
Electrochemical synthesis of γ-keto sulfones containing a β-quaternary carbon center via 1,2-migration†
Wen
Xia
a,
Yawen
Yang
a,
Xiaohui
Zhang
a,
Liangzhen
Hu
*a and
Yan
Xiong
 *abc
*abc
aSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing 401331, P. R. China. E-mail: xiong@cqu.edu.cn
bState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China
cSchool of Chemical and Environmental Engineering, and Collaborative Innovation Center for High Value Transformation of Coal Chemical Process By-products, Xinjiang Institute of Engineering, Xinjiang 830091, P. R. China
First published on 13th September 2023
Abstract
An effective electrochemical rearrangement reaction, both oxidant and metal catalyst-free, is presented to construct γ-keto sulfones containing a β-quaternary carbon center in this study. In the protocol, radicals are generated from benzenesulfonyl hydrazines, which cause 1,2-migrations of starting materials, 2-methylallyl alcohol derivatives, and formation of new C–C and C–S bonds. A series of aryl-migration, alkyl-migration and ring expansion products are obtained in good yields with excellent functional group tolerance. Gram-scale reaction demonstrates that the electro-oxidative rearrangement strategy has the potential for industrial practicability. Additionally, a possible and reasonable mechanism is proposed based on existing literature reports, control experiments and cyclic voltammograms.
Introduction
Organic keto sulfones with bifunctional groups have generated a great deal of interest owing to their high biological and pharmacological activity (Fig. 1).1 For example, it has already been demonstrated that N-substituted 4-arylsulfonylpiperidine-4-hydroxamic acids and their derivatives are oral MMP (matrix metalloproteinase) inhibitors for the treatment of osteoarthritis.2 A series of β-carbonyl sulfone derivatives were also reported to show antibacterial activity against Staphylococcus aureus and Candida tropicalis.3 Research studies have also indicated that sulfones with an imidazo[1,5-c]imidazol-3-one moiety have the potential to be used as a kind of orally bioavailable coagulation enzyme factor Xa (FXa) inhibitor.4 Additionally, advanced prostate cancer has been routinely treated with the antiandrogen drug bicalutamide, which contains a γ-carbonyl sulfone component.5 Furthermore, molecules possessing both carbonyl and sulfonyl functional moieties were used as starting materials to synthesize further organic compounds or natural products.6 In recent decades, more synthetic methods have been developed for γ-keto sulfones, a typical representative category, in addition to the conventional oxidative synthesis from alcohols and thioethers.7 Unsaturated vinyl sulfones and aldehydes or acyl halides are typically used in an intermolecular coupling reaction to synthesize these compounds.8 Additionally, there are also reports that oxosulfones are prepared from α,β-enones via the Michael addition of sulfinate anions or sulfonyl radicals.9 Recently, Wu and co-workers described a novel strategy of separate-embedding into sulfonylmethylate and synthesizing γ-keto sulfones by using rongalite as both a C1 synthon and a sulfone source.10 Overall, chemists have made increasing progress in the synthesis of carbonyl sulfones.11 However, due to the use of complicated catalysis and oxidants, gentler and eco-friendly approaches are required for further exploration.12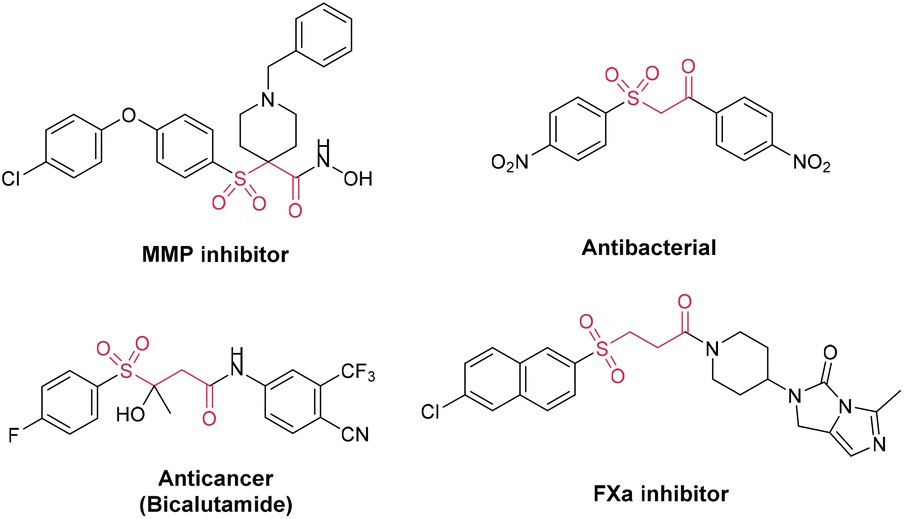 | ||
