
Electrocatalytic upcycling of plastic waste
Juhyun
Cho†
 a,
Byeongyoon
Kim†
b,
Taehyun
Kwon†
c,
Kwangyeol
Lee
*b and
Sang-Il
Choi
*a
a,
Byeongyoon
Kim†
b,
Taehyun
Kwon†
c,
Kwangyeol
Lee
*b and
Sang-Il
Choi
*a
aDepartment of Chemistry and Green-Nano Materials Research Center, Kyungpook National University, Daegu, 41566, Republic of Korea. E-mail: sichoi@knu.ac.kr
bDepartment of Chemistry and Research Institute for Natural Sciences, Korea University, Seoul, 02841, Republic of Korea. E-mail: kylee1@korea.ac.kr
cDepartment of Chemistry, Incheon National University (INU), Incheon 22012, Republic of Korea
First published on 22nd September 2023
Abstract
Plastics, which are versatile and widely used materials, are being improperly disposed of in landfills or water bodies, leading to significant environmental damage. Traditional methods for plastic waste management, such as thermal decomposition and gasification, require high energy input. Recycling plastics back into their original form is a sustainable option, but demands high purity of recycled plastics and complex pre- and post-treatments. Electrochemical upcycling has recently emerged as a new alternative, which utilizes electrochemical reactions to transform plastic waste into valuable chemical compounds. Its advantages include the ability to operate under mild conditions, the use of eco-friendly energy sources, and increased energy efficiency. This review article provides an overview of electrochemical upcycling technologies for various types of waste plastics and explores their potential for generating value-added products. It also highlights the importance of understanding reaction mechanisms, electrochemical reaction systems, and catalyst development. We believe that the integration of electrochemical upcycling and chemical depolymerization has the potential to contribute to a circular economy and mitigate the environmental impact of plastic waste.
1. Introduction
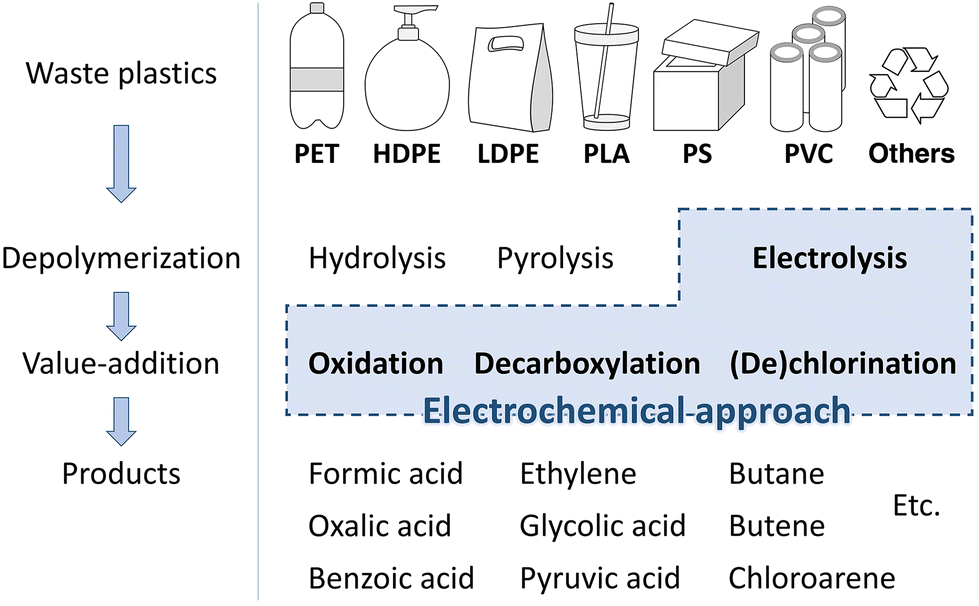
Plastics have infiltrated into every single fibre of human life and industry due to their easy production and versatility. However, the rampant usage of plastics and improper disposal of them into landfills or water bodies has engendered serious environmental pollution, which demands costly solutions.1 To address this issue, technologies have long been developed to convert plastics into energy through thermal decomposition (400–600 °C) and gasification (750 °C, over 100 atmospheres), but these technologies require significant amounts of energy and emit CO2.2,3 Another option is to recycle plastics to their original form, which has the advantage of recycling without generating CO2 emissions. However, this method requires high purity of the plastic for effective utilization, and pre-treatments such as separation and cleaning/refining are necessary before recycling, making the process complex and costly.4 In addition, recycled plastics are of lower quality than the original, making them less utilizable.5 Therefore, upcycling technologies that convert waste plastics into high-value chemicals have recently emerged as an attractive solution.6,7Electrochemical methods, which can be powered by green energy sources (wind, hydro, and solar), enable the conversion and upcycling of waste plastics with relatively low energy. Alkaline and acid hydrolysis has been known to depolymerize waste plastics under mild conditions.8–10 Despite the major drawback of the difficult extraction and recovery of monomers, recently developed electroconversion methods are capable of depolymerizing waste plastics and converting the monomers into ‘hydrogen’ energy and functional monomers. For example, polyethylene terephthalate (PET) can be hydrolysed into potassium terephthalate (TPA-K) and ethylene glycol (EG) in an alkaline electrolyte. EG can be converted into value-added chemicals through selective electro-oxidation.11–14 With conventional treatments, only 20% of PET bottles are recycled and the rest end up in landfills.15,16 However, with electrocatalytic waste plastic upcycling technology, EG can be converted into high-value formate ($600 per ton), which is much more efficient and economical than conventional waste plastic processing. However, research on the electrochemical conversion and upcycling of waste plastic is only in its infancy.17 Therefore, it is important to investigate the electrochemical treatment of various types of waste plastics to establish an energy-efficient electrochemical waste plastic upcycling system.
This review article provides an overview of electrochemical upcycling technologies for various waste plastics, such as PET, polyethylene (PE), polystyrene (PS), and polylactic acid (PLA), and explores their potential in creating value-added products (Scheme 1). First, for the electrochemical upcycling of waste plastics, it is crucial to establish a system capable of delivering charges to bulk solid plastics. This review article introduces pre-processing methods that utilize thermal and chemical depolymerization to convert plastics into small soluble molecules in electrolyte solutions. Next, the electrochemical reaction mechanisms based on the small molecular structures obtained through pre-processing were examined. Based on this, the importance of developing metal alloy catalysts that enable selective C–C bond cleavage and suppress the complete oxidation of CO2 is highlighted. Therefore, this review article will provide a way to solve environmental problems by converting waste plastics into useful materials using electrochemical methods.
 | ||
| Scheme 1 A schematic chart of the electrochemical upcycling of plastic waste. | ||
2. Electrochemical reaction systems for waste plastic upcycling
The electrocatalytic upcycling of polymers can be categorized into (1) direct electrolysis, (2) chemical depolymerization and subsequent electrochemical activation, and (3) homogeneous catalyst-mediated electrolysis. These classifications are based on the contact and electron transfer between the waste plastics and the electrodes. Since most waste plastics are solids that are insoluble in electrolytes, direct electrolysis is not valid in most cases. Therefore, this section introduces the other two methods.2.1. Chemical depolymerization and subsequent electrochemical upcycling
The ester bond in various polyesters, including PET, polytrimethylene terephthalate (PTT), polybutylene terephthalate (PBT), polyurethane (PU), and PLA, is readily hydrolysed in the presence of acids or bases, both acting as catalysts. In KOH solution, PET can be hydrolysed into TPA-K and EG. TPA-K tends to precipitate out of the solution. However, due to the high solubility (100 wt% at 20 °C) and viscosity (19.83 kg m−1 s−1 at 20 °C) of EG, energy-consuming distillation is the only viable method to separate EG (boiling point of 197.4 °C at 1 atm) from the KOH solution on a large scale. Therefore, electrochemical reactions using KOH electrolytes to convert EG into valuable products can be a favourable alternative. The representative electro-oxidation reaction pathways of EG are shown in Fig. 1.18 At the beginning of the EG oxidation reaction (EGOR), EG is adsorbed on the catalyst surface, which can be a metal such as platinum. The C–C and C–H bonds of EG are broken to generate intermediates such as glycolaldehyde (C2H4O2) or formaldehyde (CH2O), and ultimately formate (CHO2−), oxalate (C2O42−), or carbon dioxide (CO2). The reaction equations for these products are as follows. The standard reduction potentials were calculated according to the standard Gibbs energies of the reactants in the reactions.19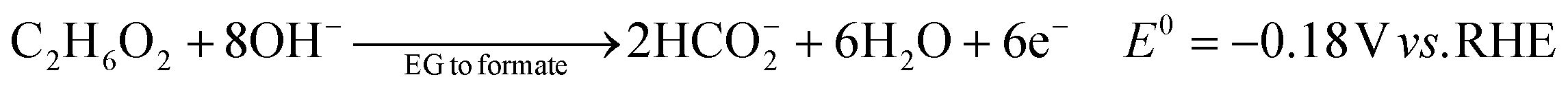
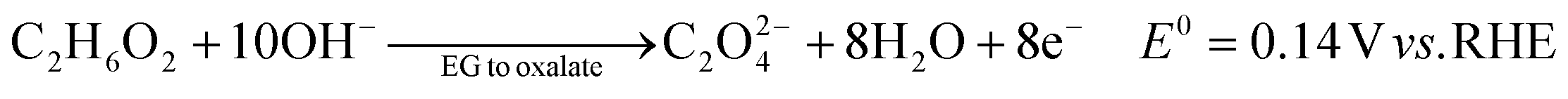
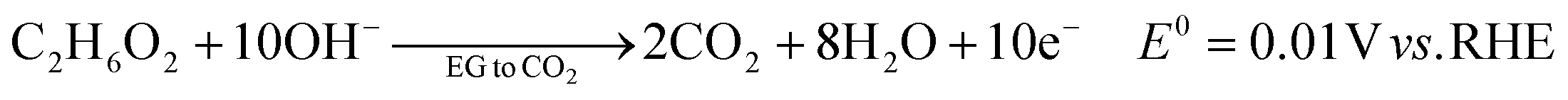
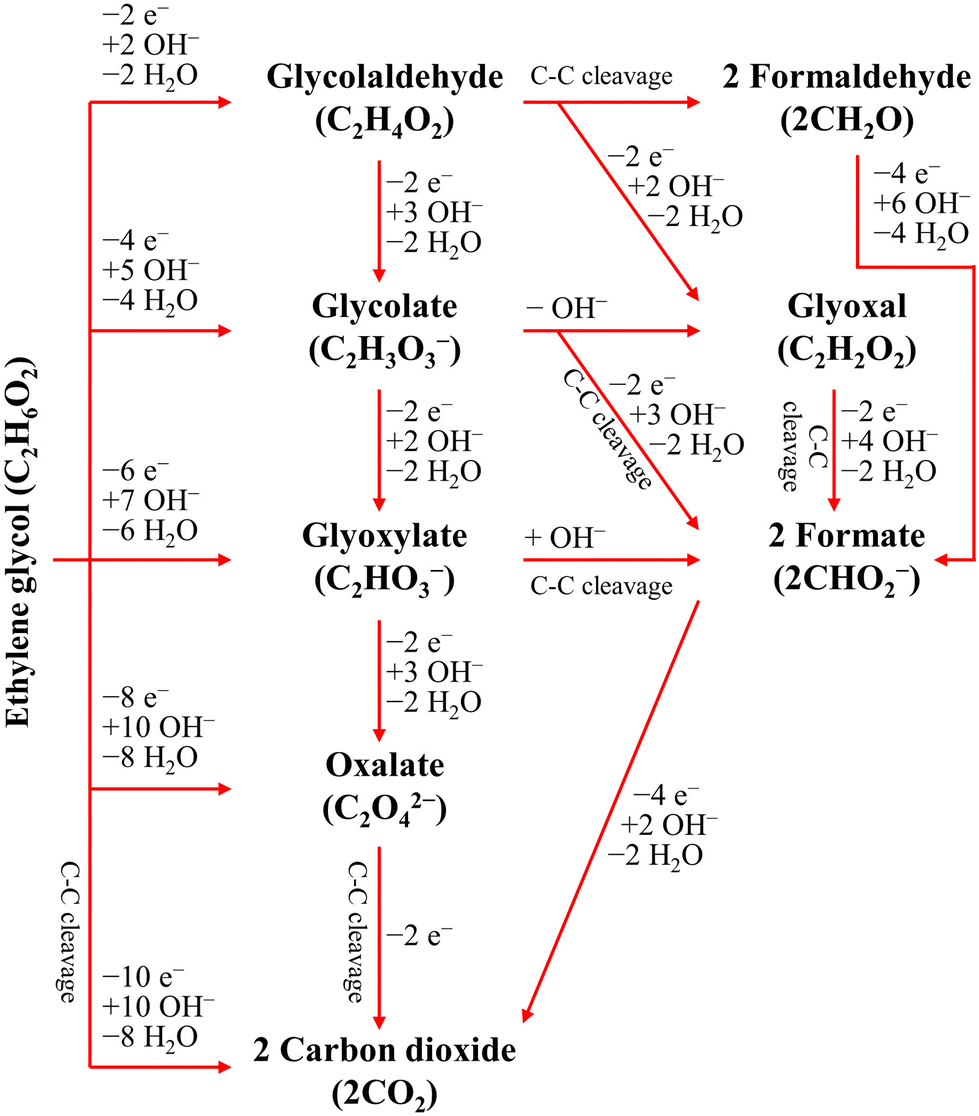 | ||
| Fig. 1 Representative EGOR pathways under alkaline conditions. Reproduced with permission from ref. 18. Copyright 2018 Elsevier. | ||
The differences in the reaction pathways depend on the reaction conditions and the catalysts used, thus the main products of interest can be produced with selectivity control. In the development of plastic electro-reforming, the focus is on obtaining value-added products such as oxalate or formate. Electrolysis, which allows for active voltage control and reaction path selection, can be accompanied by the hydrogen evolution reaction (HER) as the counter cathodic reaction.
