
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescent carbon dots from birch leaves for sustainable electroluminescent devices†
Shi
Tang
 a,
Yongfeng
Liu
ab,
Henry
Opoku
a,
Märta
Gregorsson
a,
Peijuan
Zhang
c,
Etienne
Auroux
a,
Dongfeng
Dang
c,
Anja-Verena
Mudring
d,
Thomas
Wågberg
a,
Ludvig
Edman
*ae and
Jia
Wang
*a
a,
Yongfeng
Liu
ab,
Henry
Opoku
a,
Märta
Gregorsson
a,
Peijuan
Zhang
c,
Etienne
Auroux
a,
Dongfeng
Dang
c,
Anja-Verena
Mudring
d,
Thomas
Wågberg
a,
Ludvig
Edman
*ae and
Jia
Wang
*a
aDepartment of Physics, Umeå University, SE-90187 Umeå, Sweden. E-mail: ludvig.edman@umu.se; jia.wang@umu.se
bCollege of Physical Science and Technology, Yangzhou University, Yangzhou 225002, P. R. China
cSchool of Chemistry, Xi'an Key Laboratory of Sustainable Energy Material Chemistry, Xi'an Jiao Tong University, Xi'an 710049, China
dIntelligent Advanced Materials (iAM), Department of Biological and Chemical Engineering and iNANO, Aarhus University, 8000 Aarhus C, Denmark
eWallenberg Initiative Materials Science for Sustainability, Department of Physics, Umeå University, SE-90187 Umeå, Sweden
First published on 1st November 2023
Abstract
The shift from depleting petroleum compounds to regenerating biomass as the raw material for organic semiconductors is a prerequisite if organic electronics is to become truly sustainable. Here, we report on a one-pot solvothermal synthesis of a biomass-based carbon dot (bio-CD) fluorescent semiconductor, using birch leaves as the sole raw material. These bio-CDs are highly soluble in ethanol (45 g L−1), and deliver deep-red and narrowband emission (peak wavelength = 675 nm, full width at half maximum, FWHM = 28 nm) at a high photoluminescence quantum yield of 26% in ethanol solution. Systematic structural characterization shows that molecular pheophytin a is the single fluorophore, and that this fluorophore is localized in the bulk of the bio-CD away from its polar surface. The functionality of the birch-leaf-derived bio-CDs in sustainable organic electronics is demonstrated by its employment as the printable emitter in a light-emitting electrochemical cell, which delivers narrowband deep-red luminance of 110 cd m−2, with a FWHM of 29 nm, at an external quantum efficiency of 0.29%. This study thus reveals a promising avenue for the functional benign synthesis and the practical solution-based implementation of narrowband bio-CDs in sustainable optoelectronic technologies.
Introduction
The organic light-emitting diode (OLED) is currently commercialized in advanced display applications, and it is also projected to enable high-volume transient applications in, e.g., wearable electronics,1,2 smart packaging,3 and disposable electronics.4 From a sustainability perspective, it is important that the OLED can be highly energy efficient during its light-emission operation,5 but a looming concern, in particular for the emerging transient high-volume applications, is related to the fact that its fabrication is energy intense and depends on the use of environmentally problematic materials.6 In addition, the recycling of OLEDs is difficult, and it is therefore anticipated to result in the accumulation of large amounts of electronic waste.7The heart of the OLED is the emissive organic semiconductor (OSC), but the unfortunate fact is that OSCs today almost exclusively are synthesized from non-sustainable petrochemical compounds, and in addition often comprise critical raw materials (CRMs). Thus, there is high motivation to develop non-petroleum based and CRM-free emissive OSCs.8–10 In this context, fluorescent “carbon dots” (CDs), or carbon nanoparticles, are an interesting class of emissive OSCs, which were first reported by Sun et al. in 2006.11 Since then, a variety of different CDs based on petroleum compounds12,13 or biomass14–38 as the starting material have been reported for application in bioimaging,38 sensing,39 and OLED devices.13,40–44 The biomass-derived CDs (bio-CDs) are obviously preferred from a sustainability viewpoint because of the renewable and abundant availability of their starting material.14–38
However, for emissive applications, it is often a drawback that bio-CDs commonly exhibit very broad and excitation-wavelength-dependent emission spectra, with the former being manifested in large values for the full width at half maximum (FWHM) of the emission spectrum of 100 nm or more.14 For example, Tachibana and co-workers reported on the synthesis of fenugreek-seed-derived bio-CDs, which exhibited blue-green emission with a broad FWHM of 159 nm when implemented in an OLED,45 and Sonar et al. synthesized human-hair-derived bio-CDs that delivered emission with a FWHM of 100 nm in an OLED.46 It has been demonstrated that the large FWHM and the excitation-wavelength-dependent emission spectrum often originate in the existence of a number of different emissive species or fluorophores with different optical properties within the same bio-CD.47,48 However, it is notable that this issue was recently successfully addressed by Chen and co-workers when they synthesized clover-derived bio-CDs, which delivered emission with a narrow FWHM of 25 nm in an OLED device.49
The light-emitting electrochemical cell (LEC) is similar in appearance to the OLED,50 but its unique electrochemical-doping operation enables for energy- and material-efficient fabrication of the entire device stack by printing and coating under ambient air.51–55 This promises to pave the way for a sustainable LEC fabrication, provided that the constituent materials and the process solvents51 are sustainable. A few studies on the utilization of bioderived electrolytes56,57 and substrates53,54 in LECs can be found in the scientific literature, but the corresponding reports on the investigation and implementation of biobased emissive OSCs in LECs are lacking.
Here, we report on the synthesis of emissive bio-CDs with a one-pot solvothermal method using birch leaves as the sole starting material, acetone as the extraction solvent, and a modest reaction temperature of 120 °C. The synthesized bio-CDs are highly soluble (45 g L−1) in benign ethanol and deliver wavelength-excitation-independent and narrowband deep-red light emission (FWHM = 28 nm, peak wavelength = 675 nm) with a high photoluminescence quantum yield (PLQY) of 26% in ethanol solution. We rationalize our findings by demonstrating that molecular pheophytin a is the single fluorophore, which is localized in the bulk of the bio-CD. We finally demonstrate the practical applicability of the bio-CDs by implementing them as the solution-processable emitter in an LEC device, which delivers narrowband deep-red luminance, with a FWHM of 29 nm.
Results and discussion
Fig. 1(a–d) present the key steps in the preparation and synthesis of the bio-CDs, while Fig. 1(e–h) display the derived associated morphological and chemical structures. In brief, the sole starting material for the synthesis is freshly picked birch leaves (Fig. 1a and e), which comprise a rich variety of biomolecules, such as chlorophyll a, flavonoids, glycosides, phenol carboxylic acids, fatty acids, essential oils, minerals and vitamins.58–60 The molecular structure of chlorophyll a is shown in the lower right-hand part of Fig. 1(e), while a number of the other biomolecular structures in birch leaves are displayed in Fig. S1.† The birch leaves were first heated at 100 °C for 4 h under ambient air in order to remove moisture (Fig. 1b and f). The dried birch leaves were mortared into a powder, and thereafter immersed into acetone (Fig. 1c and g). The extracted acetone-soluble part61 was transferred into an autoclave reactor for the final solvothermal carbonization reaction at 120 °C for 5 h (Fig. 1d and h). The extraction solvent, the pretreatment and solvothermal parameters were selected and optimized in order to attain a desired combination of high PLQY (see Fig. S2† for example), a narrow emission spectrum, and high solubility in a benign solvent (here ethanol). More details on our preferred bio-CD synthesis procedure can be found in the Experimental section.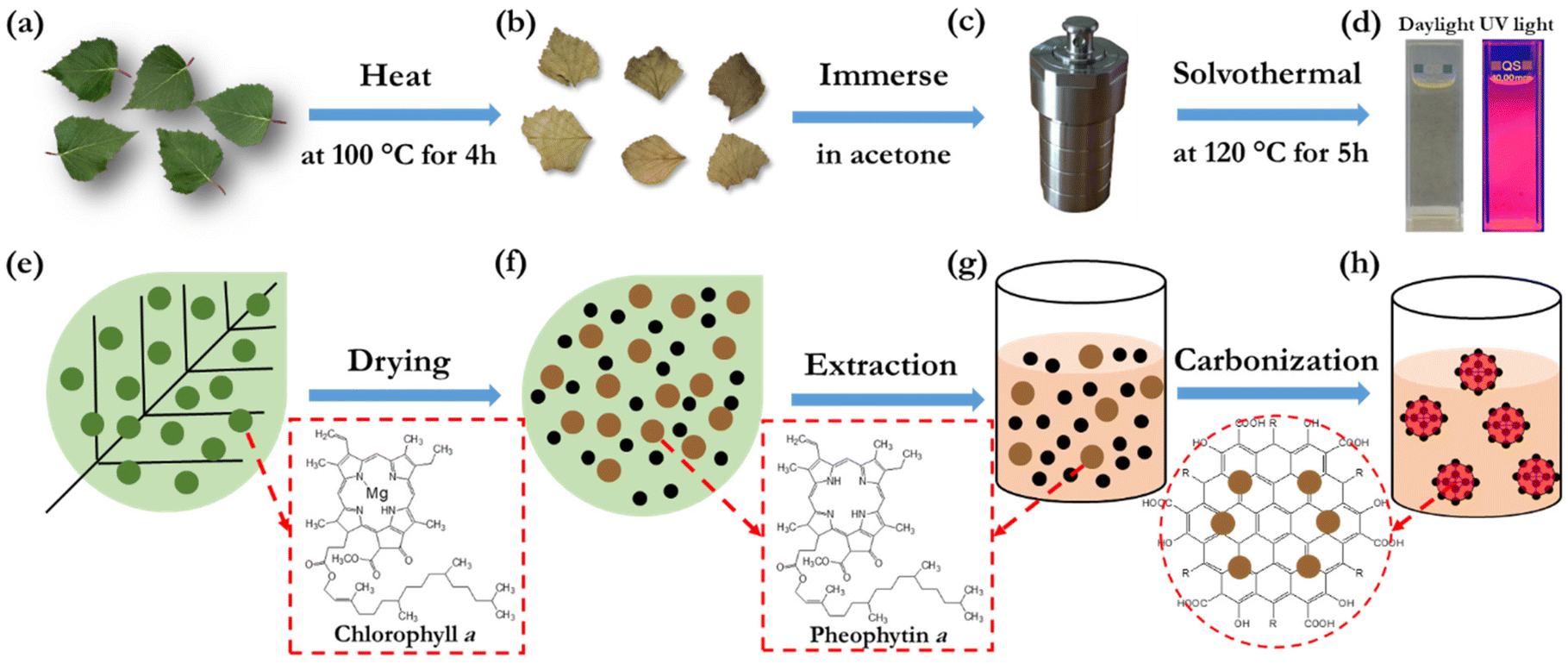 | ||
| Fig. 1 (a–d) Schematic illustration of the two preparatory heating and immersion steps, and the subsequent solvothermal synthesis in an autoclave, for the realization of the bio-CDs using birch leaves as the sole raw material. (e–h) The corresponding evolution of the morphological and chemical structures. | ||
The overall product yield of the collected bio-CDs with respect to the original dried birch-leaf raw material was ∼5%, but this low yield is not a major concern considering the natural abundance and low environmental and monetary cost of the starting material. Moreover, the collected organic rest material has value in that it, e.g., can be utilized for heat recovery by incineration without generation of net carbon-dioxide emission.
