
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid discovery of new Eu2+-activated phosphors with a designed luminescence color using a data-driven approach†
Yukinori
Koyama
 *a,
Hidekazu
Ikeno
b,
Masamichi
Harada
c,
Shiro
Funahashi
c,
Takashi
Takeda
c and
Naoto
Hirosaki
c
*a,
Hidekazu
Ikeno
b,
Masamichi
Harada
c,
Shiro
Funahashi
c,
Takashi
Takeda
c and
Naoto
Hirosaki
c
aResearch and Services Division of Materials Data and Integrated System, National Institute for Materials Science, Tsukuba, Ibaraki 305-0044, Japan. E-mail: KOYAMA.Yukinori@nims.go.jp
bDepartment of Materials Science, Graduate School of Engineering, Osaka Metropolitan University, Sakai, Osaka 599-8570, Japan
cResearch Center for Functional Materials, National Institute for Materials Science, Tsukuba, Ibaraki 305-0044, Japan
First published on 29th November 2022
Abstract
For rapid and efficient development of new phosphors, a suitable method that proposes promising candidates is expected to focus time-consuming trial-and-error experiments. A data-driven approach to discover new phosphor materials with a designed luminescence color is demonstrated in this paper. To screen compounds for a desirable luminescence color, a machine learning model has been developed for predicting emission peak wavelengths from a dataset composed of 129 Eu2+-activated phosphors. General-purpose compositional and structural features are used to represent host compounds of phosphors. Bootstrap aggregation with the gradient boosted regression trees method is adopted to obtain high predictive performance and to avoid overfitting. The predictive performance of the machine learning model is estimated to be 25 nm of mean absolute error (MAE) and 33 nm of root mean squared error (RMSE) by 10-fold cross validation. To discover new green-emitting Eu2+-activated phosphors, twenty candidate compounds have been selected to have predicted emission peak wavelengths of about 500–550 nm from a materials database, and the candidates have been synthesized and characterized by experiments. Three new Eu2+-activated phosphors, Li2Ca4Si4O13:Eu2+, Na2Ca2Si2O7:Eu2+, and SrLaGaO4:Eu2+, successfully show green or blue-green emissions as designed.
Introduction
Phosphor-converted white light-emitting-diodes (pc-wLEDs), which are composed of blue or near-ultraviolet LED chips as a primary light source, and phosphors as down-conversion luminescent materials, are one of the indispensable lighting technologies today because of their high luminous efficiency, cost effectiveness, environment-friendliness, and spectral design flexibility.1 For pc-wLED applications, phosphors have various requirements such as strong absorption of the LED light, suitable emission spectrum, high quantum efficiency, small thermal quenching/degradation, high chemical stability, and small luminance saturation. Ce3+ and Eu2+ ions are often selected as activators of the phosphors for the pc-wLEDs. These lanthanide ions utilize parity allowed 4f–5d transitions, which are often characterized by high radiative emission probability, short lifetime, and relatively broad absorption and emission spectra in contrast to parity forbidden 4f–4f transitions.2 Furthermore, because their 5d-states are strongly influenced by the host lattices, their luminescence properties can be tuned by variation of the hosts. However, it requires time-intensive trial-and-error experiments to explore and optimize new phosphors. Even though several strategies have been proposed for efficient development of new phosphors,3,4 an effective method to select candidate compounds for desirable properties is expected to focus the time-intensive experiments upon promising candidates.Recently, data-driven approaches have been reported for the rapid discovery and development of new phosphors using screening of materials databases, high-throughput density functional theory (DFT) calculations, and machine learning on luminescence properties.5–10 The emission spectrum is one of the most important characteristics of phosphors because it determines their luminescence color. The emission spectrum is often characterized by its peak top and full width at half maximum (FWHM). A relationship among host compounds, the absorption spectrum, and the emission spectrum has been investigated empirically or semi-empirically for Ce3+ and Eu2+-activated phosphors so far.11Ab initio multi-configurational quantum chemical calculations have been performed to quantitatively calculate configuration coordinate diagrams and absorption spectra.12 Constrained DFT calculations have also been conducted to evaluate absorption and emission energies.13,14 However, these theoretical methods require time-consuming calculations at both the ground and excited states. Because of the high computational cost, high-throughput theoretical calculations to screen candidate compounds are not currently feasible.
Machine learning to predict emission spectra instead of the theoretical calculations has been investigated recently.6–8 Sohn and his coworkers reported the pioneering machine-learning study on a relationship among emission peak wavelength, FWHM, and local environments of substitution sites in host lattices,5 and recently reported comprehensive machine learning to predict band gap, excitation energy, and emission energy for Eu2+-activated phosphors.6 Nakano et al. reported machine learning to predict emission peak energy from chemical compositions of the host compounds for Eu2+-activated phosphors.7 The reported prediction accuracy is not directly comparable among the theoretical calculations and the machine learning studies because they used different datasets. But the results suggest that the machine learning models6,7 have comparable prediction accuracy to the DFT calculations.14
Based on the successful machine-learning studies to date, it is expected that new phosphors with desirable luminescence properties will be developed using machine learning. Although several research groups have reported new phosphors by data-driven approaches,9 discovery of new phosphors with a designed luminescence color is still a big challenge. In this paper, we report the discovery of three new green or blue-green emitting phosphors, which a machine-learning model has proposed as green emitting phosphors. First, we developed a machine learning model to predict the emission peak wavelengths of Eu2+-activated phosphors from an in-house phosphor dataset. Next, we explored a materials database and collected candidate host compounds predicted to show green emissions by the machine learning model. Then, we synthesized and characterized the candidates, and finally discovered the three new Eu2+-activated phosphors, Li2Ca4Si4O13:Eu2+, Na2Ca2Si2O7:Eu2+, and SrLaGaO4:Eu2+. The results clearly demonstrate the power of the machine learning on the emission peak wavelength for rapid and efficient development of new phosphors with a designed luminescence color.
Methods
Data collection
Even though phosphors have been intensively investigated so far, there is no readily available dataset of phosphor materials and luminescence properties. Therefore, a dataset of host compounds and emission peak wavelengths of Eu2+-activated phosphors was collected from the literature.1,15 Only host compounds with typical oxidation states and containing Ca, Sr, or Ba elements were selected. These alkaline earth metals are considered as substitution sites for Eu2+ ions because they have the same valence and close ionic radii to Eu2+. Crystal structures of the hosts were collected from the inorganic crystal structure database (ICSD)16 and AtomWork-Adv.17 Some structure data were modified as follows. (1) Structure data with chemical compositions that deviate from the ideal compositions of the hosts, for example containing Eu2+, was corrected to have the ideal compositions of the hosts. (2) Structure data with partially occupied sites and different site occupancies were modified to have high occupancy sites only. Partially occupied sites cause ambiguity in the representation of local environments of the substitution sites. Host compounds with awkward site occupancy, which cannot be simply discretized as described above, were dropped.Emission peak wavelength is used as a target variable in this study because the emission spectra of phosphors are usually measured and reported in wavelength. The emission peak wavelengths depend on the concentrations of activators and other factors. The conditions in the literature are inconsistent, and the reported values vary more or less. If multiple emission peak wavelengths are reported for a single phosphor material and the reported values differ by more than 30 nm, the phosphor is eliminated. In our opinion, a deviation of 10 nm or more in the emission peak wavelength is conceivable due to the different conditions.
Finally, a dataset composed of 129 Eu2+-activated phosphors was prepared. The distribution and statistics of the emission peak wavelengths are respectively shown in Fig. 1a and Table 1. Constituent elements of the host compounds are summarized in Fig. 1b. Among the constituent elements, sulfur appeared as both a cation (S6+) and an anion (S2−). N, O, F, Cl, Br, and I elements were anions, and the other elements were cations.
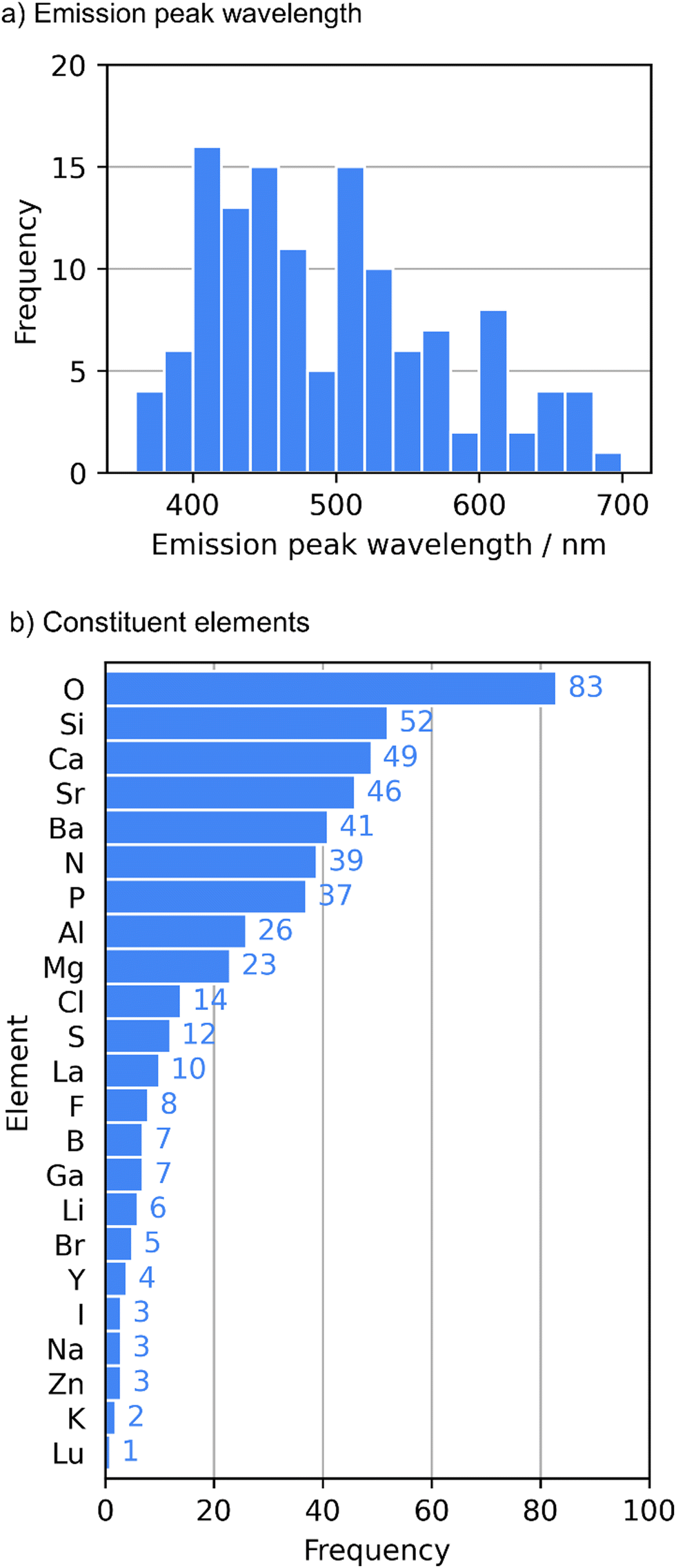 | ||
| Fig. 1 (a) Histogram of emission peak wavelengths and (b) frequency of constituent elements of Eu2+-activated phosphors used in this study. S is a cation (S6+) and an anion (S2−). N, O, F, Cl, Br, and I are anions. The other elements are cations. | ||
| Count | 129 |
| Mean (nm) | 495 |
| Median (nm) | 478 |
| Minimum (nm) | 368 |
| Maximum (nm) | 681 |
| Standard deviation (nm) | 80 |
| Mean absolute deviation (nm) | 66 |
Host representation
Two sets of features were used to represent host compounds of Eu2+-activated phosphors. The first set is a representation of chemical compositions (compositional features, hereafter), and the second set is a representation of crystal structures, particularly local environments of substitution sites for Eu2+ activators, from both geometrical and chemical aspects (structural features, hereafter).As the compositional features, general-purpose features18 were adopted. The general-purpose features were a set of statistics of elemental features to represent various aspects of chemical compositions. Nakano et al. used the same scheme for their machine learning.7 In this study, 22 elemental features and seven statistics, namely, weighted arithmetic mean, weighted geometric mean, weighted harmonic mean, weighted standard deviation, minimum, maximum, and range, were used. The elemental features and the statistics are respectively listed in Tables S1 and S2 in the ESI.† In addition to the elemental features, oxidation states were considered. As oxidation states are both positive and negative values and satisfy charge neutrality, the weighted arithmetic, geometric, and harmonic means were excluded. Instead, the seven statistics of absolute oxidation states were additionally included. As the hosts in this study are all ionic compounds, the statistics of the elemental features and the absolute oxidation states were also evaluated for each of the cations only and the anions only. The compositional features consisted of 487 features.
To represent the local environments of the substitution sites, Park et al. used geometrical and elemental features of activator-anion and activator-cation polyhedra.6 This idea was generalized, inspired by the general-purpose compositional features. The structural features used in this study consisted of three groups of features. The first group was a geometrical aspect of the substitution sites. The numbers of neighboring anions and cations, average distances to their neighboring anions and cations, distortion index,19 and bond valence sum20 were evaluated for individual Ca, Sr, and Ba sites. The neighboring anions were determined using the CrystalNN method.21 The neighboring cations were determined so that they shared neighboring anions with the substitution sites. As some of the host compounds used in this study have multiple substitution sites, the average and standard deviation of each feature among the substitution sites were evaluated and used as features of the host structures. The number of symmetrically inequivalent substitution sites was also included. The second group was analogous to the compositional features but specialized for the local environments of the substitution sites. The seven statistics of the 22 elemental features and the absolute oxidation states were calculated for the neighboring anions and the neighboring cations of individual Ca, Sr, and Ba sites. The average and standard deviation among the substitution sites were used as the features of the hosts. Besides the features of the substitution sites, density and numerical density were added as the third group. The structural features consisted of 659 features.
The features were evaluated using the Pymatgen package22 and a customized version of the XenonPy package.23
Machine learning
The general-purpose features used in this study were systematically calculated to represent various aspects of the host compounds, and thus a part of them were redundant and irrelevant to the emission peak wavelength. Therefore, feature selection was adopted before regression. First, features with low variance were dropped, and the passed features were standardized so that the means were zero and standard deviations were one. After the standardization, the features were roughly selected in the order of mutual information with the emission peak wavelength. The features were further narrowed down using recursive feature elimination (RFE) based on the importance of each feature obtained by a regression model. Finally, regression was conducted. The ridge, automatic relevance determination (ARD), random forest (RF), gradient boosted regression trees (GB), and bootstrap aggregation (bagging) of GB methods were applied for the regression. The regression method used in RFE was the same as the final regression, except for the bagging of GB regression. For the bagging of GB regression, a single GB model was used in RFE to reduce computation time. The Scikit-learn package24 was used for the machine learning.The predictive performance of the machine learning models was evaluated by 10-fold cross validation by means of the mean absolute error (MAE), root mean squared error (RMSE), and coefficient of determination (R2). The scores were averaged among the folds. The parameters of the regression models and the numbers of selected features were selected to minimize the average RMSE for the validation data. The parameter search was performed in a manner of Bayesian optimization using the Hyperopt25 and scikit-optimize26 packages with 1000 iterations for each method. Default parameters were used for the regression models used in RFE to reduce the computation time for the parameter search. The pipelines of the machine-learning models and the optimized parameters are summarized in Table S3 in the ESI.†
Experiments
Candidates of Eu2+-activated phosphors proposed by a machine learning model were synthesized and characterized by experiments. The phosphors were synthesized by a solid-state method. The starting materials (oxides or carbonates) of the host compounds were mixed with Eu2O3. The amount of Eu element was fixed at 2 at% of the substitution sites, namely, Ca, Sr, and Ba, in the hosts. The starting materials were fired in air, and then fired in a reducing atmosphere (in a carbon heater furnace filled with nitrogen). The firing temperatures and time were altered depending on the host compounds.The products were first characterized using a powder X-ray diffractometer (XRD) (Bruker, D8 ADVANCE, Cu Kα radiation) and a spectrofluorometer (JASCO, FP-8600). The powder XRD analysis indicated that some products were mixtures of the target compounds and impurity phases. As the photoluminescence (PL) spectra of the powder samples are largely influenced by impurity phases with bright luminescence, it was not clear whether the PL spectra of the mixture products were derived from the target compounds or the impurity phases. Therefore, after the first screening using the powder samples, well-crystallized particles were picked up from the products and characterized by single crystal XRD and microspectroscopy in a manner of the single-particle diagnosis approach.4 The single crystal XRD data of the picked particles were collected using a diffractometer (Bruker-AXS, SMART APEX II Ultra) with Mo Kα radiation. The data were integrated and corrected for absorption using SADABS. The crystal structures were solved and refined with SHELX. The PL spectra of the particles were obtained using a spectrometer (Otsuka electronics, MCPD7700) through a microscope (Olympus, BX51M) under 365 nm LED excitation.
Results and discussion
Comparison of regression methods
Regression methods are compared in this section. MAE, RMSE, and R2 for the training and validation data in the cross validation are summarized in Table 2. Fig. 2 illustrates predicted emission peak wavelengths with respect to the reported values in the cross validation. The ridge regression is the baseline model in this study. The R2 of the ridge regression to the validation data, 0.74, suggests that the prediction accuracy was comparable to the previous studies,6,7 although the results are not directly comparable due to the use of the different datasets.| Regression method | MAE (nm) | RMSE (nm) | R 2 | |||
|---|---|---|---|---|---|---|
| Training | Validation | Training | Validation | Training | Validation | |
| Ridge | 24 (1) | 29 (7) | 31 (1) | 36 (9) | 0.85 (0.01) | 0.74 (0.13) |
| ARD | 25 (1) | 28 (6) | 31 (1) | 34 (9) | 0.84 (0.02) | 0.77 (0.12) |
| RF | 10 (0) | 27 (7) | 14 (1) | 35 (11) | 0.97 (0.00) | 0.75 (0.19) |
| GB | 0 (0) | 24 (9) | 0 (0) | 31 (12) | 1.00 (0.00) | 0.79 (0.18) |
| Bagging of BG | 10 (0) | 25 (7) | 14 (1) | 33 (10) | 0.97 (0.00) | 0.77 (0.17) |
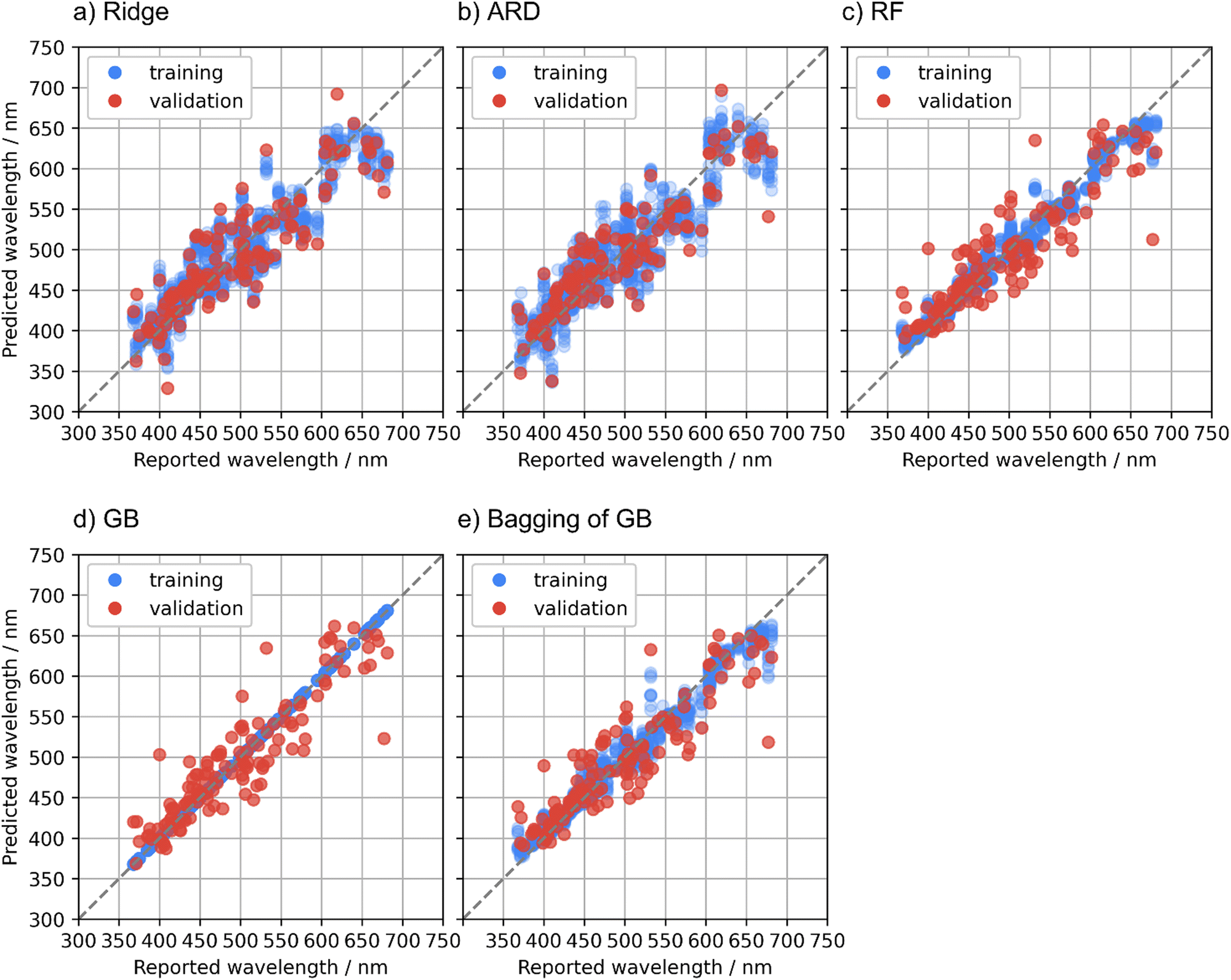 | ||
| Fig. 2 Predicted emission peak wavelengths with respect to reported values for the training (blue) and validation (red) data in the cross validation using (a) ridge, (b) automatic relevance determination (ARD), (c) random forest (RF), (d) gradient boosted regression trees (GB), and (e) bagging of GB methods. | ||
To improve the predictive performance, other regression methods were applied. The ARD regression is a Bayesian linear model with an intrinsic feature selection capability, and this method resulted in a slightly higher prediction accuracy to the validation data compared with the ridge regression. The ridge and ARD models showed relatively large fitting errors to the training data. This indicates that the relationship between the general-purpose features used in this study and the emission peak wavelength is basically nonlinear, although the general-purpose features are numerous and diverse. The small differences in the predictive performance scores between the training and validation data of these linear models imply that the obtained predictive performance almost reached the optimal of linear models.
Nonlinear regression methods were applied to obtain a higher predictive performance. The RF model showed slightly smaller MAE but larger RMSE to the validation data than the ARD model. The GB model showed much smaller MAE and RMSE to the validation data than the ARD and RF models. However, the fitting errors of the GB model to the training data were almost zero, and overfitting was concerned. To dispel the concerns about the overfitting of the GB model, the bagging technique was adopted to the GB regression. The bagging technique is also used in the RF regression and is expected to suppress the overfitting. The bagging of the GB model showed intermediate predictive performance to the validation data between the GB and RF models. The better predictive performance of the bagging of the GB model compared with the RF model is probably due to the higher predictive capacity of the GB regression as a base learner compared with that of the regression trees in the RF model.
The RF, GB, and bagging of GB models showed large prediction errors for some specific compounds in the validation folds. A plausible cause of these large prediction errors is that the phosphor dataset used in this study is not sufficiently large with respect to the diverse phosphor materials. If a host compound is unique in the dataset and is put in the validation data in a fold of the cross validation, the training data does not contain compounds like the unique host, resulting in a large prediction error. Another possible cause of the large prediction errors is the quality of the reported emission peak wavelengths. Some phosphor materials have a deviation of tens of nm or more in the reported emission peak wavelengths. Phosphors with large deviations have been eliminated from the dataset as mentioned in the Methods section, but the data might not be fully curated yet. Further investigation for the large prediction errors is beyond the scope of this study, whereas obtaining a high-quality dataset that covers diverse materials is a big issue in the data-driven materials research.
Emission peak wavelength is used as the target variable in this study, while the energy of the emission peak was used as the target variables in the previous studies.6,7 Note that in principle, correction of intensity is required to convert an emission spectrum from the wavelength to energy and vice versa, and its peak top shifts. For comparison with previous studies, the emission peak wavelengths were simply converted into energy without such intensity correction, and regression on the converted energy was conducted. The bagging of the GB method was used. The prediction accuracy and the plot of the predicted values with respect to the reported ones are shown in Table S4 and Fig. S1 in the ESI.† The present results (0.13 eV MAE, 0.16 eV RMSE) are slightly smaller (better) than those in ref. 7 (0.139 eV MAE, 0.183 eV RMSE), and slightly larger (worse) than ref. 6 (0.020 eV2 MSE corresponding to 0.14 eV RMSE). Only the features derived from the chemical composition were used in ref. 7, whereas features derived from the structure were also considered in ref. 6 and in this study. This would have resulted in the slightly poorer predictive performance in ref. 7. In ref. 6, the data were restricted to phosphors with only a single substitution site and to examples of the critical activator concentrations corresponding to concentrations showing the highest PL intensity. In contrast, some phosphors in the present dataset had multiple substitution sites and the activator concentrations depended on the literature. The restriction in ref. 6 might have suppressed the data variability and reduced the RMSE, but it also limited the coverage of the machine learning model.
Test with additional literature data
To develop new phosphor materials, the AtomWork-Adv materials database was explored and candidate host compounds of oxides, nitrides, and oxynitrides composed of main elements and containing Ca, Sr, or Ba elements were collected. Emission peak wavelengths of the collected compounds were predicted using the bagging of GB model that was rebuilt using the whole phosphor dataset with the optimized parameters. Compounds with predicted wavelengths of about 500–550 nm were selected as candidates of green-emitting phosphors. Some of the collected compounds had already been reported as Eu2+-activated phosphors, while they were not in the phosphor dataset. Therefore, an additional test was performed on the machine learning model with additional 21 Eu2+-activated phosphors.The predicted and reported emission peak wavelengths of the additional 21 phosphors are illustrated in Fig. 3, which are overlaid on the cross-validation results (Fig. 2e). MAE and RMSE to the test data were 33 nm and 42 nm, respectively. The distribution of the prediction errors looks comparable with that for the validation data in the cross validation, but the MAE and RMSE were much larger than the values estimated by the cross validation. The test data contained Sr2GeO4:Eu2+, which looked like an outlier. Sr2GeO4:Eu2+ showed the largest prediction error: 515 nm of the prediction versus 620 nm reported in ref. 27. This host compound contains Ge element, which was not in the phosphor dataset as shown in Fig. 1b. MAE and RMSE to the other 20 test data except Sr2GeO4:Eu2+ were respectively 30 nm and 37 nm, which were comparable to the results from the cross validation. These suggest that it is essential to extend the phosphor dataset to cover the diverse phosphor materials for a higher predictive performance over a wide range of candidate compounds.
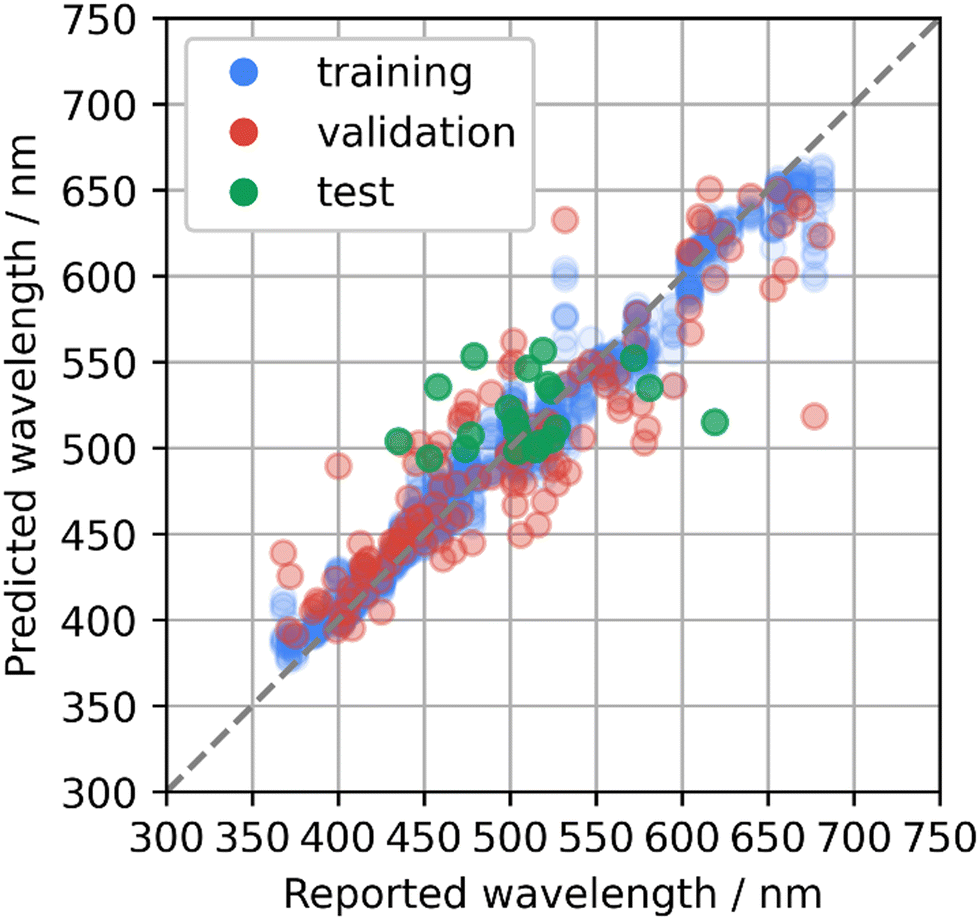 | ||
| Fig. 3 Predicted emission peak wavelengths with respect to reported values for the test data of the additionally collected Eu2+-activated phosphors (green) using the bagging of the gradient boosted regression trees method. The plot is overlaid on the cross-validation results (Fig. 2e). | ||
Exploration of new phosphor materials
As described in the previous section, oxides, nitrides, and oxynitrides composed of main elements and containing Ca, Sr, or Ba elements were collected from the AtomWork-Adv materials database to develop new phosphors. 20 candidate compounds were selected by removing high-pressure phases and selecting compounds with predicted emission peak wavelengths of about 500–550 nm. As a result, the 20 candidates were all oxides. These candidates were synthesized and characterized by experiments. Compositions, space groups, predicted wavelengths, and experimental results are summarized in Table 3. Some candidate compounds had polytypes. The prediction was done for all the polytypes. The predicted wavelengths for all the polytypes are listed together in the table. Data for the polytypes of the synthesized products are underlined.| Index | Composition | Space group | Predicted wavelength (nm) | Experimental results |
|---|---|---|---|---|
| 1 | Ba2MgGe2O7 |
P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21m (113) 21m (113) |
501 | No luminescence |
| 2 | Ba2ZnGe2O7 |
P21m (113) |
500 | No luminescence |
| 3 | Ca2Ga2GeO7 |
P21m (113) |
513 | No luminescence |
| 4 | Ca2GeO4 | P63mc (186) | 518 | No luminescence |
![[P with combining low line]](https://www.rsc.org/images/entities/i_char_0050_0332.gif) ![[n with combining low line]](https://www.rsc.org/images/entities/i_char_006e_0332.gif) ![[m with combining low line]](https://www.rsc.org/images/entities/i_char_006d_0332.gif) ![[a with combining low line]](https://www.rsc.org/images/entities/i_char_0061_0332.gif) ( (![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) ![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) ) ) |
![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif) ![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) ![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif) |
|||
| 5 | Ca2ZnGe2O7 |
P21m (113) |
510 | Low-purity products |
| 6 | Ca3Al2Ge3O12 |
Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d (230) d (230) |
486 | Eu3+ luminescence |
| 7 | Ca5Ge3O11 | C2/m (12) | 517 | Eu3+ luminescence |
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) () () |
![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) |
|||
| 8 | CaGa2O4 |
() |
|
No luminescence |
| P21/c (14) | 515 | |||
| 9 | K4BaSi3O9 | Ama2 (40) | 520 | Eu3+ luminescence |
| 10 | K4CaGe3O9 |
Pa (205) |
524 | Eu3+ luminescence |
| 11 | K4SrSi3O9 |
(![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) ) ) |
![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif) |
Eu3+ luminescence |
| Ama2 (40) | 519 | |||
| 12 | Li2Ca4Si4O13 |
P (2) |
529 | Eu2+ luminescence, 520 nm |
| 13 | Na2Ca2Si2O7 | C2/c (15) | 544 | Eu2+ luminescence, 527 nm |
| 14 | Na2SrSi2O6 |
Rm (166) |
519 | Eu3+ luminescence |
| 15 | Na4SrSi3O9 | C2 (5) | 527 | No luminescence |
| 16 | Sr2Al2GeO7 |
P21m (113) |
508 | Eu3+ luminescence |
| 17 | Sr2MgGe2O7 |
P21m (113) |
494 | No luminescence |
| 18 | Sr3Ga4O9 |
P (2) |
516 | No luminescence |
| 19 | SrGeO3 |
![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) / /![[c with combining low line]](https://www.rsc.org/images/entities/i_char_0063_0332.gif) () () |
![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif) |
No luminescence |
|
P (2) |
496 | |||
| 20 | SrLaGaO4 | I4/mmm (139) | 548 | Eu2+ luminescence, 502 nm |
The target compounds were synthesized with a purity of 70 wt% or more estimated by the powder XRD analysis, except for Ca2ZnGe2O7. Ca2ZnGe2O7 was obtained only as low-purity products, and further characterization was not conducted to it. PL spectra were measured for the powder products of the remaining 19 candidates. No luminescence was observed from the 9 products. Only sharp 4f–4f emission spectra derived from Eu3+ activators were observed from the other 7 products. Finally, emission spectra from the Eu2+ activators were observed from three products, Li2Ca4Si4O13, Na2Ca2Si2O7, and SrLaGaO4. XRD patterns of the powder products, which are shown in Fig. S2 in the ESI,† indicated that the products were mixtures of the target compounds and impurity phases. Because the PL spectra of the powder samples are largely influenced by impurity phases with bright luminescence, it is essential to verify that the observed Eu2+ luminescence derives from the target compounds. Therefore, well-grown particles were picked up from the products of these three candidates and characterized in a manner of the single-particle diagnosis.4 The single crystal XRD confirmed that the crystal structures of the picked particles were identical to Li2Ca4Si4O13,28 Na2Ca2Si2O7,29 and SrLaGaO4,30 respectively. Crystallographic information by the single crystal XRD is listed in the ESI.†Fig. 4 displays photo images and emission spectra of the picked particles of these three phosphors under 365 nm LED excitation.
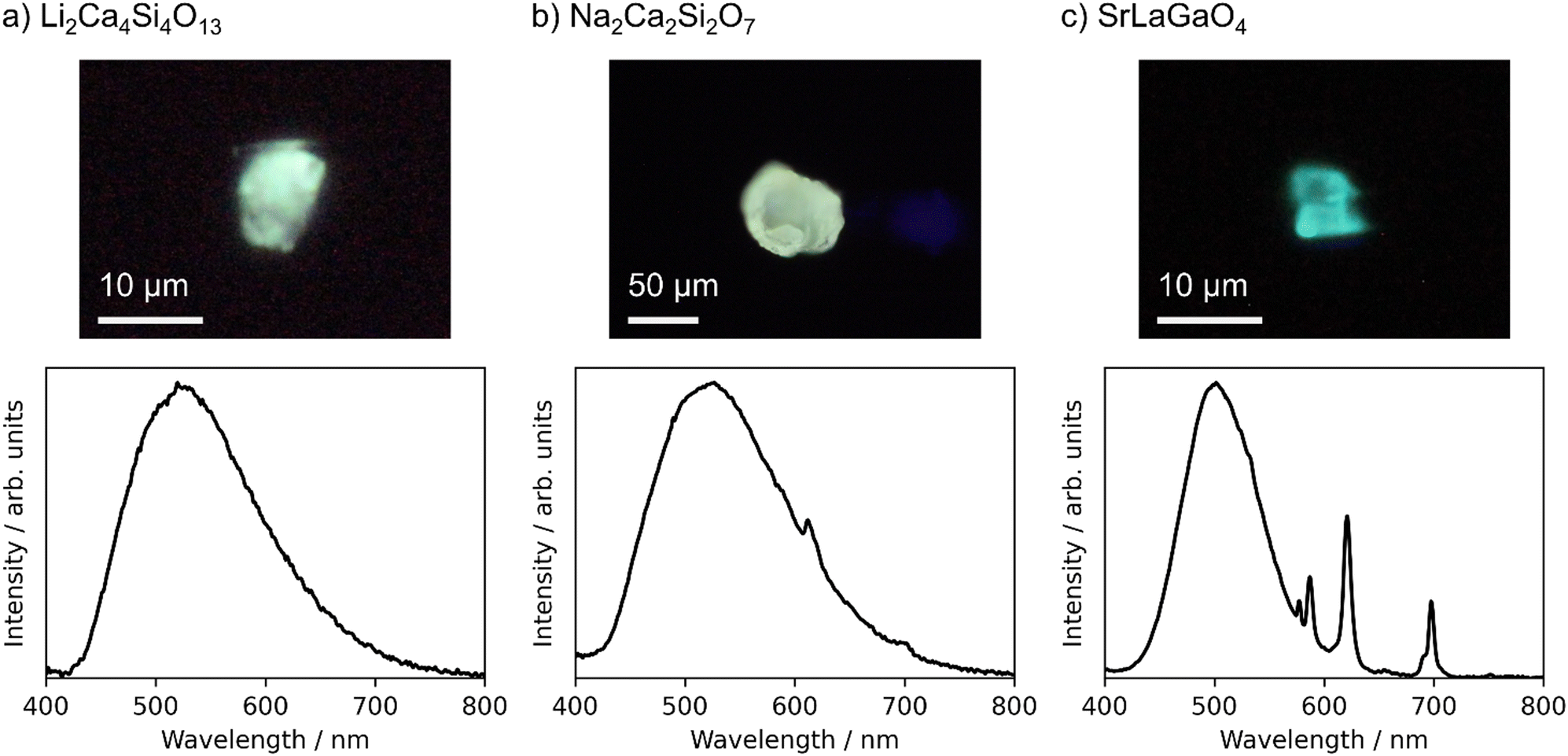 | ||
| Fig. 4 Photo images (upper panels) and emission spectra (lower panels) of particles of Eu-doped (a) Li2Ca4Si4O13, (b) Na2Ca2Si2O7, and (c) SrLaGaO4 under 365 nm LED excitation. | ||
Li2Ca4Si4O13 was predicted to have an emission peak wavelength of 529 nm, and the peak was observed at 520 nm. Na2Ca2Si2O7 were predicted to have a peak wavelength of 544 nm, and the peak was observed at 527 nm. These two phosphors show green emissions as designed, and the prediction errors were as small as 9 nm and 17 nm, respectively. In both cases, FWHMs were very large, 140 nm or more. There are many possible substitution sites for Eu2+ in both structures. The luminescence properties depend on the substitution sites, and the observed emission spectra are an integration of those from the individual sites. The machine learning prediction worked well even for such complex structures. In Na2Ca2Si2O7, weak Eu3+ luminescence was also observed. Some substitution sites might be suitable for Eu3+ even in the synthesis under a reducing atmosphere.
The Eu-doped SrLaGaO4 particle showed both a blue-green emission derived from Eu2+ activators and a characteristic red emission from Eu3+ activators. As for the Eu2+ luminescence, the predicted emission peak wavelength was 548 nm, whereas the peak was observed at 502 nm with a FWHM of 83 nm. The prediction error, 46 nm, was large, but would be acceptable with respect to the prediction accuracy of the present machine learning model. As Sr and La atoms occupy the same crystallographic site in SrLaGaO4, Eu atoms might occupy this site in a mixed valence of Eu2+ and Eu3+, resulting in the simultaneous Eu2+ and Eu3+ luminescence.
The three new Eu2+-activated phosphors were successfully discovered, but a large part of the candidates failed to show Eu2+ luminescence. The products were annealed in the reducing atmosphere to obtain Eu2+, but the reduction process seemed insufficient for some compounds. The stability of Eu2+/Eu3+ in the host lattices is attributed to the redox potential of the substituted Eu ions and the annealing conditions, whereas the annealing conditions were limited depending on the host compounds to prevent them from melting or decomposing. Even if Eu2+ is stable in the hosts, the luminescence may be quenched if the energy levels of the Eu2+ excited states overlap or are close to the conduction bands of the hosts. These are likely the reasons why many candidates have not exhibited Eu2+ luminescence. At this moment, it is hard to predict the valence and energy level of the substituted Eu ion in the host and to predict appropriate synthesis conditions to obtain Eu2+, prior to synthesis. Prediction of these factors is also important for efficient development of new Eu2+-activated phosphors and is a future task.
Conclusions
To rapidly discover new Eu2+-activated phosphors with a designed luminescence color, a machine learning model to predict emission peak wavelength was developed from the phosphor dataset composed of 129 Eu2+-activated phosphors. The general-purpose compositional and structural features were used to represent host compounds. The bagging technique with the gradient boosted regression trees method was adopted to obtain high predictive performance against the nonlinear relationship between the features and the emission peak wavelength, and to avoid overfitting with the small phosphor dataset. The predictive performance of the built machine learning model was comparable to those in previous studies.6,7 The results of the cross validation and the additional test suggest that it is essential to extend the phosphor dataset to cover the diverse phosphor materials for a higher predictive performance over a wide range of candidate compounds.Using the machine learning model, new green-emitting Eu2+-activated phosphors were searched from the AtomWork-Adv materials database. Among twenty candidate compounds predicted to have emission peak wavelengths of about 500–550 nm, three new phosphors, namely, Eu-doped Li2Ca4Si4O13, Na2Ca2Si2O7, and SrLaGaO4, were successfully synthesized. Li2Ca4Si4O13:Eu2+ and Na2Ca2Si2O7:Eu2+ showed the Eu2+ luminescence of the green color as designed. Eu-doped SrLaGaO4 showed simultaneous Eu2+ and Eu3+ luminescence, and it shows a blue-green emission derived from the Eu2+ activators. These results clearly demonstrate that the machine learning on the emission peak wavelength is useful for the rapid and efficient development of new Eu2+-activated phosphors with a designed luminescence color.
Author contributions
Y. K. and N. H. devised the basic idea of this study. N. H. collected the phosphor data. Y. K. conducted the machine learning, and H. I., T. T. and N. H. contributed to refining the models. M. H. and S. F. performed the synthesis and characterization under the guidance of T. T. and N. H. Y. K. and T. T. prepared the original draft of the manuscript. All the authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the Japan Science and Technology Agency (JST), CREST Gant Number JPMJCR19J2.References
- S. Ye, F. Xiao, Y. X. Pan, Y. Y. Ma and Q. Y. Zhang, Mater. Sci. Eng., R, 2010, 71, 1 CrossRef CAS; Z. G. Xia and Q. L. Liu, Prog. Mater. Sci., 2016, 84, 59 CrossRef; L. Wang, R. J. Xie, T. Suehiro, T. Takeda and N. Hirosaki, Chem. Rev., 2018, 118, 1951 CrossRef.
- X. Qin, X. W. Liu, W. Huang, M. Bettinelli and X. G. Liu, Chem. Rev., 2017, 117, 4488 CrossRef CAS PubMed.
- X. F. Luo and R. J. Xie, J. Rare Earths, 2020, 38, 464 CrossRef CAS; X. D. Sun, K. A. Wang, Y. Yoo, W. G. Wallace-Freedman, C. Gao, X. D. Xiang and P. G. Schultz, Adv. Mater., 1997, 9, 1046 CrossRef; E. Danielson, J. H. Golden, E. W. McFarland, C. M. Reaves, W. H. Weinberg and X. D. Wu, Nature, 1997, 389, 944 CrossRef; J. S. Wang, Y. Yoo, C. Gao, I. Takeuchi, X. D. Sun, H. Y. Chang, X. D. Xiang and P. G. Schultz, Science, 1998, 279, 1712 CrossRef PubMed; K. S. Sohn, J. M. Lee and N. S. Shin, Adv. Mater., 2003, 15, 2081 CrossRef; W. B. Park, N. Shin, K. P. Hong, M. Pyo and K. S. Sohn, Adv. Funct. Mater., 2012, 22, 2258 CrossRef.
- N. Hirosaki, T. Takeda, S. Funahashi and R. J. Xie, Chem. Mater., 2014, 26, 4280 CrossRef CAS.
- W. B. Park, S. P. Singh, M. Kim and K. S. Sohn, ACS Comb. Sci., 2015, 17, 317 CrossRef CAS PubMed.
- C. Park, J. W. Lee, M. Kim, B. D. Lee, S. P. Singh, W. B. Park and K. S. Sohn, Inorg. Chem. Front., 2021, 8, 4610 RSC.
- H. Nakano, K. Tanaka, T. Miyao, K. Funatsu, R. Shirasawa and S. Tomiya, Chem. Lett., 2017, 46, 1482 CrossRef CAS.
- S. Q. Lai, M. Zhao, J. W. Qiao, M. S. Molokeev and Z. G. Xia, J. Phys. Chem. Lett., 2020, 11, 5680 CrossRef CAS.
- J. M. Ha, Z. B. Wang, E. Novitskaya, G. A. Hirata, O. A. Graeve, S. P. Ong and J. McKittrick, J. Lumin., 2016, 179, 297 CrossRef CAS; Z. B. Wang, J. Ha, Y. H. Kim, W. B. Im, J. McKittrick and S. P. Ong, Joule, 2018, 2, 914 CrossRef; Y. Zhuo, A. M. Tehrani, A. O. Oliynyk, A. C. Duke and J. Brgoch, Nat. Commun., 2018, 9, 4377 CrossRef PubMed; S. X. Li, Y. H. Xia, M. Amachraa, N. T. Hung, Z. B. Wang, S. P. Ong and R. J. Xie, Chem. Mater., 2019, 31, 6286 CrossRef.
- S. X. Li and R. J. Xie, ECS J. Solid State Sci. Technol., 2019, 9, 016013 CrossRef.
- L. G. Vanuitert, J. Lumin., 1984, 29, 1 CrossRef CAS; P. Dorenbos, ECS J. Solid State Sci. Technol., 2013, 2, R3001 CrossRef.
- Z. Barandiarán, J. Joos and L. Seijo, Luminescent Materials: A Quantum Chemical Approach for Computer-Aided Discovery and Design, Springer; Cham, 2022 CrossRef CAS; J. L. Pascual, J. Schamps, Z. Barandiaran and L. Seijo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 104105 CrossRef CAS; Z. Barandiaran, A. Meijerink and L. Seijo, Phys. Chem. Chem. Phys., 2015, 17, 19874 RSC.
- Y. C. Jia, A. Miglio, S. Poncé, X. Gonze and M. Mikami, Phys. Rev. B, 2016, 93, 155111 CrossRef.
- Y. C. Jia, A. Miglio, S. Poncé, M. Mikami and X. Gonze, Phys. Rev. B, 2017, 96, 125132 CrossRef; Y. C. Jia, A. Miglio, S. Poncé, M. Mikami and X. Gonze, Phys. Rev. B, 2020, 101, 089902 CrossRef.
- Y. Li, M. Gecevicius and J. R. Qiu, Chem. Soc. Rev., 2016, 45, 2090 RSC; W. M. Yen and M. J. Weber, Inorganic Phosphors: Compositions, Preparation and Optical Properties, CRC Press; Boca Raton, 2004 Search PubMed.
- Inorganic Crystal Structure Database (ICSD). FIZ Karlsruhe GmbH, Germany. https://icsd.products.fiz-karlsruhe.de/.
- AtomWork-Adv. National Institute for Materials Science, Japan. https://atomwork-adv.nims.go.jp/.
- L. Ward, A. Agrawal, A. Choudhary and C. Wolverton, npj Comput. Mater., 2016, 2, 16028 CrossRef; A. Seko, H. Hayashi, K. Nakayama, A. Takahashi and I. Tanaka, Phys. Rev. B, 2017, 95, 144110 CrossRef.
- W. H. Baur, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1195 CrossRef CAS.
- M. O’keeffe and N. E. Brese, J. Am. Chem. Soc., 1991, 113, 3226 CrossRef.
- N. E. R. Zimmermann and A. Jain, RSC Adv., 2020, 10, 6063 RSC.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314 CrossRef CAS.
- XenonPy. https://github.com/yoshida-lab/XenonPy/.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825 Search PubMed.
- Hyperopt. https://hyperopt.github.io/hyperopt/.
- Scikit-optimize. https://scikit-optimize.github.io/.
- K. Fiaczyk and E. Zych, RSC Adv., 2016, 6, 91836 RSC.
- M. E. Villafuerte-Castrejón, A. Dago and R. Pomés, J. Solid State Chem., 1994, 112, 438 CrossRef.
- V. Kahlenberg and A. Hösch, Z. Kristallogr., 2002, 217, 155 CAS.
- J. F. Britten, H. A. Dabkowska, A. B. Dabkowski, J. E. Greedan, J. L. Campbell and W. J. Teesdale, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 1975 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00881e |
| This journal is © The Royal Society of Chemistry 2023 |