
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homoconjugation effects in triptycene based organic optoelectronic materials
Jai-Ram
Mistry
 ,
Stephanie
Montanaro
and
Iain A.
Wright†
*
,
Stephanie
Montanaro
and
Iain A.
Wright†
*
Department of Chemistry, Loughborough University, Loughborough, Leicestershire LE11 3TU, UK
First published on 12th January 2023
Abstract
Molecular size and shape have a commanding influence on the electronic structure and photophysical properties of organic optoelectronic molecules. Small changes in, for example, the dihedral angles between two rings in a molecule can be used to fine tune frontier orbital distributions, and make the difference between efficient or poor absorbance, luminescence, charge transport etc. as well as impacting on mechanical and thermal properties to improve processability. Recent works have made it clear that structural changes which enforce homoconjugation, the through-space overlap of frontier π-orbitals across a shared non-conjugating group, can exert even more dramatic changes on these useful functional properties. Triptycene is an archetypical homoconjugated molecule with a rigid 3D structure which presents an ideal substrate from which to learn how best to employ homoconjugation to optimise functional molecular properties. This review demonstrates this by highlighting triptycene-based molecules in the context of organic electronic materials, where homoconjugation has resulted in markedly enhanced optoelectronic properties, and provides comment on the performance and future potential of these materials in devices where possible.
Introduction
Triptycene is a classic 3D polycyclic aromatic hydrocarbon with a paddlewheel-like structure. It consists of three benzene “fins” bound via two shared sp3 bridgehead carbon atoms (structure shown in Fig. 1). This rigid structure has led to extensive study in applications including molecular gears,1,2 motors3,4 and nanovehicles,5,6 sensors,7,8 porous polymers for gas separation,9–11 photocatalysis,12–15 chiral luminescent polymers,16,17 and as a key structural component of both metal18,19 and covalent organic frameworks20 and polymer networks.21 The large internal free volume between the fins of triptycene has also been exploited to control morphology and solubility in polymers and thin films.22–27 The fins of triptycene can be readily functionalised which presents great scope for tuning of properties. While this has perhaps been most frequently been encountered as a means to exert further control over morphology in the solid state, using substituents to impart desirable optoelectronic functionality has become an increasingly visible aspect of modern triptycene chemistry as will be highlighted throughout this review.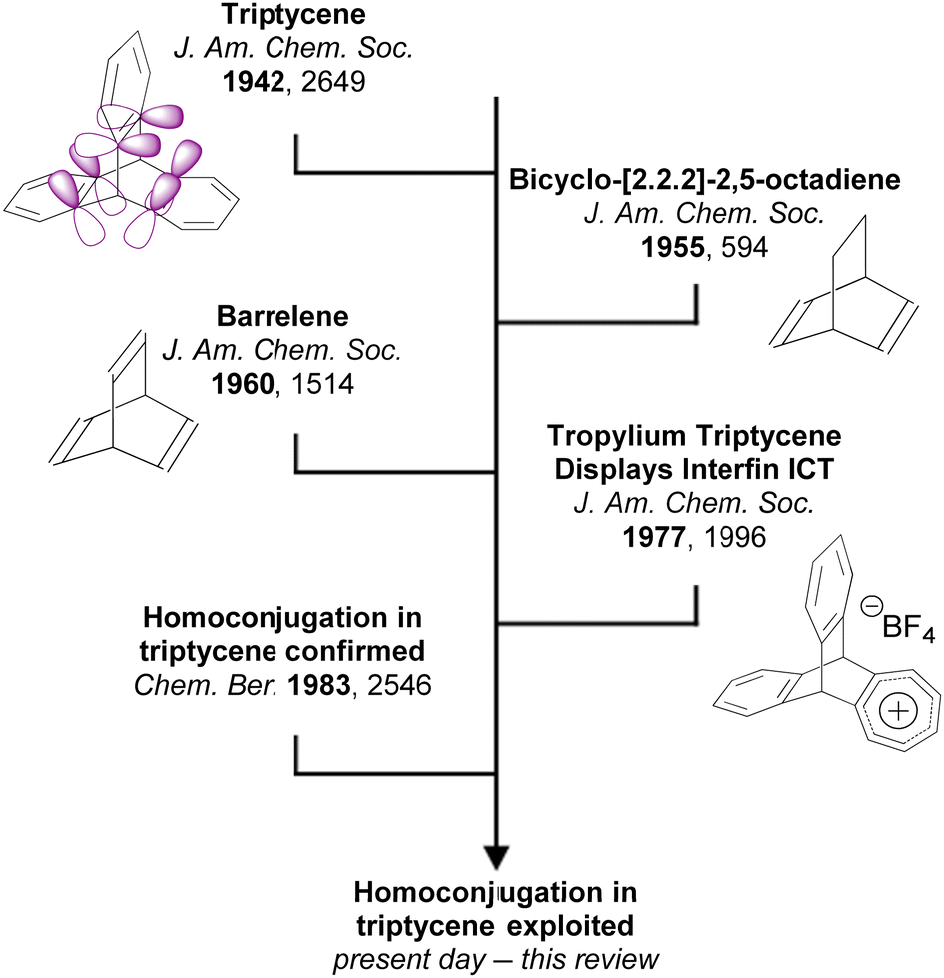 | ||
| Fig. 1 A short history of homoconjugation in triptycene. | ||
Homoconjugation in triptycene
An important characteristic of triptycene is that electronic communication can take place between the aryl fins through homoconjugation. IUPAC defines homoconjugation as “The orbital overlap of two π-systems separated by a non-conjugating group, such as CH2.”28 While homoconjugation may arise in a number of structural tropes,29 in triptycene it is largely mediated by the transannular, through-space overlap of the π-electron clouds over the aryl fins.A short history of milestones in understanding homoconjugation in triptycene is illustrated in Fig. 1. Following Bartlett's initial synthesis of triptycene in 194230 the possibility of electron transfer between the aryl fins was used to explain the differences between the UV/vis spectra of triphenylmethane and triptycene.31 The pursuit of homoconjugation underpinned the design and synthesis in 1955 of bicyclo-[2.2.2]-2,5-octadiene32 and in 1960 of bicyclo-[2.2.2]-octatriene33 which was given the informal name “barrelene” due to the barrel-like distribution of its frontier orbital density. Confirmation of homoconjugation in barrelene34–37 led to renewed interest in identifying homoconjugation effects in triptycene. As triptycene has a barrelene core it seemed intuitive that through-space electronic interactions should be occurring, but as the π-electrons in barrelene are olefinic and those in triptycene are benzenoid, the influence of any transannular interactions in triptycene remained contentious. In 1977 a triptycene bearing one cationic tropylium tetrafluoroborate fin demonstrated clear intramolecular charge transfer (ICT) character in its UV/vis spectrum which could only arise from electron transfer between fins.38 Homoconjugation in triptycene was finally proven using ultraviolet photoelectron spectroscopy in the early 1980s.39,40 Recent articles from the Mastalerz group which provides both reference to other key articles that led to this conclusion, alongside further experimental evidence of homoconjugation in triptycene derivatives.41,42
Since these studies, numerous other molecules were synthesised which clearly displayed the influence of homoconjugation, however it is only in recent years that this phenomenon has begun to be employed deliberately to exert control over the photophysical and electrochemical properties of triptycenes for optoelectronic applications. This review will focus on homoconjugation effects in triptycenes in this context. Following a short description of how to identify and modulate homoconjugation, the rest of this review consists of sections concerning organic light emitting devices (OLEDs), organic solar cells (OSCs) and charge transport/storage technologies. Specific comments will be made towards identifying the influence (or not) of homoconjugation on photophysical or electrochemical properties with the aim of obtaining a clearer picture of how best to utilise this effect in future materials development.
Observing and controlling homoconjugation in triptycene
Studies by the Rathore group provide some of the clearest pictures for identifying and understanding the influence of homoconjugation on the electronic properties of triptycene by simple visual analysis of the results of DFT calculations.43,44 A larger amount of frontier orbital density at the core of the triptycene indicates that stronger electronic coupling between the fins can be expected.In an elegant demonstration of this, they studied the influence of the regiochemistry of electron-donating alkoxy –OR substituents on the electronic structure of substituted triptycenes T23 and T14 using computational and electrochemical methods (Fig. 2a–c). In Fig. 2b it is clear that when the –OR groups are on the 1,4,5,8,11,14-peri-sites of triptycene in T14 there is more limited distribution of the HOMO over the triptycene core compared to when the –OR substituents are para- to the benzylic 9,10-bridgehead carbon atoms in the 2,3,6,7,12,13-positions in T23. This results in reduced transannular overlap and thereby diminished influence of homoconjugation in the HOMO manifold for T14. The consequences of this are directly observable in the electrochemical properties of T23 and T14 as shown in Fig. 2c. Both molecules display three sequential single-electron oxidation events corresponding to the concomitant oxidation of each fin. While for T23 three distinct and well-separated oxidation waves were observed, greatly reduced separation of these oxidations was observed for T14 demonstrating the more limited electronic coupling between fins arising from this substitution pattern.
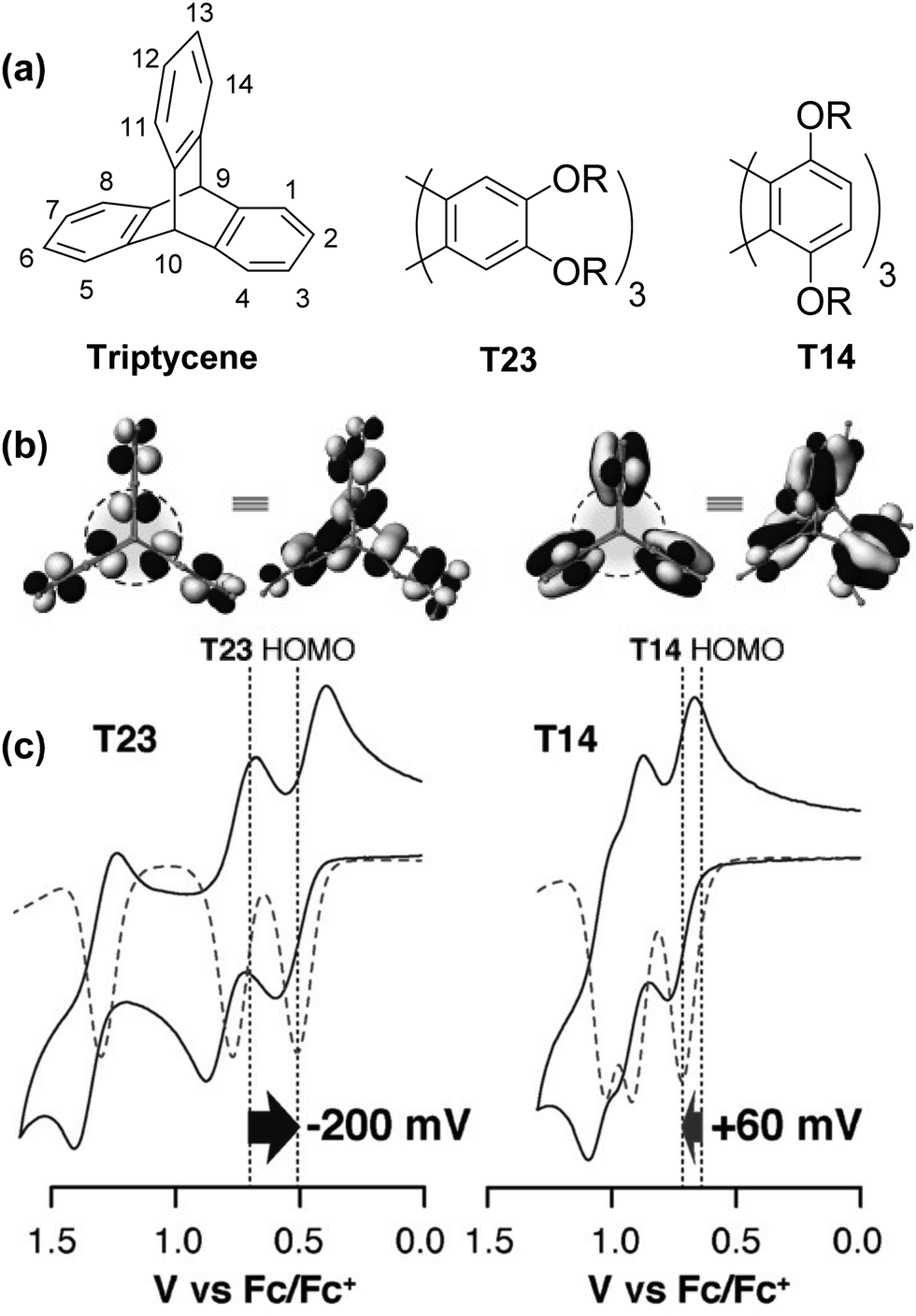 | ||
| Fig. 2 (a) Molecular structure of triptycene and alkoxy substituted triptycenes T23 and T14; (b) molecular orbital profiles for T23 and T14; (c) cyclic voltammograms for T23 and T14. Reproduced with permission from ref. 43. Copyright 2015, Wiley-VCH. | ||
As well as impacting on electrochemical behaviour, it is becoming increasingly clear that homoconjugation in triptycene derivatives can lead to increases in optical properties such as photoluminescence quantum yields (PLQY), molar extinction coefficients (ε) and oscillator strengths (fosc). These super-summative increases are greater than the threefold enhancement that might be expected in the absence of any electronic communication between the arms of the triptycene (vide infra).
Homoconjugated triptycene luminophores for OLEDs
In 2015 an initial effort to use homoconjugation to produce thermally activated delayed fluorescent (TADF) molecules was published by Kawasumi et al.45 TADF is an emission mechanism which, at a basic level, relies upon a small energy gap between the first excited singlet and triplet states (ΔEST) facilitating reverse intersystem crossing (rISC) to convert non-radiative triplet states into radiative singlet states.46 In an OLED device triplets and singlets are formed in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio so using TADF to harvest the triplets which are otherwise lost is a primary pathway to improve device efficiencies.
:1 ratio so using TADF to harvest the triplets which are otherwise lost is a primary pathway to improve device efficiencies.
Taking inspiration from established twisted donor–acceptor (D–A) TADF chromophores (whereby a large dihedral angle is imposed between D and A to ensure orthogonal separation of the HOMO and LUMO and reduce EST) this work used homoconjugation to facilitate weak orbital overlap and thereby through-space charge transfer (TSCT) between two triphenylamine D fins and a single dicyanoquinoxaline or dicyanopyrazine A fin in the triptycenes TPA-QNX(CN)2 and TPA-PRZ(CN)2 (Fig. 3a–c) for which ΔEST of 111 meV and 75 meV respectively were obtained. In the DFT results (Fig. 3d and e), homoconjugation can be observed over the sp2 carbon atoms attached to the sp3 bridgehead carbons. Solution state PLQYs were significantly reduced in the presence of air and a linear relationship between excitation and emission intensity was obtained. These two factors provide strong evidence of TADF. Yellow OLEDs with external quantum efficiencies (EQE) up to 9.4% for TPA-QNX(CN)2 (4.0% for TPA-PRZ(CN)2) were obtained. Homoconjugation therefore proved to be an effective method to facilitate TADF.
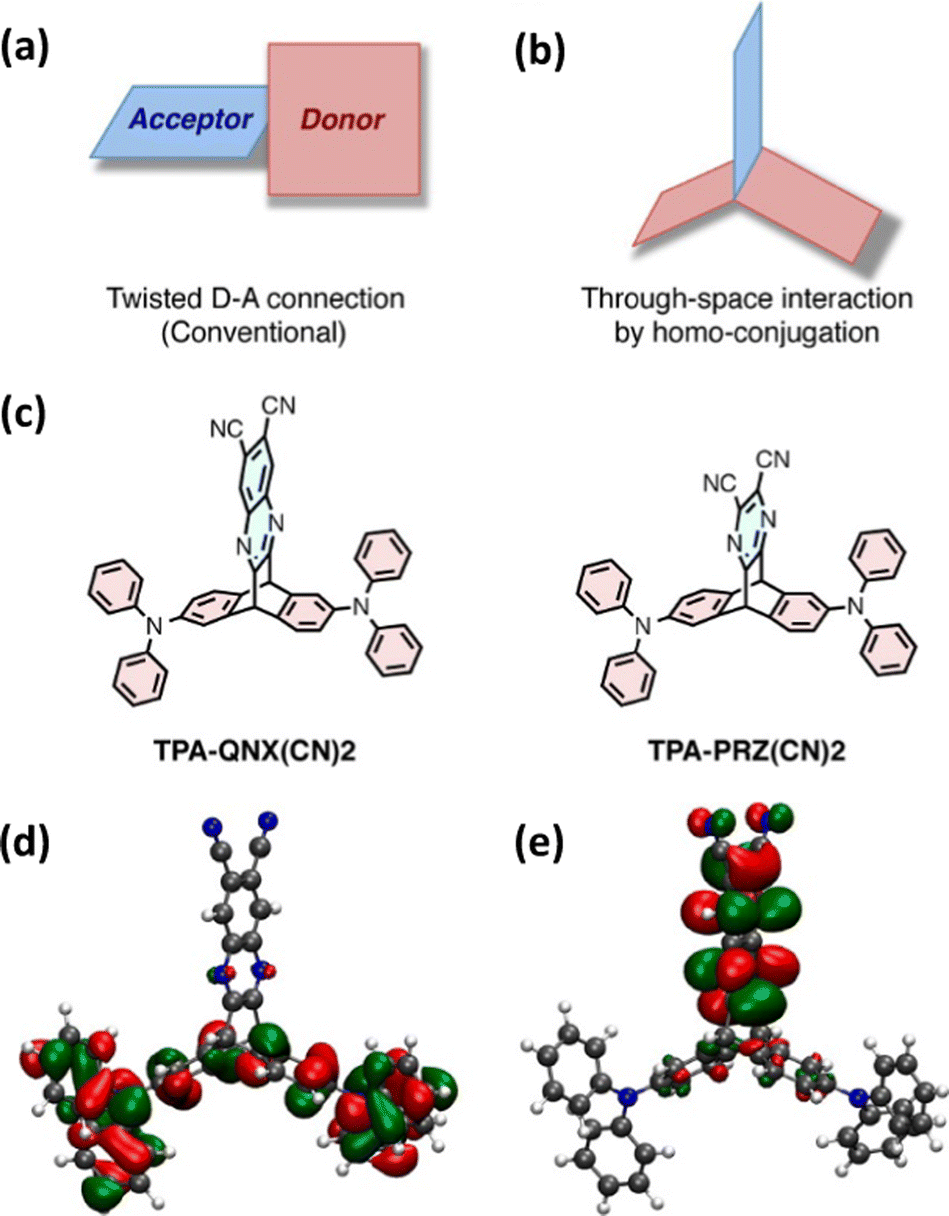 | ||
| Fig. 3 (a) Traditional and (b) homoconjugated TADF design strategy employed in ref. 45. (c) Chemical structures of TPA-QNX(CN)2 and TPA-PRZ(CN)2 and the (d) HOMO and (e) LUMO of TPA-QNX(CN)2. Adapted with permission from ref. 45. Copyright 2015, American Chemical Society. | ||
A similar design motif was exploited by Lei et al. where the acceptor fin was a simple non-functionalised quinoxaline which was used to make luminescent D–A–D and D–A–A systems (Fig. 4). These molecules exploited homoconjugation through the LUMO manifold and tuned the emission wavelengths between 329 and 625 nm by varying the fin-appended donors and acceptors.47
 | ||
| Fig. 4 Molecular structures of compounds studied in ref. 47. Copyright 2020, American Chemical Society. | ||
A subsequent report from the Swager group in conjunction with Buchwald showed that homoconjugation in a fused indole-triptycene donor TCZ used in conjunction with triazole-based acceptors exerted much more subtle control over ΔEST to improve TADF properties (structures in Fig. 5).48 The presence of the TCZ moiety led to a slight redshift in PL and a 0.03 eV reduction in ΔEST compared to a simple carbazole analogue. The bulky nature of TCZ turned on emission in a film of the molecule doped at 15 wt% into bis[2-(diphenylphosphino)phenyl] ether oxide (DPEPO). OLED devices of TCZ-TRZ and TCZ-TRZ(Me) had maximum EQE of 10.4 and 11.1% respectively but showed significant efficiency roll-off at 50 cd m−2 dropping to EQE = 3.4% and 2.0% due to degradation at high current density.
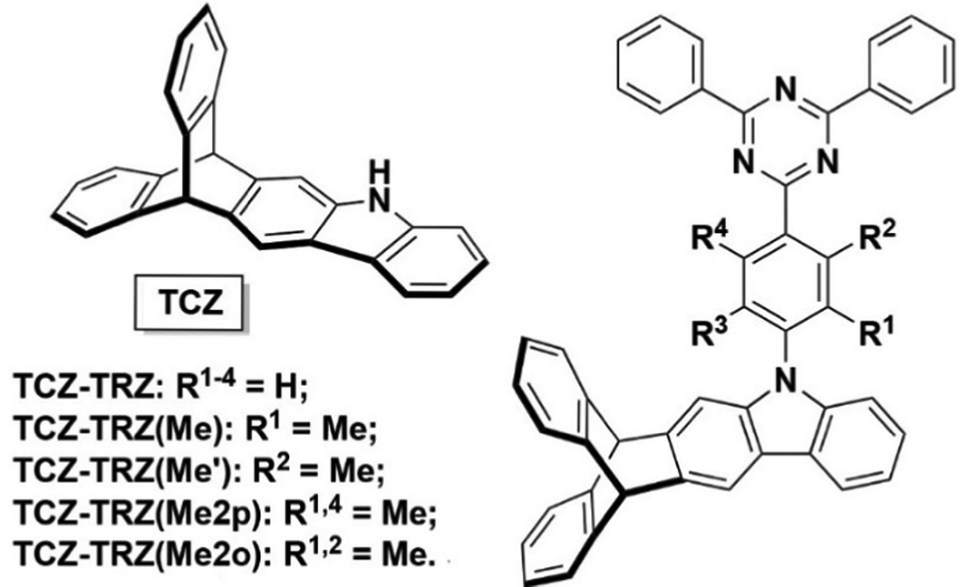 | ||
| Fig. 5 Donor–acceptor compounds of the TCZ-TRZ series from ref. 48 featuring homoconjugation on the periphery of the molecule. Adapted with permission from ref. 48. Copyright 2018, American Chemical Society. | ||
A short series of ring-fused triptycenes wherein one fin of the triptycene consisted of a coumarin-like moiety was reported by Qian et al.49DCT-1 featured a 1,4-dimethoxybenzene donor fin and was shown to undergo a homoconjugation facilitated ICT from this electron-rich fin to the electron-poor coumarin fin resulting in a charge separated excited state (structures, spectra and visual responses shown in Fig. 6). This was confirmed by the observation of positive solvatochromism and corroborated by computational insights. While the PLQY in solution was generally low, especially in comparison to the non-ICT compound, the donor–acceptor molecule displayed aggregation induced emission (AIE). The AIE was attributed to the methoxy groups hindering fin to fin stacking which leads to aggregation-caused quenching for the other molecules.
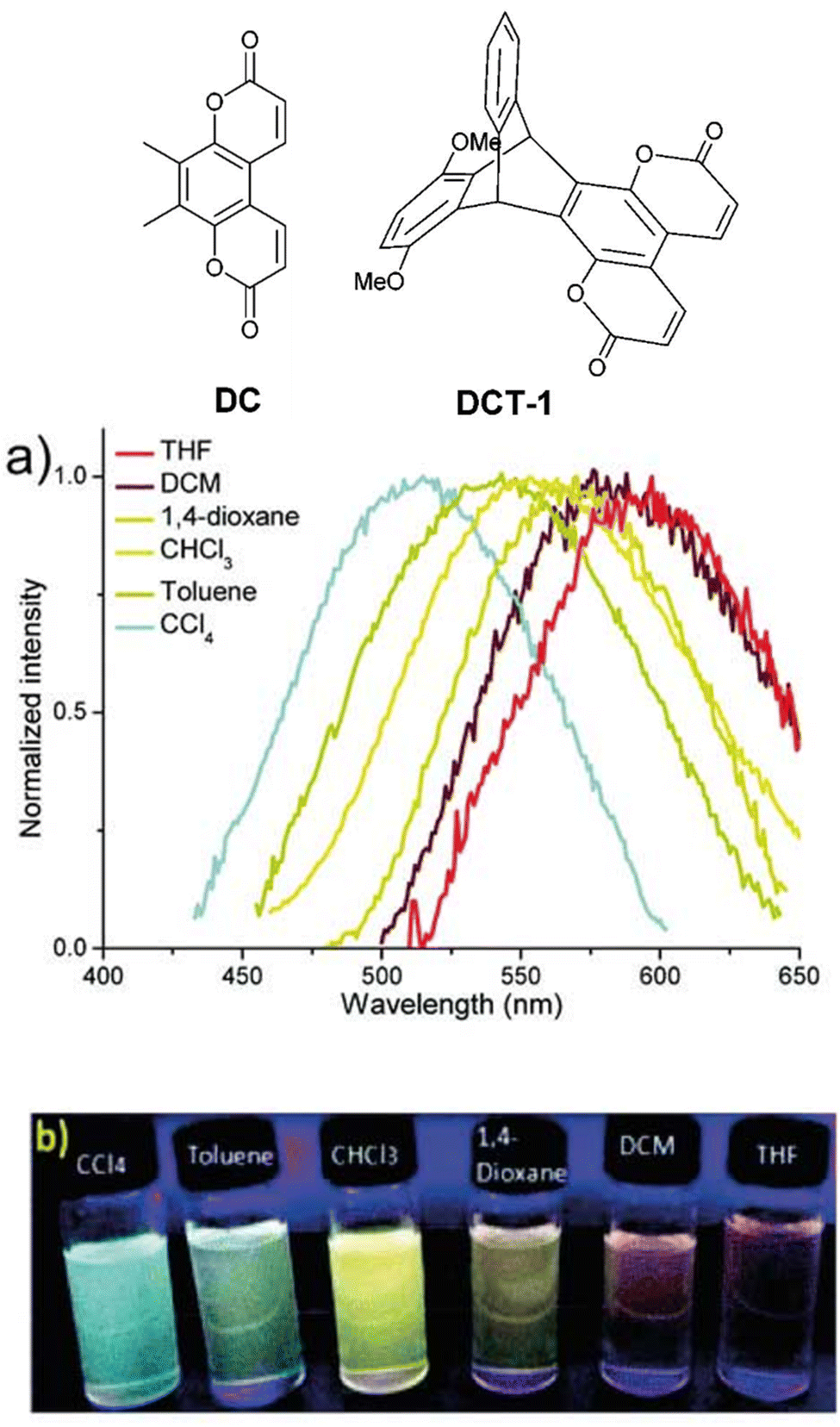 | ||
| Fig. 6 Coumarin dye DC and its homoconjugated ICT analogue DCT-1 and (a) emission spectra and (b) visual representation of the solvatochromism of DCT-1. Reproduced from ref. 49 with permission from the Royal Society of Chemistry. | ||
In 2019 Montanaro et al. presented a pyrazine fused D–π–A triptycene 3CzQ featuring six 3,6-di-tert-butylcarbazole donors bound to an electron poor pyrazine fused triptycene core through 1,4-phenylene spacers.50 In contrast with the ICT molecules discussed above where homoconjugation has facilitated TSCT between electron-rich and -poor fins, the structural motif of 3CzQ results in through-bond charge transfer (TBCT) from the donor carbazoles into the homoconjugated acceptor triptycene core. To fully evaluate the impact of this structure, the properties of 3CzQ were presented in comparison with a single-fin analogue CzQ (structures shown in Fig. 7).
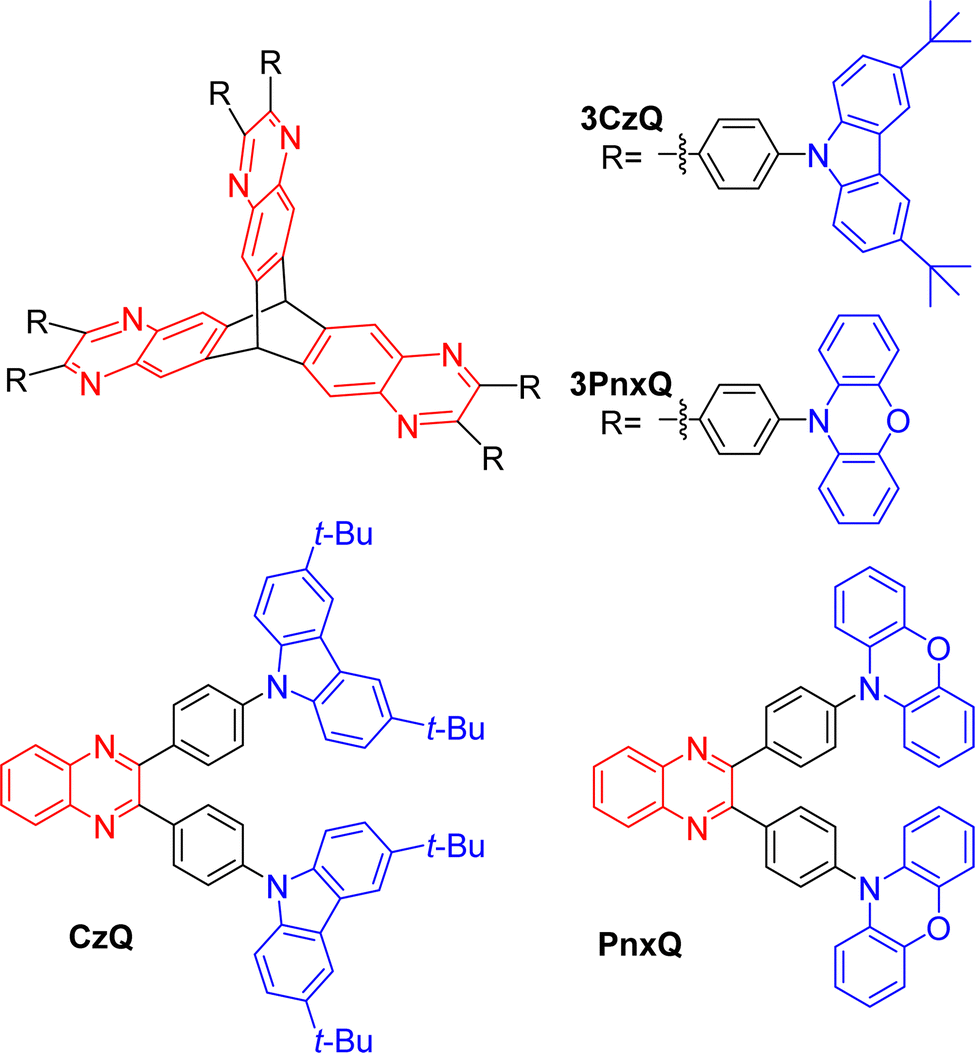 | ||
| Fig. 7 Molecular structures of triptycenes 3CzQ and 3PnxQ and their single fin analogues CzQ and PnxQ. | ||
Cyclic voltammetry supported by DFT revealed that the HOMO is distributed uniformly over the carbazole heterocycles while the LUMO is sequestered in the homoconjugated tris(quinoxline) core. The absence of significant communication between the carbazole rings of 3CzQ was evidenced by a simultaneous and reversible three-electron oxidation of the triptycene compared to the reversible single-electron oxidation observed for the single fin.
The D–π–A structure gave rise to intense ICT bands in the absorption and emission spectroscopy of both molecules (Fig. 8a and Table 1). Both also displayed blue emission with the emission maximum of the triptycene slightly red-shifted (5 nm) compared to the single fin. Due to a narrower photoluminescence full width at half maximum the Commission Internationale de L’Eclairage (CIE) coordinates of the molecules are nearly identical so colour purity remains unaffected.
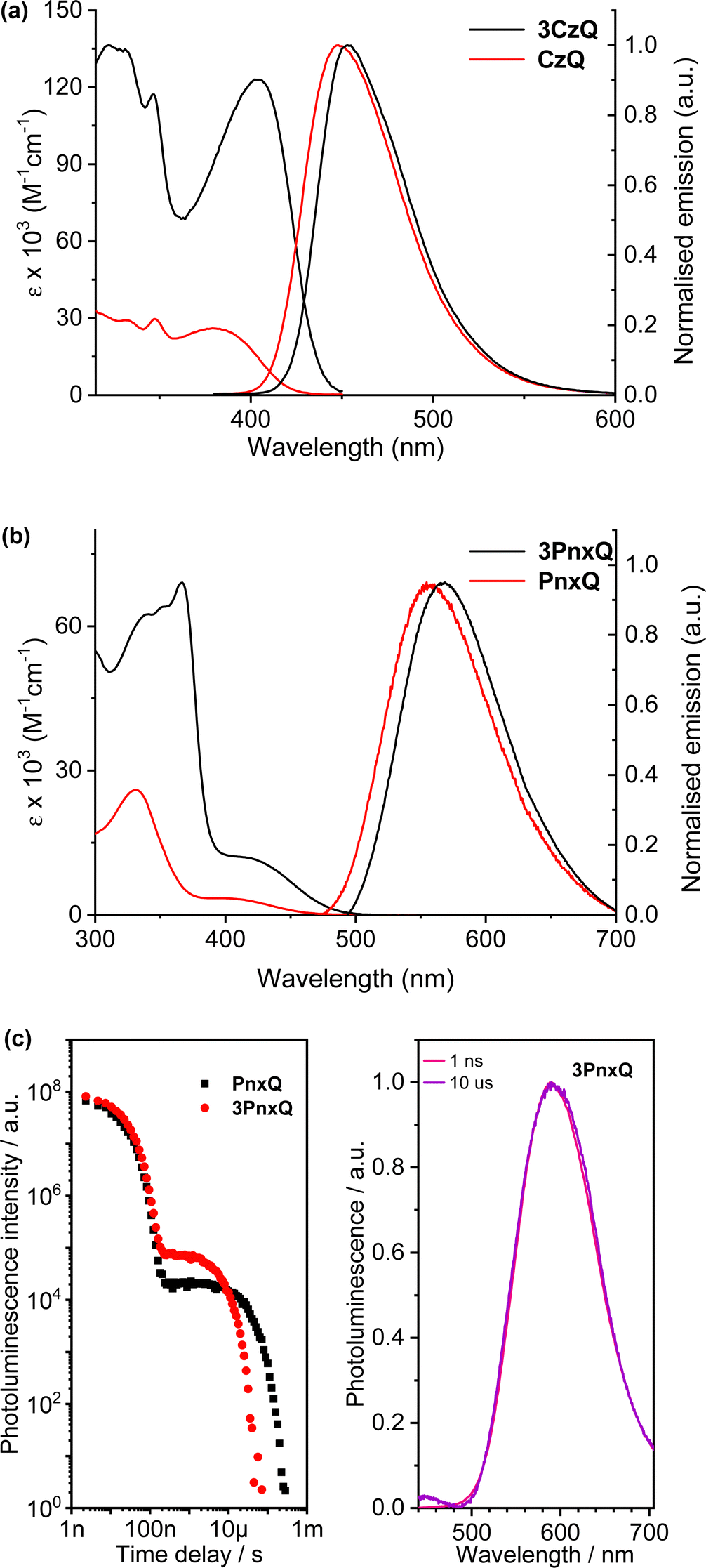 | ||
| Fig. 8 Absorption and normalised emission spectra of (a) 3CzQ and CzQ; (b) 3PnxQ and PnxQ; (c) photoluminescence decay of PnxQ and 3PnxQ (left) and prompt and delayed time-resolved spectra for 3PnxQ (right). | ||
| λ abs (nm) [ε × 103 M−1 cm−1] |
λ
maxPL (nm) |
PLQY (%) | |
|---|---|---|---|
| CzQ | 383 (26) | 448 | 35 |
| 3CzQ | 402 (123) | 453 | 56 |
| PnxQ | 398 (3.5) | 574 | 50 |
| 3PnxQ | 420 (12) | 583 | 61 |
An important observation from this study is that a near five-fold enhancement in extinction coefficient was observed for 3CzQvs.CzQ alongside a 37.5% increase in PLQY (Table 1). In contrast with the TSCT systems, TBCT is occurring from the carbazole donors into the homoconjugated tris(quinoxaline) core. TDDFT calculations revealed multiple low-lying excited states which led to a four-fold enhancement in the ICT oscillator strength 3CzQ over CzQ. This is seemingly a direct consequence of the high symmetry homoconjugated structure. Limited oxygen sensitivity was observed in the PLQY of both molecules which indicated that a triplet mediated emission process may also be at play.
The enhancement in PLQY, its apparent sensitivity to oxygen and the multiple low-lying excited states which were observed for 3CzQ presents an intriguing avenue to explore towards TADF. As described at the start of this section, TADF relies upon a narrow ΔEST to induce rISC, and efficient rISC is also dependent upon spin–orbit coupling between states. This is realised by having multiple partially delocalised triplet states with both locally-excited and charge transfer character.51–533CzQ satisfies the last of these criteria well but TADF was not observed, likely due to ΔEST being too large. By adopting a different donor ΔEST can be reduced at which point the multiple excited states facilitated by the homoconjugated iptycene will serve to enhance rISC while the super-summative increases in ε and fosc should maintain strong emission. These two properties are usually offset by one another so improving both simultaneously is a very desirable outcome.
By changing the donor moiety of 3CzQ from carbazole to the stronger donor phenoxazine 3PnxQ was realised and displayed a narrower ΔEST.54 The analogous single-fin PnxQ is a known TADF luminophore55 which was re-synthesised to permit direct comparison and validation against a known compound, the structures of both are shown in Fig. 7. Large improvements of ε and fosc were again observed for 3PnxQ alongside clear TADF emission (Fig. 8b and c, Table 1). Computational results indicated much stronger spin–orbit coupling for 3PnxQ over PnxQ and greatly increased fosc for S0 → S1 and S0 → S2. The homoconjugated structure successfully demonstrated a 250% acceleration in krISC in toluene solution and almost 100% in a solution processed 3PnxQ doped (concentration 5 wt%) PVK:PBD thin film. OLED devices of 3PnxQ in PVK:PBD showed better performance overall than those of PnxQ, particularly in terms of maximum luminance (14.8 × 103 cd m−2 for 3PnxQ; 8.9 × 103 cd m−2 for PnxQ) and charge density (220 mA cm−2 for 3PnxQ; vs. 120 mA cm−2 for PnxQ).
Homoconjugated triptycene chromophores for OSCs
In 2013 a theoretical study by Liu and Troisi highlighted that low-lying and degenerate excited states in fullerene were responsible for their reliably high performance in bulk heterojunction (BHJ) OSCs.56 They demonstrated computationally that the use of methine bridges, in other words homoconjugation, between chromophores would serve to create the degeneracy required. Yang et al. also performed DFT analysis of ladder-type NFA moieties fused to a triptycene core.57 This revealed LUMO degeneracy and homoconjugation in the HOMO. The calculations predict that more excitons will be generated from the triptycene and that the exciton binding energy is notably smaller for the 3D material due to reduced coulombic attraction. Triptycene therefore presents a promising scaffold from which to try and emulate useful properties of fullerene.A report in 2016 from the Mastalerz group was among the first to explore these findings in tandem with harnessing homoconjugation effects.58 They produced isomeric ring-fused triptycenes with electron-accepting aroylenimidazole fins C3V-D-TTAI and CS-D-TTAI (Fig. 9a). The isomers displayed similar optoelectronic properties (spectra shown in Fig. 9b) and comparable performance in BHJ devices. These molecules have optical band gaps (Eg) of 2.2 eV and ε ∼ 16 × 103 M−1 cm−1 for the lowest energy absorption band and emit orange-red light at 622 nm with a PLQY of 10–12%. While the low energy band was not formally assigned as an ICT transition, DFT results (Fig. 10) indicated that the HOMO is sited primarily over the triptycene moiety of these molecules while the LUMO and LUMO+1 are degenerate and distributed widely over the fins. This observation, alongside the absence of any of the fine-structure observed in the absorbance and emission spectra of related naphthalene diimide chromophores,59 makes some CT character seem likely. The frontier orbital distribution contrasts with the molecules 3CzQ and 3PnxQ mentioned in the previous section, where the frontier orbitals of the LUMO manifold are densely localised over the triptycene core while the HOMO is spread over the peripheral donors.
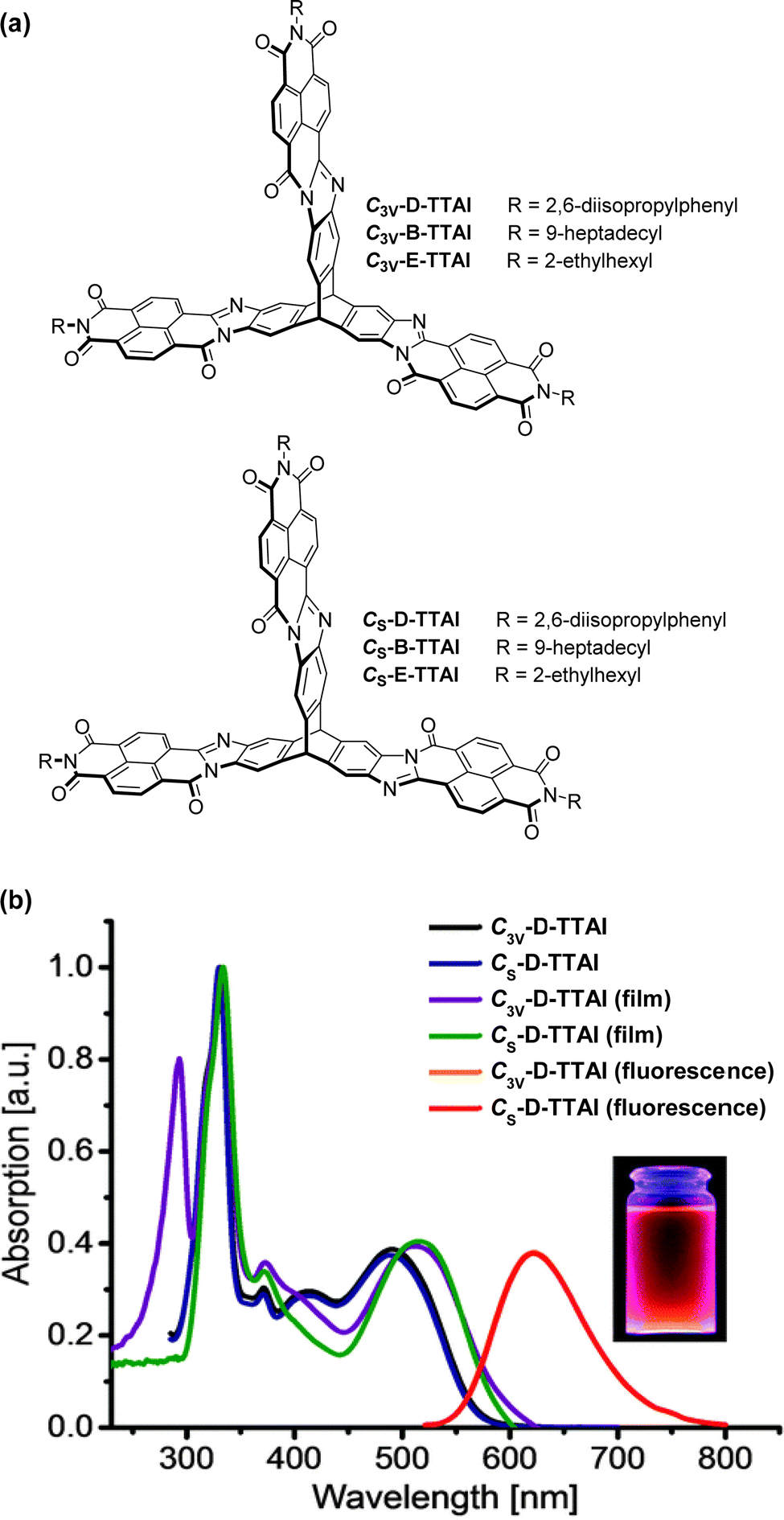 | ||
| Fig. 9 (a) Molecular structures of triptycene-trisaroylenimidazoles; (b) CH2Cl2 solution absorbance and emission spectra and thin film absorbance spectra of C3V-D-TTAI and C3V-D-TTAI. Reproduced from ref. 58 with permission from the Royal Society of Chemistry. | ||
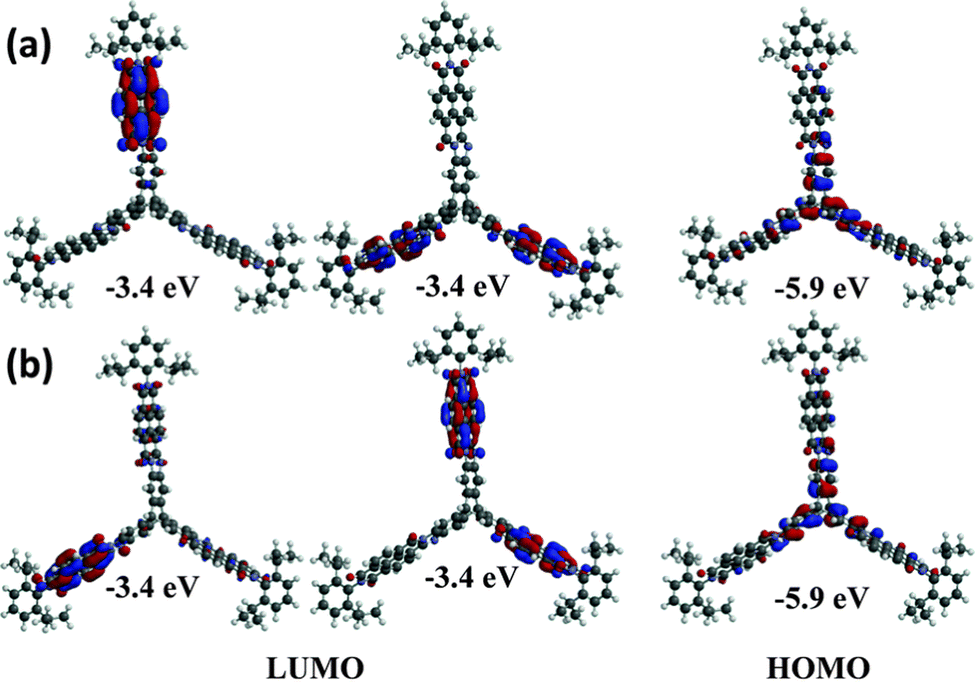 | ||
| Fig. 10 DFT-calculated frontier orbitals for (a) CS-D-TTAI and (b) C3V-D-TTAI. Reproduced from ref. 58 with permission from the Royal Society of Chemistry. | ||
BHJ OSCs of 2a-b fabricated with poly(3-hexylthiophene) (P3HT) had open circuit voltages (Voc) of 0.50 V and fill factors (FF) of 0.36 to 0.39 alongside short circuit currents (Jsc) to the order of 1 mA cm−2 were obtained. The low Jsc was attributed primarily to the poor morphology of the BHJ. The use of the more strongly electron donating polymer PTB7 increased both Voc and Jsc thereby improving the power conversion efficiency (PCE) to 1%.
To help overcome solubility issues encountered in the initial study the R-groups of the terminal diimides were changed from 2,6-di-iso-propylphenyl to 2-ethylhexyl (C3V-E-TTAI and CS-E-TTAI) and 9-heptadecyl chains (C3V-B-TTAI and CS-B-TTAI).60 The authors comment that the optical properties of the molecules do not vary upon changing the side chain groups, and indeed there is minimal change in the overall profile of the absorbance or the ultraviolet photoemission spectra (Fig. 11). However the absorbance data suggests there are significant increases in ε upon moving from an aryl to an alkyl solubilising group (Table 2).
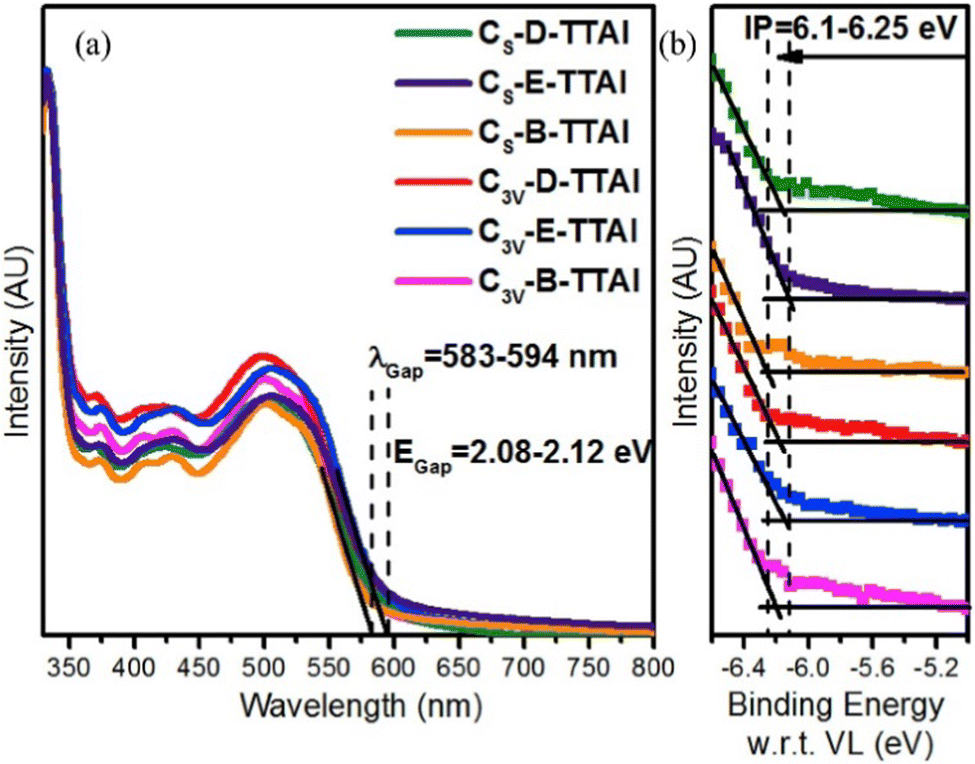 | ||
| Fig. 11 (a) Absorbance spectra of the TTAI acceptors and; (b) their ultraviolet photoemission spectroscopy indicating limited change in ionisation potential (IP). Reproduced with permission from ref. 60. Copyright 2017, Elsevier. | ||
| λ abs (nm) [ε × 103 M−1 cm−1] |
λ
maxPL (nm) |
|
|---|---|---|
| 1a | 440 (19.9) | 569 |
| 1b | 435 (19.9) | 561 |
| 1c | 442 (19.9) | 565 |
| 2a | 324 (25.1), 476 (10.0) | 624 |
| 2b | 324 (50.1), 476 (19.9) | 620 |
| 2c | 323 (31.6), 476 (12.6) | 620 |
| C 2-B-TDAI | 328 (63.1), 483 (25.1) | 615 |
| C 2-E-TDAI | 328 (50.1), 483 (15.8) | 615 |
| C S-B-TDAI | 328 (63.1), 483 (25.1) | 615 |
| C S-E-TDAI | 328 (63.1), 483 (19.9) | 615 |
| C 3V-D-TTAI | 329 (48.2), 486 (16.5) | 620 |
| C S-D-TTAI | 331 (50.1), 490 (15.8) | 618 |
| C 3V-B-TTAI | 329 (158), 484 (50.1) | 620 |
| C 3V-E-TTAI | 329 (126), 484 (50.1) | 616 |
| C S-B-TTAI | 329 (199), 484 (63.1) | 620 |
| C S-E-TTAI | 329 (126), 484 (50.1) | 620 |
In BHJ OSC with PTB7 the N-alkyl E and B analogues greatly outperformed the N-aryl D molecules, demonstrating an approximately threefold enhancement in PCE in all cases, primarily due to large increases in JSC. A maximum PCE of 3.16% was observed for CS-E-TTAI in a 1:1 D:A device. The devices still suffered from losses due to bimolecular recombination. Interestingly, the 2-ethylhexyl bearing molecules had a lower Voc than those bearing 9-heptadecyl chains despite having essentially identical frontier orbital energies. This variation in Voc was assigned to an improved microstructure in the BHJ. The 2-ethylhexyl bearing molecules also had improved Jsc over the 9-heptadecyls and therefore marginally outperformed them overall.
Subsequent papers systematically provided the properties of single fins 1a–c, single-finned triptycenes 2a–c (Fig. 12a)61 and two pairs of isomeric double-finned triptycenes C2-B-TDAI and C2-E-TDAI and CS-B-TDAI and CS-E-TDAI (Fig. 13a) where B indicates R = 9-heptadecyl and E indicates R = 2-ethylhexyl.62 For the triptycene containing 2a–c, similar frontier orbital distributions to C3V/S-D-TTAI were calculated with the HOMO localised over the triptycene core and the LUMO spread over the electron poor fins. Degenerate LUMO and LUMO+1 were again predicted for C2/S-B/E-TDAI. The non-triptycene single fins 1a–c have larger ε for the longest wavelength absorbance peak compared to 2a–c, but significantly blue-shifted absorbance (Fig. 12b). The profile of the absorbance and emission spectra of the single-finned 2a–c and double-finned TDAI compounds (Fig. 13b) are very similar to those of the triple-finned TTAI series but the triple-finned systems retain the highest ε.
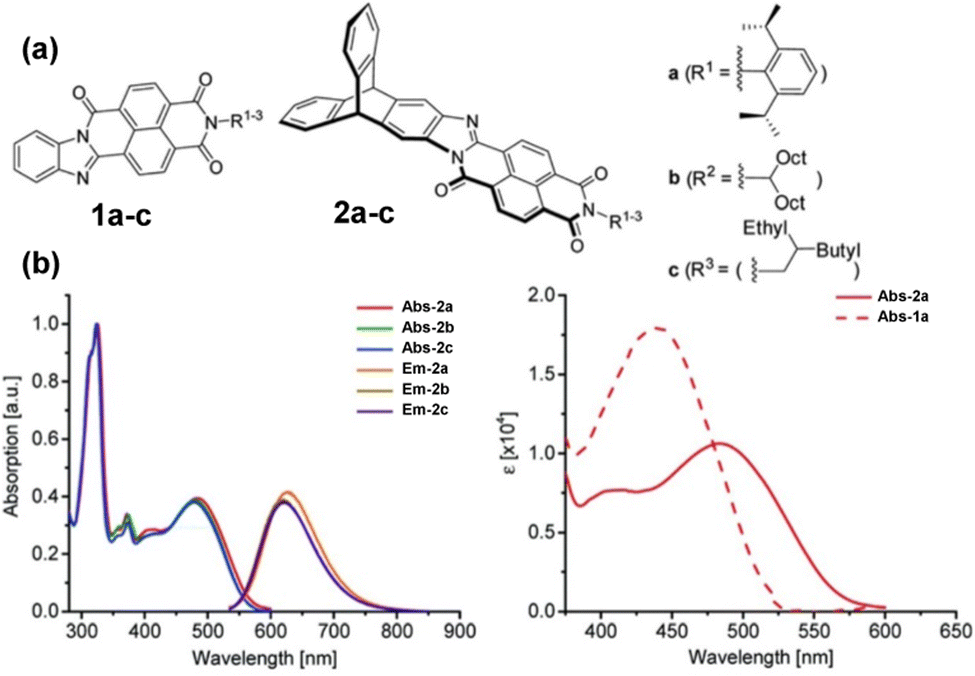 | ||
| Fig. 12 (a) Molecular structures of 1a–c and 2a–c. (b) Left: Absorbance and emission spectra of 2a–c. Right: Comparison of longest wavelength absorbance of 1a and 2a. Reproduced from ref. 61 with permission from the Chinese Chemical Society (CCS), Shanghai Institute of Organic Chemistry (SIOC), and the Royal Society of Chemistry. | ||
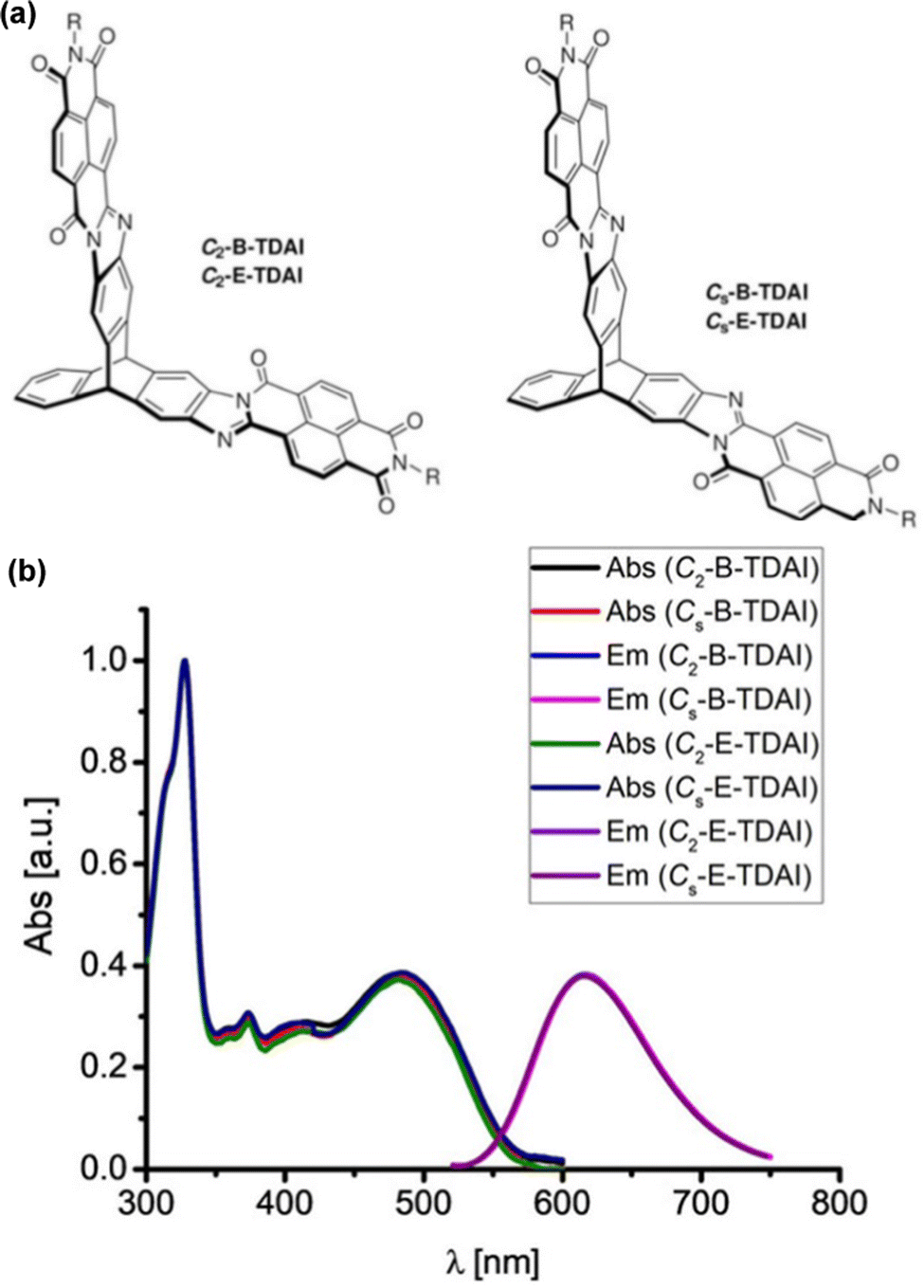 | ||
| Fig. 13 (a) Molecular structures of CS/2-B/E-TDAI; (b) solution absorbance and emission spectra for CS/2-B/E-TDAI. Adapted with permission from ref. 62. Copyright 2017, Wiley-VCH. | ||
Generally, the optical properties of the two-finned triptycenes are not improved compared to the single-finned triptycenes. They show slight enhancements in the intensity of both the highest and lowest energy bands alongside a slight blue-shift in emission and narrowing of the Stokes shift. Upon moving to a three-finned system, super-summative enhancements in both the highest and lowest energy bands are observed when comparing one to three fins for the B- and E-TTAI triptycenes. The clearest example of this is in the 2-ethylhexyl series where C3V-E-TTAI and CS-E-TTAI display a 4-fold enhancement in both the high and low energy bands compared to 2c.
While OSC performance remains modest, the presence of the triptycene moiety did lead to a higher PCE of 2.2% for 2a with PTB7 as donor (compared to 1.2% for 1a). Low FF (<42%) were observed in all devices. The double-finned triptycenes performed similarly in OSCs with CS/2-B-TDAI displaying PCE of ∼2% and CS/2-E-TDAI displaying PCE of ∼1%.
Other imide-based acceptors that have been employed with triptycene in a similar fashion are useful to make comparisons of. Perylene-3,4,9,10-tetracarboxylic acid diimides (PDIs) are an established class of NFA material. PDI is a planar molecule with a strong tendency to aggregate due to π–π stacking interactions which is a limiting factor in its utility.63 While imposing twists between adjacent PDI units has previously been used to overcome this,64,65 Liu et al. chose to exploit the internal free-volume of triptycene to hinder aggregation between electron accepting PDI fragments.66 Suzuki cross-coupling between a triptycene 2,7,14-tris(boronate) ester and a 1-bromoPDI bearing R = 2-octyldodecyl produced the molecule TP3 (Fig. 14). This greatly improved solubility and reduced aggregation.
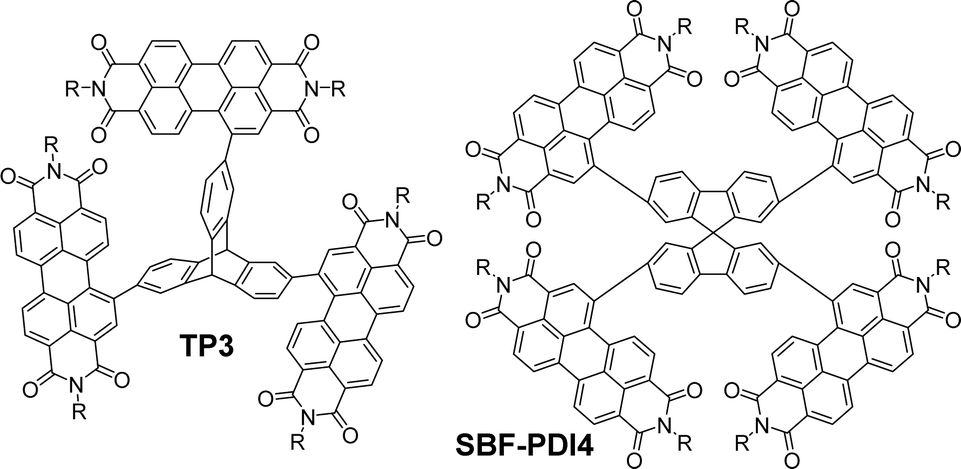 | ||
| Fig. 14 Molecular structures of non-ring-fused PDI functionalised TP3 (R = 2-octyldodecyl) and its spirocyclic SBF-PDI4 (R = 2-ethylhexyl). | ||
Degeneracy was observed in the frontier orbital manifolds as predicted by computational methods. The HOMO and LUMO manifolds were distributed almost exclusively across the pendant PDI units with only limited density over the triptycene. Experimentally, this was reflected in the electrochemistry where all three PDI moieties are reduced simultaneously, and in the UV-vis spectrum which has fine structure characteristic of PDI and a molar extinction coefficient (ε = 9.4 × 104 M−1 cm−1 at 540 nm in CHCl3) which shows only slight enhancement over unfunctionalised N,N′-bis(2-ethylhexyl)PDI molecules (ε ∼ 7 × 104 M−1 cm−1 at 525 nm in CHCl3 and ε ∼ 8.5 × 104 M−1 cm−1 at 525 nm in CH2Cl2).67,68
This study is useful to highlight as it permits comparison with the spirocyclic analogue SBF-PDI4 (Fig. 14) which has also been studied as the donor in BHJ devices.69 Spirobifluorene represents another class of 3D hydrocarbon featuring a homoconjugated sp3 carbon atom.70,71 The optical properties of SBF-PDI4 (ε = 9.3 × 104 M−1 cm−1 at 535 nm in CHCl3) are essentially identical to those of TP3, and the MO layout is similar. For these molecules, we can conclude that the triptycene or spirobifluorene core is acting as a simple scaffold and not contributing in any meaningful sense to interchromophore coupling. As such, the PDI chromophores are acting independently of each other with excitations occurring over rather than between fins.
TP3 was employed in BHJ devices with the polymer PTB7-Th displaying PCE up to 3.16%, FF = 0.39 and Voc = 0.86 V and Jsc up to 9.54 mA cm−2 with the best performing device requiring 0.5 wt% 1,8-diiodooctane (DIO) as an additive. Low FF were observed in all devices. Electron and hole mobility measured using SCLC methods were also low (10−4 cm2 V−1 s−1) albeit well-balanced. These poor attributes were ascribed to morphology issues with aggregation and phase separation still occurring despite the large IFV of the triptycene. For SBF-PDI4 a 1:1 D:A device with PTB7-Th as donor had near identical performance characteristics to those of TP3 other than the FF which was improved to 0.48. This indicates that a better heterojunction morphology is obtained using the spirocycle and, importantly, that the configuration of a 3D core can be optimised to improve device performance. The synthesis of SBF-PDI4 was also published independently by Lee et al. and incorporated in devices with polymers P4T2FBT and PV4T2FBT, obtaining PCE = 5.98% with the latter.72
An analogue of TP3 with smaller solubilising R groups (R = 6-undecyl) was presented in a separate study as an intermediate in the synthesis of the ring fused molecule T-3.73 Single (T-1) and double (T-2) PDI finned analogues were also prepared (structures and absorbance spectra shown in Fig. 15). The overall waveform is sharp and structured, very like TP3 and PDI itself. Compared to TP3, annulation of the PDI onto the triptycene core blueshifts the longest energy absorbance and the authors note a slight blueshift in the λmax and widening of the optical Eg as more PDI-fused fins are appended. This was attributed to weakening of the molecular dipole upon addition of further electron accepting fins.
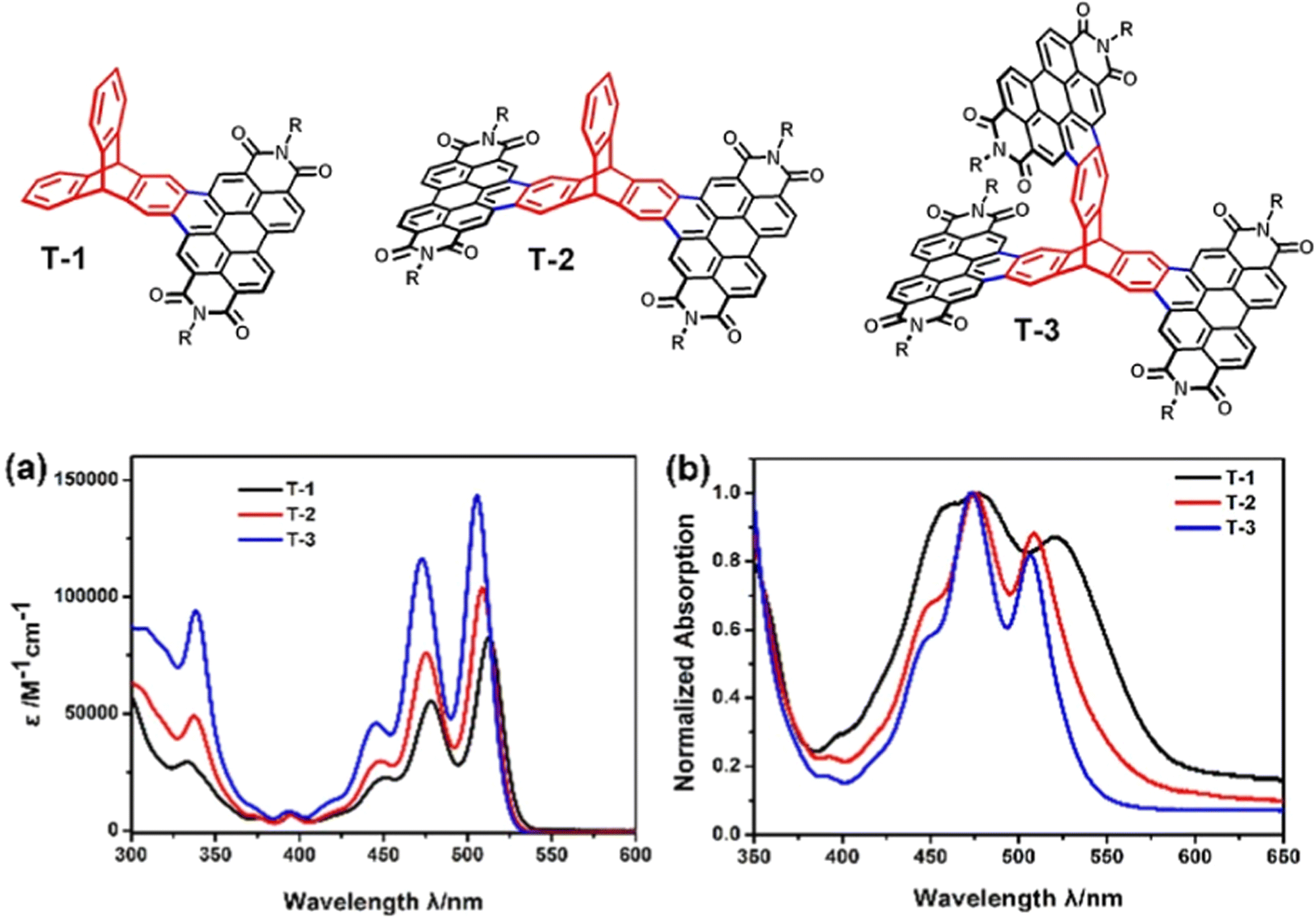 | ||
| Fig. 15 Molecular structures of PDI-fused triptycene T1–T3 (R = 6-undecyl) and their (a) CHCl3 solution absorbance spectra and (b) thin film absorbance spectra. Adapted with permission from ref. 73. Copyright 2017, Wiley-VCH. | ||
Across this series, no super-summative enhancements are observed. The ε of T-1 is comparable to that of PDI itself (and by proxy TP3) and that of T-2 is only slightly larger. T-3 displays an ε which is approximately double that of T-1. DFT results for T-3 show the HOMO is distributed over the entire molecule while the LUMO is spread uniformly over two of the PDI fins with minimal LUMO density over the triptycene. This is identical to the LUMO distribution of T-2. The frontier orbital layout for T-1 is all positioned over the PDI fin. Cyclic voltammetry confirmed that the electron accepting strength of the molecules is essentially constant across the series, which indicates that the LUMO energy has not changed as fins have been added. Oxidation processes were not observed.
In terms of device performance for this series, T-2 gave the best BHJ devices using PTB7-Th as a donor. T-2 displayed PCE of 6.15% compared to 2.28% for T-1 and 5.13% for T-3 which had comparable Jsc but slightly lower FF. Voc decreased gradually across the series as may be predicted from the blueshift in the UV spectra. T-2 and T-3 had a better balance of electron and hole mobilities which correlated with AFM results that indicated better film morphology for the T-2 and T-3 molecules.
A 2018 report by Kang et al. concerned triptycene 3, (Fig. 16a) which was shown to undergo photoinduced hole transfer with single-walled carbon nanotubes.74 The synthesis and properties of this compound alongside its first (T-3) and third (4) generation analogues followed in a subsequent contribution. Note that the smallest of these triptycenes T-3 had also been presented in ref. 73. The results concerning T-3 from these independent studies agree very well.
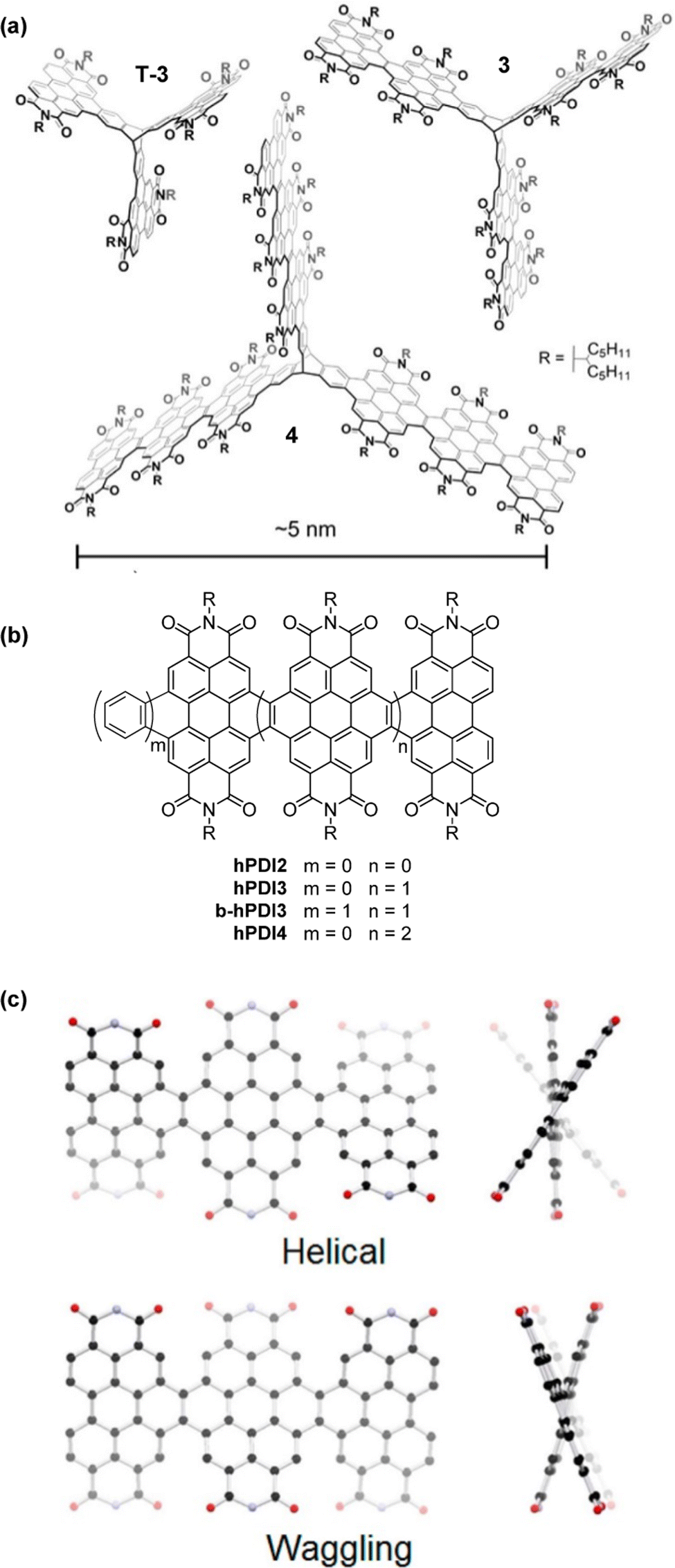 | ||
| Fig. 16 (a) Molecular structures of fin-extended PDI-fused triptycenes T-3, 3 and 4, Adapted with permission from ref. 75. Copyright 2018, American Chemical Society (b) the single-fin analogues hPDI2–4 and benzo-fused single fin analogue b-hPDI3; (c) DFT optimised Helical and Waggling geometries for hPDI3. Reproduced with permission from ref. 76. Copyright 2014, American Chemical Society. | ||
The helical PDI trimer hPDI3 and a benzene-fused analogue b-hPDI3 thereof (Fig. 16b) were employed as model compounds for comparison with 4.75hPDI3 had previously been studied by the same group in 2014 as one of a family of fused PDI oligomers alongside the dimer hPDI2 and tetramer hPDI4.76 These are helpful to consider when trying to understand the behaviour of 3 and 4.
By DFT, hPDI2–4 were shown to be non-planar due to steric repulsion between the C–H bonds of the cove regions of the PDI moieties. This results in helical or waggling geometries, or a mixture of the two in the case of hPDI4. The optimised geometries of hPDI3 are shown in Fig. 16c. The UV/vis and emission spectra for hPDI2–4 (Fig. 17) display a red-shift as the oligomer length increases. The lowest energy absorbance is assigned to a simple HOMO-to-LUMO transition over the PDI chromophores. ε of this band is essentially constant between PDI, hPDI2 and hPDI3 but then increases dramatically by ∼100% for hPDI4. This enhancement is attributed to red-shifting of the symmetry-allowed transitions which are taking place from the HOMO−2 (over the carbon–carbon double bonds which fuse the PDI moieties together) into the LUMO. This red-shifting results in overlap with the HOMO-to-LUMO transition leading to the large increase in absorption intensity.
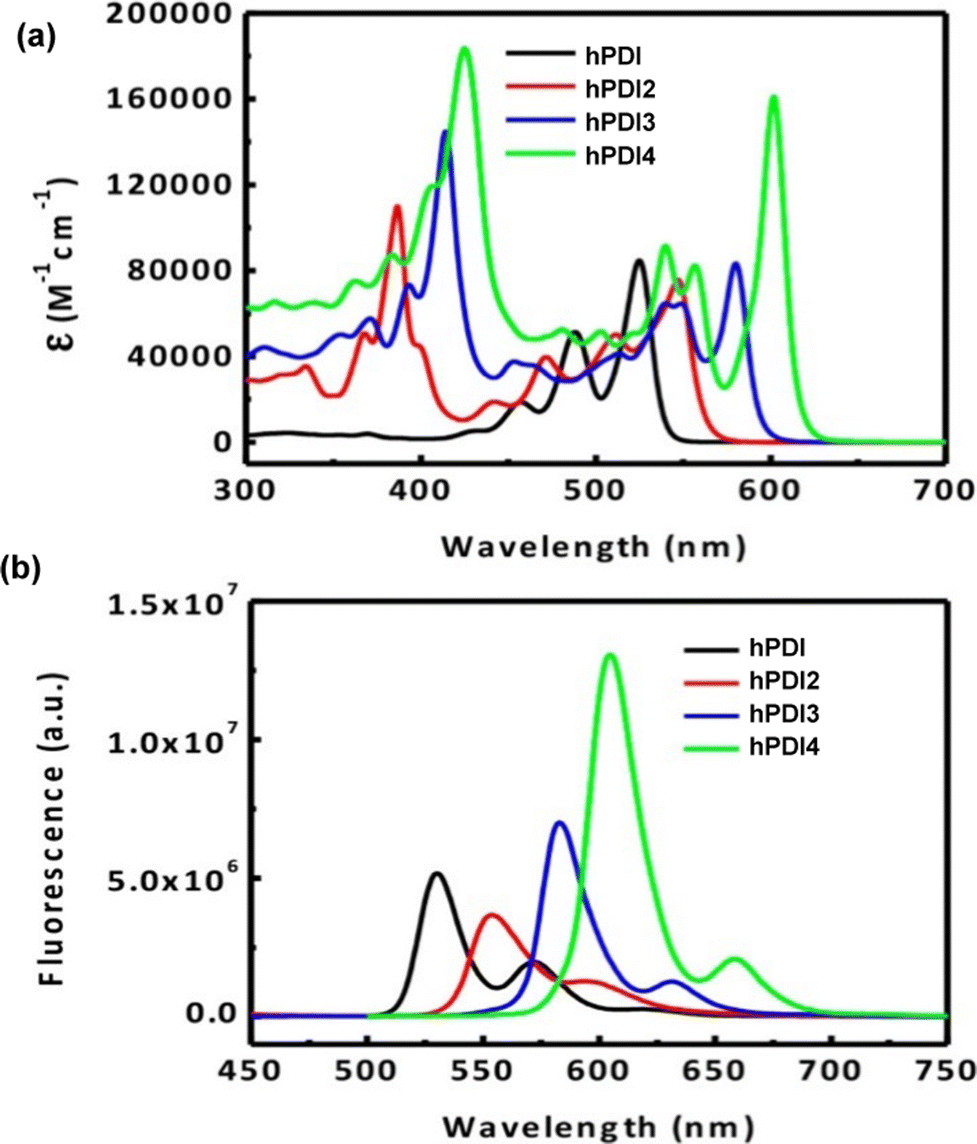 | ||
| Fig. 17 (a) UV/vis absorbance spectra for hPDI (N,N′-bis(6-undecyl)PDI) and hPDI2–4 in CH2Cl2 solution; (b) fluorescence spectra for hPDI and hPDI2–4. In CH2Cl2 solution. Reproduced with permission from ref. 76. Copyright 2014, American Chemical Society. | ||
In the UV/vis spectra of the triptycenes T-3, 3 and 4 (Fig. 18a) a similar sequential red-shift is observed for the lowest energy absorbance band with increasing fin length. An extremely high extinction coefficient (ε > 600000 M−1 cm−1) is observed for the largest derivative 4. This is over a 5-fold increase in ε compared to hPDI3. While benzene-fused b-hPDI3 also displayed a higher ε than hPDI3, the homoconjugated triptycene still displays an increase in ε of over 35% compared to three times that of b-hPDI3 (Fig. 18b).
 | ||
| Fig. 18 UV/vis absorbance spectra for (a) T-3, 3 and 4 and (b) 4 and b-hPDI3 in tetrahydrofuran solution. Adapted with permission from ref. 75. Copyright 2018, American Chemical Society. | ||
While the DFT of T-3, 3 and 4 indicated that the HOMO and LUMO have no presence over the central triptycene, with the frontier orbitals localised over the fins in a similar arrangement to hPDI2–4, TDDFT results showed that a manifold of low-lying excited states is present for 3 and 4. It is difficult to conclude whether these arise from the complex symmetry adopted by the fins themselves, homoconjugation facilitated by the central triptycene or some combination of the two. It is clear that the influence of the extended three-dimensional structure is distinct from simple ring fusion but the precise origins of super-summative enhancements in ε are challenging to pin down in these very large molecules.
Electrochemistry indicated that the oligoPDI fins were not electronically coupled. The cyclic voltammetry results revealed that each fin can be reduced simultaneously and in a stepwise fashion with each PDI moiety accepting two electrons. Therefore the largest molecule 4 is a reversible 18-e− acceptor. This is directly analogous to the multi-electron redox behaviour observed for the reduction of TP3, SBF-PDI4, and the oxidation of 3CzQ. Electron paramagnetic resonance studies of T-3 indicated that its radical anion is delocalised across all three fins which the authors attribute as being facilitated by transannular interactions over the homoconjugated triptycene core.
Triptycenes 3 and 4 were incorporated as electron transport materials in perovskite solar cells. Both outperformed PC61BM with maximum efficiencies of 16.4% and 18% respectively. Combined with the results of SCLC measurements, the authors postulate that the longer fins penetrate more effectively into the perovskite layer thereby reducing contact resistance and improving overall device performance. The use of iptycenes as interface agents to modify film surfaces is reminiscent of the use of iptycenes to exert internal control over polymer morphology. This may represent a better use for triptycene derivatives in OSCs than as bulk active components themselves.
Though not studied for any specific application, a separate series of conceptually related, laterally-expanded electron-deficient ring-fused triptycenes P1–P4 were published by Hu et al. in 2019.77 In these examples the fins are composed of pyrene N-heteroacenes with diameters of up to 10.88 nm (Fig. 19) and show similarly complex variations in optical properties as the fin length increases. The pyrene units served to disrupt conjugation along the length of the fins therefore minimal redshift with increasing conjugation length was observed due to a gradual drop in LUMO energy as determined by cyclic voltammetry and DFT.
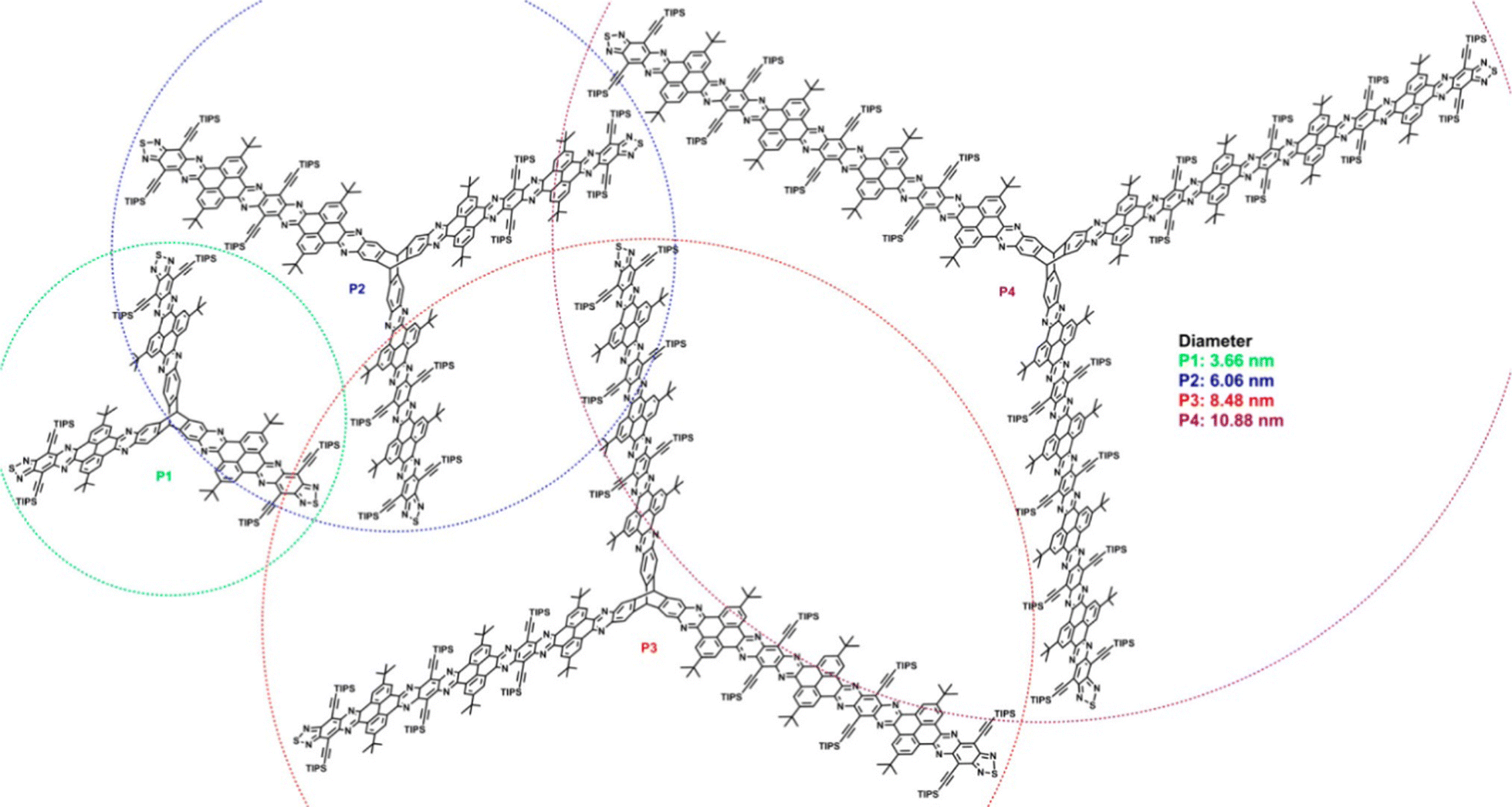 | ||
| Fig. 19 Molecular structures of nanoscale extended triptycenes P1–P4. (Reproduced with permission from ref. 77. Copyright 2019, American Chemical Society.) | ||
A theoretical study from 2019 by Sun et al. of 3D vs. 2D HTMs indicates that 3D structures have improved electron–hole dissociation characteristics which may help to improve performance, so pursuing large molecules such as 4 and P4 as charge transport materials presents an important area for further development.78
Homoconjugated charge transport and energy storage materials
In 2018, Kwon et al. disclosed the triptycene hexaketone TT and diketone TM (structures in Fig. 20) and studied TM for energy storage applications.79 Homoconjugation in the LUMO manifold was revealed from DFT analysis and cyclic voltammetry. Differential pulse voltammetry (DPV) revealed that TT could be reduced in single electron steps to the pentaanion (Fig. 20) with instabilities for the fully reduced hexaanion predicted using computational methods.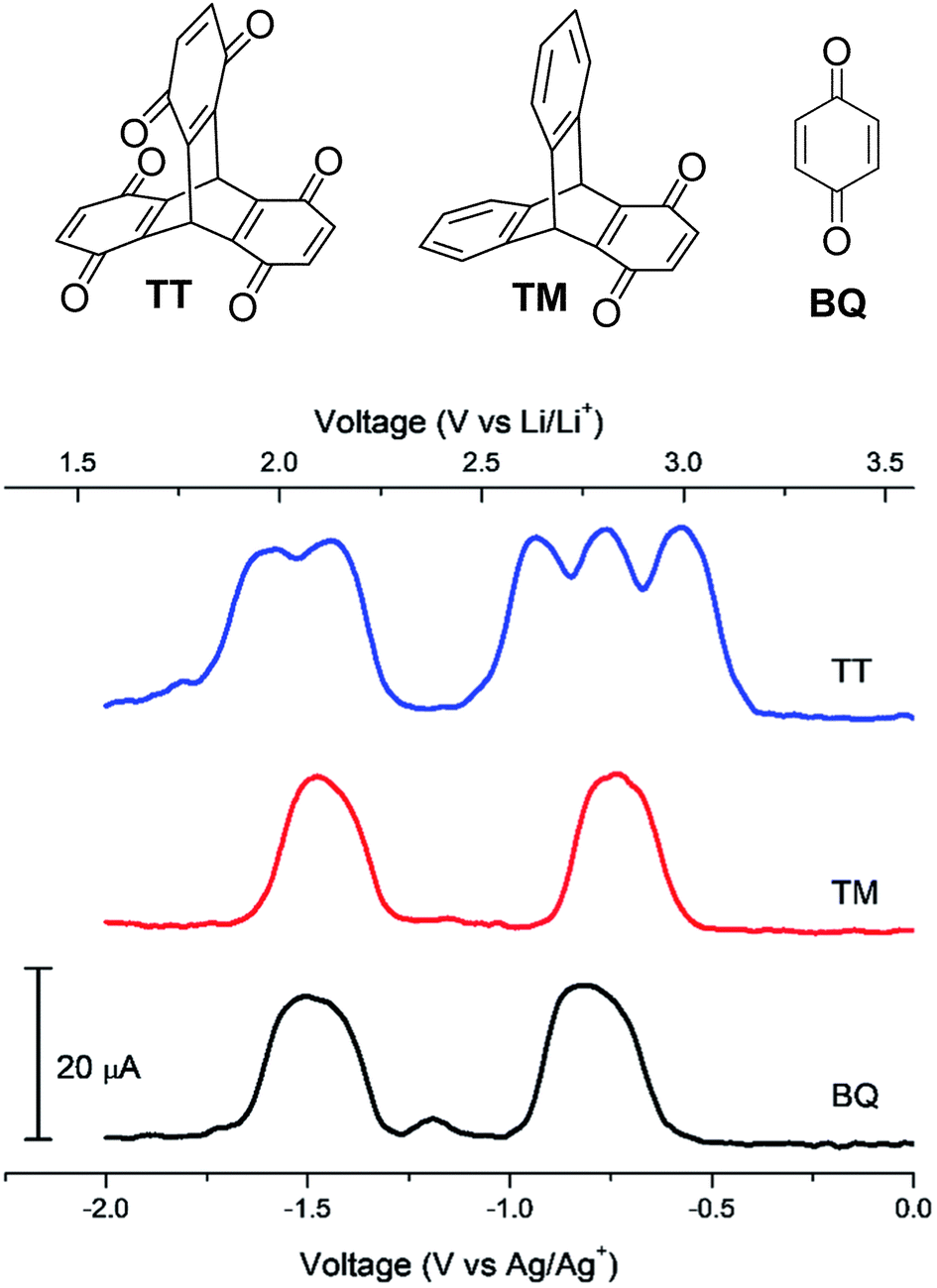 | ||
| Fig. 20 Molecular structures of TT, TM and BQ and their DPV responses. Reproduced from ref. 79 with permission from the Royal Society of Chemistry. | ||
In a Li-ion half-cell, TT displayed an initial discharge capacity of 387 mA h g−1. This approximated to 5.0 Li+ per molecule in agreement with the DPV results, although capacity had reduced by 55% after 20 charge/discharge cycles. Cycling performance could be enhanced in nanocomposite materials alongside the mesoporous carbon matrix CMK3.
Later in 2018 the Wuest group reported the isomeric triptycene hexaketone 8 which was synthesised by oxidation of 2,3,6,7,14,15-hexahydroxytriptycene 9.80 Transannular electronic effects meant that simple choice of oxidising agent permitted selective access to the partially oxidised 2,3-dione 10 and 2,3,6,7-tetraone 11 in good yields (structures in Fig. 21). The electrochemistry of 8 revealed that it could undergo reversible electrochemical reduction to a trianion in a step-wise fashion (Fig. 21a and b). It is worth highlighting the complementarity between the electrochemistry of the two electron poor tris(quinones) TT and 8 and the analogous hexa-alkoxytriptycenes studied by Rathore which were shown in Fig. 2.
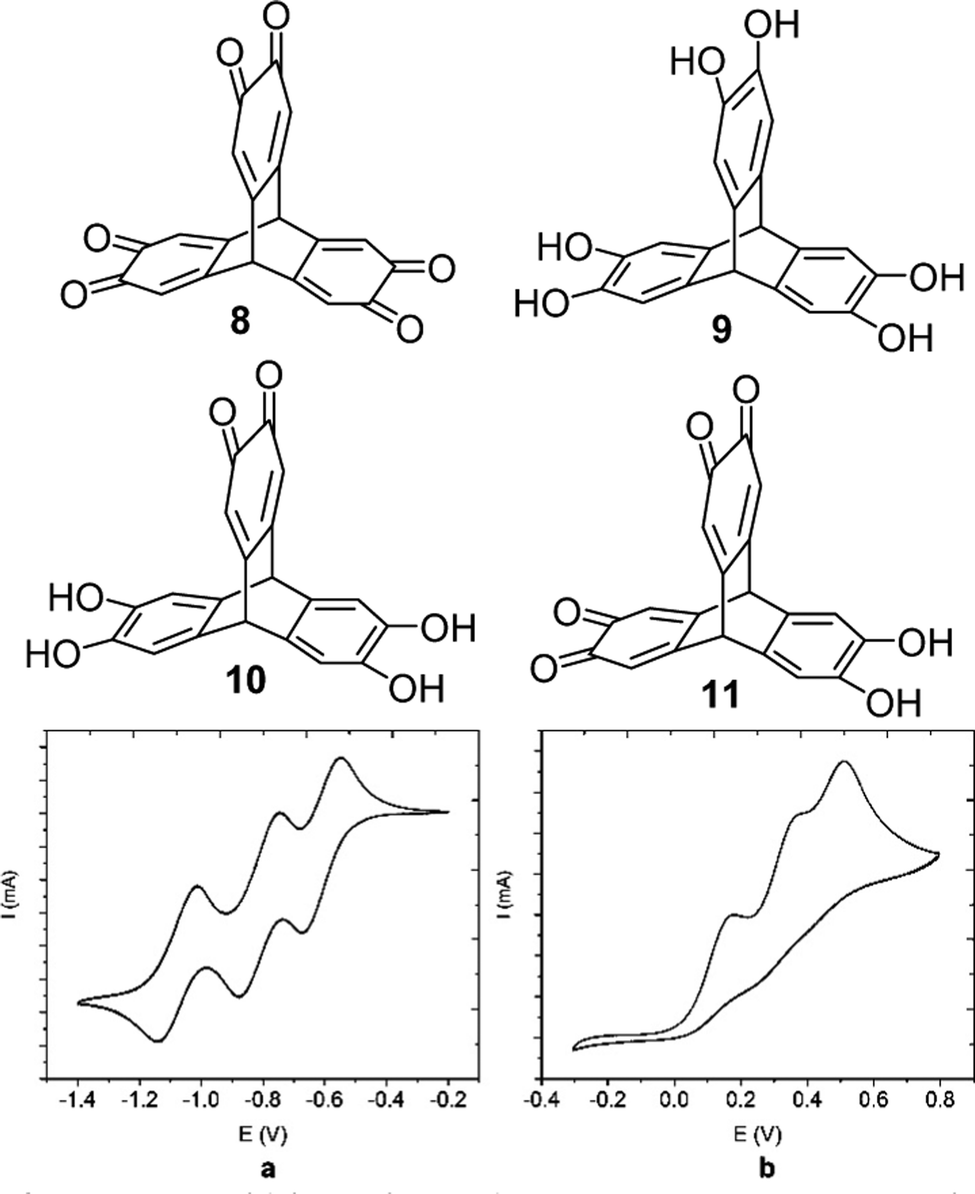 | ||
| Fig. 21 Molecular structures of 8–10 and the cyclic voltammograms of (a) trisquinone 8 and (b) hexahydroxy 9. Reproduced from ref. 80. Copyright 2018, American Chemical Society. | ||
Redox behaviour was apparent in the solid, crystalline state of dione 10 which was revealed to undergo reduction back to hexahydroxy 9 in a reversible topotactic reaction (Fig. 22). The presence of hydrazine vapours resulted in crystals of 10 changing from dark purple to colourless, this could then be reversed by exposure to HNO3 vapours. As the molecules can undergo reversible redox reactions in the solid crystalline state these molecules provide some direction towards realising rechargeable carbon-based batteries.
 | ||
| Fig. 22 Crystals of quinone 10 (a) before and (b) after exposure to N2H4 vapours. Reproduced from ref. 80. Copyright 2018, American Chemical Society. | ||
[3.3.3]Propellanes – an alternative D3h homoconjugated hydrocarbon
Finally, comparison with [3.3.3]propellanes, another class of three-finned hydrocarbon, gives some indication of how general the benefits of exploiting homoconjugation might be.In two reports authored by Lv et al.81,82peri-extension of the peripheral naphthalene units of [3,3,3]propellane 12 formed perylene monoimide (PMI) moieties on one or more fins of the propellane core. The first paper concerned tris(PMI)propellane 13 (R = 6-undecyl) and bay-annulated derivatives 14–16 in a comparative study (structures shown in Fig. 23). Frontier orbitals were distributed across the entire surface of the molecule which led to multiple clearly separated peaks in cyclic voltammetry studies. N-6-Undecyl(PMI) itself was also studied as a single-fin analogue for comparison and was shown to have a maximum ε = 33 × 103 M−1 cm−1. Each of the propellanes 13–16 displayed a greater than four-fold enhancement of ε (Table 3) in comparison with the single-fin, with benzo-fused compound 14 displaying the largest ε = 163 × 103 M−1 cm−1. Single-crystal charge-carrier mobilities in the order of 10−3 cm2 V−1 s−1 were obtained for 14 with the limited intermolecular overlap of the molecules observed in the crystal structure seeming to be a limiting factor in obtaining higher mobilities.
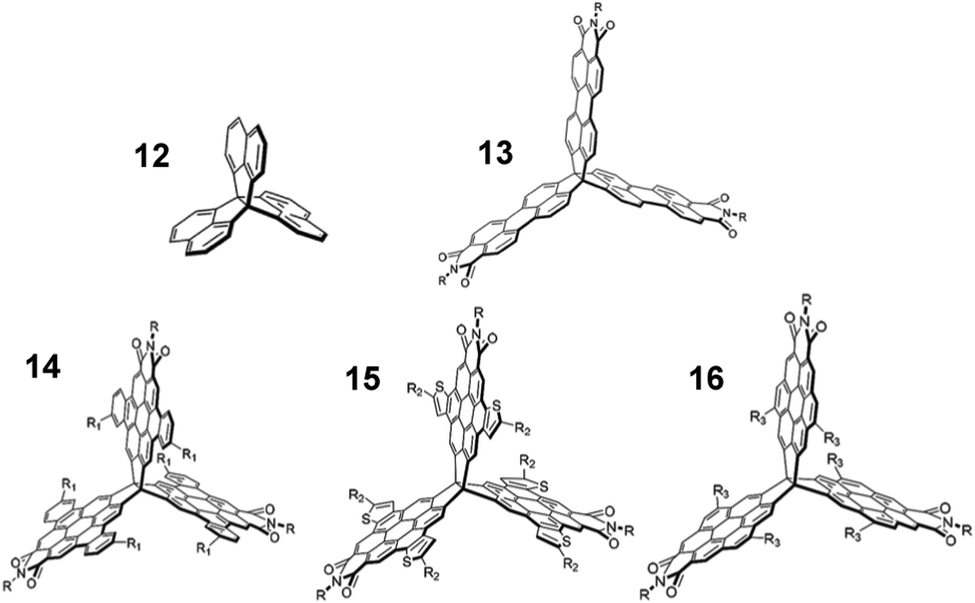 | ||
| Fig. 23 Molecular structures of propellanes 12–16 R = 6-undecyl. Reproduced from ref. 81 with permission from the Royal Society of Chemistry. | ||
| λ abs (nm) [ε × 103 M−1 cm−1] |
λ
maxPL (nm) |
PLQY (%) | |
|---|---|---|---|
| 13 | 554 (152) | 588 | 85 |
| 14 | 492 (163) | 514 | 89 |
| 15 | 486 (137) | 505 | 22 |
| 16 | 432 (122) | 481 | 32 |
| P-1 | 538 (35) | 579 | 83 |
| P-2 | 555 (81) | 585 | 89 |
| P-3 | 555 (152) | 588 | 85 |
In the second of these reports, one- (P-1), two- (P-2), and three-finned (P-3) analogues of 13 featuring a larger alkyl group (R = 2-decyltetradecyl) were also produced (Fig. 24). P-1 demonstrated very similar properties to N-2-decyltetradecyl(PMI) with only slightly red-shifted optical properties due to a narrower Eg.
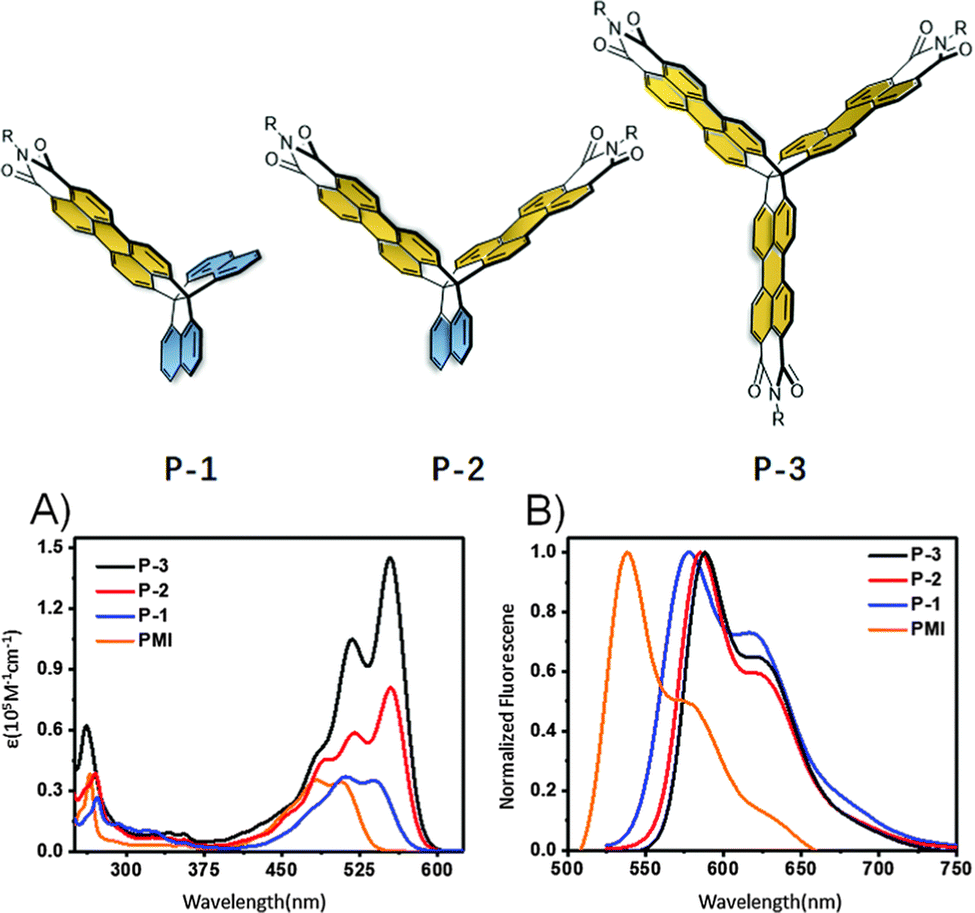 | ||
| Fig. 24 Molecular structures of P-1, P-2 and P-3, and their (A) absorbance spectra and, (B) photoluminescence spectra. Reproduced from ref. 82 with permission from the Chinese Chemical Society (CCS), Institute of Chemistry of Chinese Academy of Sciences (IC), and the Royal Society of Chemistry. | ||
The influence of homoconjugation becomes apparent in the absorbance spectra of these compounds (Fig. 24A) with the ε of P-2 (81 × 103 M−1 cm−1) being slightly more than double that of both P-1 35 × 103 M−1 cm−1) and N-2-decyltetradecyl(PMI) (34 × 103 M−1 cm−1) while the molar absorptivity almost doubles again for P-3 (ε = 152 × 103 M−1 cm−1). The fluorescence quantum yields displayed very little variation across the series staying well over 80% in all cases.
OFET devices were produced and demonstrated mobilities of 2.2 × 10−4 cm2 V−1 s−1 and 3.5 × 10−4 cm2 V−1 s−1 for P-1 and P-2 respectively, while P-3 was an entire order of magnitude lower at 2.5 × 10−5 cm2 V−1 s−1 which was ascribed to limited crystallinity in the film. P-1 and P-2 also displayed better performance in OLED devices in all parameters.
Outlook
The preceding sections highlight numerous positive effects that can be observed by exploiting homoconjugation in triptycene chromophores/electrophores. The primary benefits are:(1) Amplified molar absorptivity.
(2) Accelerated radiative decay.
(3) Generation of multiple charge carriers over small potential windows.
Considering the first two points design rules to obtain hyperchromic, potentially super-summative, enhancements in ε, and fosc can be proposed. Firstly, ensuring that the LUMO is localised on the homoconjugated core of the triptycene and straddles two or more of the fins appears to be essential. This gives rise to a manifold of low-lying excited states which interact constructively to facilitate the observed enhancements. Secondly, molecules featuring ring-fused structures and/or some dipole moment on the fins of the molecule tend to show greater enhancements than structures without an obvious dipole moment. This is intuitive and seemingly general until much longer extended chromophore fins are present.
The third benefit concerning electrochemical properties is realised in molecular designs which use electron donating or withdrawing substituents to modulate the frontier orbital density over the centre of the triptycene core. A lower amount of MO density at the homoconjugated core will lead to more redox events occuring over a smaller potential window and vice versa for molecules with large amounts of MO density over the triptycene core. Choice of substituent and regiochemistry permits control over both the magnitude of the potential window that the charges are generated over, and the nature of the charge carriers themselves.
However, challenges associated with using triptycenes also present themselves. These are usually associated with the morphology of neat films of triptycenes being poor due to the IFV of the molecules inhibiting tight packing. While this can have negative impacts on the performance of devices based upon concentrated thin films of these molecules such as BHJ OSCs or OFET devices, it is important to highlight that poor morphology is certainly not a general property of triptycenes when engineered properly. An important series of triptycenes with peri-alkoxy substituents demonstrated exceptionally good self-assembly properties which evidences this well.27,83,84
Bearing these points in mind, we can start to speculate on how to further harness the benefits of homoconjugation effects for the applications discussed.
OLED and luminescent materials
The performance of luminophores based on D–A architectures, particularly TADF emitters, can benefit greatly from the combination of very high ε and fosc which occur in the ICT bands of D–A triptycenes. This provides a realistic route to improve the effectiveness of red luminophores in particular, the efficiencies of which suffer due to the energy gap law which leads to rapid thermal NRD outcompeting radiative decay.85,86As it is the LUMO which must display homoconjugation to achieve enhancements structural modifications of the triptycene core itself will be required to further develop this concept. This could be achieved by using a wider variety of N-heterocyclic triptycenes or appending peripheral electron-withdrawing substituents onto the triptycene prior to addition of the donor moieties. In all other respects standard TADF design rules can be employed to control the emission wavelength.87–89
Morphological concerns are mitigated against in a typical doped OLED architecture as the luminophores are distributed in a small quantity throughout a bulk host matrix. As such, the main challenges presented in this context are ultimately the same as for TADF emitters in general. FWHM are too large leading to poor colour purity, however the use of multi-resonant TADF design principles may provide a route to circumventing this.90–92
OSC materials
Maximising the light absorbance across the optical band gap is one approach to obtaining higher PCEs. A higher ε results in improved absorbance of sunlight and diminished NRD thereby enhancing the VOC of a device, so hyperchromic enhancements in ε should produce better devices overall. This will be particularly important for sensitisers which have a suboptimal overlap with the solar spectrum.93–95Considering the very high ε that have been obtained in appropriately designed triptycenes such as 3Cbz, C3V-E/B-TTAI, CS-E/B-TTAI, 3 and 4, homoconjugation emerges as appealing avenue to explore for high ε materials which can be used a variety of OSC architectures. To date, studies of BHJ OSC devices using triptycene NFAs have demonstrated relatively poor performance. This is due to a combination of the structures studied to date not adhering to the LUMO-core, HOMO-shell motif required for major enhancements in ε, alongside poor BHJ morphology. The previously mentioned self-organised films of peri-alkoxy substituted triptycenes and the use of triptycene itself as a dopant to improve morphology and efficiencies in BHJ OSCs24 and other devices83,96–98 indicate that careful consideration of substitution patterns or device processing and manufacture might be used to overcome film morphology challenges.
We highlight that other photovoltaic device architectures such as dye-sensitised (DSSC) and perovskite solar cells (PSCs) may present greater opportunities for exploiting homoconjugation as well as circumventing the morphology problems encountered in BHJ devices. In PSCs 4 was used as a interfacing agent between the electron transport and perovskite layers rather than a bulk component. This produced devices with efficiency up to 18% which further demonstrates the power of triptycenes as additives. In DSSCs existing triptycene containing dyes only use triptycene as a bulky substituent rather than as a functional part of the chromophore or anchoring moeity.99,100 Triptycenes can show very strong bi-/tripodal anchoring to surfaces96,101,102 and homoconjugation can provide the high ε and tunable Eg required for harvesting solar energy. New materials designed according to the general rules proposed at the start of this Outlook section and incorporating suitable anchoring groups could thereby produce excellent new dyes for DSSCs.
Charge transport and energy storage materials
The electrochemical implications of a homoconjugated HOMO were demonstrated well by the alkoxy bearing molecules T23 and T14 which showed that by decreasing the extent of transannular orbital overlap the potential window required to assume a stable tricationic state decreased concomitantly by ∼500 mV and was obtained after just −1.2 V vs. Fc/Fc+. The LUMO can be manipulated in the same fashion to produce n-type systems. For example, 3Pxz had a homoconjugated LUMO which allowed a reversible three step reduction to take place at +0.73 V vs. Fc/Fc+ while 4 could be reversibly reduced to a remarkable octadecyl anion over a potential window of less than 1.5 V. Similar reductive behaviour was observed in quinones TT and 8. Furthermore, the triptycene core can be used indirectly to isolate the HOMO or LUMO on the periphery of a molecule leading to single-step generation of multiple charge carriers simultaneously as shown in 3Cbz and hPDI2–4.At first glance, the ability to easily generate many charge carriers from an individual organic molecule could foreseeably realise higher charge carrier density and ON/OFF ratios in charge transport devices such as OFETs. However, we would argue that what really sets the electrochemical properties of triptycenes apart is the stability of the multiply-charged states obtained. Generating very highly charged states typically causes major electrochemical stress in organic molecules but the homoconjugated materials presented to date are very robust. Building from this, triptycene-based compounds present valid alternatives to the anthraquinone systems which are under the most intense development.103 In this fashion they provide scope for use not only in traditional alkali metal systems but also for more exotic or highly charge ions such as Al3+.104,105 The 3D structure of triptycenes is well suited for incorporation into porous COF or MOF architectures which probably present the best route for further development in this context.
Conclusions
This review has aimed to provide initial insights towards harnessing homoconjugation in triptycene based molecular materials and what might be achieved by doing so.This effect can clearly be used to induce very high magnitude enhancements in useful optoelectronic properties, however there is often a trade-off between obtaining the maximum enhancement in properties and the performance of thin films of said molecules in devices. This is most notable for triptycenes in which all three fins have been functionalised. These tend to display both the biggest enhancements in properties but also the lowest quality films in devices. As such, triptycenes which display properties influenced by homoconjugation have not yet translated to notably high performance in neat or very concentrated thin film devices such as OFETs or BHJ OSCs but when used as dilute dopants in OLEDs or perovskite OSCs the three-finned triptycenes perform much better. This observation must be considered in the design and study of future homoconjugated chromophores and electrophores to obtain improved device performance. We note that few studies have deliberately set out to actively use homoconjugation, therefore a more considered approach to molecular design will lead rapidly to improved device performance from this class of materials and we have proposed some design rules towards this end.
Finally, while this review has focussed largely upon triptycenes, numerous other homoconjugated scaffolds are available. These include the spirobifluorenes and propellanes, which have been mentioned here, alongside less rigid chemical fragments such as diphenylmethanes and bisphenols. As such, there is great scope for more expansive utility of homoconjugation and the benefits it can bring in controlling functional molecular properties.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
J.-R. M. thanks the EPSRC Sustainable Hydrogen CDT (EP/S023909) and Loughborough University for support. S. M. thanks Loughborough University for a PhD studentship. I. A. W. thanks the EPSRC for support (EP/T028688/1 and EP/V048554/1).Notes and references
- D. K. Frantz, A. Linden, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 2012, 134, 1528–1535 CrossRef CAS PubMed.
- S. Toyota, K. Kawahata, K. Sugahara, K. Wakamatsu and T. Iwanaga, Eur. J. Org. Chem., 2017, 5696–5707 CrossRef CAS.
- T. R. Kelly, Acc. Chem. Res., 2001, 34, 514–522 CrossRef CAS PubMed.
- T. R. Kelly, H. De Silva and R. A. Silva, Nature, 1999, 401, 150–152 CrossRef CAS PubMed.
- C. Joachim, H. Tang, F. Moresco, G. Rapenne and G. Meyer, Nanotechnology, 2002, 13, 330–335 CrossRef CAS.
- T. Nishino, C. J. Martin, H. Takeuchi, F. Lim, K. Yasuhara, Y. Gisbert, S. Abid, N. Saffon-Merceron, C. Kammerer and G. Rapenne, Chem. – Eur. J., 2020, 26, 11913 CrossRef CAS PubMed.
- F. Bertani, N. Riboni, F. Bianchi, G. Brancatelli, E. S. Sterner, R. Pinalli, S. Geremia, T. M. Swager and E. Dalcanale, Chem. – Eur. J., 2016, 22, 3312–3319 CrossRef CAS PubMed.
- S. Barman, J. A. Garg, O. Blacque, K. Venkatesan and H. Berke, Chem. Commun., 2012, 48, 11127–11129 RSC.
- A. Fuoco, B. Comesana-Gandara, M. Longo, E. Esposito, M. Monteleone, I. Rose, C. G. Bezzu, M. Carta, N. B. McKeown and J. C. Jansen, ACS Appl. Mater. Interfaces, 2018, 10, 36475–36482 CrossRef CAS PubMed.
- M. Lee, C. G. Bezzu, M. Carta, P. Bernardo, G. Clarizia, J. C. Jansen and N. B. McKeown, Macromolecules, 2016, 49, 4147–4154 CrossRef CAS.
- X. H. Ma, Z. Y. Zhu, W. X. Shi, W. H. Ji, J. X. Li, Y. G. Wang and I. Pinnau, J. Mater. Chem. A, 2021, 9, 5404–5414 RSC.
- Q. Liang, J. Liu, Y. Wei, Z. Zhao and M. J. MacLachlan, Chem. Commun., 2013, 49, 8928–8930 RSC.
- K. Preet, G. Gupta, M. Kota, S. K. Kansal, D. B. Salunke, H. K. Sharma, S. C. Sahoo, P. Van der Voort and S. Roy, Cryst. Growth Des., 2019, 19, 2525–2530 CrossRef CAS.
- Q. Liang, G. Y. Jiang, Z. Zhao, Z. Y. Li and M. J. MacLachlan, Catal. Sci. Technol., 2015, 5, 3368–3374 RSC.
- A. M. Elewa, M. H. Elsayed, A. F. M. El-Mahdy, C. L. Chang, L. Y. Ting, W. C. Lin, C. Y. Lu and H. H. Chou, Appl. Catal., B, 2021, 285, 119802 CrossRef CAS.
- T. Ikai, T. Yoshida, S. Awata, Y. Wada, K. Maeda, M. Mizuno and T. M. Swager, ACS Macro Lett., 2018, 7, 364–369 CrossRef CAS PubMed.
- T. Ikai, T. Yoshida, K. I. Shinohara, T. Taniguchi, Y. Wada and T. M. Swager, J. Am. Chem. Soc., 2019, 141, 4696–4703 CrossRef CAS PubMed.
- Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu, D. A. Gomez-Gualdron, T. Yildirim, J. F. Stoddart and O. K. Farha, Science, 2020, 368, 297–303 CrossRef CAS PubMed.
- Z. Chen, P. Li, X. Zhang, M. R. Mian, X. Wang, P. Li, Z. Liu, M. O'Keeffe, J. F. Stoddart and O. K. Farha, Nano Res., 2021, 14, 376–380 CrossRef CAS.
- Y. Wang, C. Wu, W. Sun, Q. Pan, W. Hao, H. Liu, J. Sun, Z. Li, J. Sun and Y. Zhao, Mater. Chem. Front., 2021, 5, 944–949 RSC.
- N. Baig, S. Shetty, S. Al-Mousawi, F. Al-Sagheer and B. Alameddine, React. Funct. Polym., 2019, 139, 153–161 CrossRef CAS.
- A. Lohr and T. M. Swager, J. Mater. Chem., 2010, 20, 8107–8111 RSC.
- Z. Chen and T. M. Swager, Macromolecules, 2008, 41, 6880–6885 CrossRef CAS.
- L. Krishnan Jagadamma, L. J. McCarron, A. A. Wiles, V. Savikhin, M. T. Sajjad, M. Yazdani, V. M. Rotello, M. F. Toney, G. Cooke and I. D. W. Samuel, ACS Appl. Mater. Interfaces, 2018, 10, 24665–24678 CrossRef CAS PubMed.
- R. G. Taylor, C. G. Bezzu, M. Carta, K. J. Msayib, J. Walker, R. Short, B. M. Kariuki and N. B. McKeown, Chem. – Eur. J., 2016, 22, 2466–2472 CrossRef CAS PubMed.
- R. G. Taylor, M. Carta, C. G. Bezzu, J. Walker, K. J. Msayib, B. M. Kariuki and N. B. McKeown, Org. Lett., 2014, 16, 1848–1851 CrossRef CAS PubMed.
- N. Seiki, Y. Shoji, T. Kajitani, F. Ishiwari, A. Kosaka, T. Hikima, M. Takata, T. Someya and T. Fukushima, Science, 2015, 348, 1122–1126 CrossRef CAS PubMed.
- (a) See: https://goldbook.iupac.org/terms/view/H02842; (b) P. Muller, Pure Appl. Chem., 1994, 66, 1077–1184 CrossRef.
- R. V. Williams, Chem. Rev., 2001, 101, 1185–1204 CrossRef CAS PubMed.
- P. D. Bartlett, M. J. Ryan and S. G. Cohen, J. Am. Chem. Soc., 1942, 64, 2649–2653 CrossRef CAS.
- P. D. Bartlett and E. S. Lewis, J. Am. Chem. Soc., 1950, 72, 1005–1009 CrossRef CAS.
- J. Hine, J. A. Brown, L. H. Zalkow, W. E. Gardner and M. Hine, J. Am. Chem. Soc., 1955, 77, 594–598 CrossRef CAS.
- H. E. Zimmerman and R. M. Paufler, J. Am. Chem. Soc., 1960, 82, 1514–1515 CrossRef CAS.
- C. F. Wilcox, Jr., S. Winstein and W. G. McMillan, J. Am. Chem. Soc., 1960, 82, 5450–5454 CrossRef.
- P. Bischof, J. A. Hashmall, E. Heilbronner and V. Hornung, Helv. Chim. Acta, 1969, 52, 1745–1749 CrossRef CAS.
- E. Haselbach, E. Heilbronner and G. Schröder, Helv. Chim. Acta, 1971, 54, 153–162 CrossRef CAS.
- R. Hoffmann, E. Heilbronner and R. Gleiter, J. Am. Chem. Soc., 1970, 92, 706–707 CrossRef CAS.
- T. Nakazawa and I. Murata, J. Am. Chem. Soc., 1977, 99, 1996–1997 CrossRef CAS.
- H.-D. Martin, B. Mayer, R. Gleiter, W. Schäfer and F. Vögtle, Chem. Ber., 1983, 116, 2546–2553 CrossRef CAS.
- H.-D. Martin and B. Mayer, Angew. Chem., Int. Ed. Engl., 1983, 22, 283–314 CrossRef.
- K. Baumgartner, M. Hoffmann, F. Rominger, S. M. Elbert, A. Dreuw and M. Mastalerz, J. Org. Chem., 2020, 85, 15256–15272 CrossRef PubMed.
- S. M. Elbert, T. Kirschbaum, F. Rominger and M. Mastalerz, Org. Mater., 2021, 03, 097–102 CrossRef CAS.
- M. R. Talipov, T. S. Navale and R. Rathore, Angew. Chem., Int. Ed., 2015, 54, 14468–14472 CrossRef CAS PubMed.
- M. V. Ivanov, S. A. Reid and R. Rathore, J. Phys. Chem. Lett., 2018, 9, 3978–3986 CrossRef CAS PubMed.
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. Van Voorhis, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 11908–11911 CrossRef CAS PubMed.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- P. Lei, S. Zhang, N. Zhang, X. Yin, N. Wang and P. Chen, ACS Omega, 2020, 5, 28606–28614 CrossRef CAS PubMed.
- W. Huang, M. Einzinger, T. Zhu, H. S. Chae, S. Jeon, S.-G. Ihn, M. Sim, S. Kim, M. Su, G. Teverovskiy, T. Wu, T. Van Voorhis, T. M. Swager, M. A. Baldo and S. L. Buchwald, Chem. Mater., 2018, 30, 1462–1466 CrossRef CAS.
- R. Qian, H. Tong, C. Huang, J. Li, Y. Tang, R. Wang, K. Lou and W. Wang, Org. Biomol. Chem., 2016, 14, 5007–5011 RSC.
- S. Montanaro, D. G. Congrave, M. K. Etherington and I. A. Wright, J. Mater. Chem. C, 2019, 7, 12886–12890 RSC.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- L. S. Cui, A. J. Gillett, S. F. Zhang, H. Ye, Y. Liu, X. K. Chen, Z. S. Lin, E. W. Evans, W. K. Myers, T. K. Ronson, H. Nakanotani, S. Reineke, J. L. Bredas, C. Adachi and R. H. Friend, Nat. Photonics, 2020, 14, 636–642 CrossRef CAS.
- H. Noda, X. K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J. L. Bredas and C. Adachi, Nat. Mater., 2019, 18, 1084–1090 CrossRef CAS PubMed.
- S. Montanaro, P. Pander, J. R. Mistry, M. R. J. Elsegood, S. J. Teat, A. D. Bond, I. A. Wright, D. G. Congrave and M. K. Etherington, J. Mater. Chem. C, 2022, 10, 6306–6313 RSC.
- L. Yu, Z. Wu, C. Zhong, G. Xie, Z. Zhu, D. Ma and C. Yang, Adv. Opt. Mater., 2017, 5, 1700588 CrossRef.
- T. Liu and A. Troisi, Adv. Mater., 2013, 25, 1038–1041 CrossRef CAS PubMed.
- Y. Yang, C. Yao, L. Li, M. Bo, J. Zhang, C. Peng and J. Wang, RSC Adv., 2020, 10, 12004–12012 RSC.
- E. H. Menke, V. Lami, Y. Vaynzof and M. Mastalerz, Chem. Commun., 2016, 52, 1048–1051 RSC.
- S. Maniam, H. F. Higginbotham, T. D. M. Bell and S. J. Langford, Chem. – Eur. J., 2019, 25, 7044–7057 CrossRef CAS PubMed.
- E. H. Menke, D. Leibold, V. Lami, Y. J. Hofstetter, M. Mastalerz and Y. Vaynzof, Org. Electron., 2017, 47, 211–219 CrossRef CAS.
- E. H. Menke, D. Leibold, A. P. Ullrich, Y. Vaynzof and M. Mastalerz, Org. Chem. Front., 2017, 4, 834–838 RSC.
- E. H. Menke, D. Leibold, F. J. Berger, F. Rominger, Y. Vaynzof and M. Mastalerz, ChemPlusChem, 2017, 82, 1390–1395 CrossRef CAS PubMed.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC.
- V. Sharma, J. D. B. Koenig and G. C. Welch, J. Mater. Chem. A, 2021, 9, 6775–6789 RSC.
- X. Liu, Y. Cai, X. Huang, R. Zhang and X. Sun, J. Mater. Chem. C, 2017, 5, 3188–3194 RSC.
- S. Chen, Y. Liu, W. Qiu, X. Sun, Y. Ma and D. Zhu, Chem. Mater., 2005, 17, 2208–2215 CrossRef CAS.
- A. J. Jimenez, F. Spanig, M. S. Rodriguez-Morgade, K. Ohkubo, S. Fukuzumi, D. M. Guldi and T. Torres, Org. Lett., 2007, 9, 2481–2484 CrossRef CAS PubMed.
- J. Yi, Y. Wang, Q. Luo, Y. Lin, H. Tan, H. Wang and C. Q. Ma, Chem. Commun., 2016, 52, 1649–1652 RSC.
- T. P. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS PubMed.
- H. E. Simmons and T. Fukunaga, J. Am. Chem. Soc., 1967, 89, 5208–5215 CrossRef CAS.
- J. Lee, R. Singh, D. H. Sin, H. G. Kim, K. C. Song and K. Cho, Adv. Mater., 2016, 28, 69–76 CrossRef CAS PubMed.
- D. Meng, H. Fu, B. Fan, J. Zhang, Y. Li, Y. Sun and Z. Wang, Chem. – Asian J., 2017, 12, 1286–1290 CrossRef CAS PubMed.
- H. S. Kang, T. J. Sisto, S. Peurifoy, D. H. Arias, B. Y. Zhang, C. Nuckolls and J. L. Blackburn, J. Phys. Chem. C, 2018, 122, 14150–14161 CrossRef CAS.
- S. R. Peurifoy, E. Castro, F. Liu, X. Y. Zhu, F. Ng, S. Jockusch, M. L. Steigerwald, L. Echegoyen, C. Nuckolls and T. J. Sisto, J. Am. Chem. Soc., 2018, 140, 9341–9345 CrossRef CAS PubMed.
- Y. Zhong, B. Kumar, S. Oh, M. T. Trinh, Y. Wu, K. Elbert, P. Li, X. Zhu, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 8122–8130 CrossRef CAS PubMed.
- B. L. Hu, C. An, M. Wagner, G. Ivanova, A. Ivanova and M. Baumgarten, J. Am. Chem. Soc., 2019, 141, 5130–5134 CrossRef CAS PubMed.
- Z. Z. Sun, S. Feng, C. Gu, N. Cheng and J. Liu, Phys. Chem. Chem. Phys., 2019, 21, 15206–15214 RSC.
- J. E. Kwon, C. S. Hyun, Y. J. Ryu, J. Lee, D. J. Min, M. J. Park, B. K. An and S. Y. Park, J. Mater. Chem. A, 2018, 6, 3134–3140 RSC.
- S. Langis-Barsetti, T. Maris and J. D. Wuest, J. Org. Chem., 2018, 83, 15426–15437 CrossRef CAS PubMed.
- L. Lv, W. Sun, Z. Jia, G. Zhang, F. Wang, Z. Tan and L. Zhang, Mater. Chem. Front., 2020, 4, 3539–3545 RSC.
- L. Lv, J. Roberts, C. Xiao, Z. Jia, W. Jiang, G. Zhang, C. Risko and L. Zhang, Chem. Sci., 2019, 10, 4951–4958 RSC.
- T. Yokota, T. Kajitani, R. Shidachi, T. Tokuhara, M. Kaltenbrunner, Y. Shoji, F. Ishiwari, T. Sekitani, T. Fukushima and T. Someya, Nat. Nanotechnol., 2018, 13, 139–144 CrossRef CAS PubMed.
- T. Imaizumi, R. Takehara, Y. Yamashita, T. Yagi, F. Ishiwari, Y. Shoji, X. Wang, Y. Murakami, T. Nishino and T. Fukushima, Jpn. J. Appl. Phys., 2021, 60, 038002 CrossRef CAS.
- N. Notsuka, H. Nakanotani, H. Noda, K. Goushi and C. Adachi, J. Phys. Chem. Lett., 2020, 11, 562–566 CrossRef CAS PubMed.
- J. Eng and T. J. Penfold, Chem. Rec., 2020, 20, 831–856 CrossRef CAS PubMed.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- W. Che, Y. Xie and Z. Li, Asian J. Org. Chem., 2020, 9, 1262–1276 CrossRef CAS.
- P. de Silva, C. A. Kim, T. Zhu and T. Van Voorhis, Chem. Mater., 2019, 31, 6995–7006 CrossRef CAS.
- H. J. Kim and T. Yasuda, Adv. Opt. Mater., 2022, 10, 2201714 CrossRef CAS.
- S. Madayanad Suresh, D. Hall, D. Beljonne, Y. Olivier and E. Zysman-Colman, Adv. Funct. Mater., 2020, 30, 1908677 CrossRef CAS.
- G. Meng, L. Liu, Z. He, D. Hall, X. Wang, T. Peng, X. Yin, P. Chen, D. Beljonne, Y. Olivier, E. Zysman-Colman, N. Wang and S. Wang, Chem. Sci., 2022, 13, 1665–1674 RSC.
- M. S. Vezie, S. Few, I. Meager, G. Pieridou, B. Dörling, R. S. Ashraf, A. R. Goñi, H. Bronstein, I. McCulloch, S. C. Hayes, M. Campoy-Quiles and J. Nelson, Nat. Mater., 2016, 15, 746–753 CrossRef CAS PubMed.
- J. Yan, X. Rodríguez-Martínez, D. Pearce, H. Douglas, D. Bili, M. Azzouzi, F. Eisner, A. Virbule, E. Rezasoltani, V. Belova, B. Dörling, S. Few, A. A. Szumska, X. Hou, G. Zhang, H.-L. Yip, M. Campoy-Quiles and J. Nelson, Energy Environ. Sci., 2022, 15, 2958–2973 RSC.
- A. Markina, K.-H. Lin, W. Liu, C. Poelking, Y. Firdaus, D. R. Villalva, J. I. Khan, S. H. K. Paleti, G. T. Harrison, J. Gorenflot, W. Zhang, S. De Wolf, I. McCulloch, T. D. Anthopoulos, D. Baran, F. Laquai and D. Andrienko, Adv. Energy Mater., 2021, 11, 2102363 CrossRef CAS.
- S. Das, G. Nascimbeni, R. O. de la Morena, F. Ishiwari, Y. Shoji, T. Fukushima, M. Buck, E. Zojer and M. Zharnikov, ACS Nano, 2021, 15, 11168–11179 CrossRef CAS PubMed.
- M. Sugiyama, S. Jancke, T. Uemura, M. Kondo, Y. Inoue, N. Namba, T. Araki, T. Fukushima and T. Sekitani, Org. Electron., 2021, 96, 106219 CrossRef CAS.
- F. K.-C. Leung, F. Ishiwari, T. Kajitani, Y. Shoji, T. Hikima, M. Takata, A. Saeki, S. Seki, Y. M. A. Yamada and T. Fukushima, J. Am. Chem. Soc., 2016, 138, 11727–11733 CrossRef CAS PubMed.
- M. Yan, Q.-H. Wang, Y.-Z. Zhu, M.-L. Han, Y.-Q. Yan and J.-Y. Zheng, J. Photochem. Photobiol., A, 2021, 416, 113325 CrossRef CAS.
- M. Yan, Y.-Z. Zhu, Y.-Q. Yan, Q.-H. Wang, G.-L. Yin and J.-Y. Zheng, J. Photochem. Photobiol., A, 2022, 433, 114132 CrossRef CAS.
- F. Ishiwari, G. Nascimbeni, E. Sauter, H. Tago, Y. Shoji, S. Fujii, M. Kiguchi, T. Tada, M. Zharnikov, E. Zojer and T. Fukushima, J. Am. Chem. Soc., 2019, 141, 5995–6005 CrossRef CAS PubMed.
- S. Das, A. Asyuda, Y. Shoji, A. Kosaka, T. Fukushima and M. Zharnikov, J. Phys. Chem. C, 2021, 125, 18968–18978 CrossRef CAS.
- Y. Wu, R. Zeng, J. Nan, D. Shu, Y. Qiu and S.-L. Chou, Adv. Energy Mater., 2017, 7, 1700278 CrossRef.
- S. K. Das, S. Mahapatra and H. Lahan, J. Mater. Chem. A, 2017, 5, 6347–6367 RSC.
- D. J. Kim, D.-J. Yoo, M. T. Otley, A. Prokofjevs, C. Pezzato, M. Owczarek, S. J. Lee, J. W. Choi and J. F. Stoddart, Nat. Energy, 2019, 4, 51–59 CrossRef CAS.
Footnote |
| † Present address: School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: iain.wright@ed.ac.uk |
| This journal is © The Royal Society of Chemistry 2023 |