
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the microporosity and 3D spatial distribution of calcium phosphate cement/hydrogel biomaterials using FIB/SEM at cryogenic temperatures
Mouad
Essani
 *a,
Baptiste
Charbonnier
b,
Nicolas
Stephant
a,
Hilel
Moussi
ab,
Pierre
Weiss
c,
Jean
Le Bideau
a and
Patricia
Abellan
*a
*a,
Baptiste
Charbonnier
b,
Nicolas
Stephant
a,
Hilel
Moussi
ab,
Pierre
Weiss
c,
Jean
Le Bideau
a and
Patricia
Abellan
*a
aNantes Université, CNRS, Institut des Matériaux de Nantes Jean Rouxel (IMN), F-44000 Nantes, France. E-mail: mouad.essani@gmail.com; Patricia.Abellan@cnrs-imn.fr
bNantes Université, INSERM, UMR 1229 RMeS/ONIRIS, Regenerative Medicine and Skelton Laboratory, F-44042 Nantes, France
cNantes Université, CHU Nantes, INSERM, UMR 1229 RMeS/ONIRIS, Regenerative Medicine and Skeleton Laboratory, F-44042 Nantes, France
First published on 5th May 2023
Abstract
Considerable clinical success has been achieved ever since calcium phosphate cements (CPCs) were introduced in bone regenerative therapies. The development of novel CPC/hydrogel composites with increased mechanical and biological properties is critical for the next generation of bone substitutes. Focused ion beam/scanning electron microscopy (3D FIB/SEM) tomography combined with energy dispersive spectrometry (EDS) provide the ability to elucidate the spatial distribution of the different phases (organic/inorganic) with tens of nanometer resolution, which is crucial information for the development of biomaterials with optimized biodegradation. Characterization under cryogenic conditions by using cryo-FIB/SEM in combination with cryo-EDS allows us to resolve the sub-micrometer structural features of such composites. In this study, we present a multidimensional, low dose structural and chemical analysis of calcium deficient hydroxyapatite (CDHA) mixed with two different hydrogels: gelatin and silanized hyaluronic acid (Si-HA). We show that, depending on the chemical nature of the hydrogel, CDHA/hydrogel compounds can exhibit different properties, particularly microporosity and phase distribution at the (sub) microscale. The incorporation of Si-HA and gelatin both resulted in an increase of the amount of microporosity within the composites. While Si-HA formed a homogeneous mixture with CDHA, a separate phase of gelatin was observed inside the CDHA/gelatin compound. Our results also indicate that structural modification occurs when applying sample preparation protocols or analytical methods that are subject to altering the hydration level within these biomaterials and thus, cryogenic temperatures are necessary for their characterization using electron microscopy.
Introduction
The field of bone regeneration is currently evolving quickly and tends toward the development of ephemeral bioactive scaffolds supporting and promoting the formation of new vascularized bone tissues.1,2 Scaffolds for bone regeneration underwent a drastic evolution throughout the time, from massive bio-inert structures to hierarchically designed biomimetic scaffolds, which are today extensively investigated.3 As such, hybrid and composite biomaterials, coupling highly hydrated organic phases (mimicking the extracellular matrix) to bioactive inorganic phases (e.g., calcium phosphates), seem particularly promising.4–7 Indeed, such complex systems allow for the direct or indirect modulation of the material-induced biological response through a wide panel of architectural, physical, chemical and mechanical cues.8–11 Among them, injectable bone substitutes (IBSs) are of prime interest as the only solution for minimally invasive interventions, in addition to their relevant bioactivity and osteoconductive properties.12 Commercial IBSs are most of the time composed of either bioceramics, bioglass particles or calcium phosphate-based cements (CPCs) associated with polymeric macromolecules which enhance their intrinsic properties (e.g., mechanical resistance), their capacity to be handled (e.g., injectability, setting time, and in situ cohesivity), and material/host (e.g., cell adhesion, proliferation and differentiation) interactions.13 Particularly, the addition of gelatin or hyaluronic acid to CPCs has been extensively studied.14–16Today's IBS research is particularly focused on not only the development of innovative formulations based on tailored functionalized hydrogels and substituted CPCs to optimize bone regeneration but also the biodegradation of the material, which is one of the main current roadblocks in clinical applications. This implies that the interactions between the organic and inorganic phases should be understood to ultimately control them, which is far from being obvious due to the very complex reaction mechanisms between the two phases.17
Due to the highly hydrated soft nature of these biomimetic hybrids, little is known about their structure and evolution throughout time: their fine characterization remains quite challenging. Procedures enabling the investigation of the original structure of high water content biomaterials with the use of suitable chemical and structural characterization tools are yet to be developed. Scanning electron microscopy (SEM) combined with electron dispersive spectrometry (EDS) allow for both the structural and chemical characterization studies of biomaterials in a wide scale range, from the sub-micron to hundreds of μm length scale.18–21 Nevertheless, only surface (or near surface) analysis can be performed using SEM and EDS. Information about the real morphology, chemical distribution in depth and other structural features (e.g. 3D distribution and pore structure) is lacking in 2D characterization. With the introduction of focused ion beam (FIB) in the biological domain, the 3D structural analysis of biological samples can be achieved by combining FIB and SEM (an approach also known as FIB tomography, serial FIB/SEM or 3D FIB/SEM). This approach consists of performing serial sectioning on a region of interest (ROI) with FIB combined with SEM imaging of each newly exposed area. The process is repeated until constituting a sufficient number of sections to reconstruct a 3D volume corresponding to the multiple imaged areas. In addition to obtain information about the internal microstructure, FIB milling in combination with EDS should provide a description of the chemical composition and interactions between the different phases in depth and at buried interfaces within the sample.
Great progress has been made over the past decade with regard to the application of FIB-SEM in life sciences and a number of biological and soft-tissue materials have been studied using FIB tomography, among which are studies aiming to investigate biomaterial–tissue interfaces22 and characterize the osteocyte network in bones.23 Multiple sample preparation methods and structure preservation techniques, such as the chemical fixation, dehydration and embedding of samples in a resin, have been suggested.24 These conventional methods are however subject to altering the initial biological structure and introducing artifacts.24 Cryogenic approaches play a prominent role in addressing these problems and allow for the analysis of biological samples in their native unaltered structure. The accuracy of cryo FIB-SEM in analysing biological samples has already been proven, e.g. in the 3D visualization of cells25 and the localization of proteins within a given volume.26 Despite the widespread work on the analysis of biomaterials using FIB/SEM,22,24 few studies reported the use of FIB tomography under cryogenic conditions to observe the structure of biomaterials.
Biomaterials, such as CPC–hydrogel composites, are highly porous and contain a significant amount of water. It is therefore not unlikely that their structure and porosity vary depending on their water content. Preparation methods that include a dehydration process or any step causing a variation of the hydration level in these samples can lead to an altered structure that is not representative of the biomaterial's native state. Moreover, most of the CPC based composites are insulators (resulting in charging effects during SEM imaging), and are easily damaged under ion or electron irradiation. Optimized conditions for the FIB-SEM analysis are thus required to avoid structural damage under irradiation, while structural characterization at cryogenic temperatures would also help coping with these difficulties by increasing the tolerable dose for ionization damage. Overall, finding sample preparation and acquisition protocols that are adapted to hydrate non-conductive materials is crucial for cryo-FIB/SEM experiments of composite biomaterials.
In this work, we present multidimensional characterization of CPC composite scaffolds using FIB tomography and EDS. Structural and chemical analyses were performed on calcium deficient hydroxyapatite (CDHA) as the control group and on CDHA mixed with two different hydrogels: gelatin that is a physical crosslink hydrogel and silanized hyaluronic acid (Si-HA) that is a covalent one.27 Apart from the fact that CPC/gelatin and CPC/hyaluronic acid composites are among the most widely studied biomaterials in bone regeneration,28–33 they exhibit a highly hydrated fragile nature, which makes them typical samples of interest that could benefit from optimized protocols in cryo-FIB/SEM.
Three dimensional visualization and chemical characterization of CDHA, CDHA/gelatin and CDHA/Si-HA, at both hydrated and dehydrated states, were achieved in this study using FIB tomography and EDS, which enabled us to investigate microscale features in 3D and identify the impact of dehydration on the biomaterial's microstructure. Hydrated samples were cryo-fixed by plunge freezing in slush nitrogen and mounted on a cryostage inside the SEM chamber for analysis. The results obtained in the present paper provide an understanding of the organization of each phase inside the composite biomaterials and show that both the nature of the organic phase and the state of the biomaterials (hydrated/dehydrated) have a significant influence on the microstructure and distribution of CDHA–hydrogel composites.
Experimental
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :liquid (P:L) ratio of 2.2 g:1 mL. Hydraulic setting of the cement occurred according to the following equation:16
:liquid (P:L) ratio of 2.2 g:1 mL. Hydraulic setting of the cement occurred according to the following equation:16| 3α-Ca3(PO4)2 + H2O → Ca9(HPO4)(PO4)5(OH) |
:gel ratio (P:G) of 2.2 g:1 mL.
Medical grade gelatin (Rousselot X-pure 10B) was dissolved in 1× phosphate buffer saline at 60 °C with a concentration of 10.6% w/w. The gelatin solution was cooled a few minutes at 23 °C to reach an adapted viscosity for handling and injection and then mixed with α-TCP at a powder:gel ratio (P:G) of 2.2 g:1 mL.
Mixing gelatin and Si-HA with α-TCP was expected to provide areas where a separate organic phase could be observed. This allowed us to verify whether the cement and the hydrogel can be identified separately in the cryo-FIB/SEM data.
Both organo-mineral formulations were injected into 20 × 6 × 6 mm Teflon® moulds and were allowed to set under ambient atmosphere for 3 hours. Then, they were immersed in 0.9% w/w NaCl solution at 37 °C for a minimum of 72 hours to finalize the setting.34
Small fractions of 200 to 300 μm of the thickness of hydrated biomaterials were cryofixed by plunge freezing into liquid nitrogen slush (SN). The nitrogen freezing station and lines were pumped a priori so as to remove as much water vapor as possible before filling it with LN. In order to obtain a rapid cooling and reduce the formation of a vapor layer around the plunged specimen (Leidenfrost effect), LN was pumped inside the freezing station to form SN.35,36 Samples were further frozen by plunge-freezing into the SN few seconds after venting the freezing station. Cryo-fixed samples were further transferred to the preparation chamber, using a vacuum transfer device, and mounted on the cryo-stage. In order to provide a clean surface for FIB-SEM investigation with no ice contamination, all samples were cryo-fractured. Sputter coating by a Pt target was further carried out inside the preparation chamber (see sputtering conditions below) to ensure the sample's electrical conductivity and avoid charging effects during SEM imaging.
Two different FIB tomography protocols were adopted: one suitable for room temperature analysis and a second one adapted to cryo-experiments:
1. At room temperature, the workflow implemented using the ZEISS ATLAS software was used for the acquisition of the 3D data. The ROI for FIB milling was protected by the deposition of a Pt layer with a thickness of 1 μm along the Z direction (the direction of the ion beam) and a 15 μm × 15 μm XY area. The FIB-induced deposition was performed using an ion beam current of 0.7 nA and lasted for about 30 min. In order to obtain a clean deposition, the protective Pt layer was deposited on regions that exhibited low surface roughness. Following the typical tracking protocol provided by the manufacturer, tracking marks for the calibration of FIB serial sectioning were created on top of the Pt protective layer by milling five lines (three on the middle parallel to the X direction and two inclined lines at each side) using a low FIB current of 50 pA. The lines were further highlighted with a C deposition and a second C layer was deposited on top of the Pt layer. The comparison between Pt and C provided a better visualization of the tracking marks. A cross-section in the form of a trapezoid trench was made using a FIB current of 30 nA. In order to smoothen the face of the trapezoid trench (i.e. part exposed to the electron beam), the cross-sectioning was followed by softer milling using a FIB current of 7 nA. FIB serial sectioning was further performed using a milling current of 0.7 nA and a pixel size of 10 nm.
2. Under cryo-conditions, the FIB milling region was protected by Pt deposition. At low temperatures, the precursor gas at the exit of the Pt needle condensed immediately at the sample's cold surface, which leads to the adsorption of a massive quantity of precursor molecules (equivalent to a thickness of hundreds of μm in size) onto the surface. In order to avoid the rapid growth of the precursor layer and control its deposition, the Pt reservoir was cooled down to 28 °C, the WD for Pt deposition was increased by ∼3 mm and the needle's valve was opened manually for 10 to 20 s. Precursor molecules deposited on the sample's surface were further dissociated by irradiating the ROI by FIB continuous scanning using a current of 3 nA and 0.6 μs dwell time per pixel. The same pixel size in the X, Y plane and FIB current values for the experiments at room temperature (see above) were adopted for FIB serial sectioning and SEM imaging. Along the milling direction (Z direction), the milling distance (and thus, the nominal size in Z) was 15 nm, thus leading to a nominal voxel size of 10 nm × 10 nm × 15 nm. Another important aspect that requires attention during cryo-experiments is the duration capacity of the 30L LN Dewar that limits the milling and image acquisition time. This duration was estimated in our case to be about 8 hours before the total emptying of the Dewar. The Dewar was therefore filled with LN every 4 hours in order to ensure the continuous flow of cooled N2 gas while performing 3D acquisitions.
By choosing contrast and geometry filters, this classifier enabled us to obtain a high quality segmentation of the different phases, which allowed for a description of their distributions within each compound. The amount of porosity was expressed as the volume ratio % between the segmented “empty space” region and the rest of the material. The function “thickness meshes” implemented in Dragonfly was used to calculate each pore size from the “empty space” region, as the diameter of the largest sphere that fits within the pore.41 This enabled us to determine the mean pore size for each compound.
Results and discussion
Serial sectioning with FIB combined with SEM imaging was carried out under cryogenic conditions to investigate the microstructure of CDHA–hydrogel compounds (see the Methods section for details).CDHA/gelatin
Firstly, the composite sample consisting of the CDHA/gelatin mixture was investigated. This composite is expected to exhibit three different phases, the mineral cement, the organic matrix and an aqueous phase, filling the pores within the sample. In order to obtain an accurate evaluation of the microporosity as well as a description of the spatial distribution of both the gelatin and the cement, the phases within the mixture need to be resolved separately. In other words, SEM images must provide enough contrast to allow for the identification and segmentation of each phase. As shown in Fig. 1a, secondary electron (SE) SEM imaging did not provide a clear visualization of the phases distribution, as the aqueous phase exhibited a contrast level similar to both the CDHA and hydrogel parts of the composite. This made the segmentation of each phase difficult to perform. In order to cope with this limitation, cryo-FIB tomography was performed using backscattered electron (BSE) imaging. Because the BSEs carry information about the structure of the sample and its composition, the difference in the contrast level between the different phases was improved (Fig. 1b). The low electron energies adopted for image acquisition were practical for reducing the depth of backscattered electron emission (estimated to be ∼30–40 nm using Reimer's formula42) and obtaining good visualization of the structural details at the materials surface from the BSE images. We note that while the nominal voxel size in the FIB-SEM data is 10 nm × 10 nm × 15 nm, considering the depth at which the BSEs are emitted, the real spatial resolution is 10 nm × 10 nm × ∼30–40 nm. | ||
| Fig. 1 (a) SEM image with secondary electrons corresponding to a FIB cross-section face performed on a CDHA/gelatin compound. The aqueous phase (red circle) exhibits a close contrast level to those of CDHA (yellow circle) and gelatin. (b) Image of the same region obtained using BSE imaging. Both images were acquired using an electron energy of 1.8 keV, an electron current of 0.1 nA and a dwell time of 1.6 μs per pixel. Pixel size: 10 nm. Vertical destriping has been applied (with 10 levels and wavelet function db2) followed by CLAHE (clip 0.01, kernel size 100 and bins 256). | ||
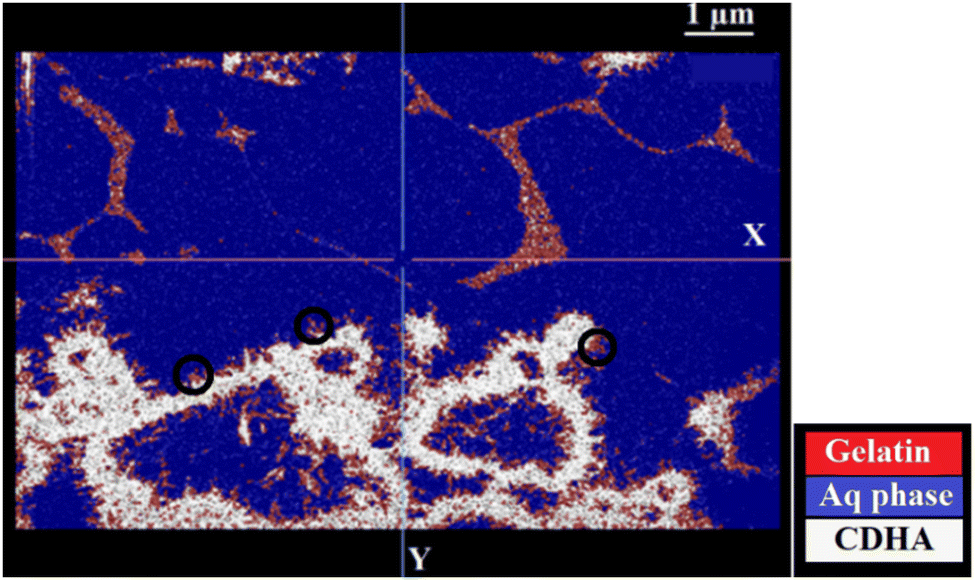
Cryo-SEM micrographs using backscattered electrons of a CDHA/gelatin compound indeed showed the presence of three different phases: water, gelatin and CDHA. Most of the cement clustered into annular agglomerates and formed needle-like particles on the sides and inside the annular spaces (Fig. 1). The contrast level of each phase can be distinguished in BSE images (Fig. 1b), which allowed us to segment the compounds separately and identify their distribution. Fig. 2 illustrates the resulting segmentation and shows the distribution of all the phases based on their contrast levels. Gelatin can be clearly identified as a separate phase inside the mixture. Additionally, the tips of the needle-like particles exhibited a contrast level close to that of gelatin. There are two possible interpretations for this observation; firstly, the segmentation may be showing the presence of gelatin on the surface of the particles (black circles in Fig. 2). Secondly, such contrast can also be a consequence of a decrease in the CDHA intensity due to the reduced amount of electrons that are backscattered from a thinner area of the particles. In order to elucidate the origin of the lower contrast level at the tips of the needle-like particles and to aid interpretation, EDS measurements were performed.
 | ||
| Fig. 2 Segmentation of the different phases in a BSE image of a CDHA/gelatin compound using the random forest trainer method incorporated in the Dragonfly® software.39 | ||
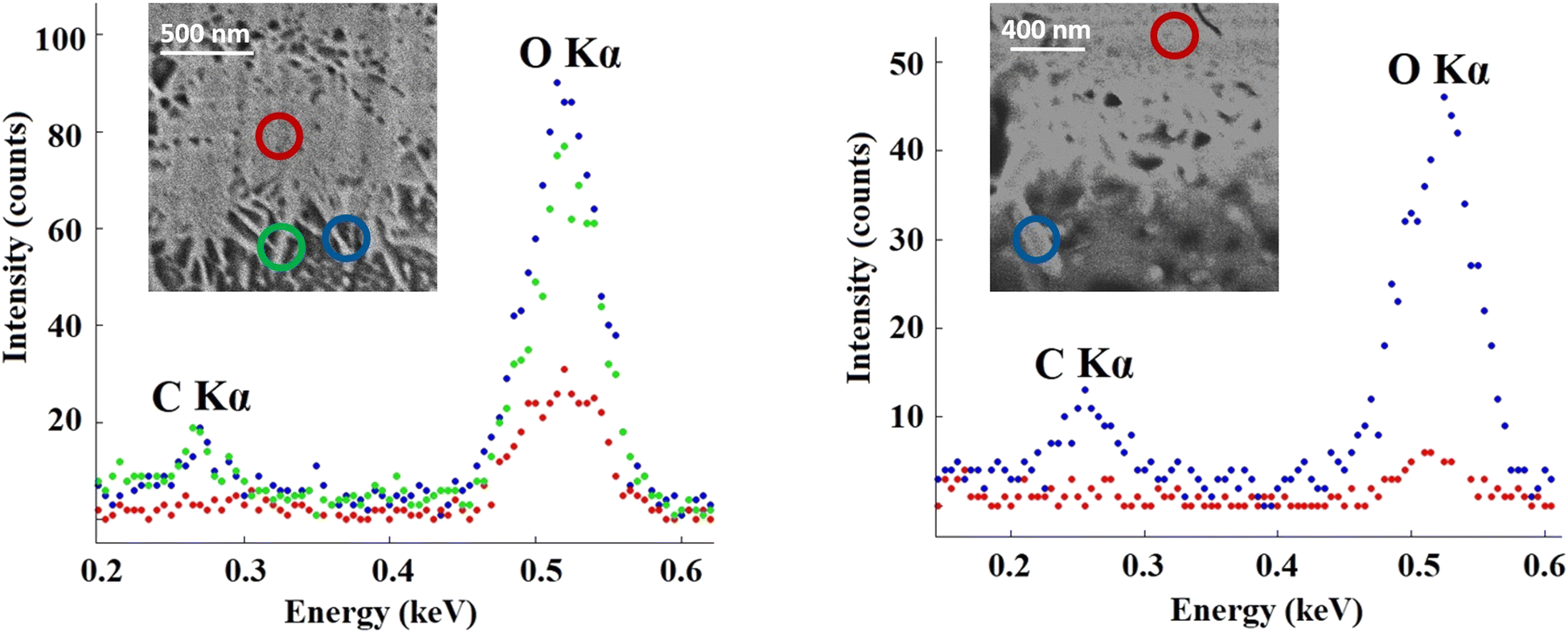
EDS measurements performed on the randomly selected CDHA/gelatin particles showed that carbon X-ray emission mostly occurred at the sides of the particles (Fig. 3) and almost no C Kα line intensity was detected in the middle. The high intensity of O Kα was observed in the spectra recorded at the sides of CDHA, resulting from the presence of the aqueous phase around the needle-like particles.
 | ||
| Fig. 3 Comparison between EDS spectra acquired at different regions in two randomly selected CDHA/gelatin particles. EDS spectra were acquired using an electron energy of 1.6 keV. | ||
Indeed, the EDS results show the presence of carbon (Fig. 3), thus gelatin, on the particle's surface and are in agreement with our first interpretation of the segmentation results shown in Fig. 2. The distribution of the organic phase would be consistent with the presence of gelatin around the needle-shaped calcium deficient hydroxyapatite particles issued from the dissolution–reprecipitation of the inorganic α-TCP precursor. Indeed, upon hydrolysis, α-TCP dissolves into calcium and phosphate ions, resulting in a supersaturated solution from which CPC crystals precipitate.43 The presence of gelatin only on the sides of the agglomerated CDHA particles and around those same regions suggests that CPC crystals precipitated in contact with the gelatin. The CDHA sites where gelatin is not present (e.g. red circles in Fig. 3) are therefore regions where the growth and hardening of the cement occurred far from the organic phase.
The gelatin phase seems to surround the agglomerated CDHA nanoneedles, but can also be observed in the aqueous phase, and this free of any calcium phosphate (Fig. 1b and 2). The phenomena driving this specific spatial organization are complex and would require further investigation. As a relative cooling of the gelatin was performed before mixing it with the inorganic cement precursor, gelatin macromolecules may have started to organize in a 3D network before the formation of the cement. It may (i) have affected the dissolution reprecipitation phenomena (and consequently formation) of the cement by disturbing sterically the local environment, (ii) have driven the nucleation of calcium phosphate phases through interactions between the carboxyl and amino moieties of the gelatin and the released calcium ions or even (iii) have limited the diffusion of calcium and phosphate ions away from the 20–40 μm precursor particles, hence calcium phosphate free gelatin macromolecules. Among others, these hypotheses could explain the microstructural differences induced by the addition of gelatin macromolecules to the hydroxyapatite calcium phosphate cement.
In order to evaluate the influence of gelatin on the microstructural properties of the cement in 3D, the CDHA/gelatin mixture was investigated by cryo-FIB tomography (see Fig. 4b) and the results were compared to those obtained for the pure inorganic cement (see Fig. 4a). For both samples, the 3D volume reconstruction was performed using the Dragonfly® software by implementing a stack of more than 700 BSE images.
 | ||
| Fig. 4 3D constructed volumes of (a) CDHA and (b) CDHA/gelatin mixture (the aqueous phase is presented in blue color). (c) 3D distribution of gelatin in a ROI inside the CDHA/gelatin compound. The random forest trainer method was used for segmentation. | ||
It can be seen from the 3D visualization of the two samples that gelatin induced a modification of the structural organization of the cement. Although needle shaped individual CDHA particles are observed in both samples, a main difference was the high presence of ring-like agglomerates of such needles as well as large “empty spaces” (of several nm up to 2300 nm within the observation area) in the CDHA/gelatin sample. While in the pure CDHA cement, the cement was more homogeneously distributed across the sample, the well separated clusters of the ring-like CDHA structures are observed in CDHA/gelatin. The 3D distribution of gelatin (Fig. 4b) confirms the previous observations and shows that gelatin has formed, in some regions, a 3D network that did not interact with the inorganic matrix (see Fig. 4c).
The microporosity of CDHA/gelatin was further identified and quantified inside the 3D constructed volume using the Random forest classifier. The amount of microporosity and the pore size in 3D were calculated and compared to those determined in hydrous CDHA (Table 1). A significant increase in both the amount of microporosity and the mean pore size was induced by incorporating gelatin into the cement. It is suggested that, by forming a separate phase, gelatin preserves a part of its highly porous structure. This acts in favor of increasing the mean compound porosity. Moreover, the annular-like arrangement of CDHA results in creating voids inside and between the agglomerated sites, which also increases the total microporosity of the cement.
| Microporosity (%) | Mean pore size (nm) | |
|---|---|---|
| CDHA | 24 ± 6 | 321 ± 62 |
| CDHA/gelatin | 58 ± 12 | 1005 ± 182 |
| CDHA/Si-HA | 43 ± 10 | 538 ± 97 |
CDHA/Si-HA
The cryo-FIB-SEM analysis of the CDHA/Si-HA mixture showed that CDHA was structured in two different ways: dense areas of the pseudo-bulk cement and star-like agglomerates with ultrafine needle-shaped particles around (Fig. 5). The ultrafine particles are also present on the surface of the compact pseudo-bulk volume. It has been reported that, in the presence of HA, the formation of such particles can be related to the precipitation of an apatitic phase.31 These particles are present more in CDHA/Si-HA than in CDHA/gelatin or the control cement, which might be the result of the enhanced precipitation of the hydroxyapatite related to the presence of HA carboxylic groups.44 These groups have been reported to favor the nucleation of apatite crystals.45 BSE micrographs shows that Si-HA seems to be forming a homogeneous-like mixture with CDHA, at least at tens of nanometer length-scale (no separate Si-HA was observed). As shown in Fig. 6, EDS spectra revealed the presence of carbon on both the dense areas and the agglomerated particles, suggesting that HA is indeed mixed with the formed CDHA clusters. Unlike in CDHA/gelatin, the precipitation of CDHA very likely preceded the cross-linking of Si-HA, which explains the fact that no separate hydrogel phase was observed within the compound. The most probable scenario is that Si-HA was adsorbed on the α-TCP particles during Si-HA/α-TCP mixing and served as a supporting substance for the precipitation of the hydroxyapatite phase. | ||
| Fig. 5 (a) BSE micrograph in the XY plane of a hydrous CDHA/Si-HA and (b) 3D visualization of the same compound. The aqueous phase is presented in blue and CDHA in white. Both images were acquired using an electron energy of 2 keV, an electron current of 0.1 nA and a dwell time of 1.6 μs per pixel. Pixel size: 10 nm in the XY plane and 15 nm in the XZ plane. | ||
 | ||
| Fig. 6 Comparison between EDS spectra acquired at different regions in two randomly selected CDHA/Si-HA particles. EDS measurements were performed using electron energies of 1.7 keV for the first particle (left hand side) and 2 keV for the second particle (right hand side). | ||
Explaining why CDHA/gelatin and CDHA/Si-HA are structured differently would require an extensive and dedicated work. However, several scenarios, alone or in combination, may be plausible. First, the gel forming kinetics may be faster for Si-HA, which directly provide an organized 3D network serving as the support for the formation of the cement. Second, and as previously evoked, calcium ions tend to interact with carboxyl groups, providing preferential nucleation sites. Both gelatin and Si-HA possess a carboxyl moiety per repeat unit; however, the Si-HA repeat unit is far smaller and with less steric hindrance than the one of gelatin. Consequently, far more nucleation sites for the precipitation of CDHA are provided, resulting ultimately in a Si-HA network embedded in a CDHA inorganic matrix. Finally, a higher concentration of phosphate ions was already available in the local environment for CDHA/Si-HA than that for CDHA/gelatin, due to the formulation process. As phosphate ions are known to dictate the precipitation of calcium phosphate phases, this may have significantly helped for the homogeneous precipitation of the inorganic phases as soon as the preferential nucleation site on the organic macromolecules were occupied by a calcium ion.
Larger empty spaces than those observed in the control cement (Fig. 4a) are present in CDHA/Si-HA (Fig. 5b), which indicates that changes in the microstructure and microporosity of the cement were induced by the hydrogel. In order to obtain a clear evaluation of these changes, the microporosity of CDHA/Si-HA was quantified from the 3D reconstructions and compared with those of CDHA/gelatin and the control cement. As presented in Table 1, the incorporation of Si-HA into the CDHA increased the amount of microporosity and the mean pore size of the cement. Given the very weak concentration of Si in the hydrogel (Si characteristic peaks not detectable with EDS. it is likely that these effects were mainly caused by the presence of HA). The increase in the amount and size of pores induced by HA was demonstrated in a previous study by Babo et al., through the 3D high resolution micro-CT analysis of cement incorporated with HA microparticles.46 Although the range of pore size reported in Babo et al.'s work (>6 μm) is different than the one investigated in our study and that the authors carried out analyses on dry samples, the results in Table 1 show that HA also affects the microporosity of the cement in a hydrated state and at a lower length scale. Another important result shown in Table 1 is that the microporosity of the cement varied differently depending on the chemical nature of the macromolecule incorporated into its formulation. At the concentration of the hydrogel used in our work, the effects on the pore structure of CDHA induced by Si-HA are less important than those induced by gelatin, especially for the modification of the pore size.
The microstructures of anhydrous CDHA and CDHA–polymer mixtures were observed using SEM (Fig. 7) and indicates the presence of microporosity at the surface. In all cases, a visual inspection of the pores reveals that these might be smaller in size than in the case of hydrous samples (Fig. 1–5). The needle-like CDHA particles can also be observed in the anhydrous compounds but the organization of the mineral phase is clearly different than in the case of hydrous samples. In the CDHA/gelatin mixture, the cement clustered into spherical-like agglomerates, whereas plate-like CDHA crystals were observed in CDHA/Si-HA. Also, the microstructure of CDHA/Si-HA appears to be more compact (Fig. 7c) than those of CDHA/gelatin (Fig. 7b) and control cement (Fig. 7a), which is probably a consequence of the cohesive properties of HA.49
 | ||
| Fig. 7 (a) SEM micrographs of (a) CDHA (b) CDHA mixed with gelatin52 and (c) CDHA mixed with Si-HA. | ||
RT FIB tomography was further performed to evaluate the microstructure and microporosity of the anhydrous compounds in 3D. In many of the acquired FIB-SEM data, imaging artifacts related to shine-through or pore-back effects38 were observed. These artifacts originate from electrons that go through the pores and produce an image of the material belonging to the subsequent section(s). Instead of observing pores with a minimal intensity in the image (i.e. black regions), a contrast characteristic of the solid material appears. This induces a modification of the pore morphology during 3D reconstructions along the milling direction. Curtaining effects, which result from the difference between the FIB-sputtering rate at the pores and that at the surrounding material, were also observed in the acquired images. An example is shown in Fig. 8 illustrating a SEM micrograph of two pores within a CDHA/gelatin sample. The elongation of pores seen in the YZ plane (Fig. 8b) is a consequence of the shine through. Curtaining effects were also observed when visualizing the pore's morphology in depth (Fig. 8b).
 | ||
| Fig. 8 (a) SEM micrograph in the XY plane showing two pores in the anhydrous CDHA/gelatin sample. (b) The same pores observed in the YZ plane after reconstructing a 3D volume from the FIB-SEM data of the CDHA/gelatin sample. | ||
An empirical approach to address the aforementioned artifacts in porous materials is by simply filling the pore's space using polymeric materials such as resins.50–53 Otherwise, minimizing or accounting for this effect in non-hydrated porous samples requires the combination of extensive image processing techniques and combination of different imaging modes simultaneously.54 In hydrogels, or other highly hydrated porous samples, the filler material is naturally water. Preserving the hydrated state through a cryo-fixation process can be optimal for avoiding these imaging artifacts. Cryo-FIB tomography is therefore advantageous for the visualization of porous structures and enabled us to limit the “shined-through” effect during our cryo-FIB-SEM acquisitions.
3D reconstructed volumes obtained from serial FIB-SEM acquisitions (Fig. 9 and 10) were further used to evaluate the microporosity of CDHA and CDHA/polymer mixtures. Table 2 presents the calculated amount of microporosity and the mean pore size of CDHA–polymer mixtures, in comparison to those characteristics of the control CDHA.
 | ||
| Fig. 9 (a) 3D reconstruction of the anhydrous CDHA sample using the Dragonfly® software.39 (b) 3D pore mesh inside the reconstructed volume. | ||
 | ||
| Fig. 10 (a) 3D reconstructed volume of the anhydrous CDH/gelatin sample using the Dragonfly® software.39 (b) 3D pore mesh inside the CDHA/gelatin reconstructed volume. (c) 3D volume of anhydrous CDH/Si-HA and its (d) 3D corresponding pores mesh. | ||
| Microporosity (%) | Mean pore size (nm) | |
|---|---|---|
| Anh CDHA | 12 ± 3 | 82 ± 12 |
| Anh CDHA/gelatin | 13 ± 4 | 110 ± 15 |
| Anh CDHA/Si-HA | 23 ± 5 | 145 ± 23 |
Firstly, both the CDHA/polymer compounds exhibited a mean pore size slightly higher than the one observed for CDHA. Secondly, the results presented in Tables 1 and 2 suggest that the microporosity is significantly affected by dehydration. Indeed, all the anhydrous compounds exhibit a mean amount of porosity and a mean pore size lower than those at the hydrated state.
Regarding the microporosity, gelatin did not have a significant influence on the amount of microporosity of the cement at the anhydrous state whereas an important increase was observed in the case of the hydrous mixture. It has been reported that the incorporation of gelatin to CPC may modify the amount of the cement porosity to a degree dependent on the gelatin contents.55 The total porosity has been shown to decrease when the amount of gelatin is lower than 5 wt% whereas the opposite effect occurs for greater gelatin concentrations.55 In our case, the unchanged amount of porosity could be related to the shrinkage of gelatin due to dehydration. By losing the liquid phase that fills the pores, after dehydration, the gelatin's internal porosity is reduced significantly. This results in a total amount of microporosity in the CDHA/gelatin close to that in CDHA. The gelatin induced however a modification of the CDHA microstructure which explains both the formation of CDHA/gelatin agglomerates (Fig. 7b) in the compound and the slight increase in the mean pore size (Table 2).
The comparison between the microporosity data obtained for CDHA and CDHA/Si-HA in both the anhydrous (Table 2) and hydrous states (Table 1) shows that the percentage factor by which Si-HA increases the porosity of the cement (∼1.8) is almost the same, regardless of the hydration level of the compound. It is therefore reasonable to assume that the difference in microporosity between hydrous and anhydrous CDHA/Si-HA compounds is mainly caused by the microstructural changes of the cement due to dehydration.
Conclusions
The structural and chemical analyses of biomaterials consisting of CDHA/hydrogel composites were performed using cryo and RT-FIB tomography. The results of this work show that both the composition of the hydrogel phase and the hydration state of CPC biomaterials during observations have a significant effect on their microstructure.As BSE imaging provided a better difference in terms of the contrast level, it allowed for the accurate quantification of the different phases and enabled us to evaluate their distribution within the CPC/hydrogel composites. 3D FIB-SEM reconstructions showed that, in CDHA/gelatin compounds, a part of gelatin exhibited a separate hydrogel phase within the mixture. Given that gelatin/α-TCP mixing was performed at ambient temperature, the gelatin partially formed a 3D network prior to mixing. Further to mixing, the regions of CDHA in contact with gelatin mainly resulted from the precipitation of CPC crystals on the organic matrix. In CDHA/Si-HA, even though Si-HA cross-linked prior to mixing it with α-TCP, it did not form a separate phase inside the hydrous CDHA/Si-HA. Si-HA was completely mixed with a cement and acted as a supporting substance for the precipitation of a newly formed hydroxyapatite phase.
The analysis of 3D reconstructions of the compounds after dehydration is consistent with a possible collapse of gelatin together with full adsorption on the CDHA surface. These observations would thus lead to wrongful interpretations of the phase distribution and structure of the compounds and are characteristic of the most common RT SEM analysis of biomaterials. CDHA/gelatin mixtures exhibited more sensitivity to hydration than CDHA/Si-HA and control cement as their mean pore size decreased by a factor of ∼9 in the dehydrated state. No significant difference between the microporosity of CDHA/gelatin and that of CDHA was observed in the case of dehydrated compounds. The incorporation of Si-HA into CDHA increased the mean pore size and the amount of porosity of the hydrated and dehydrated compounds by a similar factor (∼1.8). The difference in microporosity between hydrated and dehydrated CDHA/Si-HA is therefore believed to be a consequence of the effects of dehydration on the cement's structure.
All the differences observed between hydrous and anhydrous CPC/hydrogel composites, in terms of the phase distribution and microporosity, suggest that accurate structural characterization of highly hydrated biomaterials by electron microscopy requires the use of cryogenic temperatures, capable of preserving their hydrated state. Our results emphasize that an erroneous evaluation of the biomaterials’ properties is indeed obtained as a result of analysing a modified structure due to dehydration. Furthermore, by preserving water as a filler material inside porous samples through a cryo-fixation process, cryo-FIB-SEM enables us to avoid shine-through artifacts, thus providing a more realistic visualization of the 3D porous structure.
It is worth noting that plunge-freezing, as used in this work, can induce cryo-fixation artifacts such as the formation of ice crystals56 that can modify the internal structure of CPC/hydrogel scaffolds. These artifacts should be considered when investigating microstructural features or reporting microporosity measurements using cryo-SEM.57 Using techniques for the characterization of the crystalline structure in depth, such as micro-Raman spectrometry combined with FIB/SEM, the state of vitrification can be evaluated to optimize sample preparation.58 Sample preparation by HPF would certainly improve the vitrification state and will be explored in the future. Comparing the quantitative structural data obtained from the analysis of samples prepared by plunge-freezing under slush nitrogen, to the same samples cryo-fixed by high pressure freezing, will provide insight into the impact of the freezing method on the structural properties of biomaterials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. Abellan and M. Essani would like to acknowledge the financial support provided by the NExT initiative through the French National Research Agency (ANR) under the Programme d'Investissements d'Avenir (with reference ANR-16-IDEX-0007). The e-BRIDGE project also receives financial support from the Pays de la Loire region and Nantes Métropole. The FIB/SEM data were collected in the CIMEN Electron Microscopy Center in Nantes, funded by the French Contrat Plan État-Région and the European Regional Development Fund of Pays de la Loire.References
- G. F. de Grado, L. Keller, Y. Idoux-Gillet, Q. Wagner, A. Musset, N. Benkirane-Jessel, F. Bornert and D. Offner, J. Tissue Eng., 2018, 9, 1–18 CAS.
- B. Charbonnier, M. Hadida and D. Marchat, Acta Biomater., 2021, 121, 1–28 CrossRef CAS PubMed.
- Y. Du, J. L. Guo, J. Wang, A. G. Mikos and S. Zhang, Biomaterials, 2019, 218, 119334 CrossRef CAS PubMed.
- R. A. Mickiewicz, A. M. Mayes and D. Knaack, J. Biomed. Mater. Res., 2002, 61(4), 581–592 CrossRef CAS PubMed.
- J. Zhang, W. Liu, O. Gauthier, S. Sourice, P. Pilet, G. S. Rethore, K. Khairoun, J.-M. Bouler, F. Tancret and P. Weiss, Acta Biomater., 2016, 31, 326–338 CrossRef CAS PubMed.
- R. Zhang and P. X. Ma, Polym. Prepr., 2000, 41(2), 1618–1619 CAS.
- H. Mittelmeier and B. D. Katthagen, Z. Orthop., 1983, 121, 115 CrossRef CAS PubMed.
- J. T. Schantz, T. C. Lim, C. Ning, S. H. Teoh, K. C. Tan, S. C. Wang and D. W. Hutmacher, Oper. Neurosurg., 2006, 58, ONS-E176 CrossRef PubMed.
- H. Saijo, K. Igawa, Y. Kanno, Y. Mori, K. Kondo, K. Shimizu, S. Suzuki, D. Chikazu, M. Iino and M. Anzai, J. Artif. Organs, 2009, 12, 200–205 CrossRef PubMed.
- F. A. Probst, D. W. Hutmacher, D. F. Müller, H. G. Machens and J. T. Schantz, Calvarial reconstruction by customized bioactive implant, Handchir. Mikrochir. Plast. Chir., 2010, 42, 369–373 CrossRef CAS PubMed.
- L. Teo, S. H. Teoh, Y. Liu, L. Lim, B. Tan, J.-T. Schantz and L. L. Seah, Orbit, 2015, 34, 192–200 CrossRef PubMed.
- I. Lodoso-Torrecilla, J. J. J. P. van den Beucken and J. A. Jansen, Acta Biomater., 2021, 119, 1–12 CrossRef CAS PubMed.
- H. Moussi, P. Weiss, J. Le Bideau, H. Gautier and B. Charbonnier, Mater. Adv., 2022, 3, 6125–6141 RSC.
- A. Bigi, S. Panzavolta, L. Sturba, P. Torricelli, M. Fini and R. Giardino, J. Biomed. Sci., 2006, 78A, 739–745 CAS.
- S. J. Ding and M.-Y. Shie, Adv. Eng. Mater., 2011, 13, B246–B255 CrossRef.
- L. Schröter, F. Kaiser, S. Stein, U. Gbureck and A. Ignatius, Acta Biomater., 2020, 117, 1–20 CrossRef PubMed.
- M. C. Chang and R. DeLong, Dent. Mater., 2009, 25(2), 261–268 CrossRef CAS PubMed.
- P. Gao, H. Zhang, Y. Liu, B. Fan, X. Li, X. Xiao, P. Lan, M. Li, L. Geng, D. Liu, Y. Yuan, Q. Lian, J. Lu, Z. Guo and Z. Wang, Sci. Rep., 2016, 6(1), 14 CrossRef PubMed.
- T. Gerber, G. Holzhüter, W. Götz, V. Bienengräber, K. Henkel and E. Rumpel, Eur. J. Trauma, 2006, 32, 132–140 CrossRef.
- M. J. Coathup, K. A. Hing, S. Samizadeh, O. Chan, Y. S. Fang, C. Campion, T. Buckland and G. W. Blunn, J. Biomed. Mater. Res., Part A, 2012, 100(6), 1550–1555 CrossRef PubMed.
- R. F. De Godoy, S. Hutchens, C. Campion and G. Blunn, J. Mater. Sci., Mater. Med., 2015, 26, 54 CrossRef PubMed.
- L. Giannuzzi, D. Phifer, N. Giannuzzi and M. Capuano, J. Oral Maxillofac. Surg., 2007, 65(4), 737–747 CrossRef PubMed.
- D. M. Binkley and K. Grandfield, ACS Biomater. Sci. Eng., 2018, 4, 3678–3690 CrossRef CAS PubMed.
- K. Grandfield and H. Engqvist, Adv. Mater. Sci. Eng., 2012, 841961 Search PubMed.
- A. Schertel, N. Snaidero, H.-M. Han, T. Ruhwedel, M. Laue, M. Grabenbauer and W. Möbius, J. Struct. Biol., 2013, 184, 355–360 CrossRef CAS PubMed.
- D. Spehner, A. M. Steyere, L. Bertinettif, I. Orlova, L. Benoitg, K. Pernet-Gallayg, A. Schertel and P. Schultz, J. Struct. Biol., 2020, 211, 107528 CrossRef CAS PubMed.
- K. Flegeau, C. Toquet, G. Rethore, C. d’Arros, L. Messager, B. Halgand, D. Dupont, F. Autrusseau, J. Lesoeur, J. Veziers, P. Bordat, A. Bresin, J. Guicheux, V. Delplace, H. Gautier and P. Weiss, Adv. Healthcare Mater., 2020, 9(19), 2070068 CrossRef CAS.
- A. Bigi, P. Torricelli, M. Fini, B. Bracci, S. Panzavolta, L. Sturba and R. Giardino, Biomaterials, 2004, 27, 664–673 CAS.
- A. Bigi, S. Panzavolta and K. Rubini, Chem. Mater., 2004, 3740–3745 CrossRef CAS.
- A. Bigi, I. Cantelli, S. Panzavolta and K. Rubini, J. Appl. Biomater. Biomech., 2004, 81–87 CAS.
- S. Ahmadzadeh-Asl, S. Hesaraki and A. Zamanian, Adv. Appl. Ceram., 2011, 110, 6 Search PubMed.
- M. H. Alkhraisat, C. Rueda, F. T. Marino, J. Torres, L. B. Jerez, U. Gbureck and E. L. Cabarcos, Acta Biomater., 2009, 5, 3150–3156 CrossRef CAS PubMed.
- X. Cui, C. Huang, Z. Chen, M. Zhang, C. Liu, K. Su, J. Wang, L. Li, R. Wang, B. Li, D. Chen, C. Ruan, D. Wang, W. W. Lu and H. Pan, Bioactive Mater., 2021, 6, 3801–3811 CrossRef CAS PubMed.
- E. B. Montufar, T. Traykova, E. Schacht, L. Ambrosio, M. Santin, J. A. Planell and M. Pau Ginebra, J. Mater. Sci.: Mater. Med., 2009, 21, 863–869 CrossRef PubMed.
- B. J. Luyet and C. Kroener, The highest obtainable cooling velocities, Fourth Annual Meeting of the Biophysical Society, 1960, p. 31.
- M. J. Baker, T. T. Denton and C. Herr, Cryobiologie, 2013, 66, 43–46 CrossRef CAS PubMed.
- D. C. Joy and C. S. Joy, Micron, 1996, 27(3–4), 247–263 CrossRef.
- M. Salzer, T. Prill, A. Spettl, D. Jeulin, K. Schladitz and V. Schmidt, J. Microsc., 2015, 257, 23–30 CrossRef CAS PubMed.
- Object Research Systems (ORS) Inc., Dragonfly 3.6. Montreal, Canada, 2018. Software available at http://www.theobjects.com/dragonfly.
- B. Xu, Y. Ye and L. Nie, IEEE International Conference on Information and Automation, 2012, pp. 795–800 DOI:10.1109/ICInfA.6246927.
- C. Lanterne, S. Gueorguieva and P. Desbarats, Local Thickness Computation in 3D Meshes 679 and 3D Printability Assessment, 2016, pp. 287–292.
- L. Reimer, Scanning Electron Microscopy, Springer-Verlag, Berlin, 1998 CrossRef PubMed; J. Piños, V. Mikmeková and L. Frank, About the information depth of backscattered electron imaging, J. Microsc., 2017, 266(3), 335–342 CrossRef PubMed.
- L. Schröter, F. Kaiser, S. Stein, U. Gbureck and A. Ignatius, Acta Biomater., 2020, 117, 1–20 CrossRef PubMed.
- S.-H. Jeong, Y.-H. Koh, S.-W. Kim, J.-U. Park, H.-E. Kim and J. Song, Biomacromolecules, 2016, 17(3), 841–851 CrossRef CAS PubMed.
- K. Tadashi, K. Hyun-Min and K. Masakazu, Biomaterials, 2003, 24, 2161–2175 CrossRef PubMed.
- P. S. Babo, V. E. Santo, M. E. Gomes and R. L. Reis, Macromol. Biosci., 2016, 16(11), 1662–1677 CrossRef CAS PubMed.
- A. D-Escudero, M. Espanol, E. B. Montufar, G. Di Pompo, G. Ciapetti, N. Baldini and M.-P. Ginebra, Tissue Eng., Part C, 2017, 118–124 CrossRef PubMed.
- A. Bigi, B. Bracci and S. Panzavolta, Biomaterials, 2004, 25, 2893–2899 CrossRef CAS PubMed.
- M. H. Alkhraisat, C. Rueda, F. T. Mariño, J. Torres, L. B. Jerez, U. Gbureck and E. L. Cabarcos, Acta Biomater., 2009, 5(8), 3150–3156 CrossRef CAS PubMed , Epub 2009 Apr 7. PMID: 1940987.
- H. Moussi, B. Charbonnier, T. Rouillon, H. Terrisse, J. Véziers, M. Dutilleul, A. Schaefer, L. Chabaud, C. Lépine, M. Essani, P. Abellan, O. Gauthier, F. Tancret, J. Le Bideau, H. Gautier and V. Geoffroy, 2022, Submitted.
- R. Hegge, P. Moroni, P. Trinke, B. Bensmann, R. Hanke-Rauschenbach, S. Thiele and S. Vierrath, J. Power Sources, 2018, 393, 62–66 CrossRef.
- L. Holzer, B. Muench, M. Wegmann, P. Gasser and R. J. Flatt, J Am. Ceram. Soc., 2006, 89, 2577–2585 CrossRef CAS.
- H. Roberge, P. Moreau, E. Couallier and P. Abellan, J. Membr. Sci., 2022, 653, 120530 CrossRef CAS.
- T. Terao, G. Inoue, M. Kawase, N. Kubo, M. Yamaguchi, K. Yokoyama, T. Tokunaga, K. Shinohara, Y. Hara and T. Hara, J. Power Sources, 2017, 347, 108–113 CrossRef CAS.
- Y. Fujishiro, K. Takahashi and T. Saito, J. Biomed. Mater. Res., 2001, 54, 525–530 CrossRef CAS PubMed.
- R. Aston, K. Sewell, T. Klein, G. Lawrie and L. Grøndahl, Eur. Polym. J., 2016, 82, 1–15 CrossRef CAS.
- Z. Kaberova, E. Karpushkin, M. Nevoralová, M. Vetrík, M. Šlouf and M. D-Smrcková, Polymers, 2020, 12, 578 CrossRef CAS PubMed.
- M. Essani, J.-Y. Mevellec, B. Charbonnier, P. Moreau, H. Moussi, P. Weiss, J. Le Bideau, M. Bayle, B. Humbert1 and P. Abellan, Anal. Chem., 2022, 94(23), 8120–8125 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |