
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water-soluble nickel and iron salts for hydroxymethylfurfural (HMF) and water oxidation: the simplest precatalysts?†
Till
Kahlstorf‡
a,
J.
Niklas Hausmann‡
*a,
Indranil
Mondal
 b,
Konstantin
Laun
c,
Ingo
Zebger
c,
Tobias
Sontheimer
d and
Prashanth W.
Menezes
*ab
b,
Konstantin
Laun
c,
Ingo
Zebger
c,
Tobias
Sontheimer
d and
Prashanth W.
Menezes
*ab
aMaterial Chemistry Group for Thin Film Catalysis–CatLab, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Str. 15, 12489 Berlin, Germany. E-mail: niklas.hausmann@helmholtz-berlin.de; prashanth.menezes@helmholtz-berlin.de
bDepartment of Chemistry: Metalorganics and Inorganic Materials, Technische Universität Berlin, Straße des 17 Juni 135, Sekr. C2, 10623 Berlin, Germany. E-mail: prashanth.menezes@mailbox.tu-berlin.de
cDepartment of Chemistry, Physical Chemistry/Biophysical Chemistry, Technische Universität Berlin, Straße des 17. Juni 135, Sekr. PC14, 10623 Berlin, Germany
dStrategy Department of Energy and Information, Helmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, 14109 Berlin, Germany
First published on 21st September 2023
Abstract
Electrochemical production of large-scale chemicals and fuels is critical to reaching carbon neutrality. However, the required anodic oxidation reactions, namely the oxygen evolution reaction (OER) or the oxidation of organics into value-added products, suffer from large overpotentials. To address this challenge, researchers have been widely investigating non-water-soluble (pre)catalysts to operate in the aqueous electrolyte. On the contrary, in this work, we approach a rapid, easy, and green carbon cloth electrode preparation using merely water-soluble nitrate precursors and ethanol as chemicals and no heating steps. The drop-coated, water-soluble transition metal salts reconstruct rapidly into the respective oxyhydroxides under OER conditions, with the oxyanion acting as a beneficial sacrificial reagent. This approach is shown herein for nickel(–iron) catalysts and their successful application for the OER (220 mV overpotential at 10 mA cm−2, long-term stability of 40 h at 100 mA cm−2) and the oxidation of 5-hydroxymethylfurfural (HMF, quantitative faradaic efficiency). We compare both reactions with both electrodes closely and find that the iron-free sample is more active for the HMF oxidation in regimes where mass transport is not the main limiting factor. We anticipate that this simple electrode preparation approach can find wide application in electrocatalysis and beyond.
Introduction
Fossil fuels and petrochemicals must be replaced by carbon-neutral alternatives to stop climate change and close the carbon cycle.1 The most promising alternatives are hydrogen and hydrocarbons produced electrocatalytically through water or carbon dioxide reduction, respectively.2–4 These (cathodic) reduction reactions require protons and electrons that are supplied by their (anodic) oxidative counterpart. This anodic counterpart can be the oxygen evolution reaction (OER) or an organic oxidation reaction (OOR, hybrid water splitting).1 The OER has the advantage that only water is required as substrate; thus, it can be applied on a large scale.1,5 However, the OER produces no value-added compound. The advantage of organic oxidation reactions is that they require thermodynamically less energy and can be value-added, e.g., 5-hydroxymethylfurfural (HMF) oxidation can yield 2,5-furandicarboxylic acid (FDCA), a biomass-derived, value-added precursor for sustainable polymers.4,6,7 However, the demand for organic oxidation products is much smaller than for hydrogen and hydrocarbon fuels.1 Thus, the OOR can only partly replace the OER, and both reactions are crucial.1 Nevertheless, both reactions still suffer from large overpotentials, substantially lowering the efficiency of electrolysers.1 Thus, new catalysts that combine a high activity with a fast, green, cheap, and straightforward synthesis must be developed.So far, a prerequisite for OER and OOR (pre)catalysts has been that they are non-water-soluble, as water is the main component of the electrolyte, leading to the potential loss of the electrodes’ catalyst coating. Therefore, various non-water-soluble compounds have been investigated, such as transition metal (oxy)hydroxide,8–10 borides,11 pnictides,12 chalcogenides,13,14 alloys,15 or intermetallics.16–20 These materials comprise an active site transition metal, such as iron, cobalt, or nickel, and additional constituting elements. Under alkaline OER conditions, iron, cobalt, and nickel form oxyhydroxides, and the additional constituting elements oxidise, forming oxyanions.19,21–24 These formed oxyanions leach into the electrolyte.13,23 Thus, the additional constituents act as a sacrificial reagent, helping to create a high surface area, defect-rich, and catalytically more active iron/cobalt/nickel oxyhydroxide phase.13,19 Considering the inevitable and beneficial reconstruction process of the applied precatalysts, we projected that the simplest, commercially available transition metal salts, such as nitrates/sulfates/acetates, could also be suitable precatalysts, despite their water solubility, as they could rapidly reconstruct to the respective non-soluble oxyhydroxides while the anion acts as a beneficial sacrificial reagent.25 Furthermore, such species can be dissolved and then drop-coated, enabling a simple, rapid, and reliable deposition. Such cheap, water-soluble, readily available precatalysts could enormously simplify synthesising catalysts with various transition metals, accelerating electrocatalyst development in general.
Nickel and nickel–iron are among the promising non-noble metals for the alkaline OER and OOR, making them suitable to test our water-soluble precatalyst approach.6,15,26–31 Purely nickel-based systems are usually mediocre OER catalysts as they too strongly bind the first OER intermediate, *OH formed in the nickel(II) oxidation peak.32–34 The addition of iron substantially improves the OER activity by shifting the *OH formation to higher energies/potentials, optimising the OER intermediate binding strengths.32 For nickel- and nickel–iron-based catalysts, the OER and OOR share this first *OH reaction intermediate.32,35,36 Thus, these systems can be bifunctional for both processes. While the superiority of nickel–iron-based systems for the OER is unambiguous,37 it is controversial which system performs better for the OOR.35,36,38–40 Some of the best OOR performances have been reported for nickel–iron-based systems.38–41 However, other reports showed that monometallic, nickel-based systems could outperform bimetallic nickel–iron ones due to an easier formation of the *OH intermediate and a better suppression of the OER, an unwanted side reaction during OOR.6,32,35,36,42,43 Thus, another report comparing nickel- and nickel–iron-based systems closely for the OER and OOR is valuable.
Herein, we used simple nickel and iron nitrate precursors that we dissolved in ethanol and drop-casted on a metal-free electrode substrate, carbon cloth. This approach leads to the deposition of the respective nitrates. Under OER conditions, these nitrates reconstruct within seconds into the respective oxyhydroxides. The oxyhydroxide-loaded electrodes can be rapidly prepared, their loading straightforwardly controlled, their synthesis requires no additional chemicals besides ethanol, and they achieve an excellent OER (overpotential of 220 mV at 10 mA cm−2) and OOR performance (quantitative faradaic efficiency). Furthermore, we find that the nickel electrode performs better than the nickel–iron one for the OOR as it can sustain a quantitative faradaic efficiency until higher potentials and forms the reactive *OH intermediate more easily. However, in regimes where mass transport is strongly limiting, nickel- and nickel–iron-based systems can show the same conversion rate and the kinetic superiority of the monometallic nickel sample can be missed, explaining previous observations.
Results and discussion
Drop coating of the water-soluble nickel and iron nitrate salts
To realise the drop-coating of the water-soluble precursors, we choose carbon cloth as substrate, as it is transition-metal-free, cheap, conducting, comparably inert, has widely been applied for anodic oxidation reactions already, has a reasonably high surface area, and can soak in solutions.44 As transition metal salt, we choose nickel and iron nitrate, as they are cheap, widely applied, readily available, and can be obtained in high purities. Ethanol proved to be more suitable for drop-coating than water as it evaporates more quickly, and both nitrate salts are readily soluble inside. In conclusion, this approach requires only the electrode substrate, a pipette, metal precursors, ethanol, and not a single additional chemical, binder, or machinery.25 Furthermore, the drop coating can be performed within seconds, and no heating step is required, making this approach sustainable.45 Additionally, in contrast to most electrodeposition approaches,46,47 the loading can be controlled straightforwardly without using quartz microbalances, and this approach should be suitable for various transition metals, as it just requires a soluble precursor.
Fig. 1 shows in detail how the electrodes were prepared following these considerations. One electrode was drop-coated using pure nickel nitrate solution (NiNO3-CC) and the other using a nickel–iron nitrate solution in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio (NiFeNO3-CC). NiNO3-CC and NiFeNO3-CC were characterised by powder X-ray diffraction (PXRD, Fig. S1) infrared (Fig. S2) and Raman spectroscopy (Fig. S3†) showing that nickel(–iron) nitrate hexahydrate formed on the carbon cloth after drying at ambient conditions.
:1 molar ratio (NiFeNO3-CC). NiNO3-CC and NiFeNO3-CC were characterised by powder X-ray diffraction (PXRD, Fig. S1) infrared (Fig. S2) and Raman spectroscopy (Fig. S3†) showing that nickel(–iron) nitrate hexahydrate formed on the carbon cloth after drying at ambient conditions.
 | ||
| Fig. 1 Schematic illustration of the electrode preparation. Commercial carbon cloth (CC) was activated through acid treatment (HNO3). Ni(NO3)2 and a mixture of 80% Ni(NO3)2 and 20% Fe(NO3)3 was drop coated onto the carbon cloth and the substrate was dried under air. Subsequently, the electrodes were activated at 1.63 VRHE in 1.0 M KOH for 30 minutes to ensure full reconstruction into the respective oxyhydroxides. | ||
Reconstruction to nickel(–iron) oxyhydroxides
The next step is the reconstruction of the transition metal nitrate precursors into the respective oxyhydroxides. Here, it is essential to note that the nitrate salts are water-soluble. However, in alkaline conditions, (oxy)hydroxides are thermodynamically most stable and should form over time in accordance with the Pourbaix diagrams.48 Thus, to enable a complete and rapid reconstruction with beneficial nitrate leaching, an anodic potential of 1.63 VRHE was immediately applied when the electrodes entered the 1 M KOH electrolyte (pH 13.89).49 The current responses (Fig. S4†) show an initial peak that we assign to the precatalyst oxidation, followed by a relatively stable current that we assign to the OER. The precatalyst oxidation completes within seconds. To ensure that the material reached a steady state, we kept it for 30 min under the OER potential.The electrodes reconstructed for 30 min were washed with demineralised water and then characterised. These electrodes are called NiNO3-CC-OER and NiFeNO3-CC-OER, depending on the drop-coated transition metal. The PXRDs of both electrodes contain no sharp peaks (Fig. S1†), revealing that the crystalline nitrates have been reconstructed into X-ray amorphous phases. The infrared spectra of these amorphous phases are comparable to those of nickel(–iron) oxyhydroxides with intercalated carbonate (Fig. S2†).50 In- and ex situ Raman data (Fig. 2a and d) further supports the formation of oxyhydroxides as the in situ Raman data shows the typical bending δ(MIII-O) 470 cm−1 and stretching ν(MIII-O) 566 cm−1 vibrations of Ni(Fe)OOH. The formation of such intercalated oxyhydroxides during OER has been widely reported in the literature.12,23,29,51,52 For both phases, SEM-EDX mappings (Fig. 2a and b, for EDX spectra see Fig. S5 and S6†) disclose that nickel, (iron), oxygen, and potassium are homogeneously distributed. As reported previously, the potassium is most likely intercalated into the oxyhydroxide structure.12,23,29 For both compounds, transmission electron microscopy (TEM, Fig. 2c and f) shows porous, unordered, sheetlike morphologies. Even though both compounds did not show diffraction peaks in our PXRD setup, the selected area electron diffraction (SAED) pattern reveals that both are nanocrystalline. The diffraction rings of the SAED are typical for transition metal oxyhydroxides formed during the OER.12,53–56 The fast Fourier transforms of high-resolution TEM images confirm the lattice distances from the SAED, and the images show around 2 nm large crystallites, explaining the absence of diffraction in the PXRD. Thus, through the reconstruction of the water-soluble nitrates, porous, nanocrystalline, layered oxyhydroxides have formed.
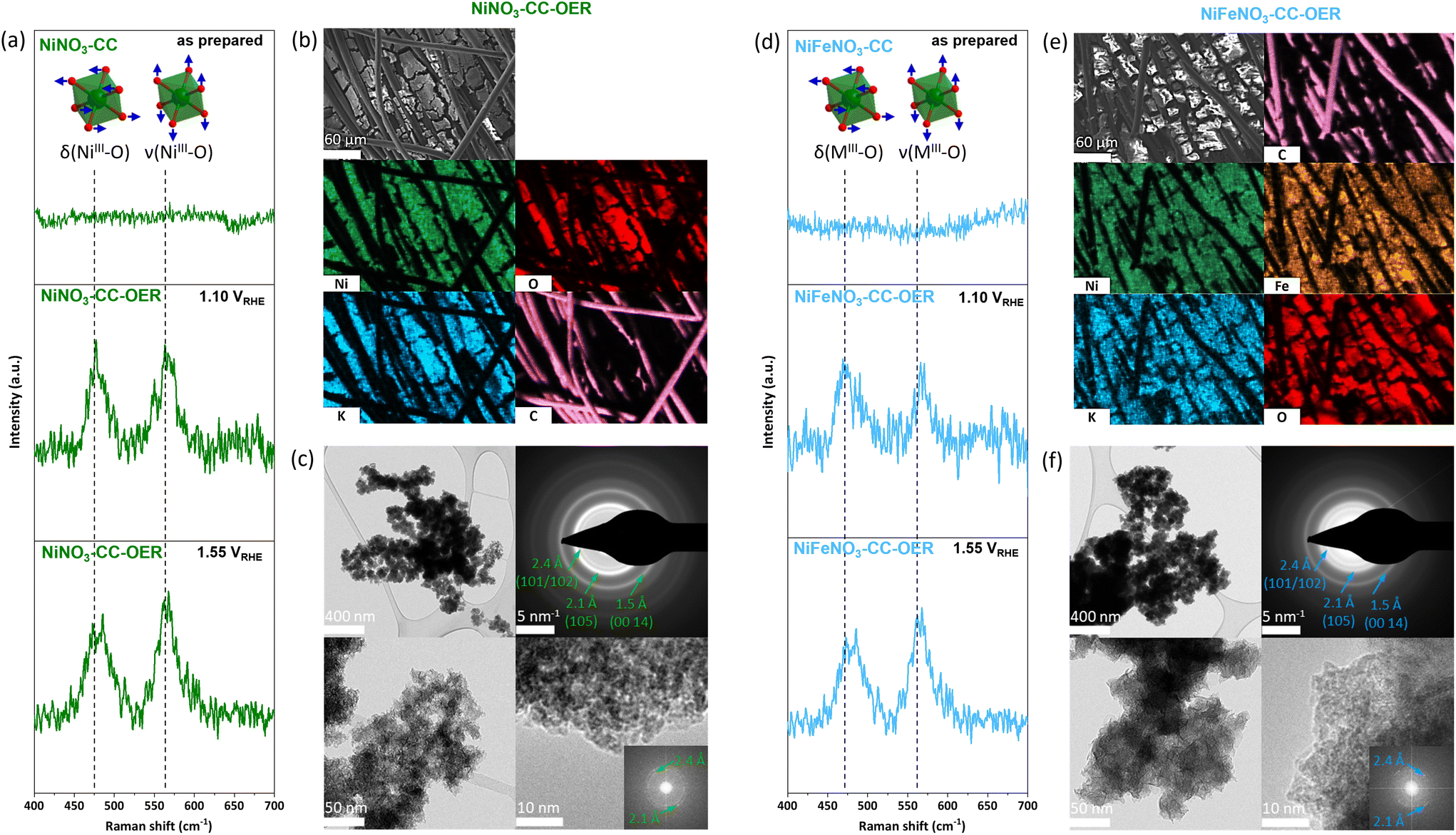 | ||
| Fig. 2 (a) and (d) in situ Raman spectra of NiNO3-CC/NiFeNO3-CC and NiNO3-CC-OER/NiFeNO3-CC-OER (see Fig. 1 for sample abbreviations) at 1.10 VRHE and 1.55 VRHE, showing the formation of oxyhydroxides. The vertical dashed lines mark the prominent vibrations of the [MO6] octahedra. The spectra of the as prepared electrodes can be found in Fig. S3.† (b) SEM image of NiNO3-CC-OER and SEM/EDX mapping of NiNO3-CC-OER showing a homogeneous distribution of nickel and oxygen as well as the incorporation of potassium from the electrolyte during activation. The EDX spectra can for the mapping can be found in Fig. S5† (c) TEM images of NiNO3-CC-OER. (e) SEM image of NiFeNO3-CC-OER and SEM/EDX mapping of NiFeNO3-CC-OER showing a homogeneous distribution of nickel, iron, and oxygen as well as the incorporation of potassium from the electrolyte during activation. The EDX spectra can for the mapping can be found in Fig. S6† (f) TEM images of NiFeNO3-CC-OER. | ||
Electrocatalytic oxygen evolution reaction (OER)
First, a loading study (0.025–4.0 mg cm−2, Fig. 3a) concerning the OER activity at 400 mV overpotential (η) was conducted for NiNO3-CC-OER and NiFeNO3-CC-OER. For both, it shows that increasing the loading to 2 mg cm−2 leads to a substantial rise in the current density. Therefore, a loading of 2 mg cm−2 have been used throughout this manuscript. For loadings above 2 mg cm−2, the current density quickly reaches a plateau, indicating a limited utilisation of the additionally loaded catalyst. Normalising the current density per loaded transition metal confirms this limited utilisation, as it keeps decreasing at higher loadings (yellow oval in Fig. 3a).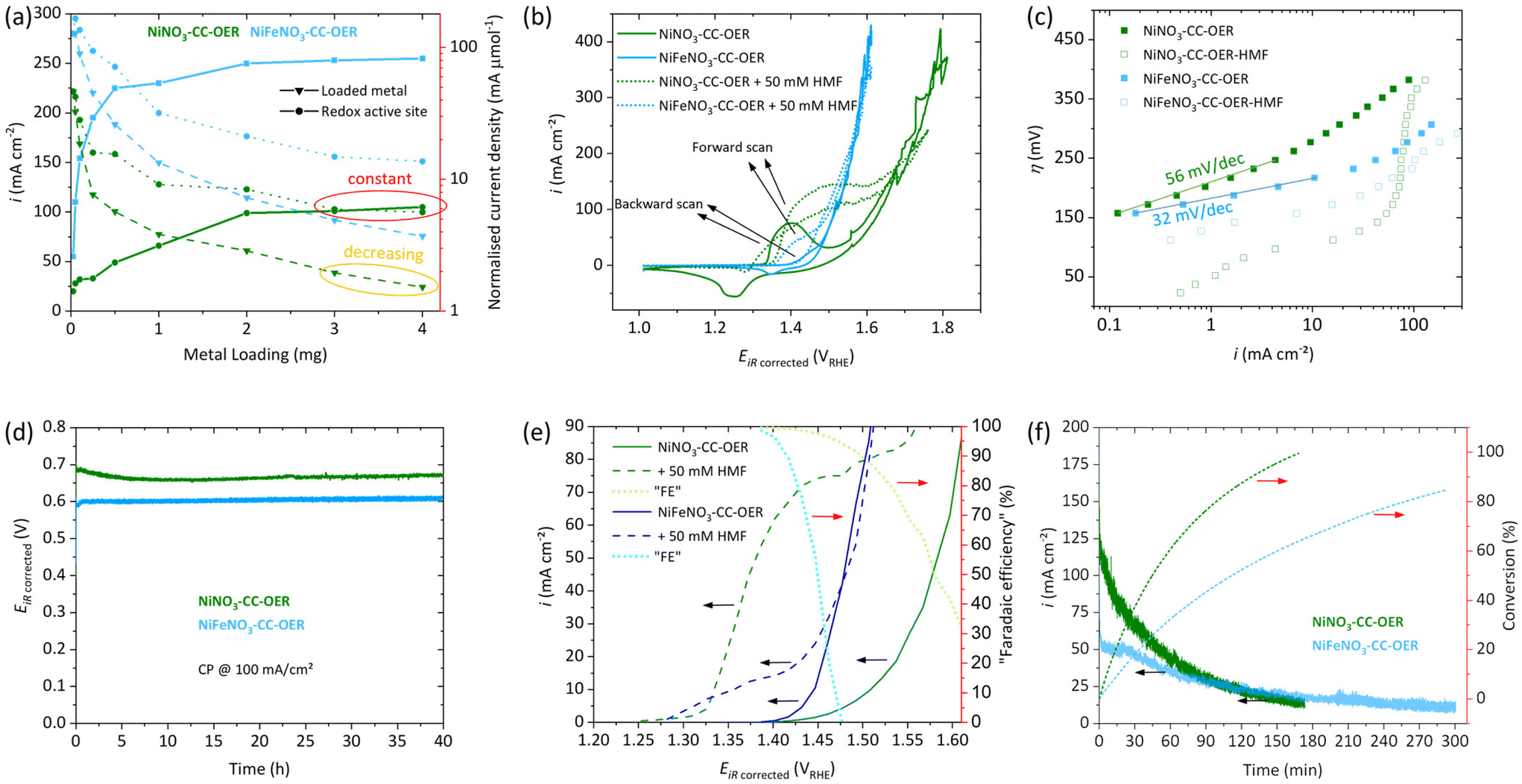 | ||
| Fig. 3 Electrochemical data, all measurements were conducted in 1.0 M KOH in a three-electrode setup with platinum as the counter electrode with Hg/HgO reference. See Fig. 1 for the naming of the samples. (a) Loading study of NiNO3-CC-OER (green) and NiFeNO3-CC-OER (blue) per geometric current density (solid lines with squares) with normalisations per loaded metal (dashed lines with triangles) and per redox active sites (dotted lines with circles) as determined in Fig. S7.† (b) Cyclic Voltammetry of NiNO3-CC-OER and NiFeNO3-CC-OER at 5 mV s−1 with and without 50 mM HMF. (c) Tafel plots of NiNO3-CC-OER and NiFeNO3-CC-OER acquired through steady-state measurements. (d) 40-hour Stability measurement of NiNO3-CC-OER and NiFeNO3-CC-OER through chronopotentiometry at 100 mA cm−2. (e) Potential versus current density through steady-state CA. “Faradaic efficiency” is the current portion that contributes to HMFOR at a given potential in per cent, assuming the OER current is identical with and without HMF. (f) CA data of a HMF bulk oxidation at 1.43 VRHE with 600 rpm stirring conversion of HMF to FDCA on the right y-axis. | ||
To understand this decrease in activity per loaded transition metal, we determined the number of redox-active sites, an often-used descriptor for OER active site availability.28,35,53 The redox activity has been measured by a previously suggested potential step method (Fig. S7†) and used to normalise the current density.57,58 The normalised current decreases initially but reaches a plateau at high loadings (3 mg cm−2) together with the geometric current density (red ovals in Fig. 3a). Thus, higher loadings lead to almost no new redox active sites and simultaneously to no increase in the geometric current density. Prerequisites for redox activity and OER availability of a site are electrolyte access and electric connection to the anode. For high loadings, the additionally loaded nickel/iron sites do not fulfil these two criteria and thus cannot participate in electrocatalysis.
We note here that double-layer capacitance measurements by cyclic voltammetry (CV) cannot be applied for these materials, as their conductivity is insufficient at potentials below the redox peak.24,30,37,59,60 For our system, previously suggested impedance-based method were not successful, as the suggested simulation models cannot reproduce our data.59,60
The electrocatalytic properties towards the OER in 1 M KOH were examined for both NiNO3-CC-OER and NiFeNO3-CC-OER. The CV reverse scans shown in Fig. 3(b) reveal an overpotential at 10 mA cm−2 (η10) of 290 mV for NiNO3-CC-OER and 220 mV for NiFeNO3-CC-OER. Both values were confirmed by chronopotentiometry measurements, maintaining these potentials for 30 minutes (Fig. S8†). These overpotentials compare favourably with previously reported ones (Table S1†). Furthermore, the incorporation of iron shifts the metal oxidation feature to higher potentials, overlapping with the onset of the OER, which has been reported previously.28,35,61 The contribution of carbon cloth was also investigated by CV, revealing current densities below 1 mA cm−2 at relevant potentials (Fig. S9†). Steady-state Tafel analysis was conducted to get insights into the kinetics of the different electrodes. Fig. 3c reveals that the estimated Tafel slope of the NiNO3-CC-OER electrode is 56 mV dec−1 and thus significantly larger than for NiFeNO3-CC-OER, which is 32 mV dec−1. Such a difference has already been reported for iron incorporation. It is consistent with the substantially better OER performance of the nickel–iron oxyhydroxides, where most likely edge-site, bimetallic, adjacent nickel–iron sites are responsible for the OER.29,30,54
We also examined the long-term stability to ensure that the simple drop-coating preparation method yielded stable electrodes. Fig. 3d reveals excellent stability over 40 hours at 100 mA cm−2. After this stability test, SEM images (Fig. S10 and 11†) with elemental mappings still show a homogeneous distribution of nickel, iron, oxygen, and potassium. Furthermore, no significant change in the morphology compared to our investigations after 30 min (Fig. 2b and e) can be seen. The XRD pattern after the 40 h stability test reveals that the materials remain amorphous (Fig. S12†).
Electrocatalytic hydroxymethylfurfural (HMF) oxidation
Besides the OER, we also investigated the electrocatalytic capabilities of our drop-coated NiNO3-CC-OER and NiFeNO3-CC-OER electrodes towards the conversion of 5-hydroxymethylfurfural (HMF) to furan dicarboxylic acid (FDCA), a promising sustainable biopolymer building block. First, cyclic voltammetry was conducted in 1 M KOH electrolyte without stirring and with the addition of 50 mM HMF (Fig. 3b). For a NiNO3-CC-OER, without HMF, a nickel(II) oxidation peak onsets at ∼1.34 VRHE; with HMF, larger current onsets around the same potential. This additional current is assigned to the oxidation of HMF.35,38,62 In the nickel(II) oxidation peak nickel(II) is oxidised to nickel(III) and NiIII+δ-O−II+(1−δ) (around 1.6 electrons per nickel are transferred and oxygen is redox non-innocent).28,35,54,63,64 Two different mechanisms for the HMFOR have been proposed in the literature, depending on the applied reaction conditions and catalysts (see Fig. S13†)6,36,40,62,65 (i) indirect oxidation, where the metal acts as a redox mediator that changes its valence state, (ii) and direct oxidation, which is potential dependent and directly oxidises HMF involving NiIII+δ-O−II+(1−δ). Nickel-based catalysts in strongly alkaline electrolytes (pH > 13) are known to favour the indirect oxidation pathway. Ni(OH)2 is oxidised to NiOOH, which is a potential-dependent step. The subsequent HMF oxidation, however, is influenced by the number of active sites that can be reduced back to Ni(OH)2 and the intrinsic catalytic activity of the catalyst.6,36,40,62,65 The enlarged oxidation peak in the forward scan of the HMF containing CVs is consistent with this hypothesis, as NiOOH is reduced to Ni(OH)2 by HMF several times in the timespan of one CV resulting in multiple oxidations of the same Ni(OH)2 site enlarging the nickel(II) oxidation peak. Contrary to this, the reduction peak in the back scan is less pronounced because the nickel(III) is reduced through the indirect HMF oxidation catalytic cycle, rather than the applied potential.6 For NiFeNO3-CC-OER, a similar behaviour can be observed, but the currents assigned to the oxidation peak and HMF oxidation are significantly smaller.Fig. 3c also includes steady-state Tafel plots of the two electrodes in HMF containing 1 M KOH. HMF oxidation begins at an overpotential below 25 mV for NiNO3-CC-OER, while for NiFeNO3-CC-OER, it starts at 107 mV. The curves have a more complex shape than the ones in HMF-free electrolyte and lack a long linear range that could be used to unambiguously assign a Tafel slope for the HMF oxidation.
