
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEtOH/H2O ratio modulation on carbon for high-Voc (1.03 V) printable mesoscopic perovskite solar cells without any passivation†
Jie
Sheng‡
,
Xiaotian
Zhu‡
,
Xiaoli
Xu
,
Jingshan
He
,
Dun
Ma
,
Jialing
Liu
and
Wenjun
Wu
 *
*
Key Laboratory for Advanced Materials, Feringa Nobel Prize Scientist Joint Research Center, Shanghai Key Laboratory of Functional Materials Chemistry, School of Chemistry and Molecular Engineering, East China University of Science & Technology, 130 Meilong Road, Shanghai 200237, P. R. China. E-mail: wjwu@ecust.edu.cn
First published on 14th February 2023
Abstract
With a work function close to that of gold, carbon is theoretically a good substitute for gold as a counter electrode in perovskite solar cells (PSCs). Recently, carbon-based printable mesoscopic perovskite solar cells (p-MPSCs) have attracted much attention on account of their all-screen-printing process, assembly in air-atmosphere and ultra-high stability. Herein, based on an improved Stöber strategy, a phenol-formaldehyde resin-based carbon (PFc) material is developed for p-MPSCs as counter electrodes. By the modulation of solvent polarity (EtOH/H2O ratio) during synthesis, PFc materials with different morphologies were obtained effectively. At the optimal ratio (EtOH/H2O = 4/3), the infiltration of the perovskite precursor in carbon electrodes was effectively improved due to the interaction between the surface functional groups (C–O/C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the prepared carbon material and Pb2+ and homogeneous porous structure. Furthermore, the open-circuit voltage (Voc) of PFc-based p-MPSCs increased from 0.97 V to 1.03 V (without any passivation treatment) with a maximum PCE of 17.64% based on the champion device.
O) of the prepared carbon material and Pb2+ and homogeneous porous structure. Furthermore, the open-circuit voltage (Voc) of PFc-based p-MPSCs increased from 0.97 V to 1.03 V (without any passivation treatment) with a maximum PCE of 17.64% based on the champion device.
1. Introduction
Owing to the increase in the power conversion efficiency (PCE) of single-junction devices for perovskite solar cells (PSCs) from 3.8%1 to 25.7%,2 they are attracting worldwide attention as one of the most promising photovoltaic devices. However, the higher cost of electrode materials (such as gold or silver) and organic hole transport materials (such as 2,2′,7,7′-tetrakis [N,N-di(4-methoxyphenyl) amino]-9-9′-spirobifluorene, spiro-OMeTAD) limit their commercialization to some extent. Carbon, with a similar work function (−5.0 eV) to that of gold (−5.1 eV),3 is theoretically a good substitute for gold as a counter electrode of PSC devices.In 2014, Prof. H. Han et al. presented firstly a new type of HTM (Hole transport Materials)-free fully printable mesoscopic perovskite solar cell (p-MPSC) based on carbon electrodes.4 It stands out due to its simplicity and low cost of preparation, coupled with its ultra-high stability. Thus far, its highest certified PCE has reached 17.7% (Voc = 0.984 V, Jsc = 24.14 mA cm−2 and FF = 74.6%).5
To further enhance their photovoltaic performances, extensive research studies have been conducted on the conductivity, energy level matching, hydrophobicity and specific surface area of carbon electrode materials.6,7 For example, Prof. H. Han et al.8 have systematically optimized the flaky graphite component in the carbon electrode consisting of graphite and carbon black and obtained PCE exceeding 11% due to the low square resistance and large pore size of graphite-based electrodes, facilitating electron/hole separation in the photoelectric conversion process. Then, Y. Yang et al.9 made the first attempt to chlorinate the graphite component for carbon electrode materials to obtain chlorinated graphite (C–Clx, x = 0.2, 0.4 and 0.6). For x = 0.4, the PCE was 1.46 times higher than that of the control group.10 In addition, the introduction of needle coke can also contribute to the wettability of perovskite precursors and inhibits charge recombination and dark current formation, enhancing the charge collection capacity of carbon electrodes.11 Recently, the use of carbonized cellulose as electrodes for p-MPSCs was found to yield a PCE value of 15.5% (Voc (open circuit voltage) = 0.937 V).12 Thus, for enhancing the photovoltaic performances of p-MPSCs, the selection and modification of carbon materials are one of the key breakthroughs.
The Stöber method involves the hydrolysis and condensation of silanol salts such as tetraethyl orthosilicate (TEOS) in an alcohol solvent (e.g., ethanol) in the presence of water and an alkaline catalyst (e.g., ammonia solution) to produce colloidal silica spheres.13 Using the Stöber method, excellent control of the particle size and a narrow particle size distribution can be achieved, resulting in spherically smooth silica, microporous silica and mesoporous silica particles. In 2011, Lu et al. prepared monodisperse resin polymer spheres with uniform and controlled submicron particle size based on the method.14 This synthesis method is a very simple and versatile way of preparing polymer balls, and is considered to be low-cost and suitable for industrial production.
In this work, phenol-formaldehyde resin-based carbon (PFc) was developed via co-assembly engineering with improved Stöber strategy (tetraethyl orthosilicate (TEOS) as the hard template, and cetyltrimethyl ammonium bromide (CTAB) as the structure guidance)15,16 (as shown in Scheme 1). Through an EtOH/H2O ratio modulation, morphological control of PFc was achieved, and its specific surface area and graphitization were enhanced. As the carbon electrode of p-MPSCs, the Voc can be increased from 0.97 V to 1.03 V (without any passivation treatment). Furthermore, the champion device obtained a PCE value of 17.64%. A novel strategy is presented for the high-Voc carbon electrode materials in the p-MPSCs.