| Fig. 1 Representative bioactive keto sulfone derivatives. | ||
The oxidative rearrangement of α,α-disubstituted allyl alcohol derivatives mediated by various sulfone sources is a new strategy for constructing such γ-keto sulfones. At the earliest opportunity, Ji proposed a two-step method for generating these compounds from α,α-diaryl allyl alcohols using benzenesulfonic acids as sulfone sources (Scheme 1a).13 The aforementioned work reported the necessity of the use of unstable hypervalent iodine oxidants in addition to operation at a high temperature. Most recently, Wang also reported a reductive radical-mediated 1,2-migration assisted by an azidyl group from 2-azidyl allyl alcohols (Scheme 1b).14 This method used sodium benzenesulfonates as the sulfone sources, took a very long time to complete and required extremely difficult operation because of the use of dangerous azides. It must be mentioned that nearly no existing methods have been able to construct a β-quaternary carbon center while generating γ-keto sulfones. Therefore, based on the findings in the literature and our earlier work,15 we developed a novel method for the synthesis of γ-keto sulfones with a β-quaternary carbon center from α,α-disubstituted 2-methyl allyl alcohol derivatives (Scheme 1c). The protocol used a new kind of benzenesulfonyl hydrazine as a sulfone source to provide sulfonyl and was efficiently capable of causing 1,2-C migration under electrochemical conditions. The raw materials of this strategy were low-cost and easily prepared. Other than the release of hydrogen and nitrogen during the oxidation rearrangement, there were no other byproducts. This oxidation protocol was accomplished via electron transfer without the utilization of hazardous and unstable oxidants or metal catalysts. All these metrics collectively demonstrated that the synthesis strategy shows atom economy and eco-friendliness, which is in line with the concept of green chemistry.16
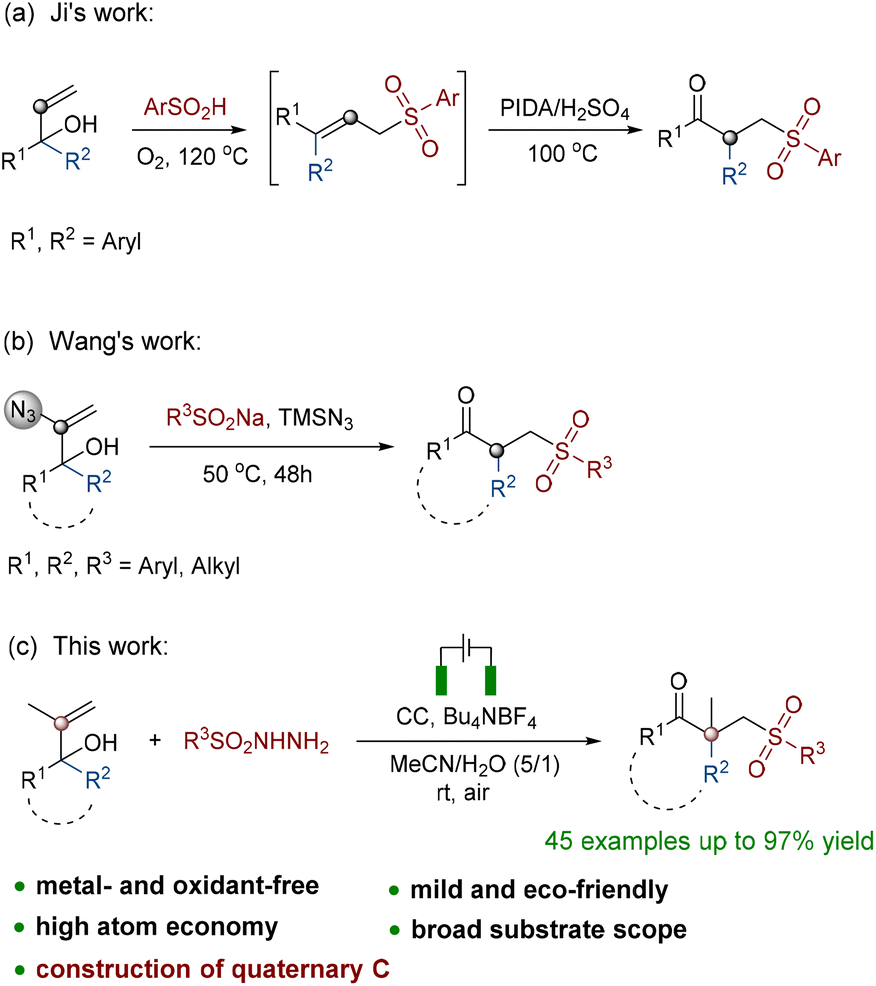 | ||
| Scheme 1 Rearrangement reactions of γ-keto sulfones. | ||
Results and discussion
We commenced the investigation by using easily prepared 2-methyl-1,1-diphenylprop-2-en-1-ol (1a) and directly purchased 4-methylbenzenesulfonhydrazide (2a) as model substrates. The electrochemical rearrangement reaction was conducted to obtain the target product 3aa in 55% yield with tetrabutylammonium hexafluorophosphate (Bu4NPF6) as the electrolyte in an undivided cell equipped with two graphite plates (Table 1, entry 1). To determine the optimal conditions, the amount of reactant 2a was initially considered (entries 1–3). The results demonstrated that two equivalents of 2a worked best, and increasing or decreasing the amounts of 2a decreased the yields. Subsequently, the influence of various electrodes on the reaction system was taken into consideration (entries 4–6). A graphite plate anode was beneficial for the generation of radicals. The porous nickel foam utilized as the cathode enabled a quick reaction with up to 68% yield due to its higher specific surface area. Conventional organic and inorganic electrolytes were also screened. The most efficient electrolyte was tetrabutylammonium tetrafluoroborate (Bu4NBF4), which increased the yield to 79% (entries 7–13). Except for the initial MeCN/H2O mixture system, it was discovered after examining the solvents that the majority of them had a detrimental effect on the electrochemical reaction (entries 14–18). The magnitude of current was ultimately taken into account in this constant current reaction (entries 19 and 20). The electrochemical procedure, which produced the target product with a 91% yield, was greatly influenced by a 15 mA current. However, experimental results showed that this strategy could not be implemented without current (entry 21).|
|
|||||
|---|---|---|---|---|---|
| Entry | Electrode | Electrolyte | Solvent | Current (mA) | Yieldb (%) |
| a Reaction conditions: graphite plate anode (10 × 10 × 3 mm), nickel foam cathode (10 × 10 × 2 mm), constant current, 1a (0.25 mmol), 2a (0.5 mmol, 2eq.), Bu4NBF4 (0.5 mmol, 2eq.), MeCN/H2O (6 mL, 5/1), rt, air, and 4 h. b Yield determined by 1H NMR using mesitylene as an internal standard. c 0.4 mmol TsNHNH2. d 0.6 mmol TsNHNH2. | |||||
| 1 | C(+)|C(−) | Bu4NPF6 | MeCN/H2O | 10 | 55 |
| 2c | C(+)|C(−) | Bu4NPF6 | MeCN/H2O | 10 | 41 |
| 3d | C(+)|C(−) | Bu4NPF6 | MeCN/H2O | 10 | 52 |
| 4 | C(+)|Pt(−) | Bu4NPF6 | MeCN/H2O | 10 | 12 |
| 5 | C(+)|Fe(−) | Bu4NPF6 | MeCN/H2O | 10 | 57 |
| 6 | C(+)|Ni(−) | Bu4NPF6 | MeCN/H2O | 10 | 68 |
| 7 | C(+)|Ni(−) | Bu4NClO4 | MeCN/H2O | 10 | 75 |
| 8 | C(+)|Ni(−) | Bu4NBF4 | MeCN/H2O | 10 | 79 |
| 9 | C(+)|Ni(−) | Bu4NI | MeCN/H2O | 10 | Trace |
| 10 | C(+)|Ni(−) | KI | MeCN/H2O | 10 | Trace |
| 11 | C(+)|Ni(−) | NaCl | MeCN/H2O | 10 | 31 |
| 12 | C(+)|Ni(−) | NaBF4 | MeCN/H2O | 10 | 49 |
| 13 | C(+)|Ni(−) | LiClO4 | MeCN/H2O | 10 | 70 |
| 14 | C(+)|Ni(−) | Bu4NBF4 | MeCN | 10 | 54 |
| 15 | C(+)|Ni(−) | Bu4NBF4 | MeOH | 10 | 36 |
| 16 | C(+)|Ni(−) | Bu4NBF4 | DMF | 10 | NR |
| 17 | C(+)|Ni(−) | Bu4NBF4 | DCM | 10 | NR |
| 18 | C(+)|Ni(−) | Bu4NBF4 | EA | 10 | Trace |
| 19 | C(+)|Ni(−) | Bu 4 NBF 4 | MeCN/H 2 O | 15 | 91 |
| 20 | C(+)|Ni(−) | Bu4NBF4 | MeCN/H2O | 20 | 73 |
| 21 | C(+)|Ni(−) | Bu4NBF4 | MeCN/H2O | 0 | NR |
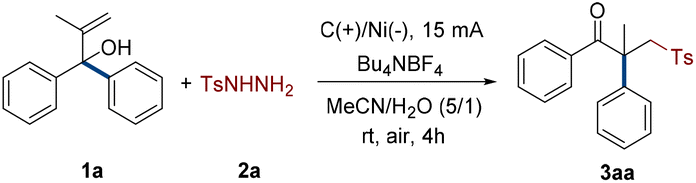
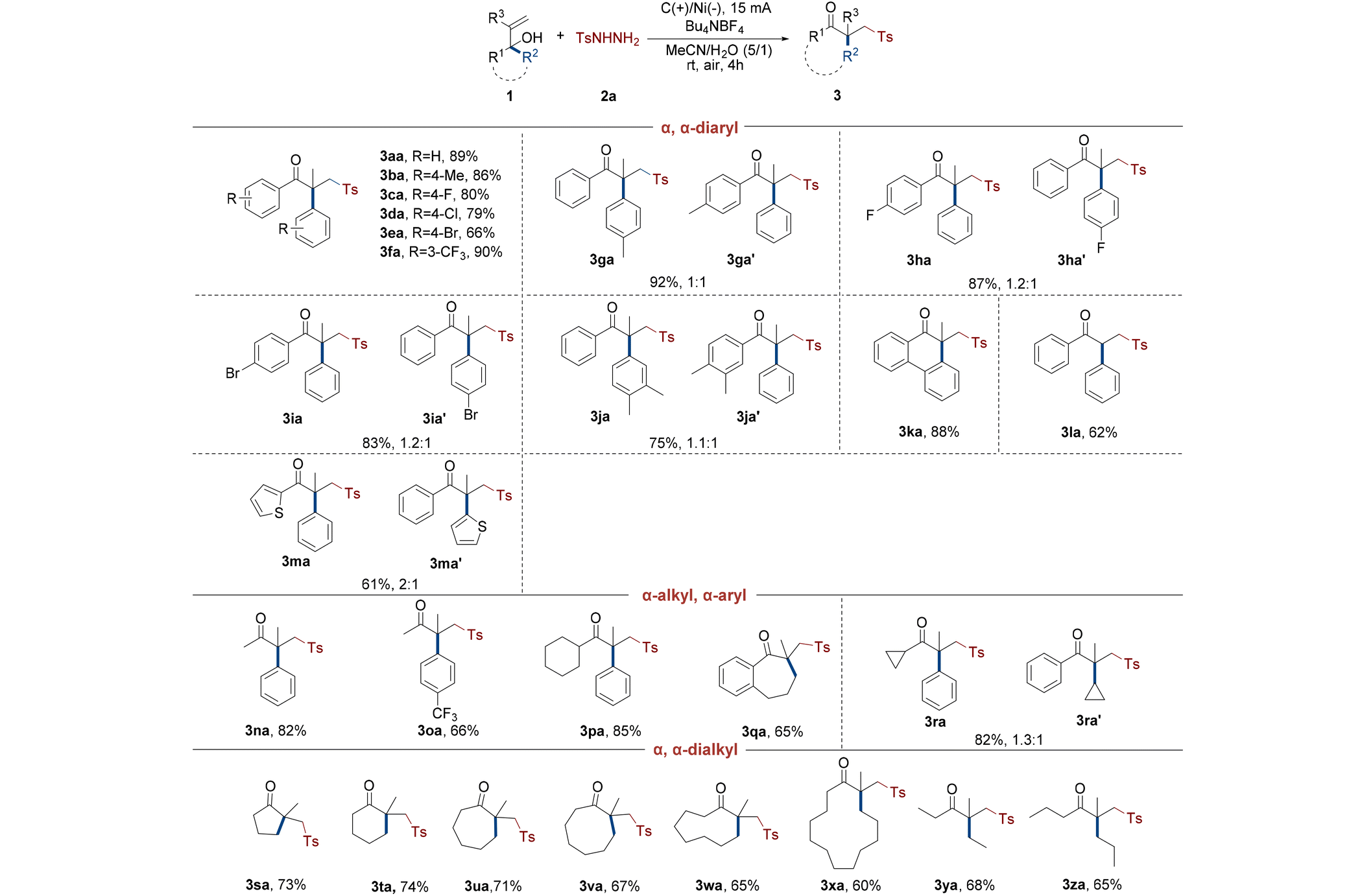
After determining the optimal reaction conditions, the scope of alcohols was investigated (Table 2). Numerous experiments indicated that α,α-diaryl allyl alcohols were capable of this electrochemical rearrangement conversion. For instance, under ideal conditions, symmetrical α,α-diaryl allyl alcohols yielded standard γ-keto sulfone 3aa with an isolated yield of 89%. The symmetrical para-halogen or electron-donating methyl groups on the allyl alcohols easily underwent aryl migration to produce the species in good yields (3ba–ea). Trifluoromethyl, a meta strong electron-withdrawing group, was well tolerated in this rearrangement process and had an excellent yield of 90% (3fa). Based on the findings, many asymmetrical α,α-diaryl allyl alcohols containing para-methyl or halogen also generated the desired sulfones in excellent total yields as high as 92% (3ga–ia, 3ga′–ia′). Using this 1,2-migration technique on asymmetrical substrates with dimethyl resulted in up to 75% overall yield of the target products (3ja and 3ja′). Given the fact that these substrates have two different aromatic groups at the α-position, their capacity to migrate was uneven due to the effect of the substituents on the benzene ring. The 1H NMR spectra of these crude products demonstrated that benzene rings substituted by electron-donating para-methyl had better migration ability than unsubstituted phenyl, while benzene rings with a para-halogen had poorer migration ability than phenyl. However, methyl and halogen had only a little effect on the capacity of aryls to migrate, and the ratio of the two isomeric products ranged from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1.2:1 (3ga–ja, 3ga′–ja′). Interestingly, the rigid and symmetrical allyl alcohol substrate underwent 1,2-migration and ring expansion under energized conditions, which produced the desired product with a yield as high as 88% (3ka). Notably, the conversion to the required γ-keto sulfone without a quaternary carbon center was successfully achieved from α,α-diphenyl ally alcohol and the yield was just 62% (3la). It was worth mentioning that diaryl allyl alcohol substituted by α-thiophene also underwent a rearrangement (3ma and 3ma′). The yield of two isomeric products was 61% overall, and 1H NMR spectra revealed a 2:1 migration ratio between phenyl and thiophene groups.
:1 to 1.2:1 (3ga–ja, 3ga′–ja′). Interestingly, the rigid and symmetrical allyl alcohol substrate underwent 1,2-migration and ring expansion under energized conditions, which produced the desired product with a yield as high as 88% (3ka). Notably, the conversion to the required γ-keto sulfone without a quaternary carbon center was successfully achieved from α,α-diphenyl ally alcohol and the yield was just 62% (3la). It was worth mentioning that diaryl allyl alcohol substituted by α-thiophene also underwent a rearrangement (3ma and 3ma′). The yield of two isomeric products was 61% overall, and 1H NMR spectra revealed a 2:1 migration ratio between phenyl and thiophene groups.
| a Reaction conditions: graphite plate anode (10 × 10 × 3 mm), nickel foam cathode (10 × 10 × 2 mm), constant current (15 mA), 1 (0.25 mmol), 2a (0.5 mmol, 2eq.), Bu4NBF4 (0.5 mmol), MeCN/H2O (6 mL, 5/1), rt, air, 4 h, and isolated yield. |
|---|
|
Following this, certain representative α-alkyl, α-aryl allyl alcohols were also tested, and the results revealed that not only the species were oxidized under electrochemical conditions, but also most of the 1,2-migrations had strong chemoselectivities. The phenyl in α-methyl, α-aryl allyl alcohol successfully migrated to generate the target products, while the methyl hardly migrated (3na and 3oa). The para strong electron withdrawing trifluoromethyl of phenyl had reduced migration ability (3oa). The benzene ring in alcohols with α-cyclohexyl also selectively migrated and transformed to 3pa, with a yield up to 85%. Moreover, an aromatic naphthenic allyl alcohol was subjected to this strategy, and only the alkyl group migrated and underwent n + 1 ring expansion due to the steric hindrance effect (3qa). It was remarkable that two α-substituents simultaneously migrated to produce isomeric mixtures during the electrochemical 1,2-migration of α-cyclopropyl, α-phenyl allyl alcohol (3ra and 3ra′). The migration ability of phenyl was stronger than that of cyclopropyl as found via verification.
More significantly, some nonaromatic naphthenic allyl alcohols were employed in this electrochemical migration to realize n + 1 ring expansion and construct cyclic γ-keto sulfones with a β-quaternary carbon center (3sa–xa). Several experimental results indicated that these alcohols transformed to the respective rearranged products with moderate to high yields, while their reactivity significantly decreased as the number of carbon atoms on the ring increased. In addition to the cyclic allyl alcohols, the chain alcohols with α,α-dialkyl substituents also efficiently underwent the 1,2-alkyl migration, with yields up to 68% (3ya and 3za).
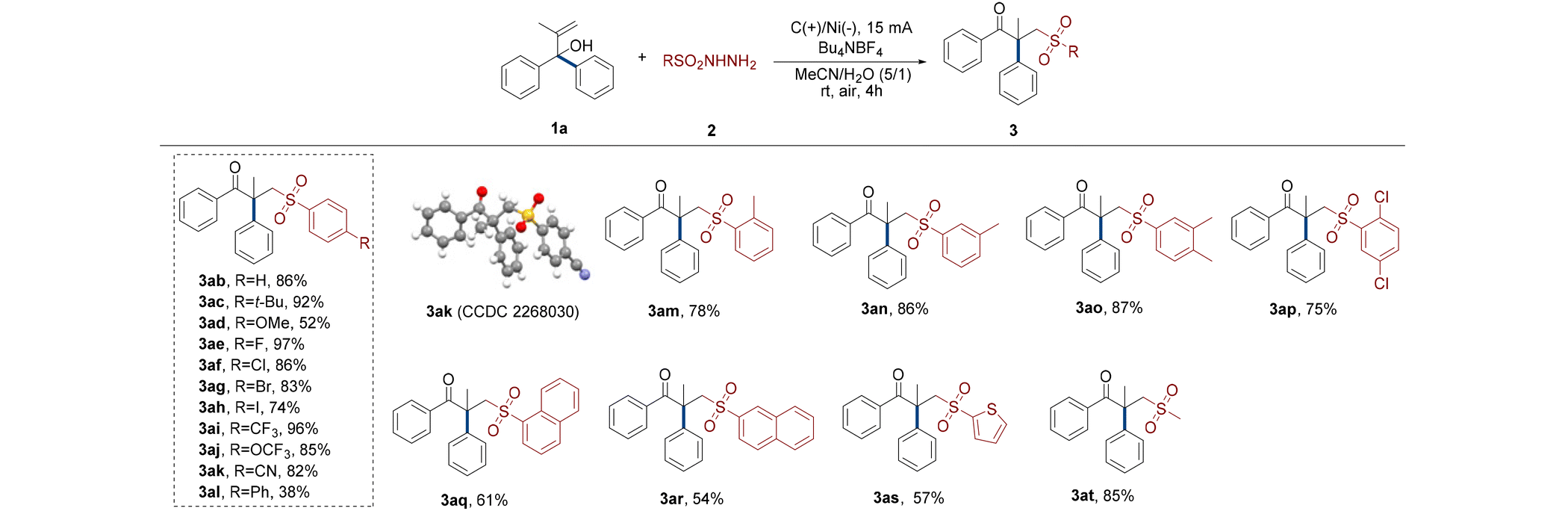
Subsequently, we tested the reaction performance of different sulfonyl hydrazides (Table 3). When the alcohol derivative 1a interacted with the benzene sulfonyl hydrazine without a substituent, product 3ab was obtained in 86% yield.
| a Reaction conditions: graphite plate anode (10 × 10 × 3 mm), nickel foam cathode (10 × 10 × 2 mm), constant current (15 mA), 1 (0.25 mmol), 2a (0.5 mmol, 2eq.), Bu4NBF4 (0.5 mmol), MeCN/H2O (6 mL, 5/1), rt, air, 4 h, and isolated yield. |
|---|
|
The electrochemical rearrangement reaction of aryl sulfonyl hydrazides containing para electron-donating groups (t-Bu and OMe) effectively occurred to produce carbonyl sulfones with yields as high as 92% (3ac and 3ad). The highest conversion yield of all para-halogenated sulfonyl hydrazide compounds to γ-keto sulfones under optimized conditions was 97% (3ae–ah). With the increase of the atomic number of halogens, the reactivities of the reactants gradually became inferior. Moreover, benzenesulfonyl hydrazine substituted by para electron withdrawing groups transformed into the required sulfones with yields up to 96% after effectively implementing this protocol (3ai–ak). X-ray diffraction analysis further confirmed the structure of 3ak (CCDC 2268030†). A clear steric effect was observed in the case of para-phenyl benzenesulfonyl hydrazine that only generated 38% of the target product (3al). The results of the investigation also demonstrated an obvious influence of methyl steric hindrance, and the order of the conversion performances of methyl benzenesulfonyl hydrazides in this electrochemical migration process was para- > meta- > ortho- (3aa, 3am and 3an). Substrates having meta- and para-dimethyl underwent the rearranged sulfonation reaction satisfactorily and the reactivity was not significantly affected by substituents (3ao). Despite the ortho-chlorine atom's steric barrier, the substrate with dichloro substituents at ortho- and para-positions was nonetheless able to accomplish a somewhat reduced rearrangement yield of 75% (3ap). In this electrochemical system, naphthalenesulfonyl hydrazines with combined steric hindrance and conjugation effects were transformed into target γ-keto sulfones with a quaternary carbon center with medium yields (3aq and 3ar). The yield of 1-naphthalenesulfonyl hydrazine from the reaction was somewhat higher than that of 2-naphthalenesulfonyl hydrazine. Intriguingly, the aromatic thiophene sulfonyl hydrazide also promoted alcohols to rearrange and obtain 57% of target sulfone under the electro-oxidation conditions (3as). Finally, the most gratifying aspect of all is that the nonaromatic methanesulfonyl hydrazide also successfully interacted and made the aryl group migrate to access target methyl sulfone derivatives with 85% isolated yield (3at).
Most interestingly, some special structures underwent different transformations under standard conditions. For instance, rigid allyl alcohols substituted by benzo cycloheptane also successfully interacted with p-toluenesulfonyl hydrazine (Scheme 2). However, the difference is that there were no 1,2-migrations. The generated sulfonyl hydrazine radicals attacked the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds and yielded sulfones with spirocarbocycle and ethylene oxide skeletons with up to 73% yield, which were very infrequent (4 and 5). These results may be caused by the effect of steric hindrance. The structure of 4 was further confirmed by X-ray diffraction analysis (CCDC 2268921†).
C double bonds and yielded sulfones with spirocarbocycle and ethylene oxide skeletons with up to 73% yield, which were very infrequent (4 and 5). These results may be caused by the effect of steric hindrance. The structure of 4 was further confirmed by X-ray diffraction analysis (CCDC 2268921†).
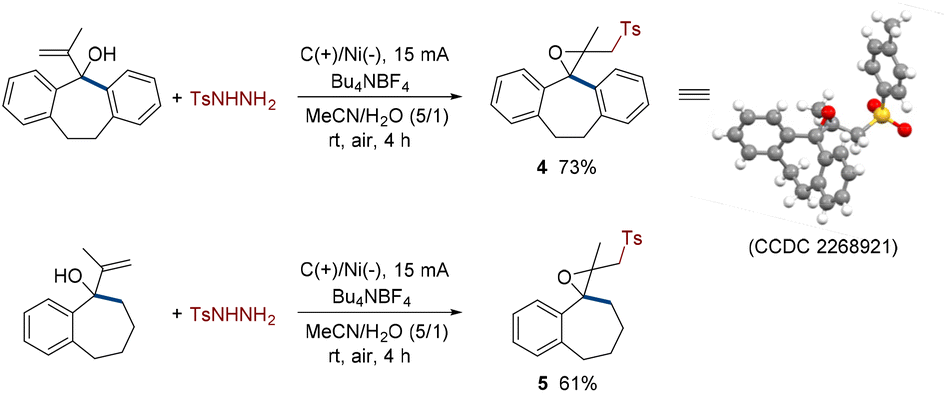 | ||
| Scheme 2 Scope of other substrates. | ||
To demonstrate the industrial practicability of this electro-oxidative rearrangement strategy, a gram-scale reaction and product transformation were performed (Scheme 3). 1a was increased to 5 mmol, and the amounts of other reactants and the volume of solvents were increased in the same proportion. The reaction was monitored after 72 hours of electrolysis. Delightfully, it produced target γ-keto sulfone 3aa in 84% isolated yield (Scheme 3a). Then, the standard product 3aa was selectively reduced to hydroxyl sulfone by sodium borohydride with a yield of 68% (Scheme 3b, 6).
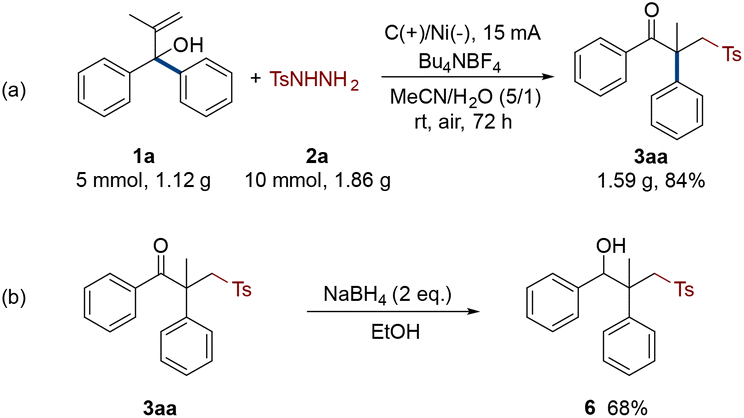 | ||
| Scheme 3 Gram-scale reaction and application. | ||
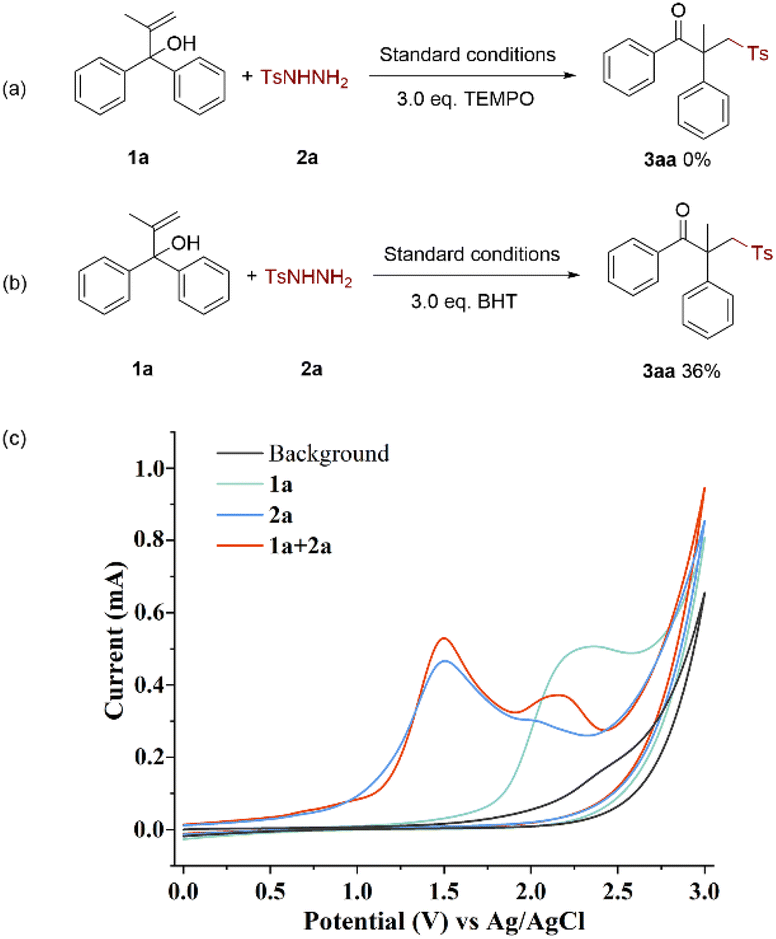
To gain insight into the details of the mechanism for the electrolysis of alcohols and sulfonyl hydrazides, several control experiments were performed (Scheme 4). The result of the initial experiment showed that 2,2,6,6-tetramethylpiperidinyl-1-oxide (TEMPO), a radical trapping agent, totally prevented the transition (Scheme 4a). Similar to that, adding butylated hydroxytoluene (BHT) greatly decreased the conversion rate of this electrochemical oxidation process (Scheme 4b). These findings showed the possibility of a radical route being engaged in the electrochemical oxidation.
 | ||
| Scheme 4 Control experiments and cyclic voltammograms. | ||
Additionally, cyclic voltammetry (CV) experiments of model substrates were conducted (Scheme 4c). It was evident that 2a was oxidized preferentially at the anode because the oxidation peak of 1a was observed at 2.36 V while that of 2a was seen at 1.50 V and 2.01 V. The oxidation peak of 1a in the mixture shifted a little, indicating that 2a interacted with 1a and to some extent inhibited the electro-oxidative process of 1a itself.
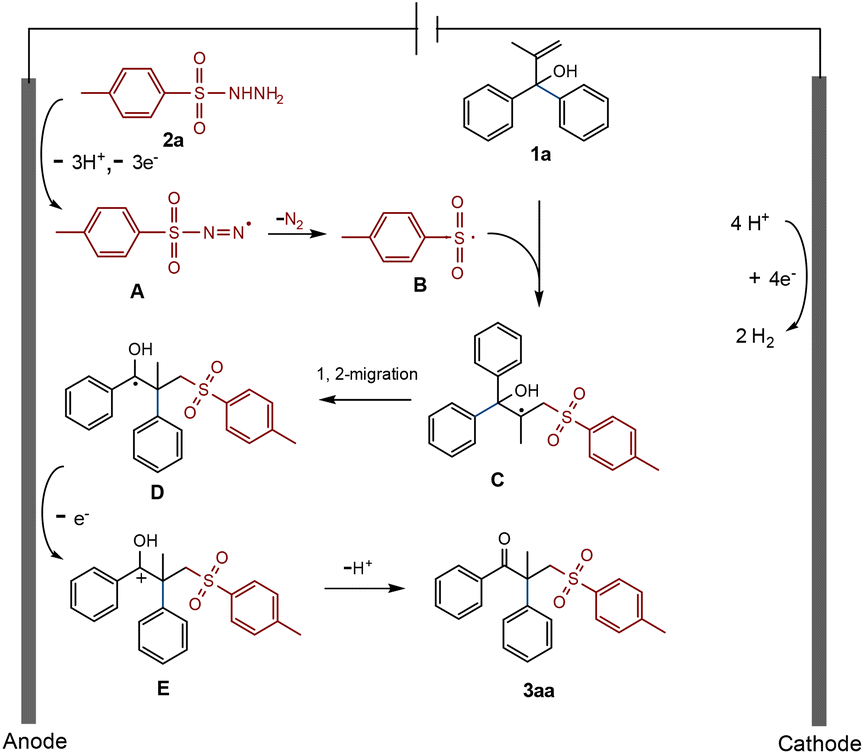
Based on the above research studies and references,17 a possible electrochemical radical-mediated rearrangement reaction mechanism is proposed here (Scheme 5). Taking the standard reaction as an example, p-toluenesulfohydrazide (2a) loses electrons and deprotonates in the anode to produce unstable A, which then releases N2 and transforms into the sulfonyl radical B. B attacks the CC bond of allyl alcohol 1a and subsequently changes into the tertiary carbon radical C. It then proceeds through a 1,2-phenyl migration to generate D, which loses an electron once more and is oxidized to cation E at the anode. Finally, E effortlessly undergoes deprotonation to produce the desired γ-keto sulfone product 3aa.
