
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbonatation of [ethylene–glycidyl methacrylate]-based copolymers with carbon dioxide as a reagent: from batch to solvent-free reactive extrusion†
Bruno
Guerdener
 abc,
Virgile
Ayzac
a,
Sébastien
Norsic
a,
Paul
Besognet
c,
Véronique
Bounor-Legaré
*b,
Vincent
Monteil
*a,
Véronique
Dufaud
*a,
Jean
Raynaud
*a and
Yvan
Chalamet
*c
abc,
Virgile
Ayzac
a,
Sébastien
Norsic
a,
Paul
Besognet
c,
Véronique
Bounor-Legaré
*b,
Vincent
Monteil
*a,
Véronique
Dufaud
*a,
Jean
Raynaud
*a and
Yvan
Chalamet
*c
aUniv. Lyon, Université Claude Bernard Lyon 1, CPE Lyon, CNRS, UMR 5128, Catalysis, Polymerization, Processes and Materials (CP2M), 43 Bd du 11 novembre 1918, 69616 Villeurbanne cedex, France. E-mail: vincent.monteil@univ-lyon1.fr; veronique.dufaud@univ-lyon1.fr; jean.raynaud@univ-lyon1.fr
bUniv. Lyon, CNRS, Université Claude Bernard Lyon 1, INSA Lyon, Université Jean Monnet, UMR 5223, Ingénierie des Matériaux Polymères, F-69622 Villeurbanne cedex, France. E-mail: veronique.bounor-legare@univ-lyon1.fr
cUniv. Lyon, CNRS, Université Claude Bernard Lyon 1, INSA Lyon, Université Jean Monnet, UMR 5223, Ingénierie des Matériaux Polymères, F-42023 Saint-Etienne cedex 2, France. E-mail: yvan.chalamet@univ-st-etienne.fr
First published on 14th July 2023
Abstract
The carbonatation of semi-crystalline [ethylene–glycidyl methacrylate]-based polymers (Lotader® grades) was achieved using carbon dioxide as a reagent and quaternary ammonium salts as organocatalysts to transform the polymers’ epoxide pendant groups into cyclic carbonate moieties. A batch reactor allowed us to assess the kinetics, dependence on a catalyst and overall potential of this carbonatation. The influence of the ammonium salt composition (anion/cation) was studied in toluene at 110 °C to circumvent the high melting temperatures of these ethylene unit-rich copolymers and obtain a homogeneous medium. The amount of catalyst, CO2 pressure and temperature were also optimized (TBAB, 5 mol% vs. epoxy content, 4.0 MPa, 110 °C) to allow for quantitative conversion of epoxides into cyclic carbonates. Subsequently, the reaction was transposed, for the 1st time, to reactive extrusion under CO2 using a dedicated co-rotating twin-screw extruder to allow for CO2 containment within the polymer melt. This solvent-free reactive process is perfectly adapted to semi-crystalline and/or high-Tg polymers. After optimization, a yield of up to 78% of cyclic carbonate, in addition to orthogonal epoxide, could be obtained with THAB (7.5 mol% vs. epoxy content, ∼30 g h−1 of cat.) at 150 °C with an industry-compliant polymer flow rate of 2 kg h−1. The respective reactivities of Lotader® grades were compared in batch and in an extruder, unveiling this trend towards carbonatation: AX8840 < AX8700 < AX8900. Sustainability and enhanced productivity of the carbonatation methodology developed herein relies on the use of CO2 as a C1 reagent for the functionalization of epoxide-bearing polymers harnessing a continuous and clean reactive extrusion process allowing, in a single operation and a few minutes, the production of functional polymers at the kilogram scale under solvent-free conditions.
Introduction
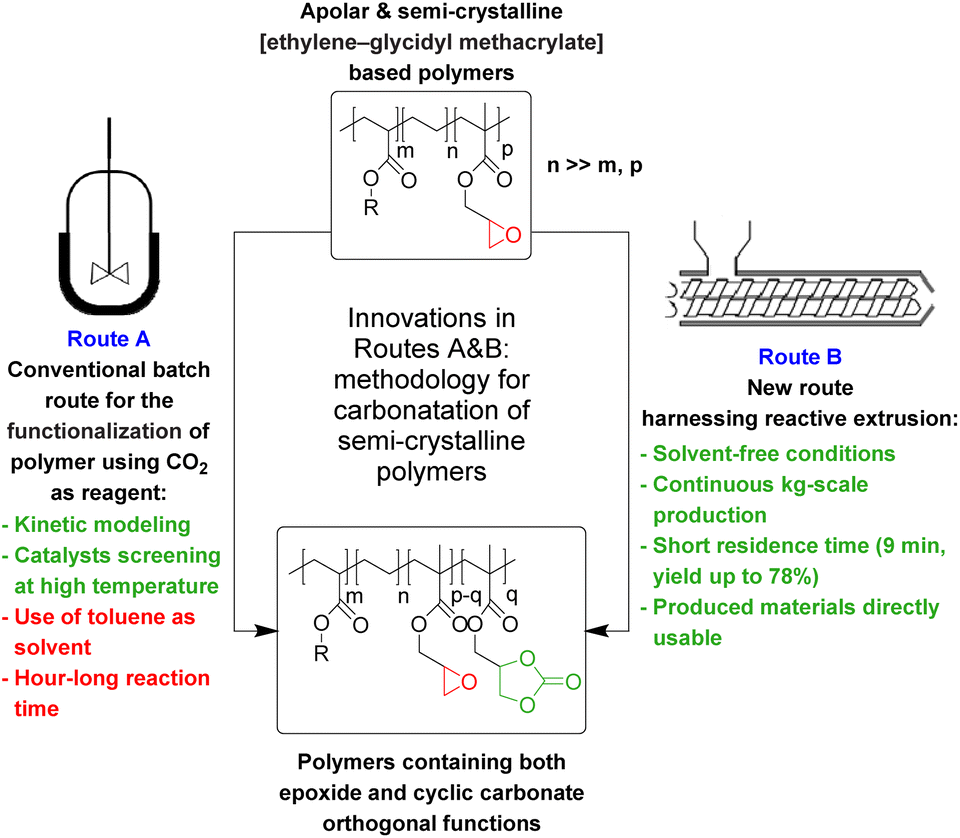
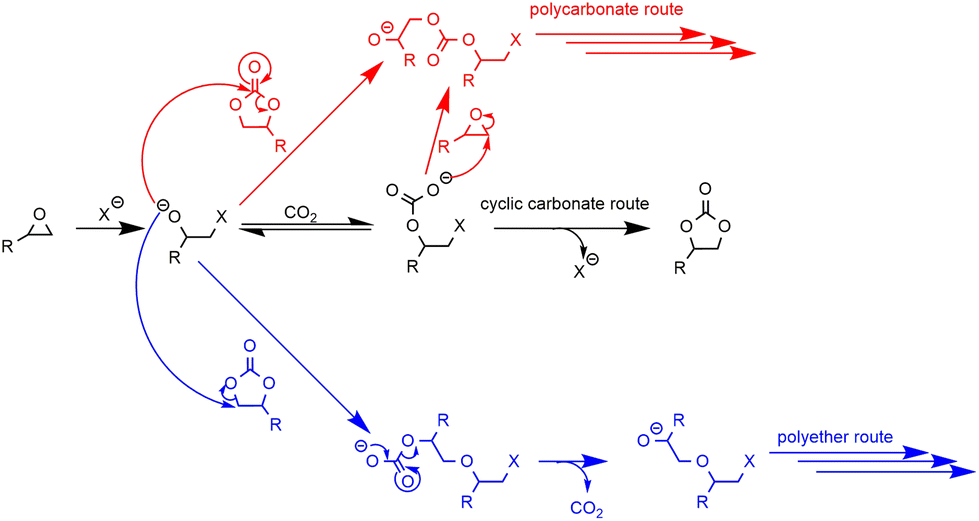
The valorization of CO2 into valuable chemical products has been extensively studied in recent years.1–5 More specifically, the synthesis of carbonates via the reaction of mostly inert CO2 with energy-rich epoxides has been the subject of great interest, due to the multiple industrial applications of cyclic carbonates6–8 and the high atom efficiency of this transformation.9Focusing on CO2 upgrading for the advantageous derivatization of epoxides into cyclic carbonates (cycloaddition) or carbonate linkages (e.g. polymerization), one can indeed manufacture useful industrial platform molecules or benefit from the direct incorporation of CO2 as a C1 building block in high-tonnage materials.10–22 When post-modification of substrates more complex than simple epoxides is considered, i.e., biosourced fatty acids,23–28 bio-based epoxides23,29–32 or synthetic polymers featuring epoxide moieties,33–40 this transformation may be relevant to improve the prospects of usability for (macro)molecules while utilizing abundant CO2. There are two possible routes for the cycloaddition of CO2 to epoxides depending on the nature of the chosen catalyst used. As catalysis is well reported and exemplified in the literature (see Schemes S1, S2 and the description of mechanisms in the ESI†),41–67 it is very easy to select and tune the solubility/reactivity/selectivity of the catalyst system to the needs for specific carbonatation (physical properties, sterics and electronics of the substrates in particular).
