
Liposome manufacturing under continuous flow conditions: towards a fully integrated set-up with in-line control of critical quality attributes†
Maryam
Sheybanifard
 a,
Luis P. B.
Guerzoni
b,
Abdolrahman
Omidinia-Anarkoli
bc,
Laura
De Laporte
bcd,
Johannes
Buyel
efg,
Rut
Besseling
h,
Michiel
Damen
h,
Ad
Gerich
h,
Twan
Lammers
a and
Josbert M.
Metselaar
*a
a,
Luis P. B.
Guerzoni
b,
Abdolrahman
Omidinia-Anarkoli
bc,
Laura
De Laporte
bcd,
Johannes
Buyel
efg,
Rut
Besseling
h,
Michiel
Damen
h,
Ad
Gerich
h,
Twan
Lammers
a and
Josbert M.
Metselaar
*a
aDepartment of Nanomedicine and Theranostics, Institute for Experimental Molecular Imaging, Uniklinik RWTH Aachen and Helmholtz Institute for Biomedical Engineering, Faculty of Medicine, RWTH Aachen University, 52074 Aachen, Germany. E-mail: jmetselaar@ukaachen.de
bDWI Leibniz Institute for Interactive Materials, Forckenbeckstrasse 50, 52074 Aachen, Germany
cInstitute of Applied Medical Engineering, RWTH University, Pauwelsstraße 20, 52074 Aachen, Germany
dInstitute for Technical and Macromolecular Chemistry, RWTH Aachen, Worringerweg 1-2, 52074 Aachen, Germany
eFraunhofer Institute for Molecular Biology and Applied Ecology IME, Forckenbeckstrasse 6, 52074 Aachen, Germany
fInstitute for Molecular Biotechnology, RWTH Aachen University, Worringerweg 1, 52074 Aachen, Germany
gInstitute of Bioprocess Science and Engineering, University of Natural Resources and Life Sciences (BOKU), Muthgasse 18, A-1190 Vienna, Austria
hInProcess-LSP, Kloosterstraat 9, 5349 AB Oss, The Netherlands
First published on 21st November 2022
Abstract
Continuous flow manufacturing (CFM) has shown remarkable advantages in the industrial-scale production of drug-loaded nanomedicines, including mRNA-based COVID-19 vaccines. Thus far, CFM research in nanomedicine has mainly focused on the initial particle formation step, while post-formation production steps are hardly ever integrated. The opportunity to implement in-line quality control of critical quality attributes merits closer investigation. Here, we designed and tested a CFM setup for the manufacturing of liposomal nanomedicines that can potentially encompass all manufacturing steps in an end-to-end system. Our main aim was to elucidate the key composition and process parameters that affect the physicochemical characteristics of the liposomes. Total flow rate, lipid concentration and residence time of the liposomes in a high ethanol environment (i.e., above 20% v/v) emerged as critical parameters to tailor liposome size between 80 and 150 nm. After liposome formation, the pressure and the surface area of the filter in the ultrafiltration unit were critical parameters in the process of clearing the dispersion from residual ethanol. As a final step, we integrated in-line measurement of liposome size and residual ethanol content. Such in-line measurements allow for real-time monitoring and in-process adjustment of key composition and process parameters.
Introduction
Across the entire arsenal of FDA/EMA-approved nanomedicines, liposomal drug products have maintained a frontrunner position.1 Liposomes are self-assembled spherical vesicles that are composed of one or more lipid bilayers surrounding an aqueous core.2 The popularity of liposomes as nanocarriers is bolstered by their biocompatibility, biodegradability, ease of manufacturing and capability of encapsulating and delivering both hydrophobic and hydrophilic therapeutic agents.3 Over the last decades, a plethora of preclinical studies on liposomal drugs have been reported. However, only a modest number of 30 investigational liposomal drug products are currently being evaluated in clinical trials.1 Moreover, only 12 parenteral liposome products have received FDA/EMA approval to enter the market.1 One reason for this purported lack of clinical success may relate to challenges pertaining to the translation of lab-scale to industrial-scale production of liposomes.4Currently, batch production is the standard industrial-scale method for liposome manufacturing.5 In this setup, a certain quantity of raw materials is mixed and processed stepwise from a crude into a refined dispersion. These processing steps are conducted discretely, sequentially, but independent of each other, with each preparation step only leading to the next step upon successful completion. At the end of the processing line, the refined dispersion is filled in dosing units, quality controlled, released and the production unit is prepared for the next batch.6,7
Besides the inefficiency and labor-intensiveness of the batch production approach, which is partly due to the necessity of having multiple discrete operational units, there are two other major limitations that need to be addressed. First, there is the risk that upon completion, a batch does not meet the release specifications and needs to be discarded, and second, this approach is sensitive to scalability issues such as batch-to-batch variation.5,8
In contrast to batch production, continuous flow manufacturing (CFM) involves a setup in which all production steps are integrated into one all-encompassing, coherent and uninterrupted system. Indeed, one single production line is created through which the formulation flows, starting as a crude and ending as a refined dispersion (i.e., an “end-to-end” system).9 CFM is not only labor efficient with all operation units now connected to operate as one system, but also delivers an adjustable production yield.10 Moreover, a CFM setup can be readily extended to allow for real-time and in-line control of key critical quality attributes. This, in turn enables achieving the desired specifications of the end product through changing key process and composition parameters while the system is running, ensuring that the final product meets the specifications.11 Lastly, CFM facilitates the scale-up simply by running the process for a longer period of time,10 which takes away the potential risks associated with scale-up in the batch production such as the use of larger vessels with more temperature variations and mixing problems as potential consequences.
To produce liposomal drug products in a CFM setup, microfluidic and millifluidic dispersion techniques have been extensively used.11–14 A microfluidic/millifluidic device typically features two or more channels that come together in a T- or Y-shape configuration. An organic lipid solution is pumped through one channel and the aqueous solution (which in many cases also includes the drug) is pumped through the other channel(s) of the device. Based on the fluid properties (e.g., viscosity, velocity, etc.) and the channel geometry, the organic and aqueous solutions mix in a laminar or turbulent flow regimen.15 Rapid mixing of the two fluids in a laminar or turbulent regimen results in an abrupt change in the polarity of the solution, decreasing the concentration of the organic solvent below the solubility of the lipid causing formation of liposomes with specific physicochemical properties such as average size and size distribution.5,15 Here, the key physicochemical characteristics of the produced liposomes are controlled by several composition and process parameters. These parameters include temperature, total flow rate (TFR), flow rate ratio between the organic stream and the aqueous stream, aqueous phase composition, lipid composition and lipid concentration.8,10,14,16
The actual liposome formation, however, is only the primary step in the manufacturing procedure and it is typically followed by other critical production steps. These steps include the removal of the solvent residues and unencapsulated drug (mostly by means of tangential flow filtration), sterile filtration, filling and quality control of the final product. In contrast to the vast number of publications on the use of microfluidic/millifluidic devices as a way to achieve continuous flow manufacturing of liposomes and other nanocarriers, the integration of subsequent production steps has not been systematically explored. Moreover, in most studies, concentrations of both the organic solvent and the lipid starting materials at the point of liposome formation are relatively low (roughly between 10–25% v/v and 5–25 mM respectively),8,13,17–21 which limits the encapsulation efficiency of hydrophilic drugs in the liposome vesicles.
In the present study, we designed and tested a CFM setup that we envision can be used for the production of liposomal drug products from raw material all the way to a refined and well-characterized liposome dispersion. To this end, we optimized the liposome formation process using a millifluidic device, evaluated a purification unit for solvent and unencapsulated drug removal and explored the feasibility of incorporating in-line measurement devices to monitor the main physicochemical properties of the liposomes such as particle size and ethanol content. Finally, the impact of several composition parameters (such as ethanol and lipid concentration) and process parameters (such as total flow rate and flow rate ratio) on the key product characteristics was explored, to allow for effective adjustments of the process while the CFM is running.
Materials and methods
Materials
Egg phosphatidylcholine (EPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-polyethylene glycol-2000 (DSPE-PEG 2000) were purchased from Lipoid GmbH (Ludwigshafen, Germany). Cholesterol (99% purity, product of USA) was obtained from Sigma-Aldrich. Phosphate buffered saline (PBS) ready-to-use tablets (ROTI Fair PBS 7.4) and ethanol were purchased from Carl Roth GmbH (Karlsruhe, Germany). An advanced programmable syringe pump PHD ULTRA was purchased from Harvard Apparatus (Holliston, USA). Minimate tangential flow filtration (TFF) cassettes with a MWCO of 100 kDa and a filtration area of 50 cm2 were acquired from Pall Corporation (Ann Arbor, USA). Hollow fibers with a MWCO of 100 kDa and a filtration area of 193 cm2 were purchased from WaterSep (USA). Regenerated cellulose MEMBRA-CEL MD10 dialysis tubing (MWCO 14 kDa) was obtained from Viskase (product of USA).Preparation of liposomes
Liposomes were prepared using a millifluidic device as explained in Costa et al. publication.10 The inner diameter (ID) of the outer channel was set at 3 mm and the ID of the inner channel was set at 0.6 mm (Fig. S1†). To form the liposomes, DSPE-PEG-2000, EPC, and cholesterol were dissolved in ethanol at a fixed molar ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60:35 respectively. Next, the ethanolic lipid solution was pumped through the inner channel of the millifluidic device, while PBS was used as the aqueous phase solution and was pumped through the outer channel, both streams come together and mix at the desired flow rate ratio to form liposomes. Formed liposomes were collected from an outlet which was the extension of the outer channel.
:60:35 respectively. Next, the ethanolic lipid solution was pumped through the inner channel of the millifluidic device, while PBS was used as the aqueous phase solution and was pumped through the outer channel, both streams come together and mix at the desired flow rate ratio to form liposomes. Formed liposomes were collected from an outlet which was the extension of the outer channel.
Characterization of liposomes
Liposomes were measured off-line using a vial module or in-line using a flow-cell module, with a 15.85 mm inner diameter flow cell. Z-ave and PDI were calculated from 10 measurements of approximately 10 seconds each for the off-line measurements and individual measurements of approximately 5 seconds each for the in-line measurements. The viscosity and refractive index (RI) of the solvents were adjusted in the NanoFlowSizer (NFS) software (XsperGo 1.11.6) in accordance with the water to ethanol ratio in the formulation and measurements were performed at ambient temperature (24–26 °C).
500–4000 cm−1 with an optical resolution of 16 cm−1.
Size adjustment using composition and process parameters obtained from design of experiment (DoE)
We implemented a design of experiment (DoE) approach to assess the effect of total flow rates (TFRs), lipid concentrations, and ethanol content on liposome size and PDI (using Design-Expert by Statease). The lipid concentrations were defined in the range of 10–100 mM total lipids dissolved in ethanol and the TFR was defined in the range of 5–70 mL min−1, resulting in Reynolds numbers in the range of 14–214 (depending on the formulation condition). An overall of 39 to 45 experimental runs (depending on the capability of the syringe pumps in providing the desired flow rates) were defined in a D-optimal design with cubic complexity of initial model. To prepare liposomes, we initially dissolved 100 mM of total lipids in ethanol. Next, to achieve the lipid concentration defined for any specific run in the DoE design, we adjusted the flow rates of the two syringe pumps coming together, one that delivers a concentrated lipid solution in ethanol (i.e., 100 mM) and one that delivers pure ethanol. The resulting diluted ethanolic lipid solution was then led to the inner channel while PBS was pumped to the outer channel of the millifluidic device with the pre-defined flow rates for that run by the DoE design. Moving from one run to another, we changed the flow rates of the streams to meet the requirements. The final ethanol content was set at 50% v/v in one set of DoE and 30% v/v in another set. Right after preparation, the size (Z-ave) and PDI of the liposomes were measured in an off-line setup using NFS device. A response surface model was built by removing non-significant terms (p > 0.05) from the design model. Exceptions were made to maintain model hierarchy and the software was used to plot the relationship between the variables and the responses.Size adjustment using the residence time of the liposomes in high ethanol environment
000 times with a mixture of PBS and ethanol (1:1 ratio). The number of particles in the dispersion as well as the videos to visualize the liposomes were recorded for 60 min using the NTA device. Another fraction of the sample was kept for continuous size measurement in the NFS device. NFS parameters (i.e., the RI and the viscosity) were set in the software to measure the sample immediately after preparation. To prevent ethanol evaporation, the sample was sealed with parafilm and then was continuously measured for 90 min. The third fraction of the dispersion was diluted with PBS (one volume of the sample with nine volumes of PBS) at defined time points of 0, 1, 2.5, 5, 10, 20, 30, 60, 90 min to fixate the size and stop the size growth. Upon completion of the 90 min continuous measurement of the second fraction, these samples were measured by NFS.
Liposome ultrafiltration: tangential flow filtration
A tangential flow filtration (TFF) system was set up using a peristaltic pump circulating the liposomal dispersion through a filtration unit. Here, we investigated the effect of two individual filtration units with different filtration areas, namely 50 cm2 and 193 cm2, on liposome filtration efficiency (see Materials section for more details). The changes in the pressure were monitored using two pressure gauges that were positioned before and after the filtration units. The retentate outlet was disconnected from the reservoir in order to have a one-time-only passage of the dispersion through the filtration unit. The inlet pressure was varied from 21 to 207 kilopascal (kPa) using different pump speeds. The outlet pressure was varied from 14 to 52 kPa by changing the filter outlet diameters (i.e., 3.2, 0.5, and 0.2 mm). The filtration efficiency was expressed as the waste volume fraction, which is calculated by dividing the filtrate volume by the sum of the filtrate and retentate volumes. Here we prepared three independent replicates.In-line measurement of the liposome size and ethanol concentration in a CFM setup
Three independent batches of liposomes were prepared as follows. Liposomes were formed as described in the section “Effect of time on liposome size as measured by NFS and NTA”. After formation and without collecting the liposomes from the outlet, one volume of the liposome dispersion (with a flow rate of 62 mL min−1) was diluted in-line with four volumes of clean PBS (with a flow rate of 248 mL min−1) using a three-way connector after which the dispersion was led through the flow-cell module of the NFS device (with inner diameter of 15.85 mm) for an in-line size and PDI measurement. We started to record NFS data immediately after turning on the pumps driving the liquid streams that generate the liposome suspension (while the flow cell was still empty). Once the syringes were emptied from the liposome precursors and the flow was stopped, the NFS flow cell was still filled with the sample, and we continued to measure liposome size and PDI without flow.After the flow cell, the dispersion was collected in a vessel in which the NIR probe was placed for ethanol content determination. The measurements were 5 seconds apart and the input stream was assessed for an overall of 4 min.
In order to evaluate the effect of lipids in the dispersion on ethanol quantification using the NIR, liposomes were prepared (70 mM of total lipid in ethanol, TFR at 62 mL min−1) and immediately dialyzed against PBS (1 L mL−1 of the ethanol per day). After 3 days of dialysis, we manually added 0.5, 1, and 5% v/v ethanol to the liposome dispersion. The known concentrations of the ethanol in the liposomal formulations were evaluated using a calibration curve. Here, we prepared three independent replicates.
Statistical analysis
Statistical analysis on non-DoE graphs was performed using the GraphPad Prism software (Prism 9 for Windows 64-bit, Version 9.1.1). Specifically, we used one-way ANOVA (in Fig. 2C and D), unpaired one-tailed student t-test (in Fig. 4C and E) and finally linear regression (in Fig. 5A). Statistical significance was defined at an alpha threshold of <0.05.Results and discussion
In the current study, we used a millifluidic device to form liposomes as the first step in the preparation of a liposomal drug product within a CFM setup. To clear the resulting liposome dispersion from the residual organic solvent, we used a tangential flow filtration (TFF) unit and noted that to make it work in an end-to-end single production line, its filtration efficiency had to be improved.28 Importantly, the integrated continuous flow manufacturing setup enables the quality control of the final product by monitoring the critical physicochemical characteristics of the liposomes in-line and while the process is running.29 To this end, we integrated the measurement devices into the CFM setup for particle size and residual ethanol analysis. The last step in the preparation of parenteral liposomal product is sterilization. This can either be achieved by sterile filtration or by ensuring aseptic processing, which is in theory possible with the entire CFM unit being a closed system30 (Scheme 1).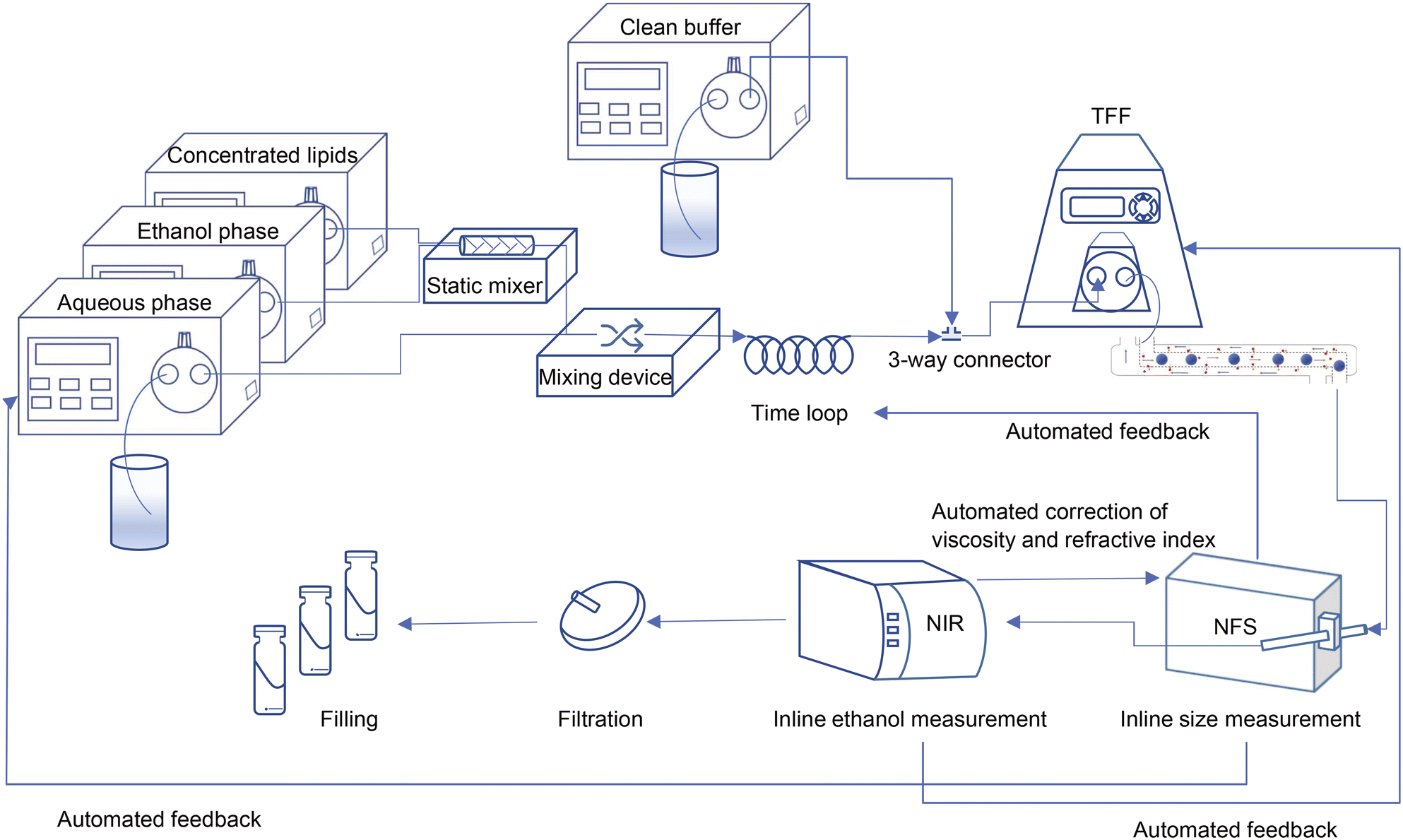 | ||
| Scheme 1 Schematic view of the ultimate design of the continuous flow manufacturing production line; from particle formation and in-line control of critical quality attributes to the filling. | ||
The effect of composition and process parameters on liposome size and PDI
As mentioned above, the primary step in production of a liposomal drug product in a CFM setup is the formation of the liposome particles. After design and assembly of the setup, we prepared more than 40 liposomal formulations and in an off-line setting, we evaluated the influence of the following process and composition parameters on liposome size and PDI: (1) total flow rate (TFR); (2) lipid concentration; (3) ethanol concentration; and (4) residence time of the liposomes in high ethanol environment after formation.At high TFRs and low lipid concentrations, we observed a decrease in the liposome size (Fig. 1A), something that is also reported by others.12 Apparently, the chance for the newly formed bilayers to merge and form larger liposomes increases upon increasing lipid concentration.31 The TFR, on the other hand, drove the mixing behaviour of the two streams. As hypothesized by Yanar et al., unlike low TFRs, in high TFRs ethanol and water mix at a faster rate and as a result, the lipids dissolved in the ethanol phase have a limited and better controlled amount of time to form phospholipid bilayers and merge into liposomes, which then remain smaller and more monodisperse.32
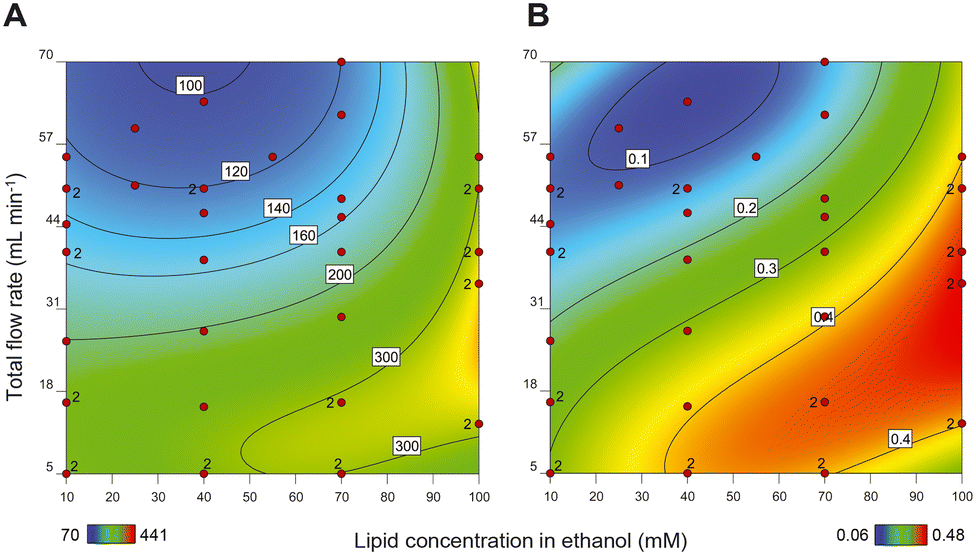 | ||
| Fig. 1 Heat maps of response surface model showing A) size (hydrodynamic diameter in nm) and B) PDI of liposomes as a result of changing composition parameter (i.e., lipid concentration) and process parameter (i.e., total flow rates (TFR)). Liposomes were formed with 50% v/v ethanol at the point of mixing and the measurements were performed in an off-line setting in the presence of the ethanol. Each red dot in the graphs represents the outcome of one specific liposomal formulation preparation. Colour bars at the bottom and values within each graph present hydrodynamic diameter (A) and PDI (B). | ||
Importantly, increasing lipid concentrations and decreasing TFR also resulted in liposomal formulations with multiple populations in the size-based graph (PDIs above 0.25) (Fig. 1B). Upon lowering the lipid concentration and increasing the TFR, the PDI of the liposomes improved and decreased to values below 0.25, with only one population of the particles. Therefore, we have considered samples with PDIs below 0.25 as monodisperse.
Our results show that the liposome size can be varied and fine-tuned between 80 to 150 nm in the region where the lipid concentration and the TFR generate liposomes with PDIs < 0.25 (Fig. 1). Although this difference in size may not seem substantial on first sight, liposomes in this size range can exhibit profound differences in in vivo behaviour in terms of pharmacokinetics and organ distribution.33 Moreover, by increasing the liposome vesicle diameter from 80 to 150 nm (1.9 times increase in diameter), the volume is actually increased by 6.6 times (as a function of d), which theoretically allows for this much more drug encapsulation.
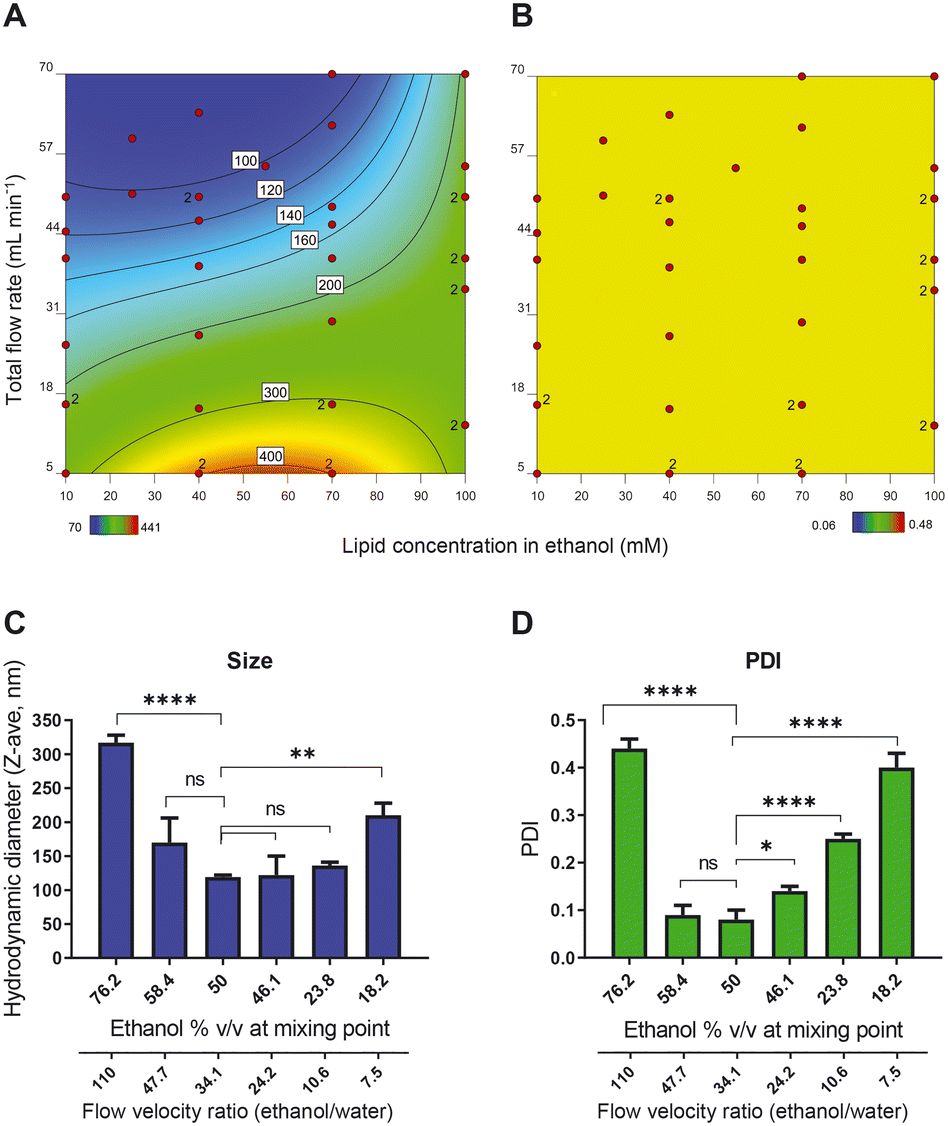 | ||
| Fig. 2 Heat maps of response surface model showing A) size (hydrodynamic diameter in nm) and B) PDI of liposomes as a result of changing composition parameter (i.e., lipid concentration) and process parameter (i.e., total flow rates (TFR)). Liposomes were formed with 30% v/v ethanol at the point of mixing and the measurements were performed in an off-line setting in the presence of the ethanol. Each red dot in the graphs represents the outcome of one specific liposomal formulation preparation. Colour bars at the bottom and values within each graph present hydrodynamic diameter (A) and PDI (B). C) and D) The effect of ethanol content at the point of mixing on C) liposome size and D) PDI. Data sets that are deemed not significantly different are shown with ns (p > 0.05) and data sets that are significantly different are shown at different levels: * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001. The liposomal dispersions are prepared at a fixed lipid concentration and a fixed TFR. | ||
To better understand the relationship between ethanol concentration at the point of mixing and the PDI, we prepared a series of samples with fixed TFR (50 mL min−1) and fixed lipid concentration (final lipid concentration of 25 mM) but with different ethanol percentages at the point of mixing. The size measurements (evaluated in an off-line setting using Malvern DLS) show that the minimum size and PDI were obtained with liposomes in 50% v/v ethanol at the point of mixing (Fig. 2C and D). Both increasing and decreasing the ethanol content led to an increase in the size and PDI of the liposomes, indicating that 50% ethanol offered the ‘sweet spot’ of working conditions. An additional advantage of working with a high ethanol percentage is the high concentration of bilayer lipids at the point of mixing, which produces a relatively concentrated vesicle dispersion. In the presence of a hydrophilic drug dissolved in the water phase, this leads to a relatively high encapsulation efficiency. It is worth noticing that to decrease the ethanol content, we decreased the flow rate of the ethanol stream relative to the aqueous phase stream and consequently, the flow velocity ratio (FVR) between ethanol and water decreased as well. This would eventually translate to an improper mixing of the organic phase and the aqueous phase.
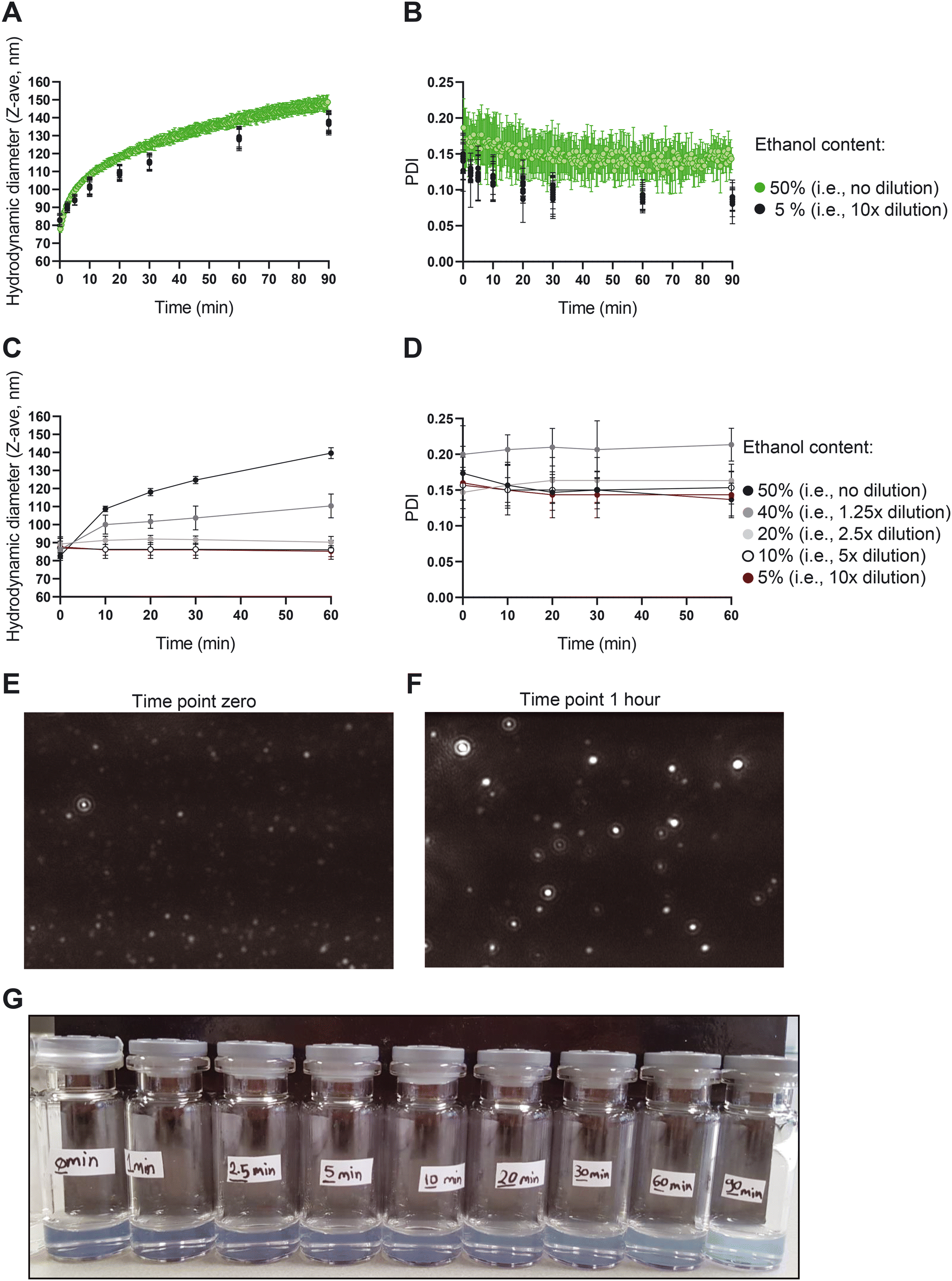 | ||
| Fig. 3 The size of liposomes in high ethanol content solutions increases over time. Changes in A) size and B) PDI of the liposomes in the presence of 50% v/v ethanol (green dots) and after size fixation through dilution at a defined time point (black dots). Changes in C) size and D) PDI of liposomes in the presence of different concentrations of the ethanol at the time point zero. E) and F) Representative NTA images of the liposomes in the presence of 50% v/v ethanol at E) time point zero and F) time point 1 hour. G) Changes in the turbidity of the liposomal formulations with time. Liposomes were formed with 70 mM lipid in ethanol and TFR 62 mL min−1 and measured in an off-line setting (n = 3). | ||
To further elucidate the mechanism behind the size growth, we recorded a one-hour video of the liposomes using NTA to monitor the size, number and motion of the particles. The data and associated particle counts showed a larger number of particles in the early time points compared to later time points (Fig. S4†). Given the increase in size, this is expected when total amount of lipids is conserved. The decreasing liposome concentration is also supported by backscatter intensity data vs. size obtained with the NFS (data not shown).