 | ||
| Scheme 5 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a novel and efficient electrochemical strategy to synthesize γ-keto sulfones. The key to this strategy is that sulfonyl radicals generated by electrooxidation promote the α-substituents of 2-methylallyl alcohol derivatives towards 1,2-migration and rearrangement synchronously. Due to the presence of 2-methyl in allyl alcohols, the constructed carbonyl sulfones are successfully placed into a stable β-quaternary carbon center. After screening, the investigations of 45 examples demonstrated an excellent functional group compatibility of the electrochemical rearrangement reaction. Scale-up experiments displayed its potential industrial application value. Additionally, a possible radical mediated oxidative rearrangement mechanism was proposed on the basis of control experiments and cyclic voltammetry curves. All research studies showed that the novel protocol was atom economic and environment friendly. Further synthesis application and pharmacological activity research of the products is underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (No. 21372265, 21350110501 and 22161045) and the Natural Science Foundation Project of CQ CSTC (cstc2018jcyjAX0155) for financial support.References
-
(a) J. Xiang, M. Ipek, V. Suri, M. Tam, Y. Xing, N. Huang, Y. Zhang, J. Tobin, T. S. Mansour and J. McKew, Bioorg. Med. Chem., 2007, 15, 4396–4405 CrossRef CAS PubMed
; (b) S. Johnson, E. Barile, B. Farina, A. Purves, J. Wei, L. Chen, S. Shiryaev, Z. Zhang, I. Rodionova, A. Agrawal, S. M. Cohen, A. Osterman, A. Strongin and M. Pellecchia, Chem. Biol. Drug Des., 2011, 78, 211–223 CrossRef CAS PubMed
- V. Aranapakam, J. M. Davis, G. T. Grosu, J. Baker, J. Ellingboe, A. Zask, J. I. Levin, V. P. Sandanayaka, M. Du, J. S. Skotnicki, J. F. DiJoseph, A. Sung, M. A. Sharr, L. M. Killar, T. Walter, G. Jin, R. Cowling, J. Tillett, W. Zhao, J. McDevitt and Z. B. Xu, J. Med. Chem., 2003, 46, 2376–2396 CrossRef CAS PubMed
- C. Curti, M. Laget, A. O. Carle, A. Gellis and P. Vanelle, Eur. J. Med. Chem., 2007, 42, 880–884 CrossRef CAS PubMed
-
(a) T. Fujimoto, M. Tobisu, N. Konishi, M. Kawamura, N. Tada, T. Takagi and K. Kubo, Bioorg. Med. Chem., 2009, 17, 7993–8002 CrossRef CAS PubMed
-
(a) P. Iversen, C. J. Tyrrell, A. V. Kaisary, J. B. Anderson, H. E. I. N. Van Poppel, T. L. Tammela and I. Melezinek, J. Urol., 2000, 164, 1579–1582 CrossRef CAS PubMed
-
(a) D. A. Evans, P. H. Carter, E. M. Carreira, J. A. Prunet, A. B. Charette and M. Lautens, Angew. Chem., Int. Ed., 1998, 37, 2354–2359 CrossRef CAS
-
(a) F. Xiao, C. Liu, D. Wang, H. Huang and G.-J. Deng, Green
Chem., 2018, 20, 973–977 RSC
-
(a) A. Bhunia, S. R. Yetra, S. S. Bhojgude and A. T. Biju, Org. Lett., 2012, 14, 2830–2833 CrossRef CAS PubMed
-
(a) X. Cheng, S. Wang, Y. Wei, H. Wang and Y.-W. Lin, RSC Adv., 2022, 12, 35649–35654 RSC
- X.-L. Chen, C.-Y. Wu, J.-T. Ma, S.-Y. Zhuang, Z.-C. Yu and Y.-D. Wu, Org. Lett., 2022, 24, 223–227 CrossRef CAS PubMed
-
(a) J. Y. Lee, Y.-T. Hong and S. Kim, Angew. Chem., Int. Ed., 2006, 45, 6182–6186 CrossRef CAS PubMed
-
(a) S. Z.-Z. Chen, S. Liu, W.-J. Hao, G. Xu, S. Wu, J.-N. Miao, B. Jiang, S.-L. Wang, S.-J. Tu and G. Li, Chem. Sci., 2015, 6, 6654–6658 RSC
- X.-Q. Chu, H. Meng, X.-P. Xu and S.-J. Ji, Chem. – Eur. J., 2015, 21, 11359–11368 CrossRef CAS PubMed
- X. Zhang, Z. Zhang, J.-N. Song and Z. Wang, Chem. Sci., 2020, 11, 7921–7926 RSC
-
(a) J. Yang, G. Li, K. Yu, B. Xu and Q. Chen, J. Org. Chem., 2022, 87, 1208–1217 CrossRef CAS PubMed
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC
-
(a) M.-W. Zheng, X. Yuan, Y.-S. Cui, J.-K. Qiu, G. Li and K. Guo, Org. Lett., 2018, 20, 7784–7789 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2268030 and 2268921. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc02128a |
| This journal is © The Royal Society of Chemistry 2023 |