
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDoes the platinum-loading in proton-exchange membrane fuel cell cathodes influence the durability of the membrane-electrode assembly?†
Ricardo
Sgarbi‡
a,
William
Ait Idir‡
b,
Quentin
Labarde
a,
Michel
Mermoux
a,
Peizhe
Wu
b,
Julia
Mainka
b,
Jérôme
Dillet
b,
Clémence
Marty
c,
Fabrice
Micoud
c,
Olivier
Lottin
b and
Marian
Chatenet
 *a
*a
aCNRS, Grenoble INP (Institute of Engineering and Management Univ. Grenoble Alpes), LEPMI, Univ. Grenoble Alpes, Univ. Savoie Mont Blanc, 38000 Grenoble, France. E-mail: marian.chatenet@grenoble-inp.fr
bLEMTA – Université de Lorraine – CNRS, Nancy, France
cCEA, LITEN, Univ. Grenoble Alpes, F-38054, Grenoble, France
First published on 11th August 2023
Abstract
MEAs with various cathode Pt loadings were elaborated and aged using a multiple-stressor accelerated stress test (AST) in a segmented PEMFC. The thinnest (lowest Pt loading) cathodes have lower initial activity, owing to larger oxygen reduction reaction hindrance and oxygen transport resistance. Although the lowest cathode Pt loadings initially degrade faster, the overall loss of ECSA at end-of-test is nearly similar whatever the cathode Pt loading, with no local heterogeneities of aging detected along the gas channels. The cathode Pt/C catalyst degrades mostly by Ostwald ripening (which seems more pronounced for lower cathode Pt loading) and nanoparticles agglomeration, owing to superficial carbon functionalization and related Pt crystallite migration: no consequent carbon corrosion is witnessed in this AST. Also, the oxidized Pt2+ ions formed by Pt corrosion diffuse/migrate roughly in a similar manner through the membrane for all cathode Pt loadings, and are re-deposited by crossover H2 close to the cathode|membrane interface. Overall, the mechanisms of Pt/C degradation are not depending on the cathode Pt loading for the chosen AST.
Keywords: Proton exchange membrane fuel cells (PEMFC); Cathode catalyst layer (CL); Platinum loading; Durability.
1 Introduction
Proton exchange membrane fuel cells (PEMFC) are considered promising for a number of applications where zero CO2 emission generators are targeted (e.g. the automotive or heavy-transportation sectors). One limitation of the PEMFCs' deployment at the large scale is their high cost (in part connected to the usage of platinum-based catalysts at their electrodes) and insufficient durability in operation.1 One easy mitigation strategy to the first point is evidently to lower the aerial loading of Pt-based catalysts; while this strategy is very feasible at the anode side (the hydrogen oxidation reaction, HOR, is fast, enabling proper anode operation at Pt loadings below 50 μg cmanode−2),2 it is much more demanding at the cathode side. Indeed, the oxygen reduction reaction, ORR, is sluggish, and oxygen is only present at 21% vol in air, hence imposing mass-transport limitations in the porous electrodes and gas diffusing media, that are further complexified by liquid water production at the cathode of PEMFCs.3–5 As a result, present state-of-the-art PEMFC cathodes employ Pt catalyst aerial loading on the order of 0.3–0.5 mgPt cmcathode−2, when one would aim at decreasing this value down to 0.1–0.2 mgPt cmcathode−2 or even below.6,7 At that stage, it must be recalled that PEMFC catalyst layers (CLs) are made from a mixture between the catalytic material (e.g. Pt nanoparticles supported on a carbon black, like Pt/Vulcan XC72) and an ionomer, that plays the role of polymeric binder, proton conducting phase and water-conducting phase.8–10 Whereas the ionomer is crucial to the electrochemical operation of the electrodes (in particular at the cathode), it also conveys adverse effects: the particular interaction between the hydrophilic (–SO3− groups) and the hydrophobic (–CF2–CF2–) backbone of usual perfluorosulfonated ionomers and the complex Pt/C surface induces the formation of a “skin” of ionomer that covers the Pt/C nanoparticles11–13 which limits the access of oxygen to the Pt sites and the evacuation of water formed upon ORR.14,15 The induced oxygen mass-transport resistance through this ionomer film has since been recognized as a major issue in PEMFC cathodes when one intends to operate at large current density,5,16 especially when the cathode Pt loading is decreased.7 As a matter of fact, the developed active area (amount of Pt sites per volume of CL or per geometric surface of membrane electrode assembly, MEA) is a key: at a given geometric current density, the local current density to each Pt site within the CL increases concomitantly with the local flux of oxygen when the amount of Pt sites decreases, which detrimentally influences the oxygen transport resistance.6,17 This increased oxygen transport resistance (hence lower cathode performance) for low cathode Pt loading is now well documented (see the previous articles cited) and this has triggered the development of highly-oxygen-permeable ionomers (HOPI) to mitigate this effect18,19 or the use of mesoporous carbon to immobilize the Pt nanoparticles in pores, preventing their direct contact with the ionomer;6,20 both materials enable (individually or combined) to improve the CL performance at low cathode Pt loading.4However, there is still very limited information about the real impact of the cathode Pt loading on the durability of the cathode (and of the whole membrane electrode assembly). The present paper aims to start filling this gap. MEAs containing different Pt loadings in the cathodic CLs are prepared using a commercial Pt/Vulcan XC72 catalyst and assembled with a given state-of-the-art membrane, given gas diffusion layers (GDL) and a given anode composition and loading. These MEAs are tested with a home-made (very degrading) accelerated stress test consisting of multiple stressors (current density/potential and relative humidity cycling and OCV hold21,22) aiming at degrading the electrodes (i.e. carbon support, catalyst, and possibly the ionomer), and the membrane. This AST protocol is used here to evaluate whether the cathode Pt loading (labeled as x-Pt/C, where x is the cathode CL Pt loading and varies in the sequence 0.05, 0.1, 0.2 and 0.3 mgPt cm−2) has an influence on the mechanisms and the extent of degradation monitored pre/during/post aging.
2 Results and discussions
MEAs with four different Pt-loadings were evaluated physicochemically and electrochemically. Four of them were submitted to electrochemical degradation tests, which consist of accelerated stress tests (ASTs), as detailed in Materials and methods section 4.3. The electrochemical performances were measured for each MEA at its beginning of test (BOT) and end of test (EOT), meaning before and after the AST, respectively. Since the physicochemical characterizations are irreversibly degrading/destructuring the MEA materials/assembly, two MEAs for each cathode Pt loading were produced, one for the electrochemical characterizations in unit PEMFC cell (section 2.1) followed by post-AST physicochemical characterizations at EOT and the other for similar characterizations on non-aged samples (as detailed in section 2.2). In the process, it was assumed that the two MEAs in each couple for a given cathode Pt loading were similar.2.1 Electrochemical characterizations in unit PEMFC
The electrochemical performances of the x-Pt/C MEAs were assessed by plotting polarization curve, measuring electrochemically active surface area (ECSA) and electrochemical impedance spectroscopy (EIS) at both BOT and EOT. Between the initial (BOT) and final (EOT) states, the MEAs experienced the tailored AST described in section 4.3.The polarization curves at BOT (Fig. 1, full lines) show an evident cell performance enhancement from the lowest (0.05-Pt/C) to the most (0.3-Pt/C) loaded cathode, that increases from the low (j < 0.5 A cm−2) to the high (j > 1 A cm−2) current density regions. This advantage of the highest cathode Pt loading is already significant in the so-called activation region (j < 0.2 A cm−2), and further increases along the polarization plot to become maximal in the so-called mass-transport limiting region (j > 1.5 A cm−2), whereas the least loaded one (0.05-Pt/C) presents the lowest cell performance throughout the voltage range. In contrast, the 0.2-Pt/C and 0.1-Pt/C MEAs start from near-superimposed polarization plots for low current densities to two entirely separated ones above 1 A cm−2. The MEA performance decay follows the decrease of cathode Pt loadings: the cell performance losses observed are more remarkable at high current densities, when all the limitations (activation, ohmic and mass-transport) do superpose, which suggests that (in the range of cathode Pt loading considered), the larger the Pt loading the smaller these overall limitations. This is further illustrated by the decrease of the values of the high-frequency resistance (Rhf), the charge-transfer resistance (Rct) and diffusion resistance (Rd) when the cathode Pt loading increases. These resistances are identified from EIS spectra measured at 1 A cm−2 (Table 1) by least-square fitting method using the equivalent electrical circuit, i.e. a modified Randles circuit for the CCL with a Warburg impedance Zw (Rd,τ) in series for the GDL, and an anode circuit used in a previous study.23
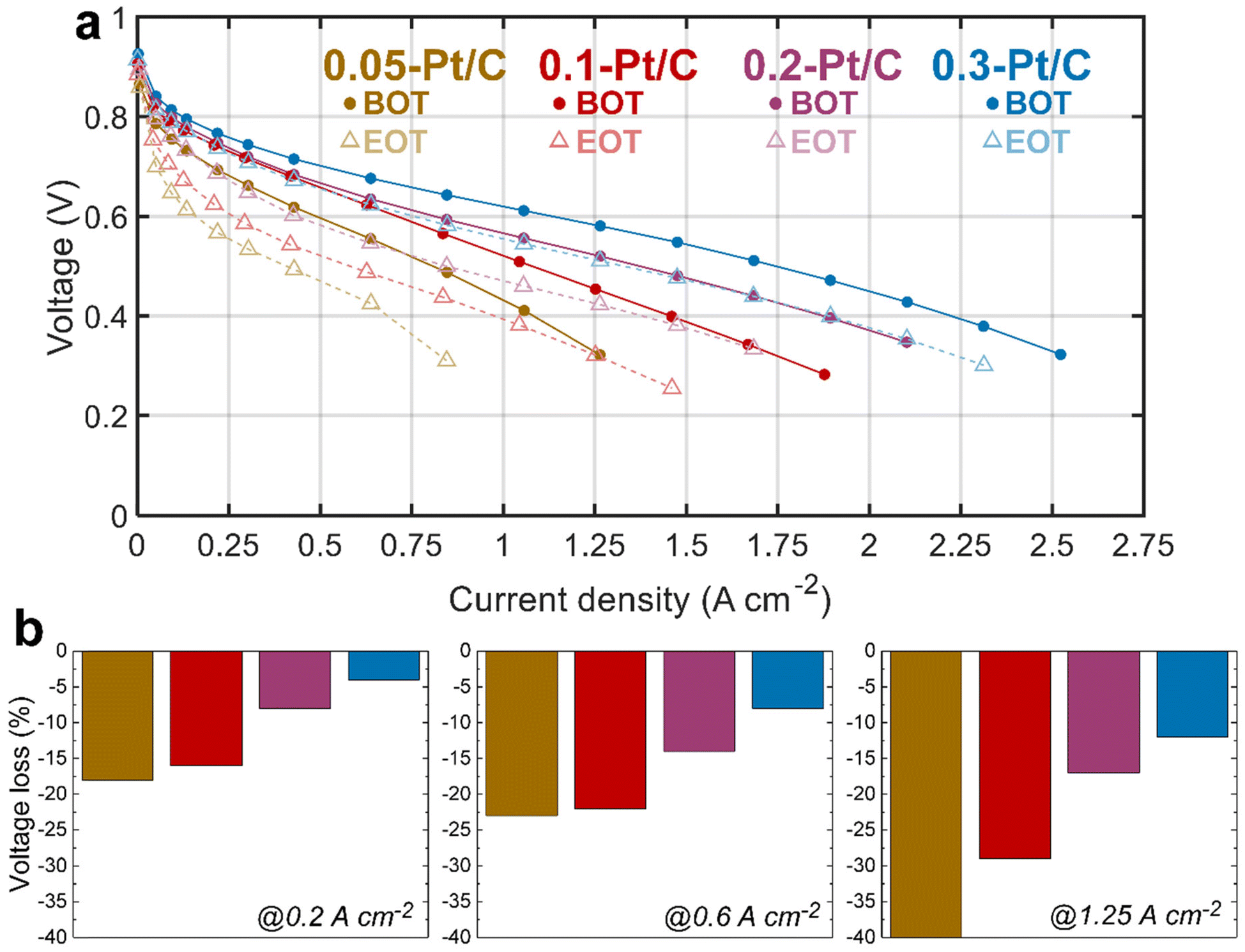 | ||
| Fig. 1 (a) Polarization curves for the MEAs comprising 0.05-Pt/C, 0.1-Pt/C, 0.2-Pt/C and 0.3-Pt/C cathode CLs at BOT and EOT; (b) voltage losses of selected points at low (0.2 A cm−2), medium (0.6 A cm−2) and high (1.25 A cm−2) current densities. | ||
| MEA | 0.05-Pt/C | 0.1-Pt/C | 0.2-Pt/C | 0.3-Pt/C |
|---|---|---|---|---|
| R hf/Ω cm2 | 0.114 | 0.109 | 0.086 | 0.070 |
| R ct/Ω cm2 | 0.213 | 0.090 | 0.087 | 0.070 |
| R d/Ω cm2 | 0.102 | 0.081 | 0.059 | 0.068 |
In particular, the pronounced mass-transport limitation observed for the smallest cathode Pt loading correlates with the increased resistance to oxygen transport within the cathode at high current densities (clearly visible in Fig. S1,† related to the EIS diagrams recorded for the four x-Pt/C MEAs at 1 A cm−2), in agreement with the literature.24–26
Comparing BOT vs. EOT, all the polarization curves exhibit significant voltage losses for all the investigated MEAs, which seems inversely correlated to the cathode Pt loading: the larger the loading the smaller the losses. It is more explicit in Fig. 1b, in which the two lowest Pt-loaded cathodes start to show a significant cell performance loss even at low current density (0.2 A cm−2) up to an early complete failure at high current density (1.25 A cm−2) for the 0.05-Pt/C MEA and ca. 30% maximum loss for 0.1-Pt/C MEA. In comparison, the 0.2-Pt/C and 0.3-Pt/C MEAs show much smaller performance losses, which means they are more robust than the MEAs with smaller cathode Pt loadings, the best results being recorded for the most Pt-loaded cathode throughout the current density range. The EIS diagrams (Fig. S1†) are totally in line with these observations. So, regardless the nature of the degradation phenomena (not known at that stage), one can already state that larger cathode Pt loading leads to better MEA robustness, at least for the present AST.
Fig. 2a presents the evolution of the cell voltage measured at (a relatively) high current density (1 A cm−2) versus time during the daily characterization step of the AST, for the four MEA compositions. As expected from the polarization plots of Fig. 1, the EIS data of Fig. S1† and Table 1, the BOT cell voltages at 1 A cm−2 decrease with the cathode Pt loading. Moreover, MEAs with small cathode Pt loading (0.05-Pt/C and 0.1-Pt/C) quickly lose cell performance (within 48 h) before exhibiting a lower time-depending degradation for longer durations. In contrast, for the largest cathode Pt loading MEA (0.3-Pt/C) this two-step performance decay versus time is much less pronounced. Comparing the slope values of cell voltage loss per hour, the order for the x-Pt/C cathodes is 0.05 > 0.1 > 0.2 ≫ 0.3 for the BOT (t < 48 h), then showing a slope inversion from 48 h to the EOT with roughly comparable values for large cathode Pt loadings (i.e. 0.2-Pt/C and 0.3-Pt/C) and low cathode Pt loadings (i.e. 0.1-Pt/C and 0.05-Pt/C) (Table 2). The cell voltage losses at 1 A cm−2 between BOT and EOT are found to drop near 50 mV for 0.3-Pt/C and 0.2-Pt/C and ≥100 mV for the two least loaded cathodes. We observe that the cell voltage drops between BOT and EOT are much smaller when measured closer to the OCV, ranging between 10–30 mV whatever the cathode Pt loading (Fig. 2a). Indeed, the OCV primarily depends on the extent of H2 crossover and this parameter is essentially determined by the quality of gas separation brought by the membrane. Here, the fluidic conditions are close for all samples with air/hydrogen flow rates being set to 170/27 standard liters per hour (sLph) for 0.3-Pt/C cathode and 110/17 sLph for the 0.05-Pt/C cathode. This shows that the membrane (i) does not significantly lose its barrier properties in the present AST procedure and (ii) does not seem more altered for smaller cathode Pt loadings. This conclusion is consistent with the absence of significant increase of hydrogen permeation currents during the AST, regardless of the cathode Pt loading employed.
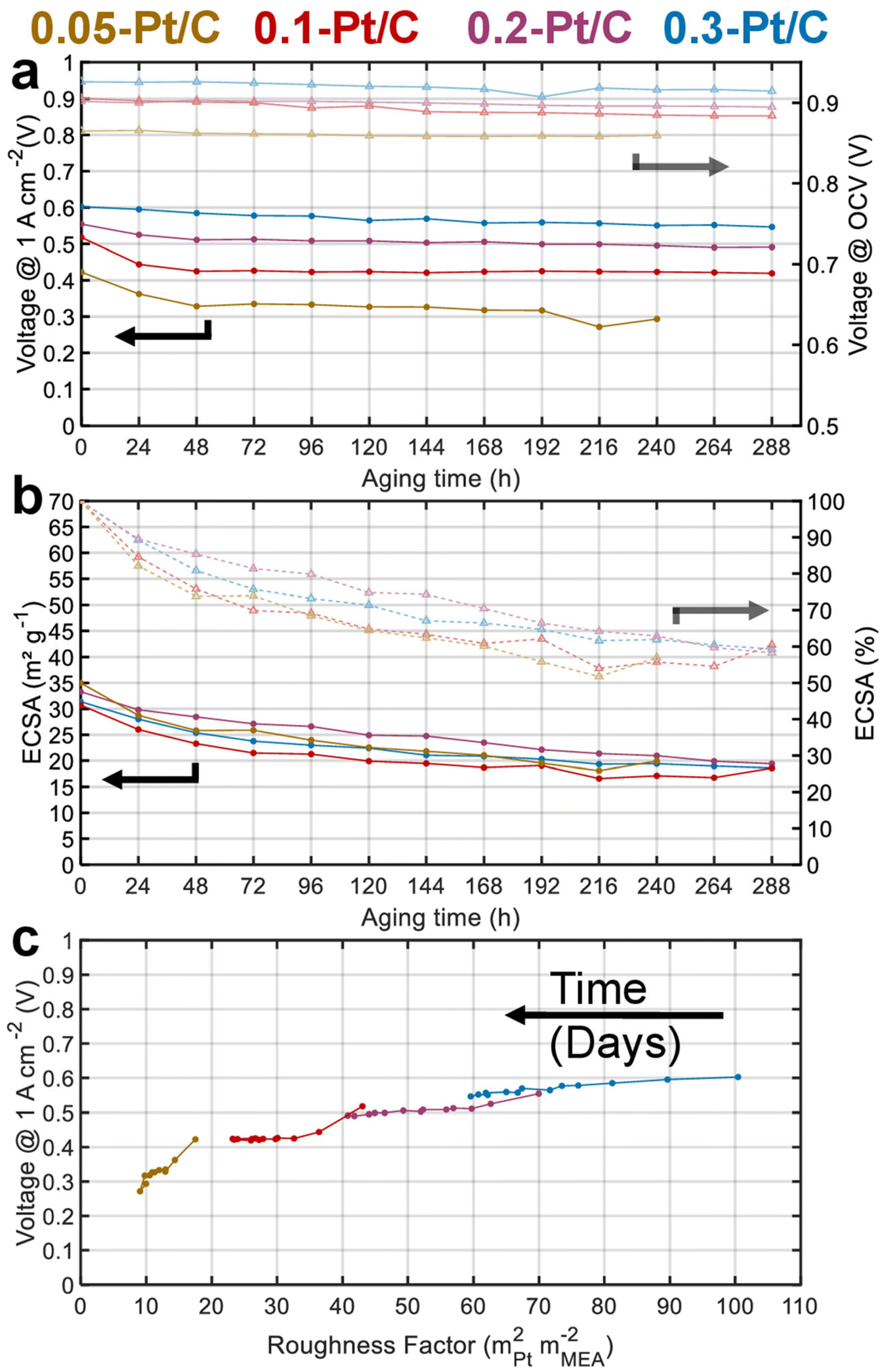 | ||
| Fig. 2 (a) Cell voltages measured at 1 A cm−2 during the daily characterization step and their OCV in the course of the AST; (b) ECSA mapping and its percentage loss; (c) cell voltage at 1 A cm−2vs. roughness factor, R.F., both values being measured in real-time along the 288 h of AST for 0.05-Pt/C, 0.1-Pt/C, 0.2-Pt/C and 0.3-Pt/C cathode CLs. | ||
| MEA | 0.05-Pt/C | 0.1-Pt/C | 0.2-Pt/C | 0.3-Pt/C |
|---|---|---|---|---|
| BOT < t < 48 h | 1962 | 1933 | 906 | 368 |
| 48 h < t < EOT | 95 | 16 | 258 | 151 |
Fig. 2b shows that all the cathode CLs have a similar electrochemical surface area (ECSA) between 30–35 m2 gPt−1 at BOT. The ECSA losses are essentially observed at the beginning of the AST for both lowest cathode Pt loading MEAs: 50% of the ECSA loss occurs within 48 h for the 0.05-Pt/C and 0.1-Pt/C MEA. This matches the cell performance losses measured at 1 A cm−2 in Fig. 2a, suggesting that ECSA losses drive the cell performance even at high current density for the low cathode Pt loading MEA. In contrast, the ECSA losses are more homogeneously distributed along the 288 h of AST for high cathode Pt loading MEAs (0.2-Pt/C and 0.3-Pt/C). Nevertheless, the cathodes lose about 40% of their ECSA throughout the AST, disregarding the initial Pt loading: the ECSA reaches ca. 20 m2 gPt−1 at EOT in all cases.
Fig. 2c plots the cell voltages measured at 1 A cm−2versus the cathode roughness factor (R.F.), both values being measured along the 288 h of AST (240 h for 0.05-Pt/C). As the roughness factor is the product of the cathode Pt loading and ECSA expressed in mPt2 mMEA−2,26 and as the latter parameter (ECSA) is near-constant for all x-Pt/C MEAs, its value decreases from the most to the least Pt-loaded CL at BOT. The roughness factor is correlated to the total oxygen transport resistance (RTotal) throughout the cell described by eqn (1), which can be broken down as the sum of the transport resistance in the flow channel (RCH), gas diffusion media (RDM), microporous layer (RMPL) and local oxygen transport in the CL (RO2).7 Given the same experimental cell conditions and similar components (except for the cathode Pt loading) for all the MEA, the oxygen transport resistance related to RCH, RDM and RMPL can be considered constant; hence, RTotal should essentially be affected by RO2 and R.F. Thus, the (bad) initial cell performance of the low Pt content cathode must be explained by the low value of R.F., possibly coupled with a large value of RO2 of the 0.05-Pt/C MEA (which is confirmed by EIS, Fig. S1† and Table 1), overall leading to a high oxygen transport resistance in the MEA at BOT. On the contrary, the high cathode Pt loading MEA (0.3-Pt/C) initially exhibits a low oxygen transport resistance at BOT; this trend overall agrees with the literature24,27 and notably the seminal study of Greszler et al.: “the electrode oxygen transport resistance is inversely proportional to platinum loading or, equivalently, platinum surface area”.7
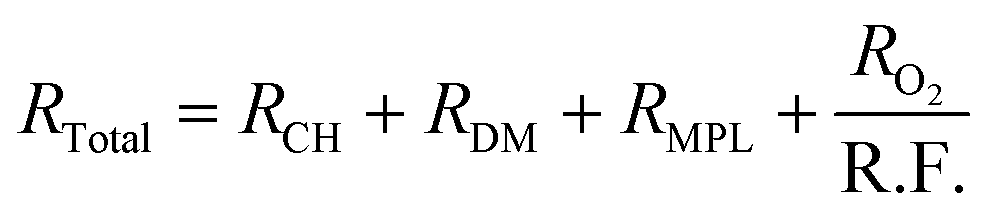 | (1) |
Upon aging, whatever the x-Pt/C MEA considered, the cathodes roughness factor continuously declines over the AST. This is ascribed to Pt losses in the membrane and growth/agglomeration of Pt nanoparticles within the CLs detected by SEM and TEM images, respectively (see section 2.2). Those effects are strongly correlated to the cell performance losses within 48 h for 0.05-Pt/C and 0.1-Pt/C, and seem more evenly distributed along the duration of the AST for 0.2-Pt/C and still more for 0.3-Pt/C. In addition, the smaller the roughness factor, the larger the slope of performance loss versus time, which explains why low-loading MEAs degrade faster than high-loading ones.
Since a segmented electrochemical cell was applied for the degradation studies,22,28–31 it was possible to track the local ECSA (and local current density) changes for all of the 20 segments along the gas channel. It is recalled here that segment #1 corresponds to the air inlet at the cathode and H2 outlet at the anode, while segment #20 corresponds to the air outlet at the cathode and H2 inlet at the anode. The evolution of degradation along the channel reactant pathway (captured by the values of local ECSA for each segment as a function of time of AST) is plotted in Fig. 3 complemented by steady-state current density (Fig. S2†). Should it be at BOT (blue curves) or EOT (red curves), the fluctuations of local ECSA observed along the gas channel are essentially similar. This means that these fluctuations likely originate from the cell design and not from the variable cathode Pt loadings tested (in general, segments close to turns in gas channels show smaller ECSA). The MEA with the lowest cathode Pt loading (0.05 mg cm−2) is an exception to this trend, though: it exhibits more pronounced heterogeneities close to the air outlet, possibly linked to its fabrication process. The cell design explanation holds for lower ECSA values obtained for the segments found close to the gas inlet (segments 1–4) and outlet (segments 17–20) compared to the average ECSA for the other segments (5 to 16). In addition to the cell design, the non-uniform distribution of O2 in the CLs32 during the AST is likely to cause non-uniform temperature and relative humidity distribution33,34 exacerbating these fluctuations during aging. For these reasons, different degrees of degradation could be found in the CLs in these segments.35 Therefore, to have a right evaluation of the Pt-loading effect in the MEA performance, the segments spotted close to the gas inlets and outlets were deliberately avoided for the physicochemical characterizations (see section 2.2). Whatever these (small) biases, these local characterizations do not seem to show any spectacular dependence of the local ECSA loss on the cathode Pt loading.
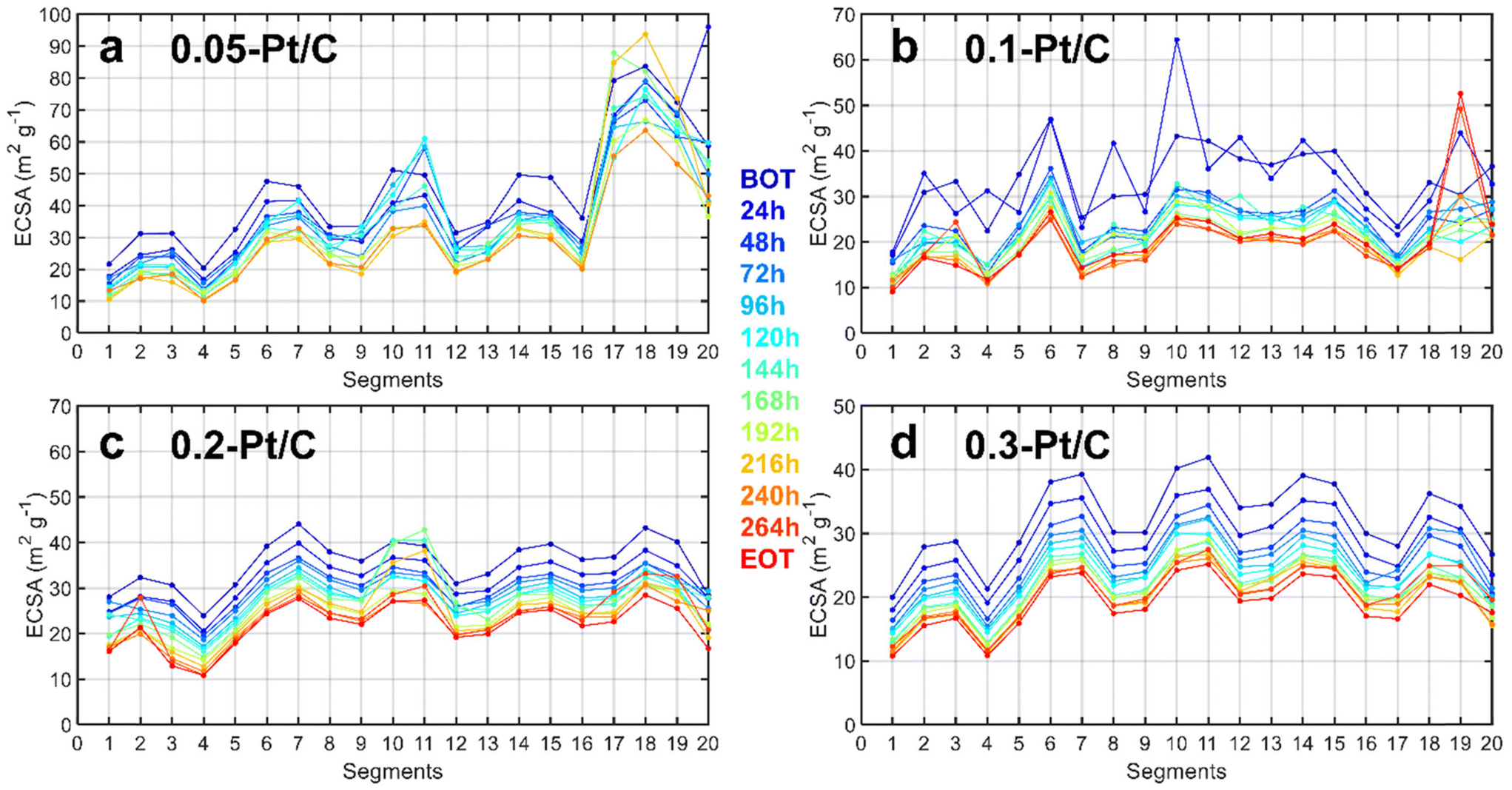 | ||
| Fig. 3 Local ECSA along the gas channel measured in a segmented cell for CLs (a) 0.05-Pt/C; (b) 0.1-Pt/C; (c) 0.2-Pt/C and (d) 0.3-Pt/C. Segment #1 corresponds to the air inlet at the cathode and H2 outlet at the anode, while segment #20 corresponds to the air outlet at the cathode and H2 inlet at the anode. | ||
2.2 Physicochemical characterizations
In this section, the MEAs with four different cathode Pt-loadings are physicochemically evaluated by TEM, GIXRD, cross-section SEM and Raman spectroscopy. As stated in the previous section, the regions analyzed correspond to segments 5 to 16 (i.e. not in the first and last pass of the channels), and it is assumed that within this “middle” area, there are no specific local heterogeneities caused by either the operating conditions or the cell design to be expected for a given sample.In terms of TEM analysis, the technique was used to assess the mean size of Pt nanoparticles and their distribution in the carbon substrate at BOT and EOT. Representative TEM images (Fig. 4) from the scrapped cathode powder reveal homogeneous spherical Pt nanoparticles distributed on the Vulcan XC72 carbon substrate with a little fraction of agglomerated and/or non-spherical nanoparticles. The average diameter of isolated and spherical Pt nanoparticles is ca. 2–3 nm, quantified in the four different x-Pt/C CLs at the BOT.
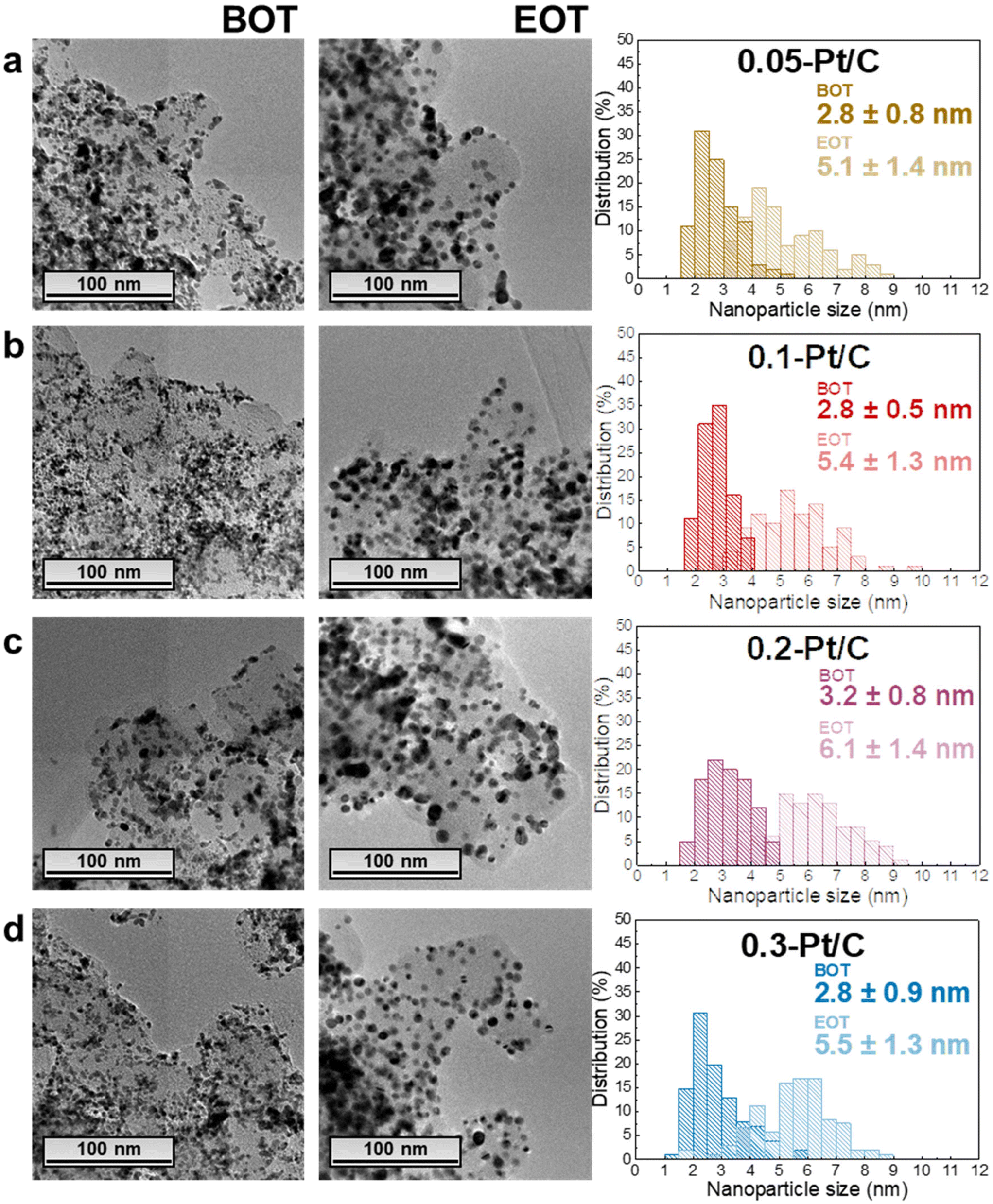 | ||
| Fig. 4 Representative TEM images, corresponding nanoparticle size distribution histograms and mean nanoparticles diameter values for the active materials scrapped from cathode CLs at BOT and EOT: (a) 0.05-Pt/C; (b) 0.1-Pt/C; (c) 0.2-Pt/C and (d) 0.3-Pt/C. | ||
The average diameter of the Pt nanoparticles grows dramatically to ca. 5–6 nm at EOT, this growth being accompanied by a widening of the nanoparticles size distribution histograms upon electrochemical aging (Fig. 4). These are typical signatures of the Ostwald ripening mechanism for Pt nanoparticles growth,36 which comprises the dissolution of the smallest nanoparticles due to their high surface energy, followed by the re-deposition of the formed Ptz+ ions onto larger nanoparticles to equilibrate the system. Hence, the small/large Pt nanoparticles become smaller/larger, broadening the particle size distributions, respectively, until all the small nanoparticles disappear. In addition, a second nanoparticle growth mechanism takes place between in-contact nanoparticles within the pre-existing agglomerates, leading to a sintering process (particles coalescence) which occurs in parallel to the Pt dissolution/re-deposition mentioned above.37 Finally, the rise in the number of agglomerates comparing EOT and BOT TEM images, shows that crystallite migration onto the Vulcan XC72 carbon support happens, likely following the gradual functionalization/corrosion of the carbon, as clearly highlighted in.38,39 In contrast, the anode CLs essentially exhibit unmodified Pt nanoparticle sizes (only measured for the 0.05-Pt/C and 0.3-Pt/C MEAs, the least- and most-loaded cathode CLs) evidencing that the procedure used in this work does not lead to significant anode damages (Fig. S3†).
Cross-sections of the MEAs were observed in SEM to evaluate whether the cathode Pt loadings influence the fate of the MEAs upon AST (Fig. 5a).
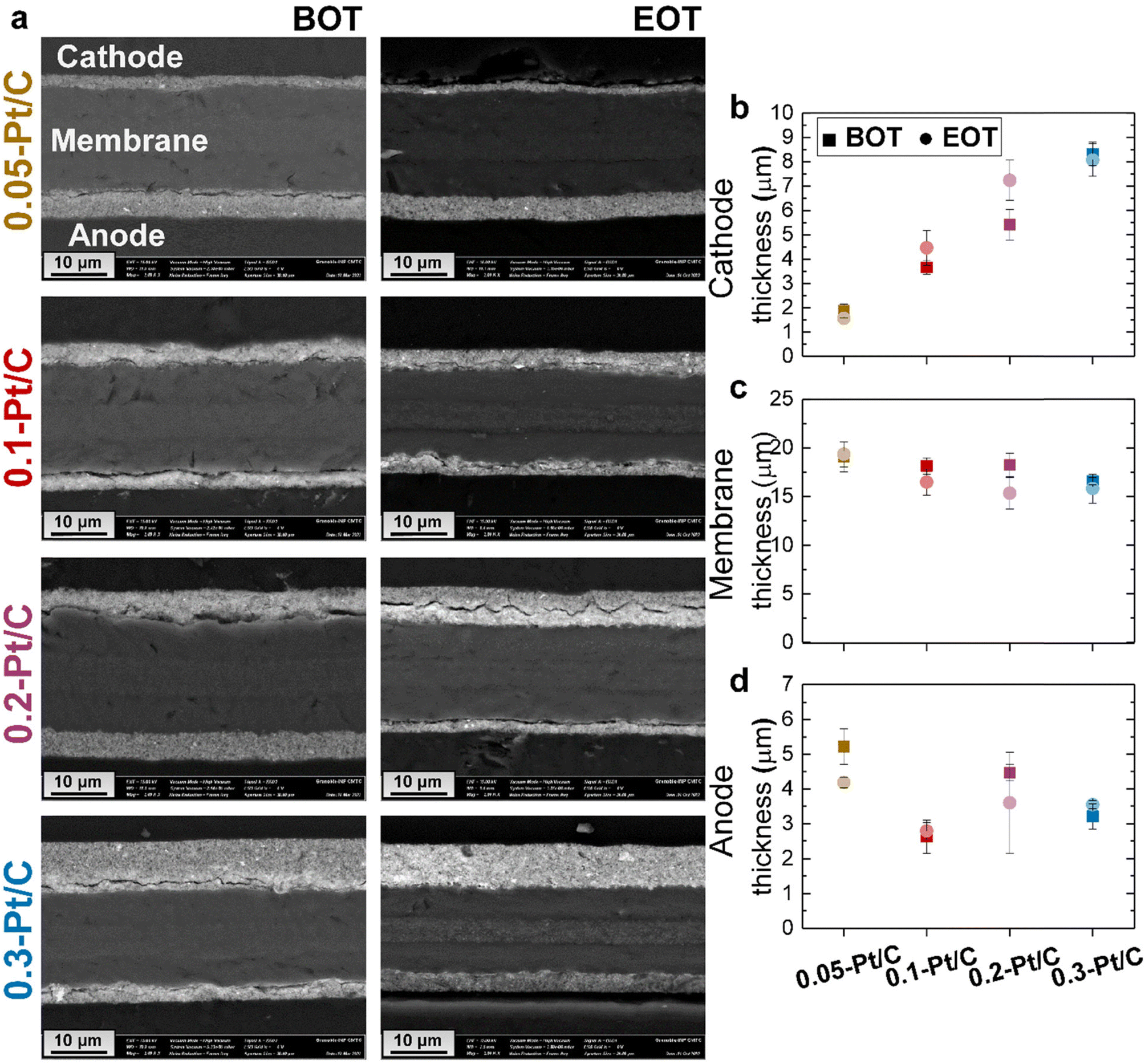 | ||
| Fig. 5 (a) Representative SEM images for the MEAs with different cathode Pt loadings: 0.05-Pt/C, 0.1-Pt/C, 0.2-Pt/C and 0.3-Pt/C at beginning of test (BOT) and end of test (EOT) with their respective (b) cathode, (c) membrane and (d) anode average thickness values. In any case, the cathode is the upper electrode and the anode is the lower electrode on the micrographs. | ||
At BOT, because the ink formulation and deposition methods were kept constant for all the x-Pt/C MEAs (given the fixed 50 wt% Pt/Vulcan XC72 catalyst and I/C ratio), increasing the cathode Pt loading results in increasing cathode CL thickness, as observed for the SEM micrographs (Mag 2 k) in Fig. 5a and the mean cathode CL values determined from two SEM images and regions in Fig. 5b: the cathode CL thickness near-linearly depends on its Pt loading, which shows that the porous texture/density of the CL is essentially maintained constant whatever the aerial Pt loading. The membrane exhibits a very slight opposite trend, smaller thickness being observed at larger cathode CL loading. One can speculate that the cathode thickness influences the degree of compression of the membrane, resulting in the decreasing trend in Fig. 5c: the thickest cathode (0.3-Pt/C) compresses the membrane up to 4 μm more than the thinnest one (0.05-Pt/C) when the MEA is assembled. The initial anode thickness has a more variable trend, in line with the variable practical loading of the anodes (respectively 0.18, 0.14, 0.11 and 0.13 mgPt cm−2 for increasing values of x in x-Pt/C MEAs).
At EOT, the cathode CLs exhibit similar mean thickness values, within the error bars compared to BOT; this means that the carbon corrosion, if any, was not significant enough in this AST to result in major CO2 generation associated with the collapse of the active layer, which had been observed in.38–40 The rather low value of the AST maximum potential (0.8 V vs. RHE, except for the plateau at OCV) explains this minor extent of carbon corrosion, the process being highly potential-dependent.41,42
Active area losses from PEMFC cathode are typically promoted by three main phenomena: Pt nanoparticles dissolution into Pt2+, re-deposition in the CLs (causing the nanoparticles' growth by Ostwald ripening), or Pt2+ migration/diffusion to the ionomer and the membrane (Pt depletion from the CL), and possibly Pt crystallites migration over the carbon support (upon its functionalization/corrosion), yielding agglomerated nanoparticles.27,36,38,43 Their growth by Ostwald ripening and agglomeration were established by the TEM images and particle size distribution histograms in Fig. 4. Pt corrosion and redeposition in the membrane is visualized by higher magnification images obtained by SEM (Mag 5 k) focusing on the membrane region (Fig. 6). The metal nanoparticles are found gathered as a band within the membrane, attesting that Pt was indeed lost to the membrane during the AST, as observed by Guilminot et al.38 Those bands of Pt nanoparticles are not cathode Pt loading-dependent for the present samples: the band is situated close to the cathode and the Ptz+ ions generated at the cathode through eqn (2)–(4)38 diffuse on roughly similar distances within the membrane for all the x-Pt/C CLs. The position of this band likely depends on the fluidics conditions of the cell (H2 and air pressures/fluxes), but also on the potential alternation (Pt2+ cations migrate to the cathode under current flow and mostly diffuse towards the anode at open-circuit).
| Pt(s) ⇌ Pt2+ + 2e− | (2) |
| Pt(s) + H2O ⇌ PtO(s) + 2H+ + 2e− | (3) |
| PtO(s) + 2H+ ⇌ Pt2+ + H2O | (4) |
 | ||
| Fig. 6 SEM images for the MEAs with different cathode Pt loadings: (a) 0.05-Pt/C; (b) 0.1-Pt/C; (c) 0.2-Pt/C and (d) 0.3-Pt/C at EOT focusing on the Pt redeposition within the membrane. | ||
GIXRD measurements were performed on the four different metal-loaded cathode CLs for the detection of the Pt crystalline phase and their crystallite sizes. The analyzed samples comprised both the cathode and anode CLs separated by the PFSA membrane (that contains a PTFE contribution). Due to the small thickness of those three components and to avoid the detection of the anode side, the grazing incidence X-ray diffraction technique was the method utilized to enable the quantification of the cathode CL without hindrance from the anode CL. Then, by positioning the X-rays source on the cathode side, the PTFE contribution from the membrane backbone is perfectly visualized in X-ray diffractograms (Fig. 7) at 2θ ≈ 18° (PDF 00-060-1504) for all samples. This means that the X-rays pass deep enough through the cathode CL and reach the membrane disregarding the cathode Pt loadings and CL thicknesses at stake.
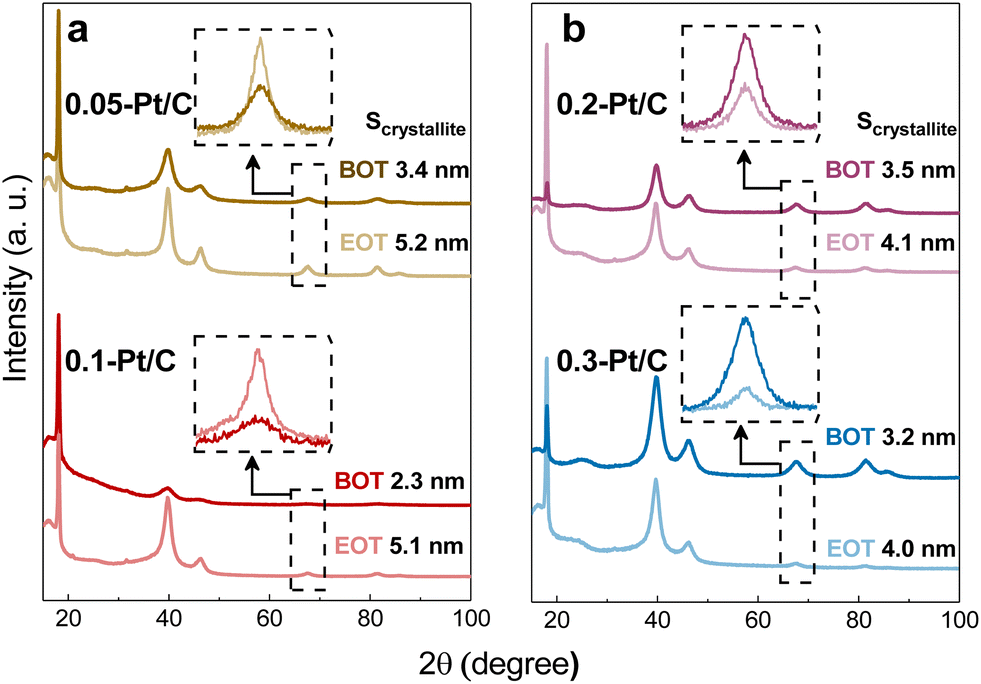 | ||
| Fig. 7 Grazing incidence X-ray diffractrograms for the cathode CLs: (a) 0.05-Pt/C and 0.1-Pt/C and (b) 0.2-Pt/C and 0.3-Pt/C at beginning of test (BOT) and end of test (EOT) with their respective crystallite size values calculated at the highlighted graph region (Pt 220; 2θ ≈ 68°). | ||
Focusing on the crystallinity of the catalyst, typical Pt/C profiles are detected, which comprise five diffraction peaks at 40, 46, 68, 82 and 86° (PDF 00-004-0802); these peaks are related to the polycrystalline Pt phases. Since the two most intense diffraction peaks are set in the range from 2θ = 40° to 50°, which is in close contact to the broad carbon peak and some PTFE contributions, as well the other two metal contributions are really close to each other at high 2θ values, the isolated peak at 2θ = 68° (Pt (220)) was chosen to determine the Pt crystallite sizes. They were determined by fitting the peak and applying Scherrer's equation following the equipment features described in section 4.1.3. The Pt crystallites have mean sizes (Scrystallite) of ca. 2–3 nm (Table 3) well matching the nanoparticles (NPs) size derived from TEM images at BOT. Also, the growth of crystallite sizes to 4–5 nm follows roughly the same trends for all samples at EOT.
| Cathode CLs | TEM: NP size (nm) | XRD: Scrystallite (nm) | ΔNP (nm) | ΔScrystallite (nm) | ||
|---|---|---|---|---|---|---|
| BOT | EOT | BOT | EOT | |||
| 0.05-Pt/C | 2.8 ± 0.8 | 5.1 ± 1.4 | 3.4 | 5.2 | +2.3 | +1.8 |
| 0.1-Pt/C | 2.8 ± 0.5 | 5.4 ± 1.3 | 2.3 | 4.6 | +2.6 | +2.3 |
| 0.2-Pt/C | 3.2 ± 0.8 | 6.1 ± 1.4 | 3.5 | 4.1 | +2.9 | +0.6 |
| 0.3-Pt/C | 2.8 ± 0.9 | 5.5 ± 1.3 | 3.2 | 4.0 | +2.7 | +0.8 |
In contrast, the observed growths (ΔNP and ΔScrystallite) from BOT to EOT do not match and may be divided into two groups regarding the value of cathode Pt loadings (and cathode CL thicknesses): the thin (0.05-Pt/C and 0.1-Pt/C) and the thick ones (0.2-Pt/C and 0.3-Pt/C) do not follow the same trend, as exhibited in Fig. 4a and b and Table 3. Considering ΔNP and ΔScrystallite as the NP and Scrystallite at EOT subtracted by their corresponding values at BOT, thin cathode CLs are characterized by ΔNP ≈ ΔScrystallite, while thick cathode CLs correspond to ΔNP > ΔScrystallite (Table 3). Here, two possible explanations can be put forth: (i) the Pt band in the membrane (composed of very large Pt crystallites) is likely more detected by the grazing X-rays for the thin cathode CLs 0.05-Pt/C and 0.1-Pt/C cathodes (because the thin CLs let more X-rays pass through the whole cathode CL thickness) resulting in overall larger crystallites, while the Pt band crystallites would be less detected when thick cathode CLs are employed, resulting in overall smaller crystallites. In short, the Pt band relative weight associated with thin CLs (containing low amounts of Pt) is thus proportionally larger in the global diffraction signal; (ii) different degrees of degradation takes place within the thin and thick cathode CLs, owing to the different potentials witnessed by cathodes of different Pt loading. Indeed, at the highest current density, large-loading cathodes experience a higher potential (better performance) than low-loading cathodes (see Fig. 1a, 2a and S4†); to be more specific, at BOT, the 0.05-Pt/C cathode potential is close to 0 V at 1.7 A cm−2, when it is only ca. 0.5 V for the 0.3-Pt/C cathode; the differences of cathode potential at OCP or at low currents is much smaller (Fig. 1a and S4†), and can be neglected. In result, the Pt2+ redeposition in the cathode CL (promoted at the lowest cathode potential) is more likely for the thinnest cathode than for the thickest one. To the authors' opinion, this second hypothesis better accounts for the larger crystallites observed for thin CLs, even though from a mass-transport point of view, Pt2+ ions should exit faster thin than thick CLs. These heterogeneities are in agreement with some related literature.36
From the results presented above, the fate of the Pt nanoparticles may only (slightly) differ according to the different potential experienced by the cathodes during the high-current phase of the AST at various cathode Pt loadings. In addition, the carbon substrate, and in particular its degree of oxidation, may also influence the catalytic activity losses. The carbon substrate oxidation likely takes place for the Pt/C catalysts.37,42 To understand carbon corrosion in the cathode CLs, Raman spectroscopy was used to investigate the carbon substrate of the cathode at BOT and EOT (Fig. 8). The Raman spectra (and related ratio of intensity of the defective and graphitic bands, Id/Ig) demonstrate negligible (or small) differences comparing the BOT vs. EOT: the Id/Ig ratio values can be considered nearly constant or within the error bar of the technique. This suggests a very small effect of carbon corrosion for the present AST, regardless the value of cathode Pt loading; this corroborates the results obtained on the SEM analyses, that showed comparable cathode CL thicknesses at BOT and EOT. The small effects caused by carbon corrosion can be related to the maximum cell voltage (nearly corresponding to the cathode potential, versus RHE) of 0.8 V applied in the AST conditions, a potential that does not severely yield complete corrosion into CO2 (stressing the electrode to E > 1 V vs. RHE would have led to massive carbon corrosion, that would be evident in Raman spectra41,42 and cross-section SEM images44). It however leads to superficial oxygenated species production,41 that could be transformed into CO2 by a Pt-assisted mechanism. In the present case, because the same Pt/C catalyst was used in all the x-Pt/C CLs, there is no reason to posit that carbon corrosion should be different depending on the cathode Pt loading, in agreement with the experimental data. As a result, the degradation observed here can be mostly attributed to the Pt nanoparticles' dissolution followed by growth (Ostwald ripening) and slight migration onto the carbon support and agglomeration, or Pt2+ transport into the membrane, where these species are reduced into a Pt-band by crossover H2.
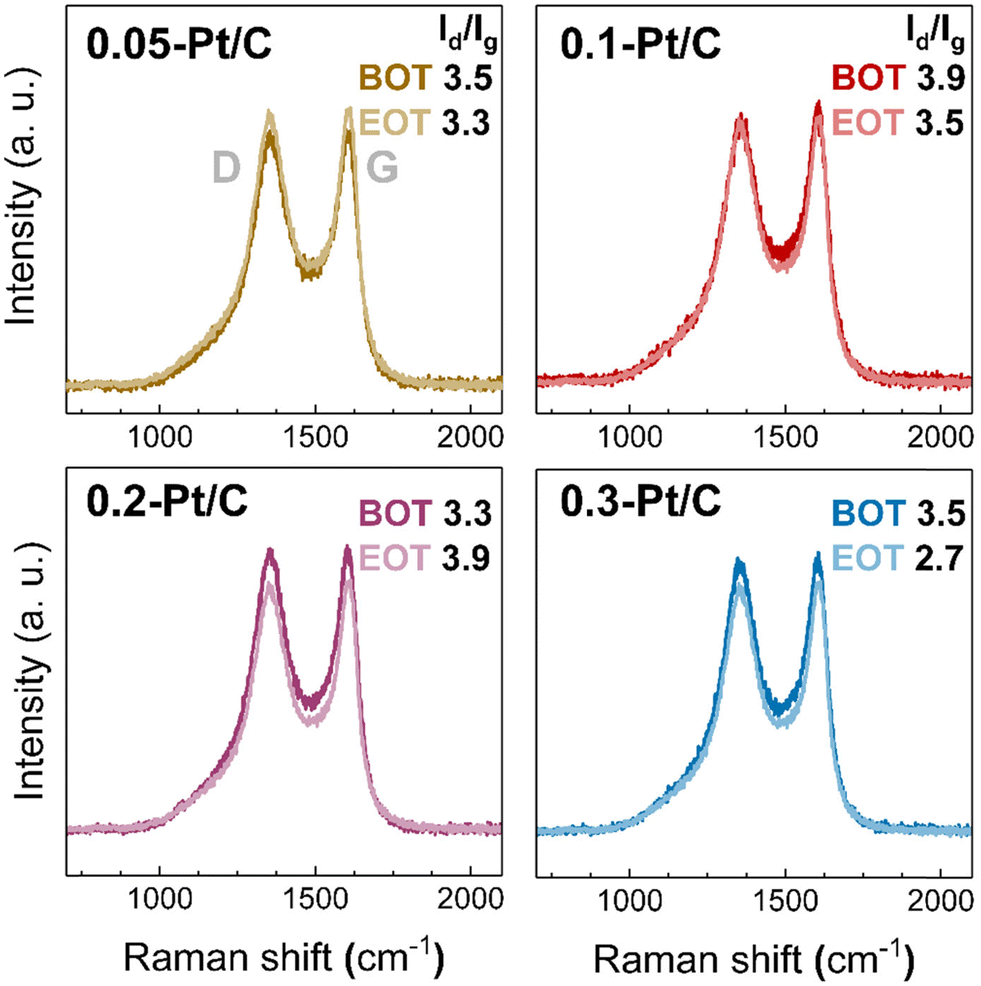 | ||
| Fig. 8 Raman spectra for 0.05-Pt/C, 0.1-Pt/C, 0.2-Pt/C and 0.3-Pt/C cathode CLs at beginning of test (BOT) and end of test (EOT) coupled to their respective Id/Ig ratio. | ||
3 Conclusions
In this contribution, MEAs varying only by their cathode Pt loading were investigated in a multiple-stressor accelerated stress test in a segmented PEMFC. These MEAs were characterized over the duration of the test in terms of polarization curves, electrochemical impedance spectroscopy; their core materials were also evaluated physicochemically, with special emphasis on the respective thicknesses of the anode, cathode and membrane layers, the definition (size/shape/degree of agglomeration) of the carbon-supported Pt catalyst nanoparticles and the extent of carbon corrosion/functionalization.The thinnest (lowest-Pt loading) cathode evidently behaves worst in terms of initial activity, being hindered by their low Pt content in the low current density (activation) region and adverse oxygen (and maybe proton) transport resistance in the high current density (mass-transport) region of the polarization curve. These trends have been fully confirmed by EIS measurements. In terms of degradation of performance during the aging, the rate is larger in the early stages (t < 48 h) of test for the lowest cathode Pt loading, and then slows down; whereas the overall loss of ECSA is in the end of the AST quite similar whatever the cathode Pt loading, smaller cathode Pt loading results in larger loss of performance.
The TEM data point towards a clear Ostwald ripening of the cathode Pt/C catalyst over the AST, that is more pronounced for the small Pt loading, owing to the lower operating potential witnessed by the materials during the large current phases of the AST that eases Pt redeposition; the anode Pt/C catalysts is essentially unharmed in the meantime, as confirmed by XRD analyzes. Some agglomeration of the cathode Pt/C nanoparticles is also witnessed, likely owing to superficial carbon functionalization and related Pt crystallite migration; indeed, no consequent carbon corrosion has been witnessed by Raman spectroscopy or by any change of the anode or cathode thickness over the AST. Also, the oxidized Pt2+ ions formed by Pt corrosion in the cathode CLs are diffused and re-deposited within the membrane close to the cathode|membrane interface. Thus, the mechanisms of Pt/C degradation (at the cathode, mostly) are essentially unchanged for the various cathode Pt loadings, only the extent of Ostwald ripening depending partly to the cathode Pt loading.
4 Materials and methods
4.1 Preparation of the membrane-electrode assemblies (MEA)
The MEAs studied in this work were home-made and based on commercial components: a reinforced PFSA membrane (MX 820.15 from Gore), gas diffusion layer (GDL) from Sigracet (ref. 22 BB) at both anode and cathode sides and Vulcan carbon (XC72; Cabot) supported Pt nanoparticles from Tanaka (50 wt% Pt, TEC10V50E) in both catalytic layers (CL) mixed with Nafion® D2020 ionomer solution. The anode and cathode CL inks were prepared similarly to a previous work of the authors:45 briefly, the electrocatalyst was mixed with ultrapure water and ethanol with 0.24 ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water mass ratio. Then, 3 mm diameter zircon beads were added and the ink was mixed overnight with a roller mill (IKA roller 10 basic) at room temperature. The Nafion® D2020 solution was incorporated afterward into the vial with an I/C ratio of 0.7 and the solution was mixed again with the roller mill for one day. The inks were finally deposited on an inert PTFE support (250 μm thick) by blade coating the next day. The electrode loading was adjusted by the height of the doctor blade and then controlled afterwards. For the lowest loading 0.05 mgPt cm−2, the ink was diluted by a factor of 2 keeping the ethanol/water ratio constant to reach the target by blade coating. The global catalyst loading was checked by weighing and comparing the bare PTFE substrate with/without dried electrodes. Additional analyses by X-ray fluorescence (Fischer XVD-SDD) were carried out on each electrode to check the local layer homogeneity, to control the global loading (in a complementary manner to weighing) and to select the most suitable layer for each targeted loading. The catalyst-coated membrane (CCM) was then finalized by decal-transfer of both electrodes onto the membrane using a hot-press at 145 °C under 1 MPa for 180 s using in-house hardware. The GDLs were pressed onto the MEA using PET gaskets (23 μm) and PTFE seals (150 μm) to control their thickness, reaching about 20% of compression.
:water mass ratio. Then, 3 mm diameter zircon beads were added and the ink was mixed overnight with a roller mill (IKA roller 10 basic) at room temperature. The Nafion® D2020 solution was incorporated afterward into the vial with an I/C ratio of 0.7 and the solution was mixed again with the roller mill for one day. The inks were finally deposited on an inert PTFE support (250 μm thick) by blade coating the next day. The electrode loading was adjusted by the height of the doctor blade and then controlled afterwards. For the lowest loading 0.05 mgPt cm−2, the ink was diluted by a factor of 2 keeping the ethanol/water ratio constant to reach the target by blade coating. The global catalyst loading was checked by weighing and comparing the bare PTFE substrate with/without dried electrodes. Additional analyses by X-ray fluorescence (Fischer XVD-SDD) were carried out on each electrode to check the local layer homogeneity, to control the global loading (in a complementary manner to weighing) and to select the most suitable layer for each targeted loading. The catalyst-coated membrane (CCM) was then finalized by decal-transfer of both electrodes onto the membrane using a hot-press at 145 °C under 1 MPa for 180 s using in-house hardware. The GDLs were pressed onto the MEA using PET gaskets (23 μm) and PTFE seals (150 μm) to control their thickness, reaching about 20% of compression.
In the study, the nature of the GDLs, anode and membrane, the cathode I/C ratio and CL deposition technique were deliberately kept constant, so to produce similar Pt density within CLs cathode loadings. The MEAs differed by their cathode Pt loadings which are 0.05, 0.14, 0.21 and 0.32 mgPt cm−2, their anode loading being roughly constant: 0.18, 0.14, 0.11 and 0.13 mgPt cm−2, respectively. The MEAs labels are defined as 0.05-Pt/C, 0.1-Pt/C, 0.2-Pt/C and 0.3-Pt/C, named after the targeted Pt loading at their cathode, which is in first approximation the only variable parameter in the study. In any case, new (non-electrochemically degraded) MEAs were fabricated without GDL to enable physicochemical comparison of their CLs and membrane with those of the corresponding aged samples.
4.2 Physico-chemical characterization
4.3 Fuel cell testing measurements and durability protocols
The durability tests were performed using the 5 × 5 = 25 cm2 segmented cell of PEMFC shown in Fig. S5 in ESI.† The flow fields of this cell are identical on both sides with 5 parallel serpentine channels (5 passes) of 0.6 mm in depth, 1 mm in width, and 1 mm ribs. Both plates were made of 316L stainless steel coated with a thin gold layer to lower the electrical contact resistance with the gas diffusion layers. The cathode plate is divided into 20 electrically-insulated segments, so that the current can be collected individually on each of them to assess the homogeneity of the MEA performance as well as local impedances, voltammograms (acquisition of the local electrochemical surface area, ECSA), and hydrogen permeation currents. Air and hydrogen were always supplied to the cell in counter-flow, with segment #1 corresponding to the air inlet at the cathode and H2 outlet at the anode, and segment #20 corresponding to the air outlet at the cathode and H2 inlet at the anode.The acquisition of local currents - including during EIS – was carried out by measuring the ohmic drop on 20 calibrated shunt resistors of 5 mΩ using a homemade electronic card and a ×600 electronic gain. In regular operating conditions (i.e. aging tests and acquisition of the impedance spectra or the polarization curves), the PEMFC-overall-current intensity was imposed by the electronic load (Armel 0–100A/0–10 V) and the cell voltage measured between the anode and cathode plates (global cell voltage). When voltammograms and hydrogen permeation currents were recorded, the cell was automatically connected to a bipolar power source (Kikusui PBZ-20-20A) to impose positive and negative voltages. A supplementary 5 mΩ shunt resistance was used to get the PEMFC overall current, while the local currents were measured with the same electronic card as in normal operation.
The cell was thermoregulated by a water cooling circuit and a thermostatic bath. The pressure was regulated by valves located at the gas outlet and inlet, and the relative humidity was controlled through the temperature of a calibrated water bubbler. More details about the segmented PEMFC characteristics and operating conditions are available in ref. 22, 29, 30 and 46 with another geometry or31 with that specific cell geometry.
The accelerated stress tests (AST) that were performed herein combine humidity-cycling, open circuit voltage (OCV) hold and load cycling; it enables to adjust independently the number of load cycles – to emphasize the stress on the electrodes-and that of humidity cycles as well as the duration of OCV hold – to emphasize the stress on the membrane and the electrode ionomer. The principle of such combined-stressor ASTs consists of keeping the gas flow rates constant, and their humidity to a low or medium value.21,28,47–49 In the present case, the gases were slightly humidified (50% RH) and their flow rates did correspond to hydrogen and air stoichiometries of 1.5 and 4, respectively, at the highest current density (between 1.15 A cm−2 and 1.8 A cm−2, depending on the MEA used). Thus, the stoichiometries increased to very high values when the current density was set to the lowest values (0.04 to 0.081 A cm−2 again, depending on the MEA used) and to infinite values during the 27 seconds OCV hold at the end of each sequence. The low and high current values were chosen to yield a cell voltage of 0.8 and 0.4 V, respectively, assuming this would induce sufficient potential cycling to accelerate the degradation of cathode materials.
Fig. S6† shows the complete AST sequence, which overall lasts 105 seconds; it consists of a 52 s hydration stage (with an alternation of 1 s at low current and 3 s at high current), followed by a dehydration stage (3 s at low current and 1 s at high current) including a 27-second OCV hold step. This AST has been used in some of our previous works,21,28 because of its ability to combine, in addition to potential cycling, a membrane chemical stress during OCV hold (through the formation of hydrogen peroxide), and a mechanical stress via the humidity cycling. Although the membrane was not targeted in this work, it has been chosen to keep the sequence (i) to induce a global stress to the MEA that remains close to real-life operation and (ii) because hydration and dehydration cycles as well as free radical formation are expected to attack the ionomer in the electrodes. Indeed, previous results of some of the authors of this work have shown a considerable increase of the electrode degradation rate21,28 when chemical and mechanical stressors were added to load, and thus potential, cycling.
The AST sequence was repeated for ten to twelve days (i.e. 10 to 12 times 24 hours), with a characterization stage performed every 24 hours (lasting about 2 hours) to monitor the decrease of the PEMFC performance and material degradation in general. This characterization stage included:
1. 60 min operation at a constant current density of 1 A cm−2.
2. Measurement of the local and global impedance spectra in galvanostatic mode at 1 A cm−2-except otherwise stated-using a peak-to-peak perturbation amplitude of 10%, with frequencies ranging from 20 mHz to 10 kHz, with 10 to 20 data points per decade.
3. Measurement of the polarization curve in two steps: a first current sweep in 60 s increments from low to high values, and then a sweep in the opposite direction. Once both scans have been completed, the average curve is then calculated using the average values of the last 30 s of each step of the forward and reverse scans. A difference between the forward and backward curve can be observed in some cases, possibly due to the formation of platinum oxides at high potential, and/or different hydration states of the cell.50
4. Finally, the hydrogen permeation current and electrode ECSA were measured, the latter on both the anode and cathode sides: the anode or cathode compartment is first flushed for 10 min with nitrogen (50% RH) while the opposite electrode is fed with hydrogen, and the hydrogen permeation current is recorded at 0.6 V. Then, the nitrogen flow is stopped and the ECSA is measured by cycling the voltage between the working and the counter/reference electrode, between 0.1 and 0.8 V, at a sweep rate of 50 mV s−1.
Before each AST, the new MEAs were subjected to a four-hour break-in stage, consisting of the repetition of potentiostatic steps at OCV (30 s), 0.6 V (45 s) and 0.3 V (60 s). The PEMFC was then operated for two hours at a constant current density of 1 A cm−2 to make sure the MEA performance was stable. Then, the average current densities corresponding to FC voltage of about 0.4 and 0.8 V were assessed and the AST was started.
Author contributions
CM elaborated the MEAs. WAI and PW performed the electrochemical (PEMFC) characterizations; RS and QL performed most of the physicochemical experiments and analyzed the corresponding data. MM performed the Raman analyses. RS and MC wrote the first version of the manuscript. OL, JM, JD, FM and MC planned the study and reviewed the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work is funded by the EIT Raw Materials ALPE project, co-funded by the European Union (grant EIT/RAW MATERIALS/SGA2020/1). The PhD thesis of QL is funded by the PEMFC95 project, funded by the “France 2030” government investment plan managed by the French National Research Agency, under the reference “ANR-22-PEHY-0005”. Some of this work has been performed within the framework of the Centre of Excellence of Multifunctional Architectured Materials “CEMAM”, Grenoble France no ANR-10-LABX-44-01. RS, QL and MC thank Stéphane Coindeau for the XRD experiments, and Francine Roussel and Camille Jourdin for the SEM images.References
- R. L. Borup, A. Kusoglu, K. C. Neyerlin, R. Mukundan, R. K. Ahluwalia, D. A. Cullen, K. L. More, A. Z. Weber and D. J. Myers, Recent developments in catalyst-related PEM fuel cell durability, Curr. Opin. Electrochem., 2020, 21, 192–200 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- O. Lottin, B. Antoine, T. Colinart, S. Didierjean, G. Maranzana, C. Moyne and J. Ramousse, Modelling of the operation of polymer exchange membrane fuel cells in the presence of electrodes flooding, Int. J. Therm. Sci., 2009, 48, 133–145 CrossRef CAS.
- K. Kodama, T. Suzuki, K. Shinozaki and R. Jinnouchi, Translating insights from experimental analyses with single-crystal electrodes to practically-applicable material development strategies for controlling the Pt/ionomer interface in polymer electrolyte fuel cells, JPhys Energy, 2023, 5, 014018 CrossRef.
- H. Iden, A. Ohma, T. Tokunaga, K. Yokoyama and K. Shinohara, Measurement of a new parameter representing the gas transport properties of the catalyst layers of polymer electrolyte fuel cells, Phys. Chem. Chem. Phys., 2016, 18, 13066–13073 RSC.
- A. Kongkanand and M. F. Mathias, The priority and challenge of high-power performance of low-platinum proton-exchange membrane fuel cells, J. Phys. Chem. Lett., 2016, 7, 1127–1137 CrossRef CAS PubMed.
- T. A. Greszler, D. Caulk and P. Sinha, The impact of platinum loading on oxygen transport resistance, J. Electrochem. Soc., 2012, 159, F831–F840 CrossRef CAS.
- M. Chatenet, L. Dubau, N. Job and F. Maillard, The (electro)catalystmembrane interface in the proton exchange membrane fuel cell: Similarities and differences with non-electrochemical catalytic membrane reactors, Catal. Today, 2010, 156, 76–86 CrossRef CAS.
- S. Woo, S. Lee, A. Z. Taning, T. H. Yang, S. H. Park and S. D. Yim, Current understanding of catalyst/ionomer interfacial structure and phenomena affecting the oxygen reduction reaction in cathode catalyst layers of proton exchange membrane fuel cells, Curr. Opin. Electrochem., 2020, 21, 289–296 CrossRef CAS.
- M. C. M. Tempelaere and M. Zimmermann, 3D-structured electrocatalysts for improved mass-transfer in proton-exchange membrane fuel cell cathodes, Curr. Opin. Electrochem., 2023, 41, 101353 CrossRef CAS.
- K. Malek, M. Eikerling, Q. Wang, T. Navessin and Z. Liu, Self-organization in catalyst layers of polymer electrolyte fuel cells, J. Phys. Chem. C, 2007, 111, 13627–13634 CrossRef CAS.
- K. Malek, T. Mashio and M. Eikerling, Microstructure of catalyst layers in PEM fuel cells redefined: A computational approach, Electrocatalysis, 2011, 2, 141–157 CrossRef CAS.
- S. A. Berlinger, A. Chowdhury, T. Van Cleve, A. He, N. Dagan, K. C. Neyerlin, B. D. McCloskey, C. J. Radke and A. Z. Weber, Impact of platinum primary particle loading on fuel cell performance: Insights from catalyst/ionomer ink interactions, ACS Appl. Mater. Interfaces, 2022, 14, 36731–36740 CrossRef CAS PubMed.
- Y. Fukuyama, T. Shiomi, T. Kotaka and Y. Tabuchi, The impact of platinum reduction on oxygen transport in proton exchange membrane fuel cells, Electrochim. Acta, 2014, 117, 367–378 CrossRef CAS.
- W. Olbrich, T. Kadyk, U. Sauter and M. Eikerling, Review—Wetting phenomena in catalyst layers of PEM fuel cells: Novel approaches for modeling and materials research, J. Electrochem. Soc., 2022, 169, 054521 CrossRef CAS.
- K. Kudo, R. Jinnouchi and Y. Morimoto, Humidity and temperature dependences of oxygen transport resistance of Nafion thin film on platinum electrode, Electrochim. Acta, 2016, 209, 682–690 CrossRef CAS.
- Y. Fukuyama, T. Shiomi, T. Kotaka and Y. Tabuchi, The impact of platinum reduction on oxygen transport in proton exchange membrane fuel cells, Electrochim. Acta, 2014, 117, 367–378 CrossRef CAS.
- K. Yamada, S. Hommura and T. Shimohira, Effect of High Oxygen Permeable Ionomers on MEA Performance for PEFC, ECS Trans., 2013, 50(2), 1495 CrossRef.
- R. Jinnouchi, K. Kudo, K. Kodama, N. Kitano, T. Suzuki and S. Minami, et al., The role of oxygen-permeable ionomer for polymer electrolyte fuel cells, Nat. Commun., 2021, 12(1), 4956 CrossRef CAS PubMed.
- S. Gottesfeld, Editors' Choice—Review—Polymer electrolyte fuel cell science and technology: Highlighting a general mechanistic pattern and a general rate expression for electrocatalytic processes, J. Electrochem. Soc., 2022, 169, 124518 CrossRef.
- S. Touhami, L. Dubau, J. Mainka, J. Dillet, M. Chatenet and O. Lottin, Anode aging in polymer electrolyte membrane fuel cells I: Anode monitoring by electrochemical impedance spectroscopy, J. Power Sources, 2021, 481, 228908 CrossRef CAS.
- S. Touhami, M. Crouillere, J. Mainka, J. Dillet, C. Nayoze-Coynel, C. Bas, L. Dubau, A. El Kaddouri, F. Dubelley, F. Micoud, M. Chatenet, Y. Bultel and O. Lottin, Anode defects' propagation in polymer electrolyte membrane fuel cells, J. Power Sources, 2022, 520, 230880 CrossRef CAS.
- S. Touhami, J. Mainka, J. Dillet, S. A. H. Taleb and O. Lottin, Transmission line impedance models considering oxygen transport limitations in polymer electrolyte membrane fuel cells, J. Electrochem. Soc., 2019, 166, F1209–F1217 CrossRef CAS.
- P. Schneider, M. Batool, A. O. Godoy, R. Singh, D. Gerteisen, J. Jankovic and N. Zamel, Impact of platinum loading and layer thickness on cathode catalyst degradation in PEM fuel cells, J. Electrochem. Soc., 2023, 170, 024506 CrossRef.
- G. Gwak, J. Lee, M. Ghasemi, J. Choi, S. W. Lee, S. S. Jang and H. Ju, Analyzing oxygen transport resistance and Pt particle growth effect in the cathode catalyst layer of polymer electrolyte fuel cells, Int. J. Hydrogen Energy, 2020, 45, 13414–13427 CrossRef CAS.
- X. Sun, H. Yu, L. Zhou, X. Gao, Y. Zeng, D. Yao, L. He and Z. Shao, Influence of platinum dispersity on oxygen transport resistance and performance in PEMFC, Electrochim. Acta, 2020, 332, 135474 CrossRef CAS.
- P. C. Okonkwo, O. O. Ige, E. M. Barhoumi, P. C. Uzoma, W. Emori, A. Benamor and A. M. Abdullah, Platinum degradation mechanisms in proton exchange membrane fuel cell (PEMFC) system: A review, Int. J. Hydrogen Energy, 2021, 46, 15850–15865 CrossRef CAS.
- S. Komini Babu, D. Spernjak, J. Dillet, A. Lamibrac, G. Maranzana, S. Didierjean, O. Lottin, R. L. Borup and R. Mukundan, Spatially resolved degradation during startup and shutdown in polymer electrolyte membrane fuel cell operation, Appl. Energy, 2019, 254, 113659 CrossRef CAS.
- T. Gaumont, G. Maranzana, O. Lottin, J. Dillet, S. Didierjean, J. Pauchet and L. Guétaz, Measurement of protonic resistance of catalyst layers as a tool for degradation monitoring, Int. J. Hydrogen Energy, 2017, 42, 1800–1812 CrossRef CAS.
- S. Abbou, J. Dillet, D. Spernjak, R. Mukundan, R. L. Borup, G. Maranzana and O. Lottin, High potential excursions during PEM fuel cell operation with dead-ended anode, J. Electrochem. Soc., 2015, 162, F1212–F1220 CrossRef CAS.
- T. Gaumont, G. Maranzana, O. Lottin, J. Dillet, L. Guétaz and J. Pauchet, In operando and local estimation of the effective humidity of PEMFC electrodes and membranes, J. Electrochem. Soc., 2017, 164, F1535–F1542 CrossRef CAS.
- X. Zhang, A. Higier, X. Zhang and H. Liu, Experimental studies of effect of land width in PEM fuel cells with serpentine flow field and carbon cloth, Energies, 2019, 12, 1–10 CrossRef.
- A. Z. Weber and J. Newman, Coupled thermal and water management in polymer electrolyte fuel cells, J. Electrochem. Soc., 2006, 153, A2205 CrossRef CAS.
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, A review of polymer electrolyte membrane fuel cells: Technology, applications, and needs on fundamental research, Appl. Energy, 2011, 88, 981–1007 CrossRef CAS.
- L. Dubau, L. Castanheira, F. Maillard, M. Chatenet, O. Lottin, G. Maranzana, J. Dillet, A. Lamibrac, J. C. Perrin, E. Moukheiber, A. Elkaddouri, G. De Moor, C. Bas, L. Flandin and N. Caqué, A review of PEM fuel cell durability: Materials degradation, local heterogeneities of aging and possible mitigation strategies, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 540–560 CAS.
- P. J. Ferreira, G. J. la O', Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, Instability of Pt/C electrocatalysts in proton exchange membrane fuel cells, J. Electrochem. Soc., 2005, 152, A2256 CrossRef.
- A. P. Soleymani, L. R. Parent and J. Jankovic, Challenges and opportunities in understanding proton exchange membrane fuel cell materials degradation using in-situ electrochemical liquid cell transmission electron microscopy, Adv. Funct. Mater., 2022, 32, 2105188 CrossRef CAS.
- E. Guilminot, A. Corcella, F. Charlot, F. Maillard and M. Chatenet, Detection of Ptz+ ions and Pt nanoparticles inside the membrane of a used PEMFC, J. Electrochem. Soc., 2007, 154, B96–B105 CrossRef CAS.
- E. Guilminot, A. Corcella, M. Chatenet, F. Maillard, F. Charlot, G. Berthomé, C. Iojoiu, J.-Y. Sanchez, E. Rossinot and E. Claude, Membrane and active layer degradation upon PEMFC steady-state operation, J. Electrochem. Soc., 2007, 154, B1106–B1114 CrossRef CAS.
- J. Dillet, D. Spernjak, A. Lamibrac, G. Maranzana, R. Mukundan, J. Fairweather, S. Didierjean, R. L. Borup and O. Lottin, Impact of flow rates and electrode specifications on degradations during repeated startups and shutdowns in polymer-electrolyte membrane fuel cells, J. Power Sources, 2014, 250, 68–79 CrossRef CAS.
- L. Castanheira, L. Dubau, M. Mermoux, G. Berthomé, N. Caqué, E. Rossinot, M. Chatenet and F. Maillard, Carbon corrosion in proton-exchange membrane fuel cells: From model experiments to real-life operation in membrane electrode assemblies, ACS Catal., 2014, 4, 2258–2267 CrossRef CAS.
- L. Castanheira, W. O. Silva, F. H. B. Lima, A. Crisci, L. Dubau and F. Maillard, Carbon corrosion in proton-exchange membrane fuel cells: Effect of the carbon structure, the degradation protocol, and the gas atmosphere, ACS Catal., 2015, 5, 2184–2194 CrossRef CAS.
- S. Helmly, R. Hiesgen, T. Morawietz, X.-Z. Yuan, H. Wang and K. Andreas Friedrich, Microscopic investigation of platinum deposition in PEMFC cross-sections using AFM and SEM, J. Electrochem. Soc., 2013, 160, F687–F697 CrossRef CAS.
- L. Dubau, M. Lopez-Haro, L. Castanheira, J. Durst, M. Chatenet, P. Bayle-Guillemaud, L. Guétaz, N. Caqué, E. Rossinot and F. Maillard, Probing the structure, the composition and the ORR activity of Pt3Co/C nanocrystallites during a 3422h PEMFC ageing test, Appl. Catal., B, 2013, 142–143, 801–808 CrossRef CAS.
- R. Riasse, C. Lafforgue, F. Vandenberghe, F. Micoud, A. Morin, M. Arenz, J. Durst and M. Chatenet, Benchmarking proton exchange membrane fuel cell cathode catalyst at high current density: A comparison between the rotating disk electrode, the gas diffusion electrode and differential cell, J. Power Sources, 2023, 556, 232491 CrossRef CAS.
- S. Komini Babu, D. Spernjak, J. Dillet, A. Lamibrac, G. Maranzana, S. Didierjean, O. Lottin, R. L. Borup and R. Mukundan, Spatially resolved degradation during startup and shutdown in polymer electrolyte membrane fuel cell operation, Appl. Energy, 2019, 254, 113659 CrossRef CAS.
- S. Subianto, M. Pica, M. Casciola, P. Cojocaru, L. Merlo, G. Hards and D. J. Jones, Physical and chemical modification routes leading to improved mechanical properties of perfluorosulfonic acid membranes for PEM fuel cells, J. Power Sources, 2013, 233, 216–230 CrossRef CAS.
- T. C. Jao, S. T. Ke, P. H. Chi, G. Bin Jung and S. H. Chan, Degradation on a PTFE/Nafion membrane electrode assembly with accelerating degradation technique, Int. J. Hydrogen Energy, 2010, 35, 6941–6949 CrossRef CAS.
- M. Marrony, R. Barrera, S. Quenet, S. Ginocchio, L. Montelatici and A. Aslanides, Durability study and lifetime prediction of baseline proton exchange membrane fuel cell under severe operating conditions, J. Power Sources, 2008, 182, 469–475 CrossRef CAS.
- M. Uchimura and S. S. Kocha, The impact of cycle profile on PEMFC durability, ECS Trans., 2007, 11, 1215–1226 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary data associated with this article can be found in the online version at DOI. See DOI: https://doi.org/10.1039/d3im00059a |
| ‡ The authors equally contributed to the work. |
| This journal is © Institute of Process Engineering of CAS 2023 |