While a large body of literature exists regarding the effects of ethanol on the behaviour of liposomes, lipid bilayers and lipid nanoparticles (LNPs), none of them report the present type of non-equilibrium behaviour with up to 50% size increase at high ethanol content. In one of the studies, results and references show liposome size reduction upon addition of up to 20% ethanol, which contrast ethanol induced growth in the area per lipid molecule referred to in that same paper,34 which points to some basic controversy in interpretation of liposome size changes with ethanol. Others reported phenomena including interdigitation and reduced membrane thickness due to ethanol,35 enhanced membrane lipid diffusion,36 and enhanced lipid disorder37 also do not offer a mechanistic explanation for the behaviour we observe. Instead, the growth we observe is likely a type of vesicle annealing driven by an Ostwald-ripening mechanism (lipid diffusion through the ethanol rich solvent drives growth of large liposomes at the expense of smaller ones), which is qualitatively supported by the fact that growth is suppressed (‘quenched’) at low ethanol concentration. However, liposome coalescence may also play a role in the growth. Initial liposome TEM images also revealed morphological changes during growth (Fig. S5†). Further detailed studies with quantitative interpretation and modelling of the growth are underway and will be subject of a future publication.
Organic solvent elimination within a CFM setup
Removal of residual organic solvent and the non-encapsulated drug is an important step during liposome production. To optimize this step for use in the CFM setup, we identified tangential flow filtration (TFF) as the best feasible option. TFF is based on repeated cycling of the liposome dispersion over a semi-permeable membrane,38 while supplementing the filtrate with clean buffer. Since cycling is only possible in batch-wise production and not an option in CFM, we designed a setup in which the liposome dispersion passes alongside the membrane only once and optimization of the TFF process was achieved by carefully gauging and adjusting the pressures during filtration unit passage.We initially studied this setup by passing water through the filtration unit. Our results show that using water, the production of filtrate relative to the sum of filtrate and retentate (which we refer to here as the ‘waste volume fraction’) slightly increased upon increasing the inlet pressure (Fig. 4B). We found that the filtration efficiency is proportional to the filter surface area; with a surface of 193 cm2 yielding an average waste volume fraction of 44%.
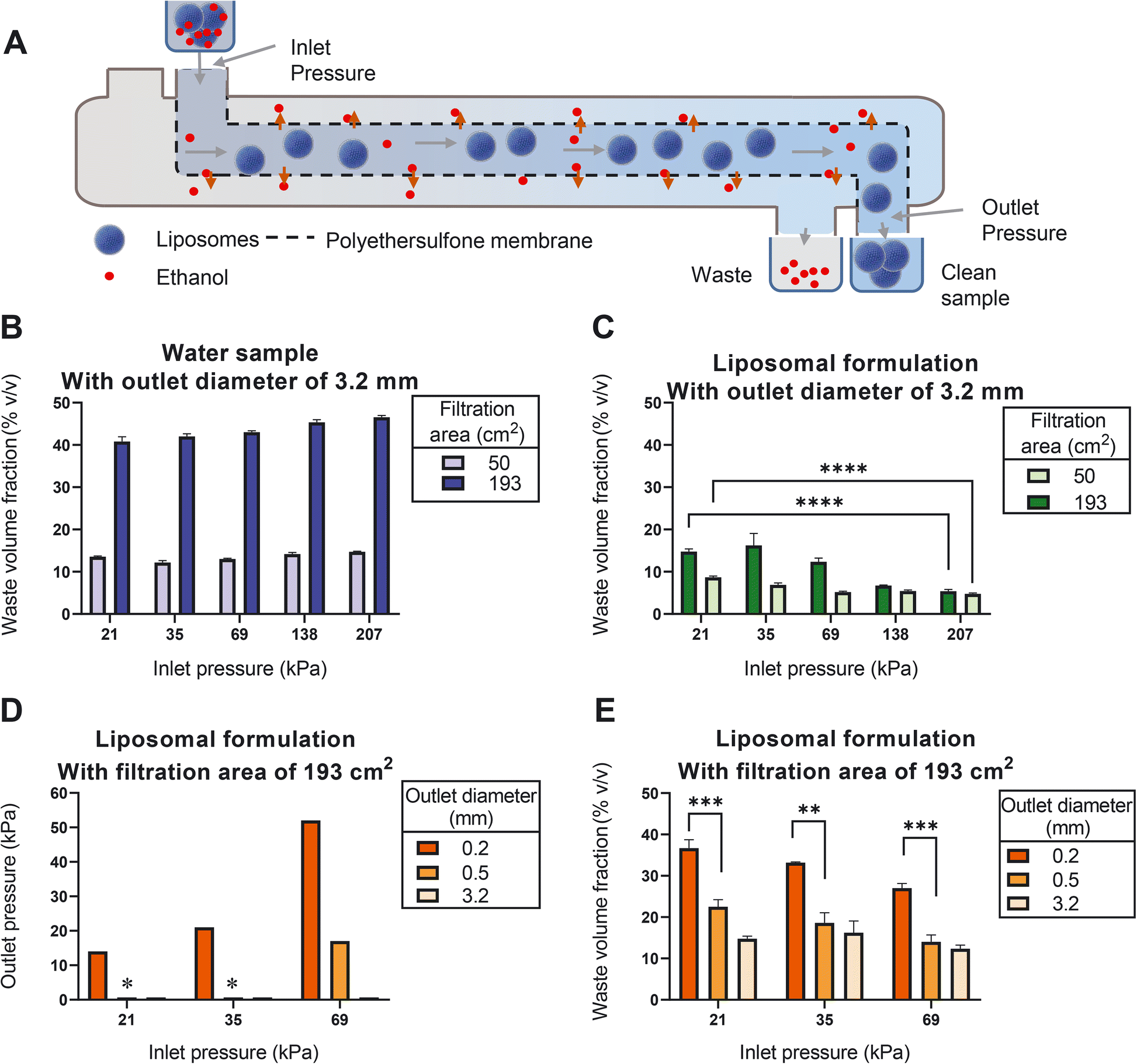 | ||
| Fig. 4 The presence of liposome dispersion, the filtration area and the pressure in the filtration unit affect the filtration efficiency of the liposomes using an open TFF system. A) Schematic view of the filtration unit. B) and C) The effect of surface area and inlet pressure on the waste volume fractions measured in B) water, and C) liposomal dispersion. D) Tightening the outlet: the outlet pressure of the filtration unit obtained at different inlet pressures. E) The effect of outlet pressure and inlet pressure on the waste volume fractions assessed in liposomes. Data sets that are deemed not significantly different are shown with ns (p > 0.05) and data sets that are significantly different are shown at different levels: * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001. Pressures with a star sign on top of them in panel D) were out-of-range measures (n = 3). | ||
Strikingly, when applying the same conditions to a liposome dispersion, the waste volume fraction was strongly reduced (Fig. 4C). We noticed that such reduction can be partially mitigated by minimizing the inlet pressure (Fig. 4C). This suggests that once the pressure is too high, liposomes are forced to enter the membrane matrix where they are clogging the pores.
Having identified a large surface area and a minimal inlet pressure as the two favorable conditions, we next evaluated the effect of the outlet pressure. We increased the outlet pressure by decreasing the outlet diameter (Fig. 4D), which resulted in an additional increase in the waste volume fraction (Fig. 4E). Most prominently, increasing the outlet pressure to 14 kPa (at inlet pressure of 21 kPa) resulted in waste volume fractions of around 36% (Fig. 4E), which is comparable to the corresponding value of water samples in Fig. 4B.
It should be noted that with the current outlet capacity of the liposome mixing device the required minimal inlet pressure at the ultrafiltration unit could not be achieved. This issue can be overcome by further increasing the surface area of the filter, using filtration units with larger inlet diameters and adjusting the dimensions of the tubing so that both production units seamlessly connect to each other with the same flow.
Real-time in-line measurement of the liposome characteristics during production
Besides easier scale up and cleaner production, the main advantage of CFM over batch-to-batch production of nanomedicines is that it allows for better control of the critical quality attributes of the product and consequently for a reduction of the chance of batch failure. This can be achieved by adjusting key process and composition parameters while compounding. It is paramount in this setup that the product characteristics can be measured real-time and in-line so that prompt intervention and correction are possible.39,40 To achieve this goal, we have benefitted from the recently developed ‘NanoFlowSizer’ (NFS) instrument, which has the unique capability to gauge size and PDI of the liposomes in-line in relevant flow conditions (from 0 to >2000 mL min−1 depending on the flow cell). In addition, a more traditional near infrared spectroscopy (NIR) device coupled to an NIR probe in the process was applied as process analytical tool to quantify the content of the ethanol during the process.Using near infrared spectroscopy to measure the ethanol content in real-time
To assess the accuracy and the precision of the NIR device in quantifying the ethanol content in the liposome dispersion, we measured known concentrations of ethanol in a liposomal dispersion. As shown in Fig. 5A, the NIR could accurately measure as low as 0.5% v/v ethanol in the presence of 35 mM total lipid. The device was then used to measure the ethanol content in an in-line setup. Liposomal formulations were prepared with 50% v/v ethanol content and diluted with PBS to an ethanol concentration of 10% v/v. The outcome of the in-line NIR of around 9% v/v ethanol was sufficient to judge this equipment fit for our purpose (Fig. 5B).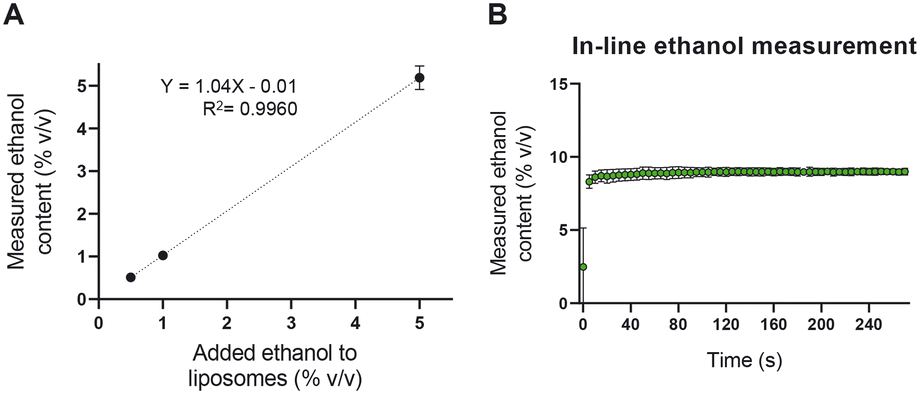 | ||
| Fig. 5 NIR spectroscopy is used as a tool to quantify the content of ethanol in an in-line setup. A) Quantification validation in the presence of the liposomes. B) Real-time measurement of the ethanol content in a CFM setup after diluting the sample (i.e., the expected ethanol content was 10% v/v). Liposomes were formed with 70 mM lipid in ethanol and TFR 62 mL min−1 (n = 3). | ||
Accurate in-line ethanol quantification is crucial in two regards. On the one hand, residual ethanol in parenteral liposome products needs to be brought below 5000 ppm (0.5%),41 something that can be achieved by optimizing the TFF module of the setup. On the other hand, the ethanol concentration defines the viscosity and RI of the formulation and thus, it influences the in-line liposome size measurements using the NFS.
In-line measurement of the dispersion's size and PDI using NanoFlowSizer
Size and PDI are critical quality attributes of a nanomedicine formulation, strongly influencing its' in vivo performance and target-site accumulation.42,43 In the CFM setup, the NFS enables us to monitor the changes in size and PDI in-line and real-time, which eventually provides the opportunity to fine tune and control these characteristics while the CFM is running. We started to prepare and measure liposomes in an off-line setup to compare size and PDI measurements obtained from the NFS with that of the conventional DLS (Fig. 6, the vial module off-line measurements of size and PDI are shown in red circles). The recorded Z-ave size and PDIs from the vial module off-line setting of the two instruments differed (85 ± 3 nm with PDIs of 0.23 ± 0.04 and 71 ± 2 nm with PDIs of 0.09 ± 0.01 using NFS and conventional DLS respectively). This is due to the fact that Z-ave and PDI represent the harmonic mean and relative harmonic variance of the scattering intensity-based size distribution. Since scatter intensity depends on the ratio of size/wavelength, for suspensions with any significant size distribution (e.g., coefficient of variation (CV) >0.1), instruments with different wavelengths of light (λ = 633 nm for standard DLS vs. λ = 1300 nm for the SR-DLS based NFS), will yield different intensity-based size distribution and thus different Z-ave and PDI, except when all particles are <0.1λ (Rayleigh regime). Conversion to volume-based liposome size distribution should give similar results for both instruments; indeed, the estimated volume averaged mean size is approximately 62 nm for both and the estimated relative variance of the volume distribution is approximately 0.08 (DLS) and 0.12 (SR-DLS).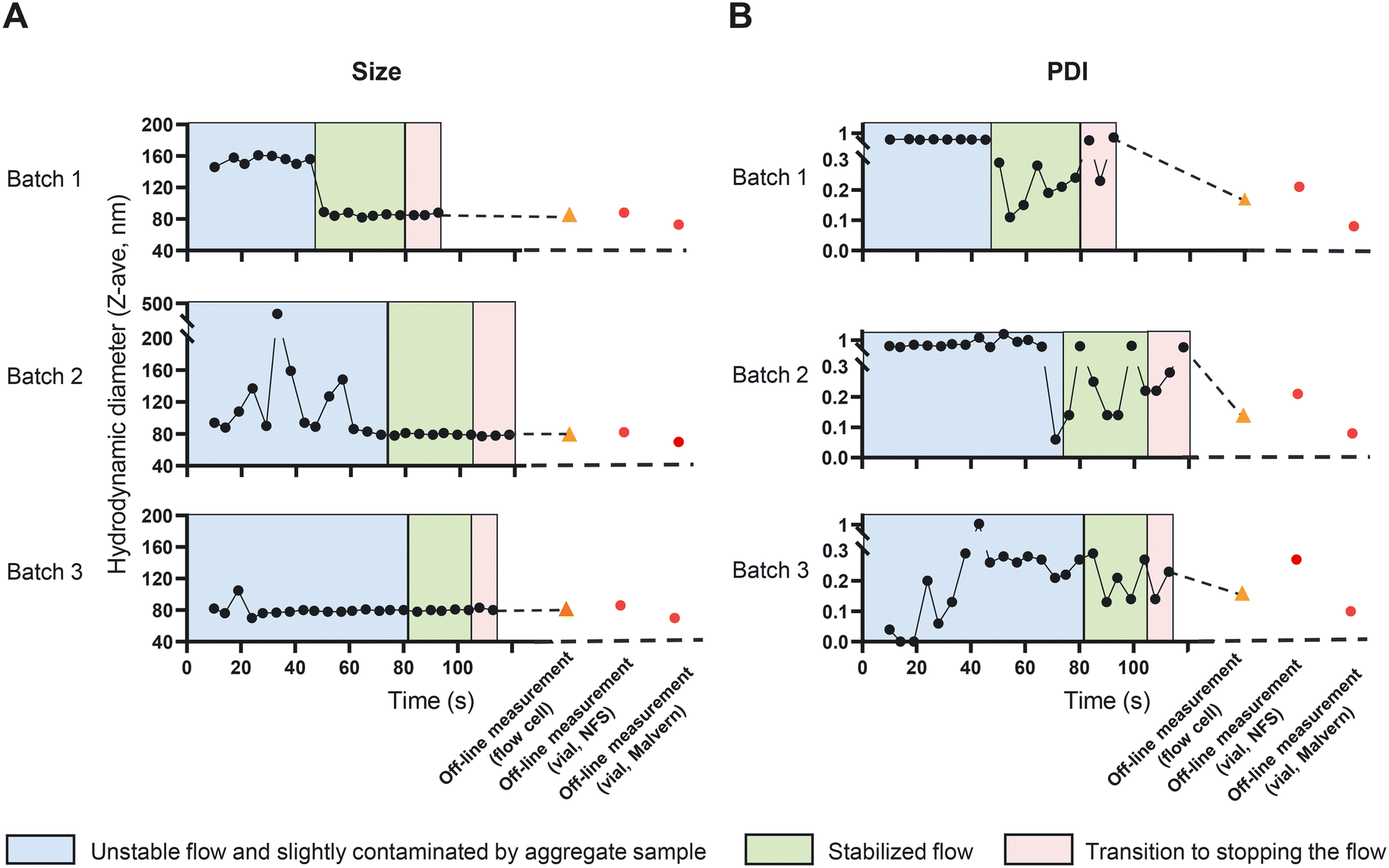 | ||
| Fig. 6 NanoFlowSizer as a tool to quantify A) the size and B) the PDI of liposome dispersions in an in-line and off-line setup. Initially, the flow exhibited large or aggregated vesicles mainly near the wall and flow was not optimally stable, causing reduced data quality (blue area). Next, both liposome size uniformity and the flow stabilized, giving optimal quality measurements (green area). After the ‘stabilized flow’ period, the pumps driving the liposome synthesis were turned off causing a transition to zero-flow in-line measurement (red area). Size and PDI in the in-line and off-line measurements of the flow-cell module (black circles and orange triangles respectively) were assessed after 5 times of dilution with PBS. Note that the NFS employs a wavelength of 1300 nm for the intensity based Z-ave size and PDI, while Malvern DLS employs a wavelength of 633 nm. Liposomes were formed with 70 mM lipid in ethanol and TFR 62 mL min−1 (n = 3). | ||
Next, we measured the dispersions in the in-line setup as described in section “In-line measurement of the liposome size and ethanol concentration in a CFM setup”. NFS in-line measurements can be affected by the stability of the flow and by the presence of large (or aggregated) vesicles that can occur typically at the start of the run. These large structures may initially stick to the wall of the measurement cell. Together these effects may reduce the quality of the flow corrections and data fits that are performed by the instrument's software. The in-line data in Fig. 6A and B were therefore categorized using a data quality indicator based on depth-uniformity of size data (diffusion), quality of the ACF flow fits and DLS cumulant fits. In the in-line runs shown in Fig. 6, an initial period of flow is visible (blue areas) in which PDI and/or mean particle size were large and/or noisy and data quality was limited. This is associated with unstable flow and/or presence of large (aggregated) vesicles, particularly near/at the flow cell wall. The latter is directly illustrated by the data in Fig. S6† showing the depth resolved liposome size during flow in the flow cell (information uniquely obtained by the NFS' SR-DLS method): the 24 second time point data in Fig. S6† in the ‘unstable’ regime of batch 2 shows the aggregates/larger vesicles near the wall. After ∼40 to 80 seconds (depending on the run), the NFS data indicated optimal quality (green), with highly stable Z-ave size and more stable, low PDI values <0.3 except for two outliers in batch 2. The depth resolved data for a stable, low PDI measurement in this regime (Fig. S6,† time point 90 seconds of batch 2) shows a uniform, depth-independent size without aggregate features. The data during this period are comparable to the off-line results (Fig. 6, orange triangle). Note that both in standard DLS and SR-DLS, the intrinsic relative variability of PDI data is typically significantly larger than for Z-ave size data, as also seen in Fig. 6. The variable timespan (40–80 s) for optimal quality is partly due to the fact that large vesicles/aggregates initially present near the wall (causing unstable flow/reduced data quality) may vary in number, size and wall interactions between batches. This causes variability in their transport through the flow cell and thus the duration of reduced data quality. The variability of the timespan is also linked to the relatively strict ‘high quality threshold’ of the quality indicator, e.g., in batch 3 (in all respects identical to batch 1 and 2) it is seen that Z-ave size and PDI data are already stable after ∼40 s.
An important point in the context of real-time process monitoring, is that continuous in-line NFS data monitors relatively large quantities of suspension passing the NFS sensor (compared to static DLS). Similar in-line observations with the NFS in other processes (unpublished data) have shown that in-line measurements can reveal infrequent but actually occurring aggregates that go undetected in off-line/static setting (when the same – few nL volume of sample in the beam is measured repeatedly). Overall, the results in Fig. 6 show the ability and the value of the NFS device as part of the CFM setup to perform in-line and real-time measurements of particle size and PDI.
We further confirmed the reproducibility of the measurements and the stability of the formed liposomes in terms of size and PDI beyond 100 seconds and till 6 minutes using a Malvern DLS instrument (Fig. S7†).
Conclusions
The goal of this study was to develop a CFM setup to allow for end-to-end manufacturing of liposomal drug products. To this end, we implemented a millifluidic device to form a liposome dispersion in a water–ethanol mixture and controlled its critical quality attributes by changing key composition and process parameters. We demonstrated that the liposome mean size is inversely proportional to the TFR and directly related to the lipid concentration and more importantly the residence time of liposomes in the high ethanol environment in the device. Notably, we formulated our liposomal dispersions in a relatively high ethanol volume percentage in comparison to most other studies in the field,8,13,17–21 allowing for an improved control of the critical quality attributes. Furthermore, high lipid content can be used in such condition, which in turn should deliver enhanced encapsulation efficiencies of hydrophilic drugs in the liposomal dispersions. With regard to the TFF process, we observed that a large filter surface area, a minimal inlet pressure and a uniformly distributed pressure in the entire filtration unit can maximize the filtration efficiency, which is required for integration of TFF in a CFM setting. Moreover, we successfully included in-line measurements of two key physicochemical characteristics of the liposome dispersions during CFM operation i.e., liposome particle size and residual ethanol content. The CFM setup that we developed here is a modular platform that allows incorporation of other desired in-process analysis. A notable idea is utilizing an in-line Raman spectrometer to quantify the total lipid content as well as the drug encapsulation efficiency. Compared to bulk production, our CFM setup has a superior scalability since large-scale production is simply achieved by running the system for a longer period. Additionally, the degree of in-process system control allows for automated feedback control of the product characteristics, which eventually allows for remote production of batches and help reduce the chance of costly batch failures. We foresee that our CFM system can further be developed into a fully integrated CFM setup to function as a versatile system for production of diverse therapeutic and diagnostic nanoparticulate medicinal products, and thereby facilitating the scale-up of more complex nanomedicinal drug products for clinical trials and eventually for the market.Conflicts of interest
R. Besseling, M. Damen and A. Gerich are the inventors of the NanoFlowSizer and shareholders of InProcess-LSP.Acknowledgements
This work was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft; DFG) in the framework of the Research Training Group 2375 “Tumor-targeted Drug Delivery” (grant number: 331065168) and the European Research Council (ERC; Meta-Targeting; grant number 864121). The authors thank Institut für Pathologie, Elektronenmikroskopische Einrichtung (EME), Universitätsklinikum Aachen for the TEM images of the liposomes.References
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2021, 6, e10246 CAS.
- G. Storm and D. J. A. Crommelin, Pharm. Sci. Technol. Today, 1998, 1, 19–31 CrossRef CAS.
- P. P. Deshpande, S. Biswas and V. P. Torchilin, Nanomedicine, 2013, 8, 1509–1528 CrossRef CAS PubMed.
- J. P. F. Longo, S. Mussi, R. B. Azevedo and L. A. Muehlmann, J. Mater. Chem. B, 2020, 8, 10681–10685 RSC.
- S. Shah, V. Dhawan, R. Holm, M. S. Nagarsenker and Y. Perrie, Adv. Drug Delivery Rev., 2020, 154–155, 102–122 CrossRef CAS PubMed.
- S. Lee, Modernizing the Way Drugs Are Made: A Transition to Continuous Manufacturing, FDA website, Published May 17, 2017 Search PubMed.
- S. L. Lee, T. F. O'Connor, X. Yang, C. N. Cruz, S. Chatterjee, R. D. Madurawe, C. Moore, L. X. Yu and J. Woodcock, J. Pharm. Innov., 2015, 10, 191–199 CrossRef.
- J.-M. Lim, A. Swami, L. M. Gilson, S. Chopra, S. Choi, J. Wu, R. Langer, R. Karnik and O. C. Farokhzad, ACS Nano, 2014, 8, 6056–6065 CrossRef CAS PubMed.
- G. Allison, Y. T. Cain, C. Cooney, T. Garcia, T. G. Bizjak, O. Holte, N. Jagota, B. Komas, E. Korakianiti, D. Kourti, R. Madurawe, E. Morefield, F. Montgomery, M. Nasr, W. Randolph, J.-L. Robert, D. Rudd and D. Zezza, J. Pharm. Sci., 2015, 104, 803–812 CrossRef CAS PubMed.
- A. P. Costa, X. Xu, M. A. Khan and D. J. Burgess, Pharm. Res., 2016, 33, 404–416 CrossRef CAS PubMed.
- Z. Chen, J. Y. Han, L. Shumate, R. Fedak and D. L. DeVoe, Adv. Mater. Technol., 2019, 4, 1800511 CrossRef.
- D. Carugo, E. Bottaro, J. Owen, E. Stride and C. Nastruzzi, Sci. Rep., 2016, 6, 25876 CrossRef CAS PubMed.
- G. Lou, G. Anderluzzi, S. Woods, C. W. Roberts and Y. Perrie, Eur. J. Pharm. Biopharm., 2019, 143, 51–60 CrossRef CAS PubMed.
- E. Kastner, R. Kaur, D. Lowry, B. Moghaddam, A. Wilkinson and Y. Perrie, Int. J. Pharm., 2014, 477, 361–368 CrossRef CAS PubMed.
- M. A. Tomeh and X. Zhao, Mol. Pharmaceutics, 2020, 17, 4421–4434 CrossRef CAS PubMed.
- J. M. Zook and W. N. Vreeland, Soft Matter, 2010, 6, 1352–1360 RSC.
- R. R. Hood, D. L. DeVoe, J. Atencia, W. N. Vreeland and D. M. Omiatek, Lab Chip, 2014, 14, 2403–2409 RSC.
- M. Jo, K.-M. Park, J.-Y. Park, H. Yu, S. J. Choi and P.-S. Chang, Colloids Surf., A, 2020, 586, 124202 CrossRef CAS.
- S. W. Z. Lim, Y. S. Wong, B. Czarny and S. Venkatraman, J. Colloid Interface Sci., 2020, 578, 47–57 CrossRef CAS PubMed.
- S. Lv, R. Jing, X. Liu, H. Shi, Y. Shi, X. Wang, X. Zhao, K. Cao and Z. Lv, Int. J. Nanomed., 2021, 16, 7759–7772 CrossRef CAS PubMed.
- M. Tiboni, M. Tiboni, A. Pierro, M. Del Papa, S. Sparaventi, M. Cespi and L. Casettari, Int. J. Pharm., 2021, 599, 120464 CrossRef CAS PubMed.
- R. Besseling, M. Damen, J. Wijgergangs, M. Hermes, G. Wynia and A. Gerich, Eur. J. Pharm. Sci., 2019, 133, 205–213 CrossRef CAS PubMed.
- N. Weiss, T. G. van Leeuwen and J. Kalkman, Opt. Express, 2015, 23, 3448–3459 CrossRef CAS PubMed.
- J. Kalkman, R. Sprik and T. G. van Leeuwen, Phys. Rev. Lett., 2010, 105, 198302 CrossRef CAS PubMed.
- C. Schuurmans, J.-P. Wijgergangs, A. Gerich and R. Besseling, Inline particle sizing in flow for demanding nanosuspension processes, AZoNano, 2022, DOI:10.13140/RG.2.2.18231.80805.
- R. Besseling, R. Arribas Bueno, R. van Tuijn and A. Gerich, Realtime droplet size monitoring of nano-emulsions during high pressure homogenization, AZoNano, 2021, DOI:10.13140/RG.2.2.30640.28166.
- C. Schuurmans, J.-P. Wijgergangs, A. Gerich, and R. Besseling, Continuous Size Monitoring of Turbid Titanium Dioxide Nanosuspensions with the Nanoflowsizer, AZoNano, 2022, https://www.azonano.com/article.aspx?ArticleID=5803, (accessed August 24, 2022) Search PubMed.
- R. D. Worsham, V. Thomas and S. S. Farid, Biotechnol. J., 2019, 14, 1700740 CrossRef PubMed.
- A. Porfire, M. Achim, C. Barbalata, I. Rus, I. Tomuta and C. Cristea, Pharmaceutical development of liposomes using the QbD approach, Liposomes Adv. Perspect, 2019, vol. 2019, pp. 1–20 Search PubMed.
- K. L. Delma, A. Lechanteur, B. Evrard, R. Semdé and G. Piel, Int. J. Pharm., 2021, 597, 120271 CrossRef CAS PubMed.
- K. Yang, J. T. Delaney, U. S. Schubert and A. Fahr, J. Liposome Res., 2012, 22, 31–41 CrossRef CAS PubMed.
- F. Yanar, A. Mosayyebi, C. Nastruzzi, D. Carugo and X. Zhang, Pharmaceutics, 2020, 12, 1001 CrossRef CAS PubMed.
- A. Nagayasu, K. Uchiyama and H. Kiwada, Adv. Drug Delivery Rev., 1999, 40, 75–87 CrossRef CAS PubMed.
- A. Pal, P. Sunthar and D. V. Khakhar, Ind. Eng. Chem. Res., 2019, 58, 7511–7519 CrossRef CAS.
- A. A. Gurtovenko and J. Anwar, J. Phys. Chem. B, 2009, 113, 1983–1992 CrossRef CAS PubMed.
- N. Kimura, M. Maeki, Y. Sato, A. Ishida, H. Tani, H. Harashima and M. Tokeshi, ACS Appl. Mater. Interfaces, 2020, 12, 34011–34020 CrossRef CAS PubMed.
- M. Patra, E. Salonen, E. Terama, I. Vattulainen, R. Faller, B. W. Lee, J. Holopainen and M. Karttunen, Biophys. J., 2006, 90, 1121–1135 CrossRef CAS PubMed.
- A. Clutterbuck, P. Beckett, R. Lorenzi, F. Sengler, T. Bisschop and J. Haas, in Continuous Biomanufacturing - Innovative Technologies and Methods, 2017, pp. 423–456, DOI:10.1002/9783527699902.ch15.
- T. De Beer, A. Burggraeve, M. Fonteyne, L. Saerens, J. P. Remon and C. Vervaet, Int. J. Pharm., 2011, 417, 32–47 CrossRef CAS PubMed.
- N. A. Helal, O. Elnoweam, H. A. Eassa, A. M. Amer, M. A. Eltokhy, M. A. Helal, H. A. Fayyaz and M. I. Nounou, Pharm. Pat. Anal., 2019, 8, 139–161 CrossRef CAS PubMed.
- I. E. W. Group, ICH guideline Q3C (R6) on impurities: guideline for residual solvents Step 5, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), 2019, pp. 1–32 Search PubMed.
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. R. Mozafari, Pharmaceutics, 2018, 10, 57 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2lc00463a |
| This journal is © The Royal Society of Chemistry 2023 |