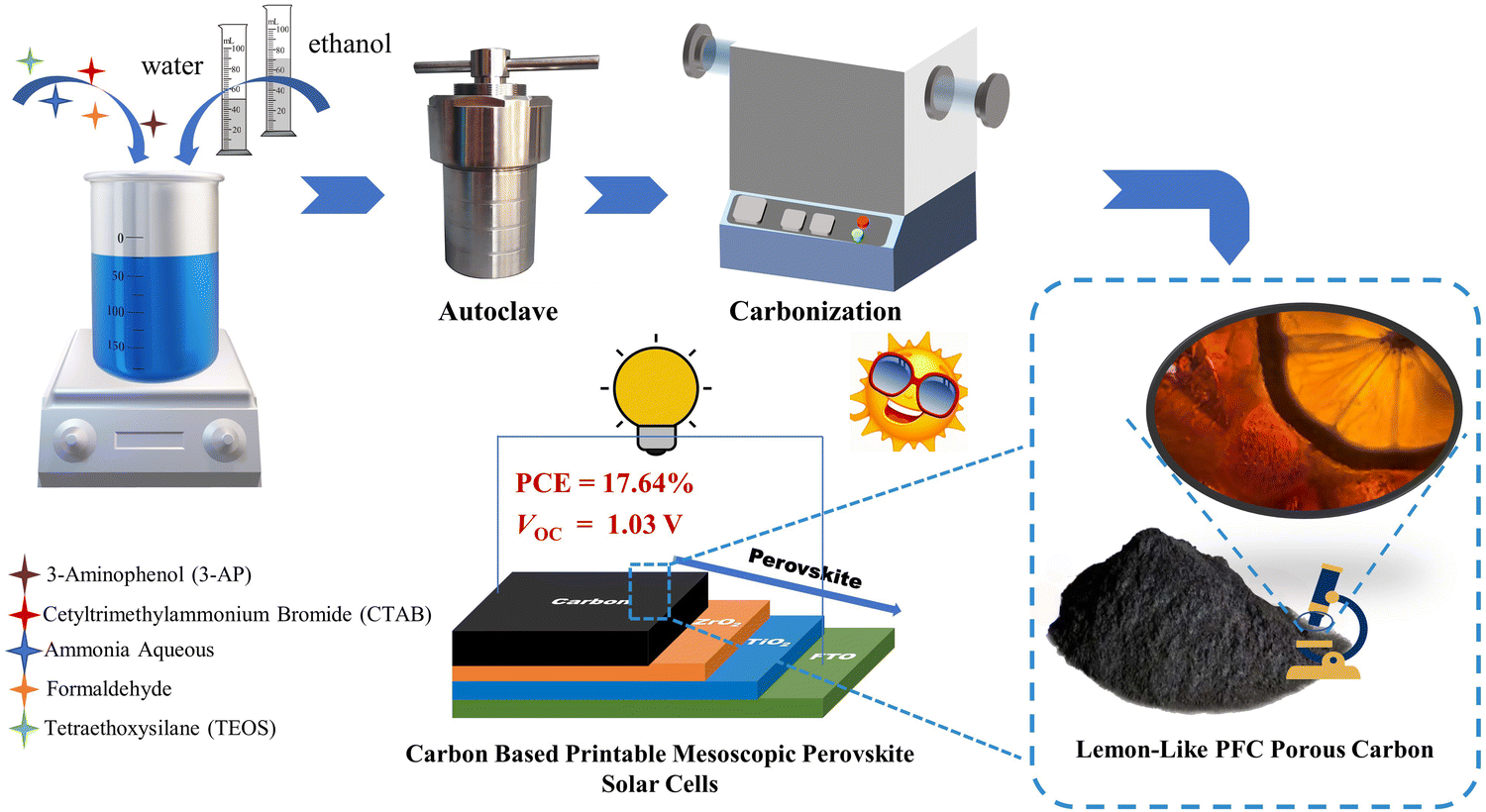 | ||
| Scheme 1 Schematic diagram of the PFc porous carbon preparation and photovoltaic application. | ||
2. Experimental section
2.1. Materials
Formaldehyde (37%), ammonia aqueous solution (NH3·H2O, 25%), 3-aminophenol (3-AP, 99%), cetyltrimethylammonium bromide (CTAB, 99%) and tetraethoxysilane (TEOS, AR) were purchased from Shanghai Titan Technology Co., Ltd. Sodium hydroxide (NaOH, AR) was purchased from Shanghai Macklin Biochemical Technology Co., Ltd. MAI (CH3NH3I, 99.5%), PbI2 (lead(II)iodide, 99.99%), hydroxypropyl cellulose and titanium diisopropoxide bis(acetylacetonate) (75 wt% in IPA) were obtained from TCI (Shanghai). MACl (CH3NH3Cl) was obtained from Alfa Aesar. DMF (N,N-dimethylformamide, 99.8%), anhydrous EtOH (99.7%), and DMSO (dimethyl sulfoxide, 99.7%) were obtained from ACROS. ZrO2 (nanometer zirconium oxide) was obtained from QingDao Huifeng Science, Inc. Graphite (99%) and α-terpineol (98%) were obtained from Adamas-beta. TiO2 paste (30NR-T) (titanium(IV)dioxide) was obtained from GreatCell Solar Materials. Unless otherwise stated, all of the aforementioned materials were used as received without further purification.2.2. The synthesis of PFc carbon
According to reference,16 0.22 g 3-AP and CTAB were dispersed in 32 mL EtOH–H2O mixture solvent with different ratios as PFc-x (x = 0, 1, 3, 4, 6 and 7, corresponding to EtOH/H2O ratios of 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, 1:6, 3:4, 4:3, 6:1, 7:0, respectively). After the solid was completely dissolved, 0.22 mL ammonia aqueous solution and 0.31 mL formaldehyde were added under continuous stirring for 20 minutes. When the mixed solution changed from a clear color to an emulsion, 1.1 mL of TEOS was added immediately and stirred with a magnetic stirrer for 24 hours. The obtained solution was transferred to a stainless-steel autoclave lined with Teflon, and reacted at 80 °C for 24 h under hydrothermal conditions. After the autoclave was cooled to room temperature, the lining was taken out, and the brick-red precursor was collected by suction filtration and washed repeatedly with deionized H2O. The precursor of PFc-x was obtained after drying at 70 °C for 30 min. It was then placed in a tube furnace and heated to 800 °C at a heating rate of 2 °C per minute, and reacted for 3 h under an argon atmosphere. After cooling to room temperature, the silica was removed by soaking in 1 M NaOH solution for 24 h, and the final PFc-x was obtained.
:7, 1:6, 3:4, 4:3, 6:1, 7:0, respectively). After the solid was completely dissolved, 0.22 mL ammonia aqueous solution and 0.31 mL formaldehyde were added under continuous stirring for 20 minutes. When the mixed solution changed from a clear color to an emulsion, 1.1 mL of TEOS was added immediately and stirred with a magnetic stirrer for 24 hours. The obtained solution was transferred to a stainless-steel autoclave lined with Teflon, and reacted at 80 °C for 24 h under hydrothermal conditions. After the autoclave was cooled to room temperature, the lining was taken out, and the brick-red precursor was collected by suction filtration and washed repeatedly with deionized H2O. The precursor of PFc-x was obtained after drying at 70 °C for 30 min. It was then placed in a tube furnace and heated to 800 °C at a heating rate of 2 °C per minute, and reacted for 3 h under an argon atmosphere. After cooling to room temperature, the silica was removed by soaking in 1 M NaOH solution for 24 h, and the final PFc-x was obtained.