Very interestingly, when CO2 is used in post-polymerization as a derivatization agent (see Scheme 1), novel polymers bearing cyclic carbonate functional groups can be created. They have found applications in different sectors. They can be used as solid electrolytes thanks to their good lithium ion conductivity,68 mixed with other polymers to create homogeneous films,35 or can play the role of reactive compatibilizers.69
 | ||
| Scheme 1 Transposition of carbonatation from batch to reactive extrusion. | ||
Moreover, cyclic carbonates and epoxides may show orthogonal reactivity: cyclic carbonates are more prone to react with amines than epoxides to produce, for example, hydroxyurethane functions,11,14,15,20,68 and epoxides are more susceptible to hydrolysis than cyclic carbonates.39
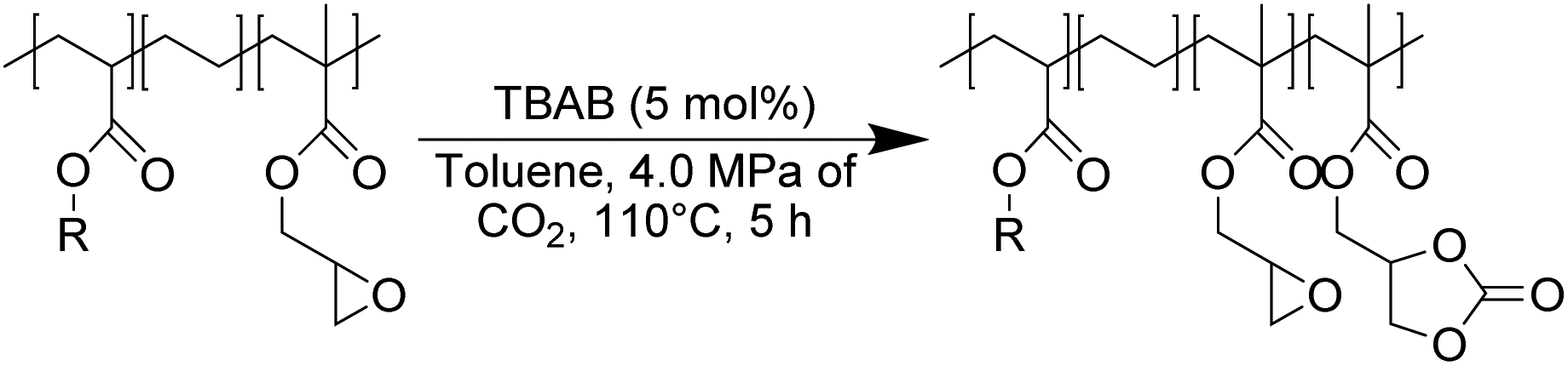
However, in most cases, functionalization with CO2 is carried out on polar polymers highly soluble in polar solvents in a batch reactor.33–40 The chemical fixation of CO2 to an apolar and semi-crystalline polymer bearing epoxide moieties is much more challenging (Scheme 1) because of the difficulties in obtaining an intimate and homogeneous mixture of the polymer, the CO2 reactant and the catalyst.
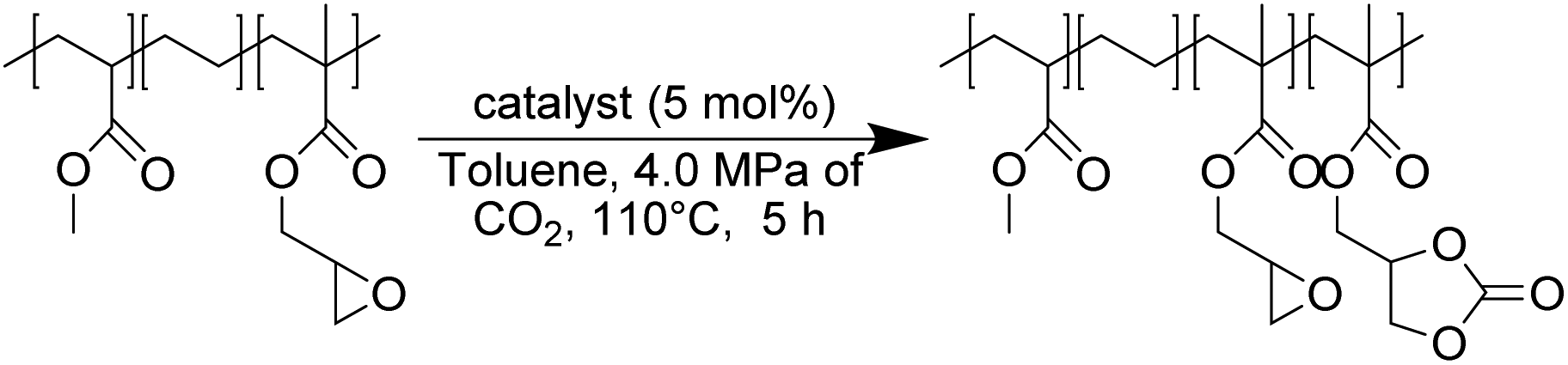
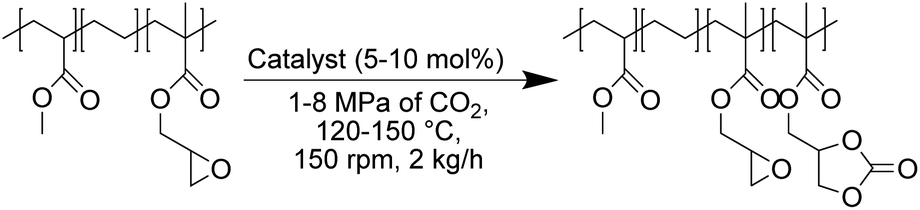
In this work (see Scheme 1), we report on the development and optimization of the conditions for converting the epoxide groups of three different ethylene-containing copolymers, thus mostly apolar copolymers – Lotader® grades AX8900, AX8700 and AX8840 (see Scheme 2 and Scheme S3 in the ESI† for more details) – into the corresponding cyclic carbonates via the organocatalyzed cycloaddition of CO2 (Scheme 3) in batch and by reactive extrusion processes. The reaction conditions were first investigated in batch for a better understanding of the influence of the different reaction parameters, and then transposed to a solvent-free reactive extrusion process in order to develop an original sustainable continuous process for the modification of epoxidized polymers with CO2. This strategy allows for the introduction of a cyclic carbonate moiety in addition to and orthogonal to the remaining epoxides, which could prove valuable for a wide variety of material applications.20,70,71
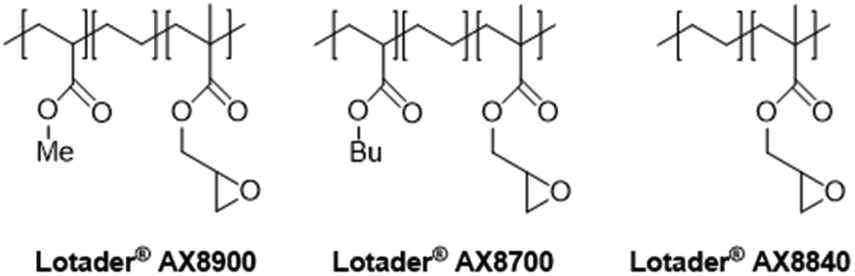 | ||
| Scheme 2 Structure of Lotader® AX8900, AX8700 and AX8840. | ||
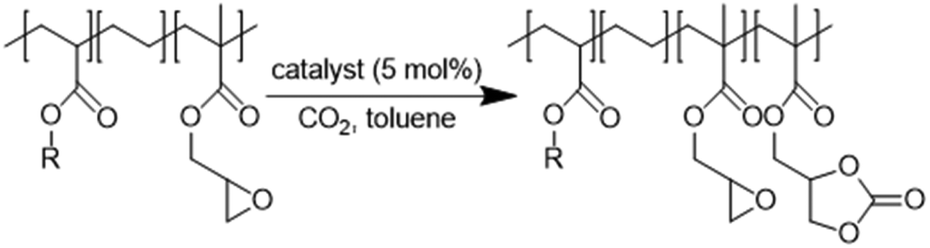 | ||
| Scheme 3 Cycloaddition of CO2 to Lotader®. | ||
We chose to pursue metal-free mediated CO2 fixation methodologies for sustainability and material compatibility considerations and to avoid problematic metallic residues in the final polymer products. Among the organocatalysts developed so far to enable the efficient transformation of molecular epoxides, one can cite onium salts with ammonium or phosphonium halides, ionic liquids and organic bases. Their catalytic behavior towards glycidyl-methacrylate based polymers will be explored in the present work (Scheme 4).
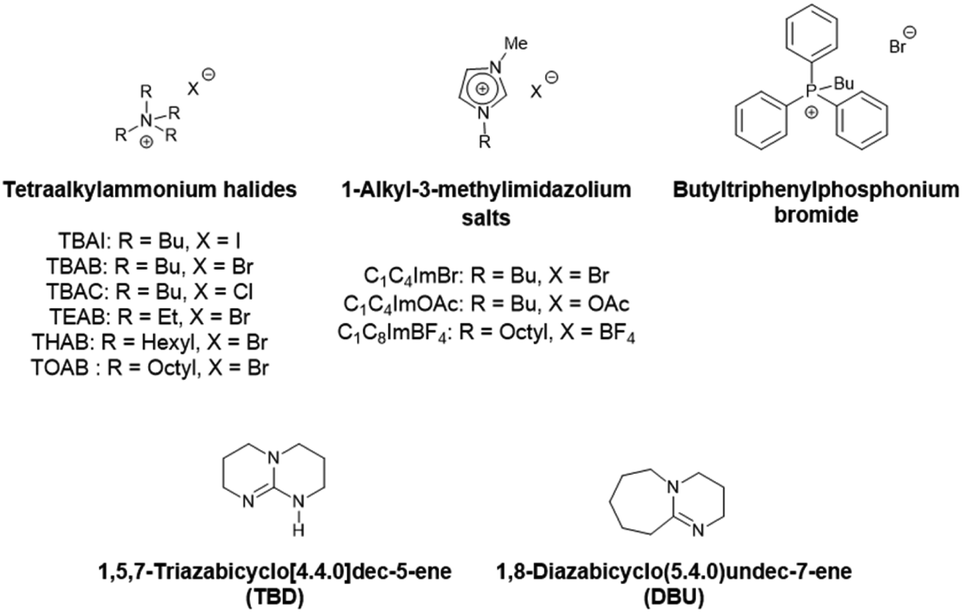 | ||
| Scheme 4 Catalysts studied in this work. | ||
Results and discussion
Screening of the catalysts
First, commercially available organic salts such as tetrabutylammonium bromide (TBAB), butyltriphenylphosphonium bromide (PPh3BuBr),1-butyl-3-methylimidazolium bromide (C1C4ImBr), 1-butyl-3-methylimidazolium acetate (C1C4ImOAc) and 1-methyl-3-octylimidazolium tetrafluoroborate (C1C8ImBF4), and organic bases such as 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were tested as catalysts for the cycloaddition of CO2 to the Lotader® AX8900 under the following conditions: 110 °C, 5 h, 4.0 MPa of CO2 and 5 mol% of catalyst. Toluene was used to solubilize the Lotader® AX8900. We did attempt to realize the transformation under “solvent-free” conditions in our batch reaction; however, very heterogeneous reaction media were obtained, even after 5 h of reaction (see Fig. S33 and Photo S1 of the ESI†), and inconsistent and low conversions in carbonates were achieved. Toluene is a good solvent for Lotader® grades and is easily accessible to most research teams. As shown in Table 1, epoxide activation with halide anions proved to be the best route to achieve satisfactory conversion (Table 1, entries 1–3), whereas only traces of cyclic carbonates were observed when using sterically hindered nucleophiles and bases (Table 1, entries 4–7). TBAB showed the best performance for the carbonatation of Lotader® AX8900 with a quantitative yield of cyclic carbonate (Table 1, entry 1). It should be noted that the imidazolium and phosphonium salts were only partially soluble in toluene at 110 °C unlike TBAB (completely soluble, even at room temperature), which may explain their low reactivity. The formation of the cyclic carbonate product was confirmed by IR-ATR (Fig. 1) and 1H NMR (Fig. 2). After 5 hours of reaction of Lotader® AX8900 with CO2 catalyzed by TBAB, the characteristic band of epoxide at 911 cm−1 disappeared with the concomitant appearance of the band at 1820 cm−1 typical of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of cyclic carbonates72 (Fig. 1). On the other hand, the 1H NMR signals g, h and i related to the glycidyl methacrylate monomers almost disappeared and new signals h′, g′ and i′ corresponding to the carbonate product appeared.
O stretching vibration of cyclic carbonates72 (Fig. 1). On the other hand, the 1H NMR signals g, h and i related to the glycidyl methacrylate monomers almost disappeared and new signals h′, g′ and i′ corresponding to the carbonate product appeared.
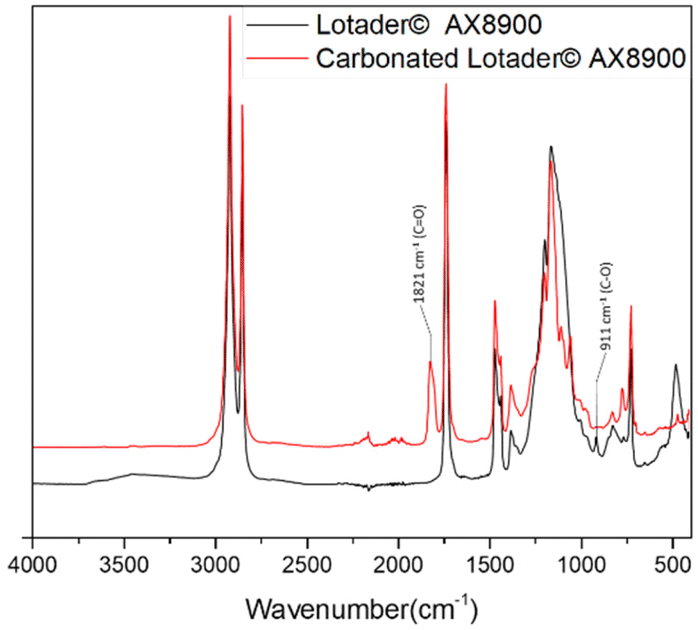 | ||
| Fig. 1 IR-ATR spectra of the Lotader® AX8900 and the modified Lotader® AX8900 with CO2 (Table 1, entry 1). | ||
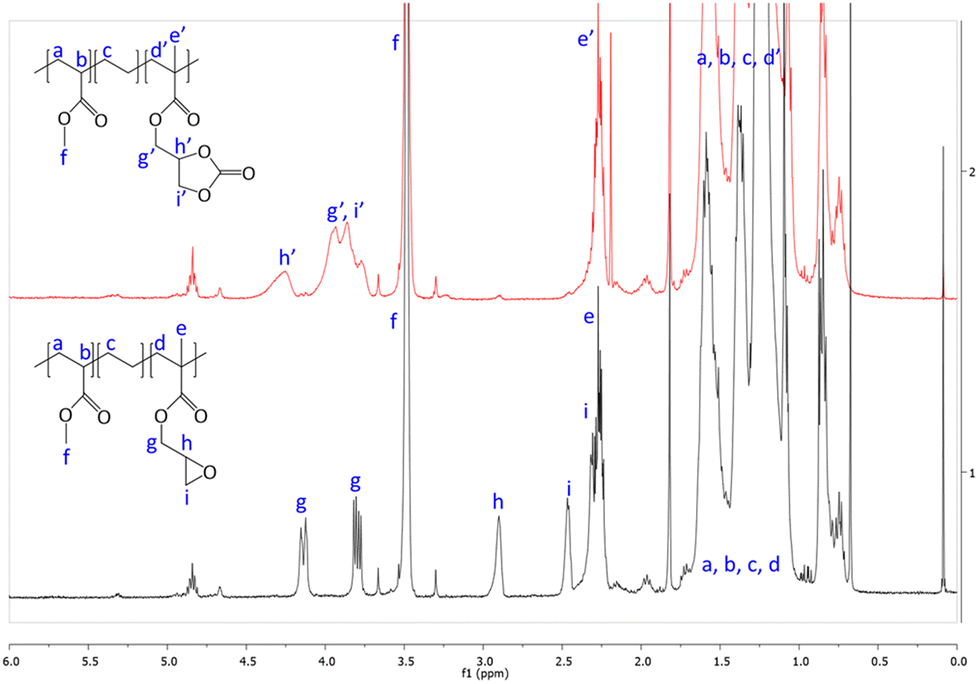 | ||
| Fig. 2 1H NMR (TCE/C6D6, 400 MHz) spectra of Lotader® AX8900 and the modified Lotader® AX8900 with CO2 (Table 1, entry 1) (see also Fig. S2 in the ESI† for further details). | ||
|
|
||
|---|---|---|
| Entry | Catalyst | Yieldb (%) |
| a Conditions: 2.0 g of Lotader® AX8900 (1.1 mmol of glycidyl methacrylate), 0.057 mmol of catalyst (5 mol%), 2.0 mL of toluene, 110 °C, 4.0 MPa of CO2, 5 h. b Yield determined by IR-ATR using a calibration curve (see Fig. S18 in the ESI†). c Yield determined by 1H NMR (see Fig. S20 in the ESI†). | ||
| 1 | TBAB | 99c |
| 2 | PPh3BuBr | 6 |
| 3 | C1C4ImBr | 24 |
| 4 | C1C8ImBF4 | 0 |
| 5 | C1C4ImOAc | 2 |
| 6 | TBD | 1 |
| 7 | DBU | 10 |
Since tetrabutylammonium bromide proved to be the best performing catalyst, other key parameters such as the structural catalyst features and reaction conditions were assessed in the following studies using tetraalkylammonium halide salts with different substituents and counteranions.
Influence of the halide
The influence of the halide anion on the reaction kinetics is a crucial parameter to take into account as it is involved in epoxide ring-opening, the first step of the catalytic cycle. The pseudo-first order kinetics of the reaction was assessed with tetrabutylammonium iodide (TBAI) and TBAB (Fig. 3). Assuming that the concentrations of CO2 and catalyst are constant in the liquid phase over time, the reaction rate could be written as: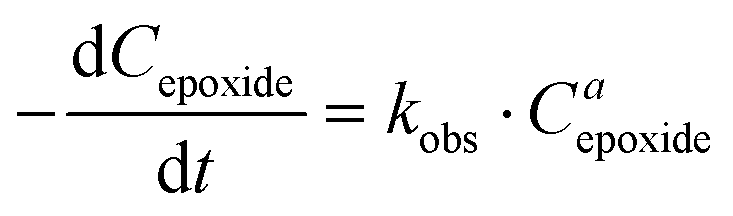 | (1) |
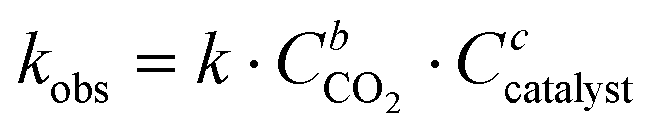 .
.
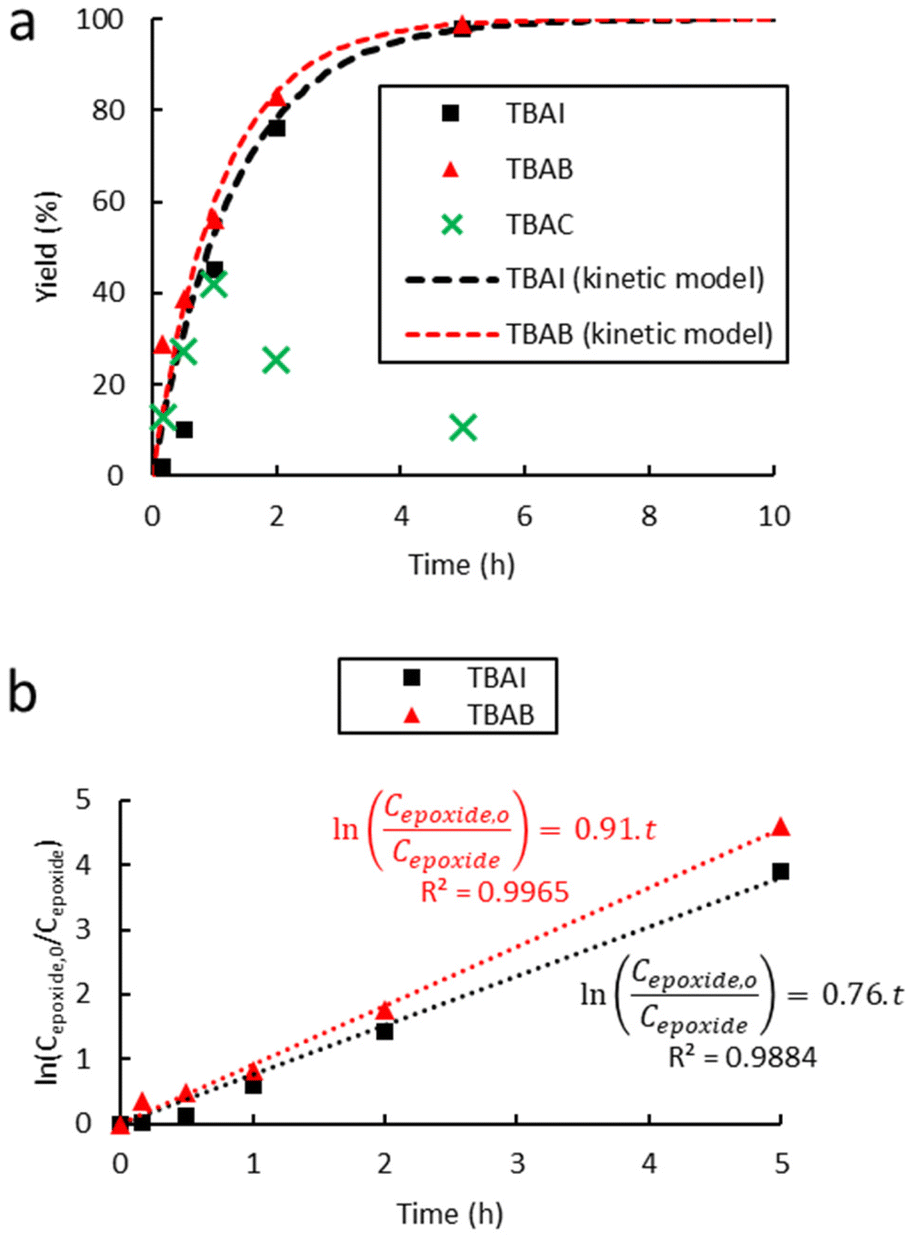 | ||
| Fig. 3 Influence of the halide anion on the reaction kinetics in the cycloaddition of CO2 to Lotader® AX8900: (a) plot of eqn (3) and (b) plot of eqn (2). Conditions: 2.0 g of Lotader® AX8900 (1.1 mmol of glycidyl methacrylate), 0.057 mmol of catalyst (5 mol%), 2.0 mL of toluene, 110 °C, 4.0 MPa of CO2. Yield was determined by IR-ATR using a calibration curve (see Fig. S18 in the ESI†). | ||
If the reaction follows the pseudo-first order kinetics, the differential equations can be integrated, and kobs can be estimated as:
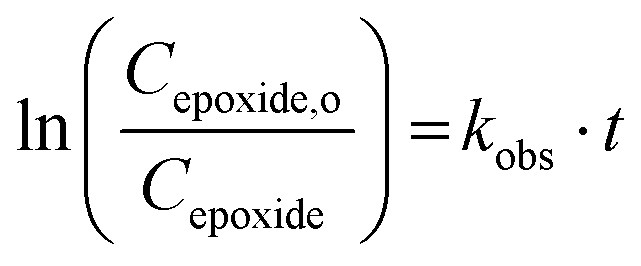 | (2) |
As no side-reaction was observed with TBAI and TBAB, the yield of the cyclic carbonate product is equal to the conversion of the epoxide moiety and can be expressed as follows:
| Yield = 1 − e−kobs·t. | (3) |
Fig. 3 shows the plots of eqn (2) and (3). From these data the rate constant kobs was estimated to be 0.76 h−1 and 0.91 h−1 for the reaction catalyzed with respectively TBAI and TBAB, suggesting that Br− has a higher catalytic activity than I−. In the case of the carbonatation of terminal molecular epoxides under neat conditions or in apolar solvents such as toluene, the order of activity follows the order of halide nucleophilicity and leaving group ability: I− > Br− > Cl− > F−.41,45,53,64,65,73–75 But in the case of the carbonatation of sterically hindered terminal epoxides, a good balance between the halide size and its leaving group ability is required.72,75 A too voluminous and polarizable halide would not favor the attack of the halide on the epoxide, which could explain why I− is less active than Br−.
In the case of tetrabutylammonium chloride (TBAC), the consumption of the formed cyclic carbonates was observed after 1 h of reaction at 110 °C (Fig. 3(a)), which could be due to a secondary reaction, leading to a polymer less or even non-soluble in toluene, likely due to crosslinking, compared to the modified polymers obtained with TBAI and TBAB. Ochiai et al. observed a similar behaviour with CO2 fixation to poly(glycidyl methacrylate-co-methyl methacrylate) catalyzed by tetrabutylammonium fluoride (TBAF) and suggested that polymerization of the sole epoxide to polyethers was favored due to the weak leaving group ability of the fluoride anion.36 Once enough cyclic carbonate species are generated, a direct nucleophilic attack of the alkoxide on the carbonate ring with subsequent decarboxylation could yield several polyether repeating units leading to some insoluble material, which was also observed in our case (see Scheme 5 and the CPMAS-NMR spectra of Fig. S24 in the ESI†).
 | ||
| Scheme 5 Potential side-reaction of epoxide with CO2. [See also ref. 36]. | ||
Influence of the alkyl chains
Ammonium bromide salts such as tetraethylammonium bromide (TEAB), TBAB, tetrahexylammonium bromide (THAB) and tetraoctylammonium bromide (TOAB) were chosen to study the influence of the alkyl chains’ length. As shown in Fig. 4, one can observe that the length does influence the catalytic activity. After 1 h of reaction at 110 °C, the following catalytic reactivity order was obtained: TEAB ≪ TOAB ≈ TBAB < THAB. These results showed that increasing the length of the alkyl chains, while improving solubility in the apolar medium, makes the ammonium cation bulkier and thus decreases the electrostatic interactions between the ion pairs. This results in greater nucleophilicity of the anion and hence enhanced reactivity.45 While the larger ammonium cation showed satisfactory activity at a short reaction time (more than 50% yield after 1 hour), TEAB was completely inactive due to its insolubility in toluene. It has also been shown that a too bulky cation should disfavor the formation of the alkoxide intermediate,72 explaining why TOAB was less active than THAB.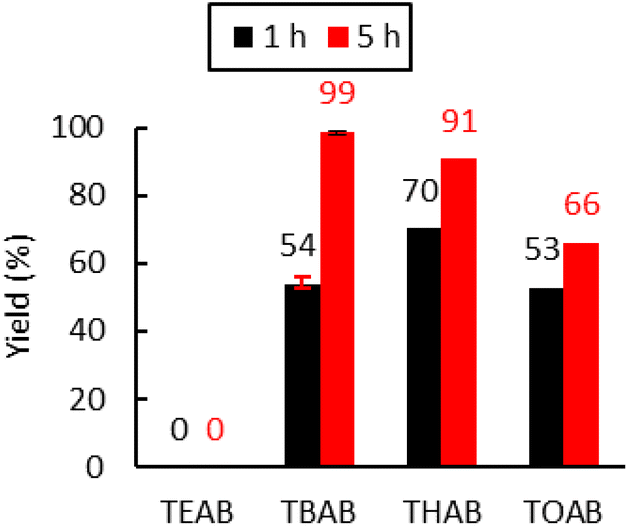 | ||
| Fig. 4 Influence of the alkyl chains on the tetraalkylammonium bromide activity. Conditions: 2.0 g of Lotader® AX8900 (1.1 mmol of glycidyl methacrylate), 0.057 mmol of catalyst (5 mol%), 2.0 mL of toluene, 110 °C, 4.0 MPa of CO2. Yield determined by IR-ATR using a calibration curve (see Fig. S18 in the ESI†). | ||
However, after 5 h at 110 °C, the catalytic reactivity order changed as follows: TEAB ≪ TOAB < THAB < TBAB. This could be explained by the fact that the bulkier the ammonium, the less stabilized the alkoxide intermediate, because of the decrease of the electrostatic interactions between the ion pairs. It would favor parallel side reactions between the alkoxides and the cyclic carbonates to produce polyether units via decarboxylation reactions, as reported before (see Scheme 5 and the CPMAS-NMR spectra of Fig. S24†).36 The overall enhanced solubility of the cation and ion pair dissociation could thus be a determining factor for improved kinetics at early stages, but a trade-off in terms of cation size would be ideal when it comes to selectivity over longer reaction times (see Fig. 4).
Influence of the reaction parameters
The dependence of the cyclic carbonate yield on the CO2 pressure was investigated at 110 °C for a period of 1 h using TBAI as a catalyst. As shown in Fig. 5(a), increasing the pressure from 2 to 4 MPa led to an increase of the yield from 15% to 45%. This could be due to a higher concentration of CO2 in the liquid phase, which may both increase the kinetic rate and reduce the viscosity of the reaction mixture via plasticization of the polymer, as already reported for other polymers.76,77 Beyond 4 MPa, the yield reaches a plateau and starts to slightly decrease from 10 MPa. This decrease in the yield with an increase of CO2 pressure has previously been observed in the case of molecular epoxides,43,45 with the supercritical state of CO2 affecting the solubility of each component in the continuous phase through dilution or even forcing precipitation in our case.78 Thus, a pressure of 4 MPa seems ideal for TBAI to operate efficiently. The influence of the amount of TBAI on catalytic performance was also studied (4.0 MPa of CO2, 110 °C, 1 h). The results in Fig. 5(b) show that increasing the concentration of TBAI from 1 to 5 mol% led to a marked improvement of the yield from 13 to 45%. A further increase of the quantity of TBAI up to 15 mol% did not lead to further activity improvement. Thus, the TBAI catalyst concentration was set at 5 mol%.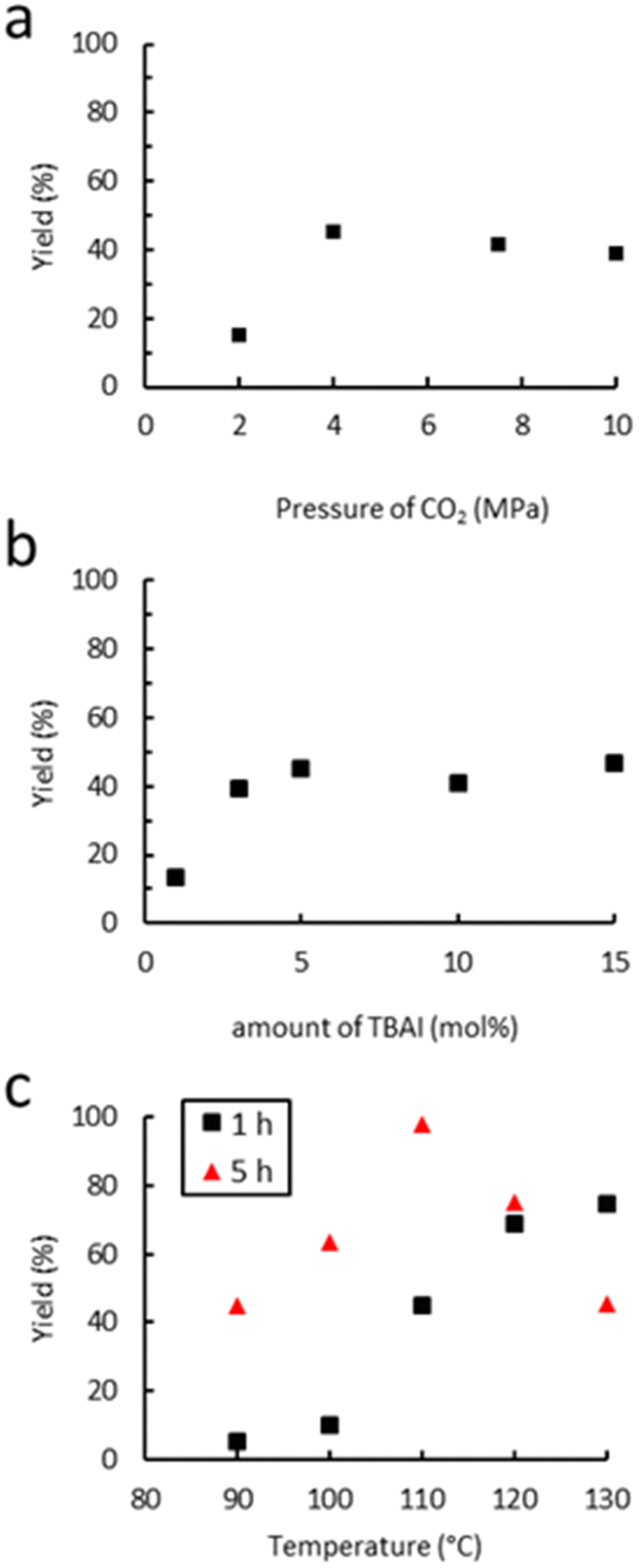 | ||
| Fig. 5 Influence of (a) the CO2 pressure, (b) amount of TBAI and (c) temperature on catalytic performance. Unless otherwise mentioned, reaction conditions: 2.0 g of Lotader® AX8900 (1.1 mmol of glycidyl methacrylate), 0.057 mmol of TBAI (5 mol%), 4.0 MPa of CO2, 2.0 mL of toluene, 110 °C, 1 h. Yield determined by IR-ATR using a calibration curve (see Fig. S18 in the ESI†). | ||
Finally, the influence of the temperature on the carbonatation reaction of Lotader® AX8900 was studied (4.0 MPa of CO2, 5 mol% TBAI) as it could greatly impact both the reaction rate and the solubility/viscosity of the reaction medium particularly when polymers are at stake. The results shown in Fig. 5(c) reveal that the reaction temperature strongly affects catalyst reactivity as very low yields were obtained after 1 hour of reaction below 100 °C. Increasing the temperature further from 100 to 120 °C led to a significant improvement in the yield of the cyclic carbonate product from 5 to 65%. A further increase of the temperature up to 130 °C did not lead to significant activity improvement. As stated above, the temperature may influence the kinetic rate and improve the gas–liquid transfer but partial or total degradation of the catalyst may also occur with the temperature increase. However, after 5 hours, an increase of the temperature from 110 °C to 130 °C led to a decrease of the yield from 99% to 45%. It could be explained by the formation of polyether repeating units and thus the consumption of the cyclic carbonate moiety via decarboxylation, as reported previously.36 Therefore, the influence of the acrylate comonomer in the polymer structure on the reaction efficiency was assessed under the following optimal conditions: TBAB (5 mol%), 4 MPa of CO2, 110 °C, 5 h.
Influence of the acrylate comonomer
The Lotader® AX8900, AX8700 and AX8840 were functionalized with CO2 under optimal conditions to study the influence of the acrylate comonomer (type or presence) on the reaction efficiency. It is notable that the three polymers are soluble in toluene at 110 °C at a concentration of 1 g of polymer per mL of toluene.Under these conditions, the factors that could influence the reaction outcome are the polarity of the polymer, the molar mass of the Lotader®, the CO2 solubility in the liquid phase and the viscosity of the liquid phase. The results presented in Table 2 show that the reactivity of the polymer increases in the following order: AX8840 < AX8700 < AX8900, which also mirrors the polarity order. AX8700 is more apolar than AX8900 because of the presence of butyl groups instead of methyl groups, and AX8840 is even more apolar because of the absence of the acrylate comonomer. This change of polarity may affect the CO2 solubility in the liquid phase. It is well known that CO2 is more soluble in relative polar media, due to its significant quadrupole moment.79 The ester groups of the acrylate comonomer may interact with CO2 and favor its solubility, thus favoring the reaction with the epoxide moieties.
|
|
|||
|---|---|---|---|
| Entry | R | Polymer | Yield (%) |
| Reaction conditions: 2.0 g of polymer (1.1 mmol of glycidyl methacrylate), 18 mg of TBAB (0.057 mmol, 5 mol%), 4.0 MPa of CO2, 2.0 mL of toluene, 110 °C, 5 h. Yield determined by 1H NMR (see Fig. S20, S21 and S22 in the ESI†). | |||
| 1 | Me | Lotader® AX8900 | 99 |
| 2 | Bu | Lotader® AX8700 | 82 |
| 3 | — | Lotader® AX8840 | 62 |
Methodology transposition to reactive extrusion
The aforementioned autoclave results showed that ammonium halides were the most active catalysts for the carbonatation of Lotader® in toluene solution. Specifically, the use of TBAB, THAB and TBAI as catalysts provided the highest yields of cyclic carbonates, up to 99% after 5 hours of reaction at 110 °C in the case of TBAB. These catalysts were therefore chosen for the carbonatation of Lotader® by reactive extrusion. Due to its flexibility in screw design and easily adjustable temperature profiles, the extrusion process has remarkable mixing and homogenization capabilities for viscous systems in the molten state. Thus, the use of an extruder as a continuous reactor makes it possible to avoid resorting to hazardous solvents, which are always difficult to extract industrially (costly devolatilization step). The reactive extrusion process is thus often considered as a promising green process, allowing for the continuous production of large quantities of modified polymers.The use of CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 79 as a reagent for the carbonatation of polymers by a reactive extrusion process80,81 has never been reported before. In contrast to radical processes, catalytic processes within reactive extruders are also extremely rare to the best of our knowledge, in particular in the realm of polymer functionalization.82,83
79 as a reagent for the carbonatation of polymers by a reactive extrusion process80,81 has never been reported before. In contrast to radical processes, catalytic processes within reactive extruders are also extremely rare to the best of our knowledge, in particular in the realm of polymer functionalization.82,83
This constitutes a significant step forward since it both offers a solvent-free alternative and a scalable process amenable to industrial eco-design requirements. The transposition of the batch process to reactive extrusion was possible thanks to the adjustments of the batch reaction conditions. A co-rotating twin-screw extruder fed with the Lotader®, the catalyst and CO2 was used to perform the experiments, at a flow rate of 2 kg h−1 and a rotational speed of 150 rpm. The overall design of this process is depicted in Scheme S4 (ESI†): it provides containment of CO2 pressure within the polymer melt for carbonatation to proceed (see also Photo S2 in the ESI†).
The minimal temperature for the extrusion of the different Lotader® was determined experimentally to be 120 °C (over the melting points of the Lotader® AX8900, AX8700 and AX8840, which are respectively 65 °C, 72 °C and 104 °C).
Furthermore, the different twin-screw extruder parameters had to be optimized to obtain good yields, as a very short average residence time of 9 minutes has been precisely determined via colorimetric measurements, compared to the hour-long reaction times in the batch process.
First, the influence of extrusion temperature and CO2 pressure was studied on the carbonatation of Lotader® AX8900 (Scheme 6). As shown in Fig. 6(a), an increase in temperature led to an increase in carbonate yield. The optimum pressure was 4.0 MPa, which is probably a good compromise between maximal CO2 solubilization/availability for reaction and the avoidance of phase separation in the melt. At this pressure, an increase of 10 °C led to an average yield increase of 15%.
 | ||
| Scheme 6 Carbonatation of Lotader® AX8900 in an extruder. | ||
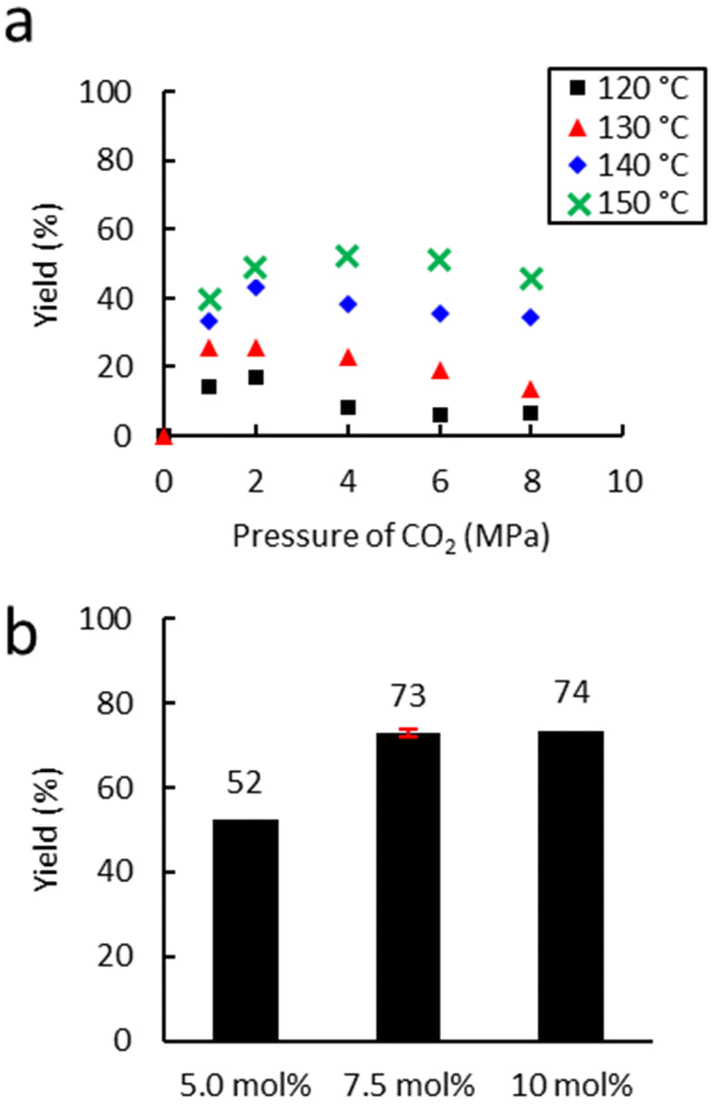 | ||
| Fig. 6 Influence of (a) temperature and (b) the amount of catalyst. Unless otherwise mentioned, reaction conditions: 2 kg h−1 of Lotader® AX8900, 18 g h−1 of TBAB (5 mol%), 150 °C, 150 rpm. Yield determined by IR-ATR using a calibration curve (see Fig. S18 ESI†). | ||
The temperature was not increased above 150 °C to avoid catalyst degradation that starts from 160 °C according to TGA analyses (see Fig. S61 and S62 in the ESI†).
Moreover, the amount of catalyst was optimized (Fig. 6(b)) with 4.0 MPa of CO2. An increase from 5.0 to 7.5 mol% led to a significant increase in yield from 53% to 72%. A further increase to 10 mol% did not lead to a significant improvement in the yield. Subsequently, the influence of the nature of the catalyst was investigated under optimal conditions (7.5 mol% catalyst, 150 °C, 150 rpm, 2 kg h−1) (Fig. 7). With TBAI, a low yield of 33% was obtained. This could be explained by the fact that the melting temperature of TBAI is 155 °C, thus it is in solid form during extrusion. The catalyst would therefore be less homogeneously dispersed within the polymer matrix, which would reduce the cyclic carbonate yield. Regarding TBAB and THAB, a similar activity to that in the batch reactor was obtained, with a yield of 72% and 78% respectively. With their melting point being at about 100 °C, their liquid state under the reaction conditions may improve their dispersion in the medium and not affect their activity.
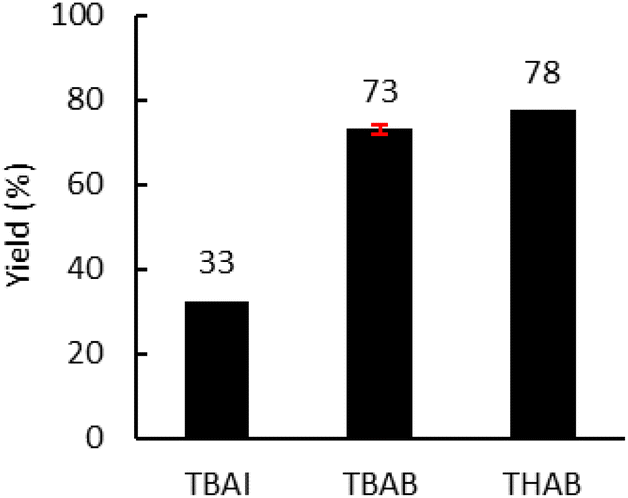 | ||
| Fig. 7 Influence of the nature of the catalyst. Unless otherwise mentioned, reaction conditions: 2 kg h−1 of Lotader® AX8900, 0.086 mol h−1 of catalyst (7.5 mol%), 150 °C, 4 MPa of CO2, 150 rpm. Yield determined by IR-ATR using a calibration curve (see Fig. S18 in the ESI†). | ||
Finally, the Lotader® AX8700 and AX8840 were also modified with CO2 in reactive extrusion under optimal conditions (TBAB (7.5 mol%), 150 °C, 150 rpm, 2 kg h−1) (Fig. 8). The same reactivity order as that obtained in batch was observed: AX8840 ≪ AX8700 < AX8840. Interestingly, as previously observed for the batch process, the integrity of the polymer chains is retained throughout the process, and even more so using the reactive extrusion process (high T °C but a low residence time vs. long shear stress stirring) as evidenced by the polymer characteristics displayed in Table S2 (ESI†).
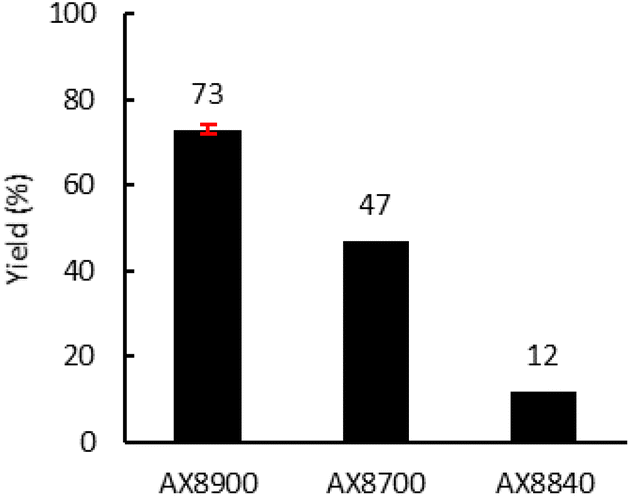 | ||
| Fig. 8 Influence of the acrylate monomer. Reaction conditions: 2 kg h−1 of Lotader®, 0.086 mol h−1 of TBAB (7.5 mol%, 27 g h−1 of cat.), 150 °C, 4 MPa of CO2, 150 rpm. Yield determined by 1H NMR (see Fig. S44, S45 and S46 in the ESI†). | ||
Conclusions
Ethylene–glycidyl methacrylate based copolymers were successfully functionalized with carbon dioxide using commercial and non-toxic tetraalkylammonium halide catalysts, leading to the formation of cyclic carbonate pendant groups on the polymer chain, in a batch reactor and an extruder. The influences of the catalyst structure and the reaction parameters were evaluated in batch and it was found that TBAI, THAB and TBAB were the most active catalysts, with TBAB achieving up to 99% yield of cyclic carbonate.The difference in reactivity between Lotader® AX8900, AX8700 and AX8840 suggested that the presence of polar groups, such as esters derived from acrylate comonomers, in the polymer structure may increase the CO2 solubility and facilitate its approach to the epoxide moieties.
The procedure developed in an autoclave has been successfully transposed to a solvent-free reactive extrusion process, with a yield of cyclic carbonate up to 78% using THAB as the catalyst on the Lotader® AX8900, allowing access to cyclic carbonates in addition to the remaining epoxides. Modifications of AX8700 and AX8840 were also successful. These modified [cyclic carbonate + epoxide]-featuring polymers may have several direct applications e.g. as intermediates for the synthesis of polyhydroxyurethanes, as reactive compatibilizers for blends of polyolefins with polyamides, polyesters, polyacids and polycarbonates, and could also display good permeability properties for membranes. The enhanced sustainability of our reactive extrusion process (kg-scale production of functionalized polymers in a continuous fashion under solvent-free conditions using CO2 as a direct C1 source) could pave the way for other industrially-relevant (co)polymers.
Experimental part
Materials and equipment
Lotader® AX8900, AX8840 and AX8700 were purchased from Arkema. CO2 was purchased from Air Liquide and the purity was 99.995%. For the batch study, 1-butyl-3-methylimidazolium acetate was purchased from Strem Chemicals. Toluene and all the other catalysts were supplied by Sigma Aldrich. For the reactive extrusion optimization, TBAB and TBAI were purchased from TCI Chemicals and THAB was purchased from Sigma Aldrich. All the chemicals were used without further purification.All batch reactions were performed in a 0.16 L stainless steel reactor.
The reactive extrusion experiments were conducted in a twin-screw extruder, a BC 21 (L/D = 36, screw diameter = 25 mm) from Clextral, using a co-rotative configuration. The extruder had 9 barrels, numbered consecutively from 1 to 9 from the feed port to the die. The die had a diameter of 3 mm and a length of 10 mm. The extruder had 9 heating zones.
Analytical methods
1H NMR spectra were recorded using a Bruker 400 Avance III, and 13C NMR spectra were recorded using a Bruker Avance II. A TCE/C6D6 (2/1) (v/v) mixture was used as a deuterated solvent. All NMR analyses were performed at 363 K. All chemical shifts were measured relative to residual 1H or 13C resonance in the deuterated solvent: 7.16 ppm for 1H and 128.06 ppm for 13C.IR-ATR measurements were performed at room temperature on Nicolet i550 FT-IR apparatus on the native and modified polymers. Characteristic bonds are cyclic carbonate CO (1820–1822 cm−1), ester CO (1734 cm−1) and epoxide C–O (911 cm−1).
General procedures (typical carbonatation processes; more details in the ESI†)
Excellent reproducibility was observed for both techniques, providing conversions within the 2–3% range of deviation between repeated experiments. This is likely due to excellent mixing both for the toluene-assisted batch process (see in particular Fig. S19, ESI†) and for the reactive extrusion (intimate mixing of the polymer melt under CO2 pressure), providing homogenised reaction media (see Fig. 6, 7 & 8 for TBAB as a catalyst: small error bars).
Author contributions
All of the experiments were conducted by B. G. and V. A., and processes were supervised by S. N. and P. B. All the authors contributed to the preparation of this manuscript. Conceptualization: V. B.-L., V. D., Y. C., V. M. and J. R.; writing-original draft, review, and editing: B. G., V. B.-L., V. D., Y. C., V. M. and J. R.Conflicts of interest
The authors are also the inventors of a French patent application deposited under the number FR2300220 and not yet published.Acknowledgements
The authors would like to thank the Institut Carnot Ingénierie@Lyon for financial support, with in particular the PhD fellowship of BG and post-doctoral contract of VA, Axel'One Campus for the NMR analyses and “La plateforme lyonnaise de caractérisation des Polymères” for the SEC analyses.References
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- X. Lim, Nature, 2015, 526, 628–630 CrossRef CAS PubMed.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- Y. Du, F. Cai, D.-L. Kong and L.-N. He, Green Chem., 2005, 7, 518 RSC.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Green Chem., 2010, 12, 2205 RSC.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed.
- L. Maisonneuve, A.-L. Wirotius, C. Alfos, E. Grau and H. Cramail, Polym. Chem., 2014, 5, 6142–6147 RSC.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- N. Kindermann, À. Cristòfol and A. W. Kleij, ACS Catal., 2017, 7, 3860–3863 CrossRef CAS.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- A. C. Deacy, E. Moreby, A. Phanopoulos and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 19150–19160 CrossRef CAS PubMed.
- S.-E. Dechent, A. W. Kleij and G. A. Luinstra, Green Chem., 2020, 22, 969–978 RSC.
- A. C. Deacy, G. L. Gregory, G. S. Sulley, T. T. D. Chen and C. K. Williams, J. Am. Chem. Soc., 2021, 143, 10021–10040 CrossRef CAS PubMed.
- X. Liu, X. Yang, S. Wang, S. Wang, Z. Wang, S. Liu, X. Xu, H. Liu and Z. Song, ACS Sustainable Chem. Eng., 2021, 9, 4175–4184 CrossRef CAS.
- L. Longwitz, J. Steinbauer, A. Spannenberg and T. Werner, ACS Catal., 2018, 8, 665–672 CrossRef CAS.
- N. Tenhumberg, H. Büttner, B. Schäffner, D. Kruse, M. Blumenstein and T. Werner, Green Chem., 2016, 18, 3775–3788 RSC.
- F. Chen, Q.-C. Zhang, D. Wei, Q. Bu, B. Dai and N. Liu, J. Org. Chem., 2019, 84, 11407–11416 CrossRef CAS PubMed.
- W. Liu, G. Lu, B. Xiao and C. Xie, RSC Adv., 2018, 8, 30860–30867 RSC.
- A. Akhdar, K. Onida, N. D. Vu, K. Grollier, S. Norsic, C. Boisson, F. D'Agosto and N. Duguet, Adv. Sustainable Syst., 2021, 5, 2000218 CrossRef CAS.
- J. Catalá, M. P. Caballero, F. de la Cruz-Martínez, J. Tejeda, J. A. Castro-Osma, A. Lara-Sánchez, J. M. García-Vargas, M. T. García, M. J. Ramos, I. Gracia and J. F. Rodríguez, J. CO2 Util., 2022, 61, 102060 CrossRef.
- H. Morikawa, M. Minamoto, Y. Gorou, J. Yamaguchi, H. Morinaga and S. Motokucho, Bull. Chem. Soc. Jpn., 2018, 91, 92–94 CrossRef CAS.
- A. Rehman, A. M. López Fernández, M. F. M. Gunam Resul and A. Harvey, J. CO2 Util., 2019, 29, 126–133 CrossRef CAS.
- F. de la Cruz-Martínez, M. Martínez de Sarasa Buchaca, J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Rodríguez-Diéguez, J. A. Castro-Osma and A. Lara-Sánchez, ACS Sustainable Chem. Eng., 2019, 7, 20126–20138 CrossRef.
- M. Navarro, L. F. Sánchez-Barba, A. Garcés, J. Fernández-Baeza, I. Fernández, A. Lara-Sánchez and A. M. Rodríguez, Catal. Sci. Technol., 2020, 10, 3265–3278 RSC.
- N. Kihara and T. Endo, Macromolecules, 1992, 25, 4824–4825 CrossRef CAS.
- J.-Y. Moon, H.-J. Jang, K.-H. Kim, S.-E. Na, D.-W. Park and J.-K. Lee, Korean J. Chem. Eng., 1999, 16, 721–724 CrossRef CAS.
- S. Y. Park, H. Y. Park, D. W. Park and C. S. Ha, J. Macromol. Sci., Part A: Pure Appl.Chem., 2002, 39, 573–589 CrossRef.
- B. Ochiai, T. Iwamoto, K. Miyazaki and T. Endo, Macromolecules, 2005, 38, 9939–9943 CrossRef CAS.
- S. Park, B. Choi, B. Lee, D. Park and S. Kim, Sep. Sci. Technol., 2006, 41, 829–839 CrossRef CAS.
- B. Ochiai, Y. Hatano and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3170–3176 CrossRef CAS.
- B. Ochiai and T. Nakayama, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5382–5390 CrossRef CAS.
- B. Liu, Y.-Y. Zhang, X.-H. Zhang, B.-Y. Du and Z.-Q. Fan, Polym. Chem., 2016, 7, 3731–3739 RSC.
- V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef PubMed.
- H. Kim, J. Catal., 2003, 220, 44–46 CrossRef CAS.
- H. Kawanami, A. Sasaki, K. Matsui and Y. Ikushima, Chem. Commun., 2003, 896–897 RSC.
- D. J. Darensbourg, S. J. Lewis, J. L. Rodgers and J. C. Yarbrough, Inorg. Chem., 2003, 42, 581–589 CrossRef CAS PubMed.
- J. Sun, S.-I. Fujita, F. Zhao and M. Arai, Appl. Catal., A, 2005, 287, 221–226 CrossRef CAS.
- H. Sugimoto and K. Kuroda, Macromolecules, 2008, 41, 312–317 CrossRef CAS.
- A. Sibaouih, P. Ryan, M. Leskelä, B. Rieger and T. Repo, Appl. Catal., A, 2009, 365, 194–198 CrossRef CAS.
- T. Seki, J.-D. Grunwaldt and A. Baiker, J. Phys. Chem. B, 2009, 113, 114–122 CrossRef CAS PubMed.
- T. Chang, L. Jin and H. Jing, ChemCatChem, 2009, 1, 379–383 CrossRef CAS.
- D. J. Darensbourg and A. I. Moncada, Macromolecules, 2010, 43, 5996–6003 CrossRef CAS.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- D. Tian, B. Liu, Q. Gan, H. Li and D. J. Darensbourg, ACS Catal., 2012, 2, 2029–2035 CrossRef CAS.
- Y. Yang, Y. Hayashi, Y. Fujii, T. Nagano, Y. Kita, T. Ohshima, J. Okuda and K. Mashima, Catal. Sci. Technol., 2012, 2, 509–513 RSC.
- T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489 RSC.
- B. Chatelet, L. Joucla, J.-P. Dutasta, A. Martinez, K. C. Szeto and V. Dufaud, J. Am. Chem. Soc., 2013, 135, 5348–5351 CrossRef CAS PubMed.
- M. A. Fuchs, T. A. Zevaco, E. Ember, O. Walter, I. Held, E. Dinjus and M. Döring, Dalton Trans., 2013, 42, 5322 RSC.
- C. J. Whiteoak, N. Kielland, V. Laserna, F. Castro-Gómez, E. Martin, E. C. Escudero-Adán, C. Bo and A. W. Kleij, Chem. – Eur. J., 2014, 20, 2264–2275 CrossRef CAS PubMed.
- T. Werner and H. Büttner, ChemSusChem, 2014, 7, 3268–3271 CrossRef CAS PubMed.
- S. Gennen, M. Alves, R. Méreau, T. Tassaing, B. Gilbert, C. Detrembleur, C. Jerome and B. Grignard, ChemSusChem, 2015, 8, 1845–1849 CrossRef CAS PubMed.
- J. Rintjema and A. W. Kleij, ChemSusChem, 2017, 10, 1274–1282 CrossRef CAS PubMed.
- S. Arayachukiat, C. Kongtes, A. Barthel, S. V. C. Vummaleti, A. Poater, S. Wannakao, L. Cavallo and V. D'Elia, ACS Sustainable Chem. Eng., 2017, 5, 6392–6397 CrossRef CAS.
- D. O. Meléndez, A. Lara-Sánchez, J. Martínez, X. Wu, A. Otero, J. A. Castro-Osma, M. North and R. S. Rojas, ChemCatChem, 2018, 10, 2271–2277 CrossRef.
- M. Hong, Y. Kim, H. Kim, H. J. Cho, M.-H. Baik and Y. Kim, J. Org. Chem., 2018, 83, 9370–9380 CrossRef CAS PubMed.
- Á. Mesías-Salazar, J. Martínez, R. S. Rojas, F. Carrillo-Hermosilla, A. Ramos, R. Fernández-Galán and A. Antiñolo, Catal. Sci. Technol., 2019, 9, 3879–3886 RSC.
- H. Ma, J. Zeng, D. Tu, W. Mao, B. Zhao, K. Wang, Z. Liu and J. Lu, Tetrahedron Lett., 2020, 61, 151593 CrossRef CAS.
- W. Natongchai, J. A. Luque-Urrutia, C. Phungpanya, M. Solà, V. D'Elia, A. Poater and H. Zipse, Org. Chem. Front., 2021, 8, 613–627 RSC.
- Q. Yao, Y. Shi, Y. Wang, X. Zhu, D. Yuan and Y. Yao, Asian J. Org. Chem., 2022, 11, e202200106 CAS.
- S. Jana, H. Yu, A. Parthiban and C. L. L. Chai, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1622–1632 CrossRef CAS.
- S. W. Hwang, D. H. Park, D. H. Kang, S. B. Lee and J. K. Shim, J. Appl. Polym. Sci., 2016, 133, 43388 CrossRef.
- J. Sun, S. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS.
- M. Blain, L. Jean-Gérard, R. Auvergne, D. Benazet, S. Caillol and B. Andrioletti, Green Chem., 2014, 16, 4286–4291 RSC.
- H.-Y. Ju, M.-D. Manju, K.-H. Kim, S.-W. Park and D.-W. Park, J. Ind. Eng. Chem., 2008, 14, 157–160 CrossRef CAS.
- L. Wang, G. Zhang, K. Kodama and T. Hirose, Green Chem., 2016, 18, 1229–1233 RSC.
- X. Wang, L. Wang, Y. Zhao, K. Kodama and T. Hirose, Tetrahedron, 2017, 73, 1190–1195 CrossRef CAS.
- C. J. Whiteoak, E. Martin, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469–476 CrossRef CAS.
- D. Gourgouillon, H. M. N. T. Avelino, J. M. N. A. Fareleira and M. Nunes da Ponte, J. Supercrit. Fluids, 1998, 13, 177–185 CrossRef CAS.
- C. Kwag, C. W. Manke and E. Gulari, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 2771–2781 CrossRef CAS.
- Footnote: some experiments were conducted using a sapphire-windowed reactor and for pressures slightly above 100 bar (∼120 bars), the entire mixture of toluene and CO2 became supercritical, which triggered the precipitation of the Lotader® copolymers. This could explain the best compromise of 40–80 bar to achieve both good solubility and enhanced availability of CO2 in the liquid toluene phase. See also: H. Zhang, Z. Liu and B. Han, J. Supercrit. Fluids, 2000, 18, 185–192 CrossRef CAS . We also tried the reaction in the absence of toluene (see Fig. S33, ESI†) and could not observe an efficient process and consistent conversions of epoxy-containing copolymers (see also Photo S1, ESI†).
- B. Rainglet, P. Besognet, C. Benoit, K. Delage, V. Bounor-Legaré, C. Forest, P. Cassagnau and Y. Chalamet, Polymers, 2022, 14, 4513 CrossRef CAS PubMed.
- L. Verny, N. Ylla, F. D. Cruz-Boisson, E. Espuche, R. Mercier, G. Sudre and V. Bounor-Legaré, Ind. Eng. Chem. Res., 2020, 59, 16191–16204 CrossRef CAS.
- C. Dubois, C. Marestin, P. Cassagnau, K. Delage, P. Alcouffe, N. Garois and V. Bounor-Legaré, Polymer, 2022, 254, 125022 CrossRef CAS.
- R. R. A. Bolt, J. A. Leitch, A. C. Jones, W. I. Nicholson and D. L. Browne, Chem. Soc. Rev., 2022, 51, 4243–4260 RSC.
- S. Martey, B. Addison, N. Wilson, B. Tan, J. Yu, J. R. Dorgan and M. J. Sobkowicz, ChemSusChem, 2021, 14, 4280–4290 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structure, properties and characterization of the commercial polymers, calibration curve for the quantitative determination of cyclic carbonates by IR-ATR, and characterization of the modified polymers with CO2 in batch and in an extruder. See DOI: https://doi.org/10.1039/d3gc01127e |
| This journal is © The Royal Society of Chemistry 2023 |