Before a closer look at the competition between the OER and HMF oxidation is taken, we investigated the effect of substrate concentration and stirring speed on the HMF oxidation. In this regard, we conducted chronoamperometry measurements (1.41 VRHE) with different stirring speeds for our system. Interestingly, without stirring, NiNO3-CC-OER does not outperform NiFeNO3-CC-OER as both electrodes only exhibit current densities under 20 mA cm−2, although one would have assumed a better performance for NiNO3-CC-OER from the CVs. However, higher stirring speeds significantly increase the current density, whereby the effect is four times stronger for NiNO3-CC-OER, increasing the current density from 17 mA cm−2 to 100 mA cm−2 (Fig. S14†). For NiFeNO3-CC-OER, even with rigorous stirring speeds, the current density does not increase to more than 40 mA cm−2. The results highlight that mass transport limitations play a vital role for the catalytic turnover. In our unstirred system, these mass transport limitations are the decisive factor. Thus, the intrinsic activity of the catalyst cannot be measured and two catalysts with different intrinsic activities might perform similarly. However, at faster stirring speeds, the electrocatalytic activity of NiFeNO3-CC-OER is limiting the overall performance, while NiNO3-CC-OER's current density is substantially higher. We suggest that these mass transport limitations might be the reason why some reports have missed the superior activity of purely nickel-based systems compared to nickel–iron-based ones.
When HMF oxidation is the desired reaction, OER is an unwanted side reaction. However, at sufficiently large potential, both processes can co-occur. To investigate these two competing reactions as a function of potential, we have acquired steady-state data (3 min chronoamperometry for each data point) in stirred, HMF-free and HMF-containing 1 M KOH electrolyte (Fig. 3e). Only catalytic processes (HMF oxidation and OER) contribute to the current density of the steady-state measurements.66 This steady-state data can be used to determine a potential at which only HMF oxidation and little OER takes place. For NiNO3-CC-OER and NiFeNO3-CC-OER, the HMF onset is around 1.3 VRHE, and the current density increases with positive potential. Then, at higher potential, the OER starts to interfere, the range between can be considered suitable for selective HMF oxidation. For NiNO3-CC-OER, this interference is less due to its 70 mV higher OER overpotential, enabling selective HMF oxidation at higher potentials and thus higher conversion rates. Additionally, using the steady-state data, we calculated and added to Fig. 3e a “faradaic efficiency” for HMF oxidation under the estimation that the OER turnover is the same with and without HMF. Consequently, Fig. 3e can be used to estimate the trade-off between turnover and faradaic efficiency for the HMF oxidation as a function of the anode potential.
Following these investigations, we choose to perform a bulk HMF oxidation with stirring and at a potential of 1.43 VRHE in 1.0 M KOH with 50 mM HMF. While higher HMF concentrations are beneficial for the current density, the reaction time will also increase. As HMF is known to be unstable under strongly alkaline conditions,67 a quick, complete conversion is vital for high yields. Furthermore, at the chosen potential, both catalysts should have a good selectivity towards HMF oxidation (Fig. 3e). NiNO3-CC-OER passed the charge necessary for 100% theoretical conversion in almost half the time (170 min) of NiFeNO3-CC-OER (300 min), as can be seen from Fig. 3f. Both reactions start at reasonable high current densities of 120 mA cm−2 and 50 mA cm−2, respectively, which steadily decline until the necessary charge was passed, due to less substrate availability. The reaction mixture was investigated by 1H-NMR before (Fig. S15) and after (Fig. S16 and S17†) the bulk oxidation. An internal standard was used to quantify the products and calculate the faradaic efficiencies for each catalyst. NiNO3-CC-OER fully converted HMF into FDCA with a faradaic efficiency of 98%, while NiFeNO3-CC-OER could only achieve 88%, indicating that the OER significantly contributed to the current density. The results fit our other findings and support that purely nickel-based systems are superior for HMF oxidation compared to those with iron incorporated.
Conclusions
We have demonstrated a rapid, simple and sustainable approach for synthesising water-soluble precursors that can be rapidly reconstructed into highly active catalysts for the oxygen evolution reaction (OER) and HMF oxidation. By drop-coating commercially available nickel and iron nitrate precursors onto carbon cloth electrodes, we achieved the deposition of the respective nitrates, which subsequently reconstructed rapidly into amorphous nickel–iron oxyhydroxide phases under OER conditions. The resulting nanocrystalline oxyhydroxide electrodes exhibited excellent OER performance, with an overpotential of 220 mV at 10 mA cm−2.The incorporation of iron in the nickel-based system shifted the oxidation of the *OH intermediate to higher energies, optimising the OER intermediate binding strengths and enhancing the OER activity. Furthermore, the nickel-based system demonstrated superior performance in the oxidation of HMF, sustaining quantitative faradaic efficiency until higher potentials and facilitating the formation of the reactive *OH intermediate. Our findings shed light on the performance comparison between nickel-based and nickel–iron-based systems for both the OER and HMFOR. While nickel–iron-based systems showed clear superiority in the OER, the OOR performance was worse. Previous reports have suggested that monometallic nickel systems could outperform bimetallic nickel–iron systems due to easier *OH intermediate formation and a better suppression of the OER during HMFOR. However, in regimes where mass transport limitations are significant, both systems may exhibit similar conversion rates, potentially overlooking the kinetic superiority of the monometallic nickel sample.
The drop-coating method utilising water-soluble precursors offers several advantages, including simplicity, a wide application range, quickness, and cost-effectiveness. It eliminates the need for additional chemicals and enables precise control over catalyst loading. Moreover, this approach can be extended to various transition metals by utilising soluble precursors, thereby accelerating the development of novel (multimetallic) catalysts for electrocatalytic conversions.
Author contributions
J. N. H. initiated the idea and developed it together with T. K. and P. W. M. T. K. prepared the electrodes, performed all electrocatalytic measurements, analysed the data. IR and TEM were measured and analysed by J. N. H. SEM experiments were conducted by I. M. K. L. and I. Z conducted in situ Raman experiments. T. S. helped in the investigation. T. K. and J. N. H. wrote the first draft. P. W. M. discussed the results, commented on the manuscript, and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funded by the German Federal Ministry of Education and Research (BMBF) in the framework of the project Catlab (03EW0015A/B). P. W. M. acknowledges the support from the BMBF project “PrometH2eus”, 03HY105C. The authors thank Prof. Dr Matthias Driess for the infrastructure.References
- T. Kahlstorf, J. N. Hausmann, T. Sontheimer and P. W. Menezes, Glob. Chall., 2023, 2200242 CrossRef PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2019, 58, 343–348 CrossRef PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed.
- J. N. Hausmann, R. Schlögl, P. W. Menezes and M. Driess, Energy Environ. Sci., 2021, 14, 3679–3685 RSC.
- Y. Yang and T. Mu, Green Chem., 2021, 23, 4228–4254 RSC.
- D. J. Chadderdon, L. Xin, J. Qi, Y. Qiu, P. Krishna, K. L. More and W. Li, Green Chem., 2014, 16, 3778–3786 RSC.
- R. Gao, J. Zhu and D. Yan, Nanoscale, 2021, 13, 13593–13603 RSC.
- R. Gao and D. Yan, Adv. Energy Mater., 2020, 10, 1900954 CrossRef CAS.
- Z. Guo, W. Ye, X. Fang, J. Wan, Y. Ye, Y. Dong, D. Cao and D. Yan, Inorg. Chem. Front., 2019, 6, 687–693 RSC.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6, 1502313 CrossRef.
- B. Dasgupta, J. N. Hausmann, R. Beltrán–Suito, S. Kalra, K. Laun, I. Zebger, M. Driess and P. W. Menezes, Small, 2023, 2301258 CrossRef CAS PubMed.
- O. Mabayoje, A. Shoola, B. R. Wygant and C. B. Mullins, ACS Energy Lett., 2016, 1, 195–201 CrossRef CAS.
- R. Gao, H. Zhang and D. Yan, Nano Energy, 2017, 31, 90–95 CrossRef CAS.
- P. W. T. Lu and S. Srinivasan, J. Electrochem. Soc., 1978, 125, 265–270 CrossRef CAS.
- I. Mondal, J. N. Hausmann, G. Vijaykumar, S. Mebs, H. Dau, M. Driess and P. W. Menezes, Adv. Energy Mater., 2022, 12, 2200269 CrossRef CAS.
- J. N. Hausmann, R. Beltrán–Suito, S. Mebs, V. Hlukhyy, T. F. Fässler, H. Dau, M. Driess and P. W. Menezes, Adv. Mater., 2021, 33, 2008823 CrossRef CAS PubMed.
- C. Walter, P. W. Menezes and M. Driess, Chem. Sci., 2021, 12, 8603–8631 RSC.
- J. N. Hausmann and P. W. Menezes, Curr. Opin. Electrochem., 2022, 34, 100991 CrossRef CAS.
- J. Masa and W. Schuhmann, ChemCatChem, 2019, 11, 5842–5854 CrossRef CAS.
- B. R. Wygant, K. Kawashima and C. B. Mullins, ACS Energy Lett., 2018, 3, 2956–2966 CrossRef CAS.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef CAS.
- J. N. Hausmann and P. W. Menezes, Angew. Chem., Int. Ed., 2022, 61, e202207279 CrossRef CAS PubMed.
- J. N. Hausmann, S. Mebs, H. Dau, M. Driess and P. W. Menezes, Adv. Mater., 2022, 34, 2207494 CrossRef CAS PubMed.
- G. Moon, M. Yu, C. K. Chan and H. Tüysüz, Angew. Chem., Int. Ed., 2019, 58, 3491–3495 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- D. A. Corrigan, J. Electrochem. Soc., 1987, 134, 377 CrossRef CAS.
- D. A. Corrigan and R. M. Bendert, J. Electrochem. Soc., 1989, 136, 723–728 CrossRef CAS.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. de Araujo, M. Gliech, D. Teschner, J. Zhu, W.-X. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed.
- F. Dionigi, J. Zhu, Z. Zeng, T. Merzdorf, H. Sarodnik, M. Gliech, L. Pan, W. Li, J. Greeley and P. Strasser, Angew. Chem., Int. Ed., 2021, 60, 14446–14457 CrossRef CAS PubMed.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Electroanal. Chem., 1971, 31, 39–49 CrossRef CAS.
- H. B. Tao, Y. Xu, X. Huang, J. Chen, L. Pei, J. Zhang, J. G. Chen and B. Liu, Joule, 2019, 3, 1498–1509 CrossRef CAS.
- H. Xiao, H. Shin and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5872–5877 CrossRef CAS PubMed.
- A. Govind Rajan, J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2020, 142, 3600–3612 CrossRef CAS PubMed.
- J. N. Hausmann, P. V. Menezes, G. Vijaykumar, K. Laun, T. Diemant, I. Zebger, T. Jacob, M. Driess and P. W. Menezes, Adv. Energy Mater., 2022, 2202098 CrossRef CAS.
- N. Heidary and N. Kornienko, Chem. Sci., 2020, 11, 1798–1806 RSC.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737–3742 CrossRef CAS PubMed.
- P. Hauke, M. Klingenhof, X. Wang, J. F. de Araújo and P. Strasser, Cell Rep. Phys. Sci., 2021, 2, 100650, DOI:10.1016/j.xcrp.2021.100650.
- W.-J. Liu, L. Dang, Z. Xu, H.-Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 CrossRef CAS.
- B. Mondal, N. Karjule, C. Singh, R. Shimoni, M. Volokh, I. Hod and M. Shalom, Adv. Energy Mater., 2021, 11, 2101858 CrossRef CAS.
- L. Wei, M. D. Hossain, M. J. Boyd, J. Aviles-Acosta, M. E. Kreider, A. C. Nielander, M. B. Stevens, T. F. Jaramillo, M. Bajdich and C. Hahn, ACS Catal., 2023, 4272–4282 CrossRef CAS.
- B. Seo, J. Woo, E. Kim, S.-H. Cheong, D. K. Lee and H. Lee, Catal. Commun., 2022, 170, 106501 CrossRef CAS.
- L. Gouda, L. Sévery, T. Moehl, E. Mas-Marzá, P. Adams, F. Fabregat-Santiago and S. D. Tilley, Green Chem., 2021, 23, 8061–8068 RSC.
- N. Cheng, Q. Liu, J. Tian, Y. Xue, A. M. Asiri, H. Jiang, Y. He and X. Sun, Chem. Commun., 2015, 51, 1616–1619 RSC.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- C. G. Morales-Guio, L. Liardet and X. Hu, J. Am. Chem. Soc., 2016, 138, 8946–8957 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- G. K. Schweitzer and L. L. Pesterfield, The Aqueous Chemistry of the Elements, Oxford University Press, Oxford, 2010 Search PubMed.
- J. N. Hausmann, B. Traynor, R. J. Myers, M. Driess and P. W. Menezes, ACS Energy Lett., 2021, 6, 3567–3571 CrossRef CAS.
- B. M. Hunter, W. Hieringer, J. R. Winkler, H. B. Gray and A. M. Müller, Energy Environ. Sci., 2016, 9, 1734–1743 RSC.
- J. N. Hausmann, E. M. Heppke, R. Beltrán–Suito, J. Schmidt, M. Mühlbauer, M. Lerch, P. W. Menezes and M. Driess, ChemCatChem, 2020, 12, 1161–1168 CrossRef CAS.
- J. N. Hausmann, R. A. Khalaniya, C. Das, I. Remy-Speckmann, S. Berendts, A. V. Shevelkov, M. Driess and P. W. Menezes, Chem. Commun., 2021, 57, 2184–2187 RSC.
- J. N. Hausmann, S. Mebs, K. Laun, I. Zebger, H. Dau, P. W. Menezes and M. Driess, Energy Environ. Sci., 2020, 13, 3607–3619 RSC.
- L. Reith, J. N. Hausmann, S. Mebs, I. Mondal, H. Dau, M. Driess and P. W. Menezes, Adv. Energy Mater., 2023, 13, 2203886 CrossRef CAS.
- P. W. Menezes, S. Yao, R. Beltrán–Suito, J. N. Hausmann, P. V. Menezes and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 4640–4647 CrossRef CAS PubMed.
- K. W. Kwan, D. Xie, R. Zhang, Z. Shan and A. H. W. Ngan, Phys. Status Solidi A, 2018, 215, 1800623 CrossRef.
- R. D. L. Smith, C. Pasquini, S. Loos, P. Chernev, K. Klingan, P. Kubella, M. R. Mohammadi, D. Gonzalez-Flores and H. Dau, Nat. Commun., 2017, 8, 2022 CrossRef PubMed.
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spöri, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Pérez-Ramírez, B. R. Cuenya, R. Schlögl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed.
- S. Watzele, P. Hauenstein, Y. Liang, S. Xue, J. Fichtner, B. Garlyyev, D. Scieszka, F. Claudel, F. Maillard and A. S. Bandarenka, ACS Catal., 2019, 9, 9222–9230 CrossRef CAS.
- S. S. Jeon, P. W. Kang, M. Klingenhof, H. Lee, F. Dionigi and P. Strasser, ACS Catal., 2023, 13, 1186–1196 CrossRef CAS.
- M. B. Stevens, C. D. M. Trang, L. J. Enman, J. Deng and S. W. Boettcher, J. Am. Chem. Soc., 2017, 139, 11361–11364 CrossRef CAS PubMed.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K.-S. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS PubMed.
- D. A. Kuznetsov, B. Han, Y. Yu, R. R. Rao, J. Hwang, Y. Román-Leshkov and Y. Shao-Horn, Joule, 2018, 2, 225–244 CrossRef CAS.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Chem. Soc., Perkin Trans. 2, 1972, 1396 RSC.
- Y. Gao, L. Ge, H. Xu, K. Davey, Y. Zheng and S.-Z. Qiao, ACS Catal., 2023, 11204–11231 CrossRef CAS.
- S. Anantharaj, S. Noda, M. Driess and P. W. Menezes, ACS Energy Lett., 2021, 6, 1607–1611 CrossRef CAS.
- M. L. Krebs, A. Bodach, C. Wang and F. Schüth, Green Chem., 2023, 25, 1797–1802 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Containing all experimental procedures and characterisation details as well as additional data. See DOI: https://doi.org/10.1039/d3gc02119j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |