
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous flow synthesis of PCN-222 (MOF-545) with controlled size and morphology: a sustainable approach for efficient production†
Alessio
Zuliani
 *,
M.
Carmen Castillejos
and
Noureddine
Khiar
*
*,
M.
Carmen Castillejos
and
Noureddine
Khiar
*
Asymmetric Synthesis and Nanosystem Group (Art&Fun), Institute for Chemical Research (IIQ), CSIC-University of Seville, 41092 Sevilla, Spain. E-mail: a.zuliani@csic.es; khiar@iiq.csic.es
First published on 21st November 2023
Abstract
Zirconium-based MOFs, such as PCN-222 (MOF-545), have gained attention due to their regular porosity, tuneable pore size, versatile structure and exceptional thermal/chemical stability. However, the synthesis of PCN-222 using traditional batch processes suffers from limitations in productivity, scalability, and control over size and morphology. This study presents a paradigm shift from batch to continuous flow (c.f.) synthesis for the efficient production of PCN-222. The optimization of batch conditions and exploration of c.f. parameters enabled the synthesis of high-quality PCN-222 with Space–Time–Yield (STY) exceeding 950 kg m−3 day−1. The c.f. approach offered advantages including improved resource efficiency, safer chemical handling, and scalability. The integration of ultrasound-assisted technique into the c.f. facilitated the shaping of PCN-222 into prolate ellipsoids, making it suitable for applications in drug delivery and catalysis. The successful substitution of hazardous solvents with greener alternatives further enhanced the sustainability of the process. Finally, the loading of a thio-N-acetylgalactosamine-PEG-sulfate ligand on PCN-222 in tandem c.f., expanded the potentialities of the flow synthetic approach. This study not only advances the field of MOF synthesis but also paves the way for efficient and sustainable manufacturing processes in c.f. synthesis of nanoparticles and functionalized nanoparticles for active drug delivery.
1. Introduction
Metal organic frameworks (MOFs) are a class of reticular materials in which metal-containing inorganic building units, i.e., metal nodes (metal ions or metallic clusters), are connected to multidentate organic building units (linkers) via coordination bonding forming two- and three-dimensional structures. MOFs have garnered significant scientific interest due to their regular porosity and tuneable pore size, multivariate structures with multiple metals (either mono-, bi-, or tri-metallic systems) and/or organic linkers, and conformationally flexible linkers and/or geometrically versatile inorganic building units.1,2 In addition, MOFs can be functionalized (both the external surface and internal pore space) by post-synthetic modifications (PSM) to install new functionalities. Up to date, more than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 different MOFs have been explored for potential applications in drug delivery, imaging, sensing, catalysis, gas (hydrogen, carbon dioxide, methane) storage and other biomedical uses, and a further half a million structures of MOFs have been hypothesized.3
000 different MOFs have been explored for potential applications in drug delivery, imaging, sensing, catalysis, gas (hydrogen, carbon dioxide, methane) storage and other biomedical uses, and a further half a million structures of MOFs have been hypothesized.3
Among all, zirconium (Zr) based MOFs are gaining prominence due to their unique combination of properties. For instance, Zr-based MOFs have exceptional thermal and chemical stability, making them ideal for high-temperature applications and in harsh chemical environments. In addition, the Zr metal center has a high coordination number and large ionic radius, which can lead to a diverse range of structures by supporting a large variety of organic ligands. A Zr-based MOF having attracted attention in the last decade is the porous coordination network-222 (PCN-222, also known as MOF-545), firstly synthesized in 2012.4–6 PCN-222 is composed of Zr(IV) clusters connected by tetrakis(4-carboxyphenyl)porphyrin (TCPP) ligands through Zr–carboxyl bonds in a three-dimensional porous network structure. The strong coordination bond between Zr and O, rising from the high charge density and bond polarization of the highly oxidated Zr(IV), results in a highly robust structure. The morphology of PCN-222 is mainly reported as micro-dimensional (>1 μm length) needle/rod-shaped crystals or, less frequently, as prolate ellipsoids (ranging from ca. 50 nm up to more than 500 nm), while the strict control of the size-distribution of PCN-222 is hardly achieved.7,8 The main characteristics of PCN-222 include high surface area of up to more than 2000 m2 g−1 and large pore volume. Thus, PCN-222 has been mainly explored for storing gases such as methane, hydrogen, and carbon dioxide and as a catalyst in organic transformations and in the immobilization of enzymes. Concerning the field of biomedicine, PCN-222 has recently attracted considerable attention due to its remarkable acid stability and biocompatibility, which offer the potential for targeted drug delivery to specific sites such as acidic tumour environments. Moreover, PCN-222 can be used as pH-sensitive drug delivery system, further enhancing its therapeutic applications.9 Additionally, PCN-222 finds potential utility in photodynamic therapy (PDT) when exposed to laser irradiation.10 The captivating biomedical applications of PCN-222 are underscored by the prospect of drug-loaded PCN-222 possessing substantial commercial value, offering opportunities for cost-effective production methods. This contrasts with other applications, such as gas storage or catalysis, where achieving economic viability can be more challenging. For example, Fairen-Jimenez et al. loaded a model drug (doxorubicin) on PCN-222 PEGylated with PEG-phosphate ligands forming a composite having excellent water dispersibility at room temperature, improved colloidal stability and delayed drug-release kinetics of the drug.11 Notwithstanding the potential of PCN-222, its synthesis has only been performed using batch processes, which can suffer from several limitations such as low yields and productivity, long reaction times, and difficulty in scaling up. Typically, the solvothermal synthesis of PCN-222 is performed under laboratory conditions by dissolving a Zr-precursor (normally Zr chloride or previously synthesised Zr6 clusters), TCPP and an acid modulator (usually benzoic acid, trifluoroacetic acid or fluorobenzyl acid) in N,N-dimethylformamide (DMF), followed by heating at 393 K under magnetic stirring from a few up to 48 hours.12–14 The purification of the so-synthesized PCN-222 is performed by centrifugation. The slow crystallization rate of PCN-222 together with heat and mass transfer issues limits the scale up of the process, restricting the production to a few mg per batch. Furthermore, the use of high volume of hazardous DMF entails important limits especially considering health and environmental issues.15 Therefore, the design of more efficient, sustainable, and scalable synthetic methods based on alternative techniques such as mechanochemistry, microwave chemistry, ultrasound-assisted synthesis and continuous flow synthesis is crucial to overpass these inefficiencies and to perform accessible production of PCN-222. The first effort for the scalable synthesis of PCN-222 reported up to date exploited microwave chemistry to produce tens of mg of the MOF with yields of 72%, within only 30 minutes.16 However, the poor control on the morphology and on the size-distribution of the MOF cannot be applied for many applications, such as drug delivery, where nanosystems must have controlled size and morphology to regulate their circulation and clearance kinetics in the bloodstream. Indeed, it has been demonstrated that specific shapes, such as prolate ellipsoidal nanoparticles exhibit lower rates of systemic elimination compared to particles with other shapes, indicating that they may circulate in the bloodstream for longer periods.17 In addition, the smaller particle size shows long in vivo circulation time, although this should be carefully checked also considering the hydrodynamic size in the blood stream. Another example for the scalable synthesis of PCN-222 has been recently reported by Yu et al. through an ultrasound-assisted method. In details, the researchers were able to synthesize PCN-222 in high crystal purity and uniform size within 30 minutes.18 Still, the use of batch synthesis limits the potential scale up of the synthesis, with important sustainable drawbacks, including low energy efficiency and high chemical risk.
In the past few years, continuous flow (c.f.) chemistry has emerged as an attractive alternative to batch synthesis methods for the efficient preparation of different chemicals and materials and has been pointed out as promising path for the industrialization of MOFs.19 C.f. chemistry offers several advantages over batch processes including resource efficiency, reduced waste generation, safer chemical handling, energy efficient, scalability and process intensification.20,21 C.f. has been also reported by the IUPAC as one of the “Top Ten Emerging Technologies” with the potential to turn the planet more sustainable.22 Furthermore, external fields such as microwave or ultrasound (US)23 can be coupled to c.f. to enhance heat transfer, facilitate the disruption of agglomerates of particles, boost the mix of the reagents and precursors in the channel and reduce the undesired deposition of solid materials on the channel wall. At the same time, US have been proved to have multiple effects such as accelerating the nucleation and growth to get monodispersed nanoparticles, influencing the crystal morphology and etching the crystals.18,24
Here, the first use of a c.f. reactor for the synthesis of PCN-222 with defined and sized-controlled prolate ellipsoid morphology (down to less than 100 nm) is reported. After optimizing the batch conditions in terms of ligand-to-metal cluster molar ratio (0.3–2.5), sequential experiments in c.f. examined the influence of ligand concentration (3.2–7.1 mM), flow rate (0.1–5 mL min−1), ligand-to-acid modulator molar ratio (0.007–0.054) and temperature (383–418 K). Considerations of some specific parameters on the sustainability of the synthesis were determined by the green metrics Environmental factor (E-factor) and Reaction Mass Efficiency (RME). The best reaction conditions allowed overpassing the challenge of preserving the quality of MOF during the rapid synthesis in c.f. The c.f. method was thus combined with US technique (20 kHz, 20–90% US amplitude) allowing the etching of the crystals and the shaping the MOF into prolate ellipsoids by variations of the dimensions of the minor axis (i.e., the equatorial axis) of the ellipsoids. Greener alternative solvents to hazardous DMF were also evaluated. The c.f. synthesis yielded high-quality PCN-222 with Space–Time–Yield (STY, one of the most common parameters representing the productivity of the synthesis) up to more than 950 kg m−3 day−1, demonstrating the robustness and scalability of the method. The potential biomedical applications of PCN-222 were finally evaluated by the loading a thio-N-acetylgalactosamine-PEG-sulfate ligand (S-GalNAc-PEG-sulfate) in tandem c.f. The ligand was incorporated onto the PCN-222 particles through Zr–sulfate interactions, contributing to the stabilization of the PCN-222 particles. Additionally, the incorporation of thio-N-acetylgalactosamine (S-GalNAc), a glycosidase-resistant analogue of natural GalNAc, holds great promise as it specifically targets the asialoglycoprotein receptor (ASGPR) overexpressed on hepatic cancer cells. This presents an exciting opportunity to efficiently develop a porphyrin-based drug delivery system using continuous flow synthesis.
2. Methods
2.1 Synthesis of Zr6 clusters
Zr6 nodes (or Zr6 clusters) were prepared according to a reported procedure without any modification.25,26 Initially, 15 mL of 80 wt% Zr(OBu)4 in n-butanol and 100 g of benzoic acid were added to 300 mL of isopropanol (iPrOH). The resulting mixture was sonicated for 20 minutes to facilitate the dissolution of benzoic acid. Sequentially, the mixture was heated under reflux with stirring overnight. The excess of iPrOH was removed by vacuum heating, leading to the formation of a white solid powder. The solid was extensively washed with iPrOH and then dried under vacuum at room temperature, yielding ca. 61%. The so-obtained Zr6 nodes capped with benzoate had a general formula Zr6(μ3-OH)4(μ3-O)4(C7H5O2)4 with molecular weight of ca. 1164 g mol−1. The presence of benzoate as capping agent was evidenced by FTIR analysis (ESI Fig. S3†).2.2 Batch Synthesis of PCN-222
PCN-222 was prepared in batch according to a reported method (with reported size of 117.8 ± 12.9 nm).11 In details, TCPP (22.5 mg, 0.028 mmol), Zr6 cluster (38 mg, 0.032 mmol) and TFA (130 μL, 1.69 mmol) were dissolved in 8 mL of DMF (US bath) in a 10 mL sealed vial. The resulting mixture was heated at 393 K for 5 hours under no mixing and then allowed to cool down to room temperature. The dark, purple-coloured MOF was collected by high-speed centrifugation (12000 rpm, 15 min), washed one time with DMF, and exchanged two times with ethanol (EtOH).
2.3 Synthesis of the thio-N-acetylgalactosamine-PEG-sulfate ligand
The thio-N-acetylgalactosamine-PEG-sulfate (S-GalNAc-PEG-sulfate) ligand was synthesized using a highly convergent approach, as depicted in Fig. 1. | ||
| Fig. 1 Synthetic approach for the synthesis of the ligand 8 (S-GalNac-PEG-sulfate). | ||
Firstly, commercially available N-acetylgalactosamine tetra-acetate 1 was treated with Lawesson's Reagent in toluene at 353 K to provide the cyclized per-O-acetylated thiazoline 2 with 71% yield. The heterocycle in compound 2 was subsequently cleaved through acid hydrolysis using TFA and water in methanol (MeOH) at 273 K.27 This process resulted in the production of thiol 3 with an 85% yield. Secondly, octaetyleneglycol 4 was treated with SOCl2 in the presence of 4-dimethylaminopyridine (DMAP) and diisopropylethylamine (DIPEA) in dichloromethane (DCM) from 273 K to r.t., affording cyclic sulfite 5 with 91% yield. Subsequently, the oxidation of 5 was carried out using NaIO4 with a Ru-catalyst in a mixture of acetonitrile (I), trichloromethane (TCM), and water, resulting in the formation of cyclic sulfate 6 with a yield of 79%.28 The condensation of thiol 3 with cyclic sulfate 6, using triethylamine (Et3N) as a base in dimethylformamide (DMF) at r.t., resulted in the formation of compound 7 with a yield of 61%. Lastly, compound 8, i.e., the S-GalNAc-PEG-sulfate ligand, was obtained in quantitative yield though Zemplen deacetylation of 7, carried out using MeONa in MeOH at r.t. Details of the synthesis as well as 1H-NMR, 13C-NMR and mass spectroscopy of the compounds are reported in the ESI.†
2.4 C.f. synthesis of PCN-222 and c.f. loading of ligands on PCN-222
Fig. 2 reports the scheme of the c.f. system for the synthesis and PSM of PCN-222 developed in this study (refer to ESI section-S1† for additional pictures). The c.f. system was divided into two sections: the first one, composed of two reactors, in which the synthesis of the PCN-222 was performed, and the second one, composed of one reactor, in which the loading of the ligand was carried out. The first section (Fig. 2a) can operate independently from the second one (Fig. 2b) by collecting the MOF after Reactor 1 or after Reactor 2. In the first section, the two feeds were composed of the Zr6 clusters together with TFA dispersed in DMF, or in the selected organic solvent, in Pump 1 (HLPC pump, Micro HPLC Pump, ThalesNano Inc., Hungary), and of tetrakis(4-carboxyphenyl)porphyrin (TCPP) dissolved in DMF, or in the selected organic solvent, in Pump 2 (single channel peristaltic pump, Cytiva Peristaltic Pump P-1, GE HealthCare Technologies Inc., USA). Before loading the mixtures in the pumps, the solids and the acid were dispersed in DMF, or in the selected organic solvent, by immersing the solutions in an US bath for 30′′, while no heating was applied before the reactor. The streams from Pump 1 and Pump 2 were mixed and entered in Reactor 1 though a T-Junction (Nylon Barb Tee 1/16′′). Reactor 1 was designed to operate between r.t. up to 438 K with a total volumetric flow ranging from 0.1 mL min−1 to 10 mL min−1 (from 0.05 mL min−1 to 5 mL min−1 in each pump). Reactor 1 was composed of a 5.6 m tube of 0.8 mm diameter and a 2.8 mL total volume (Bohlender™ PTFE tubing), twisted on itself in a ca. 60 mm height helicoid (radius of the curvature 28.5 mm) placed in a beaker filled with silicon oil. After Reactor 1, the mixture was passed through Reactor 2 where 20 kHz US can be applied with amplitude ranging from 0% to 90% (Q500 Sonicator® equipped with Qsonica CL334 converter and a titanium ½′′ probe equipped with a replaceable tip, Qsonica Llc, USA). Reactor 2 (exactly as Reactor 1) was composed of a 5.6 m tube of 0.8 mm diameter and a 2.8 mL total volume twisted on itself in a ca. 60 mm height helicoid (radius of the curvature 28.5 mm) placed in a beaker filled with water. The sonotrode was place in the beaker at a distance between the tip of the sonotrode and the bottom of the beaker corresponding to ca. one wavelength (74 mm). Concerning the second section of the system (Fig. 2b), where the loading of the ligand was performed in tandem c.f., the stream containing the synthesized PCN-222 was mixed with a stream of a DMF solution of the S-GalNAc-PEG-sulfate ligand coming from Pump 3 (infusion pump, KDS 100 legacy syringe pump, KD Scientific Inc., USA) with a T-junction and passed through Reactor 3. Reactor 3 (exactly as Reactor 1 and 2) was composed of a 5.6 m tube of 0.8 mm diameter and a 2.8 mL total volume twisted on itself in a ca. 60 mm height helicoid (radius of the curvature 28.5 mm) placed in a beaker filled with water. The collected MOF, whether etched with US or whether with/without the ligand, were purified by centrifugation at 12000 rpm for 15′ followed by one wash in DMF and two wash in EtOH. Details of the chemicals and of the characterization techniques are reported in ESI section-S2.†
 | ||
| Fig. 2 (a) Continuous flow synthesis and US-assisted etching of PCN-222 and (b) tandem loading in c.f. of the sulfate ligand on PCN-222. (c) Scheme of the preparation of PCN-222@SO3-PEG-S-GalNAc with SEM picture of PCN-222. | ||
In order to evaluate the productivity of the synthesis in c.f., the Space–Time–Yield (STY)29 was calculated according to eqn (1):
 | (1) |
Each experiment was repeated at least three times in order to determine the STY, which was thus reported with its corresponding standard deviation.
2.5 Determining the size of PCN-222
The size of PCN-222 was primarily reported as diameter of the particles. This means that, considering that PCN-222 were in the form of prolate spheroids (i.e., spheroids formed by the rotation of an ellipse about their major axis), the diameter was determined from the polar axis (see Fig. 5). At least 50 particles in various SEM images were measured for each experiment, using ImageJ, a publicly available Java image processing program developed at the Research Services Branch of the National Institute of Mental Health in Bethesda, Maryland, USA. The average size was reported along with its corresponding standard deviation (SD), the latter as an indicator of the size-distribution of the particles of PCN-222.2.6 Green metrics
Two specific green metrics, namely E-factor and reaction mass efficiency (RME), were calculated based on the established eqn (2) and (3):30,31 | (2) |
 | (3) |
The metrics were calculated using the mean mass of the synthesized PCN-222 obtained in the different runs of each experiment. As easily understandable, the lower the E-factor the lower the environmental impact of the synthesis. For example, the general E-factor found in different industrial segments varies form ca. 0.1 of oil refining, to 1–50 of fine chemicals and 50–100 of pharmaceuticals.26 On the contrary, the higher the RME, the lower the environmental impact of the synthesis. In the present study, to accentuate the disparity between batch and c.f. synthesis, green metrics were discussed without accounting for solvents. This omission was supported by the ease with which solvents can be recycled after separation from PCN-222, typically through centrifugation, rendering them potentially non-waste materials.26 Green metrics were calculated using the average mass of PCN-222 produced in each experiment. For instance, in an experiment (entry 4 in Tables S2 and S3 in the ESI†), 11.5 mg of PCN-222 (STY: 5.5 kg m−3 day−1) was produced using 22.5 mg of TCPP, 33.0 mg of Zr6 nodes, and 193.7 mg of TFA (130 μL), totalling 249.2 mg of mass of reagents (excluding the solvent used in the reaction, DMF, and the solvents for washing, DMF and EtOH). The waste mass was determined by subtracting the mass of the product and corresponded to 237.7. Thus, the E-factor was 20.7 (calculated as 237.7 divided by 11.5), and the RME was 4.6% (calculated as 11.5 divided by 249.2 and multiplied by 100). To provide a comprehensive overview, the E-factor calculated considering also the solvents can be checked in the ESI (RME does not count solvents).†
3 Results and discussion
3.1 Synthesis of Zr6 nodes (also known as Zr6 clusters)
According to the literature, the batch synthesis for the preparation of sized-controlled PCN-222 with spheroidal morphology is carried out by reacting the ligand TCPP with previously synthesized Zr6 nodes. Other methods include the preparation of Zr6 nodes in situ (generally using zirconyl chloride octahydrate as precursor), but the control on the morphology and size-distribution of PCN-222 is weak. Considering that the Zr6 nodes are prepared by a simple and scalable hydrothermal treatment using low toxic reagents, the use of previously synthesized Zr6 nodes instead of any zirconyl precursor was not affecting the main scope of the present work of designing a sustainable method for the scale up of the synthesis of PCN-222. The synthetic procedure resulted in the formation of Zr6 nodes capped with benzoate, through Zr–carboxyl bonds, having formula Zr6(μ3-OH)4(μ3-O)4(C7H5O2)4. The benzoate capping proved to be particularly advantageous, as its substitution with TCPP in the formation of PCN-222 did not release water, unlike what can potentially occur with uncapped Zr6 nodes formed in situ with the formula Zr6(μ3-OH)4(μ3-O)4(OH)4(OH2)4.32 The presence of water can indeed influence crystal growth and nucleation, ultimately leading to the production of more homogeneous and well-defined particles.333.2 Optimization of batch conditions and switching to c.f.
Initially, the reported batch synthesis of PCN-222 having spheroidal morphology was replicated and studied.11 It was observed that achieving nano-dimensional PCN-222 with controlled size-distribution resulted in low STY (below 4 kg m−3 day−1, entry 1 in Table S1 in the ESI†), considering the reaction time of 5 h. Thus, some preliminary tests were conducted by varying the ligand–metal center molar ratio (L:M) as well as the concentration of reactants and amount of acid modulator, merely focusing on the reaction STY without considering the size-distribution of the produced particles (ESI Table S1†). Reaction time was not considered to be optimized in this phase since the reactions in c.f. were sequentially always carried out in much less than 5 h. The preliminary tests highlighted that the L:M was the most relevant parameter with effect on the reaction STY. As a result, a sequence of batch experiments was performed to investigate the influence of the L:M on the STY, the length of the particles, and the size-distribution of PCN-222 (ESI Table S2†), as summarized in Fig. 3.
 | ||
| Fig. 3 (a) Variation of L:M vs. STY and size of the obtained PCN-222 particles. Reaction conditions: 393 K, 5 h, 22.5 mg TCPP (0.0284 mmol, 3.55 mM), 8 mL DMF, 130 μL TFA (L:Mod 0.017). (b) SEM images of PCN-222 obtained using L:M of 1 and L:M of 0.3, respectively, highlighting the changes in particle-size distribution in function of the amount of Zr6 clusters used. See ESI section-S3† for additional SEM images. | ||
In all the experiments, the concentration of the ligand TCPP was kept constant, while the quantity of Zr6 nodes was varied to tune the L:M from 0.3 to 2.5 (see ESI section-S3†).
Considering that the molecular formula of PCN-222 is Zr6(μ3-OH)4(μ3-O)4(C48H26N4O8)2, formed by TCPP4− ligand (C48H26N4O8) and Zr6(μ3-OH)4(μ3-O)4 nodes34 the results showed that, to increase the STY of PCN-222, it was necessary to use less quantity than the (theoretical) stoichiometric amount of the ligand TCPP (i.e., more Zr6 clusters). This was particularly evident when the L:M was equal or less than 1. At the same time, the size of the PCN-222 was observed to increase when more quantity of the metal clusters was used. This behaviour was in accordance with the reported literature related to other MOFs. In details, it is well established that a decrease of the ratio ligand–metal node influences (normally boosts) the nucleation and growth rates of MOF crystals, which in turn can also impact the size and the morphology of the final crystals.35,36 Thus, considering the STY of the synthesis, the best reaction conditions for the highest STY corresponded to the largest quantity of metal clusters. However, SEM images showed a substantial variation of the size-distribution of the PCN-222 when the L:M was ≤0.5. As a result, a L:M of 1 was selected for the shifting to c.f. synthesis, corresponding to the minimum quantity of Zr6 nodes required to obtain particles having sizes with relative SD of ca. 5% (222 ± 12 nm). A smaller L:M was discarded since it implied more consumption of metal nodes and, more importantly, larger SD (e.g., a L:M = 0.7 required 26% more metal nodes and resulted in a SD of ca. 10%). Importantly, the use of smaller L:M was also discarded considering no relevant variations in the green metrics of the synthesis. In details, when a L:M = 1 was used, E-factor = 20.7 and RME = 4.6%, while when L:M = 0.7, E-factor = 19.9 and RME = 4.8% (ESI Table S3†). The optimized batch conditions were thus selected for the shifting to c.f. synthesis. The importance of shifting the synthesis of MOFs to c.f. rises form the different benefits and advantages brought by this type of synthetic approach including improved control and reproducibility, enhanced reaction kinetics and product yield, safer and more controlled conditions, scalability, and production efficiency, reduced environmental impact, and finally versatility and process intensification. These advantages make the c.f. synthesis a promising approach to produce MOF with consistent quality and improved green characteristics and has been already applied in the synthesis of some types of MOFs such as UiO-66, MIL-100, and HKUST-1 (see ESI section-S4† for a detailed list of the most recent works related to the c.f. synthesis of MOFs). However, up to date, no report discusses the c.f. synthesis of the PCN-222/MOF-545.
3.3 Shifting the reaction from batch to c.f.: effect of the reaction parameters
Prior to perform the synthesis in c.f., the flow pattern in the reactor (Reactor 1) was investigated, giving insights into the behaviour of the flow and of the mixing in the reactor. Since the flow pattern is influenced by the velocity profile of the flow and by the stream in the cylindrical helix reactor, Reynolds number (Re) and Dean number (De) were calculated. Both Re and De were determined for the volumetric flow ranging from 0.1–10 mL min−1. The values of Re were calculated according to the eqn (4):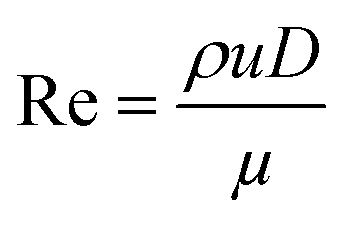 | (4) |
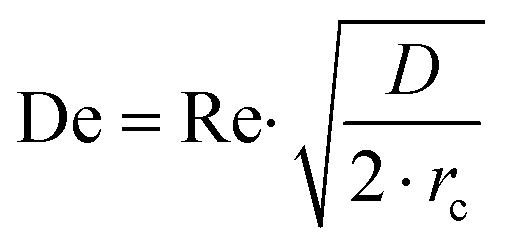 | (5) |
The reaction was thus shifted to the c.f. system by mixing in a T valve two feeds, one with the ligand TCPP dissolved in DMF and the other with the Zr6 nodes and TFA dissolved in DMF. A T-valve was preferred to a cross-valve (where TFA would have been separated from the Zr6 clusters in another feed) since the Zr6 clusters did not completely dissolve in DMF without the presence of TFA (probably TFA substitutes the benzoate in capping the Zr6 nodes, making the nodes more polar, thus more soluble in DMF). The influence of the concentration of the reagents, the volumetric flow rate, the temperature of the reaction and the amount of employed acid modulator on the STY and on the size of the PCN-222 were thus singularly determined in c.f. Initially, the reaction was shifted to c.f. starting from the optimized batch conditions, which were 393 K reaction temperature, a L:M of 1, and a concentration of 3.6 mM TCPP, using DMF as solvent. According to the results of the preliminary tests, the amount of employed TFA was initially decreased (from a ligand-to-acid modulator molar ratio (L:Mod) of 0.017 to 0.027), aiming at diminishing the size of the particles11 and, more importantly, considering that the c.f. conditions do not imply the liquid–vapour equilibrium of TFA in the head space of the reactor observed in batch conditions (thus less acid was needed to obtain particles with the same dimensions of those obtained in batch). The amount of TFA used was then optimized after studying the effects of the volume of solvent, of the flow rate, and of the temperature.
:M ratio, as shown in Fig. 4a. The lower limit of concentration was determined by too low STY (<50 kg m−3 day−1), while the upper limit was determined by the minimum amount of DMF needed to dissolve the reagents (in particular, TCPP).
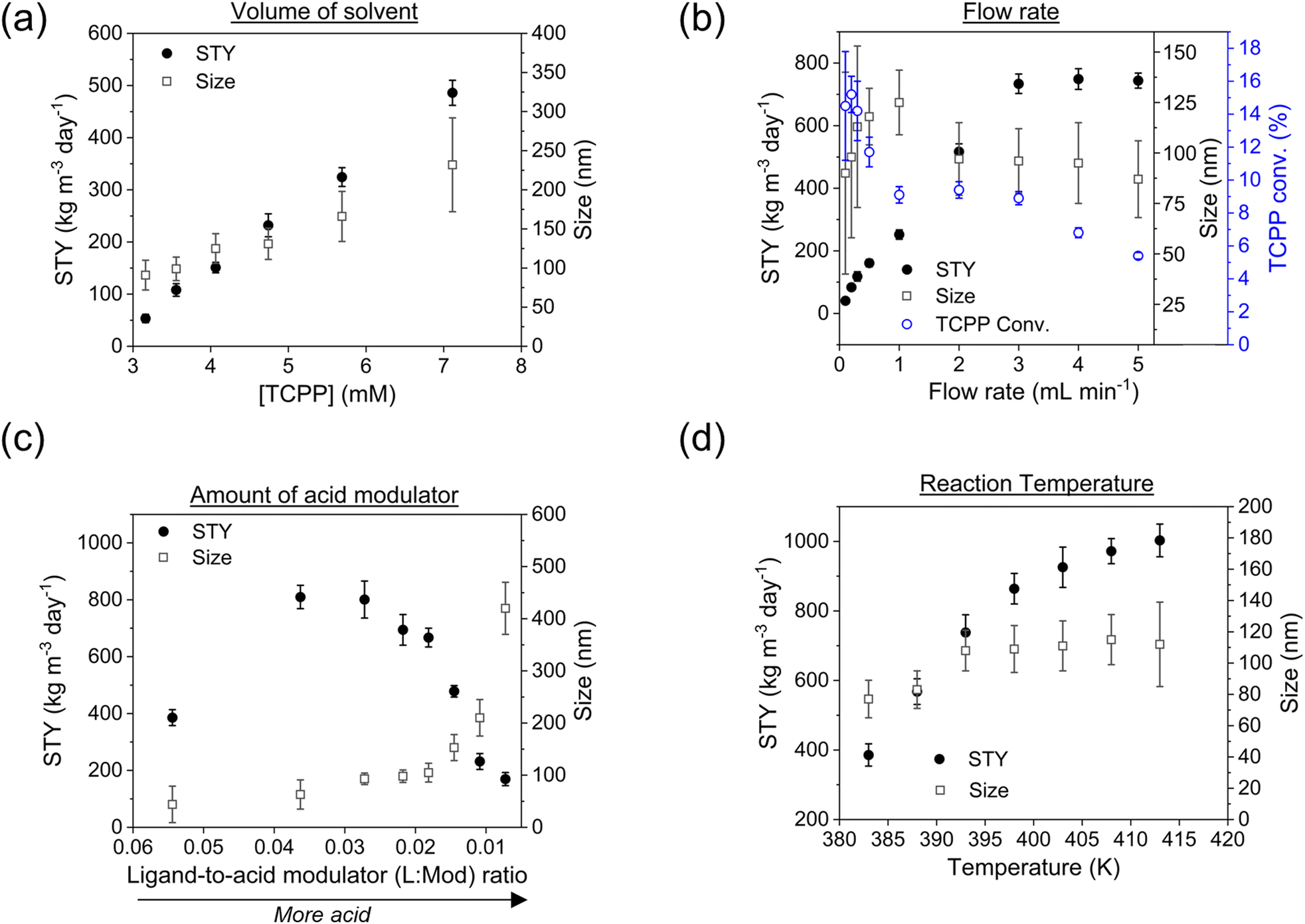 | ||
| Fig. 4 Effect of different parameters on the STY and on the size-distribution of PCN-222 synthesized in DMF. (a) Concentration of the reagents (i.e., volume of solvent), reaction conditions: 393 K, total flow 1 mL min−1, L:M = 1 and L:Mod = 0.027; (b) volumetric flow rate, reaction conditions: 393 K, L:M = 1, L:Mod = 0.027, [TCPP] = 4.7 mM; (c) amount of employed acid modulator, reaction conditions: 393 K, L:M = 1, flow rate 3 mL min−1, [TCPP] = 4.7 mM and (d) temperature of the reaction, reaction conditions: L:M = 1, L:Mod = 0.027, [TCPP] = 4.7 mM, flow rate 3 mL min−1. | ||
The results demonstrated that the STY increased linearly with the concentration of the reagents. A similar behaviour was also observed concerning the size of the particles, but with a smaller slope (see ESI section-S6† for additional details and SEM images). Nevertheless, the size-distribution of the particles showed a SD ≤15% when the concentration of TCPP was ≤4.7 mM, while higher concentration implied bigger and more disperse particles. This was in accordance with the reported data in the literature concerning the synthesis of other MOFs, where it was observed that higher reagent concentration caused the less regularity of the shape and greater mean diameter.38 A concentration of 4.7 mM TCPP was thus selected for sequential experiments. Lower values of concentration were discarded since they implied more solvent consumption and substantially increased green metrics. In details, when an amount of solvent corresponding to a 4.7 mM concentration of TCPP was used, the E-factor and the RME corresponded to 61.7 and 1.6% respectively, which varied up to 190.8 and 0.5% respectively, when the concentration of TCPP corresponded to 3.2 mM (ESI Table S8†). This was attributed to the lower STY achieved when using a lower concentration of TCPP (implying less collision of particles, thus nucleation and crystal growth), while maintaining the same amount of Zr6 clusters and TFA.
 | (6) |
Note that the molecular formula here reported is slightly different from other formulas reported in the literature (e.g., Zr6(H2O)8O8(TCPP)2)6 due to utilization of previously synthesized Zr6 nodes capped with benzoate.11 Benzoate was substituted with TCPP during the synthesis, forming PCN-222. The moles of produced PCN-222, with molecular weight ca. 2253 g mol−1 and the moles of TCPP, with molecular weight 790.8 g mol−1, fed in the c.f. were thus calculated considering the volume of the reactor of 2.8 mL. For example, the experiment carried out at 0.5 ml min−1 flow rate, resulted in a STY of 161 ± 12 kg m−3 day−1, corresponding to 0.778 ± 0.058 × 10−3 mmol of PCN-222 (reaction time was 3.89 × 10−3 day). The mmol of TCPP fed in the system (4.7 mM) were 13.16 × 10−3, thus the TCPP conversion was 11.8 ± 0.9%. As shown in Fig. 4b, the conversion of TCPP diminished by diminishing the residence time (i.e., by increasing the flow rate), while the STY increased (Fig. S6 in the ESI† repots data in the 0.1–1 mL min−1 flow rate range). In details, by increasing the flow rate from 0.1 to 3 mL min−1, the STY passed from <50 kg m−3 day−1 up to >700 kg m−3 day−1. When the flow rate was between 3 and 5 mL min−1, the STY reached a plateau, since the faster flow was balanced by lower TCPP conversion. This meant that less MOFs were produced per mL of solution primarily due to the reduced time available for crystal growth, rather than for nucleation. This observation holds even when considering the small differences in the dimension of the particles. Higher flow rates were not considered since the conversion of TCPP at 5 mL min−1 was already too low (<6%). The observation of particle size in relation to flow rate was consistent with literature findings, showing that particle size increased linearly by increasing the flow rate up to 1 mL min−1. This phenomenon was attributed to higher flow velocities facilitating the advective transport of precursor species to nucleation sites in the flow, resulting in increased crystal growth rates and larger average crystal sizes, as seen for example in the c.f. synthesis of MOF-808.39 However, when the flow rate was further increased up to 5 mL min−1, the particle size was observed to slightly decrease, reaching almost a plateau with PCN-222 particles sizing ca. 87–97 nm length. This was explained by the shorter time available for crystallization (i.e., shorter τ), especially for crystal growth rate beyond nucleation rate, which resulted in smaller crystals. Additionally, the higher flow rate led to the competitive migration of precursor species to the nucleation sites or out of the reactor, further affecting the particle size. Prospects of this observed behaviour involve studying the kinetics of crystallization, which was beyond the scope of the present work. The size distribution was observed to vary less when 0.5 mL min−1 flow rate or higher was used, while at longer reaction time, i.e., slower flow rate, larger SD of the size of the particles was observed. After evaluating the STY, the conversion of TCPP, the size, and the size-distribution of the particles, the flow rate of 3 mL min−1 was selected as the best option. Slower flow rates were discarded due to lower STY, while higher flow rates did not show any enhancement of STY and instead resulted in less conversion of TCPP. As discussed in previous section, a flow rate of 3 mL min−1 implied a theoretical τ of 56 seconds. Since the flow profile of the reactor was defined as laminar with no secondary vortices, the variance of how much fluid passed through the reactor was calculated with the plug-flow residence time distribution function E(t) in eqn (7):40
 | (7) |
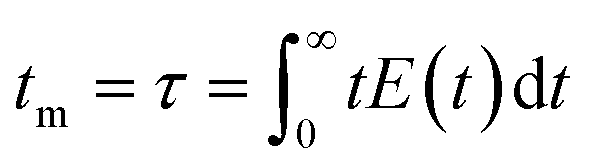 | (8) |
It was observed that the mean residence time corresponded to 55 s, in good accordance with the theoretical τ. The variance (or square of the standard deviation), σ2, was calculated according to eqn (9):40
 | (9) |
The variance resulted 148.5 s2 (this moment indicates the spread of the distribution: the greater the value the greater the spread). Lastly, the skewness, s3, was calculated with eqn (10):40
 | (10) |
Skewness resulted 0.671 s3 (this moment measures the extent that a distribution is skewed in one direction or another in reference to the mean). Additional details of the curve of the concentration of the tracker, C(t), E(t) and the data to calculate the moments of RTD (tm, σ2, and s3) are reported in the ESI section-S7.†
:Mod) was adjusted from 0.007 to 0.054 to explore the impact on particle size and STY. The results showed that particle size increased nearly linearly with the amount of acid used, while STY showed an inverse relationship, more marked, as illustrated in Fig. 4c. It is worth noting that inorganic and organic additives can be employed to control particle size by modifying crystallization. For instance, benzoic acid has been used as an additive to regulate particle size of different MOFs.38 Similarly, other acids such as HCl have been demonstrated to influence both nucleation and growth steps in the preparation of UiO-66.41 This suggests that the addition of different acids or other additives can be used to fine-tune the particle size and STY of the c.f. to achieve the desired properties for various applications. Based on the experimental results, it was found that TFA not only affected the crystal growth of PCN-222, but also the nucleation of the particles. It was also observed that the use of too much TFA led to the formation of larger particles and agglomerates with lower STY (see ESI section-S6† for additional details and SEM images). This is consistent with previous studies that have shown that the use of excessive amounts of acids can lead to the formation of large MOF particles and aggregates with poor crystallinity.42 Thus, to achieve a good balance between STY and particle size distribution, a L:Mod between 0.021 and 0.031 was found to be optimal. When operating with less acid (L:Mod 0.054), it was observed the formation of agglomerates of small particles (probably of ca. 30–40 nm). For subsequent experiments, a ligand-to-acid modulator ratio of 0.027 was thus chosen as it was found to provide a good compromise between particle size and STY with the minimum use of acid modulator.
3.4 US-assisted c.f. synthesis
After deeply investigating the reaction conditions in c.f., ultrasound (US) was employed to (re)shape PCN-222 particles into thin prolate ellipsoids (Fig. 5a). | ||
| Fig. 5 (a) Scheme of the prolate ellipsoidal shape of PCN-222 with the representation of the effect of the US and (b) effect of the US on the STY, the size-distribution, and the spheroid aspect ratio of PCN-222. Reaction conditions: 408 K, L:M = 1, L:Mod = 0.036, [TCPP] = 4.7 mM, flow rate 3 mL min−1, solvent DMF. | ||
The particle shape can significantly impact the behaviour of nanoparticles in various applications, including catalytic systems in continuous flow and drug delivery in the bloodstream. Research has indicated that prolate ellipsoids, with their elongated shape, exhibit unique hydrodynamic properties such as reduced drag and increased lateral migration in flow, enabling them to circulate for extended periods compared to spherical particles.17 This is particularly relevant in medical applications, where particles introduced into the bloodstream interact with blood components and encounter clearance mechanisms like the reticuloendothelial system, involving organs such as the liver and spleen. Controlling the spheroid aspect ratio of PCN-222 has not been reported to date, and achieving control during the synthesis step can be challenging. To address this, US was coupled with c.f. as an etching technique. Etching in the context of metal–organic frameworks (MOFs) refers to a process wherein the framework structure of a MOF is partially or completely removed using chemical or physical methods.
Various techniques, including the use of acids, bases, solvents, and external energy treatment, can achieve etching. However, the use of additional chemicals contradicts the principles of green chemistry (considering the increased amount of waste produced, or the potential use of toxic chemicals, etc.), making the treatment with external energy (e.g., heat transfer) a more sustainable option. Among these techniques, US-based etching represents a relatively recent and innovative approach for modifying MOF structures. As illustrated in Fig. 5, US was applied to the c.f. system (Reactor 2 in Fig. 2a) at amplitudes ranging from 20% to 90% (more than 90% amplitude is not recommended by the manufacturer) and its impact on particle morphology and STY was investigated (ESI section-S8†). The results revealed a linear increase in the aspect ratio of the particles with increasing US amplitude. While the mean particle length remained relatively constant, a higher SD of the size was observed. Additionally, as the US amplitude increased, the STY decreased, with the lower STY observed at 90% amplitude. These outcomes were attributed to localized pressure variations induced by US, leading to acoustic streaming and cavitation effects. Acoustic streaming, generated by the acoustic waves, involves a steady flow of fluid within the reactor. It facilitates fluid circulation, enhances mixing, and improves mass transfer and reaction kinetics. Cavitation occurs when the local pressure in the fluid drops below the vapor pressure, resulting in the formation and subsequent collapse of tiny vapor bubbles (at high local temperature). This phenomenon induces intense fluid motion, including microjets and shockwaves, which promote turbulence and enhance mixing. Two crucial observations were made. Firstly, the decrease in STY confirmed the etching of MOF crystals under the influence of US, as portions of the crystals were eroded, leading to diminished STY. Secondly, the increased SD in particle length increasing the US amplitude, indicate that the etching occurred at lower energy on the equatorial axis while at higher energy it occurred also on the polar axis (Fig. 5a). To confirm these observations, a complementary experiment was carried out by performing the US-etching with larger particles, highlighting superior changes in the aspect ratio, which increased from 2.72 ± 0.24 to 3.70 ± 0.33 (ESI Fig. S13†). The effect of the US-etching was further proven by a batch experiment redispersing, after purification, large particles of PCN-222 into fresh DMF, sequentially immersed in an US bath for 30 minutes. It was observed that the particles were etched by the US, resulting in particles with a higher aspect ratio (from 2.63 ± 0.15 to 4.22 ± 0.39, see ESI Fig. S14†). It should be noted that further investigation into US etching on PCN-222 would require computational simulations to fully investigate the breaking of the chemical bonds inside the MOFs in function of the applied US amplitude, which fall outside the scope of the present work.
3.5 Physio-chemical characterization of PCN-222
PCN-222, produced in c.f. without US-assisted etching (referred to as “PCN-222(0%US)”), and PCN-222 produced in c.f. with 60% US-assisted etching (referred to as “PCN-222(60%US)”), were compared using various analytical techniques including XRD, BET, FTIR, TGA and DLS (Fig. 6 and ESI section-S9†). Overall, except for minor variations in the textural properties, no significant differences were observed between the two samples. The analysis results aligned with the data previously reported in the literature,10,16 confirming the feasibility of producing PCN-222 using c.f. and c.f./US-assisted etching. The crystallinity and phase purity of PCN-222 were determined by comparing the experimental PXRD patterns with a predicted pattern based on single crystal data. The PXRD pattern exhibited sharp low-angle peaks, indicating a highly crystalline MOF powder (Fig. 6a). The diffraction peaks at 2.5, 4.8, 6.6, 7.1, and 9.9° were attributed to the (1 0 0), (2 0 0), (2 −1 1), (3 0 0), and (4 −2 1) crystal facets (CCDC no. 893545).43 The FTIR analysis revealed characteristic vibrational peaks in the range of 500–700 cm−1, corresponding to the asymmetric vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, N–H, CC and C–H of the pyrrole ring in TCPP (Fig. 6b).43 The peak at 1676 cm−1 in TCPP was attributed to the stretching of the CO bond, lowered in PCN-222 and indicating the coordination between –COOH and Zr atoms.43 The broad peak at ca. 3400 cm−1 corresponded to the stretching vibrations of N–H and O–H of the TCPP.44 TGA was employed to determine the phase transitions and decomposition temperatures of PCN-222 under air flow (Fig. 6c). The weight loss below 373 K was attributed to the removal of water/ethanol from the pores, followed by dehydroxylation of the Zr6 nodes between up to 723 K. The decomposition of TCPP ligands in the PCN-222 curve occurred beyond 693 K, leaving ZrO2 and carbon residues. The difference between the TGA profiles of PCN-222(0%US) and PCN-222(60%US) was determined by the same US, which generates more defect sites, resulting in a lighter material.18 PCN-222(0%US) and PCN-222(60%US) exhibited BET surface areas of 1801 m2 g−1 with pore volume of 1.69 cm3 g−1 and 1913 m2 g−1 with pore volume of 1.50 cm3 g−1, respectively. The BET surface areas were both based on the type IV N2-adsorption isotherm (Fig. 6d). The observed enhancement in the surface area parallel to the reduction in pore volume due to US treatment were consistent with the findings reported in the literature concerning the sonochemical synthesis of PCN-222.18
N, N–H, CC and C–H of the pyrrole ring in TCPP (Fig. 6b).43 The peak at 1676 cm−1 in TCPP was attributed to the stretching of the CO bond, lowered in PCN-222 and indicating the coordination between –COOH and Zr atoms.43 The broad peak at ca. 3400 cm−1 corresponded to the stretching vibrations of N–H and O–H of the TCPP.44 TGA was employed to determine the phase transitions and decomposition temperatures of PCN-222 under air flow (Fig. 6c). The weight loss below 373 K was attributed to the removal of water/ethanol from the pores, followed by dehydroxylation of the Zr6 nodes between up to 723 K. The decomposition of TCPP ligands in the PCN-222 curve occurred beyond 693 K, leaving ZrO2 and carbon residues. The difference between the TGA profiles of PCN-222(0%US) and PCN-222(60%US) was determined by the same US, which generates more defect sites, resulting in a lighter material.18 PCN-222(0%US) and PCN-222(60%US) exhibited BET surface areas of 1801 m2 g−1 with pore volume of 1.69 cm3 g−1 and 1913 m2 g−1 with pore volume of 1.50 cm3 g−1, respectively. The BET surface areas were both based on the type IV N2-adsorption isotherm (Fig. 6d). The observed enhancement in the surface area parallel to the reduction in pore volume due to US treatment were consistent with the findings reported in the literature concerning the sonochemical synthesis of PCN-222.18
 | ||
| Fig. 6 (a) PXRD patterns, (b) FTIR analysis, (c) details of TGA analysis and (d) N2 absorption/desorption isotherms of samples PCN-222(0%US) and PCN-222(60%US). Reaction conditions: 408 K, L:M = 1, L:Mod 0.036, [TCPP] 4.7 mM, flow rate 3 mL min−1, solvent DMF. | ||
3.6 Green approach of the c.f. synthesis
After completing the investigation related to switching the batch synthesis to c.f., some considerations on the green characteristics of the synthetic procedure were made. Firstly, it was observed that the c.f. synthesis itself made the preparation of PCN-222 more sustainable. This was primarily due to safer conditions that reduced chemical risks. In addition, the c.f. improved energy efficiency since the use of an oil bath in which the c.f. reaction tube is immersed allowed for enhanced heat transfer compared to a conventional oven/reactor. The STY of the synthesis also increased significantly from less than 4 kg m−3 day−1 to more than 950 kg m−3 day−1, resulting in substantially enhanced process efficiency. Considering the green aspects of the reagents used in the synthesis, it was initially considered that the Zr nodes were produced using a Zr precursor, benzoic acid, and isopropanol. Zirconium (Zr) is an element relatively abundant in the Earth's crust, and its primary ore, zircon, is widely available. Additionally, Zr can be recycled, reducing the need for extracting and processing new raw materials. However, a full investigation should be carried out for the specific applications, through a Life Cycle Assessment (LCA), to comprehensively evaluate the sustainability of Zr to produce PCN-222. On the other hand, benzoic acid is not typically considered a green chemical as it is usually produced from toluene, which generates certain byproducts, and it is not readily biodegradable. However, it is also low-toxic and has the potentialities to be industrially synthesized form biomass.45 Finally, isopropanol is generally considered a green solvent.46 Beside the Zr6 nodes, the other chemicals used in the synthesis of PCN-222, are the ligand TCPP, TFA and DMF. TCPP is normally synthesized with pyrrole and terephthalic acid although it can be potentially prepared form some natural compounds such as natural porphyrins. TFA has, according to the European Chemical Agency, reduced toxicity on various aquatic and terrestrial species and is not harmful to ecosystems. The use of large volumes of DMF for both the synthesis and purification of PCN-222 is instead considered highly hazardous.15 Therefore, a first green approach for a more environmentally friendly synthesis of PCN-222 in c.f. was carried out by substituting DMF with alternative solvents. Since the use of DMF is essential for the correct dissolution of the different chemicals, an initial approach consisted in partially substituting DMF with high boiling solvents which use is recommended, including cyclohexanone and anisole.14 However, the quality of resulting PCN-222 was too low, thus this strategy was discarded (ESI section-S10†). To completely substitute DMF, a polar aprotic solvent was needed. The only two recommended solvents of this class are acetonitrile and dimethyl sulfoxide (DMSO), although this last one is recommended with some limitations. Acetonitrile was discarded due to its low boiling point, while DMSO dissolved all reagents and was used as a substitute for DMF, successfully producing PCN-222, as shown by XRD, BET, FTIR, DLS, and TGA analysis (ESI section-S10†). The use of DMSO also allowed increasing the reaction temperature of the synthesis up to 433 K, substantially improving the STY, as shown in Fig. 7a.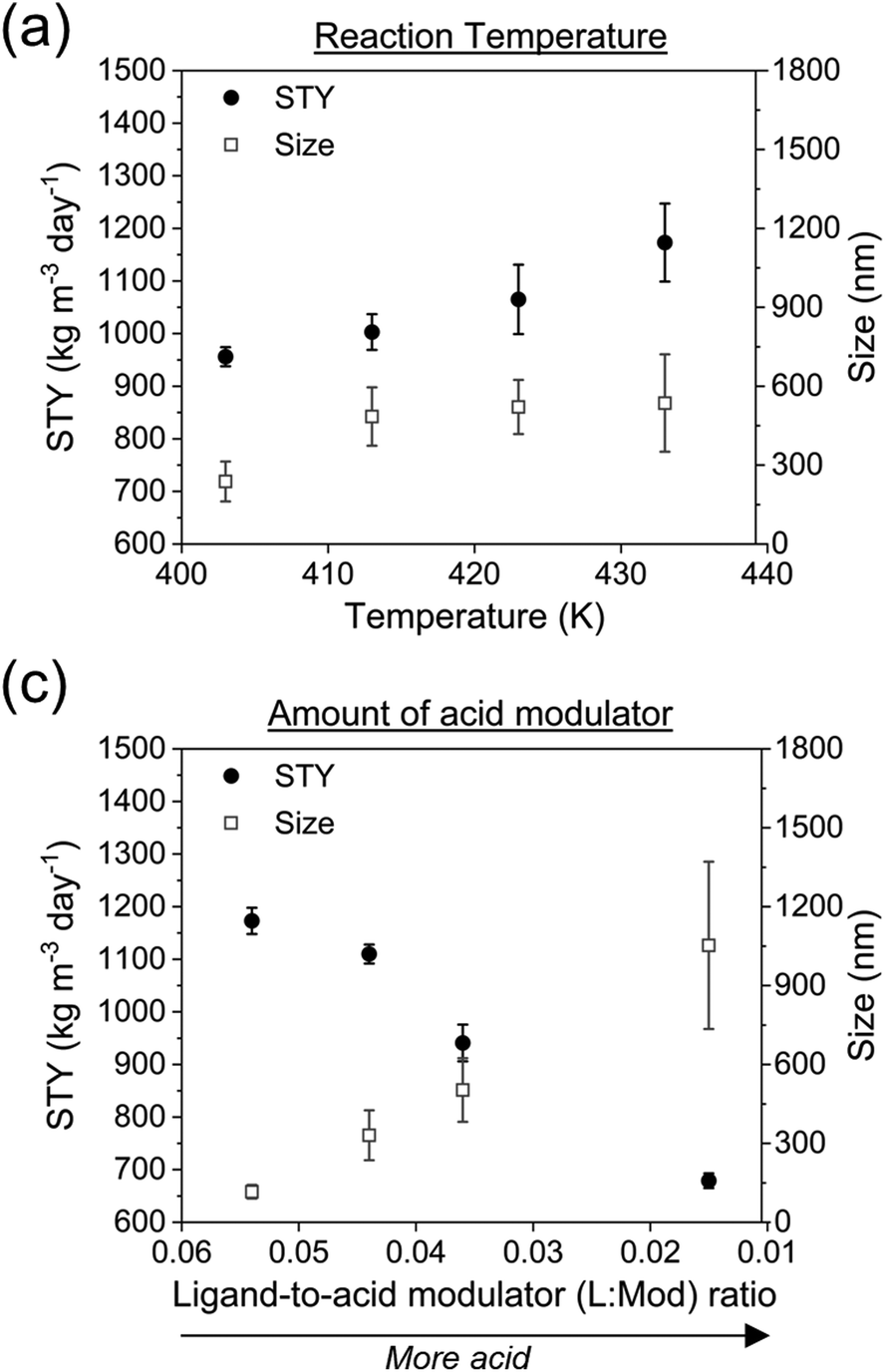 | ||
| Fig. 7 Effect of the (a) temperature of the reaction and of the (b) amount of employed acid modulator on the STY and the size-distribution of PCN-222. Reaction conditions: L:M = 1, [TCPP] = 4.7 mM, flow rate 3 mL min−1, solvent DMSO. | ||
At the same time, it was observed that the use of DMSO led to higher inhomogeneity and in bigger particles. To obtain smaller particles and, more in general, to verify the control on the dimension of the particles, a study on the effect of the acid modulator was conducted (Fig. 7b). It was found that, comparted to the particles synthesized in DMF, a reduced amount of TFA was necessary to achieve a comparable small particle size, thereby increasing the green characteristics of the synthesis. For example, the E-factor and RME for the synthesis performed in DMF to obtain particles of 113 ± 27 nm (at 413 K and L:M = 0.027, refer to ESI Table S14†) were 46.6 and 2.1, respectively. When using DMSO as the solvent, the E-factor and RME to obtain PCN-222 of 116 ± 26 nm changed to 25.4 and 3.8%, respectively. However, in general, the quality of the particles, as indicated by a larger standard deviation in size (ESI Table S17†), was much lower than that of the particles produced using DMF. Additionally, the US-etching process was less efficient when using DMSO, resulting in no changes in the aspect ratio (2.38 ± 0.26 with 0% amplitude vs. 2.44 ± 0.37 with 60% amplitude), probably due to less affinity of DMSO in dissolving the etched crystals (thus not favouring the same etching). Consequently, the c.f. green synthesis of PCN-222 with DMSO may not be suitable for applications where a strict control of the morphology and of the dimension of the particles is crucial, such as for biomedical applications, where homogeneous particles with dimension less than 100 nm are needed,47 although more investigation must be carried out. However, the c.f. green synthesis of PCN-222 here discussed shows promise for progress in other fields, such as in catalysis and gas storage applications, where MOFs particles can be used also if showing relatively high inhomogeneity in terms of particle size.48,49
3.7 Functionalization of PCN-222 with S-GalNAc-PEG-sulfate ligand
To further explore the potential of c.f. strategy for synthesizing PCN-222, a thio-N-acetylgalactosamine PEG sulfate ligand 8 (S-GalNAc-PEG-sulfate) was chosen to be loaded onto the MOF. The specific selection of the N-acetylgalactosamine ligand aimed to create a potential nanosized drug delivery system (DDS) using ligand-mediated targeting. This targeting approach is based on the fact that certain diseases, particularly cancer, involve an overexpression of receptors on the cell surface. Therefore, an active targeting strategy would involve synthesizing nanoparticles that carry specific ligands for these overexpressed receptors on their surface. This would enable an increased selective accumulation of nanoparticles in the target tissues and enhance their uptake through receptor-mediated endocytosis. In the case of liver cancer, the third most common cause of cancer death,50 it has been observed that the asialoglycoprotein receptor (ASGPR) is primarily expressed in hepatocytes and to a lesser extent in extrahepatic cells. ASGPR facilitates internalization through clathrin-mediated endocytosis and exhibits a high affinity for certain carbohydrates, including galactose,51N-acetylgalactosamine, and glucose. Studies have demonstrated that this receptor has 10 to 50 times more affinity for N-acetylgalactosamine than for galactose.52 Furthermore, ligands based on N-acetylgalactosamine have proven effective in targeting the liver via ASGPR in vivo without interference from the immune system. Therefore, ASGPR is considered a promising pathway for the selective delivery of drugs and genes to the liver using N-acetylgalactosamine as a ligand.53 The presence of a PEG chain was aimed instead to enhance the stability and biocompatibility of the nanosized system.11 The functionalization of the MOF was achieved by mixing the stream containing PCN-222 (reaction conditions: 408 K, L:M = 1, L:Mod = 0.036, [TCPP] = 4.7 mM, flow rate 3 mL min−1, 60% US) with a DMF solution containing the S-GalNAc-PEG-sulfate ligand 8 in a concentration of 5 mg mL−1 (flow rate of the ligand solution: 3 mL min−1). This process yielded the PCN-222@SO3-PEG-S-GalNAc (Fig. 2c).11 The choice of a sulfate ligand was based on previous experiments, where a phosphate ligand was used on UiO-66.54 Compared to phosphate ligands, sulfate ligands were prepared using an innovative synthetic pathway that simplified the synthesis, provided better control, and yielded higher product quantities. Additionally, this approach eliminated lengthy purification steps such as three-day dialysis or the tedious separation of phosphate ligands from phosphoric acid. The successful loading of the ligand was confirmed through different analyses, as shown in Fig. 8.
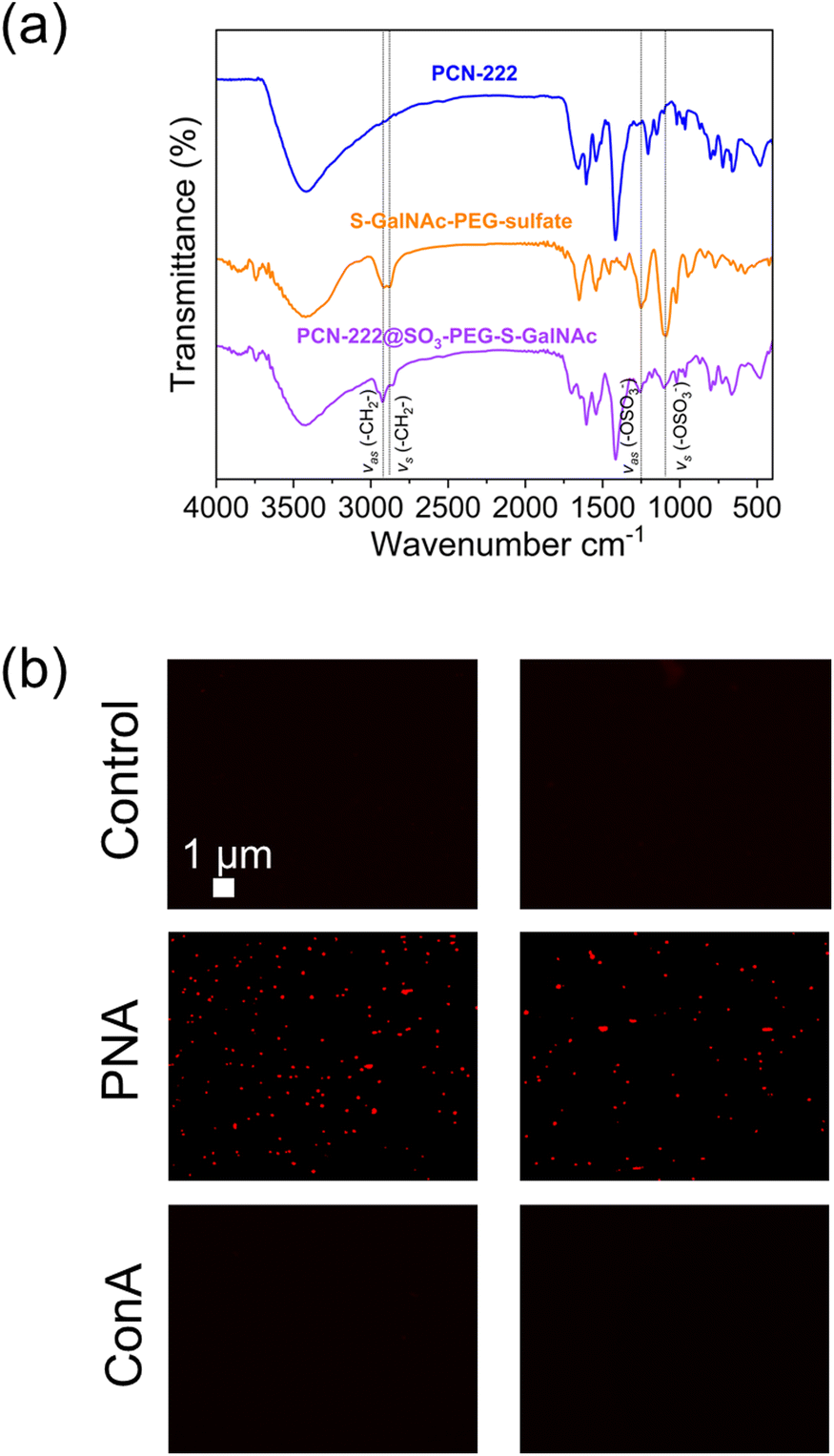 | ||
| Fig. 8 (a) FTIR analysis of PCN-222@ligand and (b) fluorescence microscopy images of the experiment of selective interaction of the PCN@SO3-PEG-S-GalNAc with lectins. | ||
FTIR spectroscopy (Fig. 8a) revealed the successful loading of the ligand on PCN-222 by the presence of characteristic peaks. Specifically, peaks observed at 2928 cm−1 and 2870 cm−1 were attributed to the asymmetric and symmetric stretching vibrations of the –CH2– group in the PEG chain,55 while peaks at 1245 cm−1 and 1135 cm−1 were attributed to the asymmetric and symmetric stretching vibrations of the sulfate (R–OSO3−) group.56,57 To further confirm the presence of the ligand bound to the MOF, a selective lectin interaction experiment was conducted (Fig. 8b, see ESI section-S11† for a scheme of the fluorescence microscopy experiment). Two variants of plant lectins were used and subsequently labelled for identification. One of them was Concanavalin A (ConA) of 26 kDa, obtained from the Canavalia ensiformis plant (jack bean), which was labelled with fluorescein isothiocyanate (FITC). The other lectin used was Peanut Agglutinin (PNA) of 110 kDa, obtained from peanuts (Arachis hypogaea), composed of four identical subunits of approximately 27 kDa each. PNA was labeled with AlexaFluor594 for identification. The sample preparation procedure is described in the ESI (section-S2†). Fluorescence was only detected in the case of the interaction with the PNA-AlexaFluor594 lectin (Fig. 8b), while it was not observed in the control experiment (PCN-222) and when ConA was used. This result confirmed the presence of thio-N-acetylgalactosamine, as the PNA lectin exhibits selective binding capability to this sugar. Additionally, the outcome suggested that PCN-222@SO3-PEG-S-GalNAc can establish specific interactions while being resistant to non-specific interactions, important for its use as drug delivery system. The literature also supports this observation, as it has been documented that Con A selectively binds to α-D-mannopyranosides, α-D-glucopyranosides, and, to a lesser extent, N-acetylglucosamines.57 On the other hand, the selectivity of the PNA lectin follows a preference trend for certain types of sugars: β-D-Gal-[1–3]-D-GalNAc > Gal > GalNAc.58 The presence of the ligand was also confirmed by TGA analysis (see ESI section-S11† for SEM images, XRD patterns and TGA profiles of PCN-222 vs. PCN-222@SO3-PEG-S-GalNAc).
The fluorescence microscopy images also unveiled the existence of agglomerates. This occurrence can be ascribed to the tetrameric nature of the PNA lectin, which enables the interconnection of various PCN-222@SO3-PEG-S-GalNAc entities, leading to the formation of agglomerates. SEM images of the PCN-222 and PCN-222@SO3-PEG-S-GalNAc showed no relevant differences (ESI section-S11†).
4. Conclusions
In conclusion, this study investigated the synthesis of PCN-222 (MOF-545) by shifting from traditional batch processes to (ultrasound-assisted) continuous flow synthesis. The aim was to overcome the limitations of batch synthesis, such as low productivity, long reaction times, and difficulty in scaling up, while achieving controlled size and morphology of the MOF. By optimizing the batch conditions and exploring various parameters in the continuous flow system, the efficient and sustainable production of PCN-222 was successfully achieved. The continuous flow approach offered numerous advantages, including improved resource efficiency, safer chemical handling, and scalability. Furthermore, the integration of ultrasound-assisted techniques allowed for the shaping, by etching, of PCN-222 into prolate ellipsoids, while the substitution of the hazardous solvent DMF with the greener alternative DMSO further enhances the sustainability of the process. Lastly, the functionalization with thio-N-acetylgalactosamine-PEG-sulfate ligands in tandem for the preparation of a nanosized drug delivery system expands the potentialities of the continuous flow strategy. This breakthrough opens doors for applications in drug delivery, catalysis, and other fields that require nanoparticles with specific size, morphology, and loaded with ligands. By demonstrating the robustness and scalability of the continuous flow synthesis, with a STY of over 950 kg m−3 day−1, the way for cost-effective and accessible production of PCN-222 has been paved.Author contributions
Conceptualization, A. Z., N. K.; methodology, A. Z., N. K.; validation, A. Z., N. K.; formal analysis, A. Z., N. K.; investigation, A. Z., M. C. and N. K.; resources, N. K.; data curation, A. Z., M. C. and N. K.; writing—original draft preparation, A. Z.; writing—review and editing, A. Z., M. C. and N. K.; funding acquisition, N. K. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Andalusian Ministry of Economy, Science and Innovation in the framework of the Plan Andaluz de Investigación, Desarrollo e Innovación (PAIDI 2020, programme 54ª “Investigación científica e innovación”, “POSTDOC_21_00594”), the Spanish Ministry of Science and Innovation (Ref: PID2020-119949RB-I00/AEI/10.13039/501100011033), the Andalusian Ministry of Economy, Science and Innovation cofinanced by the European Regional Development Fund (ERDF) from FEDER and the European Social Fund (ESF) (PY20_00882 and CV20-04221). The COST action CA-18132 “Functional Glyconanomaterials for the Development of Diagnostic and Targeted Therapeutic Probe” is also acknowledged. The authors acknowledge Dr. Carolina Carrillo-Carrión for her technical assistance in the initial batch experiments conducted for this work.References
- N. Hammi, S. Chen, A. Primo, S. Royer, H. Garcia and A. El Kadib, Green Chem., 2022, 24, 4533–4543 RSC
.
- T. Feng, S. Zhang, C. Li and T. Li, Green Chem., 2022, 24, 1474–1480 RSC
- A. Zuliani, N. Khiar and C. Carrillo-Carrión, Anal. Bioanal. Chem., 2023, 415, 2005–2023 CrossRef CAS PubMed
- P. H. Chang, R. Mukhopadhyay, B. Zhong, Q. Y. Yang, S. Zhou, Y. M. Tzou and B. Sarkar, J. Colloid Interface Sci., 2023, 636, 459–469 CrossRef CAS PubMed
- D. Feng, Z. Y. Gu, J. R. Li, H. L. Jiang, Z. Wei and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed
- M. Aghayan, A. Mahmoudi, K. Nazari, S. Dehghanpour, S. Sohrabi, M. R. Sazegar and N. Mohammadian-Tabrizi, J. Porous Mater., 2019, 26, 1507–1521 CrossRef CAS
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 8525–8529 CrossRef CAS PubMed
- X. Leng, H. Huang, W. Wang, N. Sai, L. You, X. Yin and J. Ni, Evid. Based Complementary Altern. Med., 2018, 3249023 Search PubMed
- M. He, Y. Chen, C. Tao, Q. Tian, L. An, J. Lin, Q. Tian, H. Yang and S. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 41946–41956 CrossRef CAS PubMed
- X. Chen, Y. Zhuang, N. Rampal, R. Hewitt, G. Divitini, C. A. O'Keefe, X. Liu, D. J. Whitaker, J. W. Wills, R. Jugdaohsingh, J. J. Powell, H. Yu, C. P. Grey, O. A. Scherman and D. Fairen-Jimenez, J. Am. Chem. Soc., 2021, 143, 13557–13572 CrossRef CAS PubMed
- C. Wei, H. Zhou and Q. Liu, Mater. Chem. Phys., 2021, 270, 124831 CrossRef CAS
- A. Amini, M. Khajeh, A. R. Oveisi, S. Daliran, M. Ghaffari-Moghaddam and H. S. Delarami, J. Ind. Eng. Chem., 2021, 93, 322–332 CrossRef CAS
- S. Choi, W. J. Jung, K. Park, S. Y. Kim, J. O. Baeg, C. H. Kim, H. J. Son, C. Pac and S. O. Kang, ACS Appl. Mater. Interfaces, 2021, 13, 2710–2722 CrossRef CAS PubMed
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC
- S. Carrasco, A. Sanz-Marco and B. Martín-Matute, Organometallics, 2019, 38, 3429–3435 CrossRef CAS
- E. Ben-Akiva, R. A. Meyer, H. Yu, J. T. Smith, D. M. Pardoll and J. J. Green, Sci. Adv., 2020, 6 DOI:10.1126/sciadv.aay9035
- K. Yu, Y. R. Lee, J. Y. Seo, K. Y. Baek, Y. M. Chung and W. S. Ahn, Microporous Mesoporous Mater., 2021, 316, 110985 CrossRef CAS
- W. L. Teo, W. Zhou, C. Qian and Y. Zhao, Mater. Today, 2021, 47, 170–186 CrossRef CAS
- A. Pintor, A. P. Mattey, I. Lavandera, V. Gotor-Fernández and A. Volkov, Green Chem., 2023, 25, 6041–6050 RSC
- G. Brufani, F. Valentini, G. Rossini, L. Carpisassi, D. Lanari and L. Vaccaro, Green Chem., 2023, 25, 2438–2445 RSC
- A. Zuliani, C. M. Cova, R. Manno, V. Sebastian, A. A. Romero and R. Luque, Green Chem., 2020, 22, 379–387 RSC
- C. M. Cova, L. Boffa, M. Pistocchi, S. Giorgini, R. Luque and G. Cravotto, Molecules, 2019, 24, 2681 CrossRef CAS PubMed
- H. Zhu, K. J. Wu and C. H. He, Chem. Eng. J., 2022, 429, 132412 CrossRef CAS
- H. Noh, C.-W. Kung, T. Islamoglu, A. W. Peters, Y. Liao, P. Li, S. J. Garibay, X. Zhang, M. R. DeStefano, J. T. Hupp and O. K. Farha, Chem. Mater., 2018, 30, 2193–2197 CrossRef CAS
- N. Planas, J. E. Mondloch, S. Tussupbayev, J. Borycz, L. Gagliardi, J. T. Hupp, O. K. Farha and C. J. Cramer, J. Phys. Chem. Lett., 2014, 5, 3716–3723 CrossRef CAS PubMed
- S. Knapp and D. S. Myers, J. Org. Chem., 2002, 67, 2995–2999 CrossRef CAS PubMed
- H. Zhang, X. Li, Q. Shi, Y. Li, G. Xia, L. Chen, Z. Yang and Z. X. Jiang, Angew. Chem., Int. Ed., 2015, 54, 3763–3767 CrossRef CAS PubMed
- M. Rubio-Martinez, T. D. Hadley, M. P. Batten, K. Constanti-Carey, T. Barton, D. Marley, A. Mönch, K. S. Lim and M. R. Hill, ChemSusChem, 2016, 9, 938–941 CrossRef CAS PubMed
- A. Zuliani, S. Chen and R. Giorgi, Appl. Mater. Today, 2023, 30, 101716 CrossRef
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC
- S. Nazri, M. Khajeh, A. R. Oveisi, R. Luque, E. Rodríguez-Castellón and M. Ghaffari-Moghaddam, Sep. Purif. Technol., 2021, 259, 118197 CrossRef CAS
- C. Koschnick, M. W. Terban, S. Canossa, M. Etter, R. E. Dinnebier and B. V. Lotsch, Adv. Mater., 2023, 2210613 CrossRef PubMed
- Y. Bai, Y. Dou, L. H. Xie, W. Rutledge, J. R. Li and H. C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC
- K. Suresh, D. Aulakh, J. Purewal, D. J. Siegel, M. Veenstra and A. J. Matzger, J. Am. Chem. Soc., 2021, 143, 10727–10734 CrossRef CAS PubMed
- P. Cubillas, M. W. Anderson and M. P. Attfield, Chem. – Eur. J., 2012, 18, 15406–15415 CrossRef CAS PubMed
- M. S. Alam, B. Ashokkumar and A. M. Siddiq, J. Mol. Liq., 2019, 281, 584–597 CrossRef CAS
- M. Ahmadi, S. M. Ayyoubzadeh, F. Ghorbani-Bidkorbeh, S. Shahhosseini, S. Dadashzadeh, E. Asadian, M. Mosayebnia and S. Siavashy, Heliyon, 2021, 7, e06914 CrossRef CAS PubMed
- S. D. Bagi, A. S. Myerson and Y. Román-Leshkov, Cryst. Growth Des., 2021, 21, 6529–6536 CrossRef CAS
-
H. Scott Fogler, Elements of Chemical Reaction Engineering, Pearson, 6th edn, 2021 Search PubMed
- M. J. Van Vleet, T. Weng, X. Li and J. R. Schmidt, Chem. Rev., 2018, 118, 3681–3721 CrossRef CAS PubMed
- S. M. Shaikh, P. M. Usov, J. Zhu, M. Cai, J. Alatis and A. J. Morris, Inorg. Chem., 2019, 58, 5145–5153 CrossRef CAS PubMed
- J. Jin, React. Kinet., Mech. Catal., 2020, 131, 397–408 CrossRef CAS
- G. Lidi, H. Xingfang, Q. Shili, C. Hongtao, Z. Xuan and W. Bingbing, RSC Adv., 2022, 12, 6063–6075 RSC
- E. Arceo, J. A. Ellman and R. G. Bergman, ChemSusChem, 2010, 3, 811–813 CrossRef CAS PubMed
- J. Gupta, B. W. Wilson and P. V. Vadlani, Biomass Bioenergy, 2016, 85, 313–319 CrossRef CAS
- Z. Cheng, M. Li, R. Dey and Y. Chen, J. Hematol. Oncol., 2021, 14, 85 CrossRef PubMed
- D. Lv, R. Shi, Y. Chen, Y. Chen, H. Wu, X. Zhou, H. Xi, Z. Li and Q. Xia, Ind. Eng. Chem. Res., 2018, 57, 12215–12224 CrossRef CAS
- L. J. Zhang, J. C. Yuan, L. J. Ma, Z. Y. Tang and X. M. Zhang, J. Catal., 2021, 401, 63–69 CrossRef CAS
- H. Rumgay, M. Arnold, J. Ferlay, O. Lesi, C. J. Cabasag, J. Vignat, M. Laversanne, K. A. McGlynn and I. Soerjomataram, J. Hepatol., 2022, 77(6), 1598–1606 CrossRef PubMed
- M. Negrete, E. Romero-Ben, A. Gutiérrez-Valencia, C. Rosales-Barrios, E. Alés, T. Mena-Barragán, J. A. Flores, M. C. Castillejos, P. De La Cruz-Ojeda, E. Navarro-Villarán, C. Cepeda-Franco, N. Khiar and J. Muntané, ACS Appl. Bio Mater., 2021, 4, 4789–4799 CrossRef CAS PubMed
- A. A. D'Souza and P. V. Devarajan, J. Controlled Release, 2015, 203, 126–139 CrossRef PubMed
- J. Tanaka, A. S. Gleinich, Q. Zhang, R. Whitfield, K. Kempe, D. M. Haddleton, T. P. Davis, S. Perrier, D. A. Mitchell and P. Wilson, Biomacromolecules, 2017, 18, 1624–1633 CrossRef CAS PubMed
- C. Carrillo-Carrión, V. Comaills, A. M. Visiga, B. R. Gauthier and N. Khiar, ACS Appl. Mater. Interfaces, 2023, 15, 27600–27611 CrossRef PubMed
- Z. Varga, J. Mihály, S. Berényi and A. Bóta, Eur. Polym. J., 2013, 49, 2415–2421 CrossRef CAS
- M. L. Testa, V. La Parola, F. Mesrar, F. Ouanji, M. Kacimi, M. Ziyad and L. F. Liotta, Catalysts, 2019, 9, 148 CrossRef
- J. J. Cid Martín, M. Assali, E. Fernández-García, V. Valdívia, E. M. Sánchez-Fernández, J. M. García-Fernández, R. E. Wellinger, I. Fernández and N. Khiar, J. Mater. Chem., 2016, 4, 2028–2037 Search PubMed
- B. Yoon, S. Lee and J. M. Kim, Chem. Soc. Rev., 2009, 38, 1958–1968 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Details of the c.f. system; materials and methods; preliminary tests for batch optimization; relevant publications related to the c.f. synthesis of MOFs; calculation of Re, De and residence time (τ); details of the effect of the concentration of the reagents, the volumetric flow rate, the amount of employed acid modulator, and the temperature of the reactions on the STY and on the size-distribution of PCN-222; residence time distribution and pulse experiment; US-assisted techniques; DLS analysis of the samples PCN-222(0%US) and PCN-222(60%US); green approaches of the c.f. synthesis; functionalization with S-GalNAc-PEG-sulfate ligand and 1H-NMR and 13C-NMR spectra. See DOI: https://doi.org/10.1039/d3gc02774k |
| This journal is © The Royal Society of Chemistry 2023 |