
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultimetallic Pd- and Ni-catalyzed C(sp2)–P cross-coupling under aqueous micellar conditions†
Rafael
Navrátil
 *,
Kristýna
Kellovská
and
Ondřej
Baszczyňski
*
*,
Kristýna
Kellovská
and
Ondřej
Baszczyňski
*
Department of Organic Chemistry, Faculty of Science, Charles University, Hlavova 2030/8, 128 43 Prague 2, The Czech Republic. E-mail: navratilr@natur.cuni.cz; baszczyo@natur.cuni.cz
First published on 24th October 2023
Abstract
Organophosphorus compounds containing hydrolytically and metabolically stable C(sp3)– and C(sp2)–P bonds are widely used as reagents, ligands, pesticides, herbicides, flame retardants, surface modifiers, and antiviral and anticancer drugs. These applications rely on efficient C(sp3)– and C(sp2)–P bond-forming reactions. However, currently available C(sp2)–P cross-coupling protocols require high catalyst loadings and temperatures, as well as environmentally unsustainable and harmful organic solvents (e.g., N,N-dimethylformamide, DMF). Herein, we disclose a conceptually novel strategy for performing multimetallic Pd/Ni- and dual-ligand Pd-catalyzed C(sp2)–P cross-coupling reactions in aqueous micelles under mild and environmentally friendly conditions. Micellar catalysis in water enables C(sp2)–P cross-coupling while avoiding environmentally unsustainable organic solvents, thereby reducing organic waste generation. Such micellar C(sp2)–P cross-coupling reactions tolerate various functional groups and provide access to structurally diverse (hetero)aryl (thio)phosphonates, phosphinates and phosphine oxides using inexpensive commercial materials and catalysts. Moreover, C(sp2)–P cross-coupling reactions of medically relevant substrates and drugs under late-stage functionalization settings and multistep one-pot processes highlight the potential applications of this experimental paradigm.
Introduction
Phosphonates, phosphinates, phosphine oxides and related C–P bond-containing compounds are widely applied in organic1 and pharmaceutical chemistry,2,3 agrochemistry,4 and materials science.5,6 Introducing a C–P bond into organic compounds often improves their properties, increasing water solubility, decreasing lipophilicity, and enhancing pharmacokinetics.7–9 Furthermore, C–P bonds are more metabolically stable than hydrolytically labile P–O bonds of natural phosphates.8,10 Accordingly, the increased stability of C–P bonds has proved vital for numerous applications.Isosterically replacing phosphates by more metabolically stable phosphonates has fostered the development of life-saving antiviral11 and antibacterial12 drugs and prodrugs13 because they retain the bioactivity of the original phosphates.10,14 Several bioactive phosphonates, phosphinates and phosphine oxides have also been applied in drug discovery and development,2a,3,7,15–18 yielding fosdevirine, a non-nucleoside reverse transcriptase inhibitor (Fig. 1A),19 and efonidipine, a calcium channel blocker,20 in addition to eukaryotic initiation factor 4E (eIF4E) inhibitors.21 Moreover, brigatinib, an anaplastic lymphoma kinase/epidermal growth factor receptor (EGFR) inhibitor22 with an aryl dimethylphosphine oxide motif (Ar–P(O)Me2), has been approved by the Food and Drug Administration (FDA) for cancer treatment. These applications of organophosphorus compounds rely on the development of efficient, practical, and scalable methods for the C(sp2)–P bond construction.
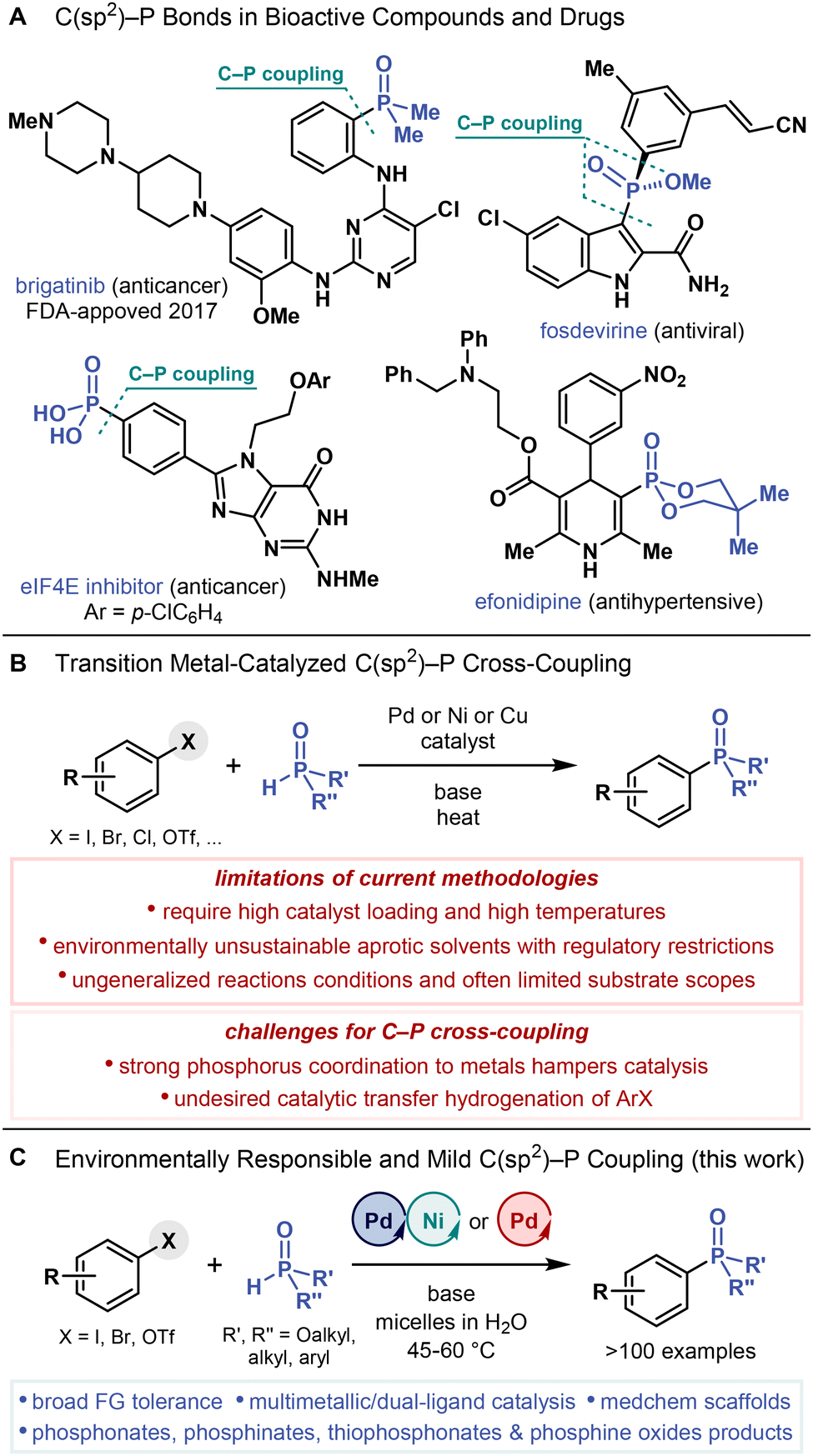 | ||
| Fig. 1 (A) Examples of bioactive organophosphorus compounds containing C(sp2)–P bonds. (B) Conventional transition metal-catalyzed C(sp2)–P cross-coupling reactions, their limitations, and obstacles to further developments. (C) C(sp2)–P cross-coupling enabled by multimetallic Pd/Ni catalysis under mild micellar conditions in water (this work). | ||
C(sp2)–P bonds are typically formed by cross-coupling (hetero)aryl and alkenyl (pseudo)halides and phenol derivatives with corresponding H–P compounds (e.g., H-phosphonates, H-phosphinates, secondary phosphine oxides, diarylphosphines) under palladium,23 nickel,24 or copper25 catalysis (also known as Hirao coupling, Fig. 1B).26,27 However, current C(sp2)–P cross-coupling methods are limited by (1) high catalyst loadings (often 10 mol %) and, in some cases, alternative procedures (slow addition of H-phosphonate)28 or phosphorus precursors (e.g., masked H-phosphonates)29 required to overcome the inhibitory effects of phosphorus nucleophiles on metal catalysts given their strong coordination properties, (2) catalytic transfer hydrogenation, whereby (hetero)aryl halides are converted into (hetero)arenes due to the undesired reducing properties of H–P compounds,30,31 (3) heating at high temperatures (often above 100 °C) to facilitate the C(sp2)–P bond-forming reductive elimination step, rendering the reactions potentially incompatible with complex substrates bearing sensitive functional groups, and (4) aprotic polar solvents, such as N,N-dimethylformamide (DMF),24,32 which is classified as toxic and hazardous33 and its use has been restricted by the European Commission.34 Further exacerbating these problems, state-of-the-art C(sp2)–P cross-coupling methods fail to meet increasing demands for environmentally responsible chemical processes with low energy costs and loadings of precious transition metal catalysts in environmentally benign solvents (e.g., water35) under mild conditions (at room temperature or mild heating) while reducing organic waste generation.
A few studies on C(sp2)–P cross-coupling in water have been reported (Fig. 2), but not without limitations. Under Pd catalysis, a single arylphosphonate has been synthesized from 4-iodotoluene and HP(O)(OEt)2 at room temperature,36 aryl phosphonates and phosphine oxides have been prepared from aryl halides and HP(O)(Oi-Pr)2 and HP(O)Ph2, respectively, albeit at 100 °C,37 and triaryl phosphine oxides have been synthesized from halobenzoic acids and HP(O)Ph2 upon microwave irradiation at an even higher temperature (180 °C).38 Under Ni catalysis, triaryl phosphine oxides have been synthesized from aryl halides and HP(O)Ph2 at 70 °C.39 Bar one, though, these methods require extensive heating. Moreover, they all show limited substrate scopes, predominantly providing triarylphosphine oxide products. Nevertheless, these precedents demonstrate that C(sp2)–P cross-coupling reactions operating under aqueous conditions are feasible. In this context, we hypothesized that the current limitations of C(sp2)–P cross-coupling methods could be overcome by performing C(sp2)–P cross-coupling reactions under mild micellar conditions in water, a concept that is often referred to as micellar catalysis.
 | ||
| Fig. 2 C(sp2)–P cross-coupling reactions operating in water and their limitations. | ||
By micellar catalysis, organic compounds can be sustainably and efficiently synthesized in water, replacing traditional organic solvents.40 In these processes, a small amount (a few wt %) of a surfactant is added into water to prepare micelles with lipophilic cores. These micelles help to solubilize otherwise water-insoluble organic compounds and serve as nanoreactors for their transformations. Several classes of organic reactions have already been adapted to micellar conditions in water, including transition metal-catalyzed cross-couplings,41,42 amide-bond coupling,43 and SNAr reactions,44 among others.40 These reactions typically operate at room temperature (or under mild heating), require significantly lower catalyst loadings, produce much less waste, and show both faster reaction rates and better purity profiles than those run in conventional organic solvents. Yet, despite all these advantages, no C(sp2)–P cross-coupling reaction has been performed under micellar conditions in water.
In this study, we describe the development of a mild, practical, general, and environmentally responsible C(sp2)–P cross-coupling method, highlighting its applications in the synthesis of complex (hetero)aryl phosphonates, phosphinates and phosphine oxides (Fig. 1C). This method is enabled by a combination of (1) Pd- and Ni-based catalysts (i.e., multimetallic catalysis), or (2) two Pd ligands (i.e., dual-ligand catalysis). Moreover, our findings demonstrate the wide-ranging feasibility of a multimetallic catalytic process under micellar conditions in water.
Results and discussion
We developed a method for performing environmentally responsible micellar C(sp2)–P cross-coupling reactions in water by screening suitable catalysts, ligands, bases and additives, as well as other reaction parameters (Tables 1–3). As a micellar catalyst, we used a well-established and commercially available “designer” surfactant TPGS-750-M (Table 1).45 TPGS-750-M is a non-toxic, inexpensive, α-tocopherol-based (vitamin E) amphiphile, which has been successfully used in numerous transition metal-catalyzed cross-coupling reactions in water.40–42,45|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R | Arl/3 ratio | Metal salt (X mol %) | Ligand (Y mol %) | Base | Deviation/additive | T | Yielda |
| a Yield was determined by 31P NMR. b Isolated yield. c Yield was determined by 1H NMR using CH2Br2 as an internal standard. d 3 equiv. ND = not detected. | ||||||||
| 1 | H | 1.2/1 | Pd(OAc)2 (5) | XantPhos (10) | Et3N | No THF | rt | 30% |
| 2 | H | 1.2/1 | Pd(OAc)2 (5) | XantPhos (10) | Et3N | 45 °C | 49% | |
| 3 | H | 1.2/1 | Pd(OAc)2 (5) | P(t-Bu)3·HBF4 (10) | Et3N | 45 °C | 53% | |
| 4 | H | 1.2/1 | Pd(OAc)2 (5) | XantPhos (10) | 2,6-Lutidine | 45 °C | 88% | |
| 5 | H | 1.2/1 | Pd(OAc)2 (1) | XantPhos (1) | 2,6-Lutidine | 45 °C | 99% (91%)b | |
| 6 | H | 1.2/1 | Pd(OAc)2 (1) | XantPhos (1) | 2,6-Lutidine | 44 hours | rt | 88% |
| 7 | Me | 1.2/1 | Pd(OAc)2 (1) | XantPhos (1) | 2,6-Lutidine | 45 °C | 84%c (79%)b | |
| 8 | Me | 1.2/1 | Pd(OAc)2 (1) | XantPhos (1) | 2,6-Lutidine | LiCI (1 equiv.) | 45 °C | 94%b |
| 9 | Me | 1/2 | Ni(XantPhos)Cl2 (5) | Et3N | No THF, Zn (2 equiv.) | rt | ND | |
| 10 | Me | 1/2 | Ni(phen)Cl2 (2.5) | 2,6-Lutidined | LiCI (1 equiv.), nano Zn (0.5 equiv.) | 45 °C | 73%c | |

|
|
|
||||||
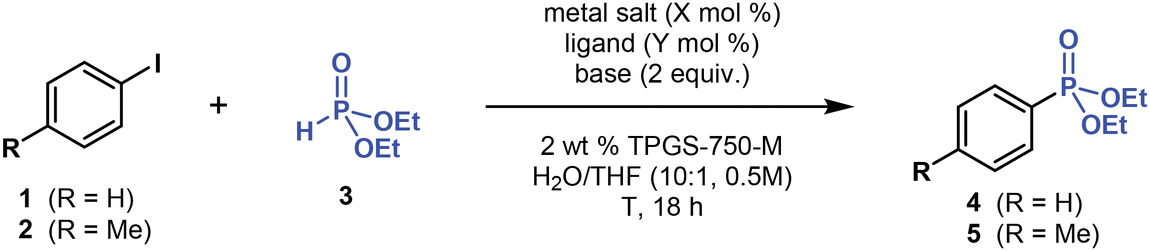
|
|
||
|---|---|---|
| Entry | Deviation from above | Yielda |
| a Yields were determined by 1H NMR using CH2Br2 as an internal standard. b Isolated yield. RSM = recovered starting material. ND = not detected. | ||
| 1 | None | 99% (94%)b |
| 2 | No [Pd], 2.5 mol% [Ni] | ND (87% RSM) |
| 3 | No [Ni], no Zn | 32% (63% RSM) |
| 4 | No Zn | ND (96% RSM) |
| 5 | Ni0(COD)DQ/phen, no Zn | 14% (83% RSM) |
|
|
||
|---|---|---|
| Entry | Deviation from above | Yielda |
| a Yields determined by 1H NMR using CH2Br2 internal standard. b Isolated yield. RSM = recovered starting material. ND = not detected. Please refer to additional optimization details in ESI.† | ||
| 1 | None | Quant (99%)b |
| 2 | Pd(OAc)2/P(t-Bu)3·HBF4, no XantPhos | 24% (67% RSM) |
| 3 | No Pd(OAc)2, no XantPhos | 47% (50% RSM) |
| 4 | No XantPhos | 13% (84% RSM) |
| 5 | No Pd[P(t-Bu)3]2 | ND (99% RSM) |
| 6 | PCy3·HBF4 or PMe(t-Bu)2·HBF4 | ND (99% RSM) |
| 7 | Multimetallic conditions (Table 2, entry 1) | 95% (full conv.) |
Micellar C(sp2)–P cross-coupling optimization
To optimize C(sp2)–P cross-coupling under micellar conditions, we initially focused our efforts on Pd-catalyzed reactions. After extensively screening reaction conditions (Table 1, please refer to ESI† for more details), we determined that the model coupling reaction of iodobenzene (1, 1.2 equivalent) with inexpensive diethyl H-phosphonate (3, 1 equivalent) provided the desired product, diethyl phenylphosphonate (4), in ∼30% 31P NMR yield when using Pd(OAc)2 (5 mol %), XantPhos (10 mol %), Et3N (2 equivalent), and 2 wt % TPGS-750-M in water (0.5 M), at laboratory temperature (∼20 °C), over 18 hours (Table 1, entry 1). Adding an organic co-solvent (10 vol %), such as THF, into the reaction mixture and raising the temperature to 45 °C increased the yield of 4 (entry 2). After screening various mono- and bidentate phosphine ligands, including dppf, dppe, dppp, DPEphos, BINAP, dtbpf, Cy-XantPhos, PCy3, PMe(t-Bu)2, APhos and Buchwald-type (JohnPhos, DavePhos, XPhos, and SPhos) ligands, among others (see ESI†), we found that XantPhos and P(t-Bu)3 (used as its bench-stable HBF4 salt, entry 3) were the most effective ligands. Furthermore, decreasing catalyst loading from 5 to 1 mol % also improved the yield of 4 (entry 5).The base used in this reaction strongly affected the yield. Switching from Et3N (pKBH+ = 11.0 in H2O) to a weaker but more lipophilic 2,6-lutidine base (pKBH+ = 6.7 in H2O) significantly improved the reaction yield (entry 4), affording 4 in 91% isolated yield. Stronger inorganic (Cs2CO3 and K3PO4) and organic (DBU, Cy2NMe, and t-BuOK) bases proved ineffective (<5% yield by 1H NMR) and more lipophilic organic base (n-Bu)3N afforded 4 in 57% yield (1H NMR). Therefore, micellar C(sp2)–P cross-coupling requires using rather weak but more lipophilic bases, such as 2,6-lutidine, to achieve high yields.
The reaction with more electron-rich 4-iodotoluene (2) yielded phosphonate 5 in a lower yield of 79% (entry 7). Nevertheless, adding 1 equivalent of LiCl into the reaction mixture restored the high reaction yields, affording 5 in 94% yield (entry 8). Most likely, LiCl enhances the yield by decreasing the concentration of 2 dissolved in water (salting-out effect), thereby increasing the concentration and reaction rate of 2 in micellar compartments. Moreover, LiCl may generally enhance cross-coupling reactions.46 As such, LiCl could have a twofold effect on micellar C(sp2)–P coupling.
Subsequently, we tested alternative catalysts, namely Cu and Ni. Despite testing several Cu catalytic systems, no product was detected, in any case. Similarly, combining Ni(XantPhos)Cl2 (5 mol %), Et3N (2 equivalent), and Zn powder (2 equivalent) provided no product (entry 9). But when combining Ni(phen)3Cl2 (2.5 mol %), 2,6-lutidine (3 equivalent), commercial nano Zn powder (0.5 equivalent), LiCl (1 equivalent) and THF co-solvent (10 vol %), we prepared 5 in 73% yield (see ESI† for more screening experiments). So at least for 2, Ni catalysis did not match the efficiency of Pd catalysis. Based on these results, we identified Pd as an efficient catalytic system for micellar C(sp2)–P coupling.
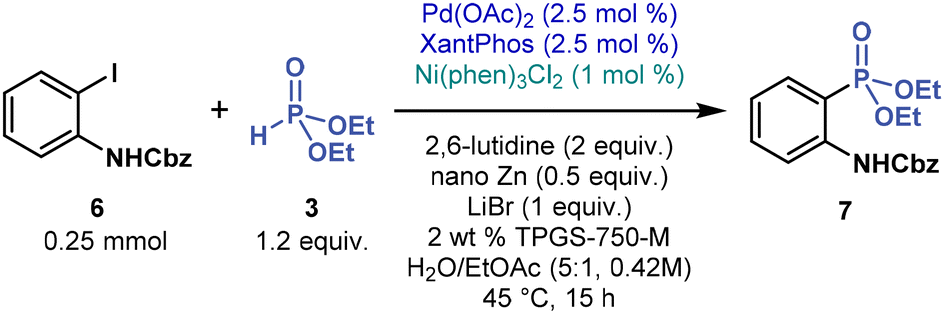
Multimetallic catalysis
When applying the best performing reaction conditions (Table 1, entries 5 and 8) to other, electronically diverse substrates (e.g., N-Boc-4-iodoaniline, 4-iodopyridine, 4-iodoanisole, and N-Cbz-2-iodoaniline), we initially observed little (<20%) to no product formation. At this point, we continued our extensive screening of ligands and reaction additives (see ESI†), eventually identifying efficient, general, multimetallic cross-coupling conditions. By combining Pd and Ni catalysts, C(sp2)–P bonds were forged in aqueous micelles (Table 2). More specifically, cross-coupling of N-Cbz-2-iodoaniline (6, 1 equivalent) with 3 (1.2 equivalent) was catalyzed by Pd(OAc)2 (2.5 mol %) with XantPhos (2.5 mol %) and Ni(phen)3Cl2 (1 mol %), in the presence of 2,6-lutidine (2 equivalent), commercial nano Zn powder (0.5 equivalent), and LiBr (1 equivalent, LiBr outperformed LiCl in most cross-couplings, see ESI†) in 2 wt % TPGS-750-M in water containing 16 vol % EtOAc co-solvent (the total concentration of 6 was 0.42 M). After stirring the reaction mixture at 45 °C, for 15 hours, phosphonate 7 was prepared in 94% yield (Table 2, entry 1). Therefore, under these reaction conditions, multimetallic catalysis promotes C(sp2)–P cross-coupling.Our control experiments confirmed that the palladium catalyst and zinc powder are essential; otherwise, no product is formed (Table 2, entries 2 and 4). The reaction without the nickel catalyst still provided 7, but in a low yield (32% by 1H NMR), with most of 6 remaining unreacted (entry 3). Initially, we suspected that zinc only reduced Ni(II) to catalytically active Ni(0); however, the experiment with an air-stable Ni(0) pre-catalyst Ni(COD)DQ47 without zinc provided 7 in only 14% yield (1H NMR). These results indicate that zinc not only is a reductant but also facilitates transmetalation between Pd and Ni species, in line with previous multimetallic catalyzed C(sp2)–C(sp2) cross-coupling reactions.48
By adding a small amount (5–20 vol %) of an organic co-solvent into a reaction run in micelles, we were able to overcome the limited solubility of some starting compounds (vide infra) and to maintain a stable emulsion throughout the reaction, thereby increasing reaction rates and yields.49 Using EtOAc as a co-solvent (H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc in 5:1 v/v ratio) proved more advantageous than THF (or any other solvent tested in this study, such as toluene, 1,4-dioxane, and acetone, among others), mainly because EtOAc (i) slightly increased the reaction yields, possibly due to the higher solubilizing power of organic compounds, and/or micelle expansion, and is (ii) ranked as a green solvent33 (iii) also used during extraction work-up.
:EtOAc in 5:1 v/v ratio) proved more advantageous than THF (or any other solvent tested in this study, such as toluene, 1,4-dioxane, and acetone, among others), mainly because EtOAc (i) slightly increased the reaction yields, possibly due to the higher solubilizing power of organic compounds, and/or micelle expansion, and is (ii) ranked as a green solvent33 (iii) also used during extraction work-up.
Our micellar C(sp2)–P coupling method is both simple and practical, and its progress can be monitored by standard TLC or LC-MS analysis. When the reaction is finished, the product is extracted by adding a small volume of EtOAc (3 × ∼1 mL for a 0.25 mmol-scale reaction) directly into the reaction vial/flask, subsequently evaporating volatiles. If desired, an excess of 3 and 2,6-lutidine can also be evaporated under high-vacuum.50 The crude product is then purified by chromatography. In some cases (vide infra), the product can be conveniently isolated in sufficient purity simply by filtering the reaction mixture or by directly injecting the reaction mixture onto a reverse-phase column chromatography column and performing the chromatography separation. The latter is particularly advantageous for isolating highly water-soluble phosphine oxide products (Fig. 5).
Some multimetallic Pd/Ni C(sp2)–P cross-couplings were problematic, particularly those with strongly coordinating phosphorus nucleophiles (e.g., secondary phosphine oxides) or with electron-rich aryl iodides and bromides. Difficulties with the latter resulted from stronger C–I and C–Br binding, which complicated oxidative addition into these bonds. Both issues proved relevant when establishing the substrate scope of aryl halides and phosphorus nucleophiles (Fig. 3–5).
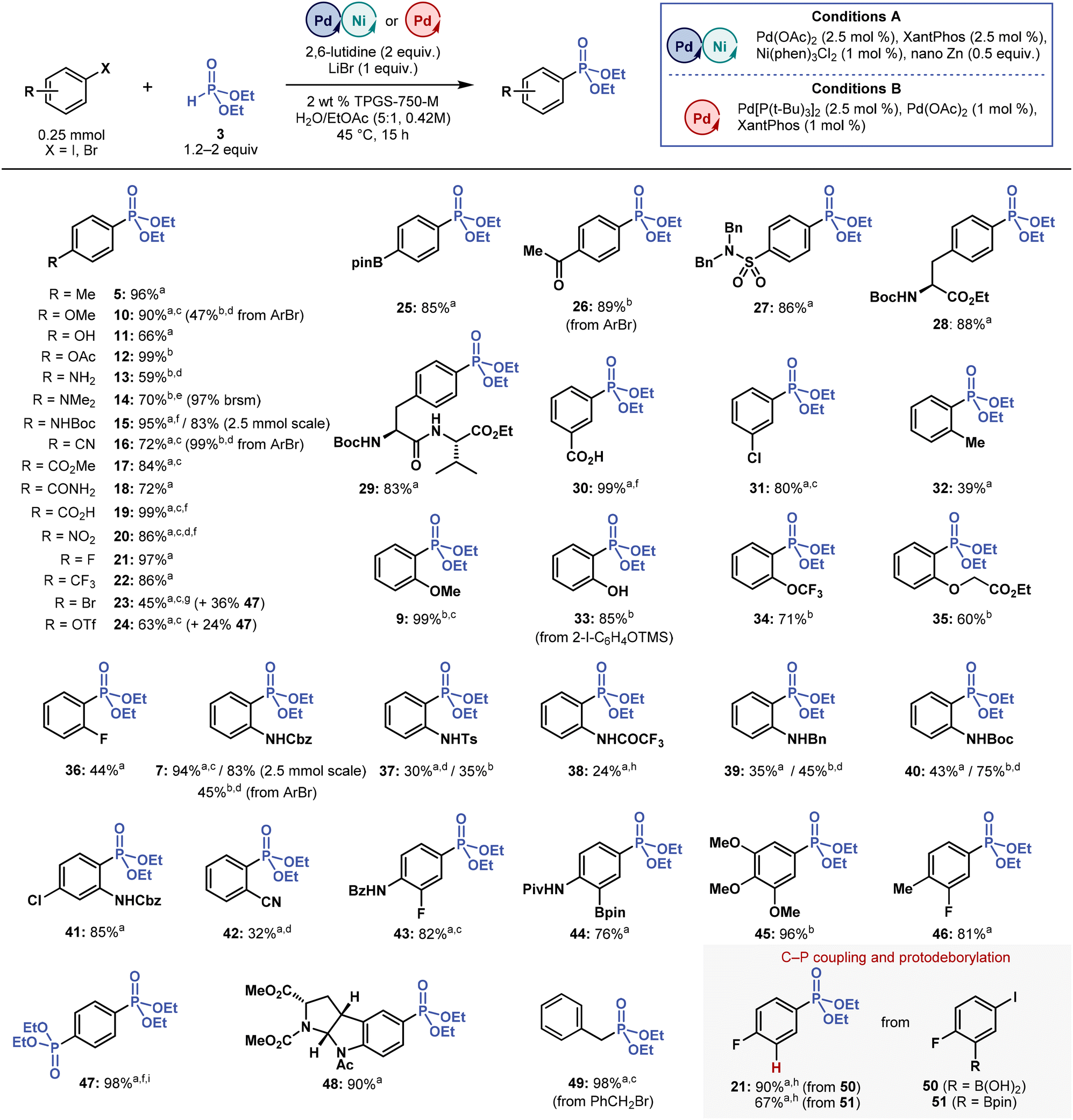 | ||
| Fig. 3 Substrate scope of multimetallic-catalysed micellar C(sp2)–P cross-coupling towards aryl halides. Isolated yields are reported, unless noted otherwise. aConditions A. bConditions B. c1.2 equivalent of 3. dPerformed at 55 °C. ePerformed at 60 °C. f3 equivalents of 2,6-lutidine. gPerformed at 35 °C. hDetermined by 1H NMR with internal standard. i3 equivalents of 3. brsm = based on recovered starting material. Please refer to ESI† for more details. | ||
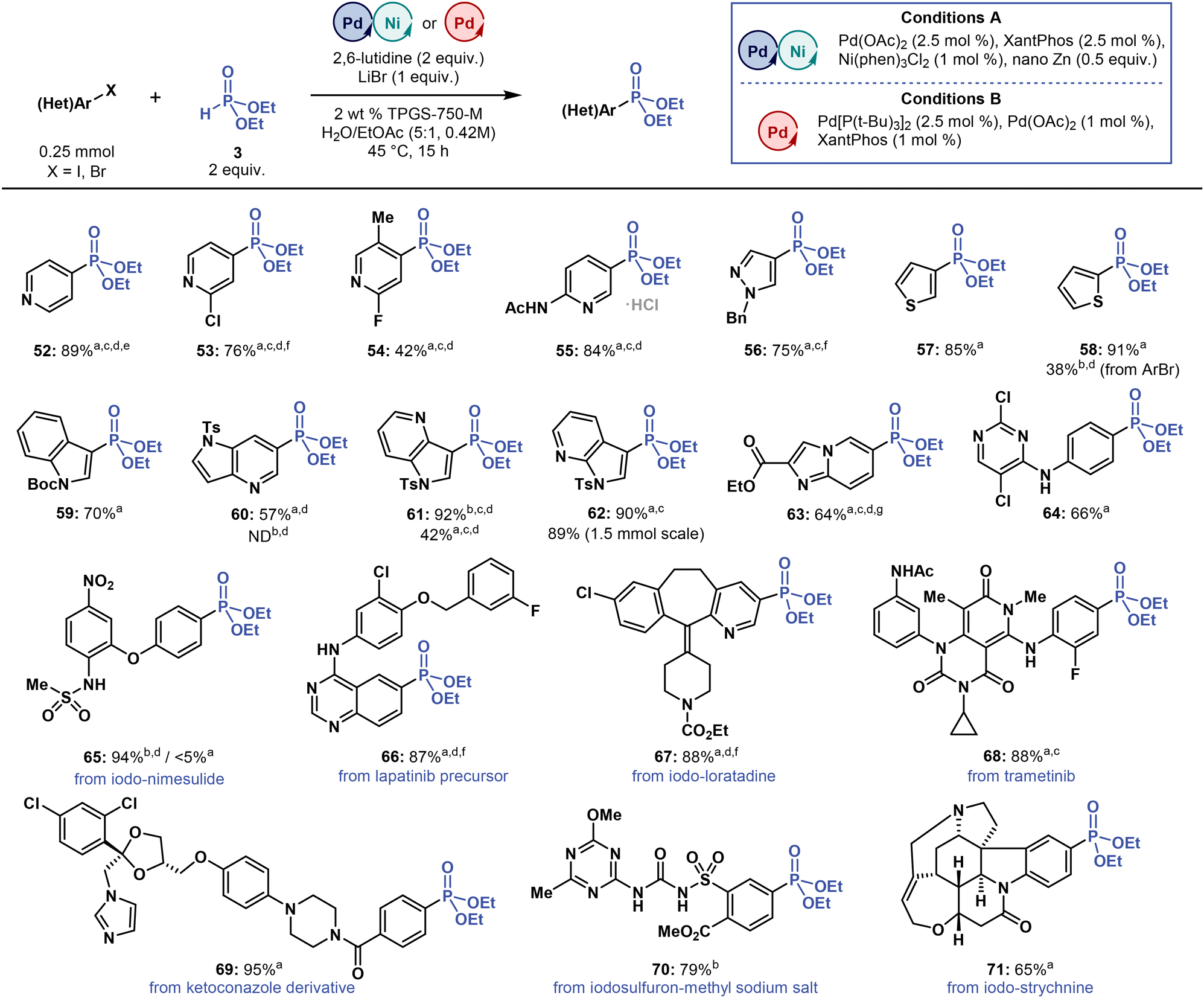 | ||
| Fig. 4 Substrate scope of multimetallic-catalysed micellar C(sp2)–P cross-coupling towards heteroaryl iodides and medically relevant compounds. Isolated yields are reported. aConditions A. bConditions B. c3 equivalent of 2,6-lutidine. dPerformed at 55 °C. e5 mol% of Ni(phen)3Cl2. f2.5 mol% of Ni(phen)3Cl2. g4 equivalents of 3. ND = not detected. Please refer to ESI† for more details. | ||
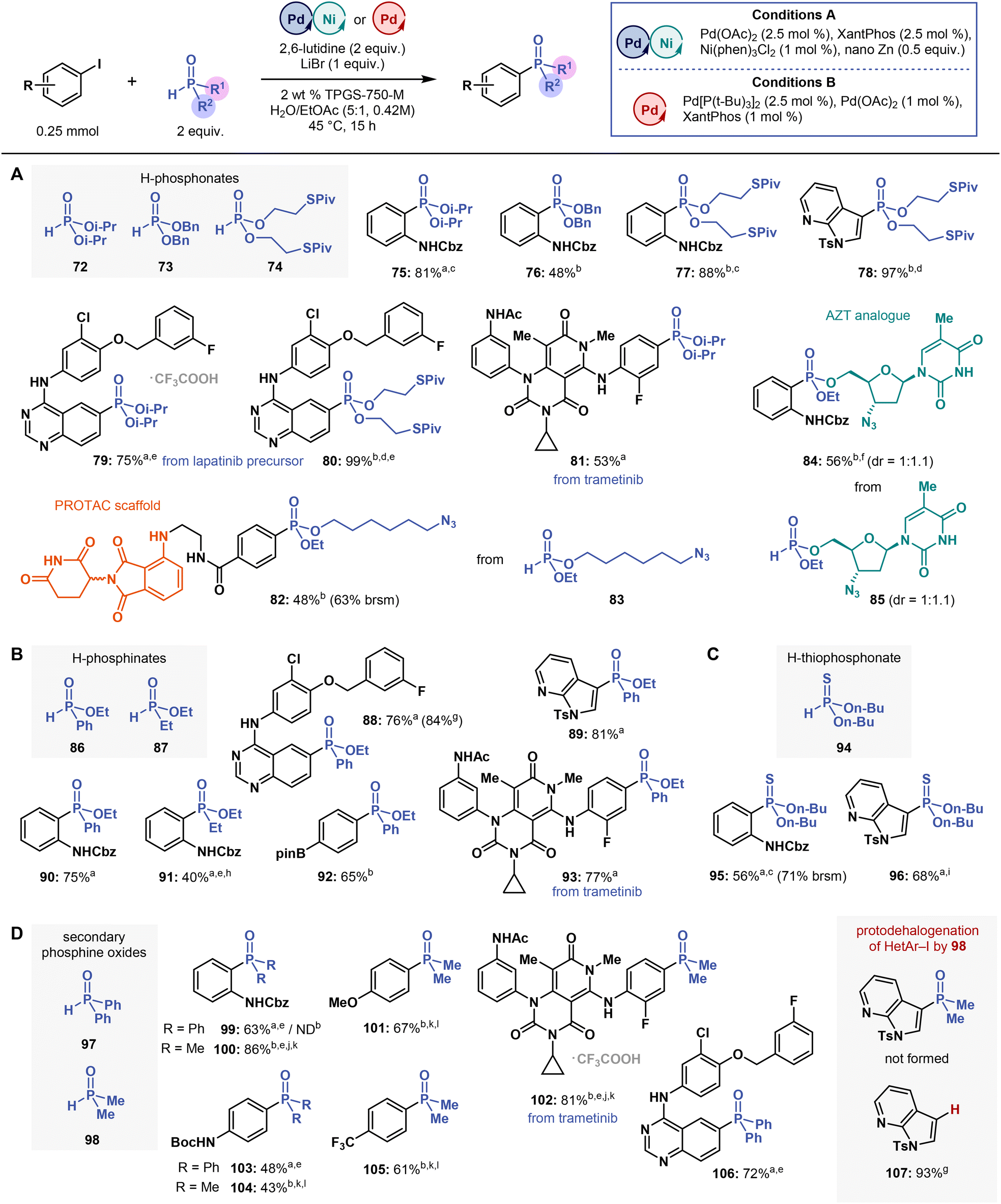 | ||
| Fig. 5 Substrate scope of H–P compounds in the multimetallic-catalysed micellar C(sp2)–P cross-coupling. Isolated yields are reported. aConditions A. bConditions B. c1.2 equivalent of H–P compound. d1.5 equivalent of 74. ePerformed at 55 °C. f1 equivalent of 85, 1.2 equivalent of 6. g,lH NMR yield using CH2Br2 as an internal standard. h2.5 mol% of Ni(phen)3Cl2, 3 equivalents of 2,6-lutidine. i2.1 equivalents of 94. j3 equivalents of 98. k3 equivalents of N-methylmorpholine instead of 2,6-lutidine. lPerformed at 60 °C. brsm = based on recovered starting material. ND = not detected. Please refer to ESI† for more details. | ||
Dual-ligand conditions
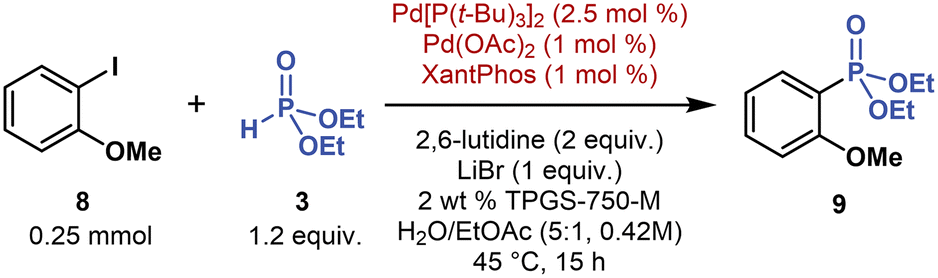
In our initial screening under single-metal Pd catalysis (Table 1, entry 3), we identified the bulky, electron-rich P(t-Bu)3 ligand as the second most efficient ligand. This ligand facilitated both oxidative addition to stronger C–halogen bonds and cross-coupling with strongly coordinating phosphorus nucleophiles, such as secondary phosphine oxides (Fig. 5, see ESI† for further experiments). To understand the effect of the P(t-Bu)3 ligand on Pd-catalyzed C(sp2)–P couplings under micellar conditions, we performed several reactions of electron-rich 2-iodoanisole (8, 1 equivalent) with 3 (2 equivalent) and Pd(OAc)2 (5 mol %) and P(t-Bu)3·HBF4 (10 mol %) (Table 3, entry 2). For a direct comparison with multimetallic conditions, we used the same base (2,6-lutidine), additive (LiBr), and co-solvent (EtOAc).The reaction between 8 and 3 using the P(t-Bu)3 ligand afforded 9 in 24% yield (1H NMR with CH2Br2 as an internal standard), whereas the reactions using other bulky, electron-rich trialkyl phosphine ligands (PCy3, PMe(t-Bu)2) provided no detectable product (Table 3, entry 6). Coupling 8 with 3 using a commercially available Pd[P(t-Bu)3]2 catalyst (5 mol %) nearly doubled the yield of 9, reaching 47% (entry 3). But when we combined Pd[P(t-Bu)3]2 (2.5 mol %) with Pd(OAc)2 (1 mol %) and XantPhos (1 mol %), 9 was formed almost quantitatively. Thus, the yield of challenging C(sp2)–P cross-coupling reactions may be optimized under dual-ligand conditions.
Under dual-ligand conditions, further control experiments showed that both P(t-Bu)3 and XantPhos ligands are essential. Without XantPhos, the yield of 9 decreased from a virtually quantitative yield to 13% (Table 3, entry 4). Without Pd[P(t-Bu)3]2, 9 was not detected (entry 5). Similar dual-ligand synergic effects have been previously described in Pd-catalyzed reactions, for example, in arene C–H functionalization,51 decarboxylative desaturation,52 and ketone α-alkylation.53
Based on our experimental data on dual-ligand C(sp2)–P coupling, oxidative addition to the C–halogen bond may be promoted by a bulky, electron-rich P(t-Bu)3 ligand. This ligand effectively competes for coordination to Pd(0) centers with nucleophilic 3, presumably in its trivalent phosphite form (P(OH)(OEt)2). In turn, reductive elimination, forging the C(sp2)–P bond, may be facilitated by the high-bite-angle (111°) bidentate XantPhos ligand,54,55 which preferentially coordinates to Pd(II) intermediates (see ESI† for plausible mechanism).56 The C(sp2)–P cross-coupling mechanism is likely more complex because acetates, derived from either Pd(OAc)2 or added KOAc (tested during screening experiments), also facilitated C(sp2)–P coupling; this positive acetate effect on C(sp2)–P cross-coupling has been previously reported, albeit in reactions performed in traditional organic solvents.23c Therefore, multimetallic and dual-ligand conditions provide complementary access to C(sp2)–P cross-coupling products (vide infra).
Substrate scope of aryl halides
We applied both optimized conditions (multimetallic Conditions A: Pd(OAc)2/XantPhos/Ni(phen)3Cl2/nano Zn; dual-ligand Conditions B: Pd[P(t-Bu)3]2/Pd(OAc)2/XantPhos) in cross-coupling reactions of several aryl iodides and bromides with H-phosphonate 3 (Fig. 3). The methods proved effective in a wide range of electron-rich and electron-poor substrates, affording phosphonate products (5, 7, 9–49) in high yields, including those with hydroxyl (11), amino (13), benzamide (18), carboxylic acid (19), ketone (26), and pinacol boronate (25, 44) groups. These results demonstrate the excellent functional group-tolerance of micellar C(sp2)–P cross-coupling.Highly crystalline substrates, such as 4-iodonitrobenzene and aryl iodide precursors of phosphonates 43 and 44, required a higher dilution (0.21 M instead of 0.42 M) because of their poor solubility. Other substrates also demanded introducing some modifications, namely using Pd[P(t-Bu)3]2 catalyst (5 mol %) with KOAc additive (0.5 equivalent) and running the reaction at a higher temperature (55 °C), to prevent potential inhibitory effects of nucleophilic (NH2 and NMe2) functional groups on cross-coupling. Under these slightly modified conditions, we prepared phosphonates 13 and 14 in 59 and 70% yields, respectively.
Aryl iodides containing Br and OTf groups, which are typically also reactive in cross-coupling reactions, provided the corresponding phosphonates 23 and 24 in 45 and 63% yields, along-side bis-phosphonate 47. The side-product 47 was formed even when we performed cross-coupling with only 1.2 equivalents of 3 and at a decreased reaction temperature (35 °C). This outcome demonstrates that the reactivity of 23 and 24 is, at least, on a par with that of corresponding starting aryl iodides. If desired, 47 can be synthesized directly by coupling 1,4-diiodobenzene to 3 (3 equivalent) in an almost quantitative yield.
Notwithstanding the results described above, micellar C(sp2)–P coupling proved highly sensitive to ortho substitution on an aromatic ring in most aryl halides. Broadly speaking, Conditions B provided higher yields of ortho-substituted phosphonates than Conditions A despite using the bulky P(t-Bu)3 ligand. Under Conditions A, only phosphonates 9 (o-OMe, 99% yield using Conditions B), 7 (o-NHCbz, 94% yield using Conditions A from ArI, 45% using Conditions B from ArBr), and 41 (85%, Conditions A) were formed in high yields.
Phosphonates with other ortho substituents (Me, OCF3, OCH2CO2Et, F and CN) were formed in low-to-good yields (32–61%), whereas some ortho substituents (OH, CO2H, CO2Me, CONH2 and NO2) were incompatible with the reaction conditions, generating products in trace amounts or in less than 20% yield (1H NMR). In addition, o-OH phosphonate 33 was accessed in 85% yield by coupling (2-iodophenoxy)trimethylsilane with 3. This finding indicates that C(sp2)–P cross-coupling shows faster kinetics than silyl ether hydrolysis under aqueous micellar conditions.
When assessing in more detail the ortho substituent effect on 2-iodoaniline derivatives, we found that 7 (o-NHCbz) was produced in 94% yield, whereas 37 (o-NHTs), 38 (o-NHCOCF3), and 39 (o-NHBn) were formed in low-to-moderate yields ranging from 24 to 45%, irrespective of the conditions (A and B). Under these cross-coupling Conditions A and B, 40 (o-NHBoc) was formed in much higher yields of 43 and 75%, respectively. But replacing hydrogen with methyl in the o-NHBoc group (o-NMeBoc) completely inhibited this reaction, possibly due to increased steric hindrance, confirming that cross-coupling reactions are affected by steric effects of ortho substituents. However, these steric effects alone do not explain the differences in the yields of phosphonates 7, 37–40. Differences in the acidity of the adjacent N–H group may also affect the efficiency of micellar C(sp2)–P coupling.
In coupling reactions with boronic acid 50 and pinacol boronate 51, the expected phosphonate products were not formed; instead, phosphonate 21 was formed in high yields, in both reactions, through a sequential C(sp2)–P coupling and an undesired base-promoted protodeborylation. Protodeborylation is a known side-reaction in the cross-coupling chemistry of base-sensitive fluorine-containing boronic acids and pinacol esters.57 To avoid this undesired reaction, we performed an additional experiment with only 1.2 equivalents of 3 and 1.2 equivalents of 2,6-lutidine, but 21 was still the main reaction product.
Substrate scope of heteroaryl halides and/or medicinally relevant compounds
Applying the best reaction conditions thus far to C(sp2)–P coupling of heteroaryl iodides and bromides with 3 (Fig. 4) required decreasing the concentration of most starting compounds to 0.21 M to improve their solubilization and increasing the reaction temperature to 55 °C to reach higher conversion rates. As a co-solvent, EtOAc was also essential, outperforming all other organic co-solvents (THF, acetone and toluene) tested in this study. Cross-coupling reactions with 4- and 3-iodopyridines, N-benzyl-3-iodopyrazole, 2- and 3-iodothiophenes, and N-Boc-3-iodoindole provided the corresponding phosphonates (52–59) in yields ranging from 70 to 91%. The exception was phosphonate 54, which was formed in only 42% yield, most likely due to steric hindrance.Reactions of medicinally relevant 4- and 7-azaindoles with 3 provided phosphonates 60–62 in yields ranging from 57 to 92%. In particular, phosphonate 61 was formed in 92 and 42% yields when we applied Conditions B (dual-ligand) and A (multimetallic), respectively, thus showcasing the strong correlation between yield and reaction conditions. The medicinally relevant, nitrogen-rich phosphonates 63 and 64 were obtained in 64 and 66% yields, respectively, and even the starting aryl iodide for 64 was also prepared under micellar conditions in water (SNAr reaction). Therefore, micellar C(sp2)–P cross-coupling provides us with access to heterocyclic phosphonates potentially relevant to the discovery and development of bioactive compounds.
When applied to drug-like scaffolds and drugs with various functional groups (Fig. 4), these micellar C(sp2)–P coupling conditions were also effective. Reactions with iodinated drugs and bioactive compounds, specifically iodo-nimesulide, iodo-loratadine, iodo-strychnine, provided the respective phosphonates 65, 67 and 71 in yields ranging from 65 to 94%. Coupling with the key lapatinib (anticancer drug) synthetic precursor58 and with bioactive iodine-containing compounds, such as trametinib (anticancer drug) and iodosulfuron-methyl sodium salt (sulfonylurea herbicide), also afforded phosphonates 66, 68, and 70 in high yield of 87, 88, and 79%, respectively. Among these drug-like scaffolds and drugs, the highest yield (95%) was achieved when preparing phosphonate 69 from an antifungal ketoconazole derivative. These findings demonstrate that micellar C(sp2)–P cross-coupling reactions can be tolerated by a wide range of functional groups of drug scaffolds, providing opportunities for late-stage modifications.
Substrate scope of H–P(O) compounds
After establishing the substrate scope for (hetero)aryl halide couplings with diethyl H-phosphonate (3), we examined cross-coupling reactions with other phosphorus partners containing H–P bonds, such as other H-(thio)phosphonates, H-phosphinates, and secondary phosphine oxides (SPOs) (Fig. 5). In solution, these compounds feature prototropic tautomerism between their electrophilic tetravalent P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)H and nucleophilic trivalent P–OH forms.59 The latter carry a lone electron pair, which is responsible for phosphorus coordination to transition metals.60 Unsurprisingly, the electron-withdrawing and -donating substituents on the phosphorus centre strongly affected the tautomerization equilibria and their rates, as previously shown by an experimental and theoretical study with a series of R1R2P(O)H compounds, wherein tautomerization rates considerably decreased in the following order: Ph2P(O)H > PhP(O)(OAlk)H > AlkP(O)(OAlk)H > (AlkO)2P(O)H (Alk = alkyl).59 As such, substituents attached to phosphorus inherently affect all reactions involving equilibria between P(O)H and P–OH tautomers in solution under both basic and acidic conditions,61 including transition metal-catalyzed reactions. This effect explains why developing a general set of conditions for cross-coupling structurally diverse phosphorus compounds can be a challenging task.
O)H and nucleophilic trivalent P–OH forms.59 The latter carry a lone electron pair, which is responsible for phosphorus coordination to transition metals.60 Unsurprisingly, the electron-withdrawing and -donating substituents on the phosphorus centre strongly affected the tautomerization equilibria and their rates, as previously shown by an experimental and theoretical study with a series of R1R2P(O)H compounds, wherein tautomerization rates considerably decreased in the following order: Ph2P(O)H > PhP(O)(OAlk)H > AlkP(O)(OAlk)H > (AlkO)2P(O)H (Alk = alkyl).59 As such, substituents attached to phosphorus inherently affect all reactions involving equilibria between P(O)H and P–OH tautomers in solution under both basic and acidic conditions,61 including transition metal-catalyzed reactions. This effect explains why developing a general set of conditions for cross-coupling structurally diverse phosphorus compounds can be a challenging task.
At first, we studied the reactivity of different H-phosphonates, more specifically di-iso-propyl H-phosphonate (72), dibenzyl H-phosphonate (73), and bis-(S-pivaloyl-2-thioethyl) H-phosphonate (74). All of them efficiently coupled with aryl halides under optimized reaction conditions, except for 73, which is hydrolytically labile and thus afforded product in low yield (Fig. 5A). Cross-coupling reactions with 74 provided direct access to phosphonates 77, 78 and 80 bearing a S-acylthioethyl (SATE, specifically S-pivaloyl-2-thioethyl group)62 prodrug moiety in high yields, ranging from 88 to 99% (Fig. 5A). Coupling reactions of the lapatinib precursor and trametinib with 72 afforded phosphonates 79 and 81, in 75 and 53% yields, respectively. Other, commercially available dimethyl and di-n-butyl H-phosphonates showed the same efficiency as 3 (data not shown; both phosphonates were used in preliminary screening experiments; see ESI†), but the sterically hindered di-tert-butyl H-phosphonate failed to afford the target products under our reaction conditions.
To further demonstrate the functional group tolerance of the micellar C(sp2)–P coupling method and its application potential in medicinal chemistry, we cross-coupled a pomalidomide derivative, frequently used in emerging proteolysis targeting chimera (PROTAC) technology,63 with a H-phosphonate 83 containing a “clickable” terminal azide (Fig. 5A). In this coupling, we prepared phosphonate 82 in 48% yield (63% brsm) using Conditions B (without any further optimization). Accordingly, 82 may be applied in PROTAC technology upon a copper-catalysed click reaction with a suitable alkyne linker connected to an inhibitor of interest. The third phosphorus substituent, the ethoxy group, may also be exchanged for an additional functionality, thereby further expanding PROTAC applications.
We also synthesized H-phosphonate 85, an analogue of the antiviral drug azidothymidine (AZT), and subjected 85 to C(sp2)–P cross-coupling with 6 (Fig. 5A). In this reaction, 1 equivalent of 85 was mixed with 1.2 equivalents of iodide 6 under Conditions B, affording the desired AZT-containing phosphonate 84 in 56% yield and in a 1:1.1 diastereomeric ratio (31P NMR). These reactions demonstrate that C(sp2)–P coupling in water may be used to synthesize potentially bioactive nucleoside analogues in medicinal chemistry.64
Despite their structural differences from H-phosphonates, two H-phosphinates, namely ethyl phenyl- (86) and ethyl ethyl-H-phosphinate (87), the latter being relevant to the synthesis of CDK9/CycT1 inhibitors,18 also reacted under micellar cross-coupling conditions. Cross-coupling reactions with 86 provided the corresponding phosphinates 88–90, 92 and 93 in yields ranging from 65 to 78% (Fig. 5B), including a trametinib derivative. But the reactions with H-phosphinate 87 were challenging, and we prepared only phosphinate 91 in 40% yield notwithstanding our efforts to optimize the reaction conditions specifically for 87. Under Conditions B or in reactions with other ligands or bases, 91 was formed in less than 20% yield, as shown by 1H NMR analysis (see ESI†). These results are in line with studies on cross-coupling reactions with 87, which also reported low yields, even under more forcing conditions (refluxing toluene).23f,g These stark differences in cross-coupling reactivity between 86 and 87 may be attributed to their different P(O)H/P–OH tautomerization rates59 and to the stronger electron-donating abilities of the ethyl group in 87. In line with these explanations, the phosphorus in 87 shows increased electron density, which strengthens its binding to metal centres, possibly inhibiting the catalytic activity of these metal centres.
Although only a few studies on cross-coupling reactions with H-thiophosphonates (containing a PS bond) have been published thus far,65 we assessed whether H-thiophosphonates, such as 94, prepared by treating di-n-butyl H-phosphonate with Lawesson's reagent, could also be coupled with aryl iodides under micellar C(sp2)–P coupling conditions. Upon coupling with aryl iodides, the desired H-thiophosphonates were formed in poor yields (<30%), but we eventually found that the excess of 94 (and possibly of thiophosphonate products as well) inhibited the reaction. When we performed the coupling reaction by adding 0.7 equivalents of 94 in three portions (2.1 equivalents in total), the thiophosphonates 95 and 96 were formed in 56% (71% brsm) and 68% yields, respectively (Fig. 5C). Under our conditions, cross-couplings with ethyl phenyl-H-thiophosphinate (prepared in a reaction of 86 with Lawesson's reagent) failed presumably because this phosphinate has strong coordination properties, which inhibit metal catalysis, similar to those of sulfur compounds. To the best of our knowledge, no metal-catalysed coupling reaction with alkyl aryl-H-thiophosphinate has been reported to date.
Lastly, we performed cross-coupling reactions with secondary phosphine oxides (SPOs) 97 and 98 (Fig. 5D). SPOs are used not only as coupling partners but also as versatile ligands for their strong binding to metal centres.66 For this reason, cross-coupling reactions with SPOs often require high catalyst loadings (10–20 mol%) and temperatures ranging between 90 to 120 °C,67 so we expected that micellar cross-coupling reactions with SPOs could be challenging. Yet, under Conditions A (Conditions B were ineffective), C(sp2)–P coupling reactions with diphenyl phosphine oxide (97) afforded the corresponding triaryl phosphine oxides 99, 103 and 106 in good yields (43–72%) (Fig. 5D). In contrast, under modified Conditions B (Conditions A were ineffective), the reactions with dimethyl phosphine oxide (98) afforded highly-polar phosphine oxides 100–102, 104 and 105 in yields ranging from 43 to 81%.
The cross-coupling reactions with 98 required using a stronger base than 2,6-lutidine (N-methylmorpholine; pKBH+ = 7.4 in H2O) and, most often, increasing the reaction temperature to 60 °C to enhance conversion. Overall, these reactions were challenging. For example, no phosphine oxide was formed from the 3-iodo-7-azaindole derivative; instead, the starting compound was cleanly converted into the corresponding protodehalogenation product 107 (93% by 1H NMR with CH2Br2 as an internal standard) (Fig. 5D). Substituting Ts for Boc as the protecting group made no difference in the reaction outcome either. So, in some reactions, 98 acts as a hydrogen donor in a Pd-catalyzed transfer hydrogenation process, as do hypophosphites.30,31 Furthermore, our results corroborate the findings of a previous report on catalytic transfer hydrogenations of alkyl and aryl halides by sodium hypophosphite under micellar conditions in water (Tween 20 surfactant).68 Similarity to H-phosphinates, the reactivity differences between 97 and 98 may stem from their distinct P(O)H/P–OH tautomerization rates,59 possibly explaining the effect of base, and from stronger binding of 98 to Pd catalyst. Therefore, micellar cross-coupling reactions with 98 require using a more strongly coordinating and bulky P(t-Bu)3 ligand and more forcing conditions (heating to 60 °C).
Scale-up experiments
We also performed reactions at 1.5–2.5 mmol scales to understand how micellar C(sp2)–P cross-couplings proceed at larger reaction scales. The yields of phosphonates 7 (83% at 2.5 mmol vs. 94% at 0.25 mmol), 62 (89% at 1.5 mmol vs. 90% at 0.25 mmol) and 15 (83% at 2.5 mmol vs. 95% at 0.25 mmol) were slightly lower in these reactions than in those at the original reaction scale (Fig. 3 and 4). Moreover, the scale-up synthesis of 7 was performed with half the original catalyst loading (1.25 mol% Pd and 0.50 mol% Ni), thus demonstrating that catalyst loadings can be reduced in cross-coupling reactions with strongly coordinating phosphorus nucleophiles such as 3.Tandem processes involving C(sp2)–P cross-coupling
Dialkyl H-phosphonates are typically prepared by mixing alcohols with toxic, moisture- and air-sensitive PCl3.69 As such, H-phosphonate syntheses are impractical and hazardous and often lead to intractable mixtures of products.70 Dialkyl H-phosphonates can also be prepared by transesterification of diphenyl H-phosphonate (108).71 In this process, 108 is mixed with an alcohol under basic (pyridine) and neat conditions, thereby completely avoiding corrosive PCl3. For this reason, we assessed whether commercial diphenyl H-phosphonate (108) can be used as an inexpensive ($0.50 per gram at Sigma-Aldrich) source of dialkyl H-phosphonates formed in situ and then directly, without isolation, converted into arylphosphonates in micellar C(sp2)–P cross-coupling, in a one-pot process.As a proof-of-concept, we treated neat, technical grade 108 (<90% purity, 2 equivalents) with EtOH (4 equivalents) and 2,6-lutidine (4 equivalents) at room temperature for 3 hours (3 formed in situ), subsequently adding 6 (1 equivalent), Pd/Ni catalyst, LiBr, 2,6-lutidine, 2 wt % TPGS-750-M in water (0.5 mL), and EtOAc (0.1 mL) into the reaction mixture (Fig. 6A). After stirring for 15 hours, at 45 °C, phosphonate 7 was formed in 80% yield from 108 and EtOH (vs. 94% from 3 with 6). This experiment demonstrates that dialkyl H-phosphonates can be prepared from inexpensive and bench-stable 108 and then directly used in micellar C(sp2)–P cross-coupling reactions. Moreover, the cross-coupling step is robust enough to tolerate phenol by-product, with only negligible decreases in reaction yields.
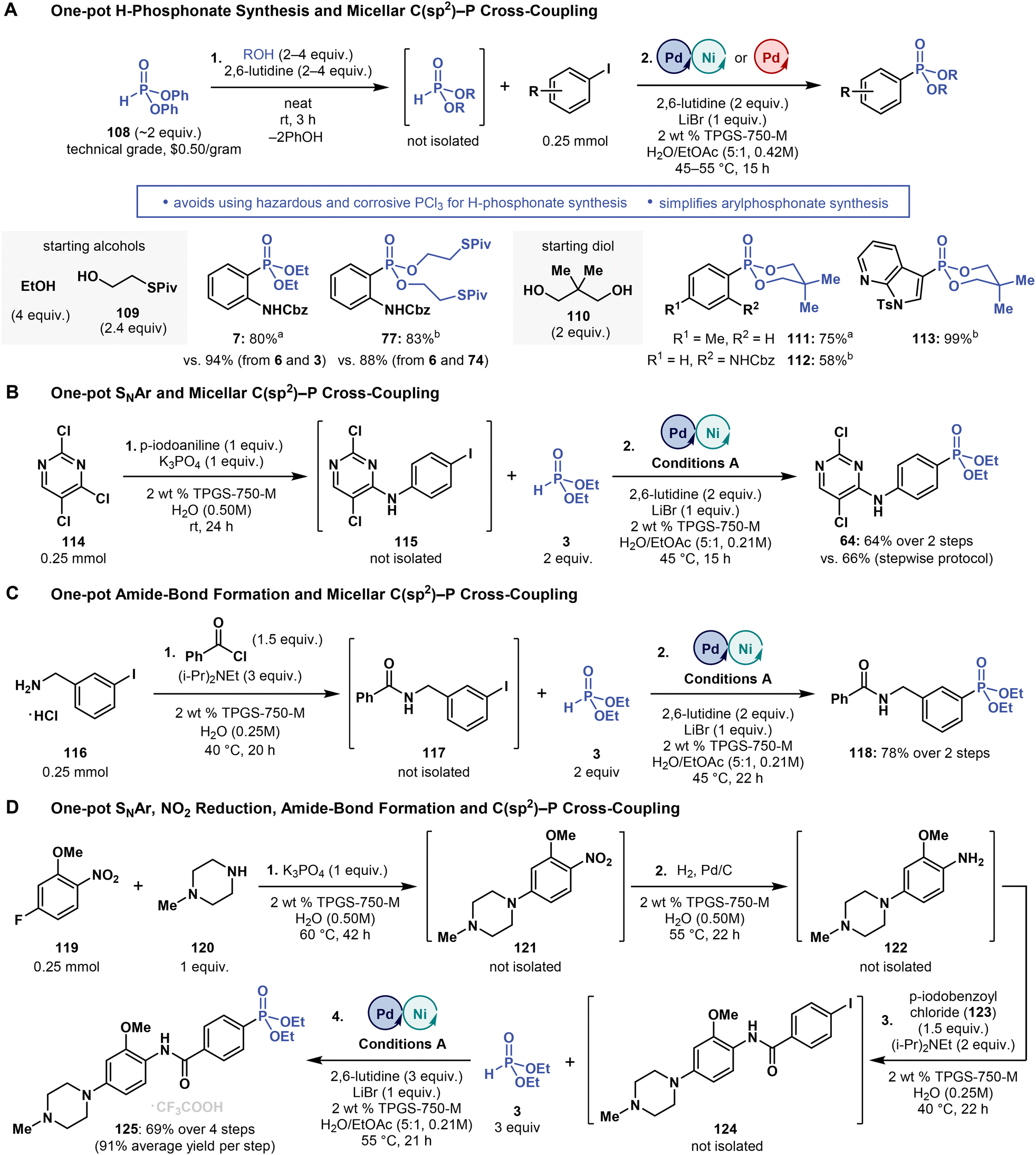 | ||
| Fig. 6 Tandem processes involving micellar C(sp2)–P cross-coupling. (A) Using diphenyl H-phosphonate (108) enables a two-step, one-pot aryl phosphonate synthesis involving the in situ formation of dialkyl H-phosphonates from alcohols. (C) Multistep one-pot processes involving C(sp2)–P cross-coupling. aConditions A. bConditions B (see Fig. 3–5). Please refer to ESI† for more details. | ||
We also prepared 77 in 83% yield (vs. 88% from 6 and 74) from 74, which was formed in situ from 108 (2 equivalent) and 109 (2.4 equivalent) (Fig. 6A). Moreover, we envisioned that this two-step one-pot protocol may provide us with a simplified access to phosphonates with the 1,3,2-dioxaphosphinane 2-oxide motif of the calcium channel blocker efonidipine20 (Fig. 1A). To this end, we synthesized structurally related phosphonates 111–113 in yields ranging from 58 to 99% using the two-step, one-pot protocol, with all reactions starting from 2,2-dimethylpropane-1,3-diol (110, 2 equivalent) and 108 (2 equivalent).
Lastly, we examined whether the micellar C(sp2)–P cross-coupling can be used in other multistep one-pot procedures. Tandem processes are commonly implemented in micellar reactions in water, significantly increasing mass balance and reducing waste, cost and labor, so they are highly attractive, particularly under process chemistry settings (e.g., pharmaceutical synthesis). To demonstrate the feasibility of micellar C(sp2)–P cross-coupling in tandem procedures, we performed three multistep, one-pot reactions (Fig. 6B–D): (i) SNAr reaction of 114 with 4-iodoaniline followed by C(sp2)–P cross-coupling with 3, (ii) amide-bond coupling between amine 126 and benzoyl chloride followed by C(sp2)–P cross-coupling with 3, and (iii) SNAr reaction of 119 with 120, followed by nitro group reduction, subsequent amine acylation and, finally, C(sp2)–P cross-coupling with 3. The two-step, one-pot reactions afforded the corresponding phosphonates 64 and 118 in 64 and 78% yield, respectively, and the four-step, one-pot reaction yielded phosphonate 125 in 69% (91% average yield per step), while skipping the work-up and product isolation of most of the reaction steps. These results highlight low barriers to adopting micellar C(sp2)–P cross-coupling reactions in tandem processes in water.
Cross-coupling of aryl triflates
Aryl triflates stand out as potential coupling partners for their excellent accessibility from ubiquitous phenols. However, their C–O bond is stronger than the C–I bond in aryl iodides, which may hamper cross-coupling by increasing energy barriers to oxidative addition. Moreover, the literature on cross-coupling chemistry of aryl triflates under micellar conditions is rather scarce.72 Despite these caveats, we tested the feasibility of C(sp2)–P cross-coupling of aryl triflates under micellar conditions in water, but many of our initial experiments failed, including multimetallic and dual-ligand conditions. Nevertheless, by screening various reaction conditions (see ESI†), we found that the dppf ligand promotes the cross-coupling of aryl triflates with 3 under modified multimetallic conditions, albeit in mediocre yields (Fig. 7). Thus, we established a small substrate scope and prepared two quinoline phosphonates 126 and 127 in 77 and 66% yield, respectively, and a phenyl alanine derivative 128 in 56% yield.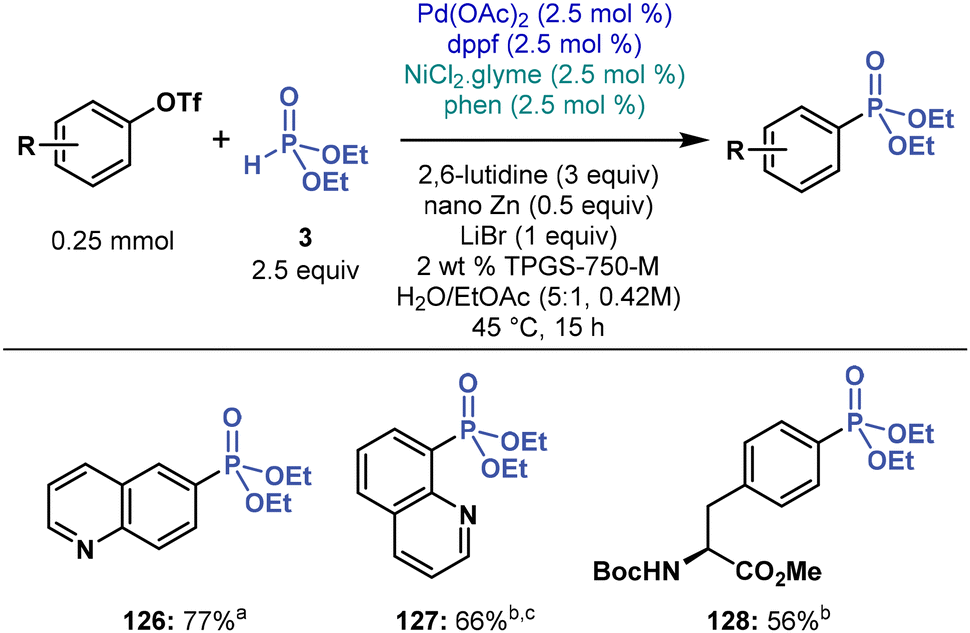 | ||
| Fig. 7 Micellar multimetallic C(sp2)–P cross-coupling of aryl triflates with 3. a5 mol% of Pd(OAc)2, 5 mol% of dppf. b1 mol% Ni(phen)3Cl2. cPerformed at 55 °C. | ||
During our screening experiments (see ESI†), we noticed that adding salts containing weakly coordinating anions (e.g., NaPF6, Mg(OTf)2, and Zn(OTf)2) inhibit C(sp2)–P cross-coupling reactions of aryl triflates. This finding is significant for Zn(OTf)2 which is continuously formed from Zn and aryl triflate. Thus, triflate anions may thwart micellar C(sp2)–P cross-coupling, stopping this reaction from a specific concentration.
Current limitations
Our method showed a broad substrate scope and functional group-tolerance. However, we encountered some limitations. In particular, ortho-substituted iodobenzenes bearing OH, NH2, NO2 and CO2H groups provided phosphonate products in poor yields (<20% by 1H NMR with CH2Br2 as an internal standard). Similarly, 2-iodopyridine and 2-iodopyrazine afforded no cross-coupling products. Despite affording the target products in serviceable yields (40–86%), cross-coupling reactions with some phosphorus nucleophiles (87, 97, and 98) still require further optimization to reach their full efficiency. Nevertheless, the recently reported surfactant Savie,73 which enhances the efficiency of many transformations under micellar conditions in water, may enable us to perform C(sp2)–P cross-coupling reactions at higher temperatures, at which the TPGS-750-M surfactant reaches its cloud point. These more forcing conditions may be the key to efficient cross-coupling reactions with poorly reactive (hetero)aryl halides and with strongly coordinating phosphorus nucleophiles (such as 87, 97, and 98). Similarly, detailed mechanistic understanding of both multimetallic and dual-ligand reaction conditions (see ESI† for plausible mechanism) may help designing more efficient micellar C(sp2)–P cross-coupling reactions and thus warrants further investigation. Our laboratory is actively pursuing these lines of research.Future perspectives
Our approach may be readily adopted by organic, medicinal and process chemists to develop more efficient micellar C(sp2)–P cross-coupling reactions of challenging substrates (e.g., 2-halopyridines, aryl triflates) while decreasing catalyst loading, ideally to ppm levels. Initial experiments performed using multimetallic (2500 ppm Pd/1000 ppm Ni) and dual-ligand (3500 ppm Pd) conditions showed that ppm level catalysis is indeed feasible but current conditions vary in reaction performance (see ESI†). Therefore, future efforts will certainly focus on identifying more highly active catalysts and may provide more efficient access to novel scaffolds containing phosphorus, such as tertiary phosphine oxides prepared from SPOs (e.g., 98), emerging, medically relevant compounds (anticancer drugs, e.g., brigatinib).Conclusion
This study establishes an experimental paradigm for mild, practical, general, and environmentally responsible C(sp2)–P cross-coupling under multimetallic Pd/Ni- and dual-ligand Pd-catalysed conditions, overcoming current limitations of C(sp2)–P cross-coupling reactions by (1) using inexpensive, widely available Pd and Ni catalyst at lower-than-typical catalyst loadings and commercially available H-phosphonates, H-phosphinates and SPOs, (2) requiring only mild heating (45–60 °C), thereby increasing functional group-tolerance and reducing energy inputs, and (3) replacing environmentally unsustainable and harmful polar aprotic solvents (e.g., DMF) with water. These reactions are easy to perform, safe and readily scalable, produce less organic waste than reactions in organic solvents (see ESI† for calculations of Process Mass Intensity, PMI, values and E factors, and their comparison with those of the literature procedures), and can be applied to synthesize over 100 (hetero)aryl phosphonates, thiophosphonates, phosphinates and phosphine oxides with a wide range of functional groups, including medically relevant substrates and drugs under late-stage functionalization settings, in addition to potentially bioactive compounds. Combined, these findings open up new opportunities for applications in medicinal chemistry and life sciences by fostering the development of environmentally friendly and mild cross-coupling reactions under micellar conditions in water, including tandem processes, while expanding the toolbox of emerging multimetallic-catalysed reactions.46a,48,74Author contributions
All authors have given approval to the final version of the manuscript. R. N. conceived the idea, designed experiments, optimized the reaction conditions, established the substrate scope, collected analytical data, and wrote the manuscript. K. K. synthesized starting compounds, helped with purification and collection of analytical data. O. B. oversaw the project and secured funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly acknowledge financial support from the Czech Science Foundation (Grant No. 23-05752S, O. B.) and the Charles University Research Centre program (Grant No. UNCE/SCI/014). We thank Dr Michal Urban and Dr Zdeněk Tošner for their assistance with NMR experiments, Kvetoslava Kertisová for HRMS analysis, and Stanislava Matějková for ICP analysis. We thank Dr Carlos V. Melo for editing the manuscript. We are also grateful to anonymous reviewers for their valuable inputs.References
- (a) V. Iaroshenko, Organophosphorus Chemistry: From Molecules to Applications, Wiley-VCH, Weinheim, 2019 CrossRef; (b) C. M. Timperley, Best Synthetic Methods: Organophosphorus(V) Chemistry, Academic Press, New York, 2015 Search PubMed; (c) L. D. Quin, A Guide to Organophosphorus Chemistry, Wiley-VCH, New York, 2000 Search PubMed; (d) C. Xie, A. J. Smaligo, X.-R. Song and O. Kwon, ACS Cent. Sci., 2021, 7, 536–558 CrossRef CAS PubMed; (e) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed; (f) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS PubMed.
- (a) S. Demkowicz, J. Rachon, M. Daśko and W. Kozak, RSC Adv., 2016, 6, 7101–7112 RSC; (b) H. Yu, H. Yang, E. Shi and W. Tang, Med. Drug Discovery, 2020, 8, 100063 CrossRef CAS.
- J. B. Rodrigueza and C. Gallo-Rodriguez, ChemMedChem, 2018, 14, 190–216 CrossRef PubMed.
- For example: (a) E. Schönbrunn, S. Eschenburg, W. A. Shuttleworth, J. V. Schloss, N. Amrhein, J. N. Evans and W. Kabsch, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1376–1380 CrossRef PubMed; (b) C. Zhou, X. Luo, N. Chen, L. Zhang and J. Gao, J. Agric. Food Chem., 2020, 68, 3344–3353 CrossRef CAS PubMed.
- (a) C. Queffélec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef PubMed; (b) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS; (c) T. Baumgartner, Acc. Chem. Res., 2014, 47, 1613–1622 CrossRef CAS PubMed.
- M. M. Velencoso, A. Battig, J. C. Markwart, B. Schartel and F. R. Wurm, Angew. Chem., Int. Ed., 2018, 57, 10450–10467 CrossRef CAS PubMed.
- M. V. Stambirskyi, T. Kostiuk, S. I. Sirobaba, A. Rudnichenko, D. L. Titikaiev, Y. V. Dmytriv, H. Kuznietsova, I. Pishel, P. Borysko and P. K. Mykhailiuk, J. Org. Chem., 2021, 86, 12783–12801 CrossRef CAS PubMed.
- P. Finkbeiner, J. P. Hehn and C. Gnamm, J. Med. Chem., 2020, 63, 7081–7107 CrossRef CAS PubMed.
- J. J. Marineau, K. B. Hamman, S. Hu, S. Alnemy, J. Mihalich, A. Kabro, K. M. Whitmore, D. K. Winter, S. Roy, S. Ciblat, N. Ke, A. Savinainen, A. Wilsily, G. Malojcic, R. Zahler, D. Schmidt, M. J. Bradley, N. J. Waters and C. Chuaqui, J. Med. Chem., 2022, 65, 1458–1480 CrossRef CAS PubMed.
- (a) G. P. Horsman and D. L. Zechel, Chem. Rev., 2017, 117, 5704–5783 CrossRef CAS PubMed; (b) R. Engel, Chem. Rev., 1977, 77, 349–367 CrossRef CAS.
- E. D. Clercq and A. Holý, Nat. Rev. Drug Discovery, 2005, 4, 928–940 CrossRef PubMed.
- For example: (a) D. L. Higgins, R. Chang, D. V. Debabov, J. Leung, T. Wu, K. M. Krause, E. Sandvik, J. M. Hubbard, K. Kaniga, D. E. Schmidt, Q. Gao, R. T. Cass, D. E. Karr, B. M. Benton and P. P. Humphrey, Antimicrob. Agents Chemother., 2005, 49, 1127–1134 CrossRef CAS PubMed; (b) Y. Cao, Q. Peng, S. Li, Z. Deng and J. Gao, RSC Adv., 2019, 9, 42204–42218 RSC.
- (a) M. Krečmerová, P. Majer, R. Rais and B. S. Slusher, Front. Chem., 2022, 10, 889737 CrossRef; (b) K. M. Heidel and C. Dowd, Future Med. Chem., 2019, 11, 1625–1643 CrossRef CAS PubMed.
- (a) T. S. Elliott, A. Slowey, Y. Ye and S. J. Conway, MedChemComm, 2012, 3, 735–751 RSC; (b) Y. Mehellou, H. S. Rattan and J. Balzarini, J. Med. Chem., 2018, 61, 2211–2226 CrossRef CAS PubMed.
- A. Mucha, P. Kafarski and Ł. Berlicki, J. Med. Chem., 2011, 54, 5955–5980 CrossRef CAS PubMed.
- (a) I. V. L. Wilkinson, K. J. Perkins, H. Dugdale, L. Moir, A. Vuorinen, M. Chatzopoulou, S. E. Squire, S. Monecke, A. Lomow, M. Geese, P. D. Charles, P. Burch, J. M. Tinsley, G. Wynne, S. G. Davies, F. X. Wilson, F. Rastinejad, S. Mohammed, K. E. Davies and A. J. Russell, Angew. Chem., Int. Ed., 2020, 59, 2420–2428 CrossRef CAS PubMed; (b) M. Chatzopoulou, E. Emer, C. Lecci, J. A. Rowley, A.-S. Casagrande, L. Moir, S. E. Squire, S. G. Davies, S. Harriman, G. M. Wynne, F. X. Wilson, K. E. Davies and A. J. Russell, ACS Med. Chem. Lett., 2020, 11, 2421–2427 CrossRef CAS PubMed.
- M. D. Sørensen, L. K. A. Blæhr, M. K. Christensen, T. Høyer, S. Latini, P.-J. V. Hjarnaa and F. Björkling, Bioorg. Med. Chem., 2003, 11, 5461–5484 CrossRef PubMed.
- G. Németh, Z. Greff, A. Sipos, Z. Varga, R. Székely, M. Sebestyén, Z. Jászay, S. Béni, Z. Nemes, J.-L. Pirat, J.-N. Volle, D. Virieux, Á. Gyuris, K. Kelemenics, É. Áy, J. Minarovits, S. Szathmary, G. Kéri and L. Őrfi, J. Med. Chem., 2014, 57, 3939–3965 CrossRef PubMed.
- C. Dousson, F. Alexandre, A. Amador, S. Bonaric, S. Bot, C. Caillet, T. Convard, D. da Costa, M. Lioure, A. Roland, E. Rosinovsky, S. Maldonado, C. Parsy, C. Trochet, R. Storer, A. Stewart, J. Wang, B. A. Mayes, C. Musiu, B. Poddesu, L. Vargiu, M. Liuzzi, A. Moussa, J. Jakubik, L. Hubbard, M. Seifer and D. Standring, J. Med. Chem., 2016, 59, 1891–1898 CrossRef CAS PubMed.
- H. Tanaka and K. Shigenobu, Cardiovasc. Drug Rev., 2002, 20, 81–92 CrossRef CAS PubMed.
- X. Chen, D. J. Kopecky, J. Mihalic, S. Jeffries, X. Min, J. Heath, J. Deignan, S. Lai, Z. Fu, C. Guimaraes, S. Shen, S. Li, S. Johnstone, S. Thibault, H. Xu, M. Cardozo, W. Shen, N. Walker, F. Kayser and Z. Wang, J. Med. Chem., 2012, 55, 3837–3851 CrossRef CAS PubMed.
- (a) W. Huang, S. Liu, D. Zou, M. Thomas, Y. Wang, T. Zhou, J. Romero, A. Kohlmann, F. Li, J. Qi, L. Cai, T. A. Dwight, Y. Xu, R. Xu, R. Dodd, A. Toms, L. Parillon, X. Lu, R. Anjum, S. Zhang, F. Wang, J. Keats, S. D. Wardwell, Y. Ning, Q. Xu, L. E. Moran, Q. K. Mohemmad, H. G. Jang, T. Clackson, N. I. Narasimhan, V. M. Rivera, X. Zhu, D. Dalgarno and W. C. Shakespeare, J. Med. Chem., 2016, 59, 4948–4964 CrossRef CAS PubMed; (b) S. Li, T. Zhang, S.-J. Zhu, C. Lei, M. Lai, L. Peng, L. Tong, Z. Pang, X. Lu and J. Ding, ACS Med. Chem. Lett., 2022, 13, 196–202 CrossRef CAS PubMed.
- (a) T. Hirao, T. Masunaga, Y. Ohshiro and T. Agawa, Synthesis, 1981, 56–57 CrossRef CAS; (b) T. Hirao, T. Maunaga, N. Yamada, Y. Ohshiro and T. Agawa, Bull. Chem. Soc. Jpn., 1982, 55, 909–913 CrossRef CAS; (c) M. Kalek and J. Stawinski, Organometallics, 2007, 26, 5840–5847 CrossRef CAS; (d) M. Kalek and J. Stawinski, Organometallics, 2008, 27, 5876–5888 CrossRef CAS; (e) M. Kalek, M. Jezowska and J. Stawinski, Adv. Synth. Catal., 2009, 351, 3207–3216 CrossRef CAS; (f) E. L. Deal, C. Petit and J.-L. Montchamp, Org. Lett., 2011, 13, 3270–3273 CrossRef CAS PubMed; (g) O. Berger, C. Petit, E. L. Deal and J.-L. Montchamp, Adv. Synth. Catal., 2013, 355, 1361–1373 CrossRef CAS; (h) M. Kalek, A. Ziadi and J. Stawinski, Org. Lett., 2008, 10, 4637–4640 CrossRef CAS PubMed; (i) Q. Dai, W. Li, Z. Li and J. Zhang, J. Am. Chem. Soc., 2019, 141, 20556–20564 CrossRef CAS PubMed; (j) J. R. Moncarz, N. F. Laritcheva and D. S. Glueck, J. Am. Chem. Soc., 2002, 124, 13356–13357 CrossRef CAS PubMed.
- (a) D. Zhu, S. Jiang, Q. Wu, H. Wang, L. Chai, H. Li and H. Li, Org. Lett., 2021, 23, 160–165 CrossRef CAS PubMed; (b) Y. Bai, N. Liu, S. Wang, S. Wang, S. Ning, L. Shi, L. Cui, Z. Zhang and J. Xiang, Org. Lett., 2019, 21, 6835–6838 CrossRef CAS PubMed; (c) Y.-L. Zhao, G.-J. Wu, Y. Li, L.-X. Gao and F.-S. Han, Chem. – Eur. J., 2012, 18, 9622–9627 CrossRef CAS PubMed; (d) C. Shen, G. Yang and W. Zhang, Org. Biomol. Chem., 2012, 10, 3500–3505 RSC.
- (a) D. Gelman, L. Jiang and S. L. Buchwald, Org. Lett., 2003, 5, 2315–2318 CrossRef CAS PubMed; (b) C. Huang, X. Tang, H. Fu, Y. Jiang and Y. Zhao, J. Org. Chem., 2006, 71, 5020–5022 CrossRef CAS PubMed; (c) H. Zhang, X. Y. Zhang, D. Q. Dong and Z. Wang, RSC Adv., 2015, 5, 52824–52831 RSC; (d) R. Beaud, R. J. Phipps and M. J. Gaunt, J. Am. Chem. Soc., 2016, 138, 13183–13186 CrossRef CAS PubMed.
- (a) A. L. Schwan, Chem. Soc. Rev., 2004, 33, 218–224 RSC; (b) F. M. Tappe, V. T. Trepohl and M. Oestreich, Synthesis, 2010, 3037–3062 CAS; (c) C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981–8006 CrossRef CAS PubMed; (d) U. S. Kanchana, E. J. Diana, T. V. Mathew and G. Anilkumar, ChemistrySelect, 2021, 6, 1579–1588 CrossRef CAS.
- For example, please refer to: (a) J. Yang, T. Chen and L.-B. Han, J. Am. Chem. Soc., 2015, 137, 1782–1785 CrossRef CAS PubMed; (b) W. C. Fu, C. M. So and F. Y. Kwong, Org. Lett., 2015, 17, 5906–5909 CrossRef CAS PubMed; (c) H. McErlain, L. M. Riley and A. Sutherland, J. Org. Chem., 2021, 86, 17036–17049 CrossRef CAS PubMed; (d) L. Liao, Y. Gui, X. Zhang, G. Shen, H. Liu, W. Zhou, J. Li and D. Yu, Org. Lett., 2017, 19, 3735–3738 CrossRef CAS PubMed.
- C.-G. Feng, M. Ye, K.-J. Xiao, S. Li and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 9322–9325 CrossRef CAS PubMed.
- (a) C. Li, T. Yano, N. Ishida and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 9801–9804 CrossRef CAS PubMed; (b) M. Min, D. Kang, S. Jung and S. Hong, Adv. Synth. Catal., 2016, 358, 1296–1301 CrossRef CAS; (c) B. Li, M. Liu, S. U. Rehman and C. Li, J. Am. Chem. Soc., 2022, 144, 2893–2898 CrossRef CAS PubMed.
- J.-L. Montchamp and Y. R. Dumond, J. Am. Chem. Soc., 2001, 123, 510–511 CrossRef CAS.
- For example, please refer to: (a) S. K. Boyer, J. Bach, J. McKenna and E. Jagdmann Jr., J. Org. Chem., 1985, 50, 3408–3411 CrossRef CAS. For reviews, refer to: (b) G. Brieger and T. J. Nestrick, Chem. Rev., 1974, 74, 567–580 CrossRef CAS; (c) R. A. W. Johnstone and A. H. Wilby, Chem. Rev., 1985, 85, 129–170 CrossRef CAS.
- Y. Belabassi, S. Alzghari and J.-L. Montchamp, J. Organomet. Chem., 2008, 693, 3171–3178 CrossRef CAS PubMed.
- (a) C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC; (b) D. Prat, J. Haylerb and A. A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- EUR-Lex (official website of European Union law) https://data.europa.eu/eli/reg/2021/2030/oj (accessed July 2023).
- (a) T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed; (b) M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 RSC.
- A. L. Casalnuovo and J. C. Calabrese, J. Am. Chem. Soc., 1990, 112, 4324–4330 CrossRef CAS.
- K. Xu, F. Yang, G. Zhang and Y. Wu, Green Chem., 2013, 15, 1055–1060 RSC.
- S. M. Rummelt, M. Ranocchiari and J. A. van Bokhoven, Org. Lett., 2012, 14, 2188–2190 CrossRef CAS PubMed.
- X. Zhang, H. Liu, X. Hu, G. Tang, J. Zhu and Y. Zhao, Org. Lett., 2011, 13, 3478–3481 CrossRef CAS PubMed.
- For reviews, please refer to: (a) B. H. Lipshutz, J. Org. Chem., 2017, 82, 2806–2816 CrossRef CAS PubMed; (b) T. Dwars, E. Paetzold and G. Oehme, Angew. Chem., Int. Ed., 2005, 44, 7174–7199 CrossRef CAS PubMed; (c) B. H. Lipshutz, S. Ghorai and M. Cortes-Clerget, Chem. – Eur. J., 2018, 24, 6672–6695 CrossRef CAS PubMed; (d) M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC.
- T. Ansari, F. Gallou and S. Handa, Coord. Chem. Rev., 2023, 488, 215158 CrossRef CAS.
- For example, please refer to: (a) A. Krasovskiy, C. Duplais and B. H. Lipshutz, J. Am. Chem. Soc., 2009, 131, 15592–15593 CrossRef CAS PubMed; (b) S. Handa, Y. Wang, F. Gallou and B. H. Lipshutz, Science, 2015, 349, 1087–1091 CrossRef CAS PubMed; (c) H. Pang, Y. Wang, F. Gallou and B. H. Lipshutz, J. Am. Chem. Soc., 2019, 141, 17117–17124 CrossRef CAS PubMed; (d) H. Pang, Y. Hu, J. Yu, F. Gallou and B. H. Lipshutz, J. Am. Chem. Soc., 2021, 143, 3373–3382 CrossRef CAS PubMed; (e) Y. Hu, X. Li, G. Jin and B. H. Lipshutz, ACS Catal., 2023, 13, 3179–3186 CrossRef CAS PubMed.
- (a) S. Hazra, F. Gallou and S. Handa, ACS Sustainable Chem. Eng., 2022, 10, 5299–5306 CrossRef CAS; (b) C. M. Gabriel, M. Keener, F. Gallou and B. H. Lipshutz, Org. Lett., 2015, 17, 3968–3971 CrossRef CAS PubMed.
- (a) N. A. Isley, R. T. H. Linstadt, S. M. Kelly, F. Gallou and B. H. Lipshutz, Org. Lett., 2015, 17, 4734–4737 CrossRef CAS PubMed; (b) N. R. Lee, F. Gallou and B. H. Lipshutz, Org. Process Res. Dev., 2017, 21, 218–221 CrossRef CAS.
- B. H. Lipshutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovskiy, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379–4391 CrossRef CAS PubMed.
- For example, please refer to: (a) K. Kang, N. L. Loud, T. A. DiBenedetto and D. J. Weix, J. Am. Chem. Soc., 2021, 143, 21484–21491 CrossRef CAS PubMed; (b) I. J. S. Fairlamb, R. J. K. Taylor, J. L. Serrano and G. Sanchez, New J. Chem., 2006, 30, 1695–1704 RSC.
- V. T. Tran, Z.-Q. Li, O. Apolinar, J. Derosa, M. V. Joannou, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 7409–7413 CrossRef CAS PubMed.
- A. M. Olivares and D. J. Weix, J. Am. Chem. Soc., 2018, 140, 2446–2449 CrossRef CAS PubMed.
- C. M. Gabriel, N. R. Lee, F. Bigorne, P. Klumphu, M. Parmentier, F. Gallou and B. H. Lipshutz, Org. Lett., 2017, 19, 194–197 CrossRef CAS PubMed.
- B.p. of 3 is 187–188 °C and b.p. of 2,6-lutidine is 143–145 °C (both at atmospheric pressure).
- For example, please refer to: (a) G. Meng, Z. Wang, H. S. S. Chan, N. Chekshin, Z. Li, P. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2023, 145, 8198–8208 CrossRef CAS PubMed; (b) S. K. Sinha, S. Panja, J. Grover, P. S. Hazra, S. Pandit, Y. Bairagi and X. L. Zhang, J. Am. Chem. Soc., 2022, 144, 12032–12042 CrossRef CAS PubMed; (c) D. Zhao, P. Xu and T. Ritter, Chem, 2019, 5, 97–107 CrossRef CAS; (d) H. Chen, P. Wedi and T. Meyer, Angew. Chem., Int. Ed., 2018, 57, 2497–2501 CrossRef CAS PubMed.
- W. M. Cheng, R. Shang and Y. Fu, Nat. Commun., 2018, 9, 5215 CrossRef PubMed.
- B. Zhao, R. Shang, G.-Z. Wang, S. Wang, H. Chen and Y. Fu, ACS Catal., 2020, 10, 1334–1343 CrossRef CAS.
- P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2769 CrossRef CAS PubMed.
- The following study showed that high-bite angle diphosphine ligands induce rapid C(sp2)–P bond-forming reductive limination: M. C. Kohler, T. V. Grimes, X. Wang, T. R. Cundari and R. A. Stockland Jr., Organometallics, 2009, 28, 1193–1201 CrossRef CAS.
- (a) M.-N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC; (b) P. W. N. M. van Leeuwen and P. C. J. Kamer, Catal. Sci. Technol., 2017, 8, 26–113 RSC.
- (a) P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156–13165 CrossRef CAS PubMed; (b) L. Chen, D. R. Sanchez, B. Zhang and B. P. Carrow, J. Am. Chem. Soc., 2017, 139, 12418–12421 CrossRef CAS PubMed.
- P. J. Medina and S. Goodin, Clin. Ther., 2008, 30, 1426–1447 CrossRef CAS PubMed.
- B. G. Janesko, H. C. Fisher, M. J. Bridle and J.-L. Montchamp, J. Org. Chem., 2015, 80, 10025–10032 CrossRef CAS PubMed.
- G. Manca, M. Caporali, A. Ienco, M. Peruzzini and C. Mealli, J. Organomet. Chem., 2014, 760, 177–185 CrossRef CAS.
- K. D. Troev, Reactivity of P–H Group of Phosphorus Based Compounds, Elsevier, London, 2017, and references therein Search PubMed.
- U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed , and references therein.
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed.
- For example, please refer to: (a) C. P. Tam, L. Zhou, A. C. Fahrenbach, W. Zhang, T. Walton and J. W. Szostak, J. Am. Chem. Soc., 2018, 140, 783–792 CrossRef CAS PubMed; (b) W. Zhang, C. P. Tam, J. Wang and J. W. Szostak, ACS Cent. Sci., 2016, 2, 916–926 CrossRef CAS PubMed.
- T. Johansson and J. Stawinski, Tetrahedron, 2004, 60, 389–395 CrossRef CAS.
- (a) T. M. Shaikh, C.-M. Weng and F.-E. Hong, Coord. Chem. Rev., 2012, 256, 771–803 CrossRef CAS; (b) P. Sutra and A. Igau, Coord. Chem. Rev., 2016, 308, 97–116 CrossRef CAS.
- For example, please refer to: (a) A. Gernet, N. Sevrain, J. N. Volle, T. Ayad, J. L. Pirat and D. Virieux, J. Org. Chem., 2020, 85, 14730–14743 CrossRef CAS PubMed; (b) H. Rao, Y. Jin, H. Fu, Y. Jiang and Y. Zhao, Chem. – Eur. J., 2006, 12, 3636–3646 CrossRef CAS PubMed; (c) M. Toffano, C. Dobrota and J.-C. Fiaud, Eur. J. Org. Chem., 2006, 650–656 CrossRef CAS; (d) Y. Xu, J. Xia and H. Guo, Synthesis, 1986, 691–692 CrossRef CAS.
- M. Oba, K. Kojima, M. Endo, H. Sano and K. Nishiyama, Green Chem. Lett. Rev., 2013, 6, 233–236 CrossRef CAS.
- J.-L. Montchamp, Acc. Chem. Res., 2014, 47, 77–87 CrossRef CAS PubMed , and references therein.
- T. M. Lord, S. L. Casino, S. E. Hartzell, K. J. Garcia, R. D. Pike and R. A. Stockland Jr., ChemistrySelect, 2016, 1, 2188–2191 CrossRef CAS.
- (a) A. D. Dal-Maso, F. Legendre, C. Blonski and P. Hoffmann, Synth. Commun., 2008, 38, 1688–1693 CrossRef CAS; (b) Q. Xiao, Y. Ju and Y. Zhao, Heteroat. Chem., 2003, 14, 208–210 CrossRef CAS.
- A few examples can be found in: (a) S. Handa, E. D. Slack and B. H. Lipshutz, Angew. Chem., Int. Ed., 2015, 54, 11994–11998 CrossRef CAS PubMed; (b) E. B. Landstrom, N. Akporji, N. R. Lee, C. M. Gabriel, F. C. Braga and B. H. Lipshutz, Org. Lett., 2020, 22, 6543–6546 CrossRef CAS PubMed; (c) S. Handa, F. Ibrahim, T. N. Ansari and F. Gallou, ChemCatChem, 2018, 10, 4229–4233 CrossRef CAS.
- J. R. A. Kincaid, M. J. Wong, N. Akporji, F. Gallou, D. M. Fialho and B. H. Lipshutz, J. Am. Chem. Soc., 2023, 145, 4266–4278 CrossRef CAS PubMed.
- (a) L. K. G. Ackerman-Biegasiewicz, S. K. Kariofillis and D. J. Weix, J. Am. Chem. Soc., 2023, 145, 6596–6614 CrossRef CAS PubMed; (b) L. K. G. Ackerman, M. M. Lovell and D. J. Weix, Nature, 2015, 524, 454–457 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc02735j |
| This journal is © The Royal Society of Chemistry 2023 |