
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineered green alga Chlamydomonas reinhardtii as a whole-cell photosynthetic biocatalyst for stepwise photoproduction of H2 and ε-caprolactone†
Vilja
Siitonen
 a,
Anna
Probst
b,
Gábor
Tóth
a,
Robert
Kourist
c,
Michael
Schroda
b,
Sergey
Kosourov
a and
Yagut
Allahverdiyeva
*a
a,
Anna
Probst
b,
Gábor
Tóth
a,
Robert
Kourist
c,
Michael
Schroda
b,
Sergey
Kosourov
a and
Yagut
Allahverdiyeva
*a
aMolecular Plant Biology, Department of Life Technologies, University of Turku, 20014 Turku, Finland. E-mail: allahve@utu.fi
bMolecular Biotechnology & Systems Biology, Rheinland-Pfälzische Technische Universität Kaiserslautern-Landau, 67663 Kaiserslautern, Germany
cInstitute of Molecular Biotechnology, Graz University of Technology, Petersgasse 14, 8010, Graz, Austria
First published on 4th July 2023
Abstract
Photosynthetic whole-cell biocatalysts are promising platforms for direct production of solar chemicals. Here, we employed the green microalga Chlamydomonas reinhardtii (hereafter Chlamydomonas) as a heterologous host for the cyclohexanone monooxygenase (CHMO) enzyme that converts exogenously added cyclohexanone to ε-caprolactone by utilising photosynthetically produced molecular oxygen (O2) and nicotinamide adenine dinucleotide phosphate (NADPH). In addition, the innate capability of Chlamydomonas to photoproduce molecular hydrogen (H2) was utilised in a one-pot stepwise production of H2 and ε-caprolactone. H2 photoproduction catalysed by innate O2-sensitive [Fe–Fe]-hydrogenase was facilitated by initial microoxic conditions and gradually declined due to accumulation of photosynthetic O2. This was accompanied by the biotransformation of cyclohexanone to ε-caprolactone by the heterologous CHMO. The optimal conditions for the formation of ε-caprolactone were the presence of acetate in the medium (mixotrophia), relatively low light intensity (26 μmol photons m−2 s−1) and addition of a low amount of ethanol [1.7% (vol/vol)]. The latter serves as a substrate inhibitor for the innate alcohol dehydrogenase (ADH) driven formation of cyclohexanol from cyclohexanone, thereby preventing competition between CHMO and ADH for the substrate, cyclohexanone. The formation of ε-caprolactone was further improved by introducing a signal sequence at the N-terminus of CHMO that directs the enzyme to the chloroplast enriched both with photosynthetic NADPH and O2, thus exploiting the compartmentalised nature of Chlamydomonas cell structure. This approach presents new opportunities for photosynthetic green chemicals production.
1. Introduction
Global challenges such as climate change and energy security are driving research towards sustainable strategies for the production of chemicals and fuels, which still often rely on fossil-based approaches. One emerging strategy is to use biocatalysts, typically enzymes isolated from living organisms, that can perform chemical transformations, thus producing relevant compounds from precursors.1 Biocatalysts offer several advantages over traditional chemical catalysts: (i) the high selectivity (chemo-, regio-, and stereoselectivity) of the enzyme-catalysed reactions; (ii) operation under milder production conditions, thus being safer for the environment; (iii) no harmful waste accumulating during synthesis, and; (iv) the use of sustainable raw materials.2 Alternatively, an organism possessing the desired enzymatic activity can be used as a whole-cell biocatalyst.3 The whole-cell biocatalyst approach offers additional advantages over isolated enzymes by providing a self-renewing source of catalysts, abolishing the need for the purification of enzymes and mitigating issues surrounding the stability of purified enzymes.4 Heterotrophic microbes (e.g. Escherichia coli and yeast) have been used as whole-cell biocatalysts for the production of various chemicals, e.g. ethanol, aromatic compounds, terpenoids, fatty acids, acrylamide, vitamins and L-amino acids.5–7 Oxygenic photosynthetic microorganisms are an emerging alternative8,9 for heterotrophs with their ability for sustainable cofactor regeneration. These microbes use solar energy to oxidise water into molecular oxygen (O2) and protons and to produce renewable reducing equivalents ferredoxin (Fd) and nicotinamide adenine dinucleotide phosphate (NADPH) to fuel cell metabolism. Heterologous expression of various oxidoreductases in photosynthetic microbes enables the exploitation of photosynthetically produced cofactors for catalysing desired biotransformations. For example, Baeyer–Villiger monooxygenases (BVMOs) utilise NAD(P)H as a cofactor to introduce an oxygen atom between a C–C bond in ketones and aldehydes, as well as to oxygenate heteroatoms.10,11 Using photosynthetic microbes as a host provides recombinant BVMO not only with renewable NADPH but also with photosynthetic O2. Indeed, successful examples of employing cyanobacterial hosts to complete the photobiotransformation have been reported.12,13It is important to note that, in cyanobacteria, photosynthesis and respiration occur in the same compartment, which creates complex conditions for controlling photosynthetic production.14,15 In contrast, eukaryotic hosts, such as the green alga Chlamydomonas, O2 production occurs in chloroplasts, the cell organelles performing photosynthesis, while respiration takes place in separate organelles, the mitochondria. Recently, wild-type Chlamydomonas cells that naturally express ene-reductases, also called Old Yellow Enzymes, were used to reduce C–C double bonds in a broad range of fed substrates.16 However, to the best of our knowledge, there are no examples of using engineered Chlamydomonas as a whole-cell biocatalyst to perform targeted oxyfunctionalisation. Importantly, Chlamydomonas can photoproduce H2via innate [Fe–Fe]-hydrogenases under anaerobic and microoxic conditions,17–19 which could bring added value to the production system.
In the present study, we engineered Chlamydomonas to express cyclohexanone monooxygenase (CHMO) from Acinetobacter20 and employed engineered Chlamydomonas to convert cyclohexanone to ε-caprolactone by introducing an oxygen atom to the carbon ring of cyclohexanone (Fig. 1). ε-Caprolactone is an important polymer precursor for the synthesis of polycaprolactone. The latter is in turn used for tissue engineering, drug-delivery systems, microelectronics, adhesives, packaging and other purposes.21 Traditional synthesis of ε-caprolactone requires harsh conditions, such as the use of peroxy acids or hydrogen peroxide.22 Our results show that recombinant Chlamydomonas can efficiently catalyse oxyfunctionalisation and that the reaction yield improves when CHMO is directed to the chloroplast, thus demonstrating the advantage of the eukaryotic cell compartmentalisation. Furthermore, we demonstrate a step-wise production of H2 and biotransformation of cyclohexanone to ε-caprolactone. These two reactions are performed under distinct levels of O2 in the system. Microoxic condition, that is important for H2 production, shifts by algal photosynthesis to fully oxic condition that is suitable for biotransformation. Thus, this principle diversifies the production of chemicals in the same photobioreactor and provides an opportunity for further engineering of industrially relevant production systems.
