
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Applicability of microwave induced plasma optical emission spectrometry for wear metal determination in lubricant oil using a multinebulizer†
Sergio J.
Abellán-Martín
 ,
Miguel Ángel
Aguirre
* and
Antonio
Canals
*
,
Miguel Ángel
Aguirre
* and
Antonio
Canals
*
Department of Analytical Chemistry and Food Science, University Institute of Materials, Faculty of Science, University of Alicante, P.O. Box 99, 03080, Alicante, Spain. E-mail: aguirre.pastor@ua.es; a.canals@ua.es
First published on 18th May 2023
Abstract
In this work, the determination of metals (i.e., Al, Cr, Cu, Mn, Ni and V) in engine lubricating oils by microwave induced plasma atomic emission spectrometry (MIP OES) has been revisited. For this purpose, a new multinebulizer in combination with the standard dilution analysis (SDA) calibration methodology has been used. The microwave plasma is very unstable and may even be extinguished when organic solvents are directly introduced. This problem can be avoided by the synergetic combination of SDA calibration and the new multinebulizer since it allows the determination of analytes in complex matrices due to the simultaneous introduction of organic and aqueous solutions, favoring plasma stabilization. Furthermore, SDA calibration also corrects the matrix effects associated with complex matrices. For the purpose of comparison, the same study has been performed using conventional calibration methodologies (i.e., external calibration (EC), internal standard calibration (IS) and standard addition calibration (SA)). To compare the calibration performances, the analytical figures of merit (i.e., limit of detection and limit of quantification) and the accuracy (i.e., trueness and precision) of the results have been evaluated. The results obtained show very similar values for SDA calibration compared to the other calibrations with a much lower consumption of reagents and resources and with a higher easiness of operation. In addition, the total analysis time has also been evaluated, which has emphasized the great advantage of the synergetic association of SDA calibration with the multinebulizer in chemical analysis using MIP OES.
1. Introduction
The concentration of metals and metalloids in fuels and oils has a direct impact on engine performance, making the detection of metals in lubricating oils extremely important.1,2 Depending on their concentrations, these species can cause a variety of issues, including engine part corrosion and reduction of the machinery lifetime. From an environmental perspective, certain elements act as catalyst poison, which increases the amount of poisonous gases and particulate matter released by vehicles.1 On the other hand, engine components suffer continuous wear under normal operating conditions, which can be minimized using lubricating oils. In addition to chemical wear, which results in organometallic species, there may be physical wear, which produces metallic particles as a result of friction, high temperatures, and pressure. Despite the presence of filters and manifolds, most of the wear metals remain in the lubricating oil and are transported through the entire system.3As a result, an increase in the concentration of certain elements in the lubricating oil following a period of activity may indicate some potential issues with engine components and their extent. The elemental analysis of used lubricating oils can be used to diagnose the condition of an engine component because some elements are related to certain engine parts. For example, elevated chromium levels may indicate excessive piston ring wear, while increases in copper, silver, aluminum, tin or lead concentrations could indicate bearing wear or corrosion. On the other hand, a high iron content indicates possible problems with the oil pump and gears or that there is rust in the system.4 Nowadays, it is a routine trend to perform an elemental analysis on used lubricating oils as part of preventive maintenance, which attempts to minimize any potential damage in the engine before irreversible damage occurs. Therefore, this analysis has economic benefits, reduces potential risks and aids in the creation of new maintenance procedures.3
The majority of analytical methods reported for this purpose were based on atomic spectrometric methods, including flame atomic absorption spectrometry (FAAS),5–7 electrothermal atomic absorption spectrometry (ETAAS),8–11 X-ray fluorescence spectrometry (XRF),12–14 laser-induced breakdown spectrometry (LIBS),15–17 inductively coupled plasma optical emission spectrometry (ICP OES),18–21 and inductively coupled plasma mass spectrometry (ICP-MS).3,22,23 Among all the mentioned techniques, ICP OES was the preferred technique for the analysis of lubricating oils.3 The capability to determine several elements is one of the key advantages of ICP-based techniques,24 especially when compared to atomic absorption spectrometry (AAS). However, ICP-based techniques have relatively high acquisition and operating costs which are one of their potential limitations. For instance, a high amount of argon is required to run an ICP analysis, and it may represent a large portion of a chemical analysis laboratory annual budget. Microwave induced plasma atomic emission spectrometry (MIP OES) is an alternative to AAS and ICP-based techniques to perform the oil analysis in terms of acquisition and operating costs.25 This technique has attracted considerable interest since a commercial instrument became available in the 2000s.26 This instrument is easy to operate, versatile, and has multi-elemental analysis capability that makes MIP OES a promising alternative. However, the main limitation in lubricant oil analysis is its matrix complexity. The organic nature of the lubricant oil itself generates matrix effects and its high viscosity makes it difficult to properly introduce the sample into the plasma. In addition, the high organic solvent load and the formation of carbon deposits on different locations of the plasma torch impose serious difficulties and practical problems. One solution to overcome these problems is the use of an external gas control module (EGCM)1,27,28 to introduce air into the plasma and contribute to maintaining its stability and minimizing background emission from organic compounds. This is because the oxygen in the air reacts with the organic matrix at high temperature to form CO and CO2, preventing the carbon emission and soot deposition on the MP torch.27 However, an MIP OES instrument equipped with an EGCM is more expensive, and therefore, the cost of analysis would increase.
A cheaper and simpler solution has been shown in previous studies29–32 where the matrix effects caused by organic matrices can be avoided by the synergistic combination of a multinebulization-based system and appropriate calibration methods. The multinebulization-based system incorporates two independent liquid inlets into a single nebulizer body with a common nebulizer gas inlet and a single outlet orifice. The nebulization mechanism is based on flow blurring technology, and as a result, it provides a great mixing efficiency.33 The main strength of the multinebulization-based system is the simultaneous introduction of organic and aqueous solutions into the plasma, mixing both immiscible solutions at the tip of the nebulizer. The introduction of water into the plasma allows the supply of oxygen necessary to carry out the complete combustion of the organic matrix in the plasma and reduce carbon deposits in the torch concentric tubes and at the tip of the injector tube. This is an important advantage over conventional liquid sample introduction systems since it does not require the continuous cleaning of the torch or the use of additional components such as cooled spray chambers or an auxiliary oxygen supply.34
On the other hand, calibration is one of the most critical steps in lubricating oil multi-element analysis. It usually requires several standard reference solutions, which can affect the sample throughput.35,36 Except when an absolute method of determination is available, it is essential to choose the most suitable calibration strategy.37 Besides, due to the lower temperatures and lower plasma robustness, MIP OES is more prone to matrix effects than ICP OES, thus typically requiring either sample treatment or the application of the standard addition method to ensure adequate accuracy when analyzing complex matrix samples. When combining the standard addition and internal standard calibrations, standard dilution analysis (SDA)35,38,39 emerges as a calibration strategy, improving the trueness of the results in complex matrix determinations.35 This calibration methodology is able to correct the matrix effects (i.e., systematic errors) caused by the sample matrix and also the fluctuations (i.e., random errors).38 Different from the traditional methods, only two solutions are required in SDA calibrations.
Therefore, the main purpose of this study was to analyze, for the first time, lubricating oil with a minimal sample pretreatment (i.e., dilution) by MIP OES. To this end, the analysis of wear metals has been investigated using a combination of the multinebulization-based system and SDA. Additionally, the method efficiency has also been checked by comparing the results obtained by means of SDA and more conventional calibration strategies (i.e., standard addition and internal standardization) in terms of accuracy (i.e., trueness and precision) of the results and productivity.
2. Experimental
2.1 Instrumentation
In the experimental work carried out, an MIP OES equipment (Model 4100, Agilent Technologies, Melbourne, Australia) was used to perform all the determinations. The sample introduction system was as follows: (i) a tube (F-4040-A, i.d. 0.76 mm, Ismatec, Switzerland) to transport organic solvent through which the samples and organic standards were introduced, (ii) a Tygon® tube (R-3607, i.d. 1.65 mm, Ismatec) to transport aqueous standards, a cyclonic spray chamber provided with an inner tube (Glass Expansion, Victoria, Australia) and a multinebulization-based system (MultiNeb®, Ingeniatrics, Seville, Spain) (Fig. 1). The multinebulizer incorporates two independent liquid inlets in a single nebulization body with a common nebulization gas inlet and a single outlet orifice.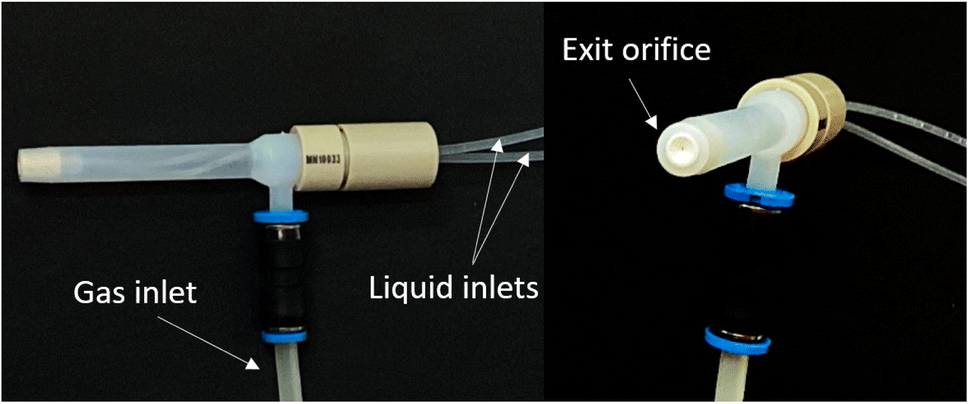 | ||
| Fig. 1 Lateral view (left) and frontal view (right) of the multinebulizer (MultiNeb®) used. | ||
The liquid streams are mixed at the tip of the nebulizer and the aerosol of the mixture of the liquids exits by the unique orifice. The new MultiNeb® provides a significant improvement since it allows mixing of two liquids in the aerosol phase inside the nebulizer at high pressure. As a result, there is an increase in the mixing efficiency and in the chemical reaction speed.
Nitrogen and compressed air were used as the source to generate the plasma and their flow rate remained constant throughout the experiments (i.e., the nitrogen for plasma generation was provided by using an external cylinder). The same nebulizer gas flow rate (i.e., 0.7 L min−1) and plasma observation position (i.e., 0.0) were used for all analytes since these were the optimized parameters for the internal standard. Since the used MIP OES collects data sequentially, SDA calibration can be strongly affected by using different conditions of the nebulizer gas flow rate and plasma observation position for different analytes.40 For comparison purposes, it was decided that the same conditions would be used for all calibration strategies. The optimum operating conditions used are shown in Table S1.†
To check the efficiency to overcome matrix effects, lubricant oil analysis was performed using different calibration methodologies. The normal mode refers to external calibration (EC), internal standardization (IS), and standard addition (SA). The continuous mode was used in calibration by standard dilution analysis (SDA), where data were acquired continuously in real time. Fig. S1† shows the experimental setup for all the calibration methodologies evaluated.
The monitored emission lines for each element are shown in Table S2.† This table also shows the yttrium emission line because it is used as an internal standard.
2.2 Samples and reagents
Samples of lubricating oils were obtained from different local car workshops. Used and unused lubricating oil were employed as real-world samples. Used lubricating oils were drained from different car engines during a routine service at a garage after usage at a certain mileage (i.e., 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 km). The samples were stored in amber glass flasks at 4 °C. The only sample treatment carried out was a gravimetric dilution (1:10) with petroleum ether (QP, Panreac, Barcelona, Spain) in order to reduce the sample viscosity. In order to evaluate the analytical figures of merit, several solutions were made depending on the calibration strategy.
000 km). The samples were stored in amber glass flasks at 4 °C. The only sample treatment carried out was a gravimetric dilution (1:10) with petroleum ether (QP, Panreac, Barcelona, Spain) in order to reduce the sample viscosity. In order to evaluate the analytical figures of merit, several solutions were made depending on the calibration strategy.
In the case of lubricating oil analysis, the concentration of the calibration standards was adjusted depending on the concentration of the analytes in the sample.
2.3 On-line SDA calibration
The conventional SDA requires the preparation of only two analytical solutions (e.g., A and B) for each sample. Solution A, containing 50% sample and 50% standard (including the internal standard), is gradually mixed with solution B, which contains 50% sample and 50% blank. As the standard is diluted (retaining the same amount of sample), many calibration points are generated over time as shown in Fig. S2.† The mathematical approach used for the calculation of the sample concentration using SDA calibration can be found elsewhere.38The on-line SDA was performed using three channels of a peristaltic pump accompanied by the instrument used. A scheme of the system is shown in Fig. S1.† Channel 1 was used to introduce organic samples. Channel 2 was used to perform the continuous dilution of the aqueous standard by the introduction of the aqueous blank. Finally, channel 3 was employed to aspirate the aqueous standards (i.e., analytes and internal standard). The three channel flows are given in Table S1.†
As shown in Fig. S1,† SDA calibration has been adapted to a flow system, where dilution is performed automatically. In this way, less solutions need to be prepared since it is not necessary to prepare solution A and solution B as mentioned above.21
When on-line SDA (Fig. S1†) and SA calibration are applied, the organic sample and the aqueous calibration standards are introduced through different channels and also with different flow rates. It is well known that the transport efficiencies of organic and aqueous samples are different. Therefore, a mathematical correction is needed for the different transport efficiencies of the nebulized organic and aqueous solutions.34 Thus, if the signal in an aqueous medium is known, the relationship between the two signals (i.e., organic and aqueous) will allow us to predict the signal in an organic medium and vice versa. To that end, the relative transport efficiency was calculated using a standard of known concentration and aqueous and organic matrix blanks. For more details, the mathematical correction has already been described elsewhere.29,34
3. Results and discussion
3.1 Limits of detection (LOD) and quantification (LOQ)
Limits of detection (LOD) were calculated according to IUPAC recommendations as 3 times the standard deviation of the blank signal (10 measurements) divided by the slope of the calibration curve.41 These recommendations were also followed to calculate the limits of quantification; in this way the LOQ is calculated as 10 times the standard deviation of the blank signal divided by the slope of the calibration curve.41Table 1 shows the limits of detection and quantification for the calibration methods used.| Element | Calibration strategy | |||||||
|---|---|---|---|---|---|---|---|---|
| EC | IS | SA | SDA | |||||
| LOD (μg kg−1) | LOQ (μg kg−1) | LOD (μg kg−1) | LOQ (μg kg−1) | LOD (μg kg−1) | LOQ (μg kg−1) | LOD (μg kg−1) | LOQ (μg kg−1) | |
| V | 3 | 9 | 2 | 6 | 1.3 | 4 | 1.8 | 6 |
| Ni | 13 | 43 | 12 | 39 | 20 | 80 | 20 | 70 |
| Cu | 3 | 9 | 2 | 6 | 5 | 15 | 4 | 13 |
| Mn | 3 | 11 | 2 | 7 | 5 | 16 | 4 | 15 |
| Cr | 1.3 | 4 | 0.9 | 3 | 1.9 | 6 | 2 | 7 |
| Al | 2 | 7 | 1.3 | 5 | 3 | 10 | 4 | 13 |
As can be seen, the LOD and LOQ values in the IS calibration were slightly better than those obtained in the EC. On the other hand, in SA calibration, a slight increase in the LOD and LOQ values was observed for most analytes because the sample matrix reduces the sensitivity. In the case of vanadium, the sample matrix increased the sensitivity and therefore caused a decrease in the LOD and LOQ. In the case of SDA calibration, the LOD and LOQ values obtained were similar to those obtained by SA calibration.
3.2 Analysis of engine lubricant oil samples
Different oil samples were chosen in order to evaluate the matrix effects under the calibration methods evaluated. Table 2 shows the concentration of each element found for the different calibration methods and the amount of standard added in spiked and non-spiked samples for used and unused lubricating oil.:10 petroleum ether) using multinebulizer/MIP OES. Recovery is shown in parentheses (%)
| Sample | Calibration method | Spiked concentration (μg g−1) | Found concentration (μg g−1) and recovery (%) | |||||
|---|---|---|---|---|---|---|---|---|
| V | Ni | Cu | Mn | Cr | Al | |||
| Unused lubricating oil sample | EC | — | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ |
| 0.97 | 1.12 ± 0.05 (114 ± 9) | 0.88 ± 0.04 (106 ± 8) | 0.85 ± 0.02 (85 ± 3) | 0.887 ± 0.007 (87.6 ± 1.1) | 0.89 ± 0.03 (91 ± 5) | 0.91 ± 0.03 (91 ± 5) | ||
| IS | — | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | |
| 0.97 | 1.15 ± 0.04 (117 ± 6) | 1.00 ± 0.08 (105 ± 14) | 0.87 ± 0.02 (86 ± 3) | 0.91 ± 0.02 (89 ± 4) | 0.92 ± 0.02 (93 ± 3) | 0.94 ± 0.03 (93 ± 4) | ||
| SA | — | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | |
| 0.97 | 1.15 ± 0.13 (103 ± 13) | 0.97 ± 0.03 (100 ± 3) | 0.93 ± 0.06 (95 ± 6) | 0.98 ± 0.18 (101 ± 16) | 0.94 ± 0.03 (97 ± 3) | 0.97 ± 0.18 (100 ± 16) | ||
| SDA | — | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | |
| 0.98 | 0.98 ± 0.09 (100 ± 9) | 1.02 ± 0.05 (105 ± 5) | 1.01 ± 0.05 (103 ± 5) | 0.92 ± 0.05 (95 ± 5) | 0.96 ± 0.06 (98 ± 6) | 0.93 ± 0.06 (95 ± 6) | ||
| Used lubricating oil sample | EC | — | 0.56 ± 0.02 | <LOQ | 2.06 ± 0.02 | <LOQ | 0.19 ± 0.04 | 0.39 ± 0.02 |
| 1.06 | 1.84 ± 0.02 (122 ± 2) | 0.86 ± 0.03 (81 ± 3) | 3.00 ± 0.03 (88 ± 3) | 0.90 ± 0.03 (85 ± 2) | 1.18 ± 0.04 (93 ± 3) | 1.38 ± 0.02 (93 ± 2) | ||
| IS | — | 0.61 ± 0.02 | <LOQ | 2.25 ± 0.03 | <LOQ | 0.21 ± 0.04 | 0.43 ± 0.02 | |
| 1.06 | 2.34 ± 0.02 (164 ± 2) | 1.03 ± 0.03 (98 ± 3) | 3.37 ± 0.04 (106 ± 4) | 1.01 ± 0.03 (96 ± 3) | 1.32 ± 0.03 (105 ± 3) | 1.55 ± 0.02 (106 ± 2) | ||
| SA | — | 0.32 ± 0.07 | <LOQ | 2.27 ± 0.4 | <LOQ | 0.29 ± 0.08 | 0.57 ± 0.13 | |
| 0.99 | 1.24 ± 0.13 (94 ± 10) | 0.97 ± 0.09 (99 ± 9) | 3.31 ± 0.3 (105 ± 11) | 1.01 ± 0.12 (102 ± 13) | 1.22 ± 0.16 (95 ± 13) | 1.53 ± 0.19 (98 ± 13) | ||
| SDA | — | 0.310 ± 0.08 | <LOQ | 2.19 ± 0.11 | <LOQ | 0.29 ± 0.02 | 0.58 ± 0.02 | |
| 1.01 | 1.38 ± 0.07 (106 ± 9) | 1.02 ± 0.09 (101 ± 9) | 3.18 ± 0.07 (99 ± 6) | 0.97 ± 0.02 (95 ± 10) | 1.36 ± 0.03 (105 ± 8) | 1.65 ± 0.06 (106 ± 6) | ||
In unused oil, when the non-spiked sample was measured, every element concentration was under the limit of quantification. On the other hand, except for Ni and Mn, the analytes were detected in used lubricating oil. The higher concentration found in the used lubricating oil sample, for the majority of analytes, confirms the change in the elemental composition of the lubricating oil after a certain usage.
The recovery value of each element was also calculated to evaluate the matrix effects by adding a known amount of analytes to each sample (Table 2). To better visualize the results obtained, Fig. S3† shows recovery values for each element and for each calibration method. As can be seen, the results obtained by using EC show that the sample matrix strongly affected the determination of the evaluated elements. The matrix effect produced a decrease in the signal, providing recovery values ranging from 81 to 93% with the exception of vanadium (i.e., 114 and 122% in unused and used lubricating oil, respectively) and nickel in unused lubricating oil (i.e., 106%). In those cases, there are still matrix effects causing an increase in the signal. Evaluating the IS calibration, recovery values ranging from 86 to 106% were obtained for all elements, except for vanadium where a recovery of 117% was obtained in the unused oil sample and 164% in the used one. These results show that yttrium was not capable of correcting the matrix effects for vanadium. Regarding the SA calibration methodology, recovery values between 94 and 105% were obtained, which means that this type of calibration corrected the matrix effects for all the analytes evaluated. It is possible to see that there were no differences for both types of samples, providing similar recovery values. Finally, the SDA methodology was evaluated, and the results were similar to those obtained with the SA calibration, obtaining recovery values in a range from 95 to 106% for all elements and without differences for both lubricating oil samples, which means that both calibration strategies can correct the matrix effects successfully. The precision of each calibration methodology was evaluated through the standard deviation of the regression equation at the calculated concentration. The standard deviations for EC and IS were similar, because in both calibration methodologies standard deviations associated with the interpolated concentration were similar. Regarding the standard deviation in the SA calibration, the results obtained ranged from 3 to 16% showing a general increase, because standard deviations associated with the extrapolated concentration are less precise than in interpolated methods. Finally, in SDA calibration, standard deviations ranged from 5 to 10%. In this case, standard deviations were slightly lower compared with those of SA calibration since the high number of calibration points in the calibration curve significantly improved the statistical parameters.
3.3 Advantages of using a multinebulizer combined with SDA calibration
The main strength of the multinebulizer is the simultaneous introduction of organic and aqueous solutions into the plasma, mixing both immiscible solutions at the tip of the nebulizer. The introduction of water into the plasma allows the supply of oxygen necessary to carry out the complete combustion of the organic matrix in the plasma and reduce carbon deposits in the concentric tubes of the torch and at the tip of the injector tube. This is an important advantage over conventional liquid sample introduction systems, since it does not require continuous cleaning of the torch or the use of additional components such as refrigerated spray chambers or an auxiliary oxygen supply.31 On the other hand, in SDA calibration, unlike EC, IS and SA calibration, only two solutions are required. This significantly reduces the consumption of reagents and the time required to perform the analysis. The time needed in each calibration method is shown in Table S3.†As can be seen, the time needed to prepared standards (sample) is lower in SDA calibration than in the rest of the calibration methodologies. The SA methodology can also avoid the matrix effects produced by the organic sample, but the time required for the analysis is 2.6 times higher than that for the SDA calibration method. Considering this, the SDA, that also corrects the matrix effects is a perfect methodology. In addition, the SDA method provides a higher sample throughput in comparison with the other calibration strategies.
Taking into account all of the above mentioned and summarizing, the most important advantages of the synergetic combination are (i) it reduces the carbon deposits at different places of the plasma torch (e.g., concentric tube and the tip of the injector tube); (ii) SDA simultaneously corrects for matrix effects (i.e., systematic errors) and for fluctuations (i.e., random errors) due to changes in instrumental parameters; (iii) it can be adapted to many chemical elements and sample types; (iv) it provides ease of operation because the system setup is simple, robust and easy to handle; (v) it significantly reduces the analysis time as less sample dilution is needed and only one standard solution is required per sample, reducing the solvent amount and the reagents needed.
4. Conclusions
It has been demonstrated that a multinebulizer together with SDA calibration is appropriate for the determination of metals (i.e., Al, Cr, Cu, Mn, Ni and V) in used engine lubricating oils by the MIP OES technique. The combination of the multinebulizer with SA and SDA calibration provided accurate results. The simultaneous introduction of organic and aqueous solutions is an easy and fast way to carry out the elemental analysis of lubricating oil samples using MIP OES. In fact, the same analysis was attempted by the direct introduction of the used lubricating oil sample, and it was not possible, as the plasma was totally unstable, and the torch deteriorated rapidly. SDA calibration provides very satisfactory results, comparable to those of SA calibration. However, the time required to prepare the calibration standards is minimal, as only one standard and the blank are required. In conclusion, taking into account all the advantages and disadvantages provided by each calibration method, SDA calibration together with the multinebulizer is a reliable, simple, fast and economical approach for the analysis of used lubricating oils using MIP OES.Conflicts of interest
All the authors declare that there are no conflicts of interest.Acknowledgements
The authors would like to thank the Spanish Ministry of Science and Innovation (PID2021-126155OB-I00) and the Regional Government of Valencia (Spain) (CIPROM/2021/062, INVEST/2022/70) for the financial support. The authors would also like to thank Agilent Technologies Inc. for the loan of the MIP OES spectrometer and Ingeniatrics for the MultiNeb® provided. This article is based upon work from the Sample Preparation Study Group and Network, supported by the Division of Analytical Chemistry of the European Chemical Society. The authors extend their appreciation to Ministry of Science and Innovation for granting the Spanish Network of Excellence in Sample Preparation (RED2022-134079-T).References
- R. S. Amais, G. L. Donati, D. Schiavo and J. A. Nóbrega, A Simple Dilute-and-Shoot Procedure for Si Determination in Diesel and Biodiesel by Microwave-Induced Plasma Optical Emission Spectrometry, Microchem. J., 2013, 106, 318–322, DOI:10.1016/j.microc.2012.09.001.
- M. G. A. Korn, D. S. S. Santos, B. Welz, M. G. R. Vale, A. P. Teixeira, D. C. Lima and S. L. C. Ferreira, Atomic Spectrometric Methods for the Determination of Metals and Metalloids in Automotive Fuels - A Review, Talanta, 2007, 73, 1–11, DOI:10.1016/j.talanta.2007.03.036.
- R. Q. Aucélio, R. M. Souza, R. C. Campos, N. Miekeley and C. L. P. Silveira, The Determination of Trace Metals in Lubricating Oils by Atomic Spectrometry, Spectrochim. Acta, Part B, 2007, 62, 952–961, DOI:10.1016/j.sab.2007.05.003.
- J. A. Palkendo, J. Kovach and T. A. Betts, Determination of Wear Metals in Used Motor Oil by Flame Atomic Absorption Spectroscopy, J. Chem. Educ., 2014, 91, 579–582, DOI:10.1021/ed4004832.
- L. Edbon, E. H. Evans, A. Fisher and S. J. Hill, Plasma Atomic Emission Spectrometry, in, An Introduction to Analytical Atomic Spectrometry, ed. E. H. Evans, John Wiley & Sons Ltd, Chichester, 1998, vol. 4, pp. 73–113 Search PubMed.
- E. J. Ekanem, J. A. Lori and S. A. Thomas, The Determination of Wear Metals in Used Lubricating Oils by Flame Atomic Absorption Spectrometry Using Sulphanilic Acid as Ashing Agent, Talanta, 1997, 44, 2103–2108 CrossRef CAS PubMed.
- I. M. Goncalves, M. Murillo and A. M. González, Determination of Metals in Used Lubricating Oils by AAS Using Emulsified Samples, Talanta, 1998, 47, 1033–1042, DOI:10.1016/s0039-9140(98)00186-6.
- S. Carballo, J. Terán, R. M. Soto, A. Carlosena, J. M. Andrade and D. Prada, Green Approaches to Determine Metals in Lubricating Oils by Electrothermal Atomic Absorption Spectrometry (ETAAS), Microchem. J., 2013, 108, 74–80, DOI:10.1016/j.microc.2013.01.002.
- R. Q. Aucélio, A. Doyle, B. S. Pizzorno, M. L. B. Tristão and R. C. Campos, Electrothermal Atomic Absorption Spectrometric Method for the Determination of Vanadium in Diesel and Asphaltene Prepared as Detergentless Microemulsions, Microchem. J., 2004, 78, 21–26, DOI:10.1016/j.microc.2004.02.018.
- K. Li, X. Wu, Z. Chen, J. Luo, X. Hou and X. Jiang, A Simple Dilution Method for the Direct Determination of Trace Nickel in Crude Oil with a Miniaturized Electrothermal Atomic Absorption Spectrometer, J. Anal. At. Spectrom., 2020, 35, 2656–2662, 10.1039/d0ja00081g.
- M. Á. Aguirre, A. Canals, I. López-García and M. Hernández-Córdoba, Determination of Cadmium in Used Engine Oil, Gasoline and Diesel by Electrothermal Atomic Absorption Spectrometry Using Magnetic Ionic Liquid-Based Dispersive Liquid-Liquid Microextraction, Talanta, 2020, 220, 121395, DOI:10.1016/j.talanta.2020.121395.
- M. Pouzar, T. Černohorský and A. Krejčová, Determination of Metals in Lubricating Oils by X-Ray Fluorescence Spectrometry, Talanta, 2001, 54, 829–835, DOI:10.1016/S0039-9140(01)00338-1.
- L. A. Meira, J. S. Almeida, F. S. Dias, P. P. Pedra, A. L. Costa Pereira and L. S. G. Teixeira, Multi-Element Determination of Cd, Pb, Cu, V, Cr, and Mn in Ethanol Fuel Samples Using Energy Dispersive X-Ray Fluorescence Spectrometry after Magnetic Solid Phase Microextraction Using CoFe2O4 Nanoparticles, Microchem. J., 2018, 142, 144–151, DOI:10.1016/j.microc.2018.06.025.
- L. A. Meira, J. S. Almeida, M. S. Oliveira, F. S. Dias, A. F. S. Queiroz, S. A. R. Soares and L. S. G. Teixeira, Biodiesel Trace Element Analysis by Energy Dispersive X-Ray Fluorescence Spectrometry Using Magnetic Solid-Phase Microextraction, Energy Fuels, 2021, 35, 510–518, DOI:10.1021/acs.energyfuels.0c02731.
- L. Zheng, F. Cao, J. Xiu, X. Bai, V. Motto-Ros, N. Gilon, H. Zeng and J. Yu, On the Performance of Laser-Induced Breakdown Spectroscopy for Direct Determination of Trace Metals in Lubricating Oils, Spectrochim. Acta, Part B, 2014, 99, 1–8, DOI:10.1016/j.sab.2014.06.005.
- P. Yaroshchyk, R. J. S. Morrison, D. Body and B. L. Chadwick, Quantitative Determination of Wear Metals in Engine Oils Using Laser-Induced Breakdown Spectroscopy: A Comparison between Liquid Jets and Static Liquids, Spectrochim. Acta, Part B, 2005, 60, 986–992, DOI:10.1016/j.sab.2005.03.011.
- P. Fichet, P. Mauchien, J. F. Wagner and C. Moulin, Quantitative Elemental Determination in Water and Oil by Laser Induced Breakdown Spectroscopy, Anal. Chim. Acta, 2001, 429, 269–278, DOI:10.1016/S0003-2670(00)01277-0.
- T. Kuokkanen, P. Perämäki, I. Välimäki and H. Rönkkömäki, Determination of Heavy Metals in Waste Lubricating Oils by Inductively Coupled Plasma-Optical Emission Spectrometry, Int. J. Environ. Anal. Chem., 2001, 81, 89–100, DOI:10.1080/03067310108044347.
- D. Prokic-Vidojevic, Z. Zivkovic and O. Barac, Application of Atomic Emission Spectrometry in Aviation Piston Engine Abnormal Wear Prevention, Sci.-Tech. Rev., 2017, 66, 23–28, DOI:10.5937/str1601023p.
- R. M. Souza, C. L. P. Silveira and R. Q. Aucélio, Determination of Refractory Elements in Used Lubricating Oil by ICPOES Employing Emulsified Sample Introduction and Calibration with Inorganic Standards, Anal. Sci., 2004, 20, 351–355, DOI:10.2116/analsci.20.351.
- M. García, M. Á. Aguirre and A. Canals, A New Multinebulizer for Spectrochemical Analysis: Wear Metal Determination in Used Lubricating Oils by On-Line Standard Dilution Analysis (SDA) Using Inductively Coupled Plasma Optical Emission Spectrometry (ICP OES), J. Anal. At. Spectrom., 2020, 35, 265–272, 10.1039/c9ja00255c.
- C. Walkner, R. Gratzer, T. Meisel and S. N. H. Bokhari, Multi-Element Analysis of Crude Oils Using ICP-QQQ-MS, Org. Geochem., 2017, 103, 22–30, DOI:10.1016/j.orggeochem.2016.10.009.
- C. Duyck, N. Miekeley, C. L. P. Silveira and P. Szatmari, Trace Element Determination in Crude Oil and Its Fractions by Inductively Coupled Plasma Mass Spectrometry Using Ultrasonic Nebulization of Toluene Solutions, Spectrochim. Acta, Part B, 2002, 57, 1979 CrossRef.
- P. Vähäoja, I. Välimäki, K. Roppola, T. Kuokkanen and S. Lahdelma, Wear Metal Analysis of Oils, Crit. Rev. Anal. Chem., 2008, 38, 67–83, DOI:10.1080/10408340701804434.
- J. Nelson, G. Gilleland, L. Poirier, D. Leong, P. Hajdu and F. Lopez-Linares, Elemental Analysis of Crude Oils Using Microwave Plasma Atomic Emission Spectroscopy, Energy Fuels, 2015, 29, 5587–5594, DOI:10.1021/acs.energyfuels.5b01026.
- W. Li, P. Simmons, D. Shrader, T. J. Herrman and S. Y. Dai, Microwave Plasma-Atomic Emission Spectroscopy as a Tool for the Determination of Copper, Iron, Manganese and Zinc in Animal Feed and Fertilizer, Talanta, 2013, 112, 43–48, DOI:10.1016/j.talanta.2013.03.029.
- G. L. Donati, R. S. Amais, D. Schiavo and J. A. Nóbrega, Determination of Cr, Ni, Pb and V in Gasoline and Ethanol Fuel by Microwave Plasma Optical Emission Spectrometry, J. Anal. At. Spectrom., 2013, 28, 755–759, 10.1039/c3ja30344f.
- S. M. Azcarate, L. P. Langhoff, J. M. Camiña and M. Savio, A Green Single-Tube Sample Preparation Method for Wear Metal Determination in Lubricating Oil by Microwave Induced Plasma with Optical Emission Spectrometry, Talanta, 2019, 195, 573–579, DOI:10.1016/j.talanta.2018.11.045.
- M. Á. Aguirre, N. Kovachev, M. Hidalgo and A. Canals, Analysis of Biodiesel and Oil Samples by On-Line Calibration Using a Flow Blurring® Multinebulizer in ICP OES without Oxygen Addition, J. Anal. At. Spectrom., 2012, 27, 2102–2110, 10.1039/c2ja30202k.
- F. D. Avila Orozco, N. Kovachev, M. Á. Aguirre Pastor, C. E. Domini, B. S. Fernández Band and A. C. Hernández, Analysis of Metals and Phosphorus in Biodiesel B100 from Different Feedstock Using a Flow Blurring® Multinebulizer in Inductively Coupled Plasma-Optical Emission Spectrometry, Anal. Chim. Acta, 2014, 827, 15–21, DOI:10.1016/j.aca.2014.04.016.
- M. García, M. Á. Aguirre and A. Canals, Determination of As, Se, and Hg in Fuel Samples by in-Chamber Chemical Vapor Generation ICP OES Using a Flow Blurring® Multinebulizer, Anal. Bioanal. Chem., 2017, 409, 5481–5490, DOI:10.1007/s00216-017-0484-6.
- J. A. V. A. Barros, M. Á. Aguirre, N. Kovachev, A. Canals and J. A. Nóbrega, Vortex-Assisted Dispersive Liquid-Liquid Microextraction for the Determination of Molybdenum in Plants by Inductively Coupled Plasma Optical Emission Spectrometry, Anal. Methods, 2016, 8, 810–815, 10.1039/c5ay02561c.
- A. M. Gañán-Calvo, Enhanced Liquid Atomization: From Flow-Focusing to Flow-Blurring, Appl. Phys. Lett., 2005, 86, 1–3, DOI:10.1063/1.1931057.
- M. García, M. Á. Aguirre and A. Canals, A New Multinebulizer for Spectrochemical Analysis: Wear Metal Determination in Used Lubricating Oils by On-Line Standard Dilution Analysis (SDA) Using Inductively Coupled Plasma Optical Emission Spectrometry (ICP OES), J. Anal. At. Spectrom., 2020, 35, 265–272, 10.1039/C9JA00255C.
- D. A. Goncalves, T. McSweeney, M. C. Santos, B. T. Jones and G. L. Donati, Standard Dilution Analysis of Beverages by Microwave-Induced Plasma Optical Emission Spectrometry, Anal. Chim. Acta, 2016, 909, 24–29, DOI:10.1016/j.aca.2015.12.046.
- H. Mark, Principles and Practice of Spectroscopic Calibration, in Chemical Analysis: A Series of Mono-Graphs on Analytical Chemistry and its Applications, ed. J. D. Winefordner and I. M. Kolthoff, Wiley-Inter-Science, New York, 1st edn, 1992, vol. 118, p. 192 Search PubMed.
- F. M. Fortunato, M. A. Bechlin, J. A. G. Neto, G. L. Donati and B. T. Jones, Internal Standard Addition Calibration: Determination of Calcium and Magnesium by Atomic Absorption Spectrometry, Microchem. J., 2015, 122, 63–69, DOI:10.1016/j.microc.2015.04.009.
- W. B. Jones, G. L. Donati, C. P. Calloway and B. T. Jones, Standard Dilution Analysis, Anal. Chem., 2015, 87, 2321–2327, DOI:10.1021/ac504152x.
- A. Virgilio, D. Schiavo, J. A. Nóbrega and G. L. Donati, Evaluation of Standard Dilution Analysis (SDA) of Beverages and Foodstuffs by ICP OES, J. Anal. At. Spectrom., 2016, 31, 1216–1222, 10.1039/c6ja00040a.
- A. G. Althoff, C. B. Williams, T. McSweeney, D. A. Gonçalves and G. L. Donati, Microwave-Induced Plasma Optical Emission Spectrometry (MIP OES) and Standard Dilution Analysis to Determine Trace Elements in Pharmaceutical Samples, Appl. Spectrosc., 2017, 71, 2692–2698, DOI:10.1177/0003702817721750.
- L. A. Currie, Nomenclature in Evaluation of Analytical Methods Including Detection and Quantification Capabilities, Pure Appl. Chem., 1995, 67, 1699–1723 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ja00093a |
| This journal is © The Royal Society of Chemistry 2023 |