The solubility of the synthesized bio-CDs in different solvents is important if they are to be practically utilized in printed electronics or employed in a liquid environment. We have therefore systematically investigated the solubility of the birch-leaf derived bio-CDs, and we find that they can be dissolved in a concentration of 16 g L−1 in acetone, more than 4 g L−1 in methanol, dimethyl sulfoxide, toluene and anisole, and an impressive 45 g L−1 in ethanol. The photographs in Fig. 1(d) show that the bio-CD-in-ethanol solution, with a solute concentration of 1.6 mg L−1, exhibits a pale-yellow appearance under daylight illumination, and that it emits with vibrant deep-red colour during exposure to UV light (λpeak = 365 nm).
Fig. 2(a) is an atomic force microscopy (AFM) image of a number of discrete and “white” bio-CDs on a Si substrate, which were deposited by spin-coating a dilute (0.1 g L−1) ethanol solution. The inset is a cross-sectional height profile along the white line in the AFM image, and it indicates the height profile of four bio-CDs. We find that the height of the bio-CD in the AFM measurement varied between 1.8 and 3.1 nm. Fig. 2(b) is a transmission electron microscopy (TEM) image of more than 50 discrete “black” bio-CDs on an ultrathin carbon-coated copper grid, which were deposited by drop-casting the dilute (0.1 g L−1) ethanol solution. The upper right inset is a representative high-resolution TEM image of one single bio-CD, and it shows that the bio-CDs feature a round shape and an ordered interior structure, with a lattice spacing of 0.20 nm. We note that this lattice spacing is similar to the characteristic d100 interplanar spacing of single-crystal graphite, which is 0.21 nm.62 The lower right inset in Fig. 2(b) is a histogram of the circular diameter derived from a collection of 50 bio-CDs, and their average diameter is determined to be 2.0 ± 0.3 nm. The combined conclusions from the TEM and AFM measurements are thus that the bio-CDs exhibit an average diameter of 2 nm, and that their interior features graphene-like order,63,64 as schematically indicated in the lower-left hand part of Fig. 1(h).
 | ||
| Fig. 2 (a) AFM image of well-separated discrete bio-CDs on a Si substrate. The white line corresponds to the height profile in the inset. (b) TEM image of more than 50 discrete bio-CDs on a carbon coated copper grid. Insets: (upper right) High-resolution TEM image indicating (by the dotted red circle) a circular-shaped bio-CD, with a diameter of 1.9 nm, and an ordered lattice structure with an interplanar spacing of 0.20 nm; (lower right) histogram of the bio-CD diameter distribution. The bio-CDs were deposited by spin-coating (a) or drop-coating (b) a 0.1 g L−1 ethanol solution. (c) The normalized PL spectrum and (d) the normalized absorption spectrum of a dilute ethanol solution comprising the bio-CDs (solid black squares), pheophytin a (open blue circles), and chlorophyll a (open red triangles). The solute concentration was 1.6 mg L−1 for all three solutions. | ||
Fig. 2(c) and (d) present the normalized PL and absorption spectra, respectively, of dilute (1.6 mg L−1) ethanol solutions of the bio-CDs, pheophytin a, and chlorophyll a, as identified in the insets. Chlorophyll a is a common chromophore in leaves and plants that contributes with their characteristic green colour, and its molecular structure is displayed in Fig. 1(e). Pheophytin a is chemically very similar to chlorophyll a, but notably lacking its central Mg atom, as shown in Fig. 1(f).
Importantly, the bio-CD solution (solid black squares) exhibits narrowband, deep-red emission, with a FWHM of 28 nm and a high PLQY of 26%, with the major PL peak located at 675 nm and a shoulder peak positioned at 720 nm. The PL spectrum of pheophytin a (open blue circles) is essentially identical, while that of chlorophyll a (open red triangles) is slightly distinguished by the weaker intensity of its shoulder peak. This difference is however larger in absorption. Fig. 2(d) shows that the bio-CD and the pheophytin a solutions feature essentially identical absorption spectra, comprising two strong absorption bands centered at 414 nm and 665 nm, while the absorption spectrum of chlorophyll a (open red triangles) is markedly different. The higher-energy absorption band is identified as the S0 → S2 Soret band, while the lower-energy band is the S0 → S1 Qy band; both bands are characteristic absorption bands of porphyrin a compounds, such as pheophytin a and chlorophyll a.65 A more detailed comparison of the chlorophyll a absorption compared to that of the bio-CDs and pheophytin a shows that its higher-energy Soret band is red-shifted by 20 nm, while its lower-energy Qy band is blue-shifted by 10 nm and also relatively more intense.
The striking similarity in both the emission and absorption spectral shape of the bio-CD solution and the pheophytin a solution suggests that the optically active species in the bio-CDs is pheophytin a, which is co-existing with the optically silent “graphene-like” constituent identified in the TEM. This conclusion is in agreement with that it has been reported that the birch-leaf abundant chlorophyll a transforms into pheophytin a during heating through the replacement of its central Mg atom with two H atoms, and that this transformation is visually manifested in a color change from dark green to brown.66,67 This color transformation is indeed observed during the first heating step in the bio-CD synthesis, as shown by the photographs in Fig. 1(a and b).
The elemental composition and the chemical structure of the bio-CDs were further investigated with X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared (FTIR) spectroscopy. Fig. 3(a) is a broad-range XPS survey scan of a thick bio-CD film, which identifies that the major elements (with the exception of hydrogen that is silent in XPS) in the bio-CD and their relative concentrations are: C (87.4%), O (12.3%), and N (0.3%). We call attention to the absence of a Mg peak at 50 eV, which confirms our earlier conclusion that the chlorophyll a in the birch-leaf starting material has been transformed into pheophytin a by the heating-induced removal of Mg. We also note with interest that the low N concentration in the bio-CDs compared to that of neat pheophytin a (7.03%) implies that the pheophytin a constituent is a minority component in the bio-CD.
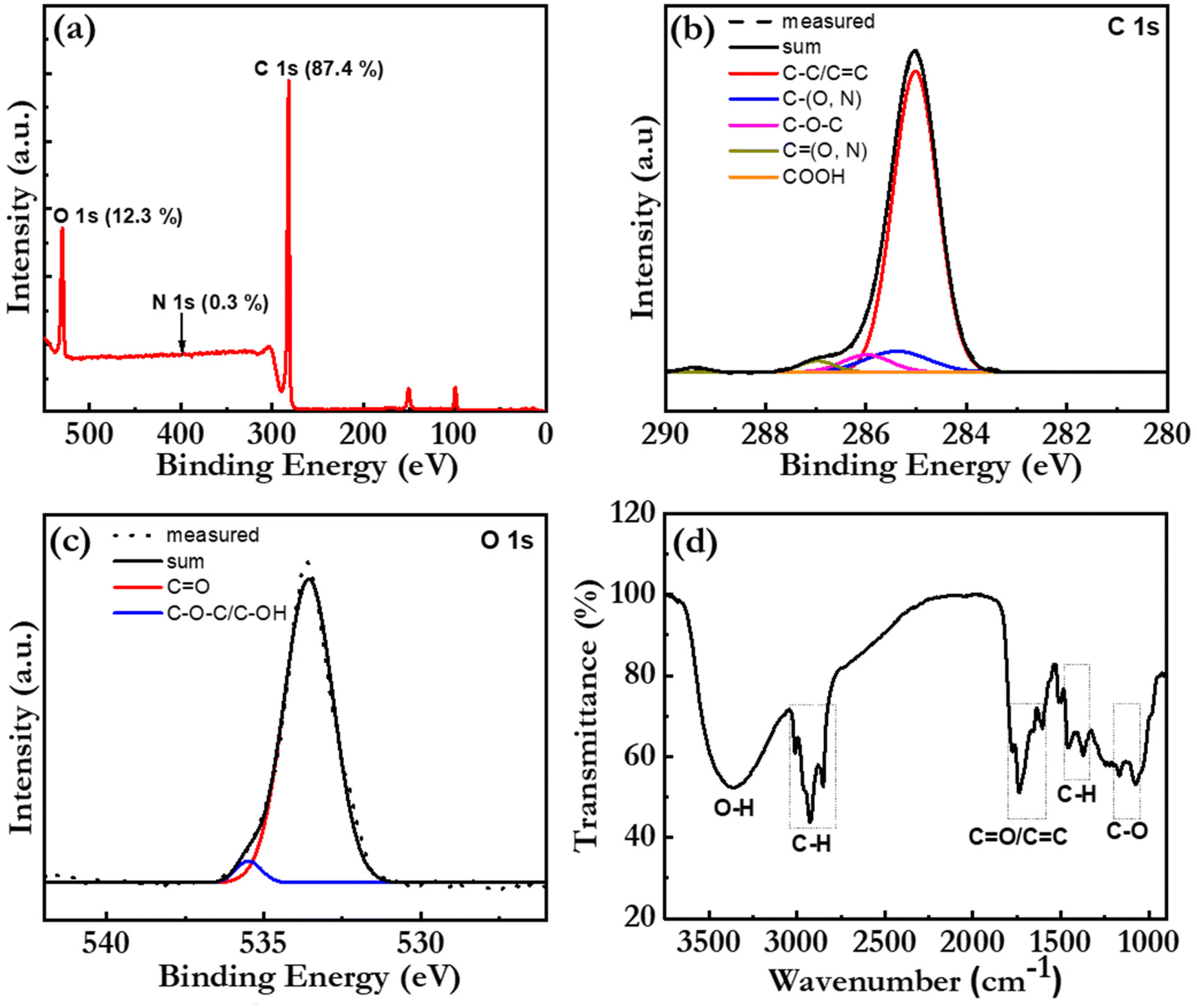 | ||
| Fig. 3 (a) The XPS survey spectrum of a 200 μm thick bio-CD film on a Si substrate. (b and c) The measured high-resolution XPS spectrum (dotted grey line) and the fitted XPS spectra in (b) the C 1s region and (c) the O 1s region. The chemical assignments and the sum of the fit spectra are identified in the insets. (d) The FTIR spectrum of a dry bio-CD powder, with the key vibrational peaks identified by the dashed black rectangles. | ||
Fig. 3(b) presents the high-resolution XPS spectrum in the C 1s region, which provides information on the chemical bonding of the carbon atoms. The successful “sum” fitting (solid black line) of the measured XPS spectrum (dotted grey line) shows that the carbon atoms are primarily involved in C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds (at 285.0 eV), but that a smaller fraction of the carbon atoms are parts of C–O/C–N bonds (285.4 eV), C–O–C bonds (286.0 eV), CO/CN bonds (287.0 eV) and COOH bonds (289.2 eV). These results are consistent with the high-resolution XPS spectrum in the O 1s region in Fig. 3(c), where the successful sum fitting reveals that a majority of the oxygen atoms are parts of CO bonds (533.5 eV), while a smaller fraction is located in C–O bonds (535.4 eV).
C bonds (at 285.0 eV), but that a smaller fraction of the carbon atoms are parts of C–O/C–N bonds (285.4 eV), C–O–C bonds (286.0 eV), CO/CN bonds (287.0 eV) and COOH bonds (289.2 eV). These results are consistent with the high-resolution XPS spectrum in the O 1s region in Fig. 3(c), where the successful sum fitting reveals that a majority of the oxygen atoms are parts of CO bonds (533.5 eV), while a smaller fraction is located in C–O bonds (535.4 eV).
Fig. 3(d) presents the FTIR spectrum of a bio-CD powder and the corresponding vibrational assignments. The identified IR-active CO and CC (peaks at ∼1730 and ∼1620 cm−1),38 C–O–C (peak at 1100 cm−1),23 C–H (peaks at 2925 and 2865 cm−1), and C–H (peaks at 1455 and 1373 cm−1)38 structures can all be found in the chemical structure of pheophytin a (Fig. 1f) and graphene. More interesting is then the observation of a strong and broad IR-active vibrational band at 3550–3100 cm−1, which is assigned to phenolic and aliphatic –OH groups,46 since these chemical groups are absent in both pheophytin a and graphene. We note however with interest that these chemical groups are common in a number of the other biomolecules that are common in the birch-leaf starting material (see Fig. S1†).68,69 Moreover, since the bio-CDs are found to be highly soluble in polar solvents, such as ethanol, we propose that these polar hydroxyl/phenolic groups are common at the bio-CD surface, whereas the more hydrophobic pheophytin a and graphene-like segments are localized in the bulk of the bio-CD. With all of the above information considered, Fig. 1(h), lower left part, presents our derived view of the bio-CD chemical structure, which essentially comprises a number of discrete pheophytin a molecules dispersed in a graphene-like bulk matrix, which is surrounded by a surface shell comprising polar hydroxyl/phenolic groups.
Fig. 4(a) presents the PL spectrum, and the corresponding PLQY (in the inset), of a bio-CD-in-ethanol solution, recorded at a wide range of excitation wavelengths. Both the PL spectral shape and PLQY are found to be independent on the excitation-wavelength, which provides support for the notion that pheophytin a is the single fluorophores in the bio-CD. We note that this observation is in sharp contrast to the strongly wavelength-excitation-dependent PL of earlier reported bio-CDs, which therefore presumably comprise several different fluorophores.27,31–38Fig. 4(b) presents the measured PL transient of the bio-CD-in-ethanol solution, following optical excitation that ends at time = 0 ns. The measured PL transient is with high accuracy fitted with a mono-exponential function (and a constant background), and the derived emissive lifetime is 6.6 ns. The mono-exponential emission is yet again in agreement with the existence of a single well-defined (i.e., pheophytin a) emitter, while the ns-short emissive lifetime shows that the bio-CDs emit by the process of fluorescence.