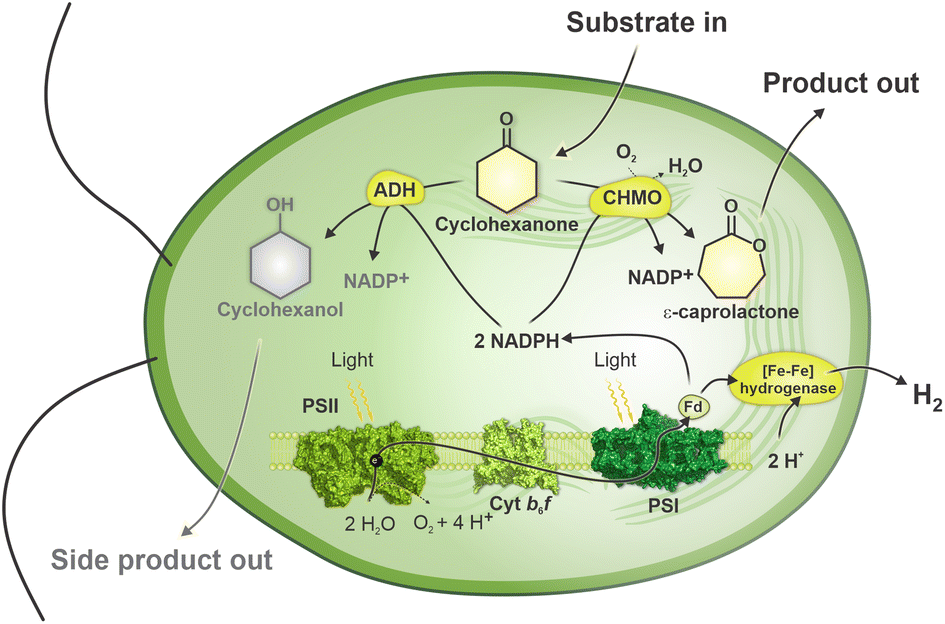 | ||
| Fig. 1 Concept of the study. Engineered Chlamydomonas that expresses CHMO converts fed cyclohexanone to ε-caprolactone by utilising photosynthetically produced O2 and reductants. Innate alcohol dehydrogenases (ADHs) compete for the substrate, cyclohexanone, and convert it to cyclohexanol. Innate [Fe–Fe]-hydrogenases photoproduce H2 under microoxic conditions utilising photosynthetically reduced ferredoxin (Fd). | ||
2. Results and discussion
2.1. Construction and characterisation of the production strains
For the heterologous expression of CHMO in Chlamydomonas, the amino acid sequence of Acinetobacter NCIB 9871 CHMO was reverse translated with the optimal Chlamydomonas codon usage and interrupted by the three introns of the Chlamydomonas RBCS2 gene to ensure high-level transgene expression.23 The CHMO coding sequence was synthesised, cloned as a standardised level 0 MoClo part24 (Fig. S1†) and assembled into a level 1 module with sequences coding for a C-terminal 3×HA tag under the control of the strong, constitutive HSP70A-RBCS2 promotor and the RPL23 terminator (Fig. S2A†). This construct was then combined with the aadA cassette, conferring resistance to spectinomycin, into a level 2 device. After transformation into the UVM4 recipient strain,25 24 spectinomycin-resistant transformants were tested for the expression of CHMO-3×HA by immunoblotting (Fig. S2B†). 13 out of 24 transformants expressed CHMO-3×HA and four of these to high levels (Fig. S2B†).2.2. Engineered Chlamydomonas converts cyclohexanone to ε-caprolactone
Chlamydomonas UVM4/CHMO was used as whole-cell biocatalyst to catalyse the light-driven conversion of 5 mM of the substrate, cyclohexanone, to ε-caprolactone. This substrate concentration was selected based on a toxicity test of substrate and product on cell growth and oxygen evolution (Fig. S3†). By the end of the photobiotransformation after 48 h, when the substrate was completely consumed, the side product, cyclohexanol comprised 85% of the products. As a consequence, only 15% ε-caprolactone was produced (Fig. 2, HL). The formation of the product matched typical course of an enzymatic reaction with the highest velocity at the beginning of the reaction. Side product formation followed a sigmoidal trend, in which initial formation of the compound experiences a lag phase accelerating to exponential rise before reaching saturation (Fig. S4A†). The specific product and side product formation activity; 0.008 units (U) per mg of chlorophyll (Chl) and 0.031 U mgChl−1, respectively, was determined by the maximal velocity of the reaction (Fig. 2A, HL). Since the turnover to ε-caprolactone was modest under the conditions used, we aimed to direct the reaction towards the desired product, ε-caprolactone, instead of the side product, that is likely to be produced by innate alcohol dehydrogenases (ADHs). Chlamydomonas harbours multiple ADHs26 that could compete with CHMO for the same substrate, cyclohexanone, that ADH converts to cyclohexanol (Fig. 1). As the next step we applied fomepizole, a known inhibitor of ADH, also known as 4-methylpyrazole.27 We found that while accumulation of cyclohexanol was slowed down by more than half to 0.013 U mgChl−1 by the addition of 30 mM fomepizole, the ratio still remained disadvantageous towards the target reaction while less substrate was consumed during the same reaction time, since after 48 h there was still a substantial proportion of substrate left (Fig. 2A and B, HLF). This implies that fomepizole could act as a competitive inhibitor not only for ADH,28 but also for CHMO, as the specific activity of ε-caprolactone formation was also slowed down to 0.006 U mgChl−1 (Fig. 2A, HLF). BVMOs are known to be promiscuous29 and fomepizole is known to act as a nonselective inhibitor.30 Interestingly, under these conditions the formation of both, the product and the side product, followed a sigmoidal trend (Fig. S4B†). At a higher concentration of fomepizole, 50 mM, the cells bleached (Fig. S5A†) and at a lower concentration, 15 mM, the inhibitory effect was less pronounced than with 30 mM of fomepizole (Fig. S6†). To conclude, the inhibition of ADH by fomepizole was found to be insufficient and lacked specificity in abolishing side product formation. The formation of ε-caprolactone was hindered by the toxicity of the compounds and the potential inhibitory effect on CHMO.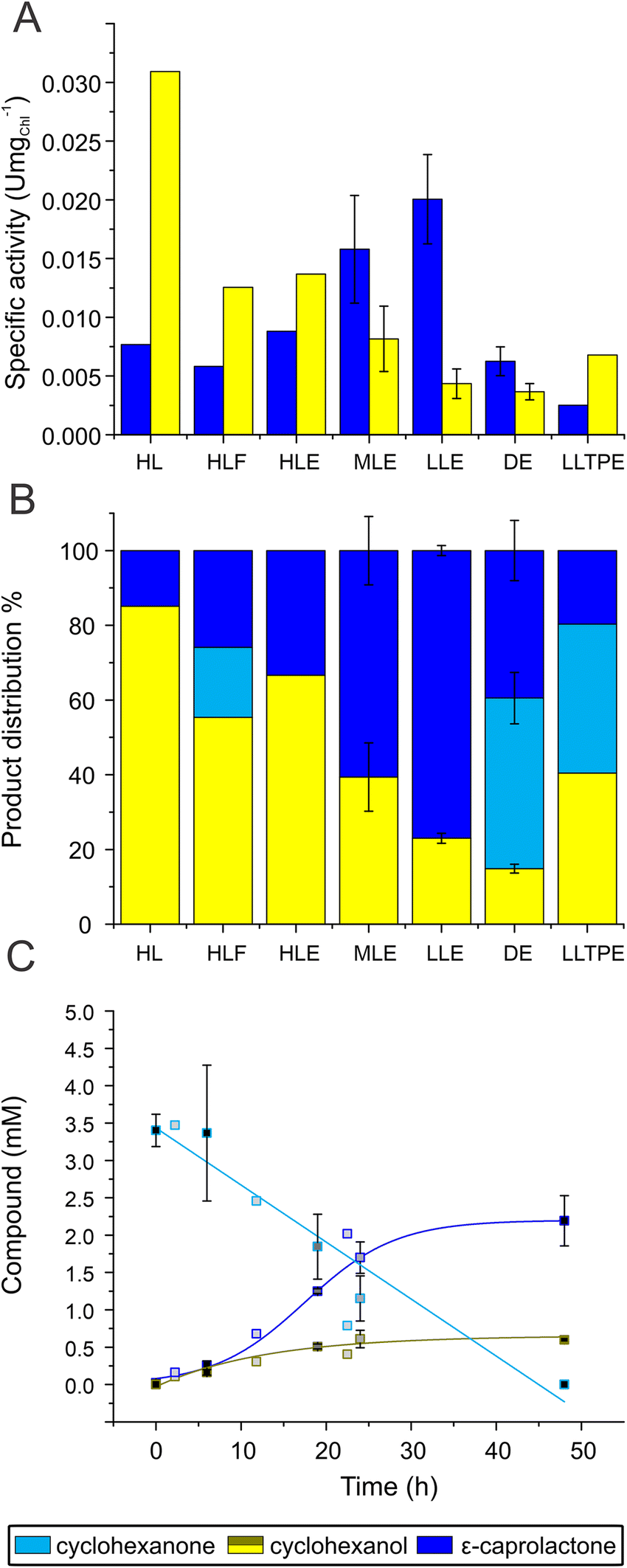 | ||
| Fig. 2 Evaluation of the biotransformation reaction of UVM4/CHMO. (A) Specific activity of product formation (blue) and side product formation (yellow) normalized to initial Chl. The reaction was performed under high light (165 μmol photons m−2 s−1) without additives (HL), with 30 mM fomepizole (HLF), with 1.7% (vol/vol) ethanol (HLE), in medium light (55 μmol photons m−2 s−1) with 1.7% (v/v) ethanol (MLE), in low light (26 μmol photons m−2 s−1) with 1.7% (v/v) ethanol (LLE), in darkness with 1.7% (v/v) ethanol (DE), in low light (26 μmol photons m−2 s−1) with 1.7% (v/v) ethanol under phototrophic conditions with 1% CO2 (LLTPE). (B) Product distribution 48 h after the reaction was started, substrate (cyan), side product (yellow), and product (blue), same conditions as in (A).( C) Time course of the reaction in LLE. The standard deviation presented in A–B are from 2–3 independent replications. For (C) N for different time points light grey 1, dark grey 2 and black 3. | ||
Next we attempted to inhibit ADHs with their natural product ethanol. Ethanol is one of the fermentation products formed by ADH in Chlamydomonas.31 Ethanol functions as a substrate inhibitor in the ADH-driven formation of cyclohexanol from cyclohexanone. As ethanol is added, ADH will exhibit a preference for utilizing ethanol instead of cyclohexanone. Indeed, ethanol accelerated ε-caprolactone production by ∼15% to 0.009 U mgChl−1, but the ratio of the produced compounds still remained favourable to side product formation (Fig. 2A and B, HLE). In the presence of ethanol, both the formation of the product and the side product followed a sigmoidal trend (Fig. S4C†). However, unlike the reaction with fomepizole, the presence of ethanol led to the complete consumption of all substrate within 48 h (Fig. 2, HLE). The results indicate that the linear compound, ethanol, did not inhibit CHMO to the same extent as the cyclic compound, fomepizole. This observation aligns with the expected behavior, as CHMO has been previously reported to exhibit lower activity towards linear substrates compared to cyclic compounds.32 Thus, supplementation of ethanol was beneficial for the desired photobiotransformation reaction by inhibiting side product formation. However, the turnover to the desired compound was still moderate, indicating that there is room for further improvement.
2.3. Mixotrophy under low light favours CHMO-driven photobiotransformation
To improve the formation of ε-caprolactone, we analysed the biotransformation under different light intensities. In addition to the initially used high light (HL, 165 μmol photons m−2 s−1), moderate (ML, 55 μmol photons m−2 s−1) and low light intensities (LL, 26 μmol photons m−2 s−1) were tested. Lowering light to 55 μmol photons m−2 s−1 accelerated CHMO-catalysed conversion by 80% compared to HL, reaching 0.016 ± 0.005 U mgChl−1 and leading to ε-caprolactone making up 61% of the products formed after 48 h (Fig. 2A and B, MLE). The best result was obtained at low light, where ε-caprolactone made up to 77% of the product formed after 48 h with 0.020 ± 0.004 U mgChl−1 specific activity (Fig. 2A–C, LLE). The fact that the reaction is working better under low light might be linked to the establishment of a high NAD(P)H to ATP ratio in algal cells due to limitation of mitochondrial respiration by O2 and low photosynthetic O2 production in dense algal cultures (100 mg Chl L−1).33,34 A high NAD(P)H to ATP ratio would favour the reactions leading to the utilisation of the excessive reducing power. Indeed, the NADPH cofactor is important for formation of the reduced state of the enzyme that becomes capable of reacting with O2 and stepping further into the biotransformation of cyclohexanone.35 Besides, low light conditions are known to decrease CO2 fixation and formation of biomass,36 which otherwise would compete with the CHMO-driven photobiotransformation.Ethanol had a similar effect on the specific activities in ML and LL as in HL (Fig. S7A†) and it was crucial to attain a majority of the desired compound also under lower light conditions as omitting ethanol led to over 56% of side product at LL with a small amount of substrate left and 85% of side product at ML with no substrate left (Fig. S7B†). To show that the biotransformation relies on light, we performed experiments in darkness. While the biotransformation proceeded to produce both products, reaction kinetics were much slower, with the specific activity of product formation being 3 times slower than in LL (0.006 ± 0.001 U mgChl−1). Side product formation was reduced by 18% compared to LL to 0.004 ± 0.0007 U mgChl−1 such that the reaction did not go to completion within 48 h, leading to 39% of ε-caprolactone and 15% cyclohexanol, while there was still 46% of substrate remaining (Fig. 2, DE).
All the reactions discussed so far were performed in TAP medium that contains acetate as an organic carbon source. This implies, that the algal cultures were subjected to mixotrophic conditions. Algal cultures can also be cultivated photoautotrophically, where algae use only photosynthesis for growth and CO2 is provided as a sole carbon source. In this context, we changed the carbon source from acetate to carbon dioxide. The cells were grown in minimal medium (TP) lacking acetate under air atmosphere supplemented with 1% carbon dioxide. Cells grown photoautotrophically (subsequentially the biotransformation performed under these conditions) led to a higher proportion of side product and overall slower reaction kinetics, as the reaction did not complete within 48 h (Fig. 2, LLTPE). Using ML or omitting ethanol did not substantially affect the specific activity for product formation, the average of which was 0.003 ± 0.0005 U mgChl−1 under all conditions, but the specific activity for side product formation accelerated twofold from 0.007 ± 0.00008 U mgChl−1 in LL with ethanol to an average of 0.015 ± 0.001 U mgChl−1 in ML with or without ethanol or LL without ethanol (Fig. 2A LLTPE and S7†). A possible explanation for the positive effect of acetate as carbon source under these conditions can be an increase of NAD(P)H production by catabolic pathways. In line with this, addition of D-glucose has been shown to accelerate biotransformation in Synechocystis sp. PCC 6803 expressing a heterologous ene-reductase.37 Mixotrophic conditions are attractive to improve algal growth, to extend the exponential phase, and to protect cells from photo-oxidative damage.38
2.4. Chloroplast targeting of CHMO improves the formation of ε-caprolactone
Despite the positive effects of low light and acetate on reaction kinetics, the reaction remained slow and a considerable portion of the final yield consisted of side product. To further enhance the reaction, we decided to physically associate the CHMO enzyme with photosynthetically derived NADPH. This was achieved by introducing the chloroplast transit peptide of PSAD (subunit of photosystem I complex)39 to CHMO. This modification allowed us to target the expressed protein to the chloroplast (CHMO_PSAD) (Fig. S2D and E†). Here we used a different strain, UVM11-CW, which is derived from a crossing of UVM11 (cw15, mt+) and CC-124 (CW15, mt−)40 as host to generate UVM11/CHMO_PSAD (Fig. S2D†). In contrast to UVM4, UVM11-CW contains a cell wall and therefore is more robust than UVM4, allowing its cultivation in large-scale photobioreactors for commercial purposes. Moreover, UVM11-CW has cilia and can mate, thus allowing crossing of strains with strong CHMO-expression levels and mutants harbouring properties that could boost photobiotransformation even further. For comparison, CHMO was expressed in UVM11-CW also without the transit peptide (UVM11-CW/CHMO) (Fig. S2C†).After transformation, 24 spectinomycin-resistant clones each were screened for the expression of CHMO-3×HA. The screening of UVM11-CW/CHMO revealed eleven out of 24 clones expressing CHMO-3×HA and four expressing it to high levels. For UVM11-CW/CHMO_PSAD, eleven clones expressed CHMO-3×HA to a detectable level with six clones showing high expression levels (Fig. S2C and E†).
Unexpectedly, the new strain UVM11-CW/CHMO, expressing CHMO without the chloroplast transit peptide, exhibited a significant improvement in the reaction compared to UVM4/CHMO (P = 0.03). The specific activity was boosted about 1.7 times, increasing from 0.020 ± 0.004 U mgChl−1 to 0.034 ± 0.006 U mgChl−1. Furthermore, the proportion of the desired product in the final product distribution after 48 h increased from 77% to 85% (Fig. 3A and S8†). The observed effect is most likely due to higher CHMO expression levels in UVM11-CW/CHMO compared to UVM4/CHMO (Fig. S2C†).
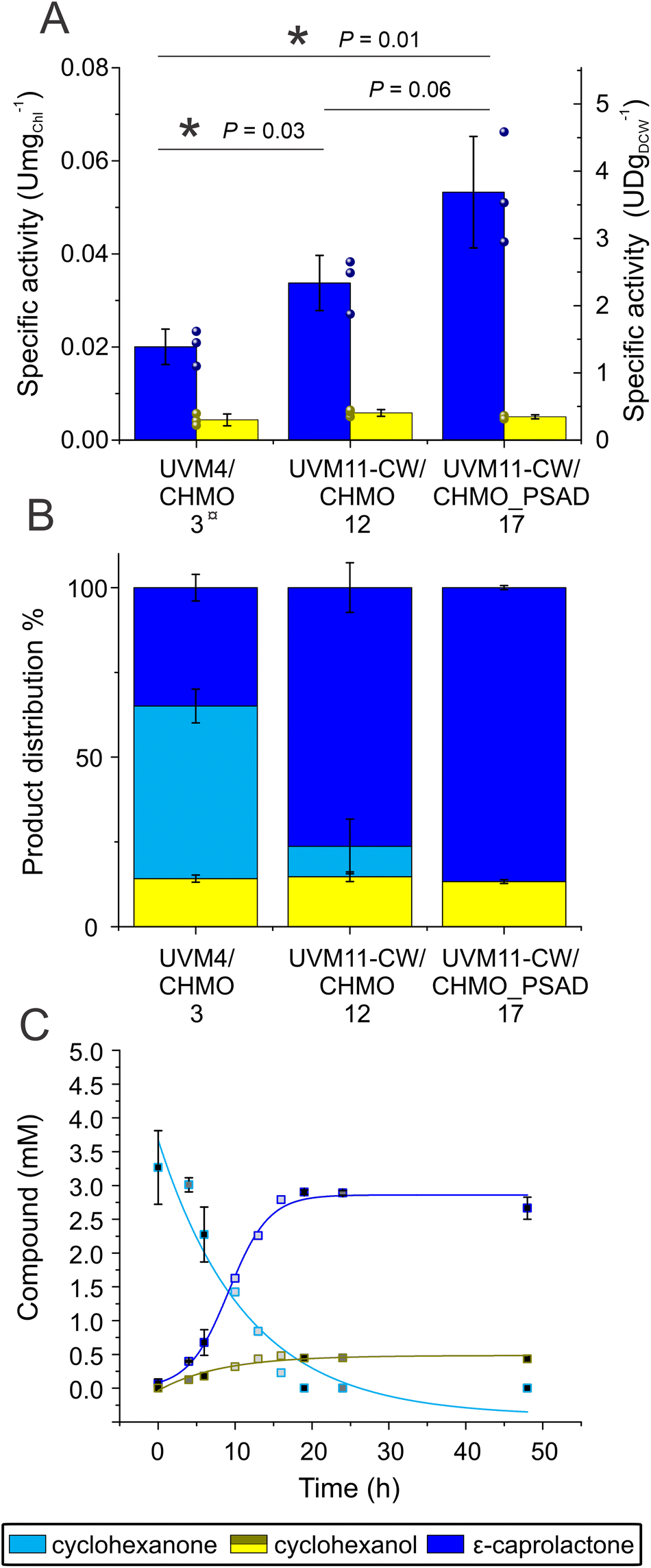 | ||
| Fig. 3 Comparison of biotransformation in UVM4/CHMO clone 3, UVM11-CW/CHMO clone 12 and UVM11-CW/CHMO_PSAD clone 17. (A) The specific activity normalized to initial Chl and DCW derived from the correlation curve (Fig. S9†). (B) Ratios of the compounds present after 19 h biotransformation. (C) Time course of the reaction of UVM-CW/CHMO_PSAD. Standard deviations are from three biological replications, except the product distribution for UVM4/CHMO which is derived from two (A and B). For (C) N for different time points light grey 1, dark grey 2 and black 3. ¤ Same data as in Fig. 2A LLE for ease of comparison. * Indicates a significant difference P < 0.05. P-values were calculated using a two-tailed type II Student's t-test. | ||
The constructed UVM11-CW/CHMO_PSAD strain exhibited even better performance: its specific activity for product formation was 0.053 ± 0.01 U mgChl−1, which is 2.7 times and 1.6 times better than UVM4/CHMO and UVM11-CW/CHMO, respectively. The strain also demonstrated faster reaction completion after 19 h (Fig. 3C) with 87% of the desired product and 13% of the side product (Fig. 3B), while UVM4/CHMO and UVM11-CW/CHMO still had substrate remaining at that time point (Fig. 3A and B). To ensure that the selected clone expressing CHMO with chloroplast transit peptide is randomly better than other strains, we tested 3–4 clones of UVM4/CHMO, UVM11-CW/CHMO and UVM11-CW/CHMO_PSAD for the conversion. Despite some variation among the clones, a consistent trend towards better conversion was observed with the clones expressing CHMO with the chloroplast transit peptide (Fig. 4A).
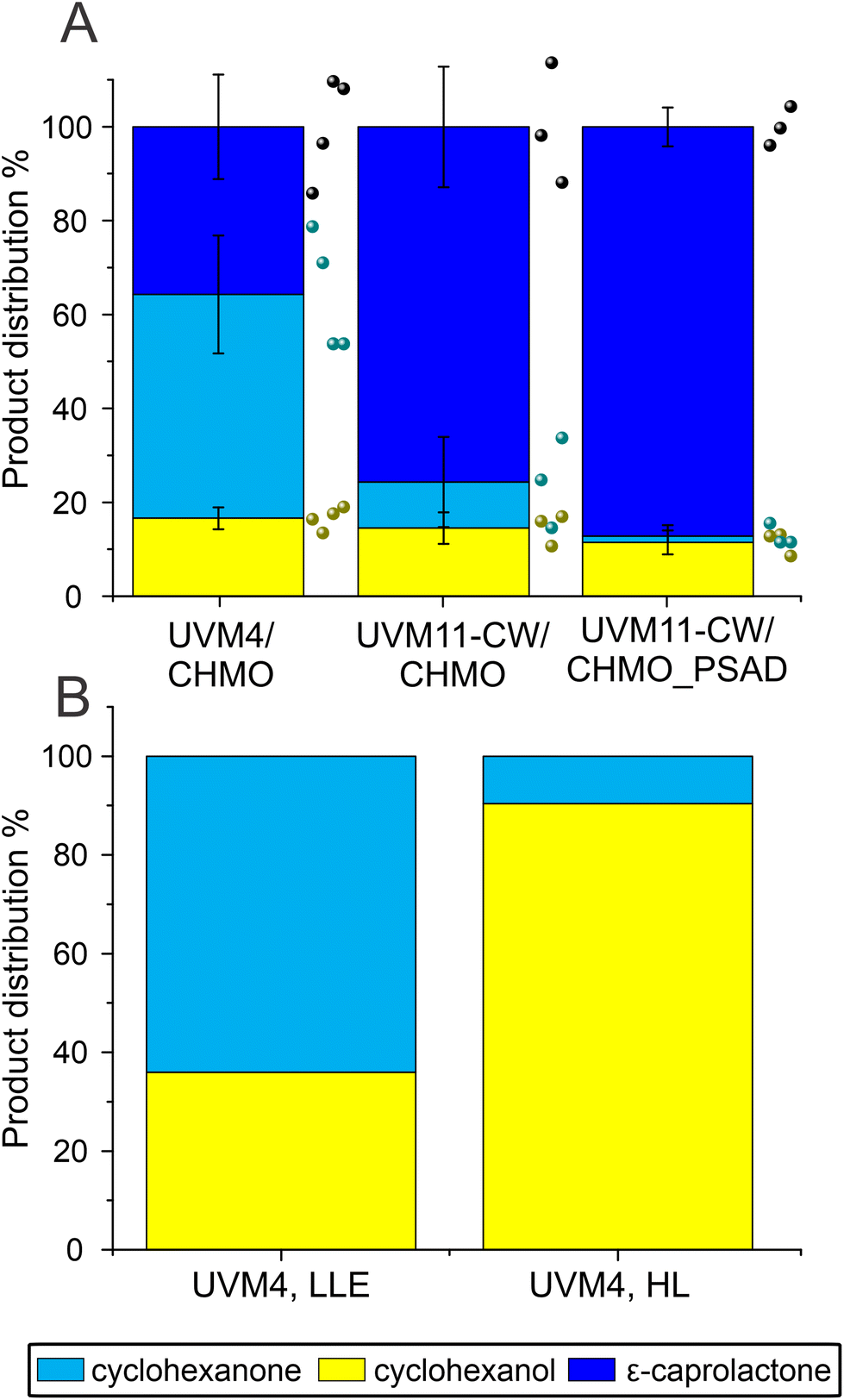 | ||
| Fig. 4 Comparison of biotransformation in UVM4/CHMO, UVM11-CW/CHMO and UVM11-CW/CHMO_PSAD. (A) The ratios of the compounds present after 19 h biotransformation, by 4 different UVM4/CHMO (clones 3, 4, 5, 15), 3 different UVM11-CW/CHMO (clones 12, 18, 23) and 3 different UVM11-CW/CHMO_PSAD (clones 6, 10, 17) strains. (B) The background strain in UVM4 (LLE) 26 μmol photons m−2 s−1 light with 1.7% EtOH (v/v) and UVM4 (HL) 165 μmol photons m−2 s−1 light without additives after 24 h of biotransformation. The presented standard deviations are from 3–4 different clones used in the study. | ||
The PSAD transit peptide was shown to efficiently target heterologous proteins to the chloroplast.41 Since in the chloroplast the NADPH supply stems directly from photosynthesis, a more effective use of it is facilitated. The exogenously added cyclohexanone is not directed to the chloroplast thus enabling ADH to convert part of the substrate to the side product in the cytosol.
Additionally, the background strain UVM4 produced only side product, while no product was detectable, which is in accordance with the activity of the endogenously expressed ADH by Chlamydomonas (Fig. 4B).
The specific activity obtained for UVM11-CW/CHMO_PSAD [3.6 ± 0.8 U per dry cell weight (DCW) (U gDCW−1, Fig. 3A)] is in the same order of magnitude as reported for Synechocystis sp. PCC 6803 (hereafter Synechocystis) with 2.3–4 U gDCW−1, which is comparable with rates in E. coli.42 However, in our study Chlamydomonas reached a better proportion of product versus side product: for Synechocystis side product was reported to constitute 50% of the products while we observed only 13% of side product in Chlamydomonas. Note, however, that the biotransformation conditions we employed differed from those reported for Synechocystis; e.g. no ethanol was used, applied light intensity was higher and the cell and substrate concentration was not identical.12,13 We conclude that Chlamydomonas is a suitable chassis for oxyfunctionalisation with final product distribution being superior to the values reported for Synechocystis. To further improve oxyfunctionalisation in Chlamydomonas, several strategies can be applied: (i) to use faster enzymes, such as the recently discovered BVMO from Burkholderia xenovorans which has shown 10-fold higher specific activity than CHMO in Synechocystis;12 (ii) to enhance the photosynthetic electron flow towards BVMOs by, for example, disrupting flavodiiron proteins that work as a ‘release valve’ for excessive electrons during sudden increase in light intensity12,14,43 There are examples of disrupting flavodiiron proteins in Synechocystis and indeed, it improved biotransformation reactions relying on NADPH12,44 (iii) Redesign photosynthetic light reactions for enhanced photosynthetic O2 production; (iv) knock out the ADH genes to completely avoid the accumulation of the side product. Chlamydomonas harbours multiple ADH genes26 that could be involved in converting cyclohexanone to cyclohexanol. Nevertheless, there is evidence that a single enzyme, ADH1, can be considered the main player in ethanol biosynthesis in Chlamydomonas.31
2.5. Engineered Chlamydomonas is capable of a stepwise photoproduction of H2 and ε-caprolactone
One advantage of employing Chlamydomonas as a host is the ability to produce molecular hydrogen, (H2).17,18,45 The reaction occurs under anaerobic conditions and depends on the activity of the O2-sensitive [Fe–Fe]-hydrogenase.46,47 When anaerobically grown algal cells are exposed to light, the enzyme accepts electrons from photosynthetically-reduced Fd, thus connecting H2 production with water oxidation by photosystem II via the photosynthetic electron transport chain.48,49 Typically, H2 photoproduction in an anaerobic alga does not proceed long due to simultaneous evolution of O2 produced by the water-oxidising complex of photosystem II.Produced O2 accumulates in the algal chloroplast and inactivates the [Fe–Fe]-hydrogenase.50 To take advantage of the fact that CHMO consumes O2, we aimed to establish a protocol for prolongation and/or semi-simultaneous H2 photoproduction and photobiotransformation. For this purpose, the biotransformation reaction was initiated under anaerobic conditions. The anaerobic UVM4/CHMO produced H2 for ∼6 h while keeping a microoxic environment. Hydrogen production plateaued upon O2 accumulation, which started after ∼6 h. As a result, the final H2 yield was close to 3 mmol L−1 of culture. Under microoxic conditions, no ε-caprolactone was detected, but as soon as the O2 started to accumulate in vials, ε-caprolactone appeared in the cultures (Fig. 5A). If the reactions were initiated under air, ε-caprolactone formation started earlier as O2 was present from the start of the reaction. To check whether CHMO activity affects H2 photoproduction, the background strain, UVM4, was assessed for H2 production. The results showed no significant difference between these strains, both produced around 3 mmol H2 L−1 of culture (Fig. 5B). Thus, the CHMO activity does not affect the H2 production, which is not surprising considering that these two activities are temporarily separated and occur under different O2 levels.
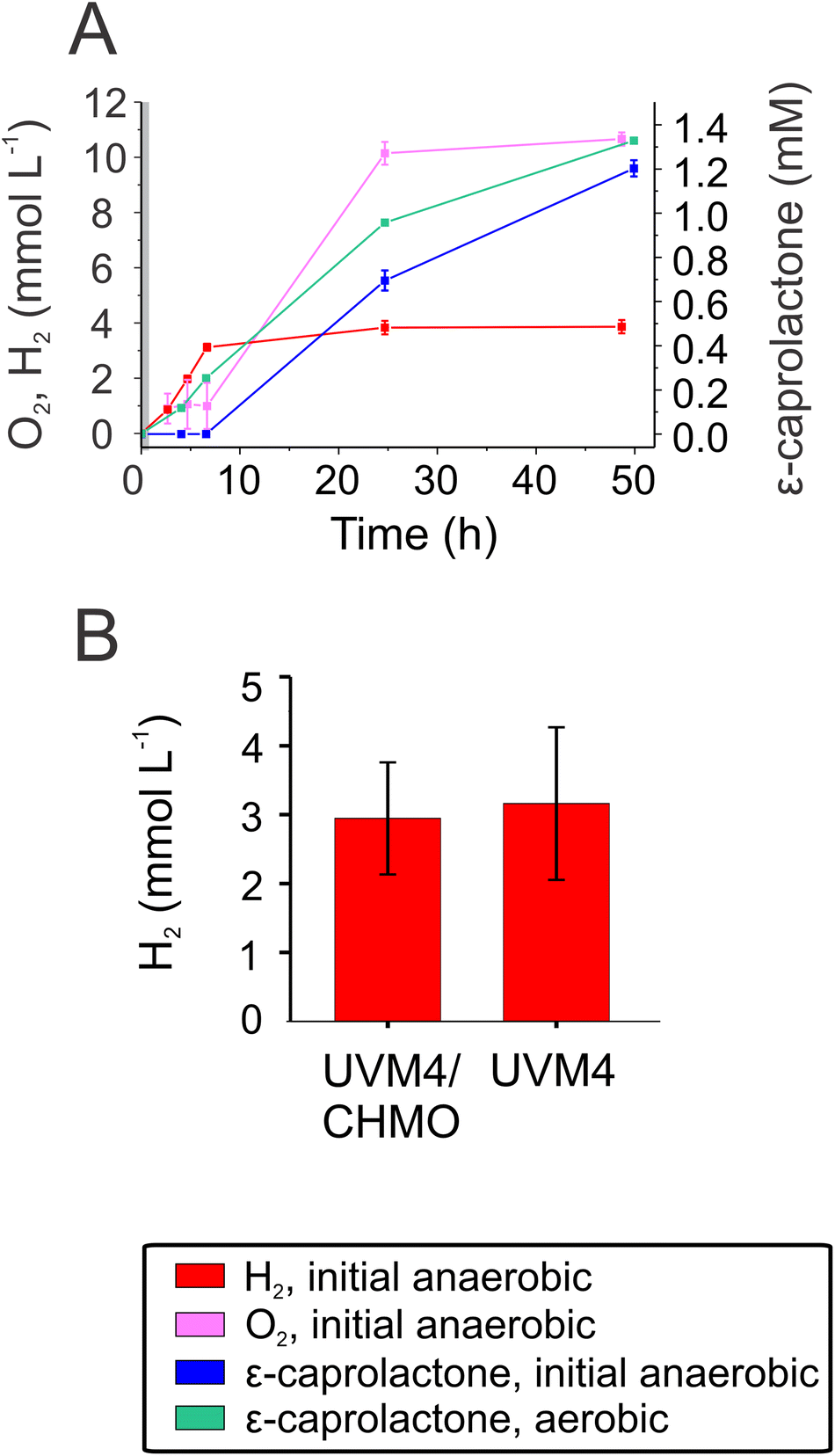 | ||
| Fig. 5 Stepwise production of H2 and ε-caprolactone. (A) Production of H2 (red), O2 (pink) and ε-caprolactone (blue) under microoxic (initial anaerobic) conditions by the UVM4/CHMO clone 15. To compare the effect of anaerobiosis on biotransformation, the production of ε-caprolactone under an air atmosphere is preseneted (see the green curve). The grey area in the beginning signifies the time when the vials were under ambient light (2–4 μmol photons m−2 s−1 before moving them to the experimental light conditions. (B) Accumulation of H2 by UVM4/CHMO clone 15 and by the background strain UVM4 after 48 h of incubation. The panel A is a representative experiment with technical repeats. The panel B shows 2–3 biological replications including three technical repeats. | ||
The green alga Chlamydomonas that we employed is a model organism for studying mechanisms of photosynthetic H2 production. Coupling H2 and ε-caprolactone formation shows that we can use a one-pot stepwise production system and it improves the value of the process, and paves way for many applications with the same principle. Scaling the reaction to an industrial level, however, presents challenges that must be carefully considered, including discrepancies in mixing, heat and mass transfer, as well as the higher expenses associated with the use of more complex and larger equipment.
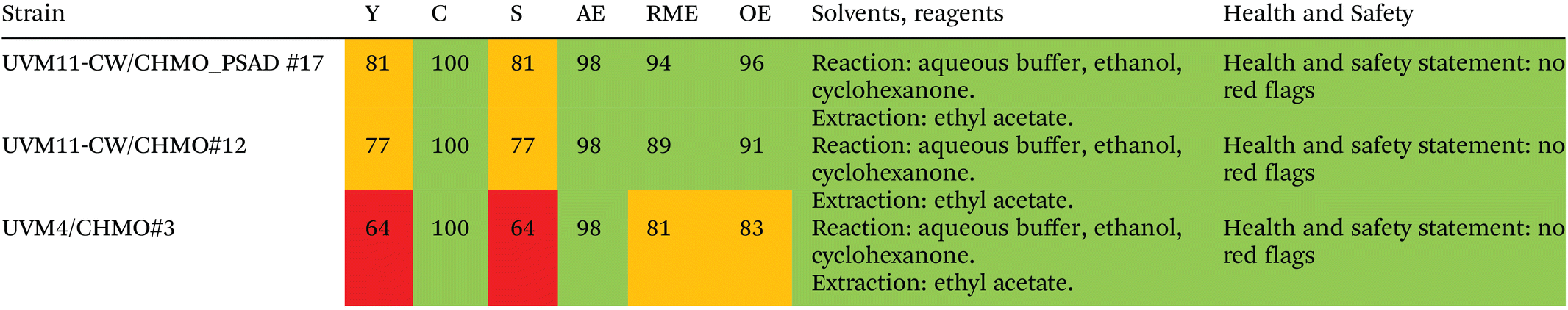
2.6. Evaluation of the greenness of the photobiotransformation
To assess the greenness of the whole-cell photosynthetic biotransformation approach, we applied the Zero Pass CHEM21 metric toolkit.51 The metrics presented in Table 1 demonstrate that both UVM11-CW strains outperform UVM4 in most parameters, resulting in green flags (>89%) and amber (70–89%) flags. Consequently, the UVM4 strain yields red flags (<70%) for both yield and selectivity. These metrics indicate that the step-by-step engineering of the green alga Chlamydomonas using synthetic biology approaches improves the overall performance of the photobiotransformation, instilling hope for further advancements in the field. Additionally, our approach advocates for the use of green solvents in algae culturing and compound extraction. The only solvent considered “recommended, problematic” is cyclohexanone, which serves as the substrate in the biotransformation to ε-caprolactone and, therefore cannot be considered a solvent in our setup. Even the most environmentally promising synthesis routes for producing ε-caprolactone rely on oxidizing agents such as peracids or oxone,52,53 whereas our oxidation is derived from water.| All values are percentages. Y = yield, C = conversion, S = selectivity, AE = atom economy, RME = reaction mass efficiency, OE = optimum efficiency. The values are means of 2–3 biological replicates, after the reaction has gone to completion. |
|---|
|
3. Conclusions
This study demonstrates the potential of Chlamydomonas, non-toxic, eukaryotic photosynthetic microorganism, for engineering the simultaneous photoproduction of H2 and ε-caprolactone in a one-pot, stepwise process. The efficiency of the reaction is enhanced when the nuclear encoded CHMO is targeted to the chloroplast. We provide evidence that intrinsic ADH activities can be inhibited by the addition of low concentrations of ethanol, favoring the production of ε-caprolactone over the formation of the side product, cyclohexanol. Our results show that mixotrophy and low light are preferred conditions for the production of ε-caprolactone. This work opens up possibilities for employing Chlamydomonas as a photosynthetic chassis for various photobiotransformations in industrially relevant processes. Furthermore, it highlights the potential for combining the production of valuable compounds like ε-caprolactone with H2 photoproduction. Thus the work extends the number of organisms that can be engineered to be used as a chassis for whole-cell biotransformations by the most important model microalga, Chlamydomonas.4. Experimental
4.1. Strains and culture conditions
For maintaining Chlamydomonas strains, cells were grown in Tris-acetate-phosphate (TAP)54,55 pH 7.2 supplemented with 100 μg mL−1 spectinomycin (Sigma) in case of the CHMO-expressing strains. Cells were grown in liquid medium and on plates with 1.5% bacto agar (Difco). Liquid cultures were incubated at 25 °C at 120 rpm orbital shaking (MIR-S100, Sanyo, Japan) under continuous light of 30–60 μmol photons m−2 s−1 using fluorescent light (Philips Master TL-D 36 W/865, the Netherlands), under ambient atmosphere or, when using TP medium (TAP medium without acetic acid), with 1% CO2 atmosphere (MLR-351 versatile environmental test chamber Sanyo, Japan). The experimental cultures were started from diluted late exponential phase pre-cultures and grown in 200 mL medium in 500 mL Erlenmeyers to mid-late exponential phase without addition of antibiotics using 35 μmol photons m−2 s−1 fluorescent light. Nuclear transformation of Chlamydomonas strains UVM4 (cw15, mt+) and UVM11-CW (CW15, mt+)25,40 was performed using the glass beads method.56 Transformants were selected on TAP-agar containing 100 mg L−1 spectinomycin (Merck S4014).4.2. Chl concentration, OD750, DCW and correlations
Chl was extracted by resuspending pelleted cells in 96% (vol/vol) ethanol. The samples were incubated in darkness so that the chl was completely dissolved and centrifuged for 1 min to obtain a white pellet. The supernatant was measured at 649 nm, 665 nm and 720 nm by using the UV-1800 spectrophotometer (Shimadzu, Japan). The total chl was calculated as described by Harris57 in addition the 720 nm absorbance values were subtracted from the 665 nm and 649 nm values to disregard the possible particles that would contribute to the absorbance values. OD750 was determined by using the UV-1800 spectrophotometer (Shimadzu, Japan) and measuring the cell suspension or appropriate dilutions. DCW was determined by taking 5–20 ml samples of cell suspension depending on the density of cells. The suspension was filtered through a glass microfiber filter (Whatman GF/F, 0.7 μm) using a vacuum pump. The filters were dried at ∼95 °C overnight and cooled down prior to weighing. For determining the correlation between chl and OD750, as well as Chl and DCW cells were adjusted to a concentration of 0.5–1 mg L−1 of chl and grown to ∼28–35 mg L−1 Chl. On the final day of the growth the cells were concentrated to 100 mg L−1, 150 mg L−1 and 200 mg L−1 of Chl and the DCW was determined from the concentrated cells.4.3. Cloning
The amino acid sequence of Acinetobacter NCIB 9871 CHMO (Uniprot number BAA86293) was reverse translated using the optimal Chlamydomonas codon usage. Chlamydomonas RBCS2 introns one, two and three were each inserted once with the flanking site CAG/intron/G resulting in four exons with a maximum length of 670 bp. Two internal BsaI recognition sites were eliminated by exchanging the codon for threonine (ACC) with ACG. To enable synthesis by IDT (Coralville, Iowa, USA), the sequence was divided into three GeneBlocks CHMO-up, CHMO-middle and CHMO-down. All fragments were flanked by BbsI recognition sites, introducing the overhangs AATG and TTCG for position B3–B4 of level 0 parts according to the MoClo syntax for plants24,58 and ligation in a defined order upon digestion with BbsI. The synthesized fragments were assembled into pAGM1287![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 58 upon digestion with BbsI (NEB) and simultaneous ligated as described previously59 resulting in level 0 part pMS596. Level 1 transcriptional units were generated by combining pMS596 with A1-B1-pCM0-011 [HSP70A-RBCS2 promoter + 5′-untranslated region (UTR)]; A1-B2 pCM0-017 (HSP70A-RBCS2 promoter + 5′-UTR); B2-pCM0-052 (PSAD chloroplast transit peptide (CTP); pMS640 (CDJ1 CTP); B5-pCM0-100 (3×HA); B6-C1-pCM0-119 (RPL23 3′-UTR + terminator) from the Chlamydomonas MoClo toolkit24 and the level 1 destination vector pICH4774258via digestion with BsaI (NEB) and ligation with T4 DNA ligase (NEB). Level 1 transcriptional units were combined with pCM1-01 (PSAD promotor + aadA gene + PSAD terminator)24,60 and end-linker pICH41744 into level 2 destination vector pAGM467358via digestion with BbsI and ligation with T4 DNA ligase.
58 upon digestion with BbsI (NEB) and simultaneous ligated as described previously59 resulting in level 0 part pMS596. Level 1 transcriptional units were generated by combining pMS596 with A1-B1-pCM0-011 [HSP70A-RBCS2 promoter + 5′-untranslated region (UTR)]; A1-B2 pCM0-017 (HSP70A-RBCS2 promoter + 5′-UTR); B2-pCM0-052 (PSAD chloroplast transit peptide (CTP); pMS640 (CDJ1 CTP); B5-pCM0-100 (3×HA); B6-C1-pCM0-119 (RPL23 3′-UTR + terminator) from the Chlamydomonas MoClo toolkit24 and the level 1 destination vector pICH4774258via digestion with BsaI (NEB) and ligation with T4 DNA ligase (NEB). Level 1 transcriptional units were combined with pCM1-01 (PSAD promotor + aadA gene + PSAD terminator)24,60 and end-linker pICH41744 into level 2 destination vector pAGM467358via digestion with BbsI and ligation with T4 DNA ligase.
The resulting strains UVM4/CHMO, UVM11-CW/CHMO and UVM11-CW/CHMO_PSAD were used for the biotransformation of cyclohexanone to ε-caprolactone. Four separate clones of UVM4/CHMO (3, 4, 5 and 15), and three separate clones of UVM11-CW/CHMO (12, 18 and 23) and UVM11-CW/CHMO_PSAD (6, 10 and 17) strains were used in the study. If not otherwise stated UVM4/CHMO clone 3, UVM11-CW/CHMO clone 12 and UVM-CW/CHMO_PSAD clone 17 (Fig. S2†) were used in the biotransformations.
4.4. Screening of CHMO overexpressing lines
Spectinomycin-resistant transformants were grown to stationary phase in TAP medium.61 Sample preparation and determination of chl content was performed as described in Hammel et al., 2020.62 Whole-cell protein corresponding to 2 μg chl was separated on 12% SDS-polyacrylamide gels. Expression of CHMO-3×HA was analysed via immunoblotting using a mouse anti-HA antibody (Merck H9658, 1:10000). Anti-mouse IgG–HRP (Santa Cruz Biotechnology sc-2031, USA) was used as secondary antibody. Chemiluminescence signals were detected via enhanced chemiluminescence using the Fusion-FX7 device (PEQLAB Biotechnology GmbH, Germany) or ECL ChemoStar (INTAS Science Imaging Instruments GmbH, Germany).
4.5. Determining tolerance towards substrate and product
UVM4 cells were grown starting from 2 μg mL−1 of Chl in the presence of different concentrations of substrate (0–20 mM) and product (0–20 mM) and growth was followed by measuring OD750 and Chl concentration. After 23 days of growth, oxygen evolution was determined by using a Clark-type electrode (Oxygraph plus system, Hansatech, Great Britain). Net oxygen evolution was measured in the presence of 5 mM NaHCO3 and under 810 μmol photons m−2 s−1 illumination (Fiber-Lite DC-950, Dolan-Jenner, USA).4.6. Biotransformation
Cells were collected by centrifugation (2000–5000g) for 5 minutes at 25 °C, resuspended in fresh medium, and incubated in the growth chamber under continuous fluorescent light (Osram Lumilux L 15W/865, Germany). The chl concentration was adjusted to 100 μg mL−1 for the experiments and cells were added to the reaction mixture. The reaction mixture contained TAP or TP, 25 mM MOPS-buffer, 5–10 mM cyclohexanone and 0–1.7% (vol/vol) ethanol or 0–50 mM fomepizole. The ethanol concentration was selected so that it would not inhibit the growth of Chlamydomonas by more than 50%.63 The concentration of fomepizole was selected based on trials, because no examples were found of use of fomepizole in Chlamydomonas cells. The flasks used were closed with rubber caps with a crimped metal ring. The rubber caps had been treated with ethanol and autoclaved twice, first in the presence of water and then in a dry cycle, prior to use. If TP medium was used, flasks were closed and 1% CO2 was added to the headspace. The flasks were kept on the side on the shaker, shaking with 90 rpm and light intensities adjusted with neutral filters (209 0.3 N Lee filters, Panavision, USA) when needed. First and last samples were taken with a pipette. Time points in between were taken using a syringe with a needle through the cap and the sample volume taken out was replaced by air. Typically, samples were frozen in liquid nitrogen and stored at −80 °C for later extraction. Extractions were made by adding 0.06% (vol/vol) HCl and half of the sample volume of ethyl acetate containing 2 mM acetophenone, followed by mixing with a multivortexer for 3 minutes. The phases were separated by a table top centrifuge at full speed for 1 min prior to collecting the ethyl acetate phase. This was done twice. The ethyl acetate phases were combined, dried with anhydrous MgSO4 and centrifuged for 4 min prior to moving to the GC vial.4.7. Analysis of reactions
Analysis of reaction products was done with a GC2010 Pro gas chromatography machine (Shimadzu, Japan). The device was equipped with a flame ionisation detector (FID), AOC-20s auto sampler, AOC-20i auto injector and a HP-5MS 30 m × 0.25 mm (5%-phenyl)-methylpolysiloxane column (19091S-133, Agilent, USA). Nitrogen was used as carrier gas. The compounds were identified by retention times and quantified by areas. Calibration of the instrument was done by preparing a mixture of cyclohexanone, ε-caprolactone and cyclohexanol at varying concentrations (0.312, 1.25, 2.5 and 10 mM with constant concentration of the internal standard acetophenone (2 mM) (all compounds from Sigma-Aldrich) in ethyl acetate. Specific reaction rates were determined by calculating the initial slope of the formation of the compound (μmol in min) and normalized to the initial chl amount (in mg) in the reactions. In cases where the curves followed a typical Michaelis–Menten enzyme reaction, the slope was determined by using the initial velocity v0. For sigmoidal curves, the maximal velocity was determined by fitting the data to slogistics2 using Origin 2015 (OriginLab Corporation, USA) and determining the Wmax. For normalizing to DCW, the correlation curve was used as explained above (Fig. S8†). Significance tests were done by two-tailed type II Student's t-test using Excel Version 2303 (Microsoft corporation, USA).4.8. Stepwise photosynthetic H2 production and biotransformation
Cells were handled in a similar manner as described for biotransformation reactions. After the reaction mixture containing UVM4/CHMO clone 15 or UVM4 was ready, the flasks were placed under ambient light with an intensity of 2–4 μmol photons m−2 s−1. To create an anaerobic environment suitable for the O2-sensitive [Fe–Fe]-hydrogenase to function, the atmosphere in the flaks was replaced with argon using needles to allow the air escape. The vials were then placed under a light intensity of 26 μmol photons m−2 s−1 and H2, O2 and ε-caprolactone production was recorded for 48 h with a 2 h interval in the beginning followed by measurements at 24 h and 48 h. Samples were taken with a 250 μL air-tight syringe (Hamilton, Merck, Germany) filled with 150 μL argon, which was replaced with a sample from the headspace and injected to the GC (Clarus 500, PerkinElmer, USA) equipped with a thermal conductivity detector and a molecular sieve 5 Å column (60/80 mesh). Argon was used as carrier gas. Air was used as a standard for oxygen and 3% hydrogen gas as a standard for H2. Biotransformation samples were taken with an air-tight syringe.4.9. Evaluation of photobiotransformation using the CHEM21 metric toolkit
To assess the greenness of our approach, we utilised the CHEM21 metric toolkit at Zero Pass. The percentage yield (Y), percentage conversion (C), percentage selectivity (S), atom economy (AE), reaction mass efficiency (RME) and optimum efficiency (OE) were calculated for the laboratory scale reaction described by McElroy et al., 2015.51 Furthermore, we followed the greenness of solvents and hazardous statements as specified by the authors. In the calculation of AE, both the substrate and water were considered as reactants for the reaction.Author contributions
Conceptualization YA; methodology VS, AP, RK, MS, SK, YA; formal analysis VS, AP; investigation VS, AP, GT; resources RK, MS, YA; writing – original draft VS; writing – review & editing all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NordForsk Nordic Center of Excellence “NordAqua” (no. 82845 to YA), the EU FET Open project “FuturoLEAF” (grant agreement no. 899576 to YA and RK), the Academy of Finland “AlgaLEAF” (project no. 322754 to YA), the Novo Nordisk Foundation project “PhotoCat” (project no. NNF20OC0064371 to YA), and by the Deutsche Forschungsgemeinschaft (SFB/TRR175, project C02; SPP1927, project Schr 617/11-1 to MS). The studies (except strain construction) were conducted in the PhotoSYN Finnish Infrastructure for Photosynthesis Research. The authors would like to acknowledge Dr Guido Durian for assisting in hydrogen measurements and Tiia Siivola for preparing Fig. S9 as well as helpful discussions with Dr Lenny Malihan-Yap.References
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12, 2859–2881 CrossRef CAS PubMed.
- B. Lin and Y. Tao, Microb. Cell Fact., 2017, 16, 106 CrossRef PubMed.
- P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2011, 15, 266–274 CrossRef CAS.
- A. Valle and J. Bolívar, Escherichia coli, the workhorse cell factory for the production of chemicals, Academic Press, 2021, pp. 115–137 Search PubMed.
- C. C. C. R. de Carvalho, Microb. Biotechnol., 2017, 10, 250–263 CrossRef PubMed.
- S. Wang, F. Zhao, M. Yang, Y. Lin and S. Han, Crit. Rev. Biotechnol., 2023, 1–28, DOI:10.1080/07388551.2022.2153008.
- J. Toepel, R. Karande, S. Klähn and B. Bühler, Curr. Opin. Biotechnol, 2023, 80, 102892 CrossRef CAS PubMed.
- J. Jodlbauer, T. Rohr, O. Spadiut, M. D. Mihovilovic and F. Rudroff, Trends Biotechnol., 2021, 39, 875–889 CrossRef CAS PubMed.
- H. Leisch, K. Morley and P. C. K. Lau, Chem. Rev., 2011, 111, 4165–4222 CrossRef CAS PubMed.
- C. Tolmie, M. S. Smit and D. J. Opperman, Nat. Prod. Rep., 2019, 36, 326–353 RSC.
- E. Erdem, L. Malihan-Yap, L. Assil-Companioni, H. Grimm, G. D. Barone, C. Serveau-Avesque, A. Amouric, K. Duquesne, V. de Berardinis, Y. Allahverdiyeva, V. Alphand and R. Kourist, ACS Catal., 2022, 12, 66–72 CrossRef CAS PubMed.
- S. Böhmer, K. Köninger, Á. Gómez-Baraibar, S. Bojarra, C. Mügge, S. Schmidt, M. M. Nowaczyk and R. Kourist, Catalysts, 2017, 7, 240 CrossRef.
- L. Nikkanen, D. Solymosi, M. Jokel and Y. Allahverdiyeva, Physiol. Plant., 2021, 173, 514–525 CrossRef CAS PubMed.
- C. W. Mullineaux, Biochim. Biophys. Acta, 2014, 1837, 503–511 CrossRef CAS PubMed.
- S. Böhmer, C. Marx, Á. Gómez-Baraibar, M. M. Nowaczyk, D. Tischler, A. Hemschemeier and T. Happe, Algal Res., 2020, 50, 101970 CrossRef.
- S. Kosourov, M. Böhm, M. Senger, G. Berggren, K. Stensjö, F. Mamedov, P. Lindblad and Y. Allahverdiyeva, Physiol. Plant., 2021, 173, 555–567 CrossRef CAS.
- S. Z. Tóth and I. Yacoby, Trends Biotechnol., 2019, 37, 1159–1163 CrossRef.
- K. E. Redding, J. Appel, M. Boehm, W. Schuhmann, M. M. Nowaczyk, I. Yacoby and K. Gutekunst, Trends Biotechnol., 2022, 40, 1313–1325 CrossRef CAS PubMed.
- N. A. Donoghue, D. B. Norris and P. W. Trudgill, Eur. J. Biochem., 1976, 63, 175–192 CrossRef CAS PubMed.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- G. R. Krow, Org. React., 2004, 251–798 Search PubMed.
- M. Schroda, Cells, 2019, 8, 1534 CrossRef CAS PubMed.
- P. Crozet, F. J. Navarro, F. Willmund, P. Mehrshahi, K. Bakowski, K. J. Lauersen, M. E. Pérez-Pérez, P. Auroy, A. Gorchs Rovira, S. Sauret-Gueto, J. Niemeyer, B. Spaniol, J. Theis, R. Trösch, L. D. Westrich, K. Vavitsas, T. Baier, W. Hübner, F. De Carpentier, M. Cassarini, A. Danon, J. Henri, C. H. Marchand, M. De Mia, K. Sarkissian, D. C. Baulcombe, G. Peltier, J. L. Crespo, O. Kruse, P. E. Jensen, M. Schroda, A. G. Smith and S. D. Lemaire, ACS Synth. Biol., 2018, 7, 2074–2086 CrossRef CAS PubMed.
- J. Neupert, D. Karcher and R. Bock, Plant J., 2009, 57, 1140–1150 CrossRef CAS.
- S. S. Merchant, S. E. Prochnik, O. Vallon, E. H. Harris, S. J. Karpowicz, G. B. Witman, A. Terry, A. Salamov, L. K. Fritz-Laylin, L. Maréchal-Drouard, W. F. Marshall, L. H. Qu, D. R. Nelson, A. A. Sanderfoot, M. H. Spalding, V. V. Kapitonov, Q. Ren, P. Ferris, E. Lindquist, H. Shapiro, S. M. Lucas, J. Grimwood, J. Schmutz, I. V. Grigoriev, D. S. Rokhsar, A. R. Grossman, P. Cardol, H. Cerutti, G. Chanfreau, C. L. Chen, V. Cognat, M. T. Croft, R. Dent, S. Dutcher, E. Fernández, H. Fukuzawa, D. González-Ballester, D. González-Halphen, A. Hallmann, M. Hanikenne, M. Hippler, W. Inwood, K. Jabbari, M. Kalanon, R. Kuras, P. A. Lefebvre, S. D. Lemaire, A. V. Lobanov, M. Lohr, A. Manuell, I. Meier, L. Mets, M. Mittag, T. Mittelmeier, J. V. Moroney, J. Moseley, C. Napoli, A. M. Nedelcu, K. Niyogi, S. V. Novoselov, I. T. Paulsen, G. Pazour, S. Purton, J. P. Ral, D. M. Riaño-Pachón, W. Riekhof, L. Rymarquis, M. Schroda, D. Stern, J. Umen, R. Willows, N. Wilson, S. L. Zimmer, J. Allmer, J. Balk, K. Bisova, C. J. Chen, M. Elias, K. Gendler, C. Hauser, M. R. Lamb, H. Ledford, J. C. Long, J. Minagawa, M. D. Page, J. Pan, W. Pootakham, S. Roje, A. Rose, E. Stahlberg, A. M. Terauchi, P. Yang, S. Ball, C. Bowler, C. L. Dieckmann, V. N. Gladyshev, P. Green, R. Jorgensen, S. Mayfield, B. Mueller-Roeber, S. Rajamani, R. T. Sayre, P. Brokstein, I. Dubchak, D. Goodstein, L. Hornick, Y. W. Huang, J. Jhaveri, Y. Luo, D. Martínez, W. C. A. Ngau, B. Otillar, A. Poliakov, A. Porter, L. Szajkowski, G. Werner and K. Zhou, Science, 2007, 318, 245–251 CrossRef CAS PubMed.
- R. T. Gentry, Alcohol, 1985, 2, 581–587 CrossRef CAS PubMed.
- H. Theorell, T. Yonetani and B. Sjöberg, Acta Chem. Scand., 1969, 23, 255–260 CrossRef CAS PubMed.
- M. D. Mihovilovic, B. Müller and P. Stanetty, Eur. J. Org. Chem., 2002, 3711–3730 CrossRef CAS.
- L. Di, A. Balesano, S. Jordan and S. M. Shi, AAPS J., 2021, 23, 20 CrossRef CAS PubMed.
- L. Magneschi, C. Catalanotti, V. Subramanian, A. Dubini, W. Yang, F. Mus, M. C. Posewitz, M. Seibert, P. Perata and A. R. Grossman, Plant Physiol., 2012, 158, 1293–1305 CrossRef CAS PubMed.
- L. Bretschneider, I. Heuschkel, A. Ahmed, K. Bühler, R. Karande and B. Bühler, Biotechnol. Bioeng., 2021, 118, 2719–2733 CrossRef CAS PubMed.
- B. Degrenne, J. Pruvost and J. Legrand, Bioresour. Technol., 2011, 102, 1035–1043 CrossRef CAS PubMed.
- A. Scoma, L. Durante, L. Bertin and F. Fava, New Phytol., 2014, 204, 890–900 CrossRef CAS PubMed.
- D. Sheng, D. P. Ballou and V. Massey, Biochemistry, 2001, 40, 11156–11167 CrossRef CAS PubMed.
- Y. Maltsev, K. Maltseva, M. Kulikovskiy and S. Maltseva, Biology, 2021, 10, 1060 CrossRef CAS PubMed.
- G. D. Barone, M. Hubáček, L. Malihan-Yap, H. C. Grimm, L. Nikkanen, C. C. Pacheco, P. Tamagnini, Y. Allahverdiyeva and R. Kourist, Biotechnol. Biofuels Bioprod., 2023, 16, 4 CrossRef CAS PubMed.
- A. K. Patel, J. M. Joun, M. E. Hong and S. J. Sim, Bioresour. Technol., 2019, 282, 245–253 CrossRef CAS PubMed.
- V. P. Chitnis, A. Ke and P. R. Chitnis, Plant Physiol., 1997, 115, 1699–1705 CrossRef CAS PubMed.
- J. Neupert, S. D. Gallaher, Y. Lu, D. Strenkert, N. Segal, R. Barahimipour, S. T. Fitz-Gibbon, M. Schroda, S. S. Merchant and R. Bock, Nat. Commun., 2020, 11, 1–17 CrossRef PubMed.
- O. D. Caspari, Front. Plant Sci., 2022, 13, 1603 Search PubMed.
- S. Schmidt, C. Scherkus, J. Muschiol, U. Menyes, T. Winkler, W. Hummel, H. Gröger, A. Liese, H.-G. Herz and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2015, 54, 2784–2787 CrossRef CAS PubMed.
- M. Jokel, X. Johnson, G. Peltier, E. M. Aro and Y. Allahverdiyeva, Plant J., 2018, 94, 822–835 CrossRef CAS PubMed.
- L. Assil-Companioni, H. C. Büchsenschütz, D. Solymosi, N. G. Dyczmons-Nowaczyk, K. K. F. Bauer, S. Wallner, P. MacHeroux, Y. Allahverdiyeva, M. M. Nowaczyk and R. Kourist, ACS Catal., 2020, 10, 11864–11877 CrossRef CAS PubMed.
- E. Touloupakis, C. Faraloni, A. M. S. Benavides and G. Torzillo, Energies, 2021, 14, 7170 CrossRef CAS.
- M. L. Ghirardi, A. Dubini, J. Yu and P. C. Maness, Chem. Soc. Rev., 2009, 38, 52–61 RSC.
- S. T. Stripp, G. Goldet, C. Brandmayr, O. Sanganas, K. A. Vincent, M. Haumann, F. A. Armstrong and T. Happe, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17331–17336 CrossRef CAS PubMed.
- A. Sawyer and M. Winkler, Photosynth. Res., 2017, 134, 307–316 CrossRef CAS PubMed.
- S. Kosourov, V. Nagy, D. Shevela, M. Jokel, J. Messinger and Y. Allahverdiyeva, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29629–29636 CrossRef CAS PubMed.
- Y. Milrad, S. Schweitzer, Y. Feldman and I. Yacoby, Plant Physiol., 2018, 177, 918–926 CrossRef CAS PubMed.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- V. Bertolini, R. Appiani, M. Pallavicini and C. Bolchi, J. Org. Chem., 2021, 86, 15712–15716 CrossRef CAS PubMed.
- M. Sitko, A. Szelwicka, A. Wojewódka, A. Skwarek, D. Tadasiewicz, L. Schimmelpfennig, K. Dziuba, M. Morawiec-Witczak and A. Chrobok, RSC Adv., 2019, 9, 30012–30018 RSC.
- E. H. Harris, The Chlamydomonas Sourcebook : a Comprehensive Guide to Biology and Laboratory Use, Elsevier Science, 1989, pp. 25–63 Search PubMed.
- D. S. Gorman and R. P. Levine, Proc. Natl. Acad. Sci. U. S. A., 1965, 54, 1665–1669 CrossRef CAS PubMed.
- K. L. Kindle, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 1228–1232 CrossRef CAS PubMed.
- E. H. Harris, The Chlamydomonas Sourcebook : a Comprehensive Guide to Biology and Laboratory Use, Elsevier Science, 1989, pp. 575–641 Search PubMed.
- E. Weber, C. Engler, R. Gruetzner, S. Werner and S. Marillonnet, PLoS One, 2011, 6, e16765 CrossRef CAS PubMed.
- A. M. Kiefer, J. Niemeyer, A. Probst, G. Erkel and M. Schroda, Front. Plant Sci., 2022, 13, 3556 CrossRef PubMed.
- J. Niemeyer, D. Scheuring, J. Oestreicher, B. Morgan and M. Schroda, Plant Cell, 2021, 33, 2935–2949 CrossRef PubMed.
- J. Kropat, A. Hong-Hermesdorf, D. Casero, P. Ent, M. Castruita, M. Pellegrini, S. S. Merchant and D. Malasarn, Plant J., 2011, 66, 770–780 CrossRef CAS PubMed.
- A. Hammel, F. Sommer, D. Zimmer, M. Stitt, T. Mühlhaus and M. Schroda, Front. Plant Sci., 2020, 11, 868 CrossRef PubMed.
- Y. Jiang, P. Xiao, Q. Shao, H. Qin, Z. Hu, A. Lei and J. Wang, Biotechnol. Biofuels, 2017, 10, 1–16 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01400b |
| This journal is © The Royal Society of Chemistry 2023 |