
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeteroatom-doped transition metal hydroxides in energy storage and conversion: a review
Yaqi
Qin†
a,
Guoping
Lu†
 *a,
Feng
Yang
a,
Chunhua
Xu
a,
Shuaijie
Jiang
a,
Yuqiu
Wang
a,
Yuxin
Tang
*b and
Pengcheng
Wang
*a
*a,
Feng
Yang
a,
Chunhua
Xu
a,
Shuaijie
Jiang
a,
Yuqiu
Wang
a,
Yuxin
Tang
*b and
Pengcheng
Wang
*a
aSchool of Chemistry and Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: glu@njust.edu.cn; alexwpch@njust.edu.cn
bInstitute of Applied Physics and Materials Engineering, University of Macau, Macau, P. R. China. E-mail: yxtang@um.edu.mo
First published on 20th January 2023
Abstract
High performance transition metal hydroxides (TMHs) are promising energy storage materials due to their simple and low-cost preparation process, high surface area, easy tunable composition, and so on. Heteroatom doping is an extensive approach adopted to tailor, both physically and chemically, the properties of TMHs, such as their lattice structure, electronic structure, lattice defects and diffusion behavior, so as to alter their catalytic performance for various redox reactions. Heteroatom doping can also improve the overall performance of TMH electrode materials in terms of specific capacitance and charge–discharge rate. Recently, the applications of heteroatom doping engineering in developing various TMHs/TMH-based composite materials with specific structures or functions in the fields of energy conversion and storage have been extensively explored. To this end, we review the latest developments in the heteroatom doping of TMHs in the fields of energy conversion and storage via metal doping, non-metal doping, and co-doping engineering. Then, the principles and effects of heteroatom doping in TMHs are discussed and generalized. Finally, the challenges and opportunities of heteroatom doping in TMHs are disclosed to provide insights regarding the further development of this research.
 Yaqi Qin | Yaqi Qin is currently a doctoral candidate at Nanjing University of Science and Technology, and she is engaged in the design, synthesis and application research of electrocatalytic materials under the guidance of Professor Pengcheng Wang. At present, she has published an article on the design and synthesis of supercapacitor electrodes. |
 Guoping Lu | Guo-Ping Lu received his PhD degree in Chemical Engineering and Technology from Nanjing University of Science & Technology in 2013. He is currently an associate professor in the School of Chemistry and Chemical Engineering at Nanjing University of Science & Technology. His research interests focus on heterogeneous catalysis for green organic synthesis and biomass conversion. |
 Yuxin Tang | Yuxin Tang is a professor in the College of Chemical Engineering at Fuzhou University. He obtained his BS and MS degrees at the Nanjing University of Aeronautics and Astronautics in 2006 and 2009, respectively, and graduated from Nanyang Technological University with a PhD in materials Science (2013). His research interests include solid-state and fast charging energy storage devices and the development of real-time electrochemical reaction monitoring techniques. |
 Pengcheng Wang | Pengcheng Wang obtained his BSc in 2008 and PhD in 2013 at Nanjing University of Science and Technology (NJUST) and was a postdoc at the National Institute of Advanced Industrial Science and Technology (AIST) from 2013 to 2014. He joined NJUST in 2014 and became a professor in 2020. His research interest includes energetic materials, energy storage materials and electrochemical reaction. |
1. Introduction
With the energy shortage and increasingly serious energy-related pollution, various energy storage devices and clean energy sources (including metal batteries and supercapacitors (SCs), H2O splitting, fuel cells and CO2 conversion) have received extensive attention from researchers.1–3 As representatives of modern energy storage equipment, SCs and battery devices have been extensively used in daily life to supply power for various portable electronic devices.4,5 As a clean energy source, H2 with high efficiency and low emission is the best alternative to fossil fuels. H2O splitting to generate H2 and O2 is one of the most economical and environmentally friendly methods.6 Meanwhile, when the urea oxidation reaction (UOR) is used as the anode reaction for water splitting to generate H2, its theoretical voltage (0.37 V vs. RHE) is much lower than the traditional H2O electrolysis voltage (1.23 V vs. RHE), which can produce H2 with lower energy consumption, providing a prospect for the purification of urea-rich wastewater.7 The electrocatalytic reduction of CO2 reaction (CO2RR) to produce a liquid fuel can not only alleviate the energy crisis but also solve the environmental problems caused by the greenhouse effect.8However, numerous obstacles remain in the development of energy storage devices. Normally, conventional Li batteries have relatively low ionic resistance, leading to some drawbacks such as safety issues and insufficient lifetime.2 The disadvantage of SCs regarding low energy density (SCs: 1–10 W h kg−1vs. batteries: 10–100 W h kg−1) has also been identified as a major challenge for their development.9 The anodic oxygen evolution reaction (OER) of H2O splitting requires a high overpotential, which increases energy consumption.7 In essence, the performance of energy storage devices is limited by the inherent properties of active materials. Therefore, various electrode materials and electrocatalysts with specific structures or functionalities are rationally designed to improve the development of energy devices.10–12
Traditional transition metal hydroxides (TMHs) as a promising class of electrode materials are mainly composed of transition metal cations (such as Co2+, Ni2+, Mn2+, and Cr3+) and OH−.9,13–15 They have been employed in various improved energy devices owing to their simple synthetic process, cheapness, unique structure and properties.16,17 TMHs can provide high capacitance from reversible faradaic reactions between electrode materials and electrolyte ions, and can store much more energy for SCs than carbon-based materials and achieve better electrochemical stability than polymer materials.9 The unique eg orbital occupancy of transition-metal OER electrocatalysts makes them highly active to absorb OER intermediates to strengthen electrocatalytic activities.18,19 Unfortunately, the low inherent conductivity of TMHs limits their diffusion rate of electrons, and their structures easily collapse in an alkaline medium. These issues have seriously hindered their applications in energy storage and conversion. Therefore, numerous studies about the structure optimization of TMHs have been reported to overcome these issues.15 There are four main strategies for their modification (Fig. 1) including morphology and nanostructure engineering,20 defect manufacturing,21 conductive substrate introducing,18 and heteroatom doping.22 The construction of 2D nano-sheets/wires using TMHs could increase their specific surface area.23,24 The combination of TMHs with conductive matrixes/carriers (such as conductive polymers and carbon-based materials) may avoid structural accumulation, reducing the “dead surface” generated in the process of conventional preparation electrodes.20,25 Creating defects can produce more active sites, which more easily exposes TMH matrix to reduce the contact resistance.26 However, these modification strategies are essential to increase the contact area between the electrode and the electrolyte and promote the charge transmission capacity of TMHs. In addition, doping heteroatoms can regulate the electronic structures of metal ions or produce new active metal sites in TMHs.27 It has been proved that the doping of active hetero-metal ions into singly active metal hydroxide structures may help widen their interlayer space and reduce their mechanical stress during the charge/discharge process, thereby leading to an obvious increase in their capacitive performance.28
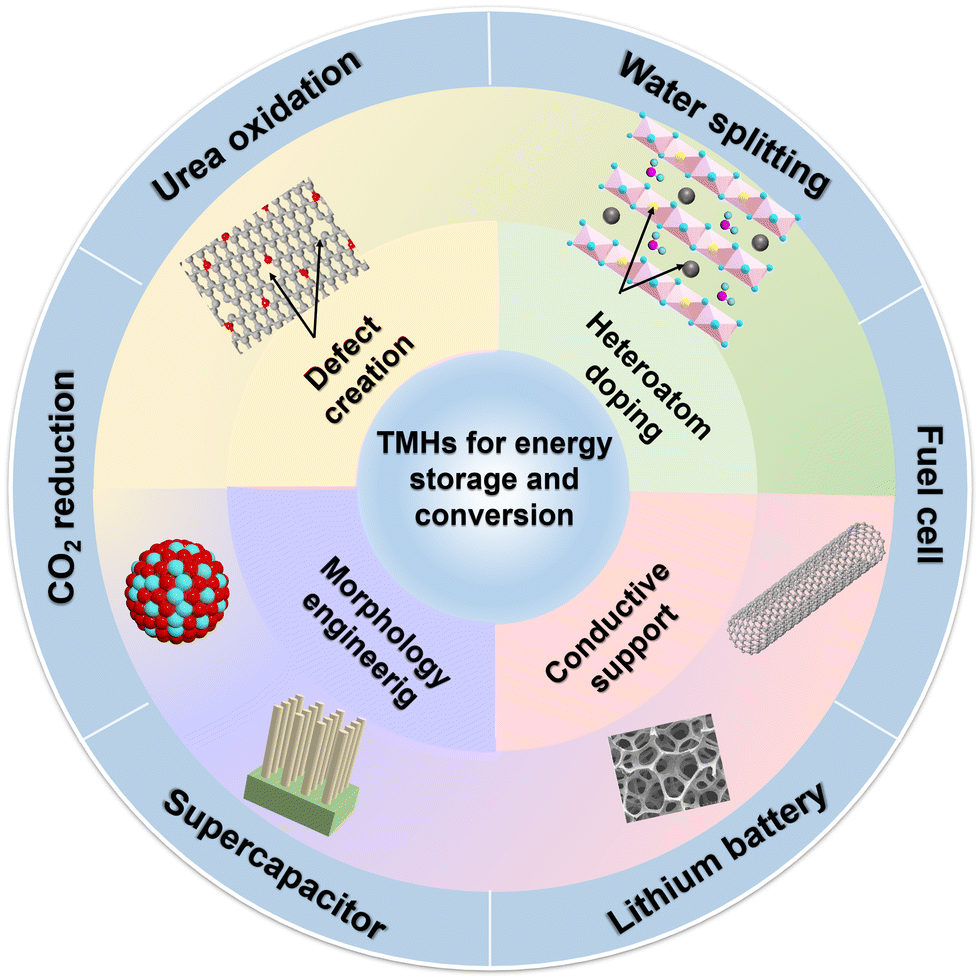 | ||
| Fig. 1 The modification and related energy applications of TMHs. | ||
Compared with other approaches, doping engineering is a simple and efficient method to improve the composition and performance of TMHs. Doping engineering can be divided into metal doping and non-metal doping based on the type of doped ions.29,30 Metal doping involves the introduction of objective metal cations outside the main body. Since there are electronic interactions among multiple metals/elements, the performance of multi-metal TMHs is superior to that of single-metal TMHs. Non-metal doping involves the introduction of non-metal anions with high solubility constants to completely or partially replace the existing anions of TMHs or doping them into the host lattice of the TMHs.28,31 The synergic effects of doped metal ions may influence the electronic structure of TMHs, improving their inherent electrochemical performance, activity, and conductivity. Co-doping involves selecting two or more dopants and combining a variety of doping principles to enhance the advantages of heteroatom doping.32 At the same time, the choice of various dopants not only considers the doping principle but also considers the mutual balance among the plurality of dopants, thereby producing a positive effect.
Generally, there are four functions of heteroatom doping on TMHs (Fig. 2): (1) heteroatom doping can form multi-component TMHs to adjust the metal valence ratio; (2) it could regulate and increase active metal sites; (3) the doped heteroatoms could tune electronic states, thereby improving the inherent conductivity of these materials; and (4) it could increase the interlayer spacing to expand the ion transport pathway, decreasing contact resistance between transition metal elements and OH−.33 More critically, heteroatom doping provides numerous crucial possibilities and application prospects in the development of multi-element TMHs owing to the wide variety and compatibility of heteroatoms.34,35
 | ||
| Fig. 2 The functions of heteroatom-doping on TMHs. | ||
So far, the applications of heteroatom-doped TMHs in energy storage and conversion devices are increasing, but few attempts systematically summarize their applications in this field. In order to witness the important development of heteroatom-doped TMHs in the energy field, we provide this in-time review by classifying heteroatom-doped TMH technologies into metal doping, non-metal doping and co-doping according to the compositional characteristics of TMHs. The applications of heteroatom-doped TMHs in H2O splitting, fuel cell and lithium battery catalysis as well as supercapacitor energy storage are also summarized. Furthermore, the principles and effects of doping are also introduced in detail. Finally, we summarize both challenges and opportunities of heteroatom doping in TMHs to demonstrate the future development direction of the heteroatom doped TMH strategy, aiming to stimulate more new and efficient investigations in this field.
2. Doping principles and effects
It is well known that the electrochemical properties of TMH materials depend primarily on certain structural characteristics: (1) the specific surface of TMHs can be controlled, such as constructing core–shell, linear, cluster, granular and nanostructures, which is beneficial to increase the capacitance and reduce the contact resistance of capacitors. (2) Modification of TMHs at the molecular level can increase the synergistic effects between the active sites and the components, thereby improving the catalytic reaction kinetics and accelerating the HER/OER catalytic process. (3) The regulation of the electronic environment affects the interfacial charge transfer and inherent conductivity and improves the internal electrocatalytic activity of TMHs.Therefore, heteroatom doping engineering enabling the structure of TMHs will be effectively modified, thereby producing an intrinsic change in their electrochemical properties. Here, we summarized the doping principles of various element types and the correlation between doped elements and the electrochemical properties of TMHs. Specifically, in order to better discuss the principle and function of doping, we divide the doped elements into the following categories: metal doping and non-metal doping (Fig. 3(a)).
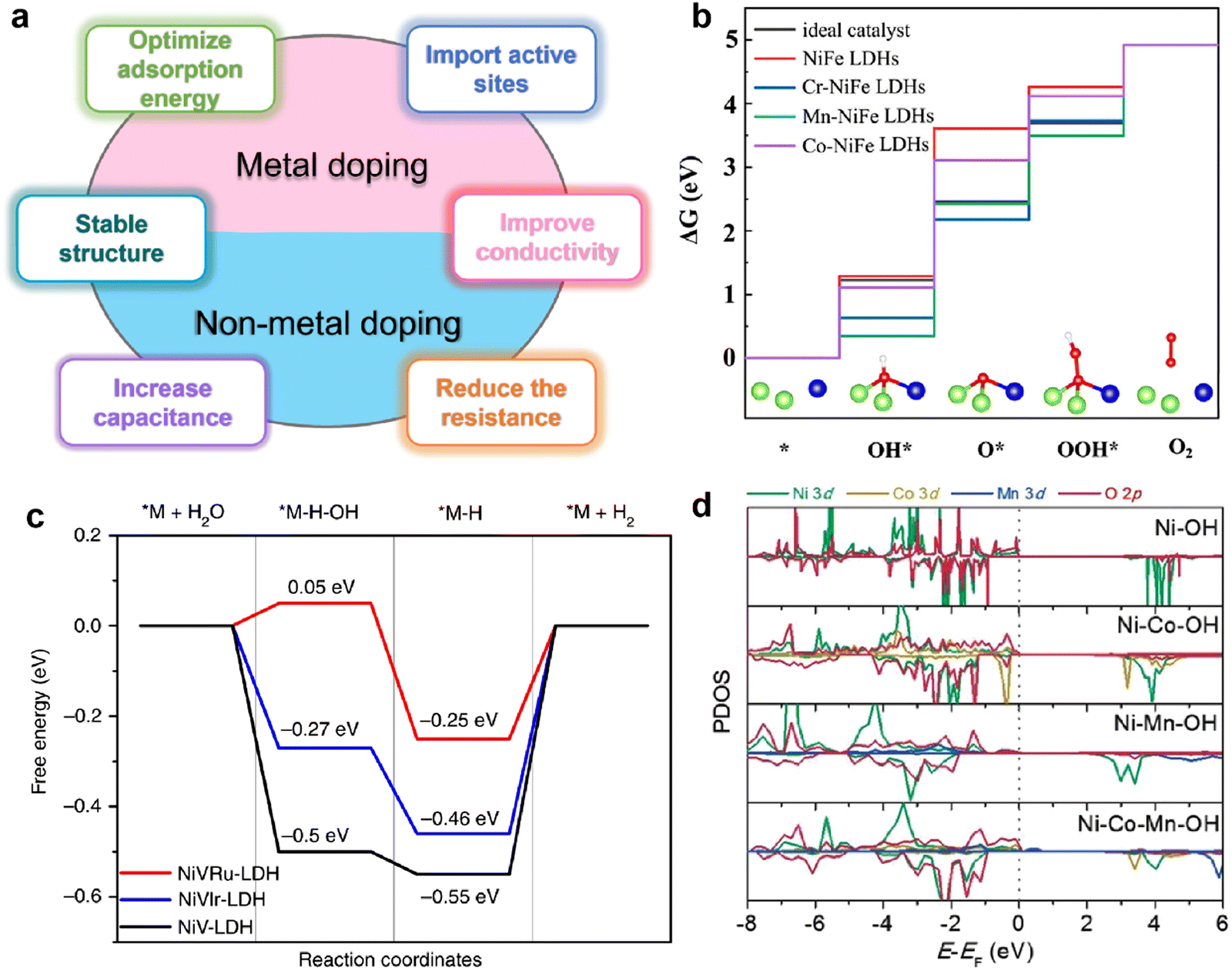 | ||
| Fig. 3 (a) Schematic diagram of the advantages of doping-modified TMHs. (b) The free energy diagram of the OER on un-doped and doped NiFe LDHs. Reproduced with permission from ref. 38 © 2020 Elsevier Inc. (c) The free energy diagram of the HER on the NiV-LDH, NiVRuLDH, and NiVIr-LDH catalysts. Reproduced with permission from ref. 39 © 2019 nature. (d) Partial density of state diagrams for Ni, Ni–Co, Ni–Mn, and Ni–Co–Mn hydroxide systems. Reproduced with permission from ref. 40 © 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
2.1. Metal doping
Generally, metal doping can significantly improve the electrochemical properties of TMHs. First, inactive metal ion (such as Mn2+ and Al3+) doping can cause the orbital hybridization of relatively low valence metal ions, which may stabilize the layer structure of the TMHs to effectively increase the capacity.Second, doping of active metal ions such as Ni2+, Co2+, Fe2+/Fe3+, and Zn2+ can introduce new active sites in TMHs, which may accelerate charge transfer and electron conductivity to enhance the electrocatalytic properties of the HER and the OER.
Finally, Cr6+, Ta5+ and V5+ having high valence metal ion doping may introduce changes in the electronic structure and overcome the H2O adsorption in the oxidation process.
2.2. Non-metal doping
From the perspective of non-metal doping modified TMHs, the doping of elements (S, P and N) may regulate material morphology and produce more defects, edge sites, and more empty d orbitals. These may contribute to exposing TMH based materials to reduce the contact resistance.In addition, high electronegativity atom doping, F and Cl, may form stable M–F bonds in TMHs, resulting in changes in the charge density and the production of active centers to increase conductivity and stability.
Furthermore, when the inorganic (NO3−, CO32−, SO42−, PO43−) or organic anions (AcO−, C17H35COO−) with a large radius are embedded in the LDH layer, the layer spaces may be expanded, and the micro-variation of the lattice is changed. Accordingly, these may cause expansion and lattice strain of the intermediate layer in the LDHs to attenuate the resistance of the interaction between the TMHS active component and OH− and increase the transmission capacity of ions.
2.3. Density functional theory
A recent discovery using density functional theory (DFT) calculations and theoretical computational models provides new guidelines for designing innovative catalyst materials and the electronic structure of the catalyst. In addition, the scaling relationship between the properties of catalytic materials and reaction intermediates can expand our understanding of the reaction mechanism at the molecular level.36 Therefore, DFT calculations provide a simple and efficient method to establish the relationship between the structure and performance of heteroatom-doped TMHs.The HER and OER are two and a half reactions in the electrocatalysis process, and high-efficiency electrocatalysts are needed to reduce the overpotential caused by the slow transfer of multiple proton-coupled electrons in these two reactions.37 The proposed OER process under alkaline conditions consists of four elementary stages: M*, M–OH, M–O, and M–OOH. Gao et al.38 designed a series of transition metal (Cr, Mn and Co) doped NiFe-LDHs to study the doping effect in the OER. The energy profiles of the OER on the un-doped and doped NiFe-LDHs are shown in Fig. 3(b), the largest energy uphill of each catalyst is the potential-determining step. When NiFe-LDHs doped with other transition metals, the Gibbs free energies of O*, OH* and OOH* intermediates calculated by DFT decrease significantly, indicating that the guest metal atoms play a role in active NiFe LDH, resulting in stronger binding of intermediates.
Yang et al.39 reported a method of site-selective incorporation of a noble metal (Ru or Ir) into NiV-LDH to investigate the original relationship between the activity of the HER and the doping effect at atomic levels. In Fig. 3(c), Ru and Ir doping can accelerate both Volmer and Heyrovsky steps in alkaline media, and Ru is more conducive to the HER. This demonstrates that doping of noble metals can reduce the energy barrier in the Volmer and Heyrovsky steps of the HER.
In order to rationally design nickel hydroxide-based electrode materials for fast energy storage, it is necessary to fully understand the structural evolution, charge storage mechanism and the contribution of the redox reaction to the capacity of the electrode. Liu et al.40 deeply explored the reasons why cation doping can improve the performance of Ni(OH)2 electrode materials in supercapacitors. The electrochemical performance of nickel hydroxides potentially depends on their deprotonation energies, which are calculated by DFT using a structural slab. The calculated Gibbs free energy change after deprotonation determines the driving force for the charge–discharge process and potentially affects the electrochemical properties. Depending on the partial density of state (PDOS) diagrams in Fig. 3(d), it is found that Co or Mn substituted in Ni(OH)2 diminish band gaps by filling the very top of the valence band near the Fermi level, which reveals that cation doping can improve the electronic conduction and excitation of electrode materials. The difficulty of charge transfer between metal cations and O in the absence of Mn in hydroxide could result in poor conductivity and sluggish charge–discharge kinetics.
DFT allows us to examine the essential influence of heteroatom doping on TMHs, check all hypotheses, and select suitable candidate heteroatoms to combine them with existing TMHs, to adjust the details of the atomic scale. DFT calculations have been successfully applied to analyze activation barriers, adsorption energies and reaction thermodynamics, which can quantify the deprotonation energy, and provide a basis for the design and prediction of heteroatom-doped TMH electrode materials for fast energy storage.
3. Metal engineering
3.1. Single-metal TMHs (LSHs)
Single metal hydroxides (LSHs) are generally divided into two categories: M(OH)2 and M(OH)(2−x)Axm−·nH2O. M(OH)2 is the original brucite phase (e.g., β-Ni(OH)2 and β-Co(OH)2),41 wherein the divalent metal cations in the octahedral structure are coordinated to six OH− ions and share edges, with no ion insertion in the neutral layer.42 M(OH)(2−mx)Axm−·nH2O is a hydrotalcite phase (such as α-Ni(OH)2 and α-Co(OH)2), where Am− represents an intercalated anion, replacing OH− in the interlayer channel.43Recently, LSHs with various morphological characteristics have been applied as electrode materials in various energy storage devices. However, α-M(OH)2 exhibits poor cycling stability and is easy to convert β-M(OH)2 and lost crystal H2O in alkaline electrolytes, resulting in structural degradation. Therefore, incorporation of metals with different valence states (such as Mn2+, Zn2+ and Al3+ Fe2+/Fe3+) into LSHs is a general strategy to enhance their intrinsic electrocatalytic activities and structural stability.34,44
| Entry | The doped heteroatom | Electrodes | Specific capacitance (F g−1) | Rate performance | Cycle performance (5 A g−1) | Highest energy density: (W h kg−1) | Highest power density: (W kg−1) | Electrolyte | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Mn | Mn doped Co(OH)2 | 1915.88 (1 A g−1) | 63.18% (20 A g−1) | 93.5% (5000) at 10 A g−1 | — | — | 2 M KOH | 45 |
| 2 | Mn | NiMn-LDH | 1498 C g−1 (2 A g−1) | 61.08% (50 A g−1) | 96.5% (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) at 10 A g−1 000) at 10 A g−1 |
62.7 | 375.2 | 6 M KOH | 46 |
| 3 | Zn | Zn doped Ni(OH)2@CNTs | 750.5 C g−1 (0.5 A g−1) | 72.9% (10 A g−1) | 115.8% (5000) at 6 A g−1 | 51.3 | 409.6 | 2 M KOH | 46 |
| 4 | Zn | NiZn−OH/rGO | 615.4 C g−1 (1 A g−1) | 62.3% (30 A g−1) | 89.7% (10000) |
53.7 | 825.1 | — | 48 |
| 5 | Al | Al-doped Ni(OH)2 | 2606 (1 A g−1) | 44.5% (20 A g−1) | 46.5% (1200) at 10 A g−1 | — | — | 6 M KOH | 50 |
| 6 | Al | Al3+-doped α-Ni(OH)2 | 1750 (1 A g−1) | 92% (10 A g−1) | 72% (2000) at 16 A g−1 | 49.6 | 573.8 | 6 M KOH | 51 |
| 7 | Zn | NiCoZn–LDH | 1742 (1 A g−1) | 77% (10 A g−1) | 89% (40000) at 10 A g−1 |
37.2 | 362 | 3 M KOH | 69 |
| 8 | S | NiV-S | 2270.4 (2 A g−1) | 51.3% (20 A g−1) | 91.9% (10000) at 25 A g−1 |
51 | 1600 | 1 M KOH | 84 |
| 9 | S | NiCo-LDH-S7.5 | 2417.7 (1 A g−1) | 81.8% (20 A g−1) | 90.19% (5000) at 2 A g−1 | 73.57 | 375 | 2 M KOH | 85 |
| 10 | S | S-doped Ni2(OH)2CO3 @MWCNTs | 1158 (1 A g−1) | 83% (5 A g−1) | 98% (20000) |
45 | 400 | 6 M KOH | 87 |
| 11 | F | F-doped α-Ni(OH)2 | 1503 (1 A g−1) | 48.7% (20 A g−1) | 82% (9000) | 67.4 | 400 | 2 M KOH | 101 |
| 12 | Cl | Co(CO3)0.35Cl0.20(OH)1.10 | 9893.75 (0.5 A g−1) | — | 75% (10000) at 0.5 A g−1 |
41.66 | 19.5 | 1 M KOH | 104 |
| 13 | N | Ni(OH)2–N | 434.7 mA h g−1 (5 A g−1) | 91.7% (20 A g−1) | 51% (1000) at 50 A g−1 | — | — | 1 M KOH | 107 |
| 14 | Cl− | α-Ni(OH)2-DS NBHMs | 1494 (1 A g−1) | 30.3% (8 A g−1) | 90.9% (500) at 4 A g−1 | — | — | 1 M KOH | 108 |
| 15 | SO42− | CoMn-LDH-SO4 | 582.07 mC cm−2 (2 mA cm−2) | 66.2% (50 mA cm−2) | 89% (18000) at (10 mA cm−2) |
0.096 mW h cm−2 | 1.5 mW cm−2 | 1 M KOH | 109 |
| 16 | NO3− | Co–Al-LDH–NO3− | 2230 (1 A g−1) | 60% (20 A g−1) | — | — | — | 1 M KOH | 110 |
| 17 | BO2− | PMNC g−1-2 | 1890 (0.5 A·g−1) | 84% (40 A g−1) | 83% (10000) |
41 | 216 | 6 M KOH | 111 |
| 18 | SDS | NiMn LDH-2 | 325 mA h g−1 (1 A g−1) | 64.3% (20 A g−1) | 89.65% (2000) at 20 A g−1 | 46.5 | 688.3 | 1 M KOH | 112 |
| 19 | OA | NA-LDH-OA | 1.040 C cm−2 (1.68 mA cm−2) | — | 94.5% (2000) at 44.1 mA cm−2 | 40.26 | 943 | 6 M KOH | 113 |
| 20 | [CoOx2]2− | I-Co(OH)2 NSs | 880 mA h g−1 (1 A g−1) | 64.3% (5 A g−1) | 99% (250) at 1 A g−1 | — | — | 1 M LiPF6 | 114 |
| 21 | Cu/Ni | Cu-Ni(OH)2 | 1832.5 mA h g−1 (0.2 A g−1) | — | 72.5% (800) at 1 A g−1 | — | — | — | 117 |
| 22 | Al/Co | Al–Co co-doped α-Ni(OH)2/GNS | 2257 (2 mV s−1) | 77.5% (50 mV s−1) | 77% (1000) | — | — | 6 M KOH | 118 |
| 23 | S/P | NiCo LDH-SP | 3384.8 (3 A g−1) | 66% (20 A g−1) | 81.3% (5000) at 8 A g−1 | 74.5 | 800 | 6 M KOH | 120 |
| 24 | C/N | C/N–Ni(OH)2/NixSy | 1731.2 (0.5 A g−1) | — | 134.6% (10000) |
38.98 | 404.36 | 1 M KOH | 122 |
| 25 | K/Cl | [K]+/[Cl]− doped Co-(OH)2 | 112.1 F cm−2 (5 mA cm−2) | 60.7% (50 mA cm−2) | 98% (5000) at 50 mA cm−2 | 39.8 | 478 | 6 M KOH | 123 |
Yin et al.46 proposed a novel strategy to improve the stability of the Ni(OH)2 electrode and modulate the electronic configuration and the layer stacking mode of α-Ni(OH)2 through controlling the Mn doping level and the occupied site (Fig. 4(a)–(c)). Due to the presence of Mn4+ ions, more anions are accommodated in the interlayers to increase the spacing along the c direction. Finally, the optimal NiMn-LDH has a high capacity (1498 C g−1 at 2 A g−1), great rate performance and excellent cycle performance (almost 100% capacity retention after 30000 cycles at 50 A g−1) (Table 1, entry 2).
 | ||
| Fig. 4 (a) Side view of the optimized supercell model with an interlayer distance of 7.9 Å (blue: Ni; yellow: Mn; red: O; white: H). (b) Supercell model and side view of the optimized second layer. (c) The total electronic densities of state (TDOS) of NiMn-LDH materials. Reproduced with permission from ref. 46 © 2021 Elsevier B.V. (d) Abstract of Fe3+ doping cobalt-based hydroxide nanosheets (e) Co 2p and Fe 2p XPS spectra of Fe3+ doping cobalt-based hydroxide nanosheets. Reproduced with permission from ref. 55 © 2021 American Chemical Society. | ||
Zhao et al.48 used a heterogenous Zn element doped in nickel-based hydroxide, which greatly optimized the inherent electronic structure and conductivity. Besides, the introduction of porous reduced graphene oxide (rGO) substrate with excellent conductivity can not only greatly accelerate the electron transfer between the collector and hydroxide but also inhibit the agglomeration of hydroxide. Thanks to the introduction of a sandwich structure, rGO and Zn doping, this Ni–Zn hydroxide/rGO material has obtained abundant contact sites and high conductivity. This SC material features suitable capacitance (615.4C g−1 at 1 A g−1), appropriate stability (87.5% after 8000 cycles), and superior capacity rate property (62.3% retention at 30 A g−1) (Table 1, entry 4).
Chen et al.49 demonstrated that the robust and high output Zn doped NiOOH–FeOOH (Zn–FexNi(1−x)OOH@NF) catalyst can be reconstructed by Zn-doping and electrooxidation. Dynamic reconstruction of Zn–(Ni/FeOOH)@NF under the oxidizing condition was revealed by a series of in situ Raman and ex situ characterizations. Zn modification to Ni only occurred in the form of the NiZn alloy rather than doping to NiO. In addition, the electron transfer from Zn to Ni can optimize the electronic property of Ni nanoparticles, improving their catalytic performance. Consequently, the reconstructed Zn–FexNi(1−x)OOH@NF electrode exhibited a low overpotential for the high-output OER (η1000= 330 mV, and η1500= 347 mV) and showed tiny performance degradation after a 1000 h stability test at 1000 mA cm−2 (Table 2, entry 1).
| Entry | The doped heteroatom | Catalyst/substrate | Electrochemical | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Overpotential (mV) (@10 mA cm−2) | Cell voltage (@10 mA cm−2) | Tafel slope (mV dec−1) | Stability (@10 mA cm−2) | |||||
| HER | OER | HER-OER (mV dec−1) | ||||||
| 1 | Zn | Zn–(Ni/FeOOH)@NF | — | 269 (100 mA cm−2) | — | 33 | 1000 h at 1000 mA cm−2 | 49 |
| 1 | Fe | Fe-doped Ni(OH)2/NF | 53.8 | 208 | 1.54 V | 79.37–45.54 | 20 h at 20 mA cm−2 | 54 |
| 2 | Fe | Co/Fe LDH/ | — | 285 | — | 44.6 | 4 h | 55 |
| 3 | Fe | NiFe-LDH@NF | — | 328 (50 mA cm−2) | — | 31.1 | 10 h at 50 mA cm−2 | 56 |
| 4 | Mn | Mn-doped NiFe(OH)2 | — | 194 | — | 52 | — | 61 |
| 5 | Ce | Fe:Ni(OH)2/FeCe:Ni–(OH)2/NF | — | 201 | — | 42.4 | 50 h | 63 |
| 6 | Ce | Ni–Fe–Ce-LDH | — | 242 | — | 34 | 24 h | 64 |
| 7 | Zn | PA-ZnFeCo LDH | — | 221 | — | 58.73 | 10000 s |
67 |
| 8 | Zn | Zn; NiFeOxHy | — | 250 | — | 28.3 | 24 h | 68 |
| 9 | Fe | Fe–CoNi LDHs | — | 260 | — | 49 | 12 h | 70 |
| 10 | Fe | Fe-doped NiCo-LDH | — | 285 | — | 62 | 16 h | 71 |
| 11 | Fe | Fe–CoMo UH | — | 245 | — | 37 | 90 h | 72 |
| 12 | Fe | Ni3V1Fe1-LDH | — | 269 | — | 67 | — | 73 |
| 13 | W | (NiFeW-LDHs | — | 211 | — | 36.44 | 120 h | 74 |
| 14 | Ta | Ta–NiFe LDH | — | 260 (50 mA cm−2) | — | 58.95 | 20 h at 100 mA cm−2 | 75 |
| 15 | Cr | CoFeCr LDH/NF | — | 202 | — | 83 | 20 h | 76 |
| 16 | Cr | Cr-CoFe LDHs/NF | — | 238 | — | 107 | 20 h | 77 |
| 17 | V | PV-NiFe LDH NSA | 19 | 295 (100 mA cm−2) | 1.43 V | 38–58 | 1000 h | 78 |
| 18 | V | Co2Fe0.5V0.5 | 242 | — | 41.4 | 20 h | 79 | |
| 19 | V | NiFeV LDHs. | — | 195 (20 mA cm−2) | — | 42 | 18 h 98% | 80 |
| 20 | V | NiFe-V | — | 254 | — | 37 | 20 h | 81 |
| 21 | S | S-doped Ni4/5Fe1/5-LDHs | — | 257 | — | 61.5 | 30 h | 86 |
| 22 | P | P-doped MoO3/FeCo-LDH/NF | — | 225 | — | 87.4 | 80 h | 91 |
| 23 | N | N-CoFe LDHs | — | 233 | — | 40.03 | — | 95 |
| 24 | F | F-doped NiFe-LDH | — | 225 | — | 79 | 10 h | 99 |
| 25 | F | F–NiAl LDH NF | — | 330 | — | 53 | 12 h | 100 |
| 26 | F | F-doped α-Ni(OH)2 | — | 325 | — | 31.89 | 30 h | 101 |
| 27 | F | F-NHO | — | 280 | — | 107 | 24 h 91.3% at 20 mA cm−2 | 102 |
| 28 | Cl | Cl-doped Co(OH)2 | — | 330 | — | 98 | 12 h | 103 |
| 29 | NO3− | Co–Al LDH–NO3− | — | 223 | — | 102 | — | 110 |
| 30 | DS− | DS− doped Co(OH)2 NCs | — | 561 | — | 172 | — | 42 |
| 31 | [MoS4]2− | MoS4-LDH/NF | 91 | — | 1.37 V | 125 | 24 h | 115 |
| 32 | POM | NiFe LDH-POM/NF | 200 | 156 | 1.6 V | 86–67 | 20 h | 116 |
| 33 | B/P | NiVFe-B-P LDH@NF | 117 | — | — | 68 | 24 h | 121 |
| 34 | Y/P | YP-Co(OH)F | 55 | 238 | 1.54 V | 52–67 | 300 h | 124 |
| 35 | Mn/F | Mn–F/Ni(OH)2–NF | — | 233 (20 mA cm−2) | — | 56.9 | 10 h at 20 mA cm−2 | 125 |
In contrast, Al3+-doped α-Ni(OH)2 has a better capacitance since Al doping can cause low crystallinity and high crystal defects. Yang et al.51 successfully synthesized another Al3+-doped α-Ni(OH)2 with flower-like morphology and porous structure through a simple one-pot hydrothermal method. This material displays an ultra-high specific capacitance of 1750 F g−1 at 1 A g−1 and a good electrochemical stability of 72% after 2000 cycles of operation. In addition, the assembled asymmetric supercapacitor with this material as the positive electrode exhibits a high energy density (49.6 W h kg−1 at 573.8 W kg−1) (Table 1, entry 6).
Wang et al.54 introduced Fe-doped α- and β-Ni(OH)2 for H2O splitting. Compared with Fe-doped β-Ni(OH)2, Fe-doped α-Ni(OH)2 is more conducive to forming the higher valence Fe sites, which are the major active centers in the OER. In contrast, Fe doping in the β phase may enhance the electron density of Fe sites, thereby facilitating the adsorption of H atoms to accelerate the HER process. Only a 53.8 mV overpotential for Fe-doped β-Ni(OH)2-240 is required to achieve a current density of 10 mA cm−2 (Table 2, entry 2). Therefore, Fe-doped β and α-Ni(OH)2 are used as the cathode and anode, respectively, in battery hydrolysis, which has high H2O splitting efficiency.
Liu et al.55 reported that Co2+ occupancy and coordination in cobalt-based LDH nanosheets can be tuned via Fe3+ doping. (Fig. 4(d)). It is found that the appropriate incorporation of Fe3+ can greatly activate the originally inactive octahedral coordinated Co2+ centers, which promotes the overall electrocatalytic activity of Co-based LDHs (Fig. 4(e)). Density functional theory (DFT) calculations also clarify that Fe3+ not only regulates the configuration of Co2+, but also acts as additional catalytic active sites. Therefore, the optimized LDH nanosheets (Co:Fe molar ratio is 5:1) show the lowest overpotential of 285 mV and the smallest Tafel slope of 44.6 mV dec−1 (Table 2, entry 3).
Dong et al.56 prepared Fe doped NiFe-LDH nanosheets and investigated in depth the influence mechanism of Fe doping by controlling the Ni/Fe ratio and the Fe valence state. Fe doping can control the structure morphology and promote the oxidation of Ni2+, making the electrode have a low overpotential of 382 mV at 50 mA cm−2, and the low gradient of Tafel is 31.1 mV dec−1 (Table 2, entry 4).
In order to improve the overall performance of nickel hydroxide and cobalt hydroxide in terms of specific capacitance and charge–discharge rate, mixed metal hydroxide seems to be better than single metal hydroxide. Doping foreign elements in LSHs can introduce an intermediate band, which has a profound impact on the band gap and electron energy of LSHs. Meanwhile, the molar ratio of active divalent metal ions to trivalent metal ions has a sensible impact on the structure, morphology and capacitive performance of LSHs.
3.2. Double metal-hydroxides (LDHs)
LDHs are types of 2D anionic clay with the structural formula [M(1−x)2+Mx3+(OH)2]x+ [(An−)x/n·yH2O]x. In the presence of OH−, the divalent cation M2+ in the M(OH)6 brucite-like structure is partially replaced by the trivalent metal cation M3+, forming a positively charged 2D host layer, while the anion An− balances the charge in the middle of the layer (Fig. 5).27 The structure of LDHs can be greatly modified by changing the M2+/M3+ molar ratio or the category of An−. Meanwhile, the nanostructures of LDHs offer greater functions by having hierarchical structures with large active surface areas, tailoring the distribution of materials homogeneously along the structure, the interconnected structure, or combining the advantages of tunable organic–inorganic components and controllable thickness at the nanoscale-all for the goal of maximizing the properties.57,58 For example, in 2010 Yan et al. assembled a positively charged LDH monolayer with a cationic functional molecule (BNMA) by using polyanions (PVS) as intermediates. This method allows fine-tuning and ordered assembly of functional cations and LDH monolayers for designing and achieving novel organic–inorganic ultra-thin films.17 However, the original LDHs show low conductivity and severe accumulation, which reduce active sites and hinder the transport of electrolyte ions.29 In addition, the 2D structure of LDHs is often deteriorated under harsh electrochemical conditions, which ultimately limits their electrochemical activity. | ||
| Fig. 5 The structure of carbonate intercalation LDHs with different M2+/M3+ molar ratios. Metal hydroxide octahedrons are stacked along the crystalline c-axis, and water and anions are present in the interlayer region. Reproduced with permission from ref. 27 © 2014 The Royal Society of Chemistry. | ||
To overcome these problems, doping active metal cations (such as Mn2+, Ce3+ Fe2+/Fe3+, Zn2+, Ta5+, V5+, and Cr3+) into the LDHs structure can increase the inherent conductivity to significantly improve the electrochemical activity. Furthermore, the doped additional metal cation can stabilize the valence of the primary metal ions of LDH and optimize their electronic structure, which protects a single component from oxidation to enhance the structure stability of LDH.59,60
 | ||
| Fig. 6 (a) DFT+U calculation for the oxidation of Ni(II) and Ni(III) ions. Reproduced with permission from ref. 61 © 2020 The Royal Society of Chemistry. (b) Schematic illustration of the synthesis of hollow Ni–Fe–Ce-LDH microcapsules mediated by Ce doping. Reproduced with permission from ref. 64 © 2020 The Royal Society of Chemistry. (c) Schematic of preparing process of Ta–NiFe LDH; (d) The XRD patterns of NiFe LDH and Ta–NiFe LDH. Reproduced with permission from ref. 75 © 2020 Elsevier B.V. (e) The adsorption energies for H2O on top of Fe sites and Co sites of CoFe LDH and Cr and Co sites of CoFeCr LDH. Reproduced with permission from ref. 76 © 2020 Elsevier B.V. (f) Schematic diagram of the structure of PV-NiFe LDH NSA. Reproduced with permission from ref. 78 © 2020 Elsevier B.V. | ||
Wu et al.62 synthesized Mn doped Ni(OH)2 for urea electrooxidation (UOR). They confirmed by Raman spectroscopy that Mn dopants promoted the formation of electroactive NiOOH at low potential and extended the Ni–O bond in NiOOH, resulting in increased phase structure disorder. Mn doping can reduce the starting potential of nickel hydroxide oxidation, which is conducive to the generation of electrochemically active NiIIIOOH, making it have excellent UOR performance.
Yan et al.64 reported hollow 3D LDH microcapsules (Ni–Fe–Ce-LDH) for the OER by Ce ion doping (Fig. 6(b)). Ce ions possess excellent properties of multivalence, flexible coordination, and high affinity for hard oxygen donors. Therefore, Ce ion doping can play a key role in stabilizing a particular crystalline phase and modifying the electronic properties. Because, from the perspective of energy, the Ce dopant is likely to occupy the MIL-88A aperture structure and enhances electronic transfer by the Ce-4f band modulation with d–f couplings. These results show that the Ce dopant plays a vital role in structural transitions and effective OER. Consequently, Ni–Fe–Ce-LDH has outstanding OER activity, with an overpotential of 242 mV at 10 mA cm−2 and a long-term durability of ≥24 h (Table 2, entry 7).
Cao et al.65 designed a La/Ce doping strategy to improve the OER performance of CoFe LDH. Because the ionic radii of La3+ and Ce3+ are much larger than Fe3+ and have unique electronic structures, La3+ or Ce3+ partially replaces Fe3+, CoFe LDH system has more defects, which elevates electrocatalytic activity and abates oxygen evolution potential. Moreover, the partial replacement of Fe3+ will affect the electronic structure of CoFe LDH, enhancing the electronic coordination in the system and showing excellent OER performance.
Duan et al.66 utilized Ce doping to modulate the electronic structure of CoFe LDH and improve the conductivity in order to improve the performance of the exchange membrane water electrolyzer. At the same time, excessive Ce doping leads to the formation of an active interface between CeO2 and CoFeCe-LDH, forming a self-assembled heterostructure CeO2/CoFeCe-LDH interface, further enhancing the OER activity and increasing the turnover frequency.
000 s (Table 2, entry 8).
Zn can be used as a strong Lewis acid that causes partial charge transfer and electron delocalization around Fe sites. Nam et al.68 disclosed a Zn-doped NiFeOxHy catalyst (Zn–NiFeOxHy) for H2O oxidation. As a dopant, therefore, Zn–NiFeOxHy exhibits superior electrocatalytic activity and stability compared with NiFeOxHy. At a current density of 10 mA cm−2, the overpotential of Zn–NiFeOxHy is increased to 250 mV (Table 2, entry 9).
Zheng et al.69 doped a small amount of inactive monovalent element Zn into NiCo LDH through two consecutive electrodepositions. Slight doping Zn2+ significantly improves the cycle performance of LDH, and the polyaniline (PANI) nano layer is used as the intermediate layer to enhance the interface interaction between LDH and the collector. The composite electrode achieved the best performance with a high specific capacitance of 1749 F g−1 and an ultralong life span with 89% capacitance retention after 40000 charge–discharge cycles (Table 1, entry 7).
Yamauchi et al.71 used Ni–Co glycerate spheres as a self-template to induce the formation of porous Fe-doped NiCo-LDH nanosheets, confirming Fe doping could accelerate the hydrolysis rate of the spheres. These porous nanosheets with high specific surface areas increase active sites, and the synergistic effects of doped Fe and the main metal (Ni and Co) may improve the OER kinetics of this catalyst. Therefore, the Fe-doped NiCo-LDH exhibits a low overpotential of 285 mV and a low Tafel slope of 62 mV dec−1 at a current density of 10 mA cm−2 in OER (Table 2, entry 11).
Gu et al.72 reported an effective strategy for synthesizing amorphous Fe-doped cobalt-molybdenum hydroxide (named Fe–CoMo UH) nanosheets. Benefiting from the ultrathin amorphous structure and multi-metal regulation, the Fe–CoMo UH nanosheets display outstanding OER performance, with a low overpotential (245 mV), a low Tafel slope (37 mV dec−1) at 10 mA cm−2, and excellent stability (Table 2, entry 12).
Li's team73 reported the preparation of Fe3+ doped NiV-LDH ultra-thin nanosheets by one-step coprecipitation. Fe doping can adjust the valence state of Ni sites, giving Ni3+ a higher oxidative capability. In addition, the presence of Fe3+ in NiV-LDH effectively promotes its charge transmission. Hence, Ni3V1Fe1 LDH shows excellent catalytic activity in the OER process. In 1.0 M KOH solution, the ultrathin Ni3V1Fe1-LDH nanosheet only needs a low-output level of 269 mV to achieve a current density of 10 mA cm−2 and also has a cycle stability of 1000 times as the electrode (Table 2, entry 13).
Zhang et al.75 employed high-valence Ta to dope a NiFe LDH catalyst to enhance H2O oxidation (Fig. 6(c)). According to XRD analysis in Fig. 6(d), Ta doping causes LDH lattice expansion. XPS analysis indicates that there is electronic interaction between Fe and Ta (electron transfer from Fe to Ta). Meanwhile, Ta doping is conducive to the adsorption of OH− on the Ta sites to reduce overpotential and improve the electrocatalytic activity in the OER. The optimized Ta–NiFe LDH displays excellent OER activity, with a low overpotential of 260 mV and a small Tafel slope of 58.95 mV dec−1 at a current density of 50 mA cm−2 (Table 2, entry 15).
Similarly, Li et al.77 prepared a kind of OER catalyst by introducing Cr dopants into CoFe LDHs (Cr-CoFe LDHs/NF) using a simple one-step hydrothermal method. DFT calculations indicate that Cr dopants could improve the electron-donation ability owing to the electronegativity differences between Cr and Co/Fe, which significantly tune the adsorption energy among four oxygen intermediates, increasing the activity of OER. This Cr-CoFe LDHs/NF catalyst has a superior overpotential of 238 mV at 10 mA cm−2 and high electrochemical durability for 20 h (Table 2, entry 17).
Zhang's group79 applied V doped CoFe LDH to improve the OER activity. DFT calculations indicate that V doping could increase the M–O bond energy within the LDH and promote charge transfer from O to metals. The Co2Fe0.5V0.5 catalyst exhibits an overpotential of 242 mV at 10 mA cm−2 in the OER under alkaline conditions, with a Tafel slope of 41.4 mV dec−1 (Table 2, entry 19).
Sun et al.80 synthesized a series of V doped NiFe LDH nanosheets array (denoted as NiFeV LDHs) via a one-step hydrothermal method for OER electrocatalysis. DFT+U simulation suggests that the high catalytic activities of the NiFeV LDHs are mainly attributed to the V doping, which alters the electronic structure and narrows the band gap, thereby improving conductivity and facilitating electron transfer. A small overpotential of only 195 mV to drive a current of 20 mA cm−2, with a low Tafel slope of 42 mV dec−1, in a 1.0 M KOH solution (Table 2, entry 20).
Liu et al.81 introduced different V contents in the process of synthesizing NiFe LDH to adjust the surface electronic structure and valence state of NiFe LDH, enhancing the electron transfer and electrochemically active surface area of LDH to further improve the intrinsic electrocatalytic activity. Compared with undoped NiFe LDHs, the oxygen evolution overpotential of NiFe-V decreases by 37 mV, and the Tafel slope decreases by 43.37 mV 10 mA cm−2 (Table 2, entry 21).
After the above discussion, we found that doping additional redox flexible metals in LDH can increase the number of active centers of the OER by improving the synergy between the nearest adjacent metal atoms. Therefore, compared with mono-active metal or bi-active metal hydroxides, doping metal elements to construct multi-active metal hydroxides may also provide a better utilization of the electroactive sites due to their homogeneous elemental distribution.
4. Non-metal doping
Recently, anion engineering received widespread attention as a strategy to modify the electrical activities of TMHs.30 As shown in Fig. 7(a), the cation provides a strong positive electric field when the anion is not polarized, which favors OH− adsorption but not O2 adsorption. Conversely, the polarized anion exhibits the covalent character of allowing electrons to enter the empty orbital of the cation, promoting the interaction. However, when over-polarized, the positive electric field is weakened, and the hydroxyl group is not completely adsorbed. Therefore, the regulation of the electronic structure to generate appropriate polarization can promote covalent and ionic interactions between cations and anions, thereby promoting electron transfer, adsorption, and desorption.82 | ||
| Fig. 7 (a) Schematic of anion regulations by optimizing the electronic structure of active sites toward water oxidation. Reproduced with permission from ref. 82 © 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (b) Crystal structure and schematic image of fluorine substitution of a brucite-like layer of LDHs. Reproduced with permission from ref. 83 © 2019 American Chemical Society. (c) Gradual changes in the morphology of the LDH-S electrode material and corresponding electrochemical properties. (d) XRD patterns of the LDH-S. Reproduced with permission from ref. 85 © 2020 American Chemical Society. (e) Schematic illustration of the synthetic of the Ni4/5Fe1/5-LDHs-S-2. Reproduced with permission from ref. 86 © 2020 Dalian Institute of Chemical Physics, Chinese Academy of Sciences. | ||
In addition, the anion engineering may also influence the hydrogen bonding nature in the interlayer space of TMHs. As a representative example (Fig. 7(b)),83 the embedding of high negative atom F− may weaken the hydrogen bond between the OH− structural group and the host ions in the interlayer space. In addition, the anions with larger diameters perhaps to expand the intermediate layer space of LDHs, which may expose more active sites and promote electron transport.
4.1. S doping
The NiV-LDH prepared by Huang's group displayed a high specific capacitance, but poor stability.84 Therefore, they further proposed an effective strategy to improve the stability of NiV-LDH by S doping. Compared with that of the original NiV-LDH, the cycle stability of NiV-S is much higher (NiV-S exhibited a retention rate of 98.5% after 10000 cycles) (Table 1, entry 8).
To further simplify the S-doping process, Zhu et al. designed a one-step hydrothermal method for preparing S-doped NiCo-LDH as a high-performance asymmetric capacitor.85 As shown in Fig. 7(c), the morphology of NiCo-LDH is changed by controlling the amount of S. S incorporated into the crystal lattice may decrease the crystallinity of the material and induce low-oxidation-state Ni species (Fig. 7(d)). In addition, the introduced S impurities may effectively tune the electronic structure of NiCo-LDH, increasing its inherent conductivity. Compared with that of the original NiCo-LDH, S-doped NiCo-LDH-S7.5 exhibits a unique nanosheet spherical structure, thus exposing more active sites and improving contact with the electrolyte. The assembled supercapacitor electrode exhibits an extremely high energy density of 73.54 W h kg−1 at a power density of 375 W kg−1 and great cycle stability (Table 1, entry 9).
Li et al.86 reported a simple method to construct an S-doped NiFe-LDH electrocatalyst for the OER, as shown in Fig. 7(e). The S doping could regulate the adsorption energy of OH* and O* on the Fe sites, accelerating the OER process. Moreover, the synergy of S and Fe sites improves the activity of the catalyst, and the amount of S doping also influences the OER activity of the NiFe-LDHs. The optimal S-doped Ni4/5Fe1/5-LDH catalyst displays excellent OER performance with an overpotential of 257 mV at 10 mA cm−2, a Tafel slope of 61.5 mV dec−1, and excellent stability (Table 2, entry 22).
Niu et al. prepared a kind of S-doped Ni2(OH)2CO3@MWCNT nanocomposites through a one-step hydrothermal method.87 XPS analysis illustrates that there is an electronic interaction between S and Ni2(OH)2CO3, so the assembled asymmetric supercapacitor (ASC) displays an energy density of 45 W h kg−1 and wonderful cycle stability (Table 1, entry 10).
4.2. P doping
The electronegativity of P is lower than those of O and S. Doping low-electronegativity species may also increase the conductivity of semiconductor catalysts, leading to faster electrochemical kinetics.88,89For example, P-doped CoNi-LDH can optimize the conductivity and electronic structure, which improves the catalytic performance in the methanol oxidation reaction (MOR).90 Xiao et al. used NaH2PO2 as the P source to generate P-CoNi-LDHs with different forms coupled with reduced graphene oxide (rGO) via cyclic voltammetry deposition. The structure–activity relationship analysis demonstrates that P doping improves the electronic structure and conductivity of this material and increases the MOR activity which is twice that of the original CoNi-LDH.
Huan et al.91 prepared a 3D P-doped MoO3/FeCo-LDH/NF ultrathin nanosheet heterostructure electrocatalyst for a high-efficiency OER. The heterostructure between P-MoO3 and FeCo LDH not only optimizes the electronic structure but also induces electron transfer. Moreover, the substitution of Mo6+ in MoO3 with P can stabilize the O vacancies, thereby providing superior conductivity and more active sites. P-MoO3/FeCo-LDH/NF exhibits significant OER catalytic activity in alkaline media, with a very low overpotential of 225 mV at a current density of 10 mA cm−2, and an excellent durability of ≥80 h (Table 2, entry 23).
Mei et al.92 synthesized P-doped NiCoZn LDH/NF through typical hydrothermal and calcination methods. P doping can reduce the adsorption energy of H, promoting desorption, and improving the corresponding catalytic activity. When PeNiCoZn LDH/NF-15% is used as the anode and cathode for urea-water electrolysis, the voltage is 1.479 V for urea-water electrolysis at 100 mA cm−2.
4.3. N doping
Plasma technology is a powerful method for surface etching and N doping.26,93,94 Wang et al. utilize N2 plasma treatment to peel off a large amount of CoFe-LDH, producing N-doped ultrathin CoFe-LDH nanosheets.95 N doping can change the electron density of adjacent metal atoms, which favors the formation of adsorption intermediates in the OER. Simultaneously, the exfoliated ultrathin CoFe-LDH nanosheets possess more atomic-sized holes and edge regions, thus exhibiting outstanding OER performance with overpotentials of 233 mV at a current density of 10 mA cm−2 (Table 2, entry 24).Kang et al. exfoliated a NiAl-LDH layer using N2 plasma to obtain mono or dual layer nanosheets.96 Nitrogen plasma can remove CO32− anion layers and form abundant oxygen vacancies on the surface of LDH. N doping can replace a portion of oxygen vacancies in the lattice to form metal–N and N–O bonds. In addition, with the help of doping nitrogen, oxygen vacancies promote the adsorption of the oxidation intermediate. Hence, both oxygen vacancies and nitrogen doping increase charge carrier density and reduce charge transfer interface resistance, thereby promoting water oxidation kinetics.
Mei et al.97 fabricated an N-doped NiZnCu LDH with rGO on Ni foam as an electrocatalyst of the HER. First, NiZnCu-LDH/rGO is synthesized by a hydrothermal method, and then, N-doping is achieved via calcination under NH3 gas. The unique morphology of N–NiZnCu LDH/rGO exposed numerous active sites, resulting in excellent catalytic activity. For ammonia oxidation reaction (AOR), UOR and hydrazine oxidation reaction (HzOR) at current densities of 10 mA cm−2, the voltages of these materials are 0.489, 1.305, and 0.010 V, respectively, with excellent stabilities (over 3000 cyclic voltammetry cycles).
4.4. F doping
The fluoride anion has the strongest electronegativity (4.0) and a similar anion diameter with O.98 Therefore, F doping may tune the electronic structure of metal sites in the TMHs and form M–F bonds, which accelerate the inherent electron transfer rate of TMHs.For example, Feng's group found that F doping can enhance the OER performance of FeNi LDH.99 XPS spectra disclose that F doping causes the modification of the electronic structure and the formation of Fe–F bonds in the FeNi-LDHs. The conversion from the M–O bonding to the M–F bonding after F-doping is significant for the OER owing to the facile formation of metal (oxy)hydroxide over the surface. F-doped NiFe-LDH exhibits a low overpotential of 225 mV at 10 mA cm−2, with a Tafel slope of 79 mV dec−1 (Table 2, entry 25).
Similarly, Xi et al.100 synthesized an F-doped NiAl LDH as an efficient OER catalyst. The catalytic activity of F–NiAl LDH is almost 10-fold stronger than that of the undoped catalyst. This is because the strongly electronegative F− is conducive to building weak M–F bonds, which easily break to form the active species of nickel oxides/hydroxides, thereby enhancing OER catalytic performance (Table 2, entry 26).
Xu et al.101 reported a novel strategy for fabricating F-doped α-Ni(OH)2 mesoporous 2D ultrathin nanosheets for H2O splitting and multifunctional electrodes in supercapacitors (Fig. 8(a)). In Fig. 8(c), the pink strip reveals the contribution of the F atom. Compared to Ni and O atoms, the peak of the F atom is weak. However, the presence of F atoms alters the coordination environments of Ni and O and further produces a new peak near the Fermi level. In addition, several bands that appear near the Fermi level and the lower Gibbs energy of the OER prove that F doping can improve conductivity (Fig. 8(d)). Therefore, the F-doped α-Ni(OH)2 nanosheets exhibit excellent electrocatalytic activity and stability with a low onset potential (260 mV) and Tafel slope (31.89 mV dec−1) in the OER (Table 2, entry 27). In addition, this material can be used in supercapacitors with high specific capacitances of 158.75 F g−1 at 1 A g−1 (Table 1, entry 11).
 | ||
| Fig. 8 (a) Schematic illustration of the synthesis of ultrathin mesoporous F-doped α-Ni(OH)2 nanosheets and their application for efficient water splitting. (b) Density of states of NiOOH. (c) Density of states of F–NiOOH. (d) Standard free-energy diagram of the OER process of NiOOH and F–NiOOH. Reproduced with permission from ref. 101 © 2019 The Royal Society of Chemistry. (e) Schematic of Cl-doped Co(OH)2 and electrochemical properties. Reproduced with permission from ref. 103 © 2017 American Chemical Society. | ||
Jiang's team102 prepared F doped Ni oxyhydroxide (F-NHO) mesogenic microspheres as high-efficiency electrocatalysts for the OER. F− is incorporated into the Ni oxyhydroxide lattice to form Ni–F bonds with enhanced ionic properties, which stabilize the Ni(III) species, achieving a higher electronegativity difference between Ni and F. Meanwhile, F− incorporation can enhance the electrochemical durability and resistance to harsh alkali and oxidation conditions under high potential, resulting in higher catalytic efficiency in the OER during H2O electrolysis (Table 2, entry 28).
4.5. Cl doping
As shown in Fig. 8(e), Zhong et al.103 introduced Cl− into Co(OH)2 to grow Cl-doped Co(OH)2 on carbon cloth as an integrated electrode. According to the experimental results, Cl in the electrocatalyst can be oxidized to destroy the internal structure of the catalyst and introduce defects. The Cl-doped Co(OH)2 nanosheets with defective structures exhibit improved electrocatalytic OER activities owing to their larger electrochemical surface areas (Table 2, entry 29).Hou et al.104 prepared Cl-doped cobalt carbonate hydroxide (Co(CO3)0.35Cl0.20(OH)1.10) nanowires for supercapacitors. The unique nanowire structure formed by Cl doping can increase the hydrophilicity and enable deep electrolyte ion diffusion, thereby reducing internal resistance. The prepared nanowires display excellent capacitances (9893.75 F g−1 at 0.5 A g−1) and energy densities (220 W h kg−1), cycle stability up to 10000 cycles (Table 1, entry 12).
In general, when these non-oxygen anion dopants enter the TMHs lattice, they induce changes in the charge density of the metallic body of the TMHs and fine-tune the chemical interactions. The synergistic effect of the dopant produces critical changes in the physical and chemical properties. Importantly, multicomponent hybrid TMHs promote synergistic effects, which reduce the adsorption energy of adsorbates (OH*, OOH*) and enhance overall conductivity. In short, combining TMHs with other components leads to multiple oxidation states and protects the individual components to improve the electrochemical performance of TMH-based nanomaterials under long-term processes against severe corrosion conditions.
4.6. Interlayer intercalation
The “layers” of the LDHs exhibit unique properties. The host layers provide a flexible confined space to accommodate various guest anions, which result in host–guest interactions such as electrostatic interactions, hydrogen-bonding. When specific non-organic or other anionic groups are applied to insert or replace the anion An− of the LDHs, the characteristics of the “layers” are altered, which has a significant effect on the physical and chemical properties of the LDHs.105,106Kim et al.108 developed a homogeneous precipitation method to prepare an α-Ni(OH)2 intercalated with dodecyl sulfate (α-Ni–(OH)2-DS). Subsequently, anion exchange was carried out to insert desired anions (Cl−, NO3−, AcO−, SO42−) without any change in morphology. α-Ni(OH)2 embedded with a smaller radius Cl− exhibits the largest specific capacitance in this series (Table 1, entry 14). Because Cl− ions are singly charged and fabulous hydrophilic, the exchange and transport of OH− ions are more facile. However, the doubly charged anions such as SO42− prevent OH− ion and water to enter the surface of Ni(OH)2, consequently, leading to low specific capacitance.
Wang et al.109 reported that Co-MOF as a template composited with different manganese salt solutions to form CoMn-LDH with different anion intercalations (Fig. 9(a)). Due to the effects of different anion metal salts, the synthesized LDH contains different layer spacing. At the same time, the MOFs can better control the 3D porous structure and increase the specific surface area. Among these LDHs, CoMn-LDH-SO4 has the largest lattice spacing and highest surface area, which exhibits the optimal supercapacitor performance, with an area capacity of 582.07 mC cm−2 and an energy density of 0.096 mW h cm−2 at a power density of 1.5 mW cm−2 (Table 1, entry 15).
 | ||
| Fig. 9 (a) Schematic of the synthesis of CoMn-LDH. Reproduced with permission from ref. 109 © 2019 American Chemical Society. (b) Schematic diagram for defect and stacking control of LDH NSs. (c) XRD pattern, (d) PDOS of Co–Al-LDH with an oxygen defect and (e) without an oxygen defect. When a water molecule develops a close interaction with metals for the Co–Al-LDH system with an oxygen defect, the PDOS of water is significantly broadened, in particular, for the occupied states. Reproduced with permission from ref. 110 © 2021 American Chemical Society. | ||
Kim et al. prepared CoAl-LDH nanosheets (LDH NSs) with variable stacking and pore structures by adjusting the size of the intercalator.110 A series of Na salts with different anions (Cl−, Br−, I−, and NO3−) are applied to restack the LDH NSs. The restacking and the embed of anions lead not only to an increase in the substrate spacing but also to significant elastic deformation of the peeled LDH NSs in Fig. 9(b) and (c). Lattice engineering simultaneously controls the number of stacks and O vacancies by tuning the size of the intercalator. DFT calculations reveal that the increase in the layer spacing will change the electrostatic potential in the lattice, which modifies the reducibility of Co3+ sites and directly affects the nearby O vacancy formation (Fig. 9(d) and (e)). Therefore, the defect-rich Co–Al-LDH-NO3− nanohybrid with a small stacking number displays excellent performance as an OER electrocatalyst (Table 2, entry 30) and a supercapacitor electrode (Table 1, entry 16).
Guo's team111 suggests that the structural stability of α-(Ni/Co)(OH)2 can be enhanced by introducing the inherent column effect of metaborate. The embedded metaborate pillars can be firmly bonded in the intermediate layer of the Ni–Co hydroxide, which stabilizes the structure of the electrodes, resulting in excellent cycle performance. At a current density of 5 A g−1, the average capacitance decay rate of the metaborate-stabilized α-(Ni/Co)(OH)2 is only ∼0.0017% after 10000 cycles (Table 1, entry 17).
All in all, anion insertion may expose more active sites, while the unique interlayer spaces in the catalytic process also avoid the loss of these active substances, thereby effectively improving the activity of the catalytic system. More importantly, the insertion of the guest anion into the LDH intermediate layer generates an attraction between the cation and guest anion, which may alter the stacking number or stack thickness or porosity of the layers.
Yang et al.112 studied the effects of the average interlayer spacing on the capacitance of NiMn LDHs. The layer spacing of NiMn LDHs is controlled (from 7.38 to 28.41 Å) by adding different amounts of sodium lauryl sulfate, while the morphology and composition are unchanged. The capacitance correlates positively with the average layer spacing of NiMn-LDH. NiMn-LDH4 with the largest interlayer spacing exhibits the highest specific capacity of 325 mA h g−1 at a current density of 1 A g−1 (Table 1, entry 18).
Qiu et al.42 enhanced the activity of layered cobalt hydroxide nanosheets as the H2O oxidation catalyst by adjusting the interlayer spacing. At first, dodecyl sulfate (DS−) inserted cobalt hydroxide is prepared. NO3− or CH3COO− inserted cobalt hydroxide NCs are then synthesized via anion exchange. When these three cobalt hydroxides are used as OER electrocatalysts in neutral phosphate-buffered saline, DS− inserted cobalt hydroxide induces the largest interlayer spacing and highest activity (Table 2, entry 31).
Zhang et al.113 used sodium oleate as a surfactant and an intercalant to prepare NiAl-LDH (NA-LDH-OA) nanosheets. The enlarged interlayer space and 2D frame structure could provide an effective pathway for ions and electron transmission, enhancing the electrochemical activity. Therefore, the assembled ASC device (NA-LDH-OA-2//AC) exhibited a high energy density of 40.26 W h kg−1 at a power density of 943 W kg−1 and maintained a good cycle performance of 94.5% after 5000 cycles (Table 1, entry 19).
Yan et al.114 used excess Na2C2O4 as an oxalate source and an intercalator to introduce cobalt oxalate ions [CoOx2]2− into the middle layer of Co(OH)2via a one-step solvothermal method to synthesize I-Co(OH)2 NSs. The embedded column ions maintain the excellent structural stability of the nanosheets, thereby enhancing the electrical conductivity and accelerating Li+ diffusion. As shown in Fig. 10(a)–(d), when the prepared I-Co(OH)2 NSs are used as the liquid lithium battery anode material, they exhibit superior rate performance (566 mA h g−1 at 5 A g−1) than that of undoped β-Co(OH)2 NSs and excellent cycle stability (870 mA h/g at 1 A g−1 after 250 cycles) (Table 1, entry 20). Fig. 10(e) shows the optimized atomic structures of the I-Co(OH)2 and [CoOx2]2− interlayers. The functional density calculation manifests that the intercalation of [CoOx2]2− leads to a higher dispersion of the band structure in Co(OH)2, thereby improving the Li+ diffusion kinetics and achieving high conductivity (Fig. 10(f)–(h)).
 | ||
| Fig. 10 Lithium-ion storage properties and electronic structure calculations of I-Co(OH)2 and β-Co(OH)2 NSs. (a and b) CV curves for the first four cycles. (c) Cycling performance and (d) rate capabilities. (e) Schematic illustration highlights the structural benefits of the anode during Li-ion diffusion. (f) Optimized atomic configurations, (g) density of states, and (h) charge-density difference. Reproduced with permission from ref. 114 © 2018 American Chemical Society. | ||
Kurungot et al.115 reported that [MoS4]2− was embedded into the interlayer space of NiCo-LDH via exchange with NO3−, forming an efficient catalyst material for urine to direct H2 production. X-ray photoelectron energy spectroscopy and electrochemical analysis illustrate that the embedded [MoS4]2− not only adjusts the interlayer spacing but also alters the overall electronic structure of NiCo-LDH, thereby reducing the internal potential and resulting in favorable kinetics for the HER. When this catalyst is used as a cathode and anode to produce a urea electrolytic cell, only ∼1.37 V cell potential is required to generate sufficient H2 and maintain long-term catalytic efficiency by reaching the benchmark 10 mA cm− in 1 M KOH/0.33 M urea. (Table 2, entry 32).
Liu's team116 applied K8[SiW11O39]·13H2O heteropoly acid as a hydrolysis and structural orientation agent to insert into NiFe LDHs, forming a 3D NiFe LDH-POM nano-flower-like structure, which was an efficient dual-functional catalyst for overall H2O splitting. Compared with the original NiFe LDH, W6+ alters the electronic structure of the active center, which minimizes the adsorption energy barrier of HO*, thereby improving the kinetics of the OER. The as-prepared catalyst achieves overall water splitting current density of 10 mA cm−2 at low overpotentials (OER: ∼200 mV; HER: ∼156 mV) in 0.1 M KOH over a period of 20 h operation (Table 2, entry 33).
Hence, LDHs are ideal supports for the immobilization of active species such as the large diameter active ions. Anions with larger embedded radii may expand the interlayer spaces of the LDHs, which can form an appropriate layer spacing to attenuate the resistance of the LDHs’ active component interaction with OH−.83 The facile electron transfer will facilitate the deprotonation/protonation reaction, achieving fast kinetics and thus high electrochemical performance.
5. Co-doping
A suitable dopant should show an ideal modulation effect on the host material, but sometimes a single dopant does not satisfy all requirements. Generally, further co-/multi-doping is required to enhance the advantages of heteroatom doping.32 Thus, for the co-doping strategy, we should not only consider the physical characteristics and theoretical requirements of the co-dopant but also confirm whether it may have a positive synergistic effect in TMHs under non-equilibrium growth conditions.5.1. Metal co-doping
The first choice of co-dopant may be two cations. Various multi-metal hydroxides can be prepared by two cations. Yuan et al.117 prepared Cu2+ and Ni3+ co-doped Ni(OH)2, using a simple one-step co-precipitation, which is an anode material of high-capacity, long-life Li-ion batteries. Cu2+ doping changes the nucleation conditions of Ni(OH)2 and increases the interlayer spacing, thereby promoting the migration of Li+ electrons and increasing the stability of the layered structure. Simultaneously, Cu2+ promotes the in situ generation of Ni3+, which improves the capacitances of Ni(OH)2. Therefore, the co-doped Ni(OH)2 exhibits an excellent lithium storage capability with an ultra-high first-discharge capacity of 1832.5 mA h g−1 at 0.2 A g−1. The reversible capacities reach 1230.1 mA h g−1 and 942.5 mA h g−1 after 800 and 1000 cycles at a rate of 1 A g−1 and 2 A g−1, respectively (Table 1, entry 21).Fan et al.118 prepared α-Ni(OH)2/graphene with co-doped metal ions (Al3+ and Co2+) by a co-precipitation approach. According to XRD analysis, Al3+ doping causes the phase to transform from β-Ni(OH)2 to α-Ni(OH)2 while Co2+ doping makes the interlayer distance of α-Ni(OH) increase. Due to its large interlayer distance, mesoporous structure and highly conductively graphene, the Al–Co co-doped α-Ni(OH)2/GNS (Al–Co–Ni/GNS) displays excellent specific capacitance (2257 F g−1 at 2 mV s−1) (Table 1, entry 22).
5.2. Non-metal co-doping
The co-doping strategy of two or more anion ions is also very common in the modification of TMHs.119 Kim et al. developed an S and P co-doping strategy to enhance the electrocatalytic activity and supercapacitor performance of NiCo-LDH.120 S and P co-doping can regulate the morphology of NiCo-LDH nanoarray (NiCo-LDH-SP). The heterogeneous porous structure effectively reduces charge transfer resistance, which achieves a rapid and efficient Faraday reaction. The assembled NiCo LDH-SP//AC supercapacitor behaves high energy density of 74.5 W h kg−1 at a power density of 0.8 kW kg−1 and maintains approximately 81.3% of the initial specific capacitance after 5000 cycles (Table 1, entry 23).Chen's group121 prepared a self-supporting B and P co-doped NiVFe LDHs@NF electrocatalyst for the HER. B-P co-doping formed numerous defects and amorphous regions on the nanosheets to ensure effective active sites and increase active surface area. In addition, the self-supporting structure enhances superhydrophilic and superhydrophobicity, which facilitates the electrolyte to enter the electrocatalyst, thereby promoting the occurrence of the HER. NiVFe-B-P LDH@NF exhibits excellent HER performance, with a low overpotential of 117 mV and a Tafel slope of 68 mV dec−1 at a current density of 10 mA cm−2 in a 1 M KOH electrolyte solution (Table 2, entry 34).
Zhu et al.122 employed egg whites as a heteroatom-doping precursor to prepare a sandwich structure of C and N co-doped nickel hydroxide/nickel sulfide (C/N–Ni(OH)2/NixSy). C-doping may improve the conductivity and cycle stability, and N-doping may expose more active sites, which enhances interactions between electrolytes and electrodes. The unique sandwich structure effectively prevents the reunion of Ni(OH)2 nanosheets and NixSy nanoparticles. C/N–Ni(OH)2/NixSy has an excellent capacitance of 1731.2 F g−1 at a current density of 0.5 A g−1. The assembled C/N–Ni(OH)2/NixSy//rGH supercapacitor displays a long-cycle performance and maintains 134.6% of the initial capacitance after 10000 cycles at the current density of 5 A g−1 (Table 1, entry 24).
5.3. Metal and non-metal co-doping
At the same time, metal and non-metal co-doping can improve the selectivity of the TMHs doping strategy. To change the electronic and thermodynamic properties of Co(OH)2, Xie et al. developed a K+/Cl− doped Co(OH)2.123 DFT calculations reveal that the substitution of OH− with Cl− can increase the adsorption energy for OH− and H2O to reduce the penalty originating from the coulombic repulsive interaction between hydrogen atoms of OH− and of Co(OH)2. At the same time, K replaces Co in the framework, which reduces the band gap to increase the conductivity (Fig. 11(a) and (b)). Electrochemical characterization confirms that the capacitances of K+/Cl−-doped Co(OH)2 are significantly enhanced (Fig. 11(c)–(e)). Compared with that of the original Co(OH)2, the capacitances of K+/Cl−-doped Co(OH)2 are increased by 139.6%, and the contained supercapacitor presents an energy density of 39.8 W h kg−1 at a power density of 478.6 W kg−1. (Table 1, entry 25). | ||
| Fig. 11 (a) Schematic illustration view of K+/Cl−-doped Co(OH)2. (b) Cl− doping can reduce the repulsive interaction between the inserted OH− with Co(OH)2 layers and the band gap properties of the K+/Cl−-doped Co(OH)2. Electrochemical performance of Co(OH)2 with different modifications. (c) CV curves at 5 mV s−1; (d) GCD curves at 5 mA cm−2; (e) areal capacitances at different current densities. Reproduced with permission from ref. 123 © 2021 American Chemical Society. | ||
Yang et al.124 reported the synthesis of phosphorus and yttrium co-doped Co(OH)F (YP-Co(OH)F) nanorod arrays on nickel foam for overall water splitting. Experimental results demonstrated that the Y and P co-doping (YP-Co(OH)F) achieves the tuning of the electronic environment, which effectively reduces the charge transfer resistance in the catalytic reaction. C-doping Y and P also induces more active centers to be exposed, thereby the properties of catalysts in H2O splitting are improved. The YP-Co(OH)F electrodes only required an overpotential of 238 mV to reach a current density of 10 mA cm−2 and exhibited an overpotential of 55 mV in the HER. When YP-Co(OH)F was used as the anode and cathode in a two-electrode structure, it only demands a cell potential of 1.54 V at 10 mA cm−2 and maintains stable water splitting for 300 h (Table 2, entry 35).
Li's team125 successfully introduced Mn(III) into 2D F-doped Ni(OH)2 to implement Mn and F co-doping strategies. The F atom has a high electrical negative property, and the doping can stabilize the structure of Ni(OH)2. At the same time, the presence of F causes minimal changes in the Ni (OH)2 internal structure, resulting in the loss of nearby oxygen atoms and generating vacancies on the surface of F/Ni(OH)2. Then, with the further introduction of Mn(III), the electron repulsion of the nickel hydroxide changes, and the oxygen-deficient content increases. DFT calculations further suggest that the introduction of O defects can significantly improve the OER catalytic performance. Mn and F co-doped Ni(OH)2–NF on Ni foam (Mn–F/Ni(OH)2–NF) proves to be an efficient catalyst for the OER. In 1 M KOH electrolyte, Mn–F/Ni(OH)2–NF has 233 mV overpotential at a current density of 20 mA cm−2, and the Tafel slope is 56.9 mV dec−1 (Table 2, entry 36).
6. Summary and outlook
In summary, this study reviews the recent progress of heteroatom doping in TMHs with regard to energy conversion and storage applications. According to the structural characteristics of TMHs, the engineering of heteroatom doping is divided into metal doping, non-metal doping, and co-doping. The doping principles and effects of different elements are discussed. As emphasized in this review, this heteroatom doping strategy results in a great improvement in the electrochemical applications of TMHs. (1) Heteroatom doping can change the structure and morphology of TMHs, increasing the activity specific surface area and the stability of the capacitors. (2) It can regulate the electronic coordination environments of THMs, and improve the charge transfer kinetics of catalytic reactions. (3) It increases metal active sites to optimize electrocatalytic adsorption energy.However, there are still several challenges limiting the doping engineering of TMH materials for energy conversion and storage applications: (1) there are no clear indicators on how to select appropriate dopants to achieve the desired effect. (2) Whether diverse doping methods will have a hold on the role of dopants, and how to choose doping methods. (3) It is difficult to control the doped techniques to achieve positive effects, and it may also disrupt the original crystal structure, resulting in the performance degradation of TMH activity. (4) The basic understanding of the structure and performance relationship of doped TMHs is still not clear enough. (5) The effect of dopants on electrode performance is not clear enough so in-depth analysis and investigation of the doping effect and principles are highly demanded.
The existence of these challenges also provides the following opportunities for researchers in this field:
(1) The feasible dopants can be classified according to their own characteristics, such as low/high valence atoms, low/high electronegativity atoms, etc.
(2) According to the characteristics of selected dopants and substrate materials, appropriate doping methods are selected and the effects are compared. Calcination doping can produce more vacancies, and hydrothermal doping can expand the interlayer spacing.
(3) Researchers should perform doping engineering without interfering with the inherent activity of TMHs. The categories, quantities and positions of dopants should be carefully controlled to ensure the controllability of the doping technique to achieve positive effects. From the characterization perspective, synchronous radiation and aberration-corrected high resolution electron microscopy can provide more information on the categories, quantities and positions of dopants.
(4) Researchers should also pay attention to the relationship between the dopant and the structure of TMH itself. The mechanism of action of doped atoms varies with the structure and composition of TMHs. They should pay more attention to how to obtain real-time information on the doped sites during catalytic processes through in situ methods. Numerous in situ technologies (such as in situ XPS, in situ Raman, in situ FTIR, in situ XRD, in situ TEM–SEM) can be employed to disclose the reaction mechanism and structure–activity relationship, which can be further confirmed by DFT calculations.
(5) Researchers should follow with interest the role of dopants in electrochemical performance. After understanding the coordination mechanism of dopants to TMHs, we should compare their practical application as an electrode. The causation for the breakthrough of the electrochemical performance of doped TMHs was explored. Thereby, an optimal doping method is explored through minor changes to prepare the most effective heteroatom doped TMHs.
Author contributions
The manuscript was written by all the authors. All authors approved the final version of the manuscript. Guoping Lu, Yuxin Tang and Pengcheng Wang designed the outline and revised the manuscript. Yaqi Qin, Feng Yang, Chunhua Xu, Shuaijie Jiang, Yuqiu Wang are responsible for searching the literature, writing, and modifying the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Natural Science Foundation of China [Grant No. 11972195].References
- M. A. M. Khan and Y. I. Go, Energy Storage, 2020, 3, 221–225 Search PubMed.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2020, 19, 1151–1163 CrossRef CAS PubMed.
- P. Ragupathy, S. D. Bhat and N. Kalaiselvi, WIREs Energy Environ., 2022, 11, 464 Search PubMed.
- C. Cao, F. Liang, W. Zhang, H. Liu, H. Liu, H. Zhang, J. Mao, Y. Zhang, Y. Feng, X. Yao, M. Ge and Y. Tang, Small, 2021, 17, 2102233 CrossRef CAS PubMed.
- L. Yao, N. Zhang, Y. Wang, Y. Ni, D. Yan and C. Hu, J. Power Sources, 2018, 374, 142–148 CrossRef CAS.
- Y. Gong, H. Zhao, D. Ye, H. Duan, Y. Tang, T. He, L. A. Shah and J. Zhang, Appl. Catal., A, 2022, 643, 118745 CrossRef CAS.
- M. Li, K. Yang, M. Abdinejad, C. Zhao and T. Burdyny, Nanoscale, 2022, 14, 11892–11908 RSC.
- R. Liu, A. Zhou, X. Zhang, J. Mu, H. Che, Y. Wang, T.-T. Wang, Z. Zhang and Z. Kou, Chem. Eng. J., 2021, 412, 128611 CrossRef CAS.
- R. Gao and D. Yan, Adv. Energy Mater., 2019, 10, 1900954 CrossRef.
- S. Fang, D. Bresser and S. Passerini, Adv. Energy Mater., 2020, 10, 1902485 CrossRef CAS.
- D. Wei, L. Chen, L. Tian, S. Ramakrishna and D. Ji, J. Chem. Technol. Biotechnol., 2022, 97 DOI:10.1002/jctb.7294.
- R. Patel, J. T. Park, M. Patel, J. K. Dash, E. B. Gowd, R. Karpoormath, A. Mishra, J. Kwak and J. H. Kim, J. Mater. Chem. A, 2018, 6, 12–29 RSC.
- L. Wan and P. Wang, Int. J. Hydrogen Energy, 2021, 46, 8356–8376 CrossRef CAS.
- J. M. Gonçalves, M. I. da Silva, H. E. Toma, L. Angnes, P. R. Martins and K. Araki, J. Mater. Chem. A, 2020, 8, 10534–10570 RSC.
- P. Li, T. Zhang, M. A. Mushtaq, S. Wu, X. Xiang and D. Yan, Chem. Rec., 2021, 21, 841–857 CrossRef CAS PubMed.
- D. Yan, J. Lu, L. Chen, S. Qin, J. Ma, M. Wei, D. G. Evans and X. Duan, Chem. Commun., 2010, 46, 5912–5914 RSC.
- Z. Guo, W. Ye, X. Fang, J. Wan, Y. Ye, Y. Dong, D. Cao and D. Yan, Inorg. Chem. Front., 2019, 6, 687–693 RSC.
- R. Gao, J. Zhu and D. Yan, Nanoscale, 2021, 13, 13593–13603 RSC.
- W. Hu, L. Chen, M. Du, Y. Song, Z. Wu and Q. Zheng, Electrochim. Acta, 2020, 338, 135869 CrossRef CAS.
- J. Sun, N. Guo, Z. Shao, K. Huang, Y. Li, F. He and Q. Wang, Adv. Energy Mater., 2018, 8, 1800980 CrossRef.
- J. Lv, X. Yang, H.-Y. Zang, Y.-H. Wang and Y.-G. Li, Mater. Chem. Front., 2018, 2, 2045–2053 RSC.
- J. Yang, C. Yu, X. Fan and J. Qiu, Adv. Energy Mater., 2014, 4, 1400761 CrossRef.
- J. Wang, Y. Zhang, J. Ye, H. Wei, J. Hao, J. Mu, S. Zhao and S. Hussain, RSC Adv., 2016, 6, 70077–70084 RSC.
- X. Gao, Y. Zhao, K. Dai, J. Wang, B. Zhang and X. Shen, Chem. Eng. J., 2020, 384, 70077–70084 CrossRef.
- R. Liu, Y. Wang, D. Liu, Y. Zou and S. Wang, Adv. Mater., 2017, 29, 1701546 CrossRef PubMed.
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC.
- X. Li, D. Du, Y. Zhang, W. Xing, Q. Xue and Z. Yan, J. Mater. Chem. A, 2017, 5, 15460–15485 RSC.
- C. Jing, B. Dong and Y. Zhang, Energy Environ. Mater., 2020, 3, 346–379 CrossRef CAS.
- R. Boppella, J. Tan, J. Yun, S. V. Manorama and J. Moon, Coord. Chem. Rev., 2021, 427, 213552 CrossRef CAS.
- Y. Liu, W. Wang, X. Xu, J.-P. M. Vederb and Z. Shao, J. Mater. Chem. A, 2019, 7, 7280 RSC.
- J. Zhang, K. Tse, M. Wong, Y. Zhang and J. Zhu, Front. Phys., 2016, 11, 117405 CrossRef.
- S. Ghosh, S. Barg, S. M. Jeong and K. Ostrikov, Adv. Energy Mater., 2020, 10, 2001239 CrossRef CAS.
- J. Li, M. Zhang, H. Zang, B. Yu, Y. Ma and Y. Qu, ChemCatChem, 2019, 11, 4998–5012 CrossRef CAS.
- Y. Li, M. Chen, B. Liu, Y. Zhang, X. Liang and X. Xia, Adv. Energy Mater., 2020, 10, 2000927 CrossRef CAS.
- S. Y. Jung, K. M. Kim, S. Mhin, Y.-K. Kim, J. H. Ryu, E. Enkhtuvshin, S. J. Kim, N. T. T. Thao, E. Choi, T. Song and H. Han, Int. J. Energy Res., 2022, 46, 11972–11988 CrossRef CAS.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. D. Araujo, M. Gliech, D. Teschner, J. Zhu, W.-X. Li, J. Greeley, B. R. Cuenya and P. Strasse, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed.
- X. Liu, X. Fan, H. Huang, H. Lin and J. Gao, J. Colloid Interface Sci., 2021, 587, 385–392 CrossRef CAS PubMed.
- D. Wang, Q. Li, C. Han, Q. Lu, Z. Xing and X. Yang, Nat. Commun., 2019, 10, 3899 CrossRef PubMed.
- B. Zhao, L. Zhang, Q. Zhang, D. Chen, Y. Cheng, X. Deng, Y. Chen, R. Murphy, X. Xiong, B. Song, C. P. Wong, M. S. Wang and M. Liu, Adv. Energy Mater., 2017, 8, 1702247 CrossRef.
- M. Ulaganathan, M. M. Maharjan, Q. Yan, V. Aravindan and S. Madhavi, Chem. – Asian J., 2017, 12, 2127–2133 CrossRef CAS PubMed.
- R. Qin, H. Wan, X. Liu, G. Chen, N. Zhang, R. Ma and G. Qiu, Inorg. Chem. Front., 2019, 6, 1744–1752 RSC.
- M. G. Siebecker and D. L. Sparks, J. Phys. Chem. A, 2017, 121, 6992–6999 CrossRef CAS PubMed.
- C. J. Oluigbo, Y. Xu, H. Louis, A. B. Yusuf, W. Yaseen, N. Ullah, K. J. Alagarasan, M. Xie, E. E. Ekpenyong and J. Xie, Appl. Surf. Sci., 2021, 562, 150161 CrossRef CAS.
- C. Zhao, S. Liang, Y. Jiang, F. Gao, L. Xie and L. Chen, Mater. Lett., 2020, 270, 127751 CrossRef CAS.
- Z. Zhang, H. Huo, L. Wang, S. Lou, L. Xiang, B. Xie, Q. Wang, C. Du, J. Wang and G. Yin, Chem. Eng. J., 2021, 412, 128617 CrossRef CAS.
- X. Ren, Z. Gan, M. Sun, Q. Fang, Y. Yan, Y. Sun, J. Huang, B. Cao, W. Shen, Z. Li and Y. Fu, Electrochim. Acta, 2022, 414, 140208 CrossRef CAS.
- Y. Du, G. Li, L. Ye, C. Che, X. Yang and L. Zhao, Chem. Eng. J., 2021, 417, 129189 CrossRef CAS.
- X. Zhang, H. Yi, M. Jin, Q. Lian, Y. Huang, Z. Ai, R. Huang, Z. Zuo, C. Tang, A. Amini, F. Jia, S. Song and C. Cheng, Small, 2022, 18, 2203710 CrossRef CAS PubMed.
- W. Ge, W. Peng, A. Encinas, M. F. Ruiz and S. Song, Chem. Phys., 2019, 521, 55–60 CrossRef CAS.
- J. Wei, D. Qiu, M. Li, Z. Xie, A. Gao, H. Liu, S. Yin, D. Yang and R. Yang, RSC Adv., 2019, 9, 10237–10244 RSC.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- L. J. Enman, M. S. Burke, A. S. Batchellor and S. W. Boettcher, ACS Catal., 2016, 6, 2416–2423 CrossRef CAS.
- Q. Cao, M. Luo, Y. Huang, Q. Liu, X. Kong, J. Lei, Z. Jiang and J. Wang, Sustainable Energy Fuels, 2020, 4, 1522–1531 RSC.
- Y. Zheng, R. Gao, Y. Qiu, L. Zheng, Z. Hu and X. Liu, Inorg. Chem., 2021, 60, 5252–5263 CrossRef CAS PubMed.
- F. Huang, B. Yao, Y. Huang and Z. Dong, Int. J. Hydrogen Energy, 2022, 47, 21725–21735 CrossRef CAS.
- Hanyue Ma, Rui Gao, Dongpeng Yan, J. Zhao and M. Wei, J. Mater. Chem. C, 2013, 1, 4128 RSC.
- Y. Li, L. Zhang, X. Xiang, D. Yan and F. Li, J. Mater. Chem. A, 2014, 2, 13250–13258 RSC.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- S. Li, C. Xi, Y.-Z. Jin, D. Wu, J.-Q. Wang, T. Liu, H.-B. Wang, C.-K. Dong, H. Liu, S. A. Kulinich and X.-W. Du, ACS Energy Lett., 2019, 4, 1823–1829 CrossRef CAS.
- Y. Zhang, C.-Q. Cheng, C.-G. Kuai, D. Sokaras, X.-L. Zheng, S. Sainio, F. Lin, C.-K. Dong, D. Nordlund and X.-W. Du, J. Mater. Chem. A, 2020, 8, 17471–17476 RSC.
- X. Yang, H. Zhang, W. Xu, B. Yu, Y. Liu and Z. Wu, Catal. Sci. Technol., 2022, 12, 4471–4485 RSC.
- Q. Zhang, S. Zhang, Y. Tian and S. Zhan, ACS Sustainable Chem. Eng., 2018, 6, 15411–15418 CrossRef CAS.
- H. Xu, C. Shan, X. Wu, M. Sun, B. Huang, Y. Tang and C.-H. Yan, Energy Environ. Sci., 2020, 13, 2949–2956 RSC.
- M. Rong, H. Zhong, S. Wang, X. Ma and Z. Cao, Colloids Surf., A, 2021, 625, 126896 CrossRef CAS.
- Y. S. Park, F. Liu, D. Diercks, D. Braaten, B. Liu and C. Duan, Appl. Catal., B, 2022, 318, 121824 CrossRef CAS.
- J. Han, J. Zhang, T. Wang, Q. Xiong, W. Wang, L. Cao and B. Dong, ACS Sustainable Chem. Eng., 2019, 7, 13105–13114 CrossRef CAS.
- S. Y. Lim, S. Park, S. W. Im, H. Ha, H. Seo and K. T. Nam, ACS Catal., 2019, 10, 235–244 CrossRef.
- W. Hu, L. Chen, X. Wu, M. Du, Y. Song, Z. Wu and Q. Zheng, ACS Appl. Mater. Interfaces, 2021, 13, 38346–38357 CrossRef CAS PubMed.
- Y. Shi, J. Li, B. Zhang, S. Lv, T. Wang and X. Liu, Appl. Surf. Sci., 2021, 565, 150506 CrossRef CAS.
- N. L. W. Septiani, Y. V. Kaneti, Y. Guo, B. Yuliarto, X. Jiang, Y. Ide, N. Nugraha, H. K. Dipojono, A. Yu, Y. Sugahara, D. Golberg and Y. Yamauchi, ChemSusChem, 2020, 13, 1645–1655 CrossRef CAS PubMed.
- L. Zeng, B. Cao, X. Wang, H. Liu, J. Shang, J. Lang, X. Cao and H. Gu, Nanoscale, 2021, 13, 3153–3160 RSC.
- Z. Wang, W. Liu, Y. Hu, L. Xu, M. Guan, J. Qiu, Y. Huang, J. Bao and H. Li, Inorg. Chem. Front., 2019, 6, 1890–1896 RSC.
- H. Li, C. Zhang, W. Xiang, M. A. Amin, J. Na, S. Wang, J. Yu and Y. Yamauchi, Chem. Eng. J., 2023, 452, 139104 CrossRef CAS.
- X. Wang, Y. Tuo, Y. Zhou, D. Wang, S. Wang and J. Zhang, Chem. Eng. J., 2021, 403, 126297 CrossRef CAS.
- Z. Wang, W. Liu, Y. Hu, M. Guan, L. Xu, H. Li, J. Bao and H. Li, Appl. Catal., B, 2020, 272, 118959 CrossRef CAS.
- L. Wen, X. Zhang, J. Liu, X. Li, C. Xing, X. Lyu, W. Cai, W. Wang and Y. Li, Small, 2019, 15, 1902373 CrossRef PubMed.
- Y. Tanga, Q. Liua, L. Dongb, H. B. Wua and X.-Y. Yu, Appl. Catal., B, 2020, 266, 118627 CrossRef.
- Y. Yang, Y. Ou, Y. Yang, X. Wei, D. Gao, L. Yang, Y. Xiong, H. Dong, P. Xiao and Y. Zhang, Nanoscale, 2019, 11, 23296–23303 RSC.
- P. Li, X. Duan, Y. Kuang, Y. Li, G. Zhang, W. Liu and X. Sun, Adv. Energy Mater., 2018, 8, 1703341 CrossRef.
- Y. Kong, Y. Wang, W. Chu and Z. Liu, J. Alloys Compd., 2021, 885, 160929 CrossRef CAS.
- B. Q. Li, S. Y. Zhang, C. Tang, X. Cui and Q. Zhang, Small, 2017, 13, 1700610 CrossRef PubMed.
- T. Sudare, M. Dubois, N. Louvain, M. Kiyama, F. Hayashi and K. Teshima, Inorg. Chem., 2020, 59, 1602–1610 CrossRef CAS PubMed.
- P. Zhou, C. Wang, Y. Liu, Z. Wang, P. Wang, X. Qin, X. Zhang, Y. Dai, M.-H. Whangbo and B. Huang, Chem. Eng. J., 2018, 351, 119–126 CrossRef CAS.
- Z. Shi, J. Zhu, Z. Li, Q. Xiao and J. Zhu, ACS Appl. Energy Mater., 2020, 3, 11082–11090 CrossRef CAS.
- S. Li, J. Liu, S. Duan, T. Wang and Q. Li, Chin. J. Catal., 2020, 41, 847–852 CrossRef CAS.
- J. Zheng, X. Lian, M. Wu, F. Zheng, Y. Gao and H. Niu, Diamond Relat. Mater., 2021, 116, 108451 CrossRef CAS.
- K. Li, B. Zhao, J. Bai, H. Ma, Z. Fang, X. Zhu and Y. Sun, Small, 2020, 16, 2001974 CrossRef CAS PubMed.
- W. Liu, Y. Chen, Y. Wang, Q. Zhao, L. Chen, W. Wei and J. Ma, Energy Storage Mater., 2021, 37, 336–344 CrossRef.
- S. Luo, L. Qian, M. Liao, X. Hu and D. Xiao, RSC Adv., 2017, 7, 45294–45303 RSC.
- C. Huang, J. Nie, Z. Xu, X. Zhang, J. Tang, B. Wang, J. Huang, C. Du and J. Chen, Int. J. Hydrogen Energy, 2021, 46, 12992–13000 CrossRef CAS.
- Z. Yang, Y. Zhang, C. Feng, H. Wu, Y. Ding and H. Mei, Int. J. Hydrogen Energy, 2021, 46, 25321–25331 CrossRef CAS.
- L. Han, L. Diao, K. Qin, J. Li, J. Sha, L. Ma and N. Zhao, Mater. Lett., 2020, 263, 127162 CrossRef CAS.
- S. Dou, L. Tao, J. Huo, S. Wang and L. Dai, Energy Environ. Sci., 2016, 9, 1320–1326 RSC.
- Y. Wang, C. Xie, Z. Zhang, D. Liu, R. Chen and S. Wang, Adv. Funct. Mater., 2018, 28, 1703363 CrossRef.
- K.-H. Kim, J. W. Choi, H. Lee, B. C. Moon, D. G. Park, W. H. Choi and J. K. Kang, J. Mater. Chem. A, 2018, 6, 23283–23288 RSC.
- S. Hu, Y. Tan, C. Feng, H. Wu, J. Zhang and H. Mei, J. Power Sources, 2020, 453, 1803358 CrossRef.
- R. S. Mulliken, J. Chem. Phys., 1934, 2, 782–793 CrossRef CAS.
- C. Pei, Y. Gu, Z. Liu, X. Yu and L. Feng, ChemSusChem, 2019, 12, 3849–3855 CrossRef CAS PubMed.
- Y. Tong, H. Mao, P. Chen, Q. Sun, F. Yan and F. Xi, Chem. Commun., 2020, 56, 4196–4199 RSC.
- N. Hussain, W. Yang, J. Dou, Y. Chen, Y. Qian and L. Xu, J. Mater. Chem. A, 2019, 7, 9656–9664 RSC.
- L. Chen, J. Chang, Y. Zhang, Z. Gao, D. Wu, F. Xu, Y. Guo and K. Jiang, Chem. Commun., 2019, 55, 3406–3409 RSC.
- Y. Kou, J. Liu, Y. Li, S. Qu, C. Ma, Z. Song, X. Han, Y. Deng, W. Hu and C. Zhong, ACS Appl. Mater. Interfaces, 2018, 10, 796–805 CrossRef CAS PubMed.
- N. Mahmood, M. Tahir, A. Mahmood, J. Zhu, C. Cao and Y. Hou, Nano Energy, 2015, 11, 267–276 CrossRef CAS.
- L. Lv, Z. Yang, K. Chen, C. Wang and Y. Xiong, Adv. Energy Mater., 2019, 9, 803358 Search PubMed.
- G. M. Tomboc, J. Kim, Y. Wang, Y. Son, J. Li, J. Y. Kim and K. Lee, J. Mater. Chem. A, 2021, 9, 4528–4557 RSC.
- Y. Wu, J. Chen, C. Li, F. Li, K. Liu, H. Liu, S. Sang and Q. Wu, J. Phys. Chem. Solids, 2019, 124, 352–360 CrossRef CAS.
- J. W. Lee, J. M. Ko and J.-D. Kim, J. Phys. Chem. C, 2011, 115, 19445–19454 CrossRef CAS.
- X. Liu, L. Zhang, X. Gao, C. Guan, Y. Hu and J. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 23236–23243 CrossRef CAS PubMed.
- N. Kim, T. H. Gu, D. Shin, X. Jin, H. Shin, M. G. Kim, H. Kim and S. J. Hwang, ACS Nano, 2021, 15, 8306–8318 CrossRef CAS PubMed.
- Y. Chen, W. K. Pang, H. Bai, T. Zhou, Y. Liu, S. Li and Z. Guo, Nano Lett., 2017, 17, 429–436 CrossRef CAS PubMed.
- X. Wang, Z. Li, J. Zhang, H. Yan, C. Wang, F. Wu, A. Tian, X. Hong, W. Dong and S. Yang, Chem. Eng. J., 2020, 398, 125618 CrossRef CAS.
- H. Zhang, M. Usman Tahir, X. Yan, X. Liu, X. Su and L. Zhang, Chem. Eng. J., 2019, 368, 905–913 CrossRef CAS.
- C. Yan, Z. Fang, C. Lv, X. Zhou, G. Chen and G. Yu, ACS Nano, 2018, 12, 8670–8677 CrossRef CAS PubMed.
- A. Nadeema, V. Kashyap, R. Gururaj and S. Kurungot, ACS Appl. Mater. Interfaces, 2019, 11, 25917–25927 CrossRef CAS PubMed.
- C. Li, Z. Zhang and R. Liu, Small, 2020, 16, 2003777 CrossRef CAS PubMed.
- J. Zhang, S. Li, R. Hu and B. Yuan, Ionics, 2021, 27, 2053–2066 CrossRef CAS.
- X. Chen, C. Long, C. Lin, T. Wei, J. Yan, L. Jiang and Z. Fan, Electrochim. Acta, 2014, 137, 352–358 CrossRef CAS.
- H. J. Song, H. Yoon, B. Ju, G.-H. Lee and D.-W. Kim, Adv. Energy Mater., 2018, 8, 1802319 CrossRef.
- K. S. Kim, N. M. Shinde, J. M. Yun and K. H. Kim, RSC Adv., 2021, 11, 12449–12459 RSC.
- X. Ma, S. Zhang, Y. He, T. He, H. Li, Y. Zhang and J. Chen, J. Electroanal. Chem., 2021, 886, 115107 CrossRef CAS.
- Y. Zhang, L. Yu, R. Hu, J. Zhang, Y. Wang, R. Niu, X. Qian and J. Zhu, J. Mater. Chem. A, 2018, 6, 17417–17425 RSC.
- Z. Sun, Z. Cao, J. Fu, Y. Zhang, Y. Liu, S. Cheng, P. Cui and E. Xie, ACS Appl. Electron. Mater., 2021, 3, 395–405 CrossRef CAS.
- G. Zhang, B. Wang, L. Li and S. Yang, Small, 2019, 15, 1904105 CrossRef CAS PubMed.
- J. Lv, X. Yang, K. Li, X. Chen, S. Sun, H.-Y. Zang, Y.-F. Chang, Y.-H. Wang and Y.-G. Li, Nanoscale Adv., 2019, 1, 4099–4108 RSC.
Footnote |
| † These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2023 |