. A. Chowdhury, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2018, 9, 1707 CrossRef PubMed.
- M. Z. Ertem, N. Kharche, V. S. Batista, M. S. Hybertsen, J. C. Tully and J. T. Muckerman, ACS Catal., 2015, 5, 2317–2323 CrossRef CAS.
- B. AlOtaibi, S. Fan, D. Wang, J. Ye and Z. Mi, ACS Catal., 2015, 5, 5342–5348 CrossRef CAS.
- B. AlOtaibi, X. Kong, S. Vanka, S. Y. Woo, A. Pofelski, F. Oudjedi, S. Fan, M. G. Kibria, G. A. Botton, W. Ji, H. Guo and Z. Mi, ACS Energy Lett., 2016, 1, 246–252 CrossRef CAS.
- L. Li, W. Liu, H. Zeng, X. Mu, G. Cosa, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2015, 137, 8328–8331 CrossRef CAS.
- X. Sun, X. Luo, X. Zhang, J. Xie, S. Jin, H. Wang, X. Zheng, X. Wu and Y. Xie, J. Am. Chem. Soc., 2019, 141, 3797–3801 CrossRef CAS PubMed.
- L. Chai, Z. Hu, X. Wang, Y. Xu, L. Zhang, T.-T. Li, Y. Hu, J. Qian and S. Huang, Adv. Sci., 2020, 7, 1903195 CrossRef CAS PubMed.
- X. Du, F. Dong, Z. Tang and J. Zhang, Appl. Surf. Sci., 2020, 505, 144471 CrossRef CAS.
- Y.-J. Chen, Y.-R. Chen, C.-H. Chiang, K.-L. Tung, T.-K. Yeh and H.-Y. Tuan, Nanoscale, 2019, 11, 3336–3343 RSC.
- G. Hyun, J. T. Song, C. Ahn, Y. Ham, D. Cho, J. Oh and S. Jeon, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5680–5685 CrossRef CAS PubMed.
- A. Wuttig, M. Yaguchi, K. Motobayashi, M. Osawa and Y. Surendranath, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4585–E4593 CrossRef CAS.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS.
- B. Zhou, P. Ou, N. Pant, S. Cheng, S. Vanka, S. Chu, R. T. Rashid, G. Botton, J. Song and Z. Mi, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1330–1338 CrossRef CAS.
- S. Chu, P. Ou, P. Ghamari, S. Vanka, B. Zhou, I. Shih, J. Song and Z. Mi, J. Am. Chem. Soc., 2018, 140, 7869–7877 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- W. J. Dong, I. A. Navid, Y. Xiao, J. W. Lim, J.-L. Lee and Z. Mi, J. Am. Chem. Soc., 2021, 143, 10099–10107 CrossRef CAS.
- B. Zhou, X. Kong, S. Vanka, S. Chu, P. Ghamari, Y. Wang, N. Pant, I. Shih, H. Guo and Z. Mi, Nat. Commun., 2018, 9, 3856 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04107c |
| ‡ These authors contributed equally to this work. |
| § Mr. Z. Wang is currently a visiting student in the School of Mechanical Engineering at Shanghai Jiao Tong University. |
| This journal is © The Royal Society of Chemistry 2023 |