On the other hand, depolymerized EG can be used as a liquid fuel for direct fuel cells with better availability, safety, and energy density over fuel volume than H2 gas. The standard cell voltage of an EGOR/oxygen reduction reaction (ORR) fuel cell is 1.22 V, which is very similar to that (1.23 V) of a hydrogen fuel cell. The aim of using direct EG fuel cells (DEGFCs) is to maximize energy production by implementing a reaction pathway for complete oxidation to carbon dioxide. However, since the activation energy of the EGOR is significantly higher than that of the hydrogen oxidation reaction (HOR), the development of catalysts that can reduce this activation energy is the major challenge.
The above two technologies differ in terms of the counter cathodic electrode choice (HER or ORR) and targeting incomplete or complete oxidation of EG. Taking these into account, the research direction should be catalyst development aimed at improving the selectivity, activity, and durability, and the development of electrolytic cells incorporating these catalysts.
The C–C chain in polyolefins, including PS, PE, polypropylene (PP), and polyvinyl chloride (PVC), is chemically inert, making them resistant to depolymerization, unlike polyesters. Thermal depolymerization of polyolefins requires excessively high temperatures (500–1000 °C) for C–C bond activation; milder conditions for degrading polyolefins must be developed.20,21 Over the past few decades, polyolefin degradation has been attempted using nitric acid, nitric oxides, or alternatively oxygen in combination with metal catalysts in acetic acid.22–24 For instance, PS with a molecular weight (MW) of 280![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da was subjected to a gas mixture of nitrogen oxide (275 kPa), oxygen (690 kPa), and nitrogen (3170 kPa) at 170 °C for 16 h, resulting in the formation of a mixture of benzoic acid, 4- and 3-nitrobenzoic acids, and gaseous oxides of carbon.23 In another case, at high concentrations of fuming nitric acid (>95%), PE with a MW of 11000 Da can be depolymerized into dicarboxylic acid in 30–50 h at temperatures above 60 °C.25 Although the energy cost of polyolefin degradation has significantly decreased, the use of highly potent acids required for the above processes poses challenges in terms of handling and waste management. Recently, Bäckström et al. reported a noteworthy method to produce oligomeric carboxylic acids utilizing microwave-assisted oxidation of PE in diluted nitric acid solutions (0.1–0.15 g mL−1).24,26 The process required a temperature of 180 °C and microwave energy of 1200 W to obtain value-added dicarboxylic acids with carbon chain lengths of 2–4. The depolymerized oligomeric carboxylic acids can be further converted into ethylene and propylene through electrochemical methods to take advantage of simplified purification, increased product value, and hydrogen production (Fig. 2).27
000 Da was subjected to a gas mixture of nitrogen oxide (275 kPa), oxygen (690 kPa), and nitrogen (3170 kPa) at 170 °C for 16 h, resulting in the formation of a mixture of benzoic acid, 4- and 3-nitrobenzoic acids, and gaseous oxides of carbon.23 In another case, at high concentrations of fuming nitric acid (>95%), PE with a MW of 11000 Da can be depolymerized into dicarboxylic acid in 30–50 h at temperatures above 60 °C.25 Although the energy cost of polyolefin degradation has significantly decreased, the use of highly potent acids required for the above processes poses challenges in terms of handling and waste management. Recently, Bäckström et al. reported a noteworthy method to produce oligomeric carboxylic acids utilizing microwave-assisted oxidation of PE in diluted nitric acid solutions (0.1–0.15 g mL−1).24,26 The process required a temperature of 180 °C and microwave energy of 1200 W to obtain value-added dicarboxylic acids with carbon chain lengths of 2–4. The depolymerized oligomeric carboxylic acids can be further converted into ethylene and propylene through electrochemical methods to take advantage of simplified purification, increased product value, and hydrogen production (Fig. 2).27
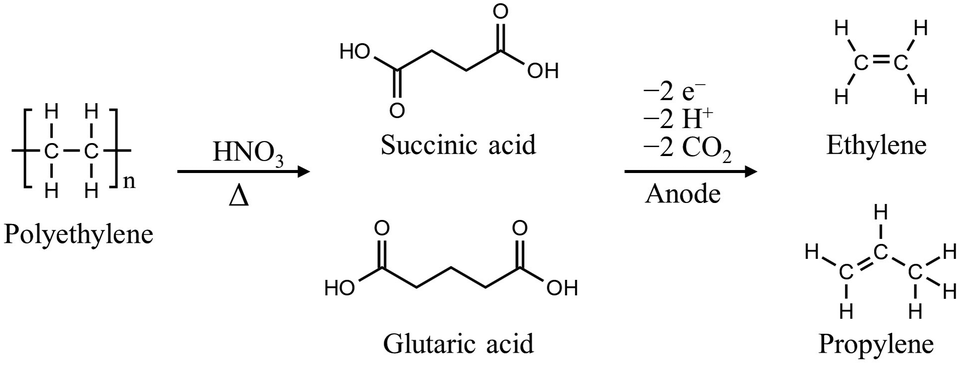 | ||
| Fig. 2 Chemical depolymerization and the electrochemical upcycling reaction scheme of PE. | ||
2.2. Homogeneous catalyst-mediated electrolysis
The harsh conditions, such as fuming nitric acid, required to break the inert chains of polyolefins discussed above, are detrimental to electrochemical devices and catalysts. Therefore, additional separation processes are necessary to convert the depolymerized oligomers into electrochemical systems, which further reduces the efficiency of the technology. There is a need for more efficient methods that operate under milder conditions and minimize the chemical pre-treatment of solid polyolefins. To meet this challenge, the use of redox-tunable molecules or radicals as homogeneous catalysts to mediate the interaction between solid-state polymers and electrodes has emerged as a promising approach for electrolytic cells. Yan et al. demonstrated an example of breaking the inert C–C bond of PS using N-hydroxyphthalimide (NHPI) as a redox mediator.28 In this approach, NHPI was oxidized to a phthalimide-N-oxyl (PINO) radical at a carbon anode and reduced back to PINO while oxidizing the –CH2– backbone of PS, thus completing the electro-redox cycle. In the presence of environmental O2, the activated –CH˙– radicals spontaneously further oxidized to peroxide species, resulting in the cleavage of C–C bonds or oxygenation of the C–H bond. They observed C–C bond cleavage products (i.e., benzaldehyde and benzoic acid) and C–H oxygenation products (i.e., 1,2-diphenylethanone and benzyl) by gas chromatography–mass spectrometry and gas chromatography–flame ionization detection from NHPI-assisted electrolysis of 1,2-diphenylethane as a model compound. As a proof-of-concept, PS (MW = 10000 Da) was electrolyzed at 1.5 V vs. Ag/AgCl until all the starting PS was converted, yielding 12% of monomers and dimers.
Electrochemical oxidation processes generate various reactive oxygen species (ROS), including hydrogen peroxide (H2O2, E° = 1.8 V vs. NHE) and hydroxyl radicals (˙OH, E° = 2.8 V vs. NHE), in aqueous electrolytes. These radicals possess significant potential to cleave the C–C backbone of polyolefin plastics.29 However, the ˙OH radical has an exceedingly short lifetime and is only dispersed within a range of 10 nm from the anode.30,31 In order to overcome this limitation, persulfate (S2O82−, E° = 2.1 V vs. NHE) and a sulfate radical (SO4˙−, E° = 2.6 V vs. NHE) were employed as mediators for electrooxidation.31–35 The weak interaction between ˙OH and the surface of boron-doped diamond (BDD) anodes results in a much higher oxygen evolution reaction (OER) overpotential, making it selectively effective for ˙OH and sulfate radical generation.36,37 As shown in Fig. 3, in 0.03 M Na2SO4 electrolyte, S2O82− anions generated at the BDD anode react with the cathode-generated ˙OH radical or H2O2 to form SO4˙−. In the presence of S2O82−, Lu et al. investigated the PS decomposition mechanisms by tracking the transformation products from the sodium dodecyl sulfate-assisted electrooxidation of PS microparticles (500 mesh).38 A total of seven transformation products, namely PS-TP1 to PS-TP7, were detected using ultra-performance liquid chromatography-Q-Exactive Orbitrap mass spectrometry, as shown in Fig. 4. The initial oxidation product identified was PS-TP4 (C17H20O4, with diphenyl rings), which could be assigned to bis(hydroxyphenyl)pentane-diol, hydroxyphenyl-phenylpentane-triol, or other isomers. The oxidative cleavage of C–C bonds occurred at the phenylethyl position, resulting in the formation of PS-TP3 (C11H20O4), and then 3-hydroxyphenyl ethylene glycol (PS-TP5, C8H10O3), hydroquinone (PS-TP6, C6H6O2), and benzoic acid (PS-TP7, C7H6O2) were formed via further oxidation. In addition, pathways involving the attack on the benzene ring also occurred, resulting in esters, aldehydes and alcohols, such as PS-TP1 (C11H20O4) and PS-TP2 (C7H14O6). Both pathways led to short-chain carboxylic acids and ultimately complete oxidation into CO2. This sodium dodecyl sulfate-assisted electrolysis of PS resulted in a significant decomposition of 42.5% of PS microparticles over a period of 72 h at a current density of 30 mA cm−2, surpassing that of 18.5% when only ˙OH radicals were operating without sulfates.
 | ||
| Fig. 3 Schematic diagram of the electrochemical in situ generation of hydroxyl and sulfate radicals for PS decomposition. | ||
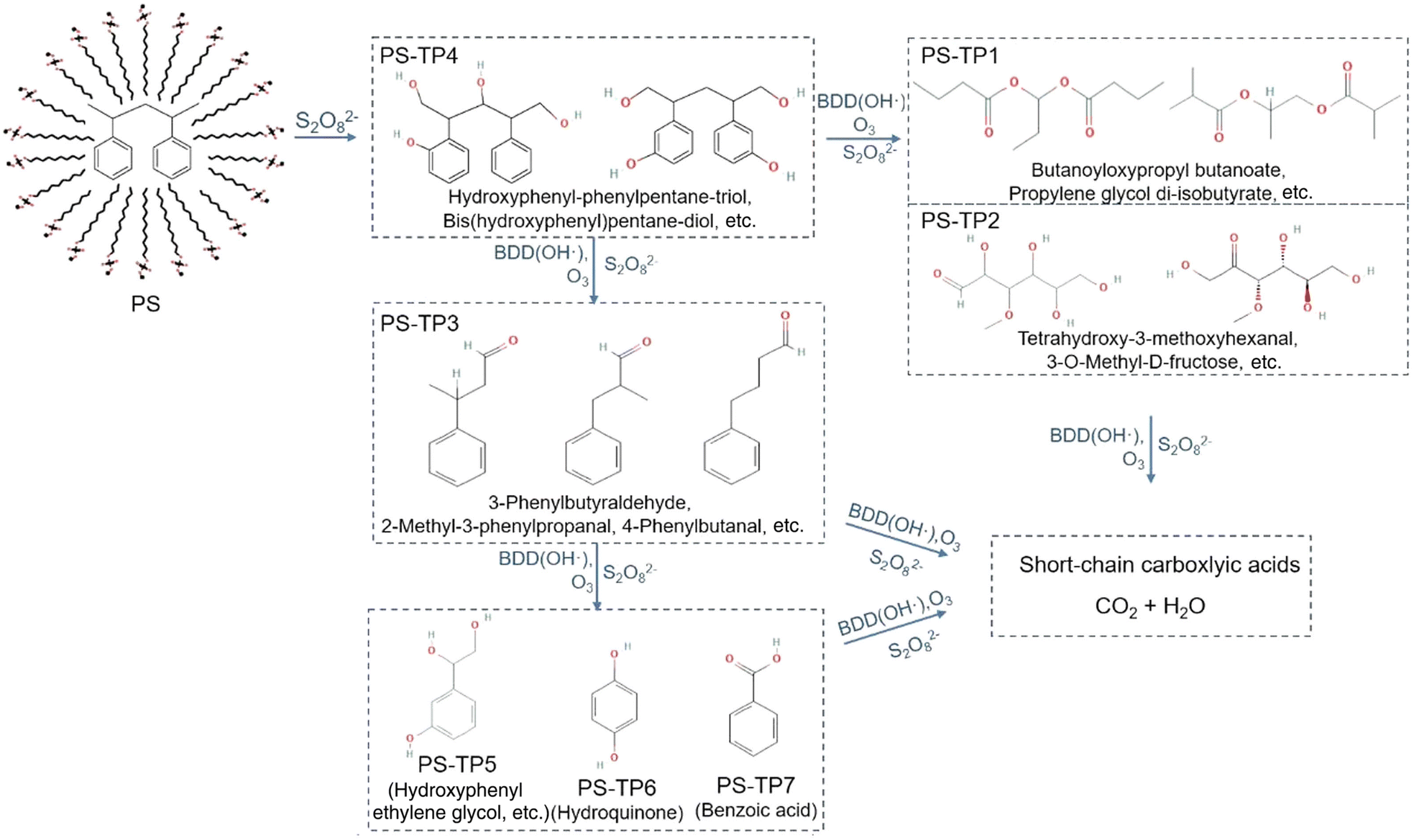 | ||
| Fig. 4 Proposed reaction pathway of the degradation of PS microparticles by sodium dodecyl sulfate-assisted electrooxidation. Reprinted with permission from ref. 38. Copyright 2022 Elsevier. | ||
Moreover, redox mediators play a pivotal role in the cleavage of functional groups from the C–C backbone. Their significance is especially pronounced in PVC recycling, where the safe removal of chlorine presents a formidable challenge. Traditional pyrolysis of PVC results in the emission of hydrogen chloride gas, which is corrosive to equipment and leads to dismal recycling rates of PVC in most countries. Consequently, recent methodologies have proposed electrochemical approaches that incorporate electrolytes or chlorine-absorbing substances to mitigate this corrosive issue. Miao et al. investigated a method whereby a TiO2 cathode dechlorinates the surface of PVC microplastics, while anode-generated ˙OH radicals depolymerize the C–C backbones, progressively exposing the chlorine from the PVC bulk solid.29 The resulting dechlorinated chains are transformed into monomeric organic acids, and the reduced chlorine is obtained as ions in a Na2SO4 electrolyte. Fagnani et al. demonstrated an electrochemical synthesis of value-added chloroarenes by di(2-ethylhexyl)phthalate (DEHP)-mediated electroreduction of waste PVC.39 DEHP and its radicals facilitate electron transfer between the cathode and PVC in an organic electrolyte composed of tetrabutylammonium tetrafluoroborate (NBu4BF4)/dimethylformamide (DMF). The released chlorine ions are then oxidized at the anode and subsequently used to chlorinate provided arene substrates. The authors emphasized that green house gas emissions can be greatly reduced by this shift from using HCl in the conventional process to utilizing PVC as a chlorine source in electrochemical upcycling for the synthesis of chloroarenes, a value-added chemical feedstock.
Other redox mediators, including active chlorine species, Ag2+ ions, and polyoxometalate, are also being considered as potential candidates for homogeneous catalysts in electrochemical plastic upcycling.40–44 These homogeneous catalysts offer the advantage of facilitating the direct management of solid plastics. Future development challenges in this application include achieving improved reaction activity as well as, for molecular catalysts, enhancing the durability of the catalyst molecules, and for radicals, achieving sufficient lifetime.
3. Electrochemical value addition of depolymerized monomers
After delving into depolymerization strategies categorized by the type of plastic backbone, the focus naturally transitions to strategies that add value to the resulting depolymerized monomers. Given the potential to derive diverse monomers from various plastics, there is a need for an approach that categorizes these monomers and their associated functional groups independently of their source plastics. This section aims to introduce the electrochemical reactions involving representative monomers.3.1. EG
PET is a widely used polyester thermoplastic with high chemical and impact resistance at room temperature.45 Two primary methods for PET recycling are mechanical and chemical approaches. While mechanical recycling is a well-established process, it has limitations, with a significant decrease in the material's ductility from 310% to 2.9% after three cycles of recycling.46–48 On the other hand, chemical hydrolysis in alkaline solutions readily extracts TPA-K and EG as monomers from PET, but it is not widely used commercially due to the complexity of the purification process, which consumes significant time and energy.49,50 To improve the purification process and achieve additional benefits, electrochemical upcycling has been proposed as an additional method of chemical hydrolysis (Fig. 5).51 Under the electrochemical upcycling process, formic acid generated by the EGOR is neutralized in a KOH electrolyte, resulting in TPA precipitation, giving a high separation yield of 94%. Further lowering the pH of the electrolyte yields a solid form of formate/potassium diformate (KDF).52 The TPA obtained through the above process can be used in the pharmaceutical industry and metal–organic skeleton synthesis, and KDF is a value-added material used as an animal feed preservative and road de-icing agent.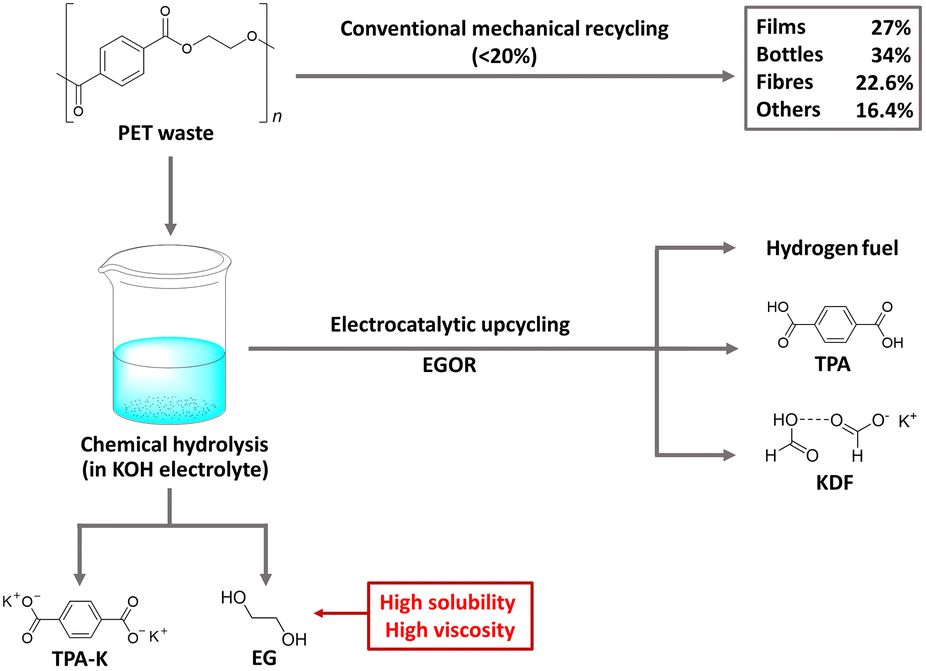 | ||
| Fig. 5 Electrocatalytic PET upcycling to commodity chemicals and H2 fuel. Reproduced with permission from ref. 51. Copyright 2021 Springer Nature. | ||
With attractive properties of high boiling point (198 °C) and energy density (4800 Ah L−1), recent research studies on EG have been focused on DEGFC and electrolysis for plastic upcycling.53–55 However, the goals of the anodic EGOR in DEGFC and electrolysis for plastic upcycling are different. In DEGFC, the objective is to completely oxidize EG to CO2 to maximize power generation, while in plastic upcycling, the goal is to obtain value-added products such as formate or oxalate. Therefore, advancing plastic upcycling requires the development of catalysts that selectively promote the desired reaction pathways.56
Xin et al. studied the reaction pathway for the selective generation of products from the EGOR using Pt/C and Au/C catalysts under alkaline conditions.57 The use of a Pt/C catalyst in the EGOR mainly produces glycolic acid, oxalic acid, and formic acid, while the use of an Au/C catalyst produces glycolic acid and formic acid. The proposed reaction pathways are illustrated in Fig. 6. In a non-C–C bond cleavage pathway, glycolaldehyde is an intermediate in the two-electron oxidation that occurs on both Pt/C and Au/C. In situ electrochemical FTIR studies confirm that the reaction intermediates are well adsorbed on the Pt/C during the EGOR. Glycolaldehyde, adsorbed on the Pt/C, can be easily oxidized to glycolic acid at 0.3 V vs. SHE without desorption. In addition, the hydroxyl group of glycolic acid is oxidized to glyoxylic acid, which is rapidly oxidized to oxalic acid at 0.6 V vs. SHE. PtOx formed from Pt/C at 0.9 V vs. SHE converts glycolic acid to oxalic acid through stepwise oxidation at 1.1 V vs. SHE. In the case of the Au/C catalyst, a positive onset potential of 0.4 V vs. SHE higher than that of the Pt/C catalyst was required for the EGOR to generate glycolic acid, but no further oxidation occurred. In a C–C bond cleavage pathway, EG was oxidized to form formic acid. This pathway also leads to the formation of CO and CO2, which ultimately produces carbonates.58–62 Pt/C produces formic acid at 0.6 V vs. SHE, and Au/C and PtOx do so at higher potentials above 1.0 V vs. SHE.
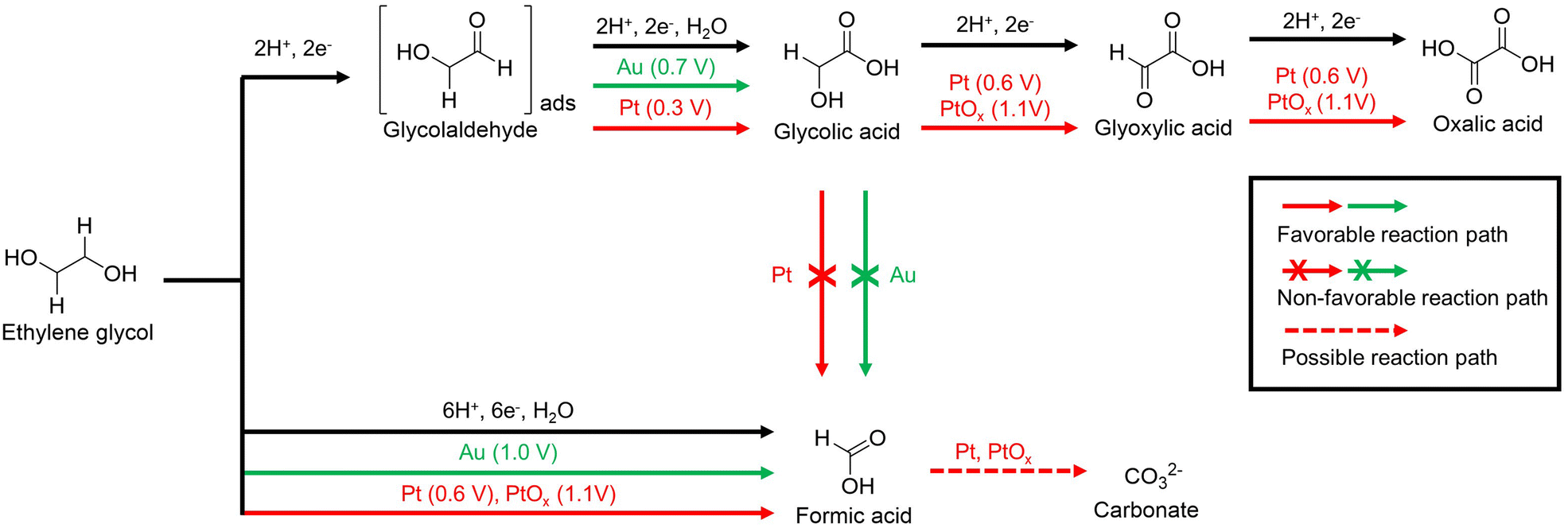 | ||
| Fig. 6 The proposed pathways for electrocatalytic oxidation of ethylene glycol on Au/C and Pt/C in alkaline media. The starting potential for each reaction pathway is indicated after the electrocatalysts. Reproduced with permission from ref. 57. Copyright 2012 Elsevier. | ||
In conclusion, direct C–C decomposition of EG is an advantageous process for producing formic acid, whereas stepwise electro-oxidation of EG is advantageous for obtaining oxalic acid. However, since oxalic acid is a toxic substance that can cause renal failure, an industrially valuable selective production process for formic acid is more attractive for the upcycling of PET. To selectively produce formic acid, the continuous electro-oxidation of hydroxyl or carboxyl groups should be promoted, therefore, necessitating the development of electrocatalysts capable of strong electro-oxidation reactions.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ˙CCH2OH species. According to the DFT study, the addition of Ag decreased the adsorption energy of the key intermediate from −2.10 to −1.96 eV, resulting in the facilitated desorption of the final product, glycolate (Fig. 7e). On the other hand, without Ag, the O˙CCH2OH intermediate was strongly adsorbed onto the Pd surface.
˙CCH2OH species. According to the DFT study, the addition of Ag decreased the adsorption energy of the key intermediate from −2.10 to −1.96 eV, resulting in the facilitated desorption of the final product, glycolate (Fig. 7e). On the other hand, without Ag, the O˙CCH2OH intermediate was strongly adsorbed onto the Pd surface.
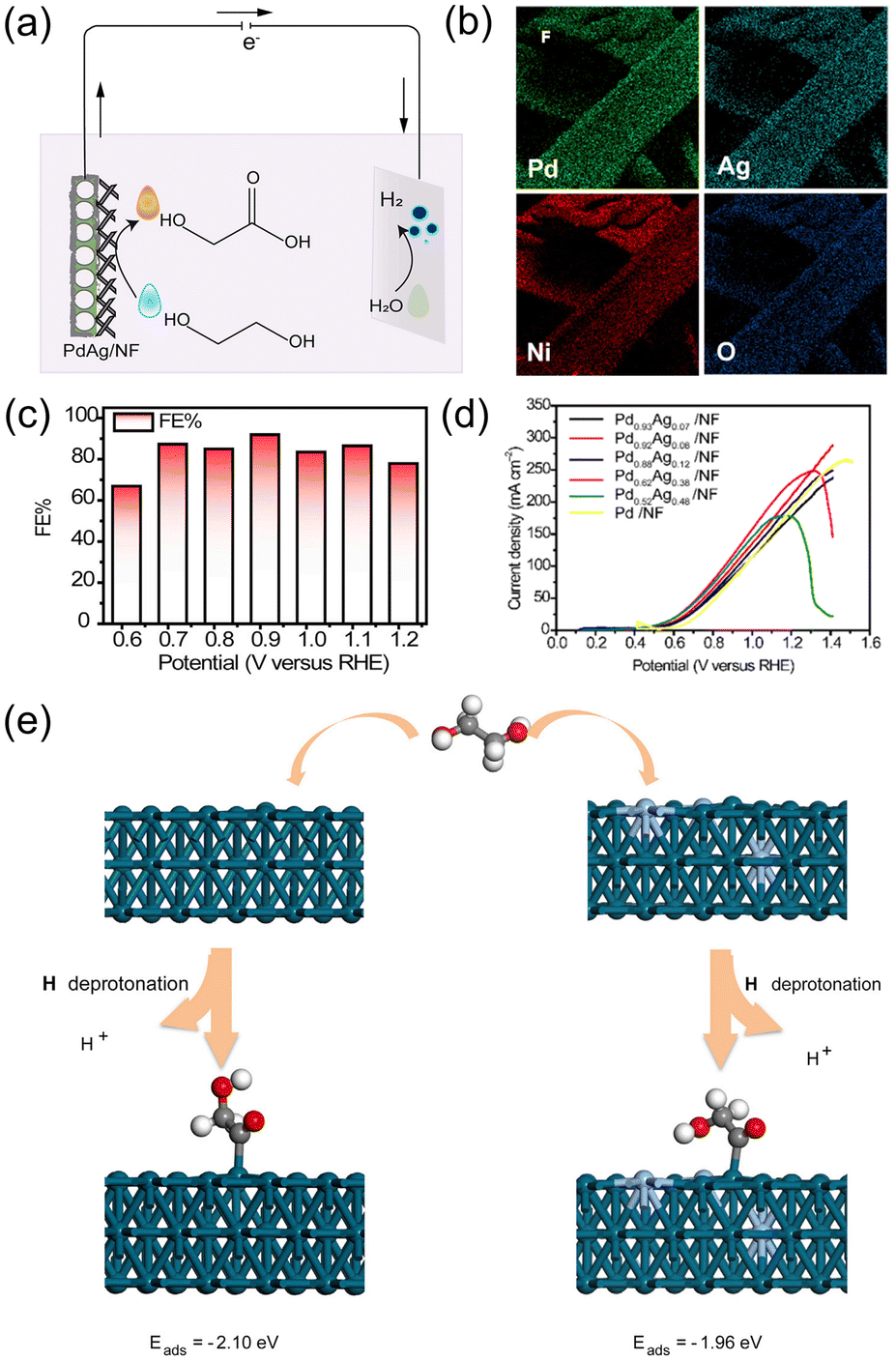 | ||
| Fig. 7 (a) Schematic illustration for the electrocatalytic glycolate production from the EGOR and counter HER in an electrolytic system. (b) SEM-EDX elemental mapping image of PdAg/NF. (c) FEs of PdAg/NF for glycolate production for 2 h of chronoamperometry at varied voltages. (d) Linear sweep voltammetry (LSV) curves of Pd0.93Ag0.07/NF, Pd0.92Ag0.08/NF, Pd0.88Ag0.12/NF, Pd0.62Ag0.38/NF, Pd0.52Ag0.48/NF and Pd/NF catalysts in 0.5 M KOH with 1 M ethylene glycol. (e) Illustration of the adsorption energy of intermediates on Pd(111) and PdAg(111). Green = Pd, Blue = Ag. Reprinted with permission from ref. 67. Copyright 2021 Elsevier. | ||
Liu et al. utilized the synergy of Pd and Ni for selective EGOR electrocatalysts to synthesize glycolate from PET-derived EG.68 The Pd–Ni(OH)2 catalyst was directly grown on a porous Ni foam (NF) by a hydrothermal method (Fig. 8a). Pd–Ni(OH)2/NF exhibited exceptional stability in the EGOR, retaining 85% of its initial current density even after 3600 seconds of operation (Fig. 8b). This remarkable performance surpassed those of Pd/C (16%) and Pd/NF (55%), conclusively confirming the superior stability of Pd–Ni(OH)2/NF in the EGOR. The FE for glycolate reached over 90% for a wide range of potentials (0.7 to 1.2 V vs. RHE) (Fig. 8c). The origin of the high glycolate-selectivity was also investigated by DFT, which revealed that the activation barrier for the desorption of O˙CCH2OH was decreased (0.68–0.51 eV) with the formation of the Pd–Ni(OH)2 interface (Fig. 8d and e).
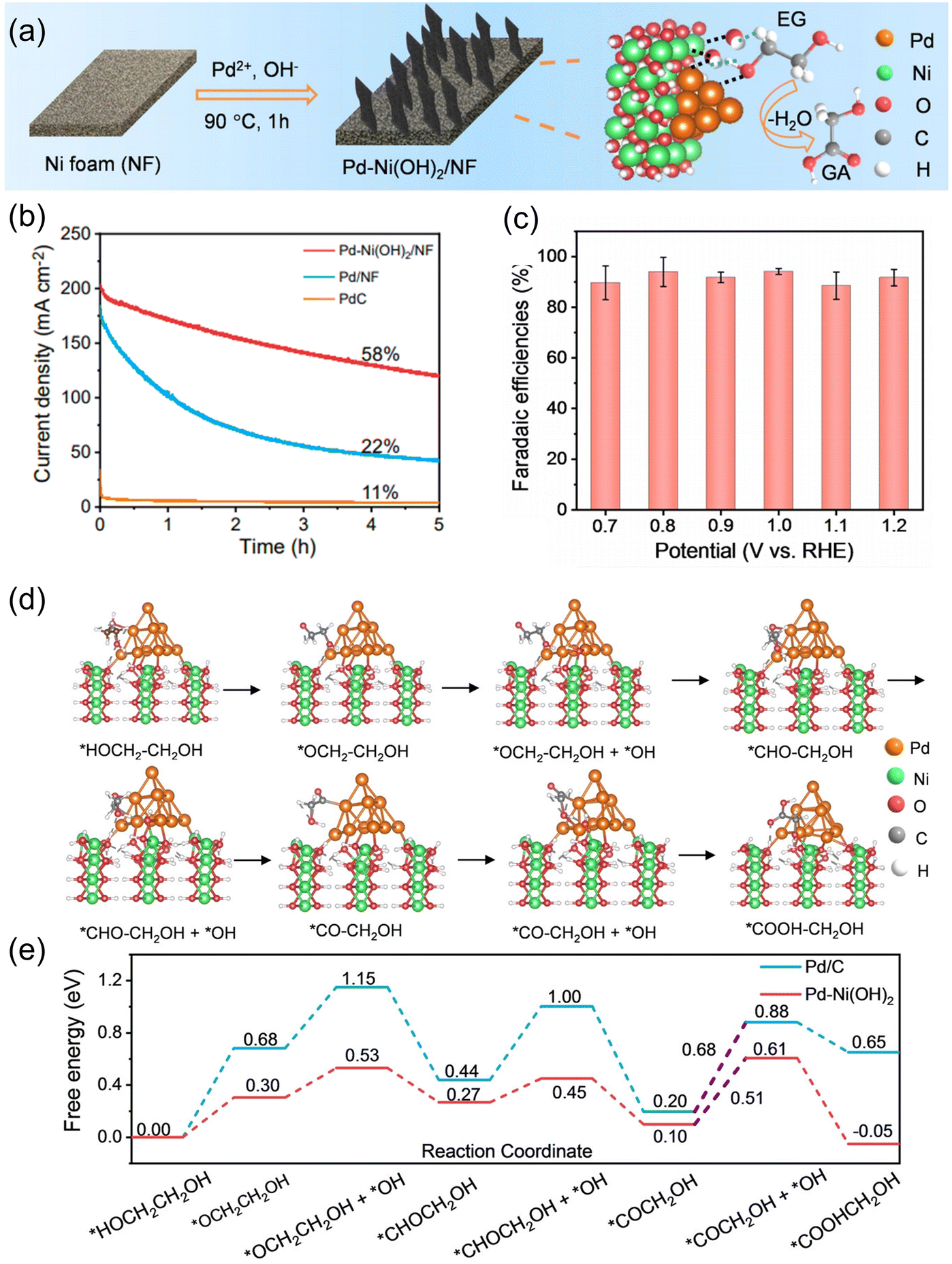 | ||
| Fig. 8 (a) Schematic illustration of the synthesis of Pd–Ni(OH)2/NF. (b) LSV curves for Pd–Ni(OH)2/NF in 1.0 M KOH with and without EG. (c) FE of Pd–Ni(OH)2/NF for glycolate production at designated voltages. (d) The DFT-optimized configurations of the EG oxidation process on Pd–Ni(OH)2. (e) Gibbs free energy diagrams for EG-to-glycolate oxidation on Pd and Pd–Ni(OH)2 (0 V vs. RHE). The numbers are the Gibbs free energies with units in eV. Reprinted with permission from ref. 68. Copyright 2023 Wiley. | ||
In addition to Pd-based catalysts, non-noble metal-based catalysts have also been investigated for the selective EGOR for the C2 product. Ozawa et al. investigated the first-principles calculation of catalytic activity and selectivity of the EGOR on Fe(001), Co(0001), and Ni(111) model surfaces.69 The calculation results showed that the order of activation energy for O–H bond dissociation forming HOCH2CHO was Co > Ni > Fe, whereas the order of activation energy for C–C bond cleavage resulting in CO2 generation was Fe > Co > Ni. Therefore, Fe is expected to be a suitable candidate for partial oxidation of EG with less CO2 generation. Matsumoto et al. prepared FeCoNi/C via a chemical reduction method.70 FeCoNi/C showed 60% oxalic acid selectivity (via 8e− oxidation) at 0.4 V vs. RHE in the EGOR under alkaline conditions (30 wt% EG + 20 wt% KOH) without the formation of CO2, whereas Pt/C showed 60% glycolic acid selectivity.
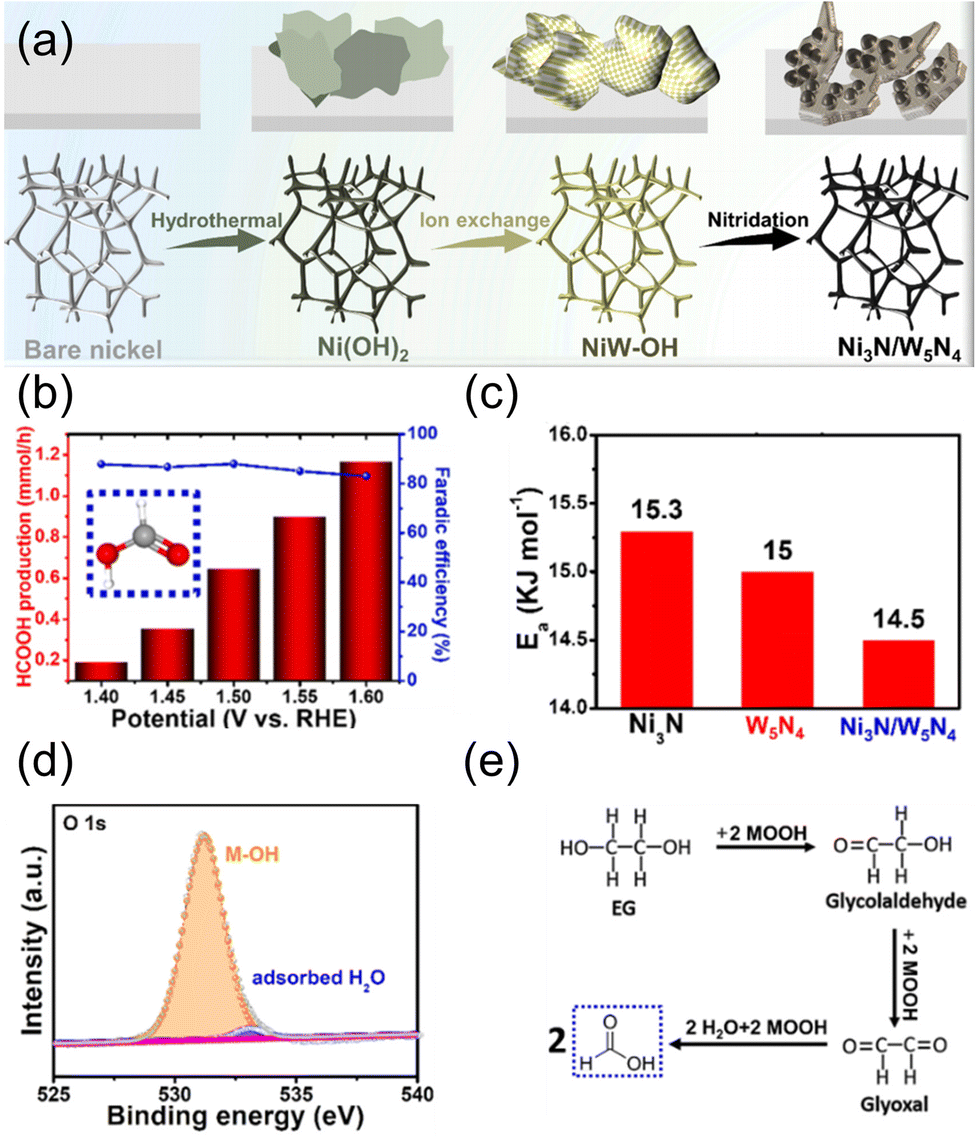 | ||
| Fig. 9 (a) Schematic illustration of the formation process of Ni3N/W5N4. (b) Formate production rate and FE under different potentials. (c) Ea value for the catalysts in DI water + plastics. (d) High-resolution XPS spectrum of O 1s (about Ni3N/W5N4). (e) The suggested reaction mechanism for the formate-selective EGOR. Reprinted with permission from ref. 72. Copyright 2022 Elsevier. | ||
| Catalyst | Electrolyte | E onset (V vs. RHE) | E electrolysis (V vs. RHE) | Current density (mA cm−2) | Mass activity (A mgM−1) | Total conversion (%) | Major product | FE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Pd/C | 2 M KOH + 5 wt% EG | 0.42 | 0.83 | 65.3 | 2.76 | 55.5 | Glycolate | 89.5 | 67 |
| Oxalate | 6.6 | ||||||||
| Pd(NiZn)/C | 2 M KOH + 5 wt% EG | 0.39 | 0.9 | 53.5 | 3.33 | 77.1 | Glycolate | 55.4 | |
| Oxalate | 37.6 | ||||||||
| PdAg/NF | 0.5 M KOH + 1.0 M EG | — | 0.91 | ∼300 | — | — | Glycolate | ∼92 | 68 |
| Pd–Ni(OH)2/NF | 1.0 M KOH + 1.0 M EG | — | 1.0 | ∼150 | — | 93.2 | Glycolate | 94.1 | 66 |
| PtRh0.02@Rh NW | 0.1 M KOH + 0.5 M EG | ∼0.5 | 0.83 | — | 1.25 | — | Glycolate | — | 70 |
| FeCoNi/C | 20 wt% KOH + 30 wt% EG | 0.34 | 0.4 | ∼10 | — | — | Oxalate | ∼60 | 73 |
| Pt/C | 20 wt% KOH + 30 wt% EG | 0.37 | 0.4 | — | — | — | Glycolate | ∼60 | |
| CoNi0.2P-uNS/NF | 1.0 M KOH + 0.3 M EG | — | 1.4 | ∼350 | — | 92 | Formate | 54 | 74 |
| Branched NiSe2 | 1.0 M KOH + 1.0 M EG | — | 1.6 | 95.23 | — | 92 | Formate | 80 | 75 |
| Glycolate | 5 | ||||||||
| Oxalate | 7 | ||||||||
| OMS-Ni1-CoP | 1.0 M KOH + 0.5 M EG | — | 1.3 | ∼10 | — | — | Formate | 96 | 76 |
| NiCo-SS-ET | 1.0 M KOH + 0.5 M EG | — | 1.43 | 300 | — | — | Formate | ∼80 | 77 |
| System | Cathode | Anode | Cell voltage (V) | Current density (mA cm−2) | Feedstock | Depolymerized product | Electrochemically upcycled products | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| MEA | Ni2P/NF | Pd–Ni(OH)2/NF | 1.2 | 100 | 100 g (PET) | 83.3 g (TPA), yield 96.3% | 23.2 g (glycolate), yield 58.5% | 20.6 L (H2), yield 98% | 68 |
| H-type | OMS-Ni1-CoP | 1.57 | 10 | 6.3 g (PET) | TPA | KDF, FE 93.2% | H2, FE ∼100% | 76 | |
| H-type | NF | Pd/NF | 0.7 | ∼400 | 2 g (PET) | TPA, yield 96% | K2CO3, FE 91% | H2, FE 98% | 14 |
| H-type | Ni3N/W5N4/NF | 1.6 | 120 | 2 g (PET) | TPA | HCOOH, FE ∼85%, 1.2 mmol h−1 cm−2 | H2 | 72 | |
| H-type | CoNi0.2P-uNS/NF | 1.24 | 50 | 0.3 g L−1 (PET) | TPA | Formate, FE 95%, 5.3 mmol h−1 cm−2 | H2 | 74 | |
| H-type | Co-Ni3N/CC | ∼1.46 | 50 | 2.1 g (PET) | TPA | Formate | H2 | 78 | |
3.2. Dicarboxylic acids
PE, which includes high-density polyethylene (HDPE) and low-density polyethylene (LDPE), and PP are major groups of polyolefin plastics and are the most important general-purpose polymers. In addition, fuels produced from PE that are oxygen-free and have high carbon and hydrogen contents have properties similar to those of fossil fuels and can be utilized as an alternative energy source.79The carboxylic acid products produced by the depolymerization of PE undergo sequential or simultaneous decarboxylation reactions at the anode, leading to the production of hydrocarbon gaseous products (such as ethane, propane, etc.) that are valuable as fuels.27 In this process, promoting simultaneous decarboxylation over sequential decarboxylation is the critical factor (Fig. 10), because the radical intermediate produced through sequential decarboxylation tends to form a dimer, which impedes the overall electrooxidation reaction. According to previous reports on related reactions, it is known that the adsorption of both carboxyl groups on the catalyst surface is preferred when a small amount of the reactant is used compared to the catalyst, and conversely, adsorption of a single carboxyl group on the catalyst surface is preferred.80 This is because reactants adsorb on confined catalytic active sites, so increasing the reactants reduces the number of catalytic active sites, resulting in the adsorption of a single carboxyl group due to the steric hindrance of the first adsorbed reactants. Therefore, further research is needed on surface modification for a high surface area and effective reaction sites of nanocatalysts.
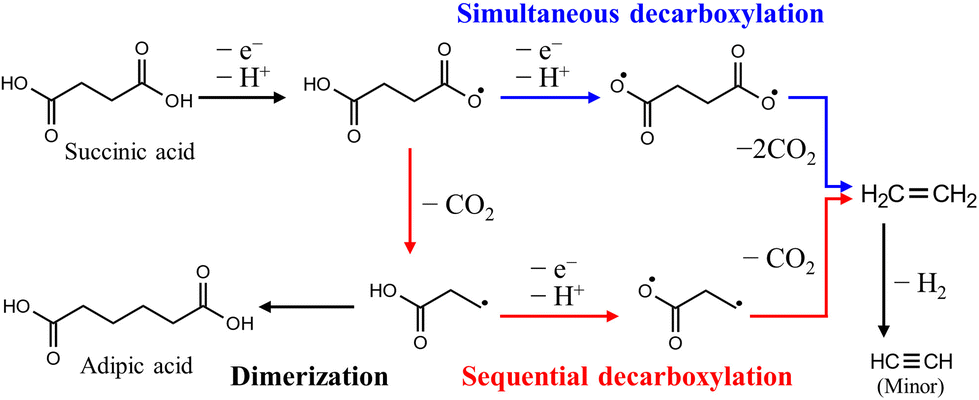 | ||
| Fig. 10 Anodic decarboxylation pathways of succinic acid produced by the depolymerization of PE. Reproduced with permission form ref. 27. Copyright 2021 ACS publications. | ||
Several studies on the decarboxylation of mono-carboxylic acids, namely, electrocatalytic decarboxylation (ECDX), have been performed to form high-value hydrocarbon molecules.78,81 It has been well-accepted that ECDX follows two types of reaction pathways: Kolbe and non-Kolbe electrolysis (Fig. 11a).38,82 The single electron oxidation and deprotonation processes form carboxyl-radical species, and further decarboxylation reaction results in the formation of alkyl-radical species. Kolbe electrolysis includes the dimerization of those alkyl radicals to form hydrocarbon products. On the other hand, non-Kolbe electrolysis includes another single electron oxidation process of alkyl radical species to form carbocation species. These carbocation species can form various products such as alkenes (by deprotonation), alcohol (by OH− addition), esters, etc.
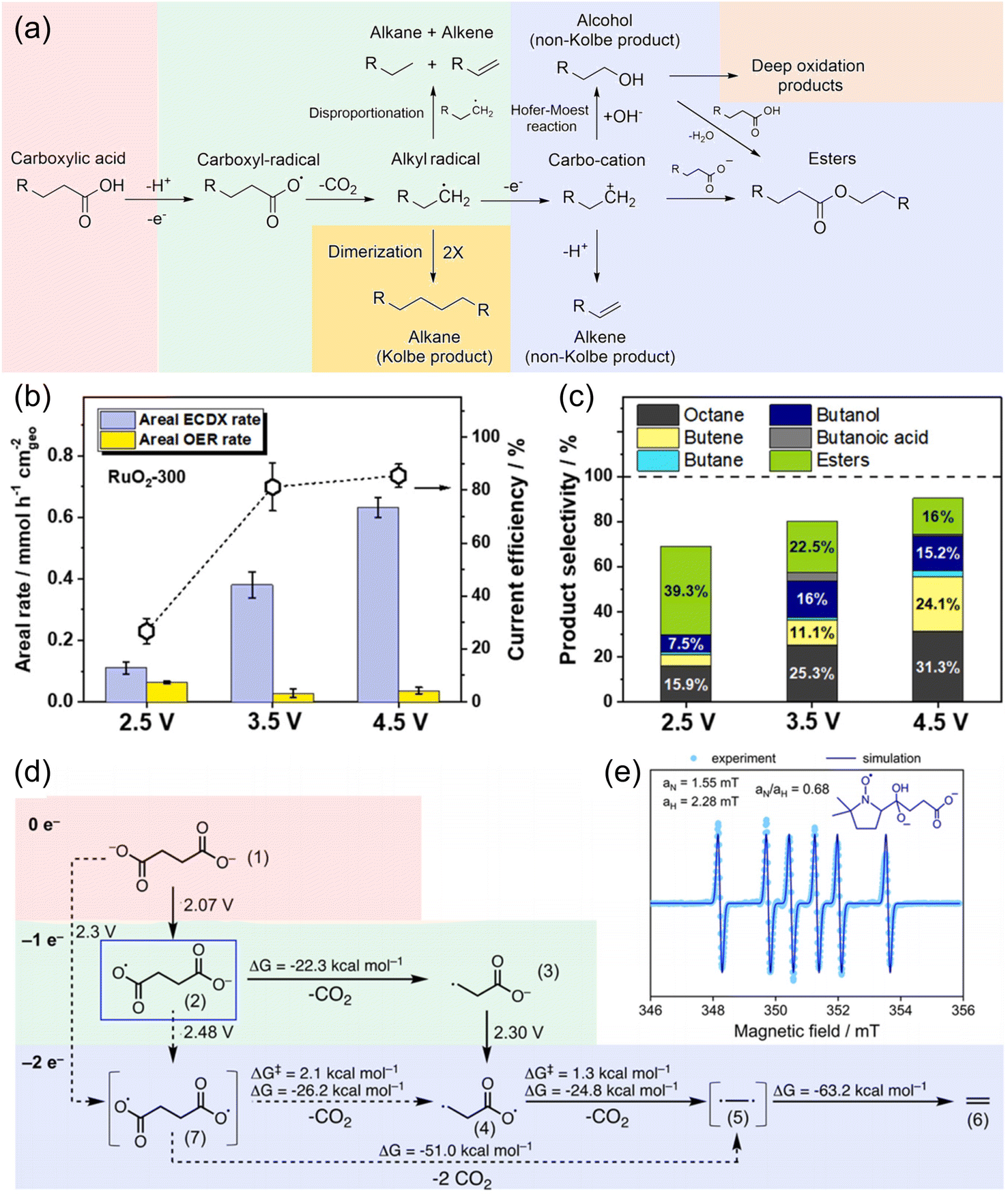 | ||
| Fig. 11 (a) Overview of the reaction mechanism for the formation of Kolbe and non-Kolbe products, esters, and deep oxidation products. (b) Areal ECDX and OER rates, and (c) product selectivity of valeric acid (VA) in the ECDX over a RuO2-300 electrode at constant potentials of 2.5, 3.5, and 4.5 V vs. RHE for 1 h. Reaction conditions: 70 mL of 0.5 M VA + 0.14 M Na2SO4 solution (pH 6) at about 0 °C. (d) Quantum chemical calculation of the reaction network. The potentials indicated are vs. RHE. (e) Spin trapping of 90 mM succinic acid in 150 mM NaOH (pH 10) with 50 mM 5,5-dimethyl-1-pyrroline N-oxide (DMPO). The adduct is trapped at 20 min reaction time (light blue dots) and the corresponding simulation (dark blue line) suggests the formation of the intermediate (2) in (d). Reprinted with permission from ref. 82 and 87. Copyright 2022 Elsevier and 2022 ACS publications. | ||
Most of the ECDX reactions were performed with noble metal-based electrocatalysts such as Pt, RuO2, and IrO2.38,81–85 For example, Xu et al. reported the selective ECDX of n-octanoic acid using Pt nanoparticles with different morphologies, such as nanospheres (NSs), nanoflowers (NFs), and nanothorns (NTs), on carbon fibre paper electrodes.83 The ECDX was conducted under alkaline conditions (0.5 M n-octanoic acid + 0.5 M KOH), and the Pt NT showed the highest yield, FE, and selectivity toward Kolbe (n-tetradecane) hydrocarbons over non-Kolbe (n-heptane and n-heptene) hydrocarbons. The DFT calculation results suggested that stronger adsorption of the ˙OH intermediate can stabilize surface free radicals, thereby inducing higher selectivity toward Kolbe products. Qiu et al. studied the ECDX of valeric acid (VA) using RuO2 nanoparticles in a 0.14 M Na2SO4 (pH 6) electrolyte.82 At a high anodic potential of 4.5 V vs. RHE (Fig. 11b and c), the selectivity of the Kolbe product (octane) increased over those of non-Kolbe products (butane, butene, butanol, etc.), compared to 2.5 V vs. RHE. Various products were separated according to their volatility and polarity. After the extraction of gas products (e.g., butane, butene, propylene, CO2, etc.), a trapping system with isopropyl alcohol was used to collect volatile products (e.g., octane, butanol, etc.). Then, dichloromethane was added to the electrolyte to extract non-volatile hydrocarbons and esters. Alcohols and residual carboxylic acids can be separated by distillation. Since majority of mono-carboxylic acids can be derived from biomass, investigations of the electrochemical reforming of related molecules by ECDX have been reported.86 Creusen et al. performed the ECDX of monoethyl succinic acid (MESA), which is one of the major products from biomass, using RuxTi1−xO2 catalysts on a Ti electrode.84 The RuxTi1−xO2 catalyst showed 74% and 58% selectivity for diethyl adipate (Kolbe product) and ethyl acrylate (non-Kolbe product), respectively.
Recently, Pichler et al. reported the ECDX of succinic acid from food waste to generate ethylene (C2H4) using a flow electrolyzer system (Fig. 11d and e).87 The use of a graphite anode resulted in the highest FE of ethylene (27.5%) at 2.8 V vs. RHE in 0.08 M succinic acid aqueous solution (pH 10). According to the DFT calculation and EPR spin-trapping experiment results, the ECDX of succinic acid undergoes sequential decarboxylation via monoalkyl-radical species; however, the formation of diradical species via direct 2e− oxidation cannot be neglected. Moreover, the flow electrolysis from microbe-digested food waste solution at 3 V and pH 6 for 2 h resulted in the production of 94.0 μmol of ethylene with 5.2% FE. However, specific investigations of the dicarboxylic acids (succinic acid and glutaric acid) from the depolymerized PE have hardly been reported. Therefore, the application of the above ECDX catalyst design strategies to dicarboxylic acid molecules and coupling them with PE depolymerization can provide a new PE upcycling technology using electrocatalysts. The overall performance of electrocatalysts for the ECDX for plastic upcycling is summarized in Table 3.
| Catalyst | Electrode | Electrolyte | E electrolysis (V vs. RHE) | Current density (mA cm−2) | Total conversion (%) | Major product | FE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| RuO2 | Ti foil | 0.14 M Na2SO4 + 0.2 M valeric acid | 4.5 | 50 | 92 | Octane | 24.5 | 38 |
| Butene | 17 | |||||||
| Butanol | 25.4 | |||||||
| Butanoic acid | 20.6 | |||||||
| IrO2 | Ti foil | 0.14 M Na2SO4 + 0.2 M valeric acid | 3.8 | 50 | 100 | Butanoic acid | 38.6 | |
| Pt-NT | CFP | 0.5 M KOH + 0.5 M n-octanoic acid | — | 250 | 34 | C7 product | ∼10 | 83 |
| C14 product | ∼24 | |||||||
| RuO2 | Pt | 0.14 M Na2SO4 + 0.5 M valeric acid | 4.5 | — | ∼80 | Octane | 31.3 | 82 |
| Butene | 24.1 | |||||||
| Butanol | 15.2 |
Currently, ECDX electrocatalyst design is still in a state of development. Because of the radical intermediates during electrocatalysis, diverse products (dimerized Kolbe product and non-Kolbe products) can be formed during the ECDX, and therefore research interest in ECDX had been focused on capturing the reaction intermediates. The selection of the catalyst material has been limited to noble metals such as Pt, IrOx, RuOx, etc. In addition, the reaction mechanism pathway by ECDX has considered the reaction intermediate molecules themselves rather than their adsorption and desorption behaviours on the surface of the electrocatalysts. Therefore, for the further development of ECDX catalyst design strategies, in addition to material synthesis and electrochemical characterization, thorough investigations should be conducted to fathom the exact ECDX mechanism considering the surface of the electrocatalyst.
3.3. Lactic acid (LA)
PLA is considered one of the most commercially promising bioplastics today. Compared to most other biodegradable plastics, PLA has better durability, transparency, and mechanical strength. Worldwide production of PLA in 2019 was 1.9 million tonnes and is expected to double every 3–4 years. However, a lot of energy is required to meet the conditions for PLA biodegradation (moisture content of 70%, temperature above 58 °C).88 As reported, PLA is capable of hydrolysis under acidic conditions, and the produced lactic acid can be used as a direct fuel instead of methanol.89 Therefore, the decomposition of PLA might become an energy-producing process with the development of relevant technologies. The schematic diagram of our proposed electrochemical upcycling of PLA is shown in Fig. 12. | ||
| Fig. 12 The schematic diagram of the electrochemical upcycling of PLA. Reproduced with permission from ref. 89. Copyright 2017 ACS publications. | ||
While complete electrooxidation of LA can result in higher electrical energy production, it also generates a significant amount of carbon dioxide. Therefore, a selective electro-oxidation technology is required to produce pyruvic acid (PA), a high-value-added product, while minimizing carbon dioxide emissions. LA, containing a carboxyl and a secondary alcohol group, converts the alcohol group into a ketone during electrooxidation to produce PA. Since the generated PA is susceptible to ˙OH radicals, during LA electrooxidation, the electrocatalyst must limit the formation of ˙OH radicals by water splitting.89,90 A recent study suggests the potential for obtaining PA through selective oxidation of LA using a specific calcined Ti foil electrocatalyst.91 Although the work presents the LA/PA coupled system of electrooxidation and reduction, it shows that LA can be selectively electro-oxidized to PA without undergoing additional oxidation and being reduced back to its original form. However, further research in the related fields is essential to establish a green hydrogen production system via the selective electrooxidation of LA, as the mechanism of the relevant reactions and the adsorption behaviour of LA are not yet clearly understood.
Some noble and non-noble metal-based electrocatalysts have been investigated for the electro-oxidation of LA. For example, electro-oxidation of LA on the Pt-based electrode results in the formation of PA, which is further oxidized to acetic acid (AA).90 Sedenho et al. compared the electrocatalytic activity for LA oxidation between Pt microparticle-modified (about 190 nm) BDD (Pt-BDD) and a polycrystalline Pt electrode.92 Compared to Pt-BDD, polycrystalline Pt showed 5-fold higher LA oxidation activity in 50 mM LA with a 0.5 M H2SO4 electrolyte. However, exact analyses of the product were not conducted. Chen et al. investigated IrOx-deposited antimony-doped tin oxide (ATO) for LA electrooxidation.89 IrOx showed selectivity toward CO2 (FE = 89%) and acetic acid (FE = 3.3%), but not PA, during electrolysis at 1.7 V vs. SHE for 760 min in the presence of 1% v/v LA in a 0.1 M KNO3 electrolyte. They proposed that the electrooxidation of LA on the IrOx surface underwent a β-hydride elimination mechanism, not a radical-assisted mechanism from PA by the H/D kinetic isotope effect (KIE) study.
Non-noble metal-based electrocatalysts have also been investigated, particularly Ni-based catalysts. Sedenho et al. prepared Ni nanoparticles and conducted electrochemical LA oxidation in an alkaline electrolyte (0.40 M LA + 1.0 M NaOH).93 Under alkaline conditions, Ni is oxidized to Ni(OH)2 species, and further electrooxidation results in the formation of NiOOH species. The performance of the LA oxidation was evaluated by comparing the ratio between anodic and cathodic peak current densities, which revealed that small nanoparticles (about 30 nm) showed higher activity than large Ni nanoparticles (about 90 nm) and bulk Ni electrodes. They suggested that the enhanced activity might have arisen from the high surface area and the thickness of surface (oxy)hydroxide species. Elakkiya and Maduraiveeran performed electro-oxidation of LA using 3-dimensional NiCo2O4 catalysts grown on a porous Ni foam electrode (3D-NiCo2O4/NF).94 The electrooxidation of LA was performed under alkaline conditions (1.0 M KOH) and showed a linear relationship between the current density and the LA concentration from 5.0 mM to 50.0 mM.
The overall performance of electrocatalysts for the EGOR for plastic upcycling is summarized in Table 4. However, the current status of the electrocatalyst design for LA conversion (particularly oxidation to PA or AA) is still in its infancy. Compared to the EGOR and ECDX, the overall reaction mechanism of LA electrooxidation is poorly understood. The selection of electrocatalyst materials has also been limited to the materials used in other electrooxidation reactions including noble metal (Pt and Ir) or transition metal (hydr)oxides (Ni and Co-based), rather than through rational design. Furthermore, the electrochemical activity of LA oxidation was evaluated only by the change in the anodic current density, rather than by product analyses. In addition to material synthesis and characterization, a thorough investigation to uncover the exact oxidation mechanism of LA has not been conducted to rationally design electrocatalysts for upcycling from anodic oxidation of LA. Therefore, the development of electrocatalysts for LA oxidation should be carried out by considering both aspects of catalytic activity and product selectivity.
| Catalyst | Electrode | Electrolyte | E onset | E electrolysis | Current density (mA cm−2) | Total conversion | Major product | FE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Pt | BDD | 0.5 M H2SO4 + 50 mM LA | ∼1.25 (V vs. SCE) | ∼1.35 (V vs. SCE) | ∼0.5 | — | — | — | 92 |
| Pt | 0.5 M H2SO4 + 50 mM LA | ∼1.2 | ∼1.38 (V vs. SCE) | ∼0.05 | — | — | — | ||
| IrOx | BDD | 0.1 M KNO3 + 1% LA | — | 1.7 (V vs. SHE) | — | 92.4 | CO2 | 89 | 89 |
| Acetic acid | 3.3 | ||||||||
| Ni | BDD | 1.0 M NaOH + 0.40 M LA | ∼0.4 (V vs. SCE) | — | — | — | — | — | 93 |
| NiCo2O4 | Ni foam | 1.0 M KOH + 25.0 mM LA | ∼0.35 (V vs. Ag/AgCl) | 0.54 (V vs. Ag/AgCl) | 15 | — | Pyruvic acid | — | 94 |
4. Conclusion and outlook
Given the wide range of plastic types used in our society, it is essential to develop plastic-specific methods for their post-use processing. Categorizing them broadly based on the types of polymer chain linkages, further subdividing them based on the molecular structure of the depolymerized units and developing tailored approaches for each plastic group would accelerate the development of plastic upcycling technology. Moreover, the direction of research and development should be determined based on the desired final products from waste plastic recycling, energy sources or value-added chemical compounds. In this regard, electrochemical recycling is proposed as a versatile technology that encompasses such applications.First and foremost, establishing a system to deliver charges to plastics is the most important task to clear. As bulk solid plastics cannot be in sufficient contact with electrodes, pre-treatment methods are primarily adopted to convert them into small molecules that can be dissolved in electrolytes through thermal and chemical depolymerization. In addition, the use of molecules or radicals capable of mediating redox reactions as homogeneous catalysts to facilitate the decomposition of solid plastics is also garnering attention.28,29,35
Next, the development of catalysts is needed to enhance the efficiency and selectivity of the electrochemical reproduction of depolymerized units. Specifically, for the main reaction of electrochemical oxidation of low MW organic compounds, methods such as designing metal alloy catalysts to modulate intermediate adsorption energy and halt the reaction at glycolate without breaking C–C bonds, or to accelerate C–C bond cleavage and formate release, thereby inhibiting the complete oxidation pathway to CO2, have been proposed.66–72
Overall, this review presents electrochemical upcycling for plastic waste management as a promising solution to address environmental challenges posed by plastic pollution. Table 5 provides a summary of the representative reaction systems corresponding to the most commonly used plastics. Electrochemical upcycling offers novel advantages, including operating under mild conditions, utilizing eco-friendly energy sources, and achieving higher energy efficiency. It also provides a pathway to generate value-added chemical products. To fully meet the societal demands, it is crucial to bring the entire process into the realm of electrochemical systems by utilizing homogeneous catalysts which can replace the costly and environmentally burdensome chemical depolymerization process. In addition, enhancing the selectivity and efficiency of the conversion process, which transforms depolymerized plastics into valuable materials such as formate, glycolate, or oxalate, remains a critical challenge. Therefore, we believe that understanding the underlying reaction mechanisms, optimizing electrochemical reaction systems, and developing catalysts with selective reaction pathways to realize the upcycling of plastics will pave the way for solving the environmental problems we are facing.
| Plastics | Polymer classes | Depolymerization | Electrochemical reaction pathways | Products | Challenges |
|---|---|---|---|---|---|
| PET | Polyester | Alkaline hydrolysis | EGOR | Formate (C1 product) or glycolate and oxalate (C2 product) | C–C cleavage control |
| PLA | Polyester | Alkaline (KOH) or acid (H2SO4) hydrolysis | Electrooxidation of LA | PA | Inhibition of complete oxidation to CO2 |
| PE | Polyolefin | Acid hydrolysis (HNO3) | ECDX | Hydrocarbon gases (alkane or alkene) | Reactive radical intermediate control |
| Homogeneous catalyst-mediated electrolysis | Short-chain carboxylic acids | Improvement of homogeneous catalysts | |||
| PS | Polyolefin | Hydroquinone, benzoic acid and short-chain carboxylic acids | |||
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Research Foundation of Korea (NRF) (Grant No. 2021R1A2C4001411, 2021R1I1A1A01052296, RS-2023-00256106, 2019R1A6A1A11044070 and 2022R1C1C2004703) and by the K-water Project Open Innovation R&D (OTSK_2022_036).References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- R. Bagri and P. T. Williams, J. Anal. Appl. Pyrolysis, 2002, 63, 29 CrossRef CAS.
- W. Kaminsky, M. Predel and A. Sadiki, Polym. Degrad. Stab., 2004, 85, 1045 CrossRef CAS.
- E. Worrell and M. A. Reuter, Handbook of Recycling: State-of-the-art for Practitioners, Analysts, and Scientists, Newnes, 2014, pp. 179–190 Search PubMed.
- K. Ragaert, L. Delva and K. Van Geem, Waste Manage., 2017, 69, 24 CrossRef CAS PubMed.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539 CrossRef CAS.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870 CrossRef CAS PubMed.
- T. Yoshioka, T. Sato and A. Okuwaki, J. Appl. Polym. Sci., 1994, 52, 1353 CrossRef CAS.
- Y. Wang, Y. Zhang, H. Song, Y. Wang, T. Deng and X. Hou, J. Cleaner Prod., 2019, 208, 1469 CrossRef CAS.
- S. Ügdüler, K. M. Van Geem, R. Denolf, M. Roosen, N. Mys, K. Ragaert and S. De Meester, Green Chem., 2020, 22, 5376 RSC.
- F. Liu, X. Gao, R. Shi, E. C. M. Tse and Y. Chen, Green Chem., 2022, 24, 6571 RSC.
- M. Du, Y. Zhang, S. Kang, C. Xu, Y. Ma, L. Cai, Y. Zhu, Y. Chai and B. Qiu, Small, 2023, 19, 2303693 CrossRef CAS PubMed.
- H. Yue, Y. Zhao, X. Ma and J. Gong, Chem. Soc. Rev., 2012, 41, 4218 RSC.
- R. Shi, K.-S. Liu, F. Liu, X. Yang, C.-C. Hou and Y. Chen, Chem. Commun., 2021, 57, 12595 RSC.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216 CrossRef CAS PubMed.
- X. Zhao, B. Boruah, K. F. Chin, M. Đokić, J. M. Modak and H. S. Soo, Adv. Mater., 2022, 34, 2100843 CrossRef CAS PubMed.
- N. L. Chauhan, V. Dameera, A. Chowdhury, V. A. Juvekar and A. Sarkar, Catal. Today, 2018, 309, 126 CrossRef CAS.
- A. Bard, R. Parsons and J. Jordan, Standard potentials in aqueous solution, CRC Press, 1985 Search PubMed.
- G. Lopez, M. Artetxe, M. Amutio, J. Alvarez, J. Bilbao and M. Olazar, Renewable Sustainable Energy Rev., 2018, 82, 576 CrossRef CAS.
- G. Lopez, M. Artetxe, M. Amutio, J. Bilbao and M. Olazar, Renewable Sustainable Energy Rev., 2017, 73, 346 CrossRef CAS.
- W. Partenheimer, Catal. Today, 2003, 81, 117 CrossRef CAS.
- A. Pifer and A. Sen, Angew. Chem., Int. Ed., 1998, 37, 3306 CrossRef CAS PubMed.
- E. Bäckström, K. Odelius and M. Hakkarainen, Ind. Eng. Chem. Res., 2017, 56, 14814 CrossRef.
- M. E. Cagiao, D. R. Rueda and F. J. Baltá Calleja, Colloid Polym. Sci., 1983, 261, 626 CrossRef CAS.
- E. Bäckström, K. Odelius and M. Hakkarainen, ACS Sustainable Chem. Eng., 2019, 7, 11004 CrossRef.
- C. M. Pichler, S. Bhattacharjee, M. Rahaman, T. Uekert and E. Reisner, ACS Catal., 2021, 11, 9159 CrossRef CAS PubMed.
- B. Yan, C. Shi, G. T. Beckham, E. Y. X. Chen and Y. Román-Leshkov, ChemSusChem, 2022, 15, e202102317 CrossRef CAS PubMed.
- F. Miao, Y. Liu, M. Gao, X. Yu, P. Xiao, M. Wang, S. Wang and X. Wang, J. Hazard. Mater., 2020, 399, 123023 CrossRef CAS PubMed.
- A. Kapałka, G. Fóti and C. Comninellis, Electrochim. Acta, 2009, 54, 2018 CrossRef.
- C. Trellu, N. Oturan, Y. Pechaud, E. D. van Hullebusch, G. Esposito and M. A. Oturan, Water Res., 2017, 118, 1 CrossRef CAS PubMed.
- D. Fabbri, A. B. Prevot and E. Pramauro, Appl. Catal., B, 2004, 49, 233 CrossRef CAS.
- H. M. Nguyen, C. M. Phan and T. Sen, Chem. Eng. J., 2016, 287, 633 CrossRef CAS.
- F. Escalona-Durán, D. Ribeiro da Silva, C. A. Martínez-Huitle and P. Villegas-Guzman, Chemosphere, 2020, 253, 126599 CrossRef PubMed.
- M. Kiendrebeogo, M. R. K. Estahbanati, Y. Ouarda, P. Drogui and R. D. Tyagi, Sci. Total Environ., 2022, 808, 151897 CrossRef CAS PubMed.
- L. Ciríaco, C. Anjo, J. Correia, M. J. Pacheco and A. Lopes, Electrochim. Acta, 2009, 54, 1464 CrossRef.
- I. Sirés, E. Brillas, G. Cerisola and M. Panizza, J. Electroanal. Chem., 2008, 613, 151 CrossRef.
- J. Lu, R. Hou, Y. Wang, L. Zhou and Y. Yuan, Water Res., 2022, 226, 119277 CrossRef CAS PubMed.
- D. E. Fagnani, D. Kim, S. I. Camarero, J. F. Alfaro and A. J. McNeil, Nat. Chem., 2023, 15, 222 CrossRef CAS PubMed.
- F. E. Durán, D. M. de Araújo, C. do Nascimento Brito, E. V. Santos, S. O. Ganiyu and C. A. Martínez-Huitle, J. Electroanal. Chem., 2018, 818, 216 CrossRef.
- A. Özcan, Y. Şahin, A. S. Koparal and M. A. Oturan, Water Res., 2008, 42, 2889 CrossRef PubMed.
- D. M. Brewis, D. Briggs, R. H. Dahm and I. Fletcher, Surf. Interface Anal., 2000, 29, 572 CrossRef CAS.
- R. F. Renneke, M. Pasquali and C. L. Hill, J. Am. Chem. Soc., 1990, 112, 6585 CrossRef CAS.
- R. S. Weber and K. K. Ramasamy, ACS Omega, 2020, 5, 27735 CrossRef CAS PubMed.
- N. E. Kamber, Y. Tsujii, K. Keets, R. M. Waymouth, R. C. Pratt, G. W. Nyce and J. L. Hedrick, J. Chem. Educ., 2010, 87, 519 CrossRef CAS.
- F. La Mantia, Handbook of plastics recycling, iSmithers Rapra Publishing, 2002 Search PubMed.
- J. M. García, Chem, 2016, 1, 813 Search PubMed.
- F. M. Lamberti, L. A. Román-Ramírez and J. Wood, J. Polym. Environ., 2020, 28, 2551 CrossRef CAS.
- V. Sinha, M. R. Patel and J. V. Patel, J. Polym. Environ., 2010, 18, 8 CrossRef CAS.
- A. B. Raheem, Z. Z. Noor, A. Hassan, M. K. Abd Hamid, S. A. Samsudin and A. H. Sabeen, J. Cleaner Prod., 2019, 225, 1052 CrossRef CAS.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed.
- S. Behera, S. Dinda, R. Saha and B. Mondal, ACS Catal., 2023, 13, 469 CrossRef CAS.
- Z. Pan, Y. Bi and L. An, Appl. Energy, 2019, 250, 846 CrossRef CAS.
- Z. Pan, Y. Bi and L. An, Appl. Energy, 2020, 258, 114060 CrossRef CAS.
- T. Raj kumar, G. Gnana kumar and A. Manthiram, Adv. Energy Mater., 2019, 9, 1803238 CrossRef.
- R. B. de Lima, V. Paganin, T. Iwasita and W. Vielstich, Electrochim. Acta, 2003, 49, 85 CrossRef CAS.
- L. Xin, Z. Zhang, J. Qi, D. Chadderdon and W. Li, Appl. Catal., B, 2012, 125, 85 CrossRef CAS.
- L. Demarconnay, S. Brimaud, C. Coutanceau and J. M. Léger, J. Electroanal. Chem., 2007, 601, 169 CrossRef CAS.
- P. A. Christensen and A. Hamnett, J. Electroanal. Chem., 1989, 260, 347 CrossRef CAS.
- M. Bełtowska-Brzezinska, T. Łuczak and R. Holze, J. Appl. Electrochem., 1997, 27, 999 CrossRef.
- S. C. Chang, Y. Ho and M. J. Weaver, J. Am. Chem. Soc., 1991, 113, 9506 CrossRef CAS.
- D. Bayer, S. Berenger, M. Joos, C. Cremers and J. Tübke, Int. J. Hydrogen Energy, 2010, 35, 12660 CrossRef CAS.
- H. Wang, Z. Jusys and R. J. Behm, Electrochim. Acta, 2009, 54, 6484 CrossRef CAS.
- Y. Yan, H. Zhou, S.-M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, J. Am. Chem. Soc., 2023, 145, 6144 CrossRef CAS PubMed.
- J. Qi, Z. An, C. Li, X. Chen, W. Li and C. Liang, Curr. Opin. Electrochem., 2022, 32, 100929 CrossRef CAS.
- A. Marchionni, M. Bevilacqua, C. Bianchini, Y.-X. Chen, J. Filippi, P. Fornasiero, A. Lavacchi, H. Miller, L. Wang and F. Vizza, ChemSusChem, 2013, 6, 518 CrossRef CAS PubMed.
- D. Si, B. Xiong, L. Chen and J. Shi, Chem Catal., 2021, 1, 941 CrossRef CAS.
- F. Liu, X. Gao, R. Shi, Z. Guo, E. Tse and Y. Chen, Angew. Chem., 2023, 135, e202300094 CrossRef.
- N. Ozawa, S. Chieda, Y. Higuchi, T. Takeguchi, M. Yamauchi and M. Kubo, J. Catal., 2018, 361, 361 CrossRef CAS.
- T. Matsumoto, M. Sadakiyo, M. L. Ooi, S. Kitano, T. Yamamoto, S. Matsumura, K. Kato, T. Takeguchi and M. Yamauchi, Sci. Rep., 2014, 4, 5620 CrossRef CAS PubMed.
- C.-Y. Lin, S.-C. Huang, Y.-G. Lin, L.-C. Hsu and C.-T. Yi, Appl. Catal., B, 2021, 296, 120351 CrossRef CAS.
- F. Ma, S. Wang, X. Gong, X. Liu, Z. Wang, P. Wang, Y. Liu, H. Cheng, Y. Dai, Z. Zheng and B. Huang, Appl. Catal., B, 2022, 307, 121198 CrossRef CAS.
- K. A. Schwarz, R. Sundararaman, T. P. Moffat and T. C. Allison, Phys. Chem. Chem. Phys., 2015, 17, 20805 RSC.
- X.-H. Wang, Z.-N. Zhang, Z. Wang, Y. Ding, Q.-G. Zhai, Y.-C. Jiang, S.-N. Li and Y. Chen, Chem. Eng. J., 2023, 465, 142938 CrossRef CAS.
- J. Li, L. Li, X. Ma, X. Han, C. Xing, X. Qi, R. He, J. Arbiol, H. Pan, J. Zhao, J. Deng, Y. Zhang, Y. Yang and A. Cabot, Adv. Sci., 2023, 10, 2300841 CrossRef CAS PubMed.
- N. Wang, X. Li, M.-K. Hu, W. Wei, S.-H. Zhou, X.-T. Wu and Q.-L. Zhu, Appl. Catal., B, 2022, 316, 121667 CrossRef CAS.
- L. Jiao, W. Wei, X. Li, C.-B. Hong, S.-G. Han, M. I. Khan and Q.-L. Zhu, Rare Met., 2022, 41, 3654 CrossRef CAS.
- X. Liu, Z. Fang, D. Xiong, S. Gong, Y. Niu, W. Chen and Z. Chen, Nano Res., 2022, 16, 4625 CrossRef.
- B. K. Sharma, B. R. Moser, K. E. Vermillion, K. M. Doll and N. Rajagopalan, Fuel Process. Technol., 2014, 122, 79 CrossRef CAS.
- X. Zhang, L. Chen, L. Yuan, R. Liu, D. Li, X. Liu and G. Ge, Langmuir, 2019, 35, 5770 CrossRef CAS PubMed.
- F. J. Holzhäuser, J. B. Mensah and R. Palkovits, Green Chem., 2020, 22, 286 RSC.
- Y. Qiu, J. A. Lopez-Ruiz, G. Zhu, M. H. Engelhard, O. Y. Gutiérrez and J. D. Holladay, Appl. Catal., B, 2022, 305, 121060 CrossRef CAS.
- S. Xu, X. Niu, G. Yuan, Z. Wang, S. Zhu, X. Li, Y. Han, R. Zhao and Q. Wang, ACS Sustainable Chem. Eng., 2021, 9, 5288 CrossRef CAS.
- G. Creusen, F. J. Holzhäuser, J. Artz, S. Palkovits and R. Palkovits, ACS Sustainable Chem. Eng., 2018, 6, 17108 CrossRef CAS.
- S. Wang, D. Ren, Y. Du, M. Zhang, N. Zhang, Y. Sun and Z. Huo, Carbon Resour. Convers., 2023, 6, 287 CrossRef CAS.
- F. J. Holzhäuser, G. Creusen, G. Moos, M. Dahmen, A. König, J. Artz, S. Palkovits and R. Palkovits, Green Chem., 2019, 21, 2334 RSC.
- C. M. Pichler, S. Bhattacharjee, E. Lam, L. Su, A. Collauto, M. M. Roessler, S. J. Cobb, V. M. Badiani, M. Rahaman and E. Reisner, ACS Catal., 2022, 12, 13360 CrossRef CAS PubMed.
- K. J. Jem and B. Tan, Adv. Ind. Eng. Polym. Res., 2020, 3, 60 Search PubMed.
- C. Chen, A. J. Bloomfield and S. W. Sheehan, Ind. Eng. Chem. Res., 2017, 56, 3560 CrossRef CAS.
- G. Horányi, J. Electroanal. Chem., 1981, 117, 131 CrossRef.
- H. Eguchi, T. Kobayashi, T. Yamada, D. S. R. Rocabado, T. Ishimoto and M. Yamauchi, Sci. Rep., 2021, 11, 13929 CrossRef CAS PubMed.
- G. C. Sedenho, P. T. Lee, H. S. Toh, C. Salter, C. Johnston, N. R. Stradiotto and R. G. Compton, Int. J. Electrochem. Sci., 2016, 11, 2166 CrossRef CAS.
- G. C. Sedenho, P. T. Lee, H. S. Toh, C. Salter, C. Johnston, N. R. Stradiotto and R. G. Compton, J. Phys. Chem. C, 2015, 119, 6896 CrossRef CAS.
- R. Elakkiya and G. Maduraiveeran, New J. Chem., 2019, 43, 14756 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |