
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTough polyurethane elastomers with high strength and rapid healing ability†
Chenghui
Qiao
a,
Xiurui
Jian
a,
Zhengguo
Gao
a,
Qingfu
Ban
*a,
Xintao
Zhang
b,
Huimin
Wang
a and
Yaochen
Zheng
 *a
*a
aDepartment of Polymer Science and Engineering, College of Chemistry and Chemical Engineering, Yantai University, 30 Qingquan Road, Yantai 264005, P. R. China. E-mail: banqingfu@163.com; zhengyaochen@163.com
bSchool of environmental and material engineering, Yantai University, 30 Qingquan Road, Yantai, P. R. China
First published on 8th March 2023
Abstract
Polymers are often susceptible to premature failure due to various physical damages. Incorporation of reversible disulfide bonds with sufficient chain diffusion in polyurethane (PU) elastomers endows them with good healable and recyclable properties, effectively prolonging their service life. However, such healable elastomers frequently exhibit poor toughness and strength. Here, a series of disulfide-containing linear poly(urea-urethane)s are facilely fabricated using 4,4′-dithiodianiline (DTDA) and adipic acid dihydrazide (AD) as chain extenders. The produced elastomer with hierarchical hydrogen bonds has a high tensile strength (60.24 MPa), strain (1489.2%) and toughness (257.24 MJ m−3), respectively. Within only 5 h, isopropyl alcohol-assisted healing at 30 °C partially regains tensile strength (44.4 MPa), strain (1254.4%) and toughness (179.52 MJ m−3). The combination of hierarchical hydrogen bonds and disulfide bonds allows the achieved elastomer to maintain high mechanical properties and a healing efficiency of approximately 80%. This study facilitates the design and fabrication of PU elastomers with high mechanical performance and healing efficiency.
Introduction
Polyurethane (PU) elastomers with regularly alternating hard domains and soft domains have been used in a variety of fields, including adhesives, coatings, textile fibers, etc.1–4 PU elastomers are often susceptible to various mechanical damages during their long-term service. Near the tip of the damage or crack, stress concentration can occur, which is a key factor in PU elastomer destruction.5,6 If such damage is restored quickly, it may not deteriorate the material's performance or reduce its service life. Therefore, self-healing is important and desired for PU elastomers.7–9Inspired by the self-healing behavior of human skin, dynamic or reversible bonds are introduced in PU chains.10 These dynamic bonds can be easily destroyed and remedied, completing self-healing.11,12 Specifically, with the assistance of temperature, ultraviolet or visible light, solvents, moisture, etc., the movements of polymer chains can be activated, promoting the reconstitution of bonds or structures within PUs.13–16 Recently, several seminal works have developed self-healing PUs. Based on ester–exchange reactions, Diels–Alder addition,17,18 disulfide bonds,19–21 B–O bonds,22,23 Schiff bases,24,25etc., such dynamic bonds have been incorporated into PUs, generating various healable materials. In general, these materials show rapid healing rates and high healing capabilities.26 Typically, dithiodiphenylamine with two benzene rings has been used to synthesize tough and self-healing PU elastomers.27 The healing efficiency can reach 75% at room temperature in 2 h. However, the mechanical properties of the fabricated PUs are poor due to these weaker dynamic bonds.28,29
Compared to PUs with weakly dynamic covalent bonds, the incorporation of reversible hydrogen bonds into the molecular backbone or side chains can produce PU elastomers with improved mechanical strengths.30–32 Adipic dihydrazide is used as the chain extender for the fabrication of PU elastomers involving abundant hydrogen bonds.33 With regularly dispersed hard and soft domains, the developed PU elastomer achieved a superhigh tensile strength (75.6 MPa) and toughness (390.2 MJ m−3), which were much greater than those of spider silk, one of the most robust natural materials. Additionally, the building block ureidopyrimidone (UPy) can be introduced into PU chains by a polycondensation reaction to improve their tensile strength and toughness by the formation of strong hydrogen bonding interactions among UPy pendant groups.34,35 Due to their reversible nature, PUs containing abundant hydrogen bonds can also self-heal at a relatively high temperature (such as ≥100 °C) and long time (≥24 h).36,37 Therefore, high strength, rapid healing ability and low processing temperature are in conflict with each other.
To address this issue, we introduce disulfides and multiple hydrogen bonds into the backbone of PUs simultaneously via the polycondensation of polytetramethylene glycol (Mn = 2000 g mol−1), isophorone diisocyanate, 4,4′-dithiodianiline and adipic dihydrazide. The weak disulfide bonds embedded in the strong hydrogen bonds of urea bonds are the hard domains of PUs. Robust urea bonds effectively mitigate premature damage to the disulfides, producing higher tensile strengths. Also, if two sulfur free radicals are sufficiently nearby, the broken disulfide bond can reform immediately. Thus, the incorporated disulfides can improve healing efficiency.38 The achieved elastomer with abundant hydrogen bonds has a high tensile strength (60.24 MPa), strain (1489.2%) and toughness (257.24 MJ m−3), respectively. At 30 °C for 5 h, the healing efficiency of PU elastomers can approach 70–80% with IPA. This study describes an easy method to trade off high strength and low healing efficiency.
Experimental
Materials
Polytetramethylene glycol (PTMEG, Mn = 2000 g mol−1) was provided by Guangzhou Haoyi Co. Ltd. Isophorone diisocyanate (IPDI, 99%), dibutyltin dilaurate (DBTDL, 95%), adipic dihydrazide (AD, 99%), 4,4′-dithiodianiline (DTDA, 98%) and anhydrous N,N-dimethylacetamide (DMAc, 99%) were all purchased from Shanghai Aladdin Co. Ltd. Absolute alcohol, isopropyl alcohol, acetone and n-hexane were purchased from Sinopharm Chemical Reagent Co. Ltd. All solvents were used as received.Synthesis of polyurethane elastomers
PTMEG (20.00 g, 10.0 mmol) was dried under vacuum at 120 °C for 2 h to remove moisture. After cooling to 70 °C, IPDI (4.49 g, 20.0 mmol), DMAc (5.0 g) and DBTDL (0.04 g) were added, and the mixture was heated under a nitrogen atmosphere at 70 °C for 3 h. Then, the viscous product was cooled to 50 °C. The chain extenders (10.0 mmol), DTDA and AD with varying mole ratios of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, 4:6, 5:5, 6:4 and 7:3, were dissolved in DMAc (50.0 g) and added into this system. The reaction was continued for another 16 h at 50 °C under nitrogen reflux. Then, the mixture was transferred into a PTFE mold and dried at 70 °C to fully remove DMAc. The obtained polyurethane samples with a thickness of approximately 0.3 mm were named PU3:7, PU4:6, PU5:5, PU6:4 and PU7:3 for later characterization.
:7, 4:6, 5:5, 6:4 and 7:3, were dissolved in DMAc (50.0 g) and added into this system. The reaction was continued for another 16 h at 50 °C under nitrogen reflux. Then, the mixture was transferred into a PTFE mold and dried at 70 °C to fully remove DMAc. The obtained polyurethane samples with a thickness of approximately 0.3 mm were named PU3:7, PU4:6, PU5:5, PU6:4 and PU7:3 for later characterization.
Measurements and characterization
Fourier transform infrared spectroscopy (FT-IR) spectra were recorded on an IR spectrometer (Shimadzu) using the KBr disk method at room temperature. Nuclear magnetic resonance (NMR) tests were performed with an NMR spectrometer (JEOL 400 MHz, Japan) at 25 °C using deuterated chloroform as the solvent. Gel permeation chromatography (GPC) tests (Waters 1515 HPLC) were performed in THF (1.0 mL min−1) to determine the molecular weight and molecular weight distribution (PDI). Ultraviolet-visible (UV-vis, Varian Cary 300) spectroscopy was used to evaluate the transparency of the sample. Differential scanning calorimetry (DSC, 200 F3, NETZSCH) analyses were used to characterize each sample's thermal properties. The samples (approximately 10 mg) were heated from −100 to 100 °C at a heating rate of 20 °C min−1. Wide angle X-ray diffraction (XRD, XRD-7000, Shimadzu) patterns were recorded from 5° to 50° with Cu-Kα radiation (λ = 0.154 nm). Small-angle X-ray scatter tests were conducted on an Anton Paar system (SAXS, Saxesess mc2), and Cu-Kα radiation (λ = 0.154 nm) was used as the X-ray source. Dynamic mechanical analysis (DMA, Q800, TA Instruments) was performed in tension mode from −100 to 120 °C at a heating rate of 5 °C min−1 at a frequency of 10 Hz. Optical microscopy (OM, Olympus DP72) images were used to evaluate healable behaviors, and tensile measurements were performed on a universal testing machine (UTM 4103, Shenzhen Sans) at a stretching speed of 50 mm min−1. Dumbbell-shaped samples were prepared with a size of 35.0 mm × 2.0 mm × 0.3 mm.Results and discussion
Synthesis strategy and characterization
A one-pot, two-step polycondensation reaction was designed and used to synthesize tough and healable PU elastomers. As shown in Fig. 1, the first condensation reaction occurred between the hydroxy group from the PTMEG diol and the isocyanate group from the IPDI monomer, generating stable carbamate hydrogen bonds and forming the soft segments of PU elastomers. Under low stress, the soft segments could undergo much larger deformations, which are related to the chain conformations and derived from the change in conformational entropy (ΔS).37 In this study, the used PTMEG diol with a moderate molecular weight (Mn = 2000 g mol−1) had a sufficiently high mobility and diffusion ability, which was a critical prerequisite for healing behavior.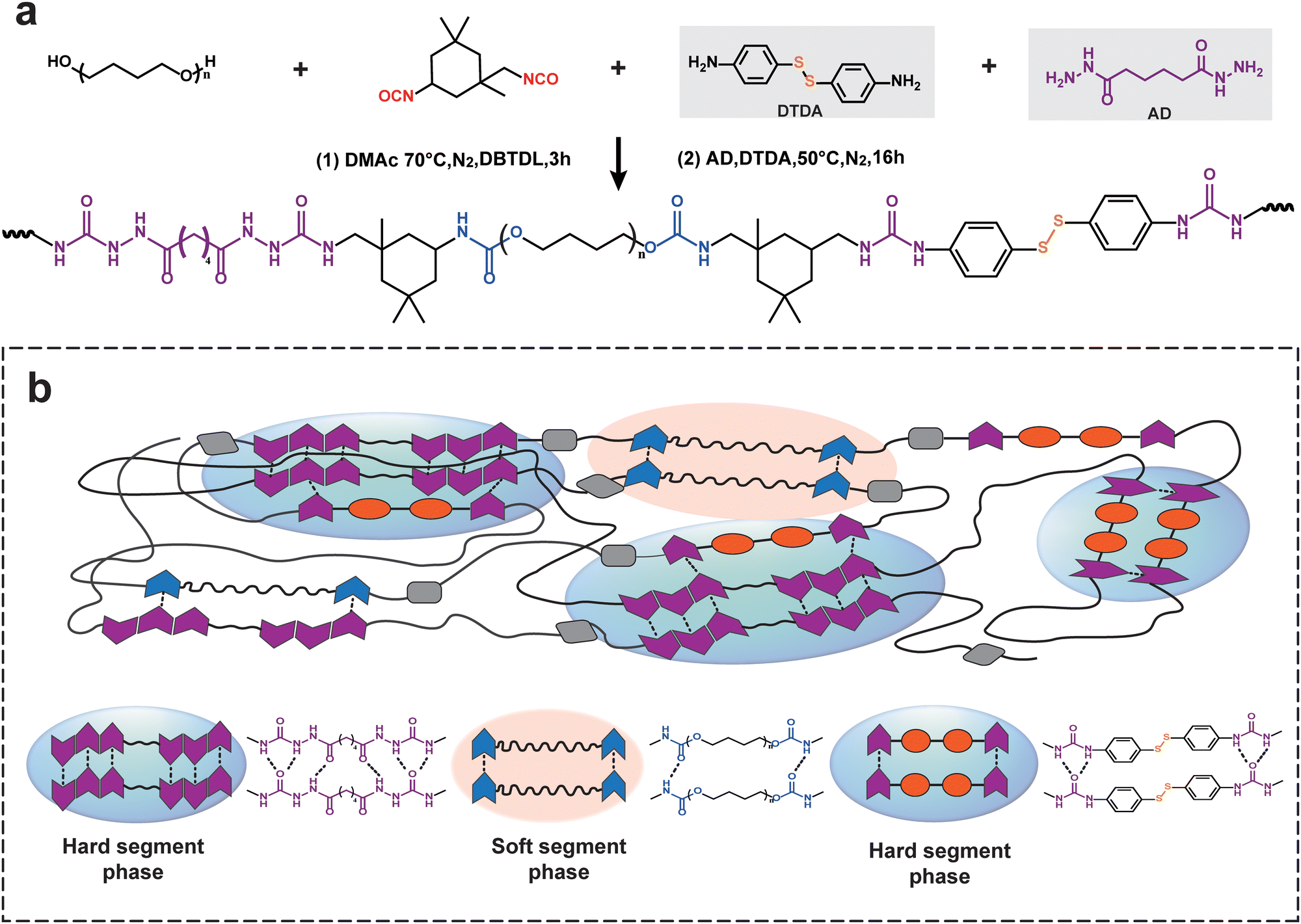 | ||
| Fig. 1 (a) Synthetic route of the PU elastomer. (b) Schematic illustration of the structure of the PU elastomer and intermolecular hydrogen bonds in the PU elastomer. | ||
In addition, the subsequent polycondensation reaction of the amino group and isocyanate group efficiently formed many urea bonds, offering active hard segments.39 These hard segment domains containing dynamic disulfide bonds and hierarchical hydrogen bonds (specifically, urea and carbamate bonds) were reversible because they are both exchangeable under the given conditions. The disulfides could recover at once as two sulfur free radicals meet each other, which confirmed the rapid healing efficiency of PUs. Moreover, dynamic disulfide bonds have been designed for and were generated in urea hydrogen bond-enriched regions. These robust hydrogen bonds protected the weak disulfides from premature failure. Concurrently, these hydrogen bonds improved the interactions of the repair surfaces, providing a higher healed tensile strength. Therefore, the resultant PU elastomer had regularly alternating soft and hard segments, exhibiting remarkable toughness and rapid healing efficiency.
For the fabrication of PU elastomers, the procedure was convenient, highly efficient and controllable. To confirm the successful synthesis of the PU elastomer, 1H NMR spectroscopy (Fig. 2a and Fig. S1, ESI†) and FTIR spectroscopy (Fig. 2b and c) were performed. The 1H NMR spectrum of the PU elastomer showed a group of characteristic signals at 1.60 and 2.23 ppm (“a” and “b”, respectively) arising from the protons of methylene belonging to the AD fragment. The signals at 6.58 and 7.29 ppm (“i” and “h”, respectively) arising from the protons of the aromatic ring belonged to the DTDA fragment. 1H-NMR resonances at 3.33–3.48 and 4.03 ppm (“e” and “e'”, respectively) could be attributed to the methylene protons of the PTMEG moiety.40
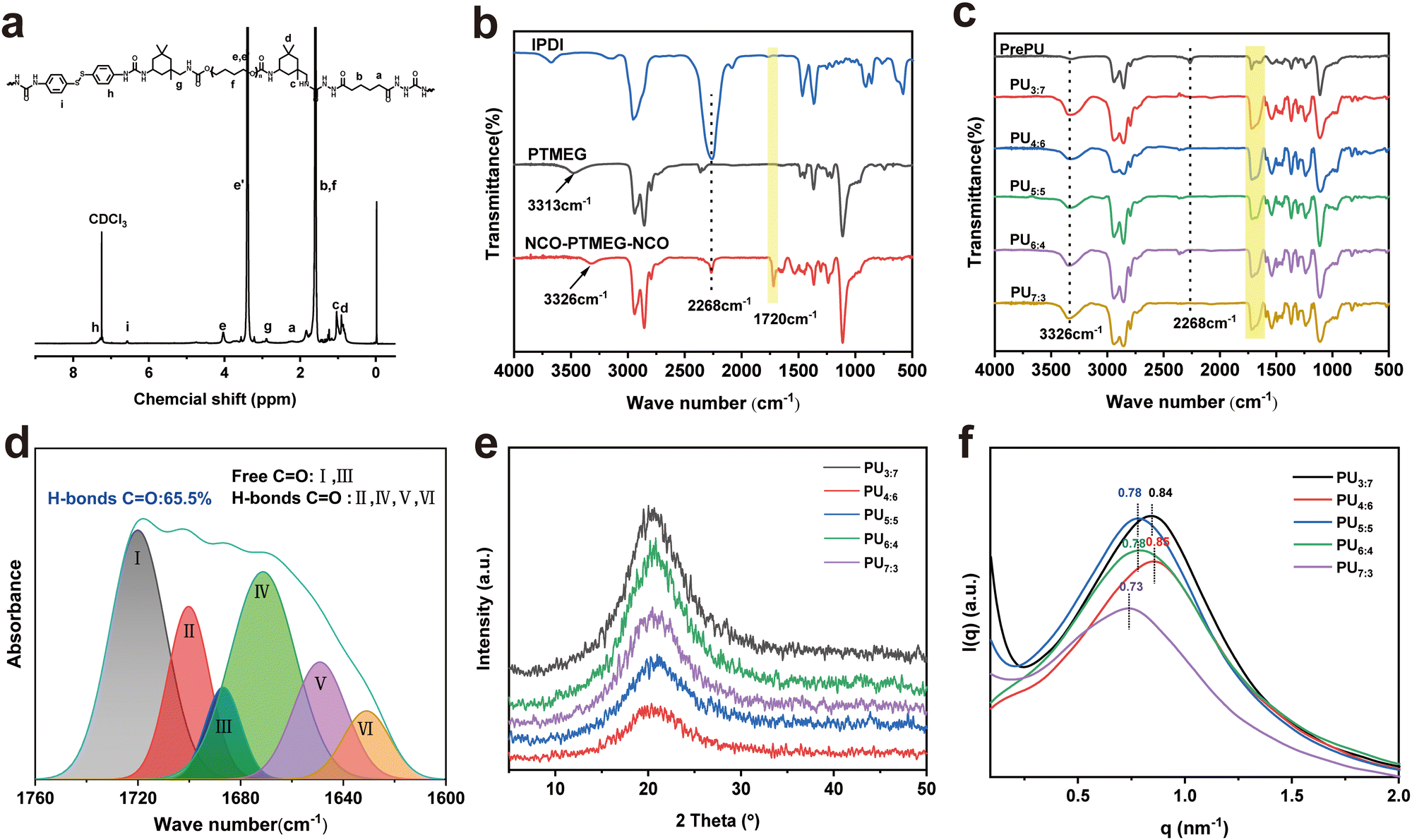 | ||
Fig. 2 (a) 1H NMR spectrum of the PU elastomer. (b) FTIR spectra of the raw materials and intermediate. (c) FTIR spectra of PU elastomers. (d) FTIR spectra of the PU3:7 elastomer in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration region (from 1760 cm−1 to 1600 cm−1). (e) WAXD patterns of PU elastomers. (f) SAXS profiles of PU elastomers. O stretching vibration region (from 1760 cm−1 to 1600 cm−1). (e) WAXD patterns of PU elastomers. (f) SAXS profiles of PU elastomers. | ||
Additionally, FTIR spectra were used to verify the successful fabrication of the PU elastomer and its intermediates. As shown in Fig. 2b, the isocyanate group of IPDI exhibited an absorption peak at 2268 cm−1. However, in the spectrum of PU elastomers (Fig. 2c), no peak appeared at 2268 cm−1, indicating that all the isocyanate groups had been consumed. The peak at 1649 cm−1 belonged to the stretching vibration of CO (υCO) in the urea segments, and the peak at 1700 cm−1 could be attributed to υCO in the urethane segments.41 By combining the results of NMR and FTIR spectra, both DTDA and AD had been introduced into the PU elastomers.
To differentiate and quantify the absorption peaks of CO caused by different interactions, the peak in the range of 1600 to 1760 cm−1 was analysed with peak fitting (Fig. 2d and Fig. S2, ESI†). The peak appearing at 1720 cm−1 was attributed to free hydrogen bonded CO in the urethane (NHC(O)O), and the peak appearing at 1700 cm−1 was attributed to ordered hydrogen bonded CO in the NHC(O)O moieties. The peak at 1686 cm−1 was attributed to free hydrogen bonds, while the peaks at 1671, 1649 and 1637 cm−1 were attributed to the free, disordered and ordered hydrogen bonded CO in the urea ((NH)2C(O)) groups.42,43 Based on the calculated integration area of the differentiated peaks, the content of hydrogen bonded CO in PU3:7 reached 65.53%. With increasing DTDA feeding, the contents of hydrogen bonded CO in PU elastomers gradually decreased (Fig. 2d). Therefore, with respect to rigid DTDA, the flexible AD monomer was verified to be beneficial for the formation of hydrogen bonds. The hydrogen bonds formed by CO groups worked as physical crosslinking points, uniformly dispersing throughout the entire PU elastomer.
To gain more insight into the structure of the PU elastomers, gel permeation chromatography (GPC) was used to determine their average molecular weights. GPC results showed that the Mns ranged from 4.28 × 104 to 9.88 × 104 g mol−1 (Fig. S3 and Table S1, ESI†).44–46 Driven by hydrogen bond interactions, the hard segments of the polymer chain were aggregated, forming “microdomains” uniformly dispersed in such soft domains. These “microdomains” worked as physical crosslinking points, limiting the activity of PU chains and reducing the size of microphase separation.
Fig. S4 (ESI†) shows the transmittance of PU films with a thickness of approximately 0.3 mm in the visible light range (400–800 nm) at room temperature. The transmittance decreased with increasing DTDA feeding because the offered PU elastomers became brownish in color (Fig. S5, ESI†). As a result, the best transmittance of the PU3:7 film was 85.3%. The structures of PU elastomers were examined by X-ray diffraction (XRD) and small-angle X-ray scattering (SAXS) (Fig. 2e and f). Fig. 2e shows that the PU elastomers had amorphous structures. These structures were obtained by the irregular packing of PU molecular chains.7,42 The SAXS data signified the microphase-separated structure of PU elastomers (Fig. 2f). The q values of PU3:7–PU7:3 were calculated to be 0.84, 0.85, 0.78, 0.78 and 0.73, respectively. Thus, the average distances between the hard domains were calculated to be 7.48, 7.66, 8.05, 8.05 and 8.61 nm, respectively. The distance between the hard domains in PU elastomers decreased with increasing AD/DTDA ratio.
Molecular chain motion behaviors
Differential scanning calorimetry (DSC) was used to evaluate the thermal properties of PU elastomers (Fig. 3a). The glass transition temperatures (Tgs) of the resultant PUs were approximately −70 °C, which approached the Tg of PTMEG. No endothermic melting peaks appeared in the DSC curves, confirming that no crystalline phase existed in the products again. In addition, with increasing DTDA loading, the Tg of PU elastomers slightly increased due to the incorporation of DTDA fragments with rigid aromatic rings.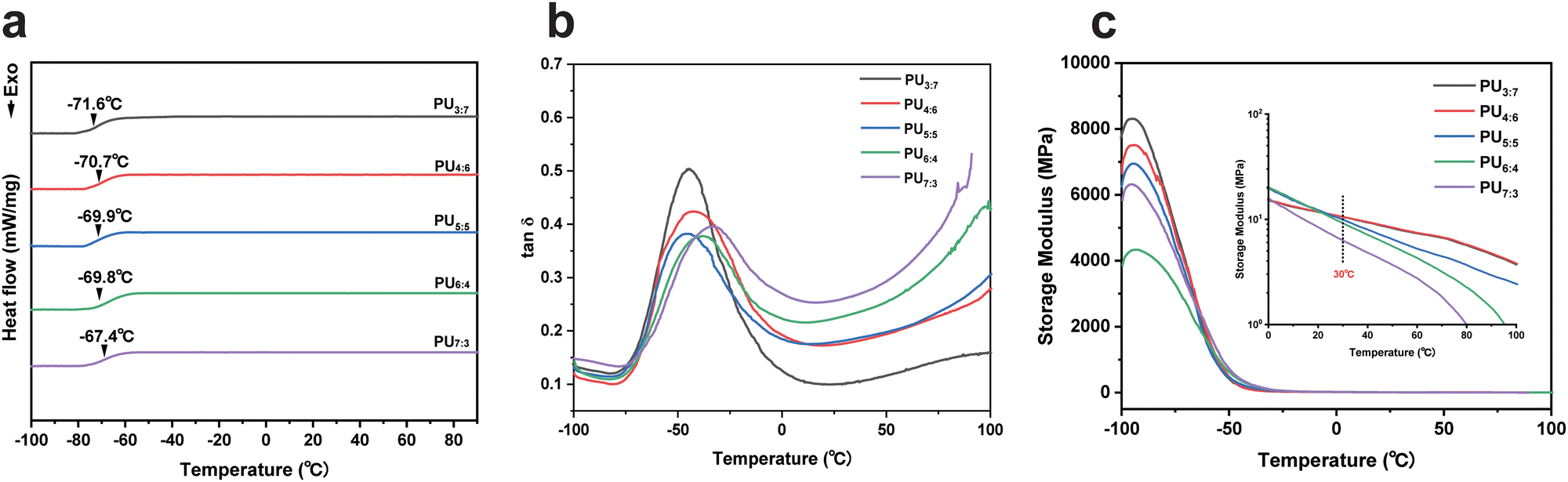 | ||
| Fig. 3 (a) DSC curves of PU elastomers. (b and c) The temperature dependence of tan (δ) and storage modulus of PU elastomers, respectively. Inset: the temperature dependence of storage modulus of PU elastomers in the temperature range from 0 °C to 100 °C. | ||
To describe the relaxation and viscoelasticity of the achieved PU elastomer chains, the temperature dependence of the storage modulus, loss modulus and tanδ were measured using dynamic mechanical analyses (DMA).47,48 The tanδ values of the PU elastomers were −46.63, −44.23, −44.66, −38.03 and −32.33 °C, respectively (Fig. 3b and Fig. S6 and S7, ESI†). Relatively low Tg made the polymer chains active and mobile at room temperature, allowing for the possibility of self-healing. Fig. 3c shows that the storage modulus of PU elastomers at −100 °C decreased with increasing DTDA content, which was consistent with the hydrogen bond contents of PU samples. As the temperature increased, the storage modulus decayed to approximately 10.0 MPa at 30 °C, meaning that the resulting PUs were flexible and mobile. These results of DSC and DMA tests implied that benzene rings within DTDA had an important effect on the Tg of PU elastomers, while the storage modulus of PU elastomers primarily relied on the hydrogen bond content.
Mechanical properties of PU elastomers
The tensile properties of PU elastomers are presented in Table S2 (ESI†). PU3:7 showed the highest tensile strength of 60.24 MPa and the shortest elongation at break of 1489.2%, giving a toughness of 92.17 MJ m−3. In contrast, the tensile strength of PU7:3 was 11.05 MPa, and the largest elongation at break of PU7:3 approached 2209.9%. The tensile strengths of PU4:6, PU5:5, and PU6:4 were 45.19 MPa, 37.53 MPa and 15.61 MPa, respectively. The largest elongations at break of PU4:6, PU5:5, and PU6:4 were 1372.9%, 1352.1% and 2115.1%, respectively. The tensile strength of PU elastomers increased with increasing AD content. The toughness of the obtained PU elastomers also gradually increased by the inclusion of AD, specifically, increasing from 92.17 MJ m−3 (PU7:3) to 257.24 MJ m−3 (PU3:7). The improvement in toughness was attributed to hydrogen bond interactions. When experiencing tensile forces, the hard domains oriented parallel to the tensile direction, preventing the chains from slipping and maintaining high strength.49 Therefore, hydrogen bonds and hard microdomains were critical to the mechanical properties of PU elastomers.
Dynamic interactions, including hydrogen-bond interactions and disulfide bonds, can dissipate energy. In this study, energy dissipation was evaluated by conducting cyclic tensile tests at a strain of 400% and five consecutive cycles.50 In the first cycle, the hysteresis loop had the largest hysteresis area (encircled by the tensile-recovery curve), indicating significant energy dissipation (4.81 MJ m−3). During tensile testing, with increasing strain, hydrogen bonds and disulfide bonds partly broke up. Then, the corrupted units, particularly those fractured disulfide bonds, were reconfigured randomly during tensile unloading. Because the broken hydrogen bonds did not have sufficient time to return to their initial states, the hydrogen bonds became weaker than the original ones.51 For the following cycle tests (Fig. S8, ESI†), the resultant hysteresis areas of all the investigated PUs decreased markedly. The primary reason for this result was that the rupture rates of hierarchical hydrogen and disulfide bonds were faster than the rates of reconstruction. From the second cycle to the fifth cycle, PUs containing fewer hydrogen bonds and more disulfides (e.g., PU6:4 and PU7:3) showed a slight change in hysteresis areas, implying disulfides with a rapid recovery rate, which is markedly faster than the reconstruction rate of hydrogen bonds and the unloading speed (50 mm min−1).
Healing behavior of PU elastomers
The diffusion of molecular chains promoted the impaired or ruptured PU elastomers to perform self-healing, prolonging service life. Generally, high temperature was able to activate the movement and diffusion of polymer chains and enhance healing efficiency.52,53 To investigate the self-healing performance of PU elastomers, the full-cut samples were closed and jointed together at 70 °C. The healing efficiency (%) and the ratio of tensile strength of healed and virgin PU samples were used to evaluate self-healing ability. As shown in Fig. S9 (ESI†), at 70 °C, the final healing efficiency of the PU samples after 12 h reached 43.0–57.5%. Except for PU7:3, the healing efficiency was enhanced with increasing loading of disulfide bonds. For healable PUs, the healing efficiency generally depends most on the reformation of hydrogen bonds and disulfide bonds. Hydrogen and disulfide bonds could partly or fully reform. There were some defects (e.g., a gap) between the cut PU samples, leading to a low healing efficiency (approximately 50%). In addition, the content of chain extenders influenced the healing efficiency. For example, PU3:7 and PU7:3 had healing efficiencies of 42.98% and 27.6%, respectively. As the AD content increased, the healing efficiency of PU elastomers gradually decreased (Fig. S9, ESI†).
To improve self-healing efficiency, two strategies – elevated temperature and solvent-assisted – were used.54 However, at a higher healing temperature (e.g., 90–100 °C), the tested sample deformed severely because the hydrogen bonds were destroyed and chains began to flow. Also, its mechanical properties were poor (e.g., nearly zero tensile strength). For the solvent-assisted strategy, polar organic solvents were used as hydrogen bond donors and acceptors, temporarily decreasing the interactions among polymer chains. Fig. 4a shows the healing efficiency of PU5:5 with the assistance of five types of solvents, including anhydrous ethanol, IPA, acetone, ethyl acetate and hexane. The cut PU5:5 elastomer exhibited a high healing efficiency (approximately 80%) with the help of polar solvents. As the IPA or ethanol molecules diffused in the PU samples, they disturbed the hydrogen bonds among –NH and CO groups, making them move toward the dissociated state.55 For these nonpolar solvents, no swelling phenomena occurred. Thus, using hexane as the assisting agent, the healing efficiency of PU was only 2.92%. Fig. 4c and Fig. S10 (ESI†) show the tensile stress–strain curves of healed PU samples with different healing times at 30 °C with the assistance of IPA. As expected, the healed efficiency and strain strength of PUs increased with longer time. When the healing time was beyond 5 h at 30 °C, both the tensile strength and healing efficiency of the healed PU5:5 tended toward stability, which were higher than those (22.35 MPa and 53.3%) healed by the thermally driven method at 70 °C for 12 h. Fig. 4d and e show the stress–strain curves and the corresponding healing efficiency achieved for virgin and healed PU samples. The healing efficiencies were in the range of 70–80% at 30 °C for 5 h, except for that of PU7:3 (49.5%). Thus, with the assistance of IPA, the healed PUs achieved exceptional mechanical performance at low healing times of 5 h. Compared to those studies that investigated healing materials at temperatures of ≤50 °C and IPA- or moisture-assisted healing, the mechanical properties obtained from healed PUs were remarkable (Fig. 4f). For example, the tensile strength, elongation at break and healing efficiency of PU3:7 after healing at 30 °C for 5 h approached 44.33 MPa, 1252.6% and 73.6%, respectively. The healed dumbbell-shaped PU sample could withstand stretching, and the elongation reached 750%, indicating the robustness of the healed PU elastomers (Fig. 4g). In addition, after the stretched PU relaxed for 2 min at room temperature, it had a strain of approximately 110%, illustrating that it was easy to recover.
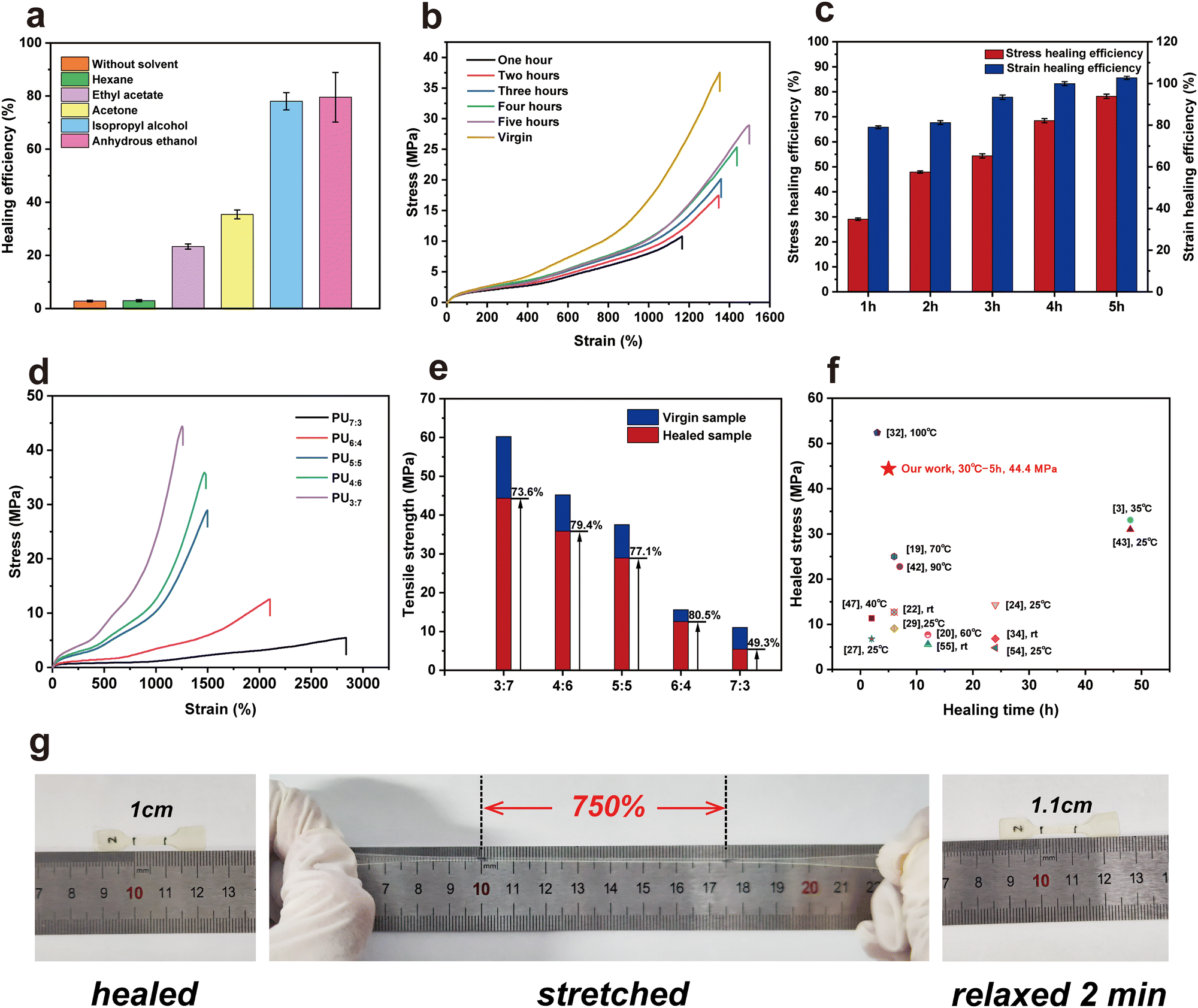 | ||
| Fig. 4 (a) Healing efficiency of PU5:5 with the assistance of five types of solvents. (b) Tensile stress–strain curves of healed PU5:5 samples with different healing times at 30 °C with the assistance of IPA. (c) Stress and strain healing efficiency of PU5:5 with different healing times at 30 °C with the assistance of IPA. (d) and (e) Healing ability of PUs, in terms of the stress and tensile strength as functions of the strain and PU samples, respectively. (f) Summarizing the properties of healed PUs versus healing times of various healing materials. (g) Photographs of the healed, stretched, and relaxed PU5:5 dumbbell-shaped sample. | ||
Interpretation of the healing behavior of PUs
In the above sections, the relationship between structure and properties was analyzed in detail. Three factors, including physical entangling among polymer chains, multihydrogen bonds and disulfide bonds, were attributed to the toughness and robustness of the PU elastomers. The hydrogen bonds and disulfide bonds were reversible in nature, making the cut samples heal under a suitable driving force. Because the PUs were subjected to physical damage, some hydrogen and disulfide bonds within PUs were destroyed. The fractured disulfide became sulfur free radicals. These sulfur free radicals trapped in hard microdomains had a long life and could be recombined to reform disulfide. Their fast recovery rate favored the closing and fusing of the fracture surface. Simultaneously, with IPA permeation, the PU sample partially swelled. The hydrogen bonds between –NH and CO of PU chains were partly replaced by those between –OH and CO. Then, PU chains were activated because they eliminated the limitation of intermolecular hydrogen bonds, promoting the fusion of the cut PU elastomer. As IPA volatilized, multiple hydrogen bonds gradually reconstituted, forming a healed PU elastomer (Scheme 1).
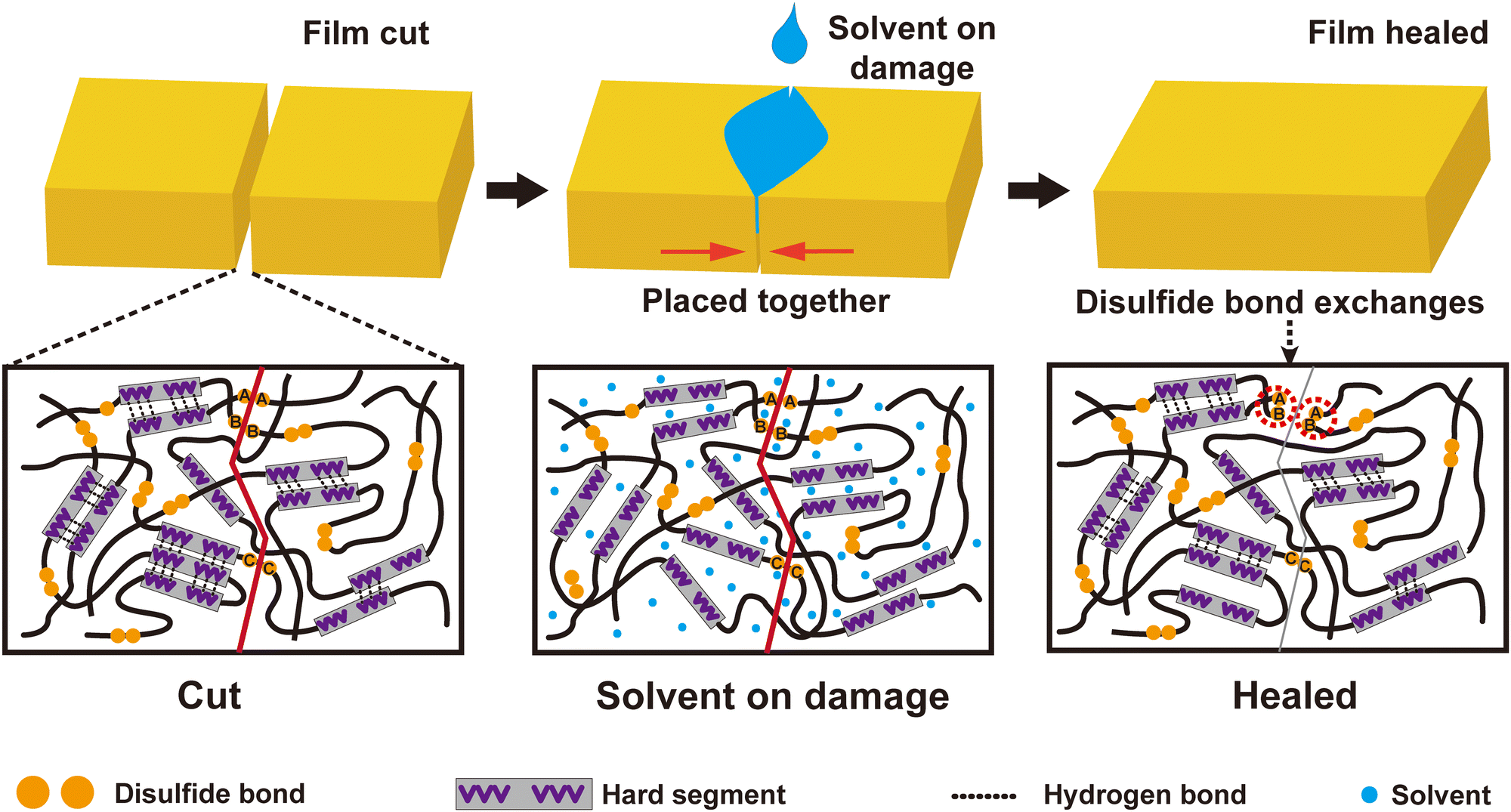 | ||
| Scheme 1 Schematic illustration of the healing process driven by IPA. | ||
The healing efficiencies of most healed PU samples approached 70–80%, indicating a 20–30% difference in tensile strength between the virgin and healed samples. The gap in tensile strength may be ascribed to two factors: (1) the imperfect recovery of hydrogen bonds and (2) the reformation of disulfides on the same cut side (see Fig. S11, ESI†). When fabricating PU samples, DMAc with a high boiling point allowed the entire PU chain to move freely, forming highly dense hydrogen bonds. Confined to the hard domains, the multiple hydrogen bonds ensured exceptional mechanical properties. In contrast, with IPA-assisted healing, the PU samples could swell, permitting only some polymer segments to move. Thus, many defects were involved in the healed PU samples, resulting in performance degradation. Concurrently, some disulfides regenerated within the same side of the cut sample, which had nearly zero contribution to the tensile strength of the PU elastomers (Scheme 1). For example, a higher content of disulfides in a PU sample (e.g., PU7:3) increased the chances of reformed disulfides on the same side of the cut sample, leading to a lower tensile strength. The aromatic disulfide metathesis reported by Odriozola and others may rarely work because most aromatic disulfides are trapped in hard microdomains, and few molecular chains can move freely at room temperature.29 Therefore, it is difficult to achieve a 100% healing efficiency using the proposed method.
Conclusions
A series of poly(urea-urethane) (PU) elastomers with reversible multiple hydrogen bonds and disulfide bonds were successfully synthesized using a facile one-pot two-step polycondensation strategy with commercially available reagents. The produced PU elastomers had strong mechanical properties and rapid self-healing abilities. Typically, the tensile strength, strain at break and toughness of PU3:7 approached 60.24 MPa, 1489% and 257.24 MJ m−3, respectively. At 30 °C for 5 h and, with the assistance of IPA, the tensile strength and strain at break of the healed PU3:7 were 44.4 MPa and 1254%, respectively. In addition, the material exhibited a healing efficiency of 73.6%. The strength gap (20–30%) can be attributed to the defective recovery of hydrogen bonds and the reformation of disulfides in the same cut side. This study thus presents a solution to easily fabricate PU elastomers with remarkable performance and high room temperature self-healing efficiency.
Author contributions
Chenghui Qiao: methodology, investigation, experimental analysis and writing – original draft. Xiurui Jian: investigation, experimental analysis and writing – original draft. Zhengguo Gao, Xintao Zhang and Huimin Wang: methodology and experimental analysis. Qingfu Ban: experimental analysis and writing – review and editing. Yaochen Zheng: conceptualization, funding acquisition, and writing – review and editing.Conflicts of interest
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 21905243) and the Natural Science Foundation of Shandong Province (No. ZR2017MEM023).References
- D. Zhao, S. Liu, Y. Wu, T. Guan, N. Sun and B. Ren, Prog. Org. Coat., 2019, 133, 289–298 CrossRef CAS.
- J. Xie, L. Fan, D. Yao, F. Su, Z. Mu and Y. Zheng, Mater. Today Chem., 2022, 23, 100708 CrossRef CAS.
- Y. Eom, S. M. Kim, M. Lee, H. Jeon, J. Park, E. S. Lee, S. Y. Hwang, J. Park and D. X. Oh, Nat. Commun., 2021, 12, 621 CrossRef CAS PubMed.
- W. Sun, N. Luo, Y. Liu, H. Li and D. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 10498–10507 CrossRef CAS PubMed.
- H. Guo, Y. Han, W. Zhao, J. Yang and L. Zhang, Nat. Commun., 2020, 11, 2037 CrossRef CAS PubMed.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- S. Utrera-Barrios, R. Verdejo, M. A. López-Manchado and M. Hernández Santana, Mater. Horiz., 2020, 7, 2882–2902 RSC.
- R. H. Aguirresarobe, S. Nevejans, B. Reck, L. Irusta, H. Sardon, J. M. Asua and N. Ballard, Prog. Polym. Sci., 2021, 114, 101362 CrossRef CAS.
- J. Kang, J. B. H. Tok and Z. Bao, Nat. Electron., 2019, 2, 144–150 CrossRef.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- Z. Wang, X. Lu, S. Sun, C. Yu and H. Xia, J. Mater. Chem. B, 2019, 7, 4876–4926 RSC.
- A. Shaabani, R. Sedghi, H. Motasadizadeh and R. Dinarvand, Chem. Eng. J., 2021, 411, 128449 CrossRef CAS.
- T. T. Truong, S. H. Thai, H. T. Nguyen, D. T. T. Phung, L. T. Nguyen, H. Q. Pham and L.-T. T. Nguyen, Chem. Mater., 2019, 31, 2347–2357 CrossRef CAS.
- H. Feng, W. Wang, T. Wang, L. Zhang, W. Li, J. Hou and S. Chen, J. Mater. Sci. Technol., 2023, 133, 89–101 CrossRef.
- S. Ji, W. Cao, Y. Yu and H. Xu, Adv. Mater., 2015, 27, 7740–7745 CrossRef CAS PubMed.
- K. Song, W. Ye, X. Gao, H. Fang, Y. Zhang, Q. Zhang, X. Li, S. Yang, H. Wei and Y. Ding, Mater. Horiz., 2021, 8, 216–223 RSC.
- C. Lin, D. Sheng, X. Liu, S. Xu, F. Ji, L. Dong, Y. Zhou and Y. Yang, Polymer, 2019, 182, 11822 Search PubMed.
- S. W. Yang, X. S. Du, S. Deng, J. H. Qiu, Z. L. Du, X. Cheng and H. B. Wang, Chem. Eng. J., 2020, 398, 11 Search PubMed.
- Y. Lai, X. Kuang, P. Zhu, M. Huang, X. Dong and D. Wang, Adv. Mater., 2018, 30, e1802556 CrossRef PubMed.
- Y. Yang, X. Lu and W. Wang, Mater. Des., 2017, 127, 30–36 CrossRef CAS.
- X. Li, R. Yu, Y. He, Y. Zhang, X. Yang, X. Zhao and W. Huang, ACS Macro Lett., 2019, 8, 1511–1516 CrossRef CAS PubMed.
- C. Y. Bao, Y. J. Jiang, H. Y. Zhang, X. Y. Lu and J. Q. Sun, Adv. Funct. Mater., 2018, 28, 1800560 CrossRef.
- S. Delpierre, B. Willocq, G. Manini, V. Lemaur, J. Goole, P. Gerbaux, J. Cornil, P. Dubois and J.-M. Raquez, Chem. Mater., 2019, 31, 3736–3744 CrossRef CAS.
- W. Fan, Y. Jin, L. Shi, R. Zhou and W. Du, J. Mater. Chem. A, 2020, 8, 6757–6767 RSC.
- Z. Zou, C. Zhu, Y. Li, X. Lei, W. Zhang and J. Xiao, Sci. Adv., 2018, 4, eaaq0508 CrossRef PubMed.
- X. Yang, S. Wang, X. Liu, Z. Huang, X. Huang, X. Xu, H. Liu, D. Wang and S. Shang, Green Chem., 2021, 23, 6349–6355 RSC.
- S. M. Kim, H. Jeon, S. H. Shin, S. A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- W. B. Ying, Z. Yu, D. H. Kim, K. J. Lee, H. Hu, Y. Liu, Z. Kong, K. Wang, J. Shang, R. Zhang, J. Zhu and R. W. Li, ACS Appl. Mater. Interfaces, 2020, 12, 11072–11083 CrossRef CAS PubMed.
- R. a Li, T. Fan, G. Chen, H. Xie, B. Su and M. He, Chem. Eng. J., 2020, 393, 124685 CrossRef CAS.
- S. Park, G. Thangavel, K. Parida, S. Li and P. S. Lee, Adv. Mater., 2019, 31, e1805536 CrossRef PubMed.
- X. Wang, S. Zhan, Z. Lu, J. Li, X. Yang, Y. Qiao, Y. Men and J. Sun, Adv. Mater., 2020, 32, 2005759 CrossRef CAS PubMed.
- Z. Li, Y. L. Zhu, W. Niu, X. Yang, Z. Jiang, Z. Y. Lu, X. Liu and J. Sun, Adv. Mater., 2021, 33, e2101498 CrossRef PubMed.
- B. Wu, Z. Liu, Y. Lei, Y. Wang, Q. Liu, A. Yuan, Y. Zhao, X. Zhang and J. Lei, Polymer, 2020, 186, 122003 CrossRef CAS.
- X. Yan, Z. Liu, Q. Zhang, J. Lopez, H. Wang, H. C. Wu, S. Niu, H. Yan, S. Wang, T. Lei, J. Li, D. Qi, P. Huang, J. Huang, Y. Zhang, Y. Wang, G. Li, J. B. Tok, X. Chen and Z. Bao, J. Am. Chem. Soc., 2018, 140, 5280–5289 CrossRef CAS PubMed.
- E. M. Foster, E. E. Lensmeyer, B. Zhang, P. Chakma, J. A. Flum, J. J. Via, J. L. Sparks and D. Konkolewicz, ACS Macro Lett., 2017, 6, 495–499 CrossRef CAS PubMed.
- C. C. Hornat and M. W. Urban, Nat. Commun., 2020, 11, 1028 CrossRef CAS PubMed.
- J. M. Matxain, J. M. Asua and F. Ruiperez, Phys. Chem. Chem. Phys., 2016, 18, 1758–1770 RSC.
- J. Chen, F. Li, Y. Luo, Y. Shi, X. Ma, M. Zhang, D. W. Boukhvalov and Z. Luo, J. Mater. Chem. A, 2019, 7, 15207–15214 RSC.
- T. Jing, X. Heng, X. Guifeng, L. Li, P. Li and X. Guo, Mater. Chem. Front., 2022, 6, 1161–1171 RSC.
- F. Sai, H. Zhang, J. Qu, J. Wang, X. Zhu, Y. Bai and P. Ye, Appl. Surf. Sci., 2022, 573, 151526 CrossRef CAS.
- Y. Cai, L. Yan, Y. Wang, Y. Ge, M. Liang, Y. Chen, H. Zou and S. Zhou, Chem. Eng. J., 2022, 436, 135156 CrossRef CAS.
- H. Wu, X. Liu, D. Sheng, Y. Zhou, S. Xu, H. Xie, X. Tian, Y. Sun, B. Shi and Y. Yang, Polymer, 2021, 214, 123261 CrossRef CAS.
- W. W. Graessley, The Entanglement Concept in Polymer Rheology, Springer Berlin Heidelberg, Berlin, Heidelberg, 1974, pp. 1–179 DOI:10.1007/BFb0031037.
- T. G. Fox and S. Loshaek, J. Appl. Phys., 1955, 26, 1080–1082 CrossRef CAS.
- T. G. Fox and S. Loshaek, J. Polym. Sci., 1955, 15, 371–390 CrossRef CAS.
- H. Bi, G. Ye, H. Sun, Z. Ren, T. Gu and M. Xu, Addit. Manuf., 2022, 49, 102487 CAS.
- Z. Shi, J. Kang and L. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 23484–23493 CrossRef CAS PubMed.
- T. Zheng, T. Li, J. Shi, T. Wu, Z. Zhuang, J. Xu and B. Guo, Macromolecules, 2022, 55, 3020–3029 CrossRef CAS.
- F. Dong, X. Yang, L. Guo, Y. Wang, H. Shaghaleh, Z. Huang, X. Xu, S. Wang and H. Liu, J. Mater. Chem. A, 2022, 10, 10139–10149 RSC.
- Y. Jia, L. Zhang, M. Qin, Y. Li, S. Gu, Q. Guan and Z. You, Chem. Eng. J., 2022, 430, 133081 CrossRef CAS.
- R. A. Li, T. Fan, G. Chen, H. Xie, B. Su and M. He, Chem. Eng. J., 2020, 393, 124685 CrossRef CAS.
- D. Wang, J. Xu, J. Chen, P. Hu, Y. Wang, W. Jiang and J. Fu, Adv. Funct. Mater., 2019, 30, 1907109 CrossRef.
- M. W. M. Tan, G. Thangavel and P. S. Lee, Adv. Funct. Mater., 2021, 31, 2103097 CrossRef CAS.
- S. Nakagawa, S. Nakai, K. Matsuoka and N. Yoshie, Polymer, 2019, 161, 101–108 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma01021f |
| This journal is © The Royal Society of Chemistry 2023 |