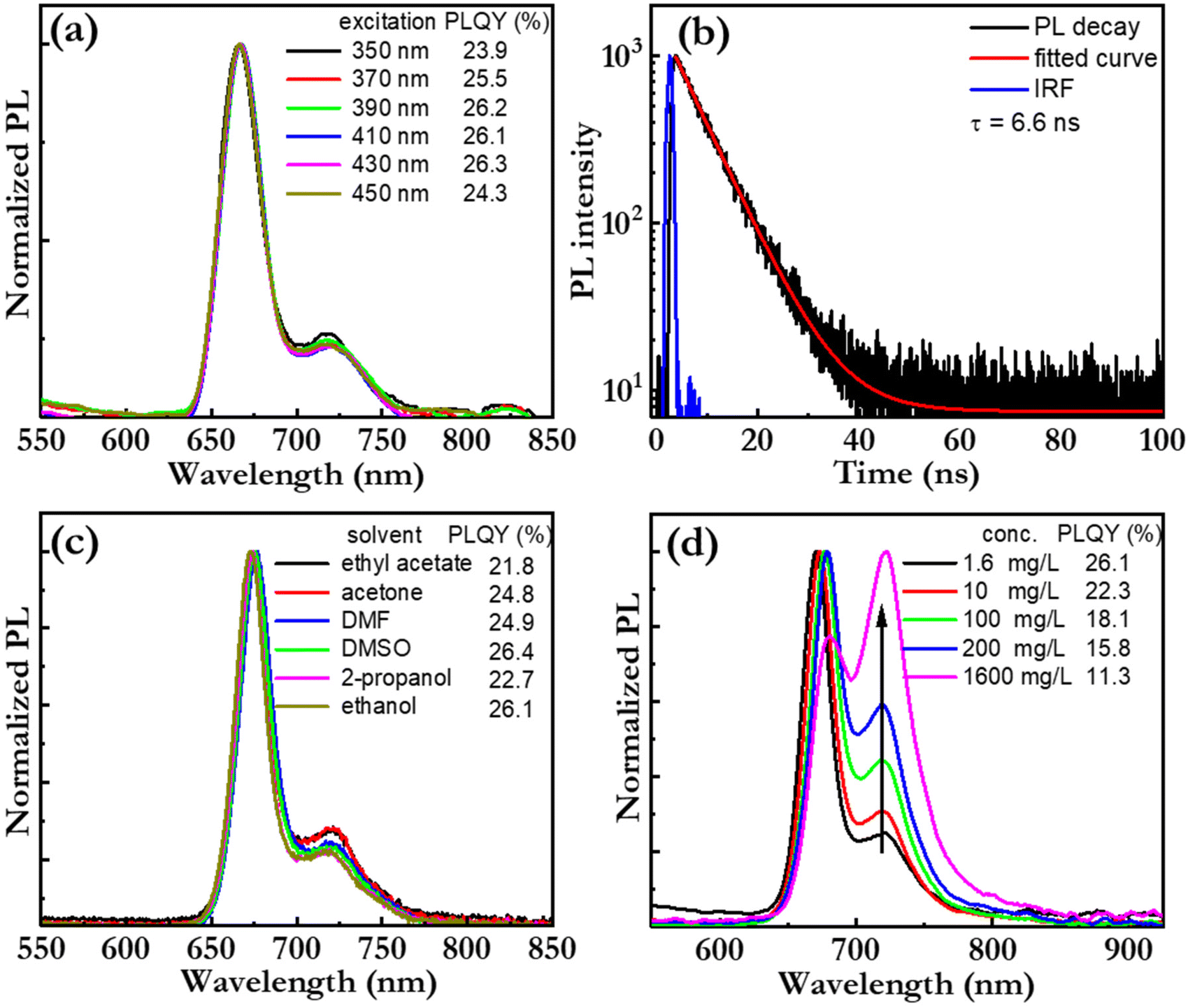 | ||
| Fig. 4 (a) The normalized PL spectra of the bio-CD-in-ethanol solution (solute concentration = 1.6 mg L−1) as a function of excitation wavelength, as specified in the inset. The inset also presents the measured values for the PLQY. (b) The measured and fitted PL transient of the bio-CD-in-ethanol solution, and the instrument response function (ÌRF). The derived emissive lifetime is presented in the inset. The measurement was performed at room temperature, and the excitation wavelength of the pulsed laser was 375 nm. (c) The normalized PL spectrum and the PLQY of a bio-CD solution as a function of the solvent, as specified in the inset. The solute concentration was 1.6 mg L−1, and the excitation wavelength was 410 nm. (d) The normalized PL spectra of a bio-CD-in-ethanol solution at different bio-CD concentration, as identified in the inset. The corresponding PLQY values are also presented in the inset, while the arrow in the graph indicates increasing bio-CD concentration. | ||
Fig. 4(c) presents the PL spectrum, and the corresponding PLQY (in the inset), of the bio-CDs in a wide variety of solvents, ranging from highly polar ethanol (dielectric constant = 24.3) to relatively non-polar ethyl acetate (dielectric constant = 6.0). The bio-CD concentration was kept constant at 1.6 mg L−1. Both chlorophyll a and pheophytin a exhibit strongly solvent-dependent photoluminescence, owing to their propensity for polarity-dependent intermolecular interactions, such as hydrogen-bond formation, with the solvent molecules.70 However, in our study, the PL spectral shape and the PLQY were found to be highly invariant to the selection and polarity of the solvent, which suggests that the interactions between the pheophytin a emissive species in the bio-CD and the surrounding solvent molecules are negligible.
We further find that the bio-CD-in-ethanol solutions stored under ambient air exhibits very good photostability during exposure to both daylight and UV light (see Fig. S3†), while both chlorophyll a and pheophytin a in contrast exhibits poor photostability under such conditions. The combination of a high solubility in polar solvents (such as ethanol), solvent-polarity-independent PL properties, and a good photostability thus provide support for that the optically active and relatively hydrophobic pheophytin a species is embedded within the bulk of the bio-CD structure, as schematically depicted in Fig. 1(h), and thereby protected from ambient solvent molecules and oxygen.71
A conclusion at this stage is that a functional biobased starting material for the synthesis of the narrowband and deep-red emitting bio-CDs must comprise chlorophyll a (that can be chemically converted to pheophytin a), while the rest of the biomolecules in the starting material (see Fig. S1†) only contribute with carbon for the graphene-like core structure and polar hydroxyl/phenolic groups for the hydrophilic surface groups (cf.Fig. 1h). We have tested this conclusion by executing an identical synthesis procedure with leaves from the plant “Clusia rosea” (a tropical and sub-tropical flowering plant), as the starting material instead of birch leaves. Fig. S4† shows that the investigated optical properties are invariant to this change in the starting material, thus yielding support for that the key requisite for the starting material indeed is that it is rich in chlorophyll a that can be chemically converted to pheophytin a.
Fig. 4(d) presents the normalized PL spectrum, and the corresponding PLQY (in the inset), as a function of the bio-CD concentration. We find that the main PL peak red shifts, that the lower-energy shoulder increases in relative magnitude, and that the PLQY decreases with increasing bio-CD concentration to reach a modest value of 3% for the neat bio-CD film. These data suggest the bio-CDs have a strong propensity for aggregation in concentrated solution and neat form, and that this aggregation facilitates for energy transfer to pheophytin a “trap units” that feature red-shifted emissive with poor efficiency. The take-home message is thus that, in order to inhibit aggregation-caused quenching and enable for efficient emission in the solid state, the bio-CDs should be dispersed into a solid-state host matrix.72
With the emissive and structural properties of the bio-CD established, we turn to the investigation of its functionality as the emissive OSC in an LEC device. The above identified issue of aggregation-caused quenching of the emission of the bio-CDs was addressed by identifying two prospective majority “host” OSCs for the effective dispersion of the bio-CDs: the small-molecule compound 2,7-bis(diphenylphosphoryl)-9,9′-spirobifluorene (SPPO13) and the high-molecular-weight conjugated polymer, poly(9,9-bis(3′-(N,N-dimethyl)-N-ethylammoinium-propyl-2,7-fluorene)-alt-2,7-(9,9-dioctylfluorene))dibromide (PFNBr). The molecular structure of the two host compounds are presented in the insets of Fig. 5(c and d). SPPO13 was selected because of its reported n-type/bipolar electrochemical doping capacity,73 but its modest solubility (15 g L−1) and low viscosity in ethanol rendered its solution-based fabrication very difficult. We therefore complemented it with p-type PFNBr, which contributes with high solubility (30 g L−1) and high viscosity in ethanol.
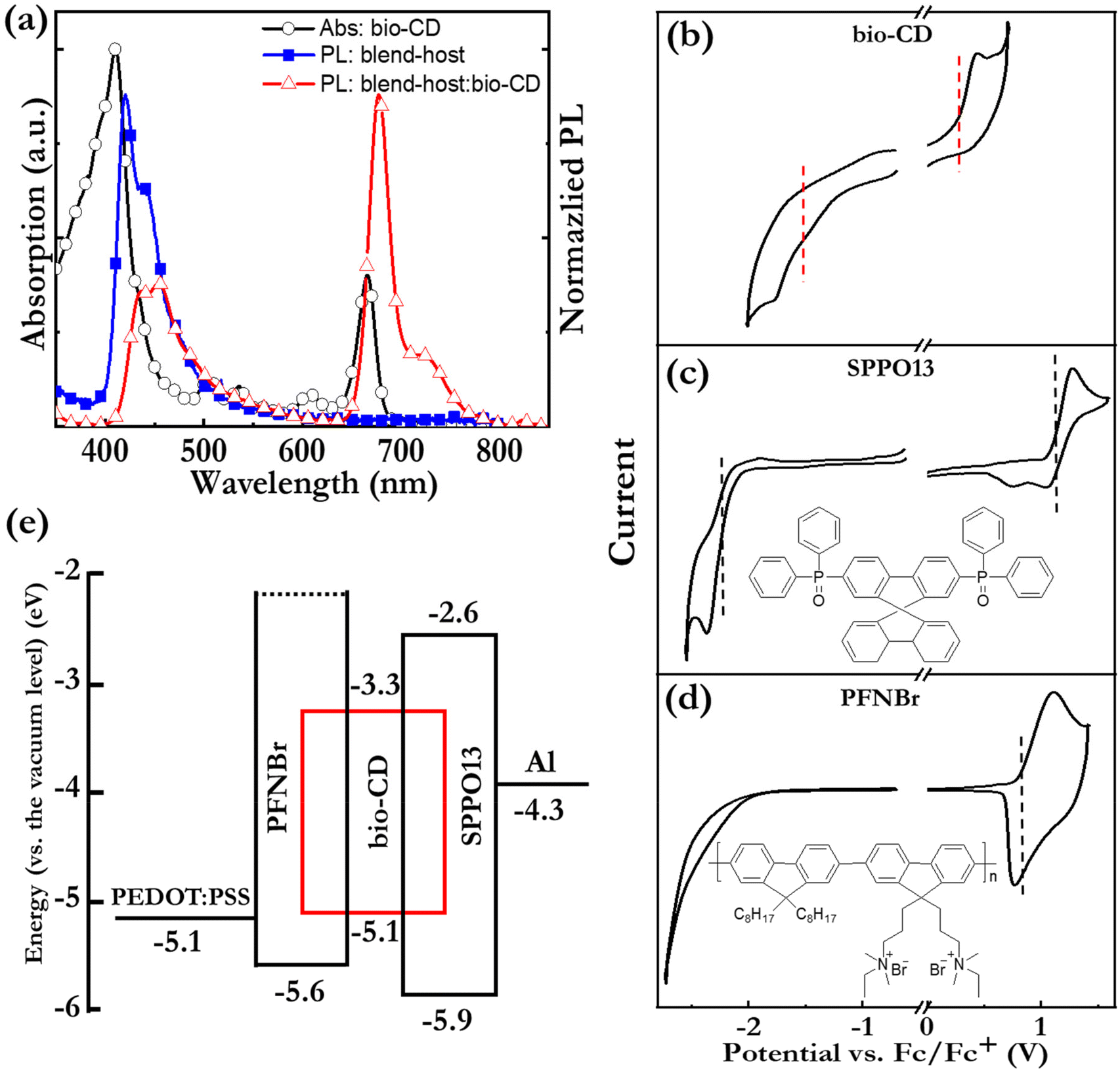 | ||
Fig. 5 (a) The PL spectra of thin films of the blend-host and the blend-host![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :bio-CD, and the absorption spectrum of the bio-CD-in-ethanol solution. The solution concentration was 1.6 mg L−1. The excitation wavelength was 300 nm. (b–d) CV traces of neat thin films of the bio-CD emitter, the SPPO13 host, and the PFNBr host. The electrolyte is 0.1 M THABF4 in acetonitrile, and the scan rate is 0.1 V s−1. The molecular structure of the two host compounds are presented in insets of (b and c). (e) The electron-energy diagram of the blend-host:bio-CD LEC. :bio-CD, and the absorption spectrum of the bio-CD-in-ethanol solution. The solution concentration was 1.6 mg L−1. The excitation wavelength was 300 nm. (b–d) CV traces of neat thin films of the bio-CD emitter, the SPPO13 host, and the PFNBr host. The electrolyte is 0.1 M THABF4 in acetonitrile, and the scan rate is 0.1 V s−1. The molecular structure of the two host compounds are presented in insets of (b and c). (e) The electron-energy diagram of the blend-host:bio-CD LEC. | ||
Fig. 5(a) shows that the PL spectrum of the SPPO13:PFNBr (1:1 by mass) “blend-host” film (solid blue squares) exhibits good spectral overlap with the absorption spectrum of a dilute (1.6 mg L−1) bio-CD solution (open black squares), which is a prerequisite for efficient Förster resonance energy transfer from the blend host to the bio-CD “guest” in a host:guest film. The PL spectrum of the blend-host:bio-CD film (100:2.7 by mass, open red triangles) confirms that the host-to-guest energy transfer is rather efficient by that the major emission peak at 675 nm originates from the minority bio-CD constituent, although the existence of a minor PL peak at 455 nm shows that the energy transfer is not complete. Nevertheless, the PLQY of the solid blend:host:bio-CD film is quite respectable at 12%.
The operational mechanism of LEC devices constitutes the in situ formation of p–n junction doping structure by electrochemical doping of the OSC(s). It is accordingly fundamental that the constituent OSC(s) in the active material of the LEC can be p-type and n-type electrochemically doped,50,74,75 and this electrochemical doping capacity can be evaluated by cyclic voltammetry (CV). Fig. 5(b–d) present CV traces of neat films of the bio-CD guest, the SPPO13 host, and the PFNBr host. The observation of (partially) reversible oxidation reactions for all three compounds and (partially) reversible reduction reactions for the bio-CD and SPPO13 imply that the bio-CD and SPPO13 feature bipolar electrochemical doping capacity whereas PFNBr can be solely p-type doped electrochemically.
The vertical dashed lines mark the derived onset potentials for the electrochemical doping reactions, VFc/Fc+ (with respect to the Fc/Fc+ reference potential); and with the aid of the equation, LUMO/HOMO = −e·(4.8 V + VFc/Fc+), we can calculate the LUMO and HOMO values of the host and guest OSCs, as summarized in the electron-energy diagram in Fig. 5(e). These data reveal that both the electron and hole will be trapped on the bio-CD guest during the operation of LEC devices.
The LEC devices were fabricated with the SPPO13:PFNBr:bio-CD active material sandwiched between an indium-tin-oxide (ITO)/poly(3,4-ethylenedioxythiophene):poly(styrene-sulfonate) (PEDOT:PSS) anode and an Al cathode. The two presented devices, LEC-low and LEC-high, were distinguished by that the mass concentration of the bio-CD guest was 2.7 mass% and 5.4 mass%, respectively. In a sustainability context, we mention that environmentally benign ethanol51 was used as the ink solvent for the spin-coating deposition of the 120 nm-thick active material. The presented LEC devices were driven by a constant current density of j = 77 mA cm−2.
Fig. 6(a and b) present the initial operation of pristine LEC-low and LEC-high devices. Both devices exhibit increasing luminance and decreasing voltage during the initial constant-current operation, which are the characteristic LEC signatures of ion-redistribution induced electric-double-layer formation and electrochemical doping of the OSCs.76 In this context, we call attention to that the only prospective mobile ion in the active material is Br−. This observation thus establishes that the Br−, anion can be liberated from the large PFN+ cation, and that it, by redistribution under the influence of an electric field, can contribute to both the formation of the electric double layers and to the electrochemical doping of the OSCs in the active material. The LEC-low device exhibits a reasonably fast turn on of less than 10 s to a luminance of 100 cd m−2, which shows that the Br− anion is quite mobile within the active material.77 Fig. S5† presents photographs of the LEC-low device in the idle off state and during light emission. This device also delivers a peak luminance of 110 cd m−2 at an external quantum efficiency (EQE) of 0.29%. Further improvements in the EQE can be attained by identifying host:bio-CD combinations that feature a higher PLQY in the active material, and the development of bio-CDs that can efficiently harvest the triplet excitons for light emission. We also speculate that LEC-common losses due to exciton-polaron quenching and poor outcoupling can be alleviated by a correlated systematic cavity investigations and device engineering.
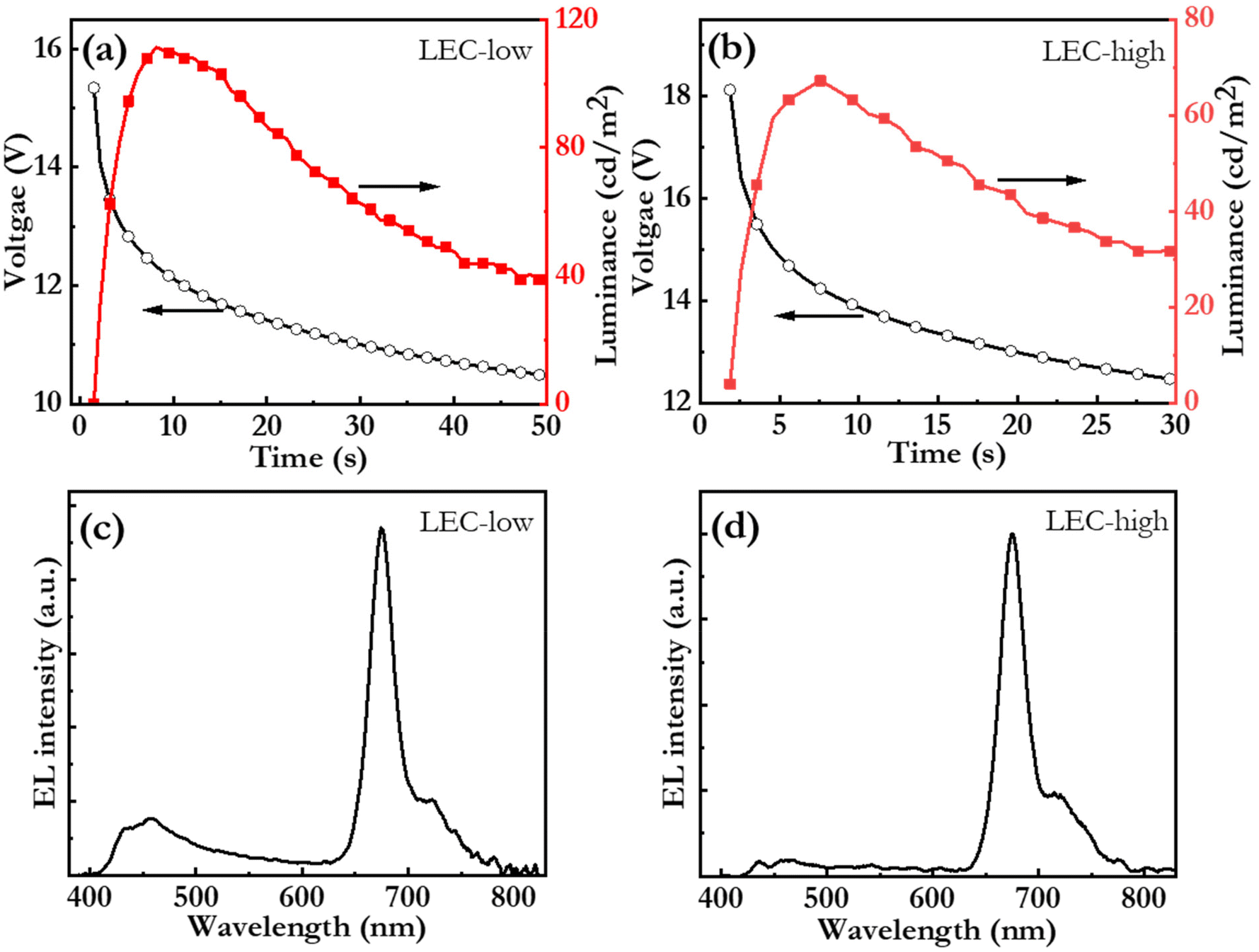 | ||
| Fig. 6 (a and b) The temporal evolution of the voltage and the luminance and (c and d) the steady-state EL spectrum for the blend-host:bio-CD LEC devices with a bio-CD concentration of 2.7 mass% (LEC-low) and 5.4 mass% (LEC-high). All of the ITO/PEDOT:PSS/blend-host:bio-CD/Al LEC devices were driven by a constant current density of 77 mA cm−2. | ||
Fig. 6(c and d) present the steady-state electroluminescence (EL) spectrum from the LEC-low and LEC-high devices, respectively. The LEC-low device exhibits a major EL peak at 675 nm with a FWHM of 29 nm, which is identified to originate from the bio-CD guest (cf.Fig. 2c), but also a minor higher-energy band, between 400 and 500 nm, which is assigned to emission from the blend-host (cf.Fig. 5a). Pure bio-CD guest emission can however be attained by increasing the bio-CD concentration to 5.4%, as demonstrated by the narrowband, and deep-red EL, with a FWHM of 28 nm and a peak wavelength of 675 nm, from the LEC-high device in Fig. 6(d).
We finally report that it is possible to spray coat a water-based PEDOT:PSS ink on top of the bio-CD based active material, and that this spray coating leaves the beneath active material structurally intact (Fig. S6a†).78–80 This demonstration paved the way for the fabrication of a semi-transparent LEC, equipped with ITO for the cathode and PEDOT:PSS for the anode, which delivers narrow-band, deep-red, light emission (FWHM = 33 nm) from both planar electrodes (Fig. S6b and c†). This effort can be considered a first step towards the realization of a truly sustainable light-emission device, which is based on biomass-derived materials, which is fabricated with environmentally sound processes and benign process chemicals, and which features an enabling form factor and practical light-emission features.
Conclusions
We report on a straightforward one-pot synthesis of bio-CDs using birch leaves as the sole raw material. These highly soluble bio-CDs exhibit excitation-wavelength-independent, narrowband, deep-red PL, with a FWHM of 28 nm, a peak emission wavelength of 675 nm, and a PLQY of 26% in ethanol solution. A systematic optical and structural characterization reveals that the single emissive species is molecular pheophytin a, which is embedded in graphene-like bulk, which in turn is surrounded by a shell comprising polar hydroxyl groups. The functionality of the birch-leaf-derived bio-CDs in sustainable organic electronics was demonstrated by their employment as the guest emitter in an LEC device, which delivers narrowband deep-red emission with a luminance of 110 cd m−2 at an external quantum efficiency of 0.29%. The demonstrated benign synthesis and processing, in combination with the promising PL and EL properties, highlight the potential of bio-CDs in future electronic applications which emphasize a sustainable lifecycle.Experimental
Materials and synthesis
The birch leaves were picked in June at the Umeå University campus, which is located in the center of Umeå city in northern Sweden. The freshly picked birch leaves were rinsed with tap water, dried at 100 °C for 4 h, and then grinded into powder. 0.5 g of the dried and grinded birch-leaf powder was mixed with 10 mL acetone (HPLC, ≥ 99.0%, Fisher Scientific). The extracted solution in acetone was transferred into a poly(tetrafluoroethylene) Teflon©-lined autoclave (25 mL) and heated in an oven at 120 °C for 5 h. After cooling to room temperature, the crude solution was centrifuged at 10000 rpm for 10 min to remove large particles. The collected supernatant was filtered through a 0.45 μm PTFE membrane, followed by purification via column chromatography with a mixture of petroleum ether (HPLC, ≥99.0%, VWR international) and ethyl acetate (HPLC, ≥99.0%, Fisher Scientific) as the eluent. The petroleum-ether:ethyl-acetate eluent volume ratio was changed from 10:1 in the beginning to 10:5 during the chromatographic separation. The collected bio-CD sample was dried under dynamic vacuum by rotary evaporation, and the resulting bio-CD powder was re-dispersed in ethanol (≥99.0%, VWR international) before further use.
Characterization
The TEM sample was prepared by drop-casting a dilute (0.1 g L−1) bio-CD-in-ethanol solution on an ultrathin carbon-coated copper grid. The morphology of the bio-CDs was measured by TEM (Talos F200X, Thermo Fisher Scientific) at an accelerating voltage of 200 kV. The AFM sample was made by spin-coating a dilute (0.1 g L−1) bio-CD-in-ethanol solution on a well-cleaned silicon substrate. The height of bio-CD was measured by AFM using a MultiMode SPM microscope operating in non-contact mode and equipped with a Nanoscope IV Controller (Veeco Metrology). The elemental composition was analysed by XPS (Axis Ultra DLD, Kratos) and the presence of functional groups determined by FTIR (IFS/v66, Bruker), respectively. The samples were fabricated on clean Si substrate and dried at room temperature for formation of ∼200 μm thick drop-cast film. The optical properties were investigated by UV-Vis absorption spectroscopy (LS45, PerkinElmer), PL spectra, PLQY (C9920-02G, Hamamatsu Photonics), and time-resolved PL (FLS1000, Edinburgh). The CV measurement was carried out with a potentiostat (Autolab PGSTAT302, driven by the GPES software). The working electrode is the drop-coated material-under-study on a Au-covered glass substrate, the counter electrode was a Pt rod, the quasi-reference electrode was a Ag wire, and the electrolyte was 0.1 M tetrahexylammonium tetrafluoroborate (THABF4, Sigma-Aldrich) in acetonitrile solution with a scan rate was 0.1 V s−1. Directly after each CV scan, a calibration scan was run with a small amount of ferrocene added to the electrolyte solution. All CV potentials are reported versus the ferrocene/ferrocenium ion (Fc/Fc+) reference potential. The reduction/oxidation onset potentials are defined as the intersection of the baseline with the tangent of the current at its half peak. The CV sample preparation and measurement were performed in a N2-filled glove box ([O2] < 2 ppm, [H2O] < 1 ppm).Device fabrication and characterization
The host compounds, poly(9,9-bis(3′-(N,N-dimethyl)-N-ethylammoinium-propyl-2,7-fluorene)-alt-2,7-(9,9-dioctylfluorene))dibromide (PFNBr, Lumtec) and 2,7-bis(diphenylphosphoryl)-9,9′-spirobifluorene (SPPO13, Lumtec), were used as received. The master solutions were prepared by dissolving respective host compound separately in methanol at a concentration of 15 g L−1, and by thereafter stirring the solution at 70 °C for >3 h. The active-material ink was prepared by blending bio-CDs and host materials in a desired mass ratio, followed by stirring on the hot plate with magnetic stirring at 70 °C for >1 h.The carefully cleaned indium-tin-oxide (ITO) coated glass substrates (20 Ω sq−1, Thin Film Devices) were spin-coated by a poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) ink (Clevios P VP AI 4083, Heraeus) at 4000 rpm for 60 s, and thereafter dried at 120 °C for 30 min. The dry PEDOT:PSS layer was spin-coated with the active-material ink at 2000 rpm for 60 s, and thereafter dried at 70 °C for 10 min. The dry thickness of the PEDOT:PSS and the active-material layers was 40 nm and 120 nm, respectively, as measured with a profilometer (DekTak XT, Bruker). A set of 100 nm thick Al cathodes were deposited on top of the active material by thermal vacuum evaporation at p < 5 × 10−6 mbar. The 8.5 × 1.5 mm2 emission area was defined by the overlap between the ITO anode and the Al cathode.
The PEDOT:PSS ink was prepared by diluting a commercial PEDOT:PSS dispersion (Clevios SV3, Heraeus, GER) with 400 volume% of methanol, followed by ≥45 min sonication in an ultrasonic bath. The PEDOT:PSS ink was spray-coated directly on top of the dry active material using the spray box, with the substrate maintained at 60 °C. The spray parameters were: N2 gas pressure = 410 kPa, ink flow rate = 4 m min−1, number of sweeps = 3, spray time = 60 s. A shadow mask defined the shape of the PEDOT:PSS top electrodes during the spray coating, and the overlap of the patterned PEDOT:PSS top electrodes and the patterned ITO bottom electrodes defined four 2 × 2 mm2 LEC devices on one substrate. Immediately after the spray-coating of the PEDOT:PSS electrodes, the devices were dried at 120 °C for ≥4 min on a hotplate. The dry thickness of the PEDOT:PSS electrode was 250 nm, as determined by a cross-sectional SEM image.
The LEC devices were measured with a computer-controlled source-measure unit (Agilent U2722A) and a calibrated photodiode, equipped with an eye-response filter (Hamamatsu Photonics), and connected to a data acquisition card (National Instruments USB-6009) via a current-to-voltage amplifier. The EL spectrum was measured with a calibrated spectrometer (USB2000+, Ocean Optics). All of the above device procedures, with the exception of the deposition of PEDOT:PSS, were carried out in two interconnected N2-filled glove box ([O2] < 2 ppm, [H2O] < 1 ppm).
Author contributions
JW, LE, and ST conceptualized the idea. ST carried out the majority of the structural, electrochemical and device experiments. YL performed the material synthesis. HO and MG carried out the photophysical characterization. PZ and DD performed the TEM measurements. EA performed the spray-coating fabrication of the semi-transparent device. All authors have discussed the results and contributed to the analysis. AVM, TW, JW and LE contributed with research funding and supervision. JW, LE, and ST wrote the first draft of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge Dr Cheng Choo Lee for technical support and valuable advice during the SEM and TEM measurements, and the Umeå Core Facility for Electron Microscopy (UCEM), the Vibrational Spectroscopy Core Facility (ViSp), and the XPS platform at Umeå University. The authors thank Dr Joan Ràfols Ribé for providing the Clusia rosea leaves for bio-CDs synthesis. The authors also acknowledge generous support from J. C. Kempes Minnes Stipendiefond (SMK-21-0015 and SMK-1956), the Swedish Energy Agency (45419-1, 46523-1, 50779-1, and P2021-00032), the Swedish Research Council (2018-03937, 2019-02345, 2020-04437, 2021-04778), Bertil & Britt Svenssons stiftelse för belysningsteknik (2021 höst-14 and 2022 höst-31), the Knut and Alice Wallenberg Foundation (WISE-AP01-D02 and KAW 2022.0381), Vinnova SIO Grafen (2022-01319), the Swedish Foundation for International Cooperation in Research, Higher Education via an Initiation Grant for Internationalization (2019-8553) and the Wallenberg Initiative Materials Science for Sustainability (WISE-AP01-D02).References
- X. Shi, Y. Zuo, P. Zhai, J. Shen, Y. Yang, Z. Gao, M. Liao, J. Wu, J. Wang, X. Xu, Q. Tong, B. Zhang, B. Wang, X. Sun, L. Zhang, Q. Pei, D. Jin, P. Chen and H. Peng, Nature, 2021, 591, 240–245 CrossRef CAS.
- W. Liu, C. Zhang, R. Alessandri, B. T. Diroll, Y. Li, H. Liang, X. Fan, K. Wang, H. Cho, Y. Liu, Y. Dai, Q. Su, N. Li, S. Li, S. Wai, Q. Li, S. Shao, L. Wang, J. Xu, X. Zhang, D. V. Talapin, J. J. de Pablo and S. Wang, Nat. Mater., 2023, 22, 737–745 CrossRef CAS PubMed.
- ‘Ultra-thin OLEDs make packaging shine’, https://www.karlknauer.com/en/innovation-and-trends/smart-packaging/oled, (accessed October 2023).
- Y. Jeon, H.-R. Choi, J. H. Kwon, S. Choi, K. M. Nam, K.-C. Park and K. C. Choi, Light: Sci. Appl., 2019, 8, 114 CrossRef CAS.
- R. Pode, Renewable Sustainable Energy Rev., 2020, 133, 110043 CrossRef.
- J. Bauri, R. B. Choudhary and G. Mandal, J. Mater. Sci., 2021, 56, 18837–18866 CrossRef CAS.
- J.-M. Yeom, H.-J. Jung, S.-Y. Choi, D. S. Lee and S.-R. Lim, Int. J. Environ. Res., 2018, 12, 479–488 CrossRef CAS.
- M. Irimia-Vladu, Chem. Soc. Rev., 2014, 43, 588–610 RSC.
- E. Fresta, V. Fernández-Luna, P. B. Coto and R. D. Costa, Adv. Funct. Mater., 2018, 28, 1707011 CrossRef.
- M. Nieddu, M. Patrian, S. Ferrara, J. P. F. Werner, F. Kohler, E. Anaya-Plaza, M. A. Kostiainen, H. Dietz, J. R. Berenguer and R. D. Costa, Adv. Sci., 2023, 10, 2300069 CrossRef CAS PubMed.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- S. Lu, L. Sui, J. Liu, S. Zhu, A. Chen, M. Jin and B. Yang, Adv. Mater., 2017, 29, 1603443 CrossRef PubMed.
- B. Zhao and Z. A. Tan, Adv. Sci., 2021, 8, 2001977 CrossRef CAS.
- Y. Lou, X. Hao, L. Liao, K. Zhang, S. Chen, Z. Li, J. Ou, A. Qin and Z. Li, Nano Sel., 2021, 2, 1117–1145 CrossRef CAS.
- P. Kaur and G. Verma, Mater. Today Sustain., 2022, 18, 100137 CrossRef.
- G. Wang, Q. Guo, D. Chen, Z. Liu, X. Zheng, A. Xu, S. Yang and G. Ding, ACS Appl. Mater. Interfaces, 2018, 10, 5750–5759 CrossRef CAS PubMed.
- L. Zhu, Y. Yin, C.-F. Wang and S. Chen, J. Mater. Chem. C, 2013, 1, 4925–4932 RSC.
- R. Liu, L. Zhang, J. Zhao, C. Hou, Y. Huang, Z. Huang and S. Zhao, Adv. Ther., 2019, 2, 1900011 CrossRef.
- X. Xu, L. Cai, G. Hu, L. Mo, Y. Zheng, C. Hu, B. Lei, X. Zhang, Y. Liu and J. Zhuang, J. Lumin., 2020, 227, 117534 CrossRef CAS.
- P. Wang, Y. Yan, Y. Zhang, T. Gao, H. Ji, S. Guo, K. Wang, J. Xing and Y. Dong, Int. J. Nanomed., 2021, 16, 2045–2058 CrossRef.
- A. Dager, T. Uchida, T. Maekawa and M. Tachibana, Sci. Rep., 2019, 9, 14004 CrossRef PubMed.
- A. Dager, A. Baliyan, S. Kurosu, T. Maekawa and M. Tachibana, Sci. Rep., 2020, 10, 12333 CrossRef CAS.
- F. Qin, J. Bai, Y. Zhu, P. He, X. Wang, S. Wu, X. Yu and L. Ren, Phys. Chem. Chem. Phys., 2023, 25, 2762–2769 RSC.
- Z. Liu, W. Jin, F. Wang, T. Li, J. Nie, W. Xiao, Q. Zhang and Y. Zhang, Sens. Actuators, B, 2019, 296, 126698 CrossRef CAS.
- H. Rao, W. Liu, K. He, S. Zhao, Z. Lu, S. Zhang, M. Sun, P. Zou, X. Wang, Q. Zhao, Y. Wang and T. Liu, ACS Sustainable Chem. Eng., 2020, 8, 8857–8867 CrossRef CAS.
- J. Liu, T. Kong and H.-M. Xiong, Adv. Mater., 2022, 34, 2200152 CrossRef CAS.
- G. Hu, L. Ge, Y. Li, M. Mukhtar, B. Shen, D. Yang and J. Li, J. Colloid Interface Sci., 2020, 579, 96–108 CrossRef CAS.
- C. Huang, H. Dong, Y. Su, Y. Wu, R. Narron and Q. Yong, Nanomaterials, 2019, 9, 387 CrossRef CAS.
- Y. Sun, S. Liu, L. Sun, S. Wu, G. Hu, X. Pang, A. T. Smith, C. Hu, S. Zeng, W. Wang, Y. Liu and M. Zheng, Nat. Commun., 2020, 11, 5591 CrossRef CAS PubMed.
- V. C. Hoang, K. N. Dinh and V. G. Gomes, J. Mater. Chem. A, 2019, 7, 22650–22662 RSC.
- A.-M. Alam, B.-Y. Park, Z. K. Ghouri, M. Park and H.-Y. Kim, Green Chem., 2015, 17, 3791–3797 RSC.
- H. Ding, Y. Ji, J.-S. Wei, Q.-Y. Gao, Z.-Y. Zhou and H.-M. Xiong, J. Mater. Chem. B, 2017, 5, 5272–5277 RSC.
- A. Tyagi, K. M. Tripathi, N. Singh, S. Choudhary and R. K. Gupta, RSC Adv., 2016, 6, 72423–72432 RSC.
- K. K. Gudimella, T. Appidi, H.-F. Wu, V. Battula, A. Jogdand, A. K. Rengan and G. Gedda, Colloids Surf., B, 2021, 197, 111362 CrossRef CAS PubMed.
- X. Tang, H. Wang, H. Yu, B. Bui, W. Zhang, S. Wang, M. Chen, L. Yuan, Z. Hu and W. Chen, Mater. Today Phys., 2022, 22, 100576 CrossRef CAS.
- R. Atchudan, T. N. J. I. Edison, M. Shanmugam, S. Perumal, T. Somanathan and Y. R. Lee, Phys. E, 2021, 126, 114417 CrossRef CAS.
- J. Zhou, Z. Sheng, H. Han, M. Zou and C. Li, Mater. Lett., 2012, 66, 222–224 CrossRef CAS.
- W. Li, S. Wu, H. Zhang, X. Zhang, J. Zhuang, C. Hu, Y. Liu, B. Lei, L. Ma and X. Wang, Adv. Funct. Mater., 2018, 28, 1804004 CrossRef.
- J. Zhao, M. Huang, L. Zhang, M. Zou, D. Chen, Y. Huang and S. Zhao, Anal. Chem., 2017, 89, 8044–8049 CrossRef CAS PubMed.
- P. Lagonegro, U. Giovanella and M. Pasini, Coatings, 2021, 11, 5 CrossRef CAS.
- C. Kang, S. Tao, F. Yang and B. Yang, Aggregate, 2022, 3, e169 CrossRef CAS.
- A. Xu, G. Wang, Y. Li, H. Dong, S. Yang, P. He and G. Ding, Small, 2020, 16, 2004621 CrossRef CAS PubMed.
- H. Jia, Z. Wang, T. Yuan, F. Yuan, X. Li, Y. Li, Z. A. Tan, L. Fan and S. Yang, Adv. Sci., 2019, 6, 1900397 CrossRef PubMed.
- Z. Wang, F. Yuan, X. Li, Y. Li, H. Zhong, L. Fan and S. Yang, Adv. Mater., 2017, 29, 1702910 CrossRef.
- N. Urushihara, T. Hirai, A. Dager, Y. Nakamura, Y. Nishi, K. Inoue, R. Suzuki, M. Tanimura, K. Shinozaki and M. Tachibana, ACS Appl. Nano Mater., 2021, 4, 12472–12480 CrossRef CAS.
- A. Singh, A. Wolff, S. D. Yambem, M. Esmaeili, J. D. Riches, M. Shahbazi, K. Feron, E. Eftekhari, K. Ostrikov, Q. Li and P. Sonar, Adv. Mater., 2020, 32, 1906176 CrossRef CAS PubMed.
- S. Khan, A. Gupta, N. C. Verma and C. K. Nandi, Nano Lett., 2015, 15, 8300–8305 CrossRef CAS.
- L. Bao, C. Liu, Z.-L. Zhang and D.-W. Pang, Adv. Mater., 2015, 27, 1663–1667 CrossRef CAS.
- R. Chen, Z. Wang, T. Pang, Q. Teng, C. Li, N. Jiang, S. Zheng, R. Zhang, Y. Zheng, D. Chen and F. Yuan, Adv. Mater., 2023, 35, 2302275 CrossRef CAS.
- Q. Pei, G. Yu, C. Zhang, Y. Yang and A. J. Heeger, Science, 1995, 269, 1086–1088 CrossRef CAS PubMed.
- C. Larsen, P. Lundberg, S. Tang, J. Ràfols-Ribé, A. Sandström, E. M. Lindh, J. Wang and L. Edman, Nat. Commun., 2021, 12, 4510 CrossRef CAS PubMed.
- A. Sandström, H. F. Dam, F. C. Krebs and L. Edman, Nat. Commun., 2012, 3, 1002 CrossRef PubMed.
- J. Zimmermann, S. Schlisske, M. Held, J.-N. Tisserant, L. Porcarelli, A. Sanchez-Sanchez, D. Mecerreyes and G. Hernandez-Sosa, Adv. Mater. Technol., 2019, 4, 1800641 CrossRef.
- J. Zimmermann, L. Porcarelli, T. Rödlmeier, A. Sanchez-Sanchez, D. Mecerreyes and G. Hernandez-Sosa, Adv. Funct. Mater., 2018, 28, 1705795 CrossRef.
- Z. Zhang, K. Guo, Y. Li, X. Li, G. Guan, H. Li, Y. Luo, F. Zhao, Q. Zhang, B. Wei, Q. Pei and H. Peng, Nat. Photonics, 2015, 9, 233–238 CrossRef CAS.
- N. Jürgensen, J. Zimmermann, A. J. Morfa and G. Hernandez-Sosa, Sci. Rep., 2016, 6, 36643 CrossRef.
- J. Zimmermann, N. Jürgensen, A. J. Morfa, B. Wang, S. Tekoglu and G. Hernandez-Sosa, ACS Sustainable Chem. Eng., 2016, 4, 7050–7055 CrossRef CAS.
- T. Costea, L. Vlase, R. Ancuceanu, M. Dinu, N. Olah, M. Popescu and C. E. Gîrd, Rom. Biotechnol. Lett., 2016, 21, 11527–11538 CAS.
- D. Kregiel, E. Pawlikowska and H. Antolak, Molecules, 2018, 23, 1664 CrossRef.
- M. S. Vladimirov, V. D. Nikolić, L. P. Stanojević, L. B. Nikolić and A. D. Tačić, Adv. Technol., 2019, 8, 65–77 CrossRef.
- B. J. Sjursnes, L. Kvittingen and R. Schmid, J. Chem. Educ., 2015, 92, 193–196 CrossRef CAS.
- A. Ananthanarayanan, X. Wang, P. Routh, B. Sana, S. Lim, D.-H. Kim, K.-H. Lim, J. Li and P. Chen, Adv. Funct. Mater., 2014, 24, 3021–3026 CrossRef CAS.
- L. Song, J. Shi, J. Lu and C. Lu, Chem. Sci., 2015, 6, 4846–4850 RSC.
- F. Liu, M.-H. Jang, H. D. Ha, J.-H. Kim, Y.-H. Cho and T. S. Seo, Adv. Mater., 2013, 25, 3657–3662 CrossRef CAS PubMed.
- R. Livingston, R. Pariser, L. Thompson and A. Weller, J. Am. Chem. Soc., 1953, 75, 3025–3026 CrossRef CAS.
- K. Östbring, M. Rayner, I. Sjöholm, J. Otterström, P.-Å. Albertsson, S. C. Emek and C. Erlanson-Albertsson, Food Funct., 2014, 5, 2157–2165 RSC.
- S. M. Milenković, J. Zvezdanović, T. Anđelković and Z. Dejan, Adv. Technol., 2012, 1, 16–24 Search PubMed.
- D. I. Arnon, Plant Physiol., 1949, 24, 1–15 CrossRef CAS PubMed.
- W. Zheng, N. Shan, L. Yu and X. Wang, Dyes Pigm., 2008, 77, 153–157 CrossRef CAS.
- B. Myśliwa-Kurdziel, K. Solymosi, J. Kruk, B. Böddi and K. Strzałka, Eur. Biophys. J., 2008, 37, 1185–1193 CrossRef PubMed.
- S. Barazzouk, L. Bekalé and S. Hotchandani, J. Mater. Chem., 2012, 22, 25316–25324 RSC.
- S. Tang, A. Sandström, P. Lundberg, T. Lanz, C. Larsen, S. van Reenen, M. Kemerink and L. Edman, Nat. Commun., 2017, 8, 1190 CrossRef.
- Y. Liu, S. Tang, X. Wu, N. Boulanger, E. Gracia-Espino, T. Wågberg, L. Edman and J. Wang, Nano Res., 2022, 15, 5610–5618 CrossRef CAS.
- P. Matyba, K. Maturova, M. Kemerink, N. D. Robinson and L. Edman, Nat. Mater., 2009, 8, 672 CrossRef CAS PubMed.
- K. Yasuji, T. Sakanoue, F. Yonekawa and K. Kanemoto, Nat. Commun., 2023, 14, 992 CrossRef CAS PubMed.
- J. Fang, P. Matyba, N. D. Robinson and L. Edman, J. Am. Chem. Soc., 2008, 130, 4562–4568 CrossRef CAS PubMed.
- J. Mindemark and L. Edman, J. Mater. Chem. C, 2016, 4, 420–432 RSC.
- P. Matyba, H. Yamaguchi, G. Eda, M. Chhowalla, L. Edman and N. D. Robinson, ACS Nano, 2010, 4, 637–642 CrossRef CAS PubMed.
- E. Auroux, A. Sandström, C. Larsen, P. Lundberg, T. Wågberg and L. Edman, Org. Electron., 2020, 84, 105812 CrossRef CAS.
- E. Auroux, A. Sandström, C. Larsen, E. Zäll, P. Lundberg, T. Wågberg and L. Edman, Adv. Electron. Mater., 2021, 7, 2100253 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical species in birch leaves, photostability, photophysics of clusia rosea derived bio-CD, photograph of LEC device, and semi-transparent LEC device. See DOI: https://doi.org/10.1039/d3gc03827k |
| This journal is © The Royal Society of Chemistry 2023 |