
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Configuration dependent photovoltaic properties of cyclometalated heavy metal complexes for organic solar cells†
Tianjian
Yang
,
Aihua
Zhou
,
Yinming
He
,
Zhichao
Yao
,
Xiaochen
Song
,
Xianwang
Tao
and
Youtian
Tao
 *
*
Key Lab for Flexible Electronics and Institute of Advanced Materials, Nanjing Tech University, 30 South Puzhu Road, Nanjing, 211816, China. E-mail: iamyttao@njtech.edu.cn
First published on 5th December 2022
Abstract
Cyclometalated heavy metal complexes as a new class of photovoltaic materials have attracted specific attention owing to their triplet feature, which is expected to be beneficial for longer exciton diffusion lengths and more efficient exciton dissociation in organic solar cells (OSCs). In this work, based on the organic linear ligand of 2-(5′′-tert-butyl-[2,2′:5′,2′′-terthiophen]-5-yl)benzo[d]thiazole (tTBz), three cyclometalated heavy metal complexes including square-planar heteroleptic Pt(II) complex tTBzPt, octahedral heteroleptic Ir(III) complex tTBzIr and octahedral homoleptic tTBz3Ir are designed as electron donor materials for OSCs. Notably, the influences of molecular spatial configuration on the optoelectronic properties and photovoltaic performances are systematically investigated. tTBz and tTBzPt show only fluorescence emission with lifetime <1 ns, while tTBzIr and tTBz3Ir exhibit a phosphorescent triplet lifetime of 176 and 276 ns, respectively. The power conversion efficiencies (PCEs) follow the order of tTBz3Ir > tTBzIr > tTBzPt > tTBz, with values from 5.71, 3.73, 1.08 to ∼0% for PCBM and 7.97, 6.75, 1.40 to 0.47% for Y6 acceptor based devices, respectively. The more significant 3D geometry of tTBz3Ir demonstrates the best photovoltaic performance owing to comprehensive factors of enhanced absorption, extended exciton lifetime, increased charge transport and optimized film morphologies. Our work not only promoted the PCE of triplet cyclometalated heavy-metal complexes from previously reported <4% to a higher level of ∼8%, but also illustrates the significance of structural geometries for the design of new organic photovoltaic materials.
Introduction
Functional metal complexes have drawn considerable attention in many areas like optics, electronics, magnetic devices, catalysis, pharmaceutics, gas storage and separation, etc.1–5 Compared to pure organic molecules, one of the unique features of metal complexes is their highly modular and diversified molecular spatial configurations. For example, by synergistically tuning the chemical structures of organic ligands and the electronic configurations of metal centers, various mono-nuclear metal complexes with linear, (square) planar, tetrahedral, octahedral and dodecahedral geometries, etc, or even multi-nuclear complexes with multi-dimensional spatial structures could be appropriately designed.6–9 Great success has been achieved by using cyclometalated six-coordinate octahedral Ir(III) complexes and four-coordinate planar Pt(II) complexes as phosphorescent triplet emitters in organic light-emitting diodes (OLEDs) from both academia and industry.10,11 Phosphorescent heavy metal complexes can convert singlet excitons into triplets through the efficient intersystem crossing (ISC) process, due to the strong spin–orbital coupling (SOC) effect induced by heavy metal centers, and thus to utilize 100% excitons in OLEDs.12 Generally, electroluminescence efficiencies are highly determined by the cyclometalated main ligand, regardless of the structural geometries of either bis-heteroleptic or tris-homoleptic Ir complexes.13 Different from OLEDs, the investigation of cyclometalated heavy metal complex-based triplet photovoltaic materials in organic solar cells (OSCs) lags far behind.14In typical organic solar cells, the mechanism involves four steps of light absorption and exciton generation at the photoactive donor (D) and/or acceptor (A) materials, exciton diffusion into D/A interfaces, exciton dissociation (charge separation) at the D/A interface, charge transport and extraction.15 Thus, the photovoltaic performance is determined by many factors. For example, the fundamental optoelectronic properties originated from the chemical structures of photoactive materials, like optical absorption capability, energy levels and charge carrier mobilities of the donor and acceptor materials.16,17 Besides, device structures and film morphologies could also significantly influence the efficiency of OSCs.18,19 In addition, one of the intrinsic issues is to increase the average exciton diffusion lengths (LD), since longer exciton diffusion lengths in OSCs are expected to facilitate exciton dissociation and reduce charge recombination.20–23 From the formula of 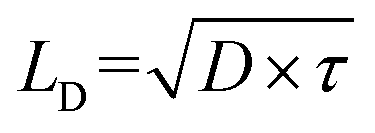 , D indicates the exciton diffusion coefficient, which is related to the charge mobilities, and τ is the exciton lifetime.14,24 Therefore, triplet excitons with 2–3 orders longer lifetime than singlet excitons are estimated to possess extended LD.25 On the other hand, previous reports revealed that similarly in OLEDs, non-geminate recombination in OSCs should also lead to the formation of 3CT (charge transfer) and 1CT states with a ratio of 3
, D indicates the exciton diffusion coefficient, which is related to the charge mobilities, and τ is the exciton lifetime.14,24 Therefore, triplet excitons with 2–3 orders longer lifetime than singlet excitons are estimated to possess extended LD.25 On the other hand, previous reports revealed that similarly in OLEDs, non-geminate recombination in OSCs should also lead to the formation of 3CT (charge transfer) and 1CT states with a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Gelinas et al. showed that a sufficiently high triplet state of the molecule can significantly reduce the thermalization of 3CT states, making them more viable for further charge dissociation.26
:1. Gelinas et al. showed that a sufficiently high triplet state of the molecule can significantly reduce the thermalization of 3CT states, making them more viable for further charge dissociation.26
In current state-of-the-art organic photovoltaics, triplet states and charge transfer triplet states have only rarely been considered to improve device efficiencies.14,27 One of the big challenges is to explore triplet photovoltaic materials that synergistically bear with sufficiently long triplet lifetime, appropriately high triplet energy levels, broad and strong light absorption and favorable charge carrier mobilities.24,25,28–31 It is noted that cyclometalated Ir or Pt complexes embedded random terpolymer donors or acceptors demonstrated a high PCE up to 17.2%,32–34 which is superior to those of metal-free organic copolymers. Unfortunately, no triplet feature was observed, and the enhanced PCE was mainly contributed from the improvement of morphology with reduced aggregation and crystalline behavior.35–37 Hence, it is more appropriate to study small molecular triplet heavy metal complexes to better understand the role and mechanism of triplet excitons in OSCs. Unfortunately, the maximum PCE for previously reported small-molecule triplet heavy metal complexes has been limited to less than 4%, with a short circuit current density (JSC) below 10 mA cm−2 and a fill factor (FF) < 0.50.24
Based on the above issues, we designed three heavy metal Pt or Ir complexes with different structural geometries by involving 2-(5′′-tert-butyl-[2,2′:5′,2′′-terthiophen]-5-yl)benzo[d]thiazole (tTBz) as the same cyclometalated main ligand. Of these, the 4-coordinate heteroleptic Pt complex tTBzPt which consists of one bidentate tTBz as the cyclometalated C^N ligand and 2,2,6,6-tetramethyl-3,5-heptanedione (thd) as the ancillary ligand shows a highly planar structure. Furthermore, the two 6-coordinate Ir complex exhibits three-dimensional octahedral geometries. The heteroleptic tTBzIr includes bis-tTBz as C^N ligands and acetylacetonate (acac) as the ancillary ligand, while the homoleptic tTBz3Ir consists of tris-identical tTBz ligands. On the basis of the linear tTBz ligand, upon cyclometalation, strong extra metal-to-ligand charge transfer (MLCT) transition absorption appears. When employed as electron donor materials for BHJ OSCs, the absorption, charge transport, exciton dissociation and the PCE demonstrated a gradient improvement from linear tTBz, planar tTBzPt, octahedral heteroleptic tTBzIr to octahedral homoleptic tTBz3Ir for both fullerene and non-fullerene acceptor based devices. tTBz3Ir with the most optimized configuration and longest triplet lifetime achieved the highest PCE, with values of 5.71 and 7.97% for PCBM and Y6 devices, respectively. To our knowledge, this is the first report to systematically discuss the relationships between molecular spatial configurations and photovoltaic properties in metal complex-based OSCs, which could provide guidelines for the design of new organic photovoltaic materials.
Results and discussion
Synthesis and characterization
The chemical structures for the related materials in this study are shown in Fig. 1. The synthetic details can be found in the ESI.† As shown in Scheme S1 (ESI†), the cyclometalated tTBz ligand, named 2-(5′′-tert-butyl-[2,2′:5′,2′′-terthiophen]-5-yl)benzo[d]thiazole was synthesized through a step of Suzuki coupling reaction. The modest conjugation length of the tTBz ligand could ensure certain light absorption and charge transport properties of the metal complexes. The Pt complex tTBzPt was obtained according to literature-reported procedures for heteroleptic Pt or Ir complexes.38 First, the tTBz ligand was directly coordinated with K2PtCl4 to form the cyclometalated Pt(II) μ-chloride-bridged dimer. Then the relevant dimer was reacted with 2,2,6,6-tetramethyl-3,5-heptanedione (thd) to achieve the final heteroleptic tTBzPt. Tert-Butyl substituted thd was selected into the planar skeleton of tTBzPt to reduce the rigidity of the compound and thus to increase the solubility, making it suitable for device fabrication and also appropriate to inhibit the adverse aggregation in square-planar Pt complexes. For the two six-coordinated Ir complexes, it is inappropriate to obtain the Ir(III) μ-chloride-bridged dimer by direct coordination between the tTBz ligand and the Ir center due to the large ligand size. Therefore, both tTBzIr and tTBz3Ir were prepared through the post coupling procedure starting from the bromo-substituted Ir complex of bis(2-(5-bromothiophen-2-yl)benzo[d]thiazole) iridium (pentane-2,4-dione) or tris(2-(5-bromothiophen-2-yl)benzo[d]thiazole) Ir (III) with 2-(5′-(tert-butyl)-[2,2′-bithiophen]-5-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane. The chemical structures of the four compounds were confirmed by 1H nuclear magnetic resonance (1H NMR) (Fig. S1–S4, ESI†), matrix-assisted laser desorption/ionization time-of-flight (MALDITOF) (Fig. S5–S8, ESI†) and elemental analysis. In addition, by performing thermogravimetric analysis (TGA) (Fig. S9, ESI†), the decomposition temperature (Td, represents for a 5% weight loss) was achieved to be 347, 273, 347 and 392 °C for tTBz, tTBzPt, tTBzIr and tTBz3Ir, respectively. Notably, the tris-homoleptic Ir complexes demonstrated the highest thermal stability.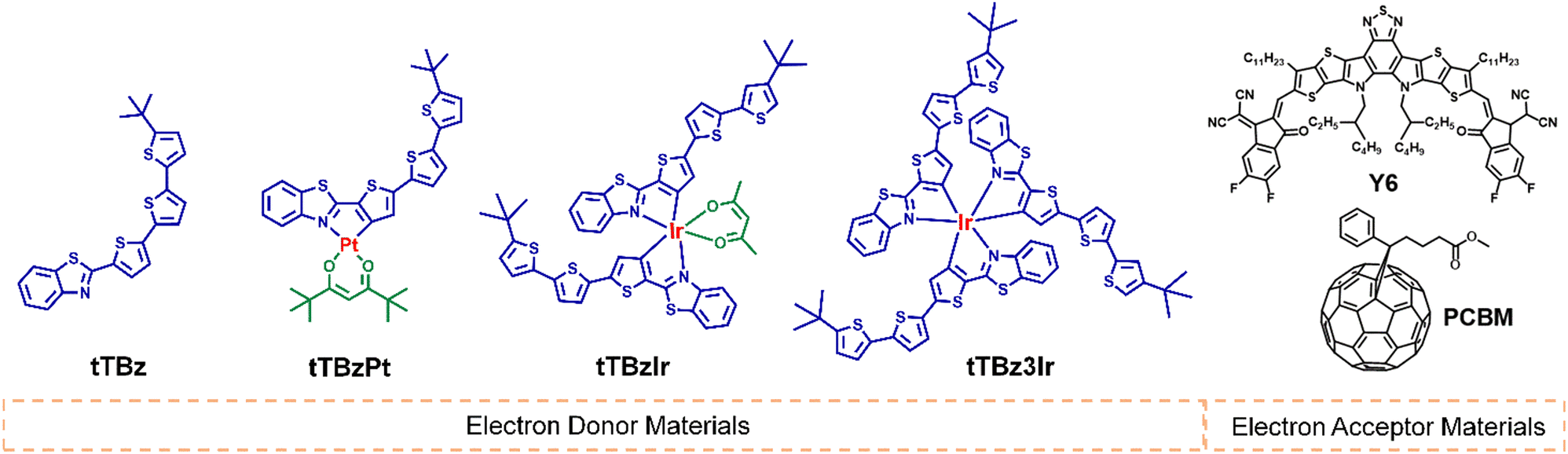 | ||
| Fig. 1 Chemical structures for related donor and acceptor materials in this work. | ||
Molecular geometries and optoelectronic properties
To explore the specific molecular configuration of the cyclometalated complexes, molecular geometry studies were carried out using density functional theory (DFT) at the B3LYP/6-31G (d) level with the tert-butyl substituents replaced by short methyl groups. As shown in Fig. 2a, the conjugated backbone of the organic tTBz ligand is considerably planar, showing a rather linear molecular skeleton. The Pt(II) complex tTBzPt displayed a rather planar geometry ranging from the backbone of the cyclometalating bidentate ligand, the metal center to the ancillary ligand. On the other hand, the Ir(III) center allows for 6-coordination, thus octahedral configurations with three-dimensional (3D) asymmetric molecular structures are allowed to form in tTBzIr and tTBz3Ir. Such spatially twisted geometries for the cyclometalated Ir complexes presumably favored not only inhibiting triplet–triplet annihilation for enhanced triplet features but also reduced intermolecular ligand–ligand and/or metal–metal interactions for facilitated phase separation in blend films, which would be more beneficial for PCE increment than the planar Pt complex in OSCs.25,39,40 More specifically, heteroleptic tTBzIr includes two bidentate tTBz as cyclometalated C^N ligands and acac as an ancillary ligand, while the homoleptic tTBz3Ir comprises three identical C^N ligands. Compared with the acac ligand in tTBzIr, the additional large conjugated tTBz ligand in tTBz3Ir is beneficial to enhance the light absorbing and charge transporting abilities. Moreover, homoleptic tTBz3Ir showed a more significant 3D structure than the heteroleptic tTBzIr, which might cause differentiated self-aggregation characteristics as well as miscibility behavior when blended with electron acceptors, and thus to affect the active-layer film morphologies and photovoltaic performances.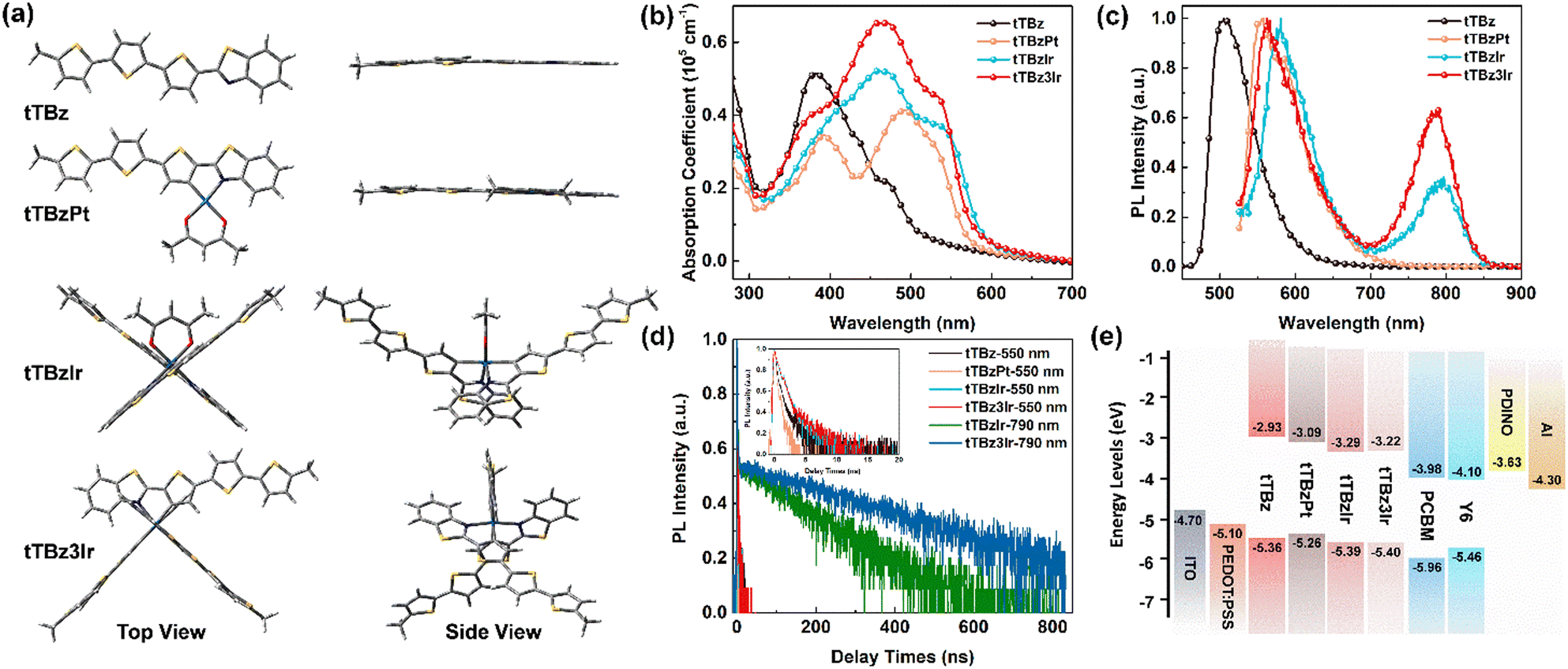 | ||
| Fig. 2 (a) Simulated chemical geometries the compounds; (b) UV-Vis absorption coefficient spectra in thin films; (c) normalized PL spectra of CHCl3 (10−5 M) and (d) corresponding transient PL decay curves recorded at 550 nm and 790 nm; (e) energy diagrams of the materials used in this work. | ||
The UV-Visible coefficient absorption spectra of tTBz, tTBzPt, tTBzIr and tTBz3Ir in the film state are shown in Fig. 2b. The corresponding absorption maximum and their extinction coefficients are summarized in Table 1. From Fig. 2b, tTBz presented obvious absorption bands from 250–500 nm, the short wavelength bands could be attributed from the typical n–π* and π–π* transition for aromatic rings, while the longer wavelength absorption with a peak located at 381 nm is assigned for intramolecular charge transfer (ICT) transition, and a further longer wavelength with a shoulder peak at 469 nm is estimated from the absorption of intermolecular interactions (aggregation) due to the highly planar structure for the ligand. After coordination with heavy-metal centers, all tTBzPt, tTBzIr and tTBz3Ir exhibited obviously enhanced absorption profiles at longer-wavelength regions. The greater increase in tTBz ligand units, the higher the absorption intensities observed for ligand–ligand (L–L) interactions at ∼460 nm. For example, the extinction coefficients were enhanced from ∼0.2 × 105 cm−1 for tTBz to 0.53 × 105 and 0.66 × 105 cm−1 for tTBzIr and tTBz3Ir, respectively. Among all four compounds, owing to the most delocalized conjugated system in the planar Pt complex of tTBzPt, a red-shifted absorption peak at 491 nm was detected, which is potentially ascribed to the ligand-to-metal charge transfer (LMCT) transition.41,42 Besides, compared with the organic tTBz ligand, the new absorption bands with shoulder peaks at around 535 nm for the three heavy metal complexes are assigned for singlet state metal-to-ligand-charge-transfer (1MLCT) transitions. Similarly, the 1MLCT absorption intensity highly relies on the number of ligands, tTBz3Ir with three identical tTBz ligands showing the strongest absorption, followed by tTBzIr with bis-tTBz ligands. The remarkably improved light-absorption within the visible region upon coordination with the heavy metal center, suggesting the stronger solar light harvesting ability for heavy metal complexes compared with the tTBz ligand, particularly for the tris-tTBz3Ir, as 43% of the total energy in the solar spectrum was concentrated on the visible region (400–700 nm), with only 5% for the ultra-violet region (300–400 nm).43
| λ abs (nm)/(105 cm−1) | λ PL (nm) | τ (ns) | τ (ns) | E g (eV) | E HOMO/ELUMO (eV) | μ h (cm2 V−1 s−1) | T d (°C) | |
|---|---|---|---|---|---|---|---|---|
| a Absorption peak (λabs) and corresponding extinction coefficients in the film state. b Emission wavelength (λPL) in CHCl3. c Average PL decay lifetime recorded at 550 nm with an excitation of 460 nm at room temperature. d Average PL decay lifetime recorded at 780 nm with an excitation of 460 nm at room temperature. e Measured from hole-only devices fitted by the SCLC method. | ||||||||
| tTBz | 381/0.51, 469/0.22 | 505 | 0.51 | — | 2.43 | −5.36/−2.93 | 4.08 × 10−6 | 347 |
| tTBzPt | 391/0.34, 491/0.41, 529/0.32 | 557 | 0.25 | — | 2.17 | −5.26/−3.09 | 3.72 × 10−5 | 273 |
| tTBzIr | 389/0.36, 460/0.53, 534/0.37 | 581, 791 | 0.78 | 116 | 2.10 | −5.39/−3.29 | 5.96 × 10−5 | 347 |
| tTBz3Ir | 381/0.41, 460/0.66, 535/0.45 | 564, 784 | 0.88 | 276 | 2.18 | −5.40/−3.22 | 8.56 × 10−5 | 392 |
On the other hand, despite encouraging achievements being made on the enhancement of absorption intensity for the heavy-metal complex, the absorption feature was only limited within 600 nm. Therefore, when applied as photoactive donor materials, the combination with the recently reported non-fullerene Y6 acceptor tends to be a more appropriate alternate than previously used typical fullerene acceptors like PCBM. PCBM exhibited very weak absorption in the range of 300–700 nm, while Y6 demonstrated a complementary absorption profile with heavy metal complexes in the wavelength of 600–1000 nm (Fig. S10, ESI†). Therefore, an enhanced Jsc of relevant OSCs is expected to be achieved in Y6-based active layer blends than PCBM due to the better coverage of the solar spectrum, as will be discussed below.
The photoluminescence (PL) spectra of the compounds in dilute CHCl3 solution are depicted in Fig. 2c, and the corresponding transient PL decay curves are displayed in Fig. 2d. Intense fluorescence emission peaking at 505 nm with a mono-modal profile was observed for the organic tTBz ligand in the range from 450–650 nm. Similar mono-modal fluorescence from 500–700 nm with emission maxima at 557 nm was determined for the square planar Pt complex tTBzPt. From time-resolved transient photoluminescence measurements, the PL lifetimes for both tTBz ligand and tTBzPt were measured at 0.51 and 0.25 ns, respectively, indicating their singlet feature. However, multi-modal emission profiles were measured for the two octahedral Ir complexes. The maximum emission peak at 564 nm for tTBz3Ir and 581 nm for tTBzIr are possibly singlet ligand centered (1LC) and/or 1MLCT dominated fluorescence, which could be conjectured by their <1 ns lifetime (Table 1). The slightly blue-shifted emission for homoleptic tTBz3Ir than heteroleptic tTBzIr is consistent with their similar shifts for 1MLCT absorption. Further longer wavelength emission bands from 700–850 nm with relatively lower intensity could only be observed for the two octahedral Ir complexes, the emission peak was measured at 791 nm for tTBzIr and 784 nm for tTBz3Ir, respectively, which could be assigned to the triplet-dominated phosphorescence according to the obviously prolonged lifetime of 116 and 276 ns (taken at 790 nm). It should be noted that the cyclometalated heavy-metal complex-based emitters in OLEDs normally exhibit an intense phosphorescence feature with completely suppressed 1LC fluorescence emission.44 However, the corresponding phenomena are quite different for cyclometalated heavy-metal complex-based photovoltaic materials. The incomplete eliminated singlet fluorescence and inadequate triplet characteristics with weak or undetected phosphorescence at lower energy were possibly ascribed to the reduced ISC rate induced by the rather large conjugated size of the tTBz ligand in the corresponding heavy metal complexes.25,45,46 In addition, partially influenced by the triplet–triplet annihilation (TTA) and aggregation caused quenching (ACQ), an absence of triplet feature was the result in the highly planar Pt complex tTBzPt.47,48 In contrast, the two Ir complexes with twisted and 3D molecular configurations commendably avoid such detriments and thereby room-temperature triplet-state phosphorescence emission as well as significantly long lifetimes could be observed. The successful utilization of triplet excitons in cyclometalated Ir complexes, especially for the homoleptic compound tTBz3Ir with increased phosphorescent intensity and longer lifetime than tTBzIr, was beneficial to promote exciton dissociation and suppress charge recombination in BHJ OSCs, which might promote the PCE improvement in the relevant photovoltaic devices.25
The electrochemical properties were characterized by cyclic voltammetry (CV), and the corresponding curves are plotted in Fig. S11 (ESI†). The energy levels of the highest occupied molecular orbitals (HOMO) were estimated from the onsets of oxidation curves against ferrocenium/ferrocene (FeCp2+/0). Besides, the energy levels of the lowest unoccupied molecular orbitals (LUMO) were calculated according to the equation of ELUMO = −(EHOMO + Eg) eV, where Eg represents for the optical bandgap calculated from the absorption edge of the film state UV-Vis spectra. As a result, the EHOMO/ELUMO values of tTBz, tTBzPt, tTBzIr and tTBz3Ir were estimated to be −5.36/−2.93 eV, −5.26/−3.09 eV, −5.39/−3.29 eV and −5.40/−3.22 eV, respectively. The energy level alignments of the materials used in this work are illustrated in Fig. 2e. Compared to the organic ligand tTBz, the deeper LUMO energy levels for cyclometalated heavy-metal complexes were enabled by the reduced Eg values owing to the existence of the MLCT absorption band located in the bathochromic region. Moreover, the heteroleptic and homoleptic Ir complexes exhibited similarly low-lying EHOMO and ELUMO than the Pt-analogue, which potentially favors a high open-circuit voltage (Voc) when coupling with related acceptors in OSCs.
As mentioned above, one of the main shortcomings of cyclometalated heavy-metal complex-based photovoltaic materials that limited the device performance was their insufficient charge transporting abilities. In this work, by using the space charge limited current (SCLC) method (Fig. S12, ESI†),49 the hole mobilities of tTBz, tTBzPt, tTBzIr and tTBz3Ir were calculated to be 4.08 × 10−6, 3.72 × 10−5, 5.96 × 10−5 and 8.56 × 10−5 cm2 V−1 s−1, respectively. The hole mobility is highly correlated with the molecular configuration, and follows on an order of linear tTBz < square-planar Pt complex < heteroleptic octahedral Ir complex < homoleptic octahedral Ir complex. It is shown that, by gradually increasing the numbers of cyclometalated tTBz ligands, a more efficient hole-transporting process could be achieved in heavy-metal complexes. Therefore, besides the conjugated ligands containing multiple aromatic heterocycles, we speculate that the charge transport channel also occurred through the metal center. tTBz3Ir employing more active ligands provides supplementary charge transporting channels in terms of both quantity and dimensionality, which provides a practical design strategy towards high-performance organometallic photovoltaic materials.
Photovoltaic performance
The photovoltaic properties of the three cyclometalated heavy-metal complexes and the corresponding organic ligand tTBz were systematically investigated by fabricating the solution-processed BHJ OSCs with a conventional device structure of (ITO)/poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS)/donor:(PCBM or Y6)/N,N-dioxide of bis(N,N′-dimethylaminopropyl) perylene diimide (PDINO)/Al, where the thin layers of the anode and cathode interface materials of PEDOT:PSS and PDINO were used to facilitate the hole and electron extraction, respectively. The desirable devices were carefully optimized by changing the donor:acceptor weight ratio and thickness for the active layers (Table S1–S6, ESI†), while other extra treatments such as solvent additives or post-annealing showed negative effects on the photovoltaic performance. The current–voltage (J–V) curves and key photovoltaic parameters of the optimal devices are summarized in Fig. 3a and Table 2, respectively.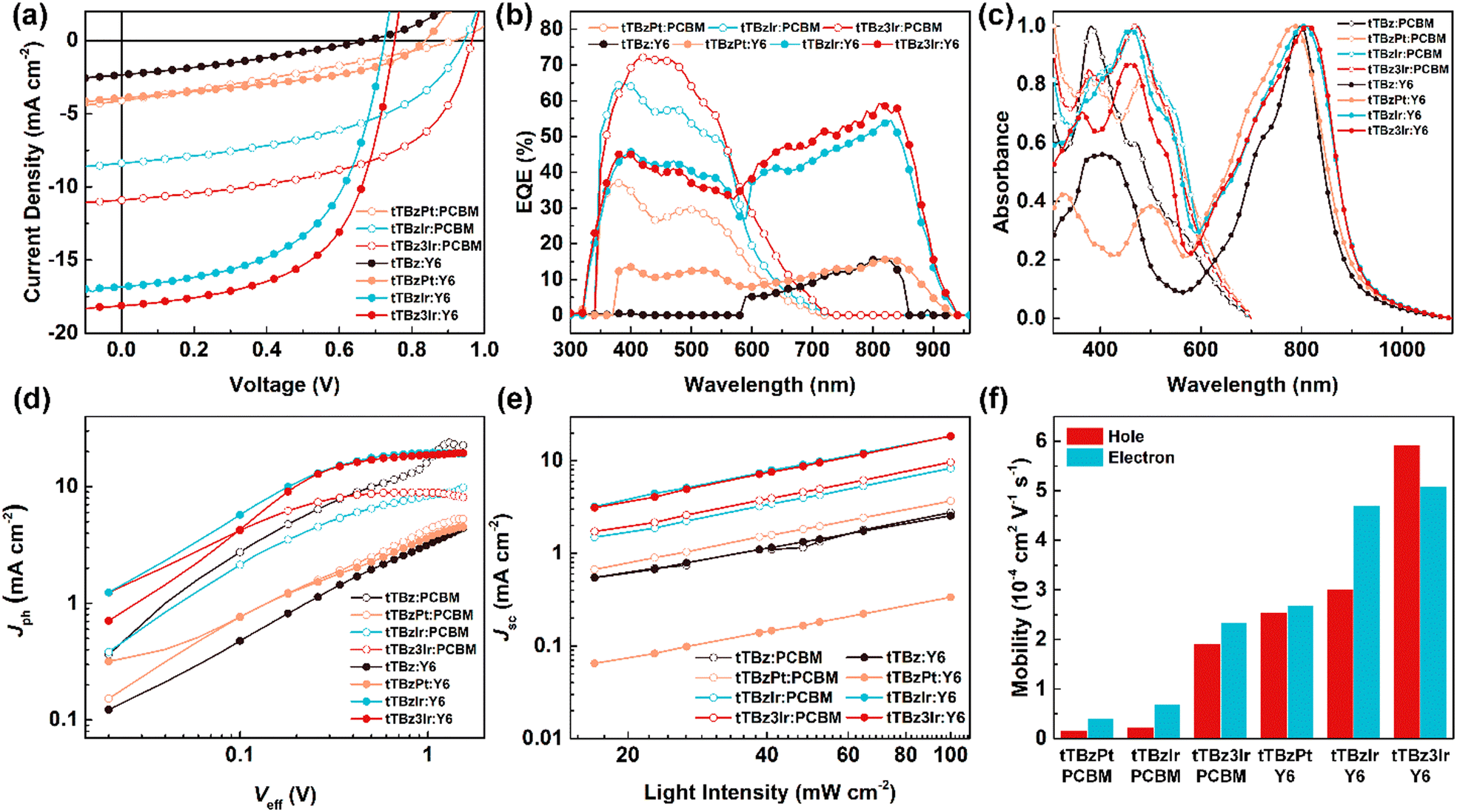 | ||
| Fig. 3 (a) J–V curves and (b) EQE curves of the OSCs based on tTBzPt:PCBM, tTBzIr:PCBM, tTBz3Ir:PCBM, tTBzPt:Y6, tTBzIr:Y6 and tTBz3Ir:Y6; (c) normalized UV-Vis absorption spectra for blended films; (d) exciton dissociation and (e) charge recombination behavior for the photovoltaic devices and (f) hole and electron mobilities for the related active layers. | ||
| Active layer | V oc (V) | J sc (mA cm−2) | FF | PCEa (%) | P (E,T) (%) | α | μ h/μe (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|---|
| a Statistical data obtained from over 12 devices. | |||||||
| tTBz:PCBM | no photovoltaic response | 0.4 | 0.90 | — | |||
| tTBzPt:PCBM | 0.90 | 4.16 | 0.29 | 1.08 (1.04 ± 0.04) | 20.3 | 0.96 | 1.58 × 10−5/4.07 × 10−5 |
| tTBzIr:PCBM | 0.93 | 8.39 | 0.48 | 3.73 (3.60 ± 0.13) | 54.9 | 0.98 | 2.34 × 10−5/6.84 × 10−5 |
| tTBz3Ir:PCBM | 0.96 | 10.91 | 0.55 | 5.71 (5.44 ± 0.27) | 83.8 | 0.99 | 1.91 × 10−4/2.34 × 10−4 |
| tTBz:Y6 | 0.67 | 2.34 | 0.30 | 0.47 (0.45 ± 0.02) | 17.5 | 0.88 | — |
| tTBzPt:Y6 | 0.84 | 3.92 | 0.42 | 1.40 (1.36 ± 0.04) | 27.8 | 0.93 | 2.55 × 10−4/4.70 × 10−4 |
| tTBzIr:Y6 | 0.73 | 16.84 | 0.55 | 6.75 (6.44 ± 0.31) | 61.0 | 0.98 | 3.01 × 10−4/2.68 × 10−4 |
| tTBz3Ir:Y6 | 0.75 | 18.12 | 0.59 | 7.97 (7.79 ± 0.18) | 88.4 | 1.00 | 5.92 × 10−4/5.10 × 10−4 |
For OSCs based on PCBM as the acceptor material, when employing tTBz as the donor, the photoelectric signal was too weak to be detected. In comparison, the obvious photovoltaic response could be achieved in the tTBzPt:PCBM-based devices with a PCE of 1.08%, a Voc of 0.90 V, a Jsc of 4.16 mA cm−2 and a FF of 0.29, respectively. Besides, when utilizing the cyclometalated Ir complexes as donor materials, further dramatically improved photovoltaic performances were attained. A maximum PCE of 3.73% was increased for TBzIr:PCBM blends, corresponding to a Voc of 0.93 V, Jsc of 8.39 mA cm−2 and FF of 0.48. The related values were further enhanced to 5.71%, 0.96 V, and 10.91 mA cm−2 with a FF of 0.55 for tTBz3Ir:PCBM based devices.
On the other hand, towards the purpose of broadening the absorption spectra of the active layers to overlap more with the solar spectrum, the replacement of acceptor materials from PCBM to Y6 resulted in encouragingly increased PCEs. Nonetheless, the photovoltaic properties of the tTBz:Y6- and tTBzPt:Y6-based OSCs were still at a very low-level, with PCEs of 0.47 and 1.40%, Voc of 0.67 and 0.84 V, Jsc of 2.34 and 3.92 mA cm−2, and FF of 0.30 and 0.42, respectively. However, for the cyclometalated Ir complex-based blends, both Jsc and FF were significantly enhanced through the replacement of acceptor materials. More specifically, devices based on tTBzIr:Y6 showed a remarkably increased PCE to 6.75%, with a Voc of 0.73 V, Jsc of 16.84 mA cm−2 and FF of 0.55, respectively. Further improved device performance to 7.97%, 0.75 V, 18.12 mA cm−2 and 0.59 was obtained for tTBz3Ir:Y6 blends, which showed the highest PCE and Jsc values for small-molecular heavy-metal complex-based OSCs. The relationships between molecular structural geometries and device PCEs demonstrated the same trends of linear tTBz < heteroleptic square-planar Pt complex < heteroleptic octahedral Ir complex < homoleptic octahedral Ir complex in both fullerene and non-fullerene based OSCs, indicating the superiority of metal complexes with an octahedral configuration by tris-identical cyclometalated ligands. To our knowledge, this is the first report to show the significance of structural geometries of organometallic compounds on the photovoltaic performance of OSCs.
It can be seen that for devices based on different tTBz derivatives in the two acceptor systems, the notably varied photovoltaic performances were undoubtedly attributed from the distinct differences in Jsc and FF. To further confirm their photovoltaic properties and explore the impact factors, the external quantum efficiency (EQE) spectra and the normalized UV-Vis absorption spectra of the blended active layers in OSCs were measured. As shown in Fig. 3b, in PCBM devices, the photoelectrical response was mainly observed from 350–600 nm. The EQE for tTBz3Ir:PCBM-based OSCs showed the highest intensity in most spectrum ranges, with the highest value over 70%, which is slightly higher than those in the tTBzIr:PCBM device. On the other hand, the tTBzPt:PCBM blends showed almost the same response range but sharply decreased EQE intensity with peak value <37%. These results agreed well with the Jsc values obtained from the J–V measurements. The EQE increments were also in accord with their improved light-absorbing abilities (Fig. 2b and 3c). After judicious selection of acceptor materials, OSCs based on Y6 possessed a supplementary photoelectric response in a wide range from 585 nm to 942 nm, which was mainly attributed from the strong optical absorption ability of the Y6 acceptor from 600 to 1000 nm. (Fig. 3c). Similarly, the two Ir complex containing devices exhibited notably higher EQE than the Pt complex and the ligand in the whole spectrum. Despite similarly broad and strong UV-Vis absorption spectra being achieved in both tTBz:Y6 and tTBzPt:Y6 blends, their extremely weakened responses in EQE spectra suggested that the fully absorbed photons were unsuccessfully converted into electrons. In particular, tTBz:Y6 based devices displayed nearly no photoelectric response from 300–580 nm. Therefore, the rather low detectable PCE originated from the photogeneration of free charges mostly by Y6. The broad and strong optical absorption but inferior photovoltaic performance especially in Y6-based devices indicated that the photon conversion process for tTBz and tTBzPt-based OSCs presumably suffered from insufficient exciton diffusion/dissociation and/or severe recombination process, which may be attributed to their inherent molecular structure for strong aggregation and the lack of triplet behavior.
Exciton dissociation and charge properties
To gain a deeper insight into the exciton dissociation properties of these cyclometalated heavy-metal complex-based OSCs, the photocurrent density (Jph) versus effective voltage (Veff) curves were plotted and are shown in Fig. 3d. In detail, Jph is defined as the difference between current density measured under illumination and dark conditions, respectively. Veff is defined as Veff = V0 − Vbias, where V0 is the voltage when Jph = 0 and Vbias represents the applied voltage bias. Generally, the Jph of the devices could reach a saturation region (Jsat) when at high Veff (≧2 V), suggesting almost all photogenerated excitons are dissociated into free carriers and extracted by electrodes with minimal recombination. In this case, the exciton dissociation probabilities (P(E,T)) of the corresponding OSCs could be evaluated using the formula P(E,T) = Jph/Jsat. The devices based on tTBz and tTBzPt exhibited relatively low P(E,T) values of 0.4%/17.5% and 20.3%/27.8% for PCBM/Y6 acceptors, respectively, implying extremely insufficient exciton dissociation in these devices. In comparison, more photo-induced excitons could be effectively dissociated in the cyclometalated Ir complex-based devices. Specifically, highly increased P(E,T) values to 54.9%, 61.0%, 83.8% and 88.4% were achieved for the tTBzIr:PCBM, tTBzIr:Y6, tTBz3Ir:PCBM and tTBz3Ir:Y6-based OSCs, respectively. The homoleptic Ir complex demonstrated a better exciton dissociation ability than the heteroleptic compound in both PCBM and Y6 based photovoltaic devices. The exciton dissociation behavior was in good agreement with their photovoltaic efficiencies, and the best P(E,T) value was obtained in tTBz3Ir:Y6 blends, which was also well consistent with its superior photovoltaic performance.Furthermore, the charge recombination mechanisms of devices were explored by measuring the dependence of Jsc on light intensity (Plight), and the corresponding Jsc − Plight curves were plotted in Fig. 3e. The relationships between Jsc and Plight could be represented using a simple formula of Jsc ∝ (Plight)α, where the exponential α indicates the degree of bimolecular recombination. Generally, if the bimolecular recombination in OSCs is regarded negligible, the α value is closer to 1. The α value of the devices for small-molecular donors tTBz, tTBzPt, tTBzIr and tTBz3Ir blended with fullerene/non-fullerene acceptors (PCBM/Y6) were obtained to be 0.90/0.88, 0.96/0.93, 0.98/0.98 and 0.99/1.00, respectively. It is found that devices based on tTBz suffered from serious bimolecular recombination, which was mainly ascribed to the existence of excessive self-aggregation behavior in blend films induced by the rigid and planar molecular structure of the linear organic ligand.25 In contrast, through coordination to metal centers they form complex structures, but intermolecular interaction (aggregation) could be greatly reduced particularly in the 3D octahedral geometries of cyclometalated Ir complexes, which induced the more drastically inhibited bimolecular recombination. In addition, the long-lived triplet excitons together with higher hole mobility for a potentially extended longer LD in tTBz3Ir containing devices, seemed to play a critical role in suppressing the charge recombination and improving the exciton dissociation in OSCs. Therefore, tTBz3Ir blended devices demonstrated the highest exciton dissociation probabilities and the least bimolecular recombination. To illustrate the influence of the molecular configuration of donor materials on the charge transport properties of the corresponding photoactive layers, the SCLC method was carried out. The hole- and electron-only devices with structures of ITO/PEDOT:PSS/active layer/Au and ITO/ZnO/active layer/PDINO/Al were fabricated to measure the hole (μh) and electron (μe) mobilities of the related active layers. The fitted J–V curves are shown in Fig. S13 (ESI†) and the histograms of the charge mobilities in different blend films are depicted in Fig. 3f. As shown, in the fullerene system, the μh/μe for the device based on tTBzPt, tTBzIr and tTBz3Ir were determined to be 1.58 × 10−5/4.07 × 10−5, 2.34 × 10−5/6.84 × 10−5 and 1.91 × 10−4/2.34 × 10−4 cm2 V−1 s−1, respectively. When employing Y6 as acceptor, both the hole and electron mobilities of all blends were significantly increased, with μh/μe of 2.55 × 10−4/4.70 × 10−4, 3.01 × 10−4/2.68 × 10−4 and 5.92 × 10−4/5.10 × 10−4 cm2 V−1 s−1, for tTBz, tTBzPt, tTBzIr and tTBz3Ir, respectively. Notably, regardless of the acceptor materials, the charge mobilities of the blend films showed an order of tTBz3Ir > tTBzIr > tTBzPt, which demonstrates the same trends with the hole mobility of neat metal complexes. It is noted that the multi-fold enhanced hole and electron mobilities in Y6 based devices compared with PCBM was not only attributed from the inherently better charge transport properties of Y6 but also better-matched film morphologies between the corresponding donor and acceptor materials, as will be illustrated below.
Blend morphology
The surface morphologies of the blended films were characterized through atomic force microscopy (AFM). As shown in Fig. 4a and i, the tTBz:PCBM blends exhibited a relatively rough surface with a root-mean-square (RMS) roughness value as high as 27.0 nm. Since fullerene acceptors are well-known for their amorphous aggregation characteristics,50 the sizeable aggregations in tTBz:PCBM blends were induced by the strong intermolecular interaction of tTBz owing to its highly planar structure, which was harmful to the generation and extraction of charges. Smoother films with reduced RMS to 2.93 nm were observed for the tTBz:Y6 blends. Despite the combination with Y6 resulting in a nanoscale phase-separated morphology, the excitons of tTBz still failed to dissociate efficiently at tTBz/Y6 interfaces, as illustrated in the EQE spectra and carrier dynamics part. Compared to the linear organic ligand, from Fig. 4b–d, all of the cyclometalated heavy-metal complexes displayed enormously better compatibility with inconspicuous D/A separation or aggregated phase for PCBM blended films. And the RMS values were almost identically small as ∼0.36–0.38 nm. As a result, the optimized morphology with avoidable excessive aggregation to form sufficient contact between donor and acceptor domains is expected to refrain charge recombination in OSCs. In contrast, involving Y6 led to a clearer and uniformed nano fibrillar texture compared to the relevant PCBM blends, which were reasonable for the distinctive crystalline behavior of Y6.16 Meanwhile, obvious phase separation with considerable domain size was achieved in Y6 blends, which contributed to improve the charge transport ability.18 The results rationally explained the big difference of the charge mobilities between different acceptor based blended films. Besides, as shown in Fig. 4f–h, enlarged domain sizes could be found from heteroleptic to homoleptic Ir complexes and Y6 blends, corresponding to the much higher RMS value of 4.68 nm for tTBz3Ir:Y6 than 2.40 nm for tTBzIr. It is noteworthy that the blended films of tTBzPt:Y6 also depicted a comparably large size of pure domain, whereas insufficient exciton dissociation and serious bimolecular recombination happened. Therefore, it is speculated that the extremely short lifetime (< 1 ns) for the Pt complex limited the exciton LD in tTBzPt:Y6 blended films. Though showing the extension of the pure domain, the restricted LD of singlet excitons in tTBzPt was not enough to meet the requirement of longer distance for diffusion to D/A interfaces, thereby resulting in charge recombination and unbalanced charge transport processes. In comparison, the triplet excitons with 2–3 orders longer lifetimes in tTBz3Ir relaxed this limitation to a certain extent. | ||
| Fig. 4 Film morphology images. (a–h) AFM height images and (i–p) AFM phase images of the corresponding blend films. | ||
Conclusions
In conclusion, based on the linear organic cyclometalated tTBz ligand, we have designed and synthesized three heavy metal Pt and Ir complexes with varied molecular spatial configurations. The relationships between materials structure and photovoltaic properties have been systematically investigated. It has been found that when employed as electron donor materials, the PCE presented the same trends of octahedral homoleptic tris-tTBz3Ir > octahedral heteroleptic bis-tTBzIr > square-planar heteroleptic tTBzPt > linear tTBz in both fullerene and non-fullerene acceptor devices. Upon cyclometalation with heavy metal centers, enhanced 1MLCT absorption, increased hole mobility and improved film morphology could be achieved. However, the triplet characteristics have only been observed in the two Ir complexes. Photovoltaic devices based on homoleptic tTBz3Ir with tris-identical cyclometalated ligands have shown the strongest light-harvest ability, the best charge transport, the most efficient exciton dissociation and the least bimolecular recombination. Therefore, PCEs of 5.71% and 7.97% were attained in tTBz3Ir:PCBM and tTBz3Ir:Y6 blended devices, which were superior to those of tTBzIr (3.60% and 6.44%), tTBzPt (1.04% and 1.36%) and tTBz (∼0 and 0.45%) based devices. Furthermore, particularly in Y6 blends, the optimized octahedral homoleptic configuration of tTBz3Ir could induce an improved morphology with promoted phase separation and increased domain size, which was beneficial to the charge transport in the BHJ active layers. More interestingly, it is abnormal in this work that the square-planar tTBzPt showed an absence of triplet feature, which resulted in insufficient exciton dissociation and adverse charge recombination in OSCs compared to Ir complexes with long-lived triplet excitons. Our work elucidates the significance of triplet behavior in organometallic complexes to organic photovoltaics. The influence of molecular spatial configuration on photovoltaic performances is expected to provide guidelines for the future design of new photoactive materials in OSCs.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the National Natural Science Foundation of China (Grant No. 61761136013 and Grant No. 91833304) for financial support.Notes and references
- A. Ramdass, V. Sathish, E. Babu, M. Velayudham, P. Thanasekanra and S. Rajagopal, Coord. Chem. Rev., 2017, 343, 278–307 CrossRef CAS.
- S. Gómez-Coca, D. Aravena, R. Morales and E. Ruiz, Coord. Chem. Rev., 2015, 289–290, 279–392 Search PubMed.
- K. T. Mahmudov, A. V. Gurbanov, F. I. Guseinov and M. F. Da. Silva, Coord. Chem. Rev., 2019, 387, 32–46 CrossRef CAS.
- É. N. Oiye, M. F. M. Ribeiro, J. M. T. Katayama, M. C. Tadini, M. A. Balbino, I. C. Eleotério, J. Magalhães, A. S. Castro, R. S. M. Silva, J. W. Júnior, E. R. Dockal and M. F. D. Oliveira, Crit. Rev. Anal. Chem., 2019, 49, 488–509 CrossRef PubMed.
- A. R. Muguruza, R. F. D. Luis, N. Iglesias, B. Bazán, M.-K. Urtiaga, E. S. Larrea, A. Fidalgo-Marijuan and G. Barandika, J. Inorg. Biochem., 2020, 205, 110977 CrossRef CAS PubMed.
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC.
- D.-Y. Wang, R. Liu, W. Guo, G. Li and Y. Fu, Coord. Chem. Rev., 2021, 429, 213650 CrossRef CAS.
- C. Qi, X. Wang, Z. Chen, S. Xiang, T. Wang, H.-T. Feng and B. Z. Tang, Chem. Front., 2021, 5, 3281 CAS.
- G. Li, D. Zhu, X. Wang, Z. Su and M. R. Bryce, Chem. Soc. Rev., 2020, 49, 765 RSC.
- C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418–4441 CrossRef CAS.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- E. V. Puttock, M. T. Walden and J. A. G. Williams, Coord. Chem. Rev., 2018, 367, 127–162 CrossRef CAS.
- A. B. Kajjam and S. Vaidyanathan, Chem. Rec., 2018, 18, 293–349 CrossRef CAS PubMed.
- Y. Jin, Y. Zhang, Y. Liu, J. Xue, W. Li, J. Qiao and F. Zhang, Adv. Mater., 2019, 31, 1900690 CrossRef PubMed.
- T. Hao, W. Zhong, S. Leng, R. Zeng, M. Zhang, L. Zhu, Y. Yang, J. Song, J. Xu, G. Zhou, Y. Zou, Y. Zhang and F. Liu, Sci. China: Chem., 2022, 65, 1634–1641 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed.
- J. Gao, S. Qu, L. Yang, H. Wang, F. Du, J. Yu and W. Tang, Chem. Eng. J., 2021, 412, 128770 CrossRef.
- M. Jiang, H. Bai, H. Zhi, L. Yan, H. Y. Woo, L. Tong, J. Wang, F. Zhang and Q. An, Energy Environ. Sci., 2021, 14, 3945 RSC.
- X. Gao, H. Wang, Z. Li, X. Tao, X. Qin, T. Yang, X. Song, L. Bu, G. Lu and Y. Tao, Chem. Eng. J., 2022, 450, 138018 CrossRef CAS.
- P. Bi, S. Zhang, Z. Chen, Y. Xu, Y. Cui, T. Zhang, J. Ren, J. Qin, L. Hong, X. Hao and J. Hou, Joule, 2021, 5, 2408–2419 CrossRef CAS.
- M. T. Sajjad, A. Ruseckas, L. K. Jagadamma, Y. Zhang and I. D. W. Samuel, J. Mater. Chem. A, 2020, 8, 15687 RSC.
- Y. Cai, Q. Li, G. Lu, H. S. Ryu, Y. Li, H. Jin, Z. Chen, Z. Tang, G. Lu, X. Hao, H. Y. Woo, C. Zhang and Y. Sun, Nat. Commun., 2022, 13, 2369 CrossRef CAS PubMed.
- Y. Zhang, M. T. Sajjad, O. Blaszczyk, A. J. Parnell, A. Ruseckas, L. A. Serrano, G. Cooke and I. D. W. Samuel, Chem. Mater., 2019, 31, 6548–6557 CrossRef CAS.
- T. Yang, Y. He, Y. Cheng, X. Gao, Y. Wu, W. Yuan and Y. Tao, Dalton Trans., 2021, 50, 9871 RSC.
- Q. Wu, Y. Cheng, Z. Xue, X. Gao, M. Wang, W. Yuan, S. Huettner, S. Wan, X. Cao, Y. Tao and W. Huang, Chem. Commun., 2019, 55, 2640 RSC.
- A. Rao, P. C. Y. Chow, S. Gélinas, C. W. Schlenker, C.-Z. Li, H.-L. Yip, A. K.-J. Jen, D. S. Ginger and R. H. Friend, Nature, 2013, 500, 435–439 CrossRef CAS PubMed.
- H. Gao, R. Yu, Z. Ma, Y. Gong, B. Zhao, Q. Lv and Z. Tan, J. Polym. Sci., 2022, 60, 865 CrossRef CAS.
- Y. Jin, J. Xue, J. Qiao and F. Zhang, J. Mater. Chem. C, 2019, 7, 15049–15056 RSC.
- H. Zhen, Q. Hou, K. Li, Z. Ma, S. Fabiano, F. Gao and F. Zhang, J. Mater. Chem. A, 2014, 2, 12390 RSC.
- L. Yang, W. Gu, L. Lv, Y. Chen, Y. Yang, P. Ye, J. Wu, L. Hong, A. Peng and H. Huang, Angew. Chem., Int. Ed., 2018, 57, 1096–1102 CrossRef CAS PubMed.
- L. Qin, X. Liu, X. Zhang, J. Yu, L. Yang, F. Zhao, M. Huang, K. Wang, X. Wu, Y. Li, H. Chen, K. Wang, J. Xia, X. Lu, F. Gao, Y. Yi and H. Huang, Angew. Chem., Int. Ed., 2020, 59, 15043–15049 CrossRef CAS PubMed.
- T. Wang, R. Sun, M. Shi, F. Pan, Z. Hu, F. Huang, Y. Li and J. Min, Adv. Energy Mater., 2020, 10, 2000590 CrossRef CAS.
- X. Xu, K. Feng, Z. Bi, W. Ma, G. Zhang and Q. Peng, Adv. Mater., 2019, 31, 1901872 CrossRef PubMed.
- X. Gao, D. Shi, M. Wang, Z. Xue, Y. Hu, Y. Tao and W. Huang, J. Mater. Chem. C, 2018, 6, 9903 RSC.
- X. Gao, Y. Liang, H. Wang, T. Yang, S. Huettner, J. Wang, F. Zhu and Y. Tao, Org. Electron., 2019, 70, 93–100 CrossRef CAS.
- Z. Xue, S. Wang, J. Yang, Y. Zhong, M. Qian, C. Li, Z. Zhang, G. Xing, S. Huettner, Y. Tao, Y. Li and W. Huang, npj Flexible Electron., 2018, 2, 1 CrossRef CAS.
- M. Qian, R. Zhang, J. hao, W. Zhang, Q. Zhang, J. Wang, Y. Tao, S. Chen, J. Fang and W. Huang, Adv. Mater., 2015, 27, 3546–3552 CrossRef CAS PubMed.
- V. N. Kozhevnikov, M. C. Durrant and J. A. G. Williams, Inorg. Chem., 2011, 50, 6304–6313 CrossRef CAS PubMed.
- S. He, Z. Lin, F. Du, X. Wang, Y. Liu and W. Tang, Chem. Eng. J., 2022, 441, 135973 CrossRef CAS.
- W. Wu, G. Zhang, X. Xu, S. Wang, Y. Li and Q. Peng, Adv. Funct. Mater., 2018, 28, 1707493 CrossRef.
- Z. Hao, F. Meng, P. Wang, Y. Wang, H. Tan, Y. Pei, S. Su and Y. Liu, Daltron Trans., 2017, 46, 16257–16268 RSC.
- H. Isci, Ö. Dag and W. R. Mason, Inorg. Chem., 1993, 32, 3909–3914 CrossRef CAS.
- D. Meng, R. Zheng, Y. Zhao, E. Zhang, L. Dou and Y. Yang, Adv. Mater., 2022, 34, 2107330 CrossRef CAS PubMed.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259 RSC.
- C.-L. Ho, B. Yao, B. Zhang, K.-L. Wong, W.-Y. Wong, Z. Xie, L. Wang and Z. Lin, J. Organomet. Chem., 2013, 730, 144–155 CrossRef CAS.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef CAS PubMed.
- M. A. Baldo, C. Adachi and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 967–977 Search PubMed.
- Y. Zhang, J. Miao, J. Xiong, K. Li and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202113718 CAS.
- X. Ding, X. Chen, Y. Xu, Z. Ni, T. He, H. Qiu, C.-Z. Li and Q. Zhang, Chem. Eng. J., 2022, 429, 132298 CrossRef CAS.
- T. Yang, X. Gao, Y. He, H. Wang and Y. Tao, J. Mater. Chem. C, 2020, 8, 5761 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00999d |
| This journal is © The Royal Society of Chemistry 2023 |