2.3. Preparation of the carbon electrode pastes
Graphite powder (4.3 g) were mixed with 0, 0.25, 0.75 and 1.0 g, PFc-x, respectively, in 15 mL terpineol. Then, 1 g nano-ZrO2 powder and 1 g hydroxypropyl cellulose were added. Finally, the mixture was processed by ball milling for 48 h to obtain the carbon paste with different contents of PFc-x.2.4. Preparation of the perovskite precursor
The perovskite precursor solution was prepared by mixing 159 mg MAI, 461 mg PbI2, 27 mg MACl, 800 μL N,N-dimethylformamide (DMF) and 200 μL dimethyl sulfoxide (DMSO), and stirred until completely dissolved in the glove box.2.5. Device fabrication
The FTO substrates were ultrasonically cleaned with detergent, deionized H2O, acetone, and anhydrous EtOH for 20 min, respectively. The compact TiO2 layer was deposited onto the conductive surface of FTO by spray pyrolysis at 500 °C with 2% (v/v%) titanium diisopropoxide bis(acetylacetonate) solution in anhydrous EtOH, and then maintained at 500 °C for 30 min. The mesoporous TiO2 (m-TiO2) was deposited onto the compact layer by screen-printing the 30NR-T, and then sintered at 500 °C for 40 min. After that, a mesoporous ZrO2 (m-ZrO2) and a carbon electrode were deposited by screen-printing layer-by-layer. Then, both m-ZrO2 and carbon layer were sintered at 400 °C for 40 min. After cooling down, the perovskite solution was dropped on the carbon layer to fill the triple mesoporous layer structure, and annealed at 100 °C for 2 h. All these processes were done in ambient air.2.6. Device characterizations
The photocurrent density–voltage characteristics were measured by a Keithley 2400 source meter under air mass 1.5 (AM1.5) illumination at 100 mW cm−2 using an Oriel 91106 solar simulator (Newport, USA) with a scan rate of 20 mV s−1. The active area of the device is approximately 0.64 cm2, and a mask with an oblong aperture (0.0875 cm2) was applied under J–V tests. The IPCE spectra were recorded by a Newport-74125 system (Newport Instruments). Electrochemical impedance spectroscopy (EIS) was measured on Zahner Ennium electrochemical workstations in the frequency range from 1 Hz to 3.92 MHz. Scanning electron microscope (SEM) images were characterized by using a Helios G4UC scanning electron microscope. Transmission electron microscopy (TEM) images were characterized by a Talos F200X. X-ray photoelectron spectroscopy (XPS) was conducted on a Thermo Scientific™ K-Alpha™+ spectrometer equipped with a monochromatic Al Kα X-ray source (1486.6 eV) operating at 100 W. Samples were analyzed under vacuum (P < 10−8 mbar) with a pass energy of 150 eV (survey scans) or 30 eV (high-resolution scans). X-ray diffraction (XRD) spectra were obtained from an X-ray diffractometer (Rigaku D/Max 2550 VB/PC). Fourier transform infrared spectroscopy (FTIR) was measured by a Thermo Nicolet Corporation (7800–350/cm 0.01/cm/6700) instrument. The work function of the perovskite film was characterized by ultraviolet photoelectron spectroscopy (UPS, Thermo Escalab 250XI) (HeI, hν = 21.2 eV). Fluorescence spectra (PL) and photoluminescence lifetimes (TRPL) were determined by an Edinburgh FLS890 spectrometer. N2 adsorption and desorption isotherms were measured using a Micromeritics ASAP-2020 instrument at liquid nitrogen temperature (77 K), and the samples were pretreated at 300 °C for 3 h. The specific surface area and pore size distribution were determined by the Brunauer–Emmett–Teller (BET) and Barret–Joyner–Halenda (BJH) methods. The contact angle measurement was done using the contact angle surface analyzer. Contact angles were obtained on a SDC-80 (SINDIN) drop shape analysis system, using water and organic solvent as probe liquids (0.5 μL). Several drops (typically three repetitions) were quickly placed on the surface, the needle was pulled back, and the drop shape was captured immediately with the camera. Images were analyzed with drop shape analysis software to determine the contact angle by the method most suitable for each given drop, usually circle fitting. The space charge limiting current (SCLC) was measured using a Keithley 2400 source meter, with the applied voltage increasing from 0 V to 2 V in 0.01 V intervals with a delay time of 20 ms.3. Results and discussion
As shown in Scheme 1, the phenolic resin precursors were synthesized by a solvothermal method in which CTAB was used as a soft template and structural guide, and silica (derived from the decomposition of TEOS) was used as a hard template to produce SiO2/phenol-formaldehyde (PF) microspheres by ammonia-catalyzed polymerisation of 3-aminophenol (3-AP) with formaldehyde. In the Stöber-like system, TEOS was used as silica oligomers from hydrolysis to form a mesoporous structure in PF. The SiO2/PF spheres were then pyrolyzed under nitrogen protection to remove the CTAB and form a porous carbon structure, and then the SiO2 core was etched with a 1 M NaOH solution. The target product PFc material was then carbonized by heating in a nitrogen atmosphere at 1073 K in a tube furnace. Furthermore, the solvent polarity of the Stöber-like system directly affects the relative rates of TEOS hydrolysis and resin polymerisation, and thus the product morphology can be effectively tuned by varying the ratio of EtOH and water (changing the polarity of the solvent mixture).17In order to analyze the variation of functional groups on the surface of PFc during the carbonization process, Fourier transform infrared spectroscopy (FTIR) characterization of PFc-4 was first performed (Fig. 1(a) and (b)). As shown in Fig. 1(a), the broad and strong peak at 3417 cm−1 before carbonization corresponds to the O–H and N–H stretching vibrations. The weak peaks at 2927 cm−1 and 2857 cm−1 correspond to the C–H stretching vibrations of the methyl and methylene groups, respectively. The strong peak at 1623 cm−1 corresponds to the CC stretching vibrations in aromatic rings, and the shoulder peak at 1243 cm−1 corresponds to the C–N stretching vibrations of aromatic amines.18 It is worth noting that the broad and strong peak at 1080 cm−1 corresponds to the antisymmetric stretching vibration of Si–O–Si. The narrow and sharp peak at 808 cm−1 corresponds to the symmetric bending vibration of O–Si–O, and the sharp and strong peak at 460 cm−1 corresponds to the bending vibration of Si–O–Si.19
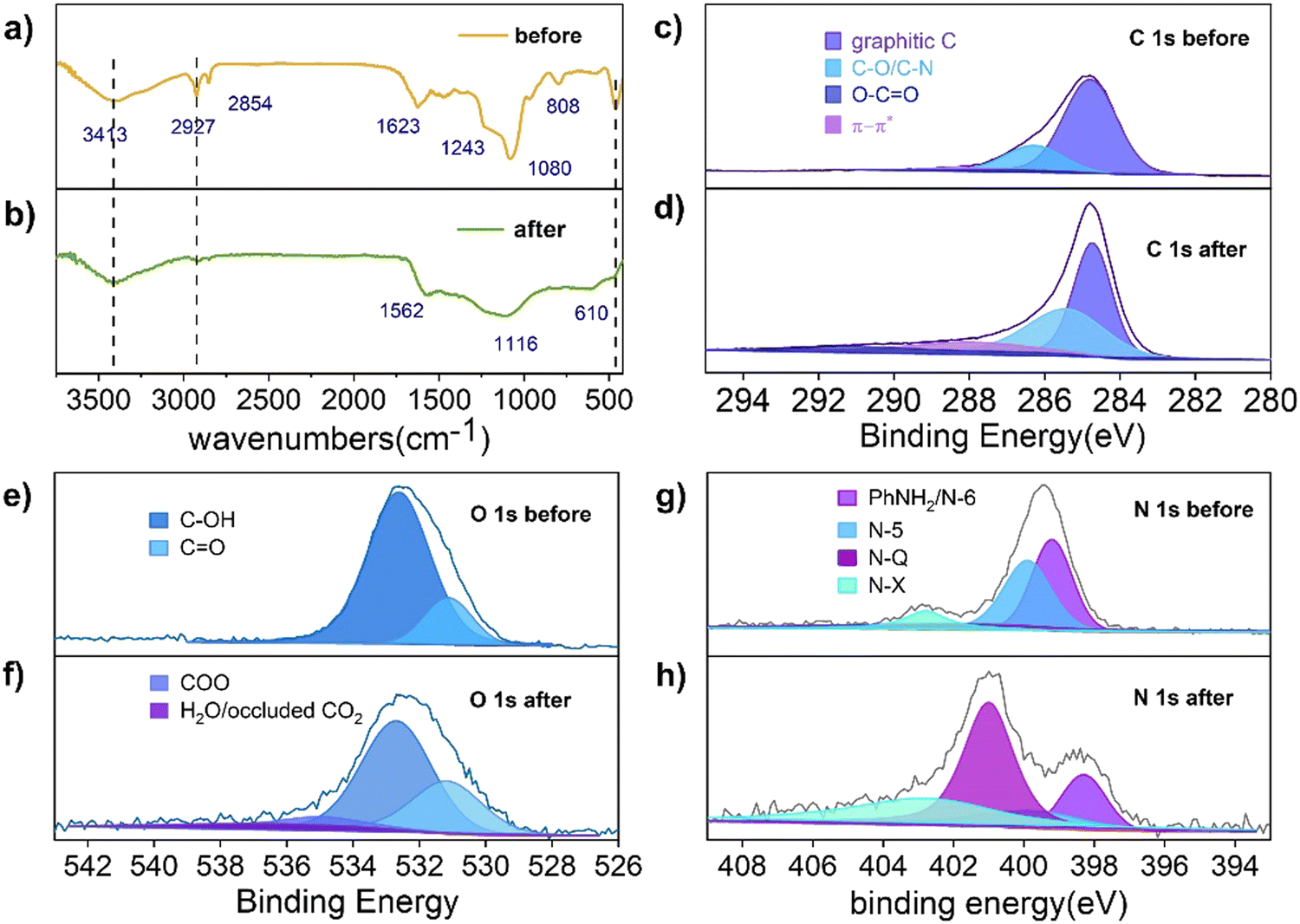 | ||
| Fig. 1 FTIR spectra of the sample (a) before and (b) after carbonization. XPS spectra of C1s, O1s, N1s of the sample (c), (e), (g) before and (d), (f), (h) after carbonization, respectively. | ||
After carbonization (Fig. 1(b)), the peaks at 2927 cm−1 and 2857 cm−1 almost disappeared, and the intensity of the peak at 3417 cm−1 is significantly weakened. This indicates that the –OH and N–H bonds still remained on the surface of PFc. The CC stretching vibration peak at 1562 cm−1 indicates that the phenolic resin was successfully carbonized.14 A significant decrease in the intensity of the C–O stretching vibration at 1116 cm−1 indicates a partial loss of the hydroxyl group during the carbonization process. In contrast, the disappearance of the O–Si–O bending vibration at 808 cm−1 indicates that the silicon oxide was successfully removed during the etching process. In addition, a new ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H bending vibration absorption peak appears at 610 cm−1, indicating that the conjugation of the material is enhanced to some extent after carbonization, which facilitates charge transfer within the electrode.20
C–H bending vibration absorption peak appears at 610 cm−1, indicating that the conjugation of the material is enhanced to some extent after carbonization, which facilitates charge transfer within the electrode.20
To further investigate the effect of the carbonization process on the material elements and their chemical state, X-ray photoelectron spectroscopy (XPS) measurements were conducted to characterise PFc before and after carbonization (Fig. 1(c)–(f)). As shown in Fig. 1(c), the C 1s spectrum of PFc before carbonization exhibits peaks at binding energies of 284.7, 286.2 and 288.3 eV, which correspond to graphitic C, C–O/C–N and CO, respectively. After carbonization (Table S1, ESI†), the graphitic carbon content decreases, and the C–O/C–N and CO peaks increase with the increase of oxygen probably from the NaOH etching process. The CO is partly from the oxidation of surface hydroxyl groups.21,22 The elevated C–N content, on the other hand, may be from the conversion of NH3 adsorbed on the surface. In contrast, the XPS spectra of O 1s (Fig. 1(e) and (f)) and N 1s (Fig. 1(g) and (h)) present the conversions between the doped forms of oxygen and nitrogen before and after carbonation. As shown in Fig. 1(e) and (f), in conjunction with Table S2 (ESI†), the content of surface hydroxyl groups is reduced, while the content of CO is increased, which is consistent with the results from Table S1 (ESI†). In addition, some hydroxyl groups shift to higher oxidation states (carboxylic acids). The peak at 537 eV can be attributed to the CO2 adsorbed on the pore surface, indicating that carbonization does increase the porosity and specific surface area of the material. From Table S4 (ESI†), the total surface oxygen content decreased from 12% to 4.57% after carbonization. As shown in Fig. 1(g) and (h), the surface N exists as pyridine N (N-6), pyrrole N (N-5), graphite N (N-Q) and oxidized N (N-X) with binding energies at 399.2, 399.9, 401.0 and 402.7 eV, respectively.20 In combination with Table S3 (ESI†), the content of N-6 and N-5 on the surface of the carbon material decreases, and that of N-Q and N-X increases substantially after carbonization. In particular, N-Q, the N atom embedded in the carbon matrix and bound to three carbon atoms, can improve the electrical conductivity of the carbon material, favouring its internal electron transfer. According to the ref. 23 and 24 the increase in surface N-X content also favours the increase in conductivity of PFc. According to Table S4 (ESI†), the surface total N content decreased from 8.38% to 4.88%, indicating that there is a loss of N during the carbonization process.
For the solvent mixture in this Stöber-like system, the effect of different EtOH/H2O ratios and subsequent polarity changes on the morphology and internal structure of PFc was investigated. The morphology of the PFc samples with different EtOH/H2O ratios was characterized using (SEM) and (TEM). As shown in Fig. S2 (ESI†), the PFc-0 and PFc-7 have completely different microporous structures, showing the determining effect of the solvent ratios on the morphology. This is because the addition of EtOH into H2O regulates the hydrolysis rate of TEOS under the bridging effect of CTAB by changing the polarity of the solution, which in turn affects the interaction with the phenolic resin.25 The electron micrographs of PFc-1, 3, 4 and 6 obtained by varying EtOH/H2O are shown in Fig. 2. As shown in Fig. 2(a) (SEM) and 2e (TEM) combined with Fig. S3a (ESI†), for the hollow carbon sphere structure of PFc-1, the particle size, shell thickness and internal pore size are 70, 14 and 25 nm, respectively. From SEM (Fig. 2(b) and (c)) and TEM (Fig. 2(f) and (g)), the particle sizes of PFc-3 and PFc-4 increased to 0.8 and 1.2 μm, respectively. As presented in Fig. 2(h) (cross section SEM), a lemon-like porous structure is found in PFc-3 and PFc-4. As the EtOH/H2O ratio continues to increase, the core–shell structure of PFc-6 increased and began to agglomerate. The larger particle size of the core–shell structure forms due to a greater amount of ethanol slowing down the hydrolysis rate of TEOS and increasing the polymerization rate of resin, so that a larger resin core is obtained.25 With the hydrolysis of TEOS, it coats the outer surface of the resin core with silica at the same time. Through etching and sintering, a uniform mesoporous structure finally formed as the PFc carbon materials. The mesoporous structure is beneficial to increase the specific surface area of PFcs, promoting the contact between the perovskite crystal and counter electrode, and improving the interfacial charge transfer properties. On the other hand, for the assembly process of p-MPSCs devices, the mesoporous carbon structure can promote the downward penetration of the perovskite precursor solution to the M-TiO2 and ZrO2 layers and reduce the internal defects of the devices.14 In addition, the elemental distribution of C (Fig. 2(j)) and O (Fig. 2(k)) was obtained from the TEM image of the sample PFc-4 (Fig. 2(i)). As shown in Fig. 2(k), the O element in PFc-4 is not only well distributed but also abundant, which will help to enhance the wettability of the perovskite precursor on the surface of the carbon electrode.
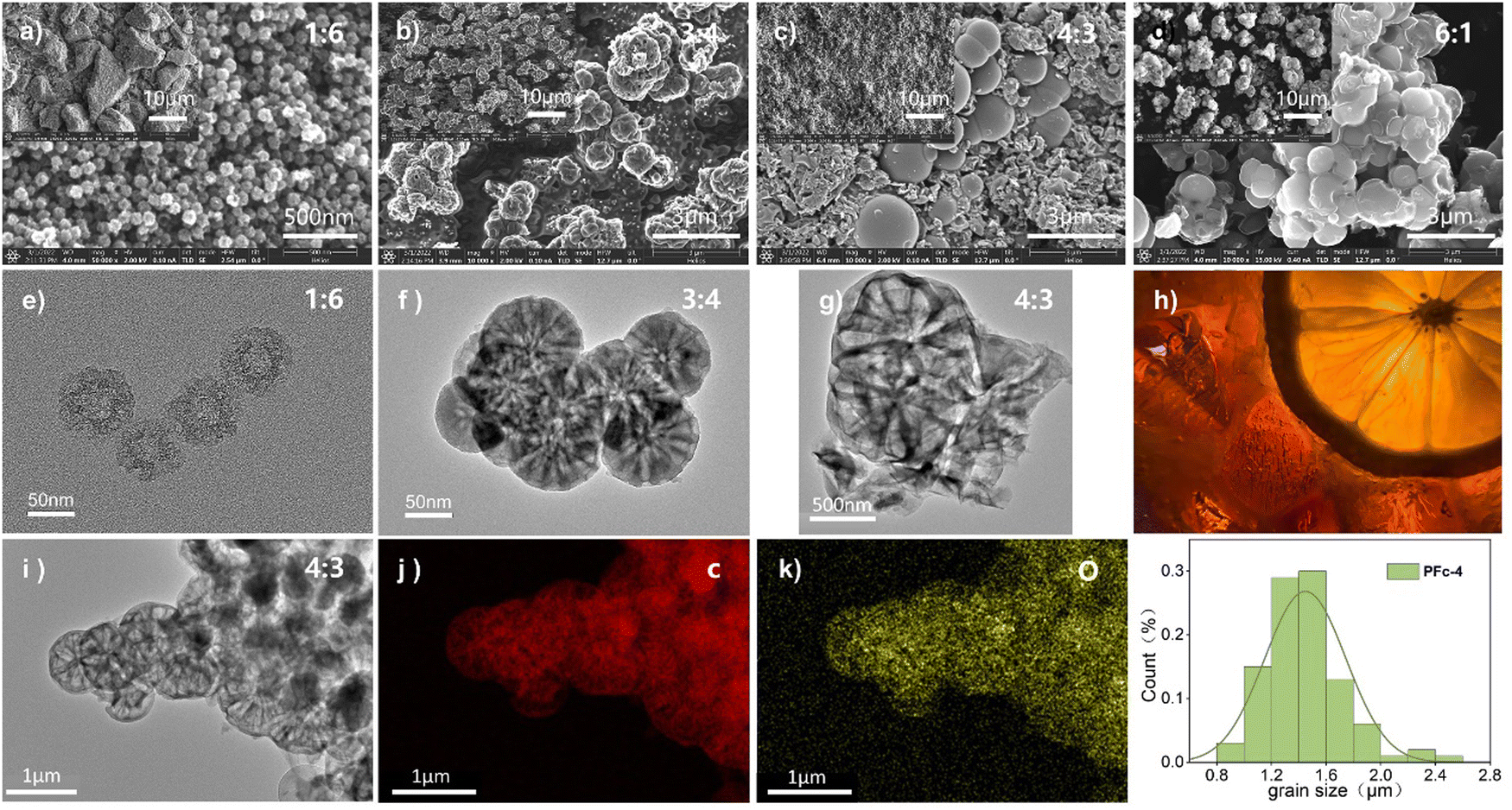 | ||
| Fig. 2 SEM and TEM images of the PFc samples: PFc-1 (a), (e), PFc-3 (b), (f), PFc-4 (c), (g), PFc-6 (d), (h) cross section of a lemon, (i)–(k) elemental mappings of PFc-4, (l) granulometric distribution of PFc-4. | ||
To confirm the effect of different morphologies with various polarities of solvent on the electrical conductivity of the carbon material, the square resistance of the films made from samples PFc-1, PFc-3, PFc-4 and PFc-6 are listed in Table S5 (ESI†). The average square resistances of the four samples are 11.72, 19.44, 16.57, 19.00 and 18.58 Ω sq−1 respectively. The square resistances do not show any regular change with the EtOH/H2O ratios. To investigate the effect of morphology with different EtOH/H2O ratios on the photovoltaic performance of p-MPSCs devices, we characterized the degree of graphitisation of the PFc samples by Raman spectroscopy (Fig. 3(a)) with an excitation wavelength of 532 nm. As shown in Fig. 3(a), two peaks near 1350 and 1580 cm−1 can be attributed to the sp3 defective site (D band) and crystalline graphite (G band), respectively.26 The peak intensity ratios of the D-band to G-band (ID/IG) for PFc-0, PFc-1, PFc-3, PFc-4, PFc-6 and PFc-7 are 0.667, 1.353, 0.434, 0.392, 0.610 and 0.637, respectively. With an increase of the EtOH/H2O ratios, the ID/IG values decrease first and then increase, and PFc-4 reaches the lowest one. The lowest ID/IG value for PFc-4 indicates its highest degree of graphitization,27 a high uniformity of particle size, a lower density of defect states and fewer dangling bonds. For p-MPSC, the high degree of graphitisation facilitates the charge transport within the carbon electrode,28 which in turn increases the open circuit voltage (Voc) and fill factor (FF) (Fig. 5(a)). In Fig. S5 (ESI†), the XRD pattern shows a higher graphite peak around 26° for PFc-4 compared to the control group, which is consistent with the test results of Raman spectroscopy.
 | ||
| Fig. 3 (a) Raman spectra of PFc-0, PFc-1, PFc-3, PFc-4, PFc-6, and PFc-7. (b) Raman spectra of PFc-4, (c) pore size distribution of PFc-4, (d) and nitrogen adsorption/desorption isotherms of PFc-4. | ||
To further detect their internal structures, nitrogen isothermal adsorption/desorption curves (Brunauer–Emmett–Teller method, BET) were used to characterise the porosity and specific surface area of the samples. As shown in Fig. 3(d) and Fig. S4a (ESI†), the PFc samples prepared with different EtOH/H2O ratios show a typical type IV isothermal circuit with clear H3 hysteresis loops in the range of relative pressure from 0.3 to 0.9, indicating a rich pore structure.29,30 The shifts of the adsorption curves towards the Y-axis at the low pressure end indicate a strong interaction between the samples and nitrogen.31 From the pore size distribution curves of PFcs (Fig. 3(c) and Fig. S4b, ESI†), all samples have a sharp peak within 10.00 nm, corresponding to the micro-pores formed by the decomposition of CTAB during the carbonization process. As shown in Table S6 (ESI†), the specific surface area and pore volume of the PFc samples obtained from the BET tests do not show much regularity. However, it demonstrates that all the samples (exception of PFc-6) exhibit excellent microporous structures with specific surface areas and pore volumes of 620–970 m2 g−1 and 0.5–1.3 cm3 g−1, respectively. The PFc-3 and PFc-4 samples have similar specific surface areas (699.948 and 703.636 m2 g−1, respectively) and pore volumes (0.585 and 0.590 cm3 g−1, respectively).
Based on the above characterization results regarding the morphology, graphitization and specific surface area, we selected PFc-3, PFc-4 and PFc-6 to replace 50% of the carbon black in the original carbon paste for the carbon electrodes of the p-MPSCs devices. As shown in Fig. S9 (ESI†) and Table 1, the relatively high photoelectric conversion efficiencies of the devices based on PFc-3, PFc-4 and PFc-6 are obtained for all three samples compared to control. The fill factor (FF) of all three samples exceeded 70% due to the improved contact properties between the perovskite crystals and carbon materials, which reduce the interfacial charge transfer resistance. As for PFc-6, due to the poor homogeneity and occurrence of agglomeration, its Jsc and Voc are lower and FF is higher than those of control. An average PCE of 14.69% is also obtained, which is higher than that of the control group (14.19%). For PFc-4 with the best morphological homogeneity and highest graphitization, the highest average PCE of 16.33% is obtained with high Voc (average value of 0.98 V) and FF (average value of 73.06%). The optimized photovoltaic parameters of the champion p-MPSC devices based on PFc-4 and control are listed in Table 2. The highest PCE for PFc-4 and control are 17.64% (Voc = 1.03 V, Jsc = 21.54 mA cm−2, FF = 79.93%) and 15.31% (Voc = 0.96 V, Jsc = 21.60 mA cm−2, FF = 73.15%), respectively. In particular, without any passivation treatment, PFc-4 gives a Voc of up to 1.03 V and a FF of 79.93%. This is close to the device's current maximum efficiency of 18.86%.32
| Electrodes | J sc [mA cm−2] | V oc [V] | FF [%] | PCE [%] | PCE max [%] |
|---|---|---|---|---|---|
| PFc-3 | 23.00 ± 1.2 | 0.94 ± 0.01 | 71.61 ± 4.5 | 15.46 ± 0.4 | 16.14 |
| PFc-4 | 22.68 ± 0.5 | 0.98 ± 0.04 | 73.06 ± 4.6 | 16.33 ± 0.5 | 17.64 |
| PFc-6 | 20.94 ± 0.1 | 0.93 ± 0.03 | 75.28 ± 3.1 | 14.69 ± 0.7 | 15.49 |
| Control | 21.35 ± 1.4 | 0.96 ± 0.06 | 61.96 ± 8.4 | 14.19 ± 1.4 | 15.31 |
| Electrodes | J sc [mA cm−2] | V oc [V] | FF [%] | PCE [%] |
|---|---|---|---|---|
| PFc-4 | 21.54 | 1.03 | 79.93 | 17.64 |
| Control | 21.60 | 0.96 | 73.15 | 15.31 |
So, the optimal amount of PFc-4 to be used for the fabrication of devices (partially replace the amount of carbon black in the carbon paste of control group) was next investigated. The amounts selected are 25%, 50%, 75% and 100%, respectively, and denoted as 25-PFc-4, 50-PFc-4, 75-PFc-4 and 100-PFc-4. The relative photovoltaic parameters obtained are shown in Fig. S10. The highest Voc, FF and PCE are obtained for 50-PFc-4. So, for all subsequent assembled devices, the amount of PFc-4 used is 50%.
To further investigate the deeper reasons for the best photovoltaic conversion performance of PFc-4, we proceeded to test the XRD spectra of the devices based on PFc-4 and the control group. As shown in Fig. 4(a), the main diffraction peaks located at 14.14° and 28.40° correspond to the (100) and (200) crystal planes of perovskite, respectively. Compared with the control group, there are no peak shifts or new diffraction peaks for PFc-4, indicating that its morphology does not enter the crystallization process of perovskite. Contrary to the expected results, the intensity of the perovskite diffraction peaks based on the PFC-4 devices was slightly lower than that of the control group. This is mainly due to the perovskite crystals being basically distributed inside the device, rather than on the surface for the specific structure of the p-MPSCs devices (Fig. S8, ESI†). From the partial magnification (Fig. 4(b)) of Fig. 4(a), all of the main crystalline peaks of perovskite based on the PFc-4 devices shift to smaller angular directions, indicating that the increase in the size and crystallinity of the perovskite grains helps to reduce the boundaries and suppress non-radiative recombination. This, in turn, can promote hole transport in the perovskite crystals, facilitating enhancement of the current density33 of the p-MPSCs device based on PFc-4 (as listed in Table 1). The increase in Jsc is also related to the work function (WF) of the carbon material. As shown in the UPS test results in Fig. S7 (ESI†), the WF of control and PFc-4 are 5.47 and 5.41 eV, respectively. The reduced WF of PFc-4 indicates a lower binding of the excited state electrons, in favor of electron transport within the carbon electrode and at the interface between carbon and perovskite. In addition, the J–V curves of the cells prepared from PFc-4 with forward and reverse scans are shown in Fig. S13 (ESI†), showing a slight hysteresis effect.
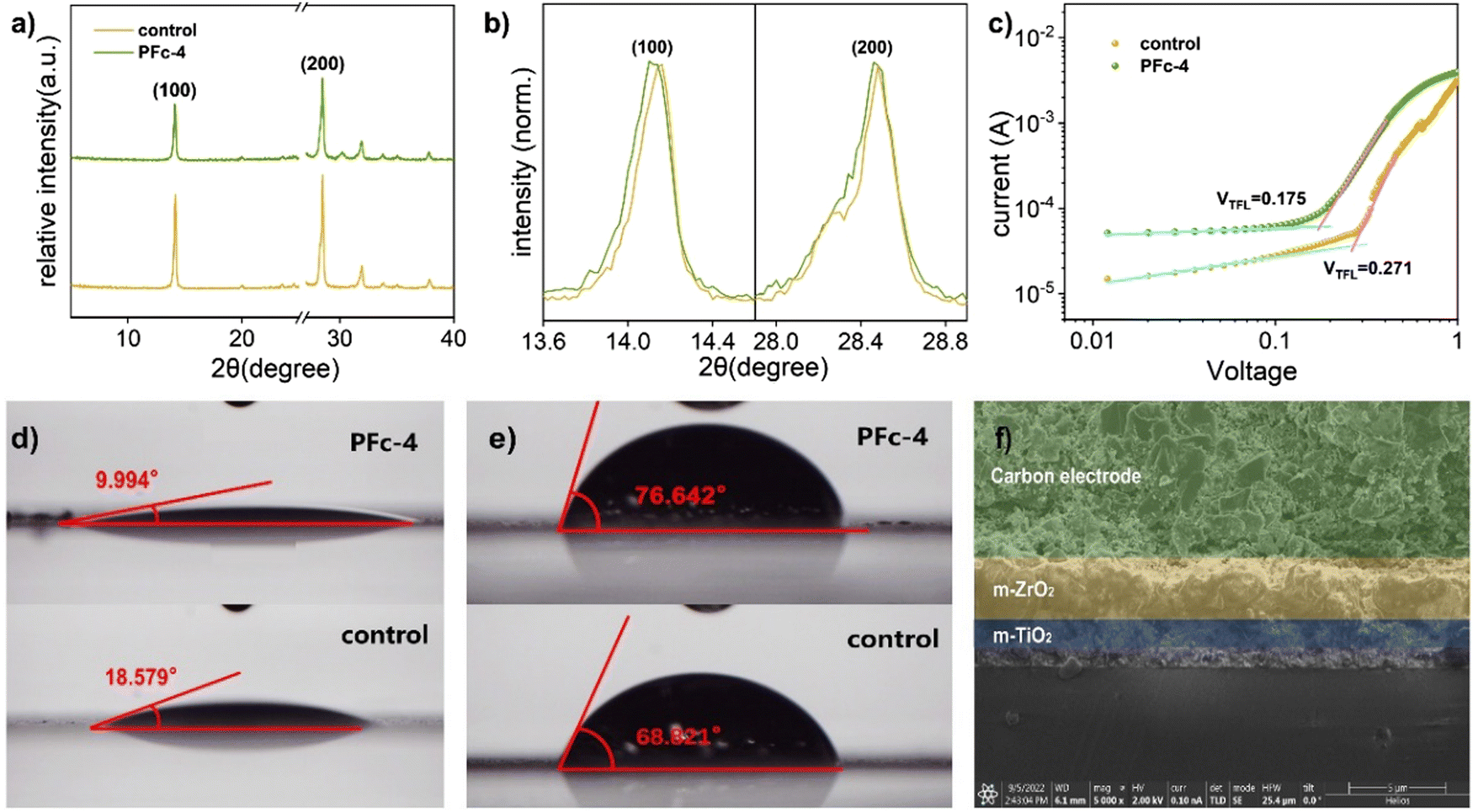 | ||
| Fig. 4 (a) and (b) XRD spectra of PFc-4 and the ontrol. (c) The SCLC plot of hole-only devices. The device structure is Glass/FTO/compact-NiO/mp-ZrO2/porous carbon, and the deposition process of perovskite is identical to that of the complete device. The contact angle of the (d) perovskite precursor solution and (e) H2O on the control and PFc-4 modified p-MPSCs. (f) Cross-section SEM image of p-MPSCs based on PFc-4. | ||
For the devices with the same structure, their Voc is mainly controlled by the defect state density of the device. Compared with control (Voc = 0.96 V), the higher Voc of PFc-4 (Voc = 1.03 V) is due to its lower density of defect states. To confirm this conclusion, the space charge-limited current (SCLC) curves were tested with the FTO/compact-NiO/mesoporous(mp)-ZrO2/carbon structure (Fig. 4(c)). For the SCLC, the voltage at the intersection of the first and second regions corresponds to the trap-filled limit voltage (VTFL). As shown in Fig. 4(c), the smaller VTFL of PFc-4 compared with the control indicates its lower density of defect states (Ndefect), which is attributed to the improvement of the crystalline quality of the perovskite and contact property between the perovskite and carbon electrode.34 According to ref. 35 and 36, Ndefect can be calculated according to eqn (1).
| Ndefect = 2ε0εrVTFL/eL2 | (1) |
To further determine the non-radiative recombination inhibition capability, photoluminescence spectroscopy (PL) (Fig. 5(b)) and time-resolved photoluminescence spectroscopy (TRPL) (Fig. 5(c)) based on the Control group and PFc-4 containing perovskite were conducted on the conductive substrates. As shown in Fig. 5(b), PFc-4 exhibits weaker PL intensity compared with the Control group, indicating its faster carrier transfer rate and higher charge extraction efficiency at the interface of carbon and perovskite. In addition, the blue shift of the PL peak of PFc-4 from 774 nm to 766 nm shows the decrease of the trap state density at the grain boundary,38 which is consistent with the result from Fig. 4(c).
 | ||
| Fig. 5 (a) J–V curves of p-MPSCs with the control and PFc-4. (b) Photoluminescence (PL) and (c) time-resolved PL (TRPL) spectra of perovskite deposited on the half devices (p-MPSCs with mesoscopic ZrO2 and carbon layers on the glass). (d) The steady-state output and current density of the corresponding devices measured at a bias of 0.708 V for the PFc-4 device under 100 mW cm−2 AM 1.5G irradiation. (e) Nyquist plots of PSCs based on the control and PFc-4 modified p-MPSCs at 600 mV under dark conditions. (f) Vocversus light intensity of PSCs based on the control and PFc-4 modified p-MPSCs. (g) Ambient stabilities of the devices based on the control and PFc-4 modified p-MPSCs by storing the unencapsulated device in ambient condition (temperature: 25 °C; relative humidity: 40–60%) for 131 days. | ||
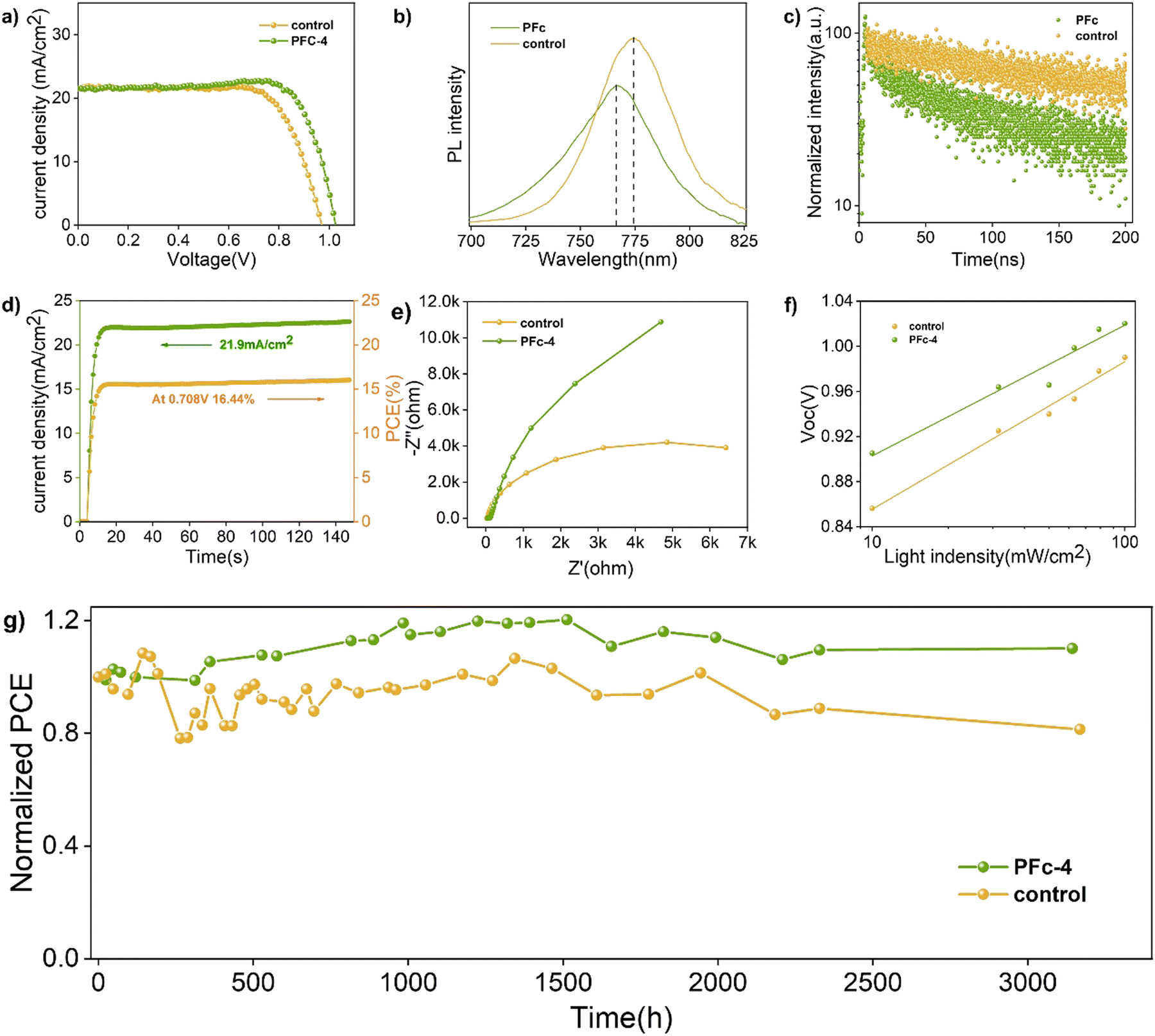
To get the charge transport lifetime inside the materials, the TRPL spectra of control and PFc-4 (Fig. 5(c)) were fitted using the double exponential decay eqn (2).39
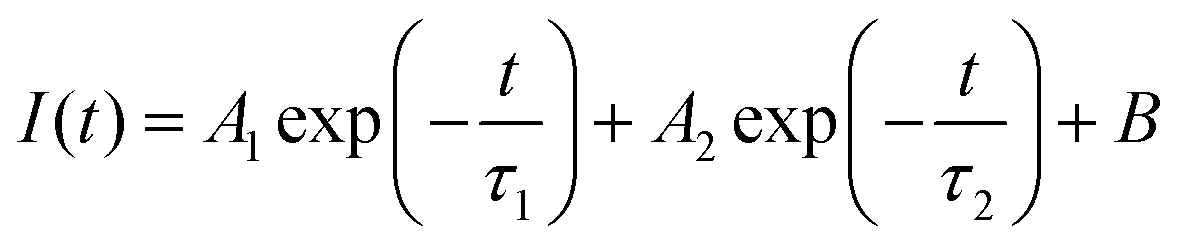 | (2) |
The average decay time constant (τave) is then calculated using eqn (3).
| τave = (A1τ12 + A2τ22)/(A1τ1 + A2τ2) | (3) |
The relevant parameters obtained from the fitting results are listed in Table S7 (ESI†). As shown in Table S7 (ESI†), the average carrier lifetime of PFc-4 decreases from 155.06 ns of the control group to 76.55 ns, indicating a faster charge transfer rate inside the PFc-4 electrode.
For the improvement of the charge transport characteristics, the role of the built-in potential (Vbi) should also be considered. Mott–Schottky is often used to elucidate the effect of the built-in potential (Vbi) on the charge transfer performance of the solar cells.38 As shown in Fig. S11 (ESI†), the straight-line portion between 0.4 and 0.8 V of the Mott–Schottky curves for the control group and PFc-4 devices can be fitted with eqn (4).
| C−2 = 2(Vbi − V)/(A2qεε0N) | (4) |
From the horizontal coordinate intercept of fitted lines, C−2 = 0, so V = Vbi. As a result, the Vbi for the control and PFc-4 groups are 0.68 and 0.71 V, respectively. The higher Vbi indicates that the device based on PFc-4 has less carrier accumulation at the interface and faster carrier transport rate, suppressing charge recombination. In Fig. 5(d), the p-MPSC devices with PFc-4 as the counter electrodes also exhibit good steady-state output Jsc (21.9 mA cm−2) and PCE (16.44%) at a bias of 0.708 V.
For the charge transfer and recombination properties of the p-MPSC devices, they can also be reflected in the electrochemical impedance spectroscopy (Nyquist plot) test results.39 As shown in Fig. 5(e), the diameter corresponding to the half-circle in the high frequency region represents the charge transfer resistance (Rct). Compared to the control group, the devices based on PFc-4 have a smaller Rct (Fig. S12, ESI†). A lower Rct results in a smaller interfacial contact resistance between carbon and the perovskite crystals, favoring interfacial charge transfer. In the low frequency region, the diameter of the semicircle corresponds to the charge recombination resistance (Rrec). The larger Rrec of PFc-4 indicates that the recombination of photoexcited electrons can be effectively suppressed, which is consistent with the Ndefect value fitted from Fig. 4(c).
In contrast, the suppression of non-radiative recombination of the device can be characterised by a Vocversus light intensity curve fitted using eqn (5).
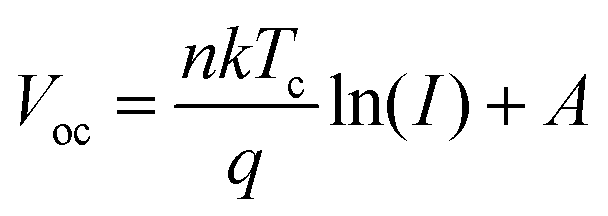 | (5) |
As the light intensity increases, more photogenerated carriers are produced, ultimately leading to a high open-circuit voltage.40,41 A value of n that is close to 1 indicates less carrier recombination. As shown in Fig. 5(f), the Voc of the PFc-4-based device is higher than that of the control-based one under any light intensity. The fitted results of the n-value for the control and PFc-4 groups are 2.20 and 1.64, respectively, exhibiting the higher non-radiative recombination suppression capability for PFc-4. This is consistent with the results from Fig. 5(e) and Fig. S12 (ESI†).
During the fabrication process of the m-PSCs devices, the perovskite precursor is added dropwise on the top of the carbon electrode and penetrates the device. Thus, the wettability of the carbon electrode to the perovskite precursor significantly impacts the device performance.
As shown in Fig. 4(d), the contact angle of the perovskite precursor on PFc-4 is reduced from 18.579° to 9.994° compared with the control group. The better wettability of PFc-4 facilitates the diffusion and distribution of perovskite within the device, and improves the contact property between carbon and perovskite. The superior wettability of PFc-4 is also evidenced by the SEM image of the device interface (Fig. 4(f)). As shown in Fig. 4(f), the perovskite is densely filled in the ZrO2 and TiO2 layers. In addition, functional groups, such as C–O/C–N and CO, are present in PFc-4 and can interact with Pb2+ to further enhance the contact and interaction between the carbon electrode and perovskite.42 The cross-sectional elementary analysis from energy dispersive X-ray spectroscopy (Fig. S8, ESI†) further supports the evidence of perovskite distribution within the device. As depicted in Fig. S8 (ESI†), the elements C, Ti and Zr represent the C, TiO2 and ZrO2 layers, respectively. Pb and I are mainly distributed in the ZrO2 and TiO2 layers and a small amount in the C layer close to the C/ZrO2 interface, showing good wettability between PFc-4 and perovskite with good permeability.
For the perovskite solar cells, H2O, which can cause decomposition of perovskite crystals, is one of the main factors affecting its stability.43 As shown in Fig. 4(e), the contact angles of H2O on control and PFc-4 groups are 68.821° and 76.642°, respectively. The high capability of PFc-4 to suppress water penetration can improve the stability of the device due to the inhibition of the perovskite crystal decomposition.
Finally, the long-term stabilities of the p-MPSCs devices based on the control and PFc-4 carbon electrodes were observed at 25 °C (RH = 40–60%). As shown in Fig. 5(g), the PCE of the control group retained 81% of the initial value after 131 days, while that of PFc-4 reached 110% of the initial efficiency44–48 due to the recrystallization process of perovskite under a certain humidity.49–54
4. Conclusion
In summary, a phenol-formaldehyde resin-based carbon (PFc) was developed via co-assembly engineering with improved Stöber strategy (tetraethyl orthosilicate (TEOS) as the hard template, cetyltrimethyl ammonium bromide (CTAB) as the structure guidance) for p-MPSCs as counter electrodes. The open-circuit voltage (Voc) of PFc-based p-MPSCs increases from 0.97 V to 1.03 V (without any passivation treatment) with a maximum PCE of 17.64%, exhibiting excellent stability by modulation of solvent polarity (EtOH/H2O ratio). By means of infrared spectroscopy, SEM, TEM, SCLC, PL, TRPL, Raman and XRD, it is found that the new PFc carbon material has good infiltrative property to the perovskite precursor, and shows superior defect state and non-radiation recombination inhibition effect. It has a promising prospect due to its low-cost, simple method and high performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by NSFC/China (grant no. 21788102, 22075083), and the Programme of Introducing Talents of Discipline to Universities (grant no. B16017).References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells, J. Am. Chem. Soc., 2009, 131(17), 6050–6051 CrossRef CAS PubMed.
- M. Kim, J. Jeong, H. Lu, T. K. Lee, F. T. Eickemeyer, Y. Liu, I. W. Choi, S. J. Choi, Y. Jo, H. B. Kim, S. I. Mo, Y. K. Kim, H. Lee, N. G. An, S. Cho, W. R. Tress, S. M. Zakeeruddin, A. Hagfeldt, J. Y. Kim, M. Gratzel and D. S. Kim, Conformal quantum dot-SnO2 layers as electron transporters for efficient perovskite solar cells, Science, 2022, 375(6578), 302 CrossRef CAS PubMed.
- F. Zhang, X. Yang, H. Wang, M. Cheng, J. Zhao and L. Sun, Structure Engineering of Hole-Conductor Free Perovskite-Based Solar Cells with Low-Temperature-Processed Commercial Carbon Paste As Cathode, ACS Appl. Mater. Interfaces, 2014, 6(18), 16140–16146 CrossRef CAS PubMed.
- A. Mei, X. Li, L. Liu, Z. Ku, T. Liu, Y. Rong, M. Xu, M. Hu, J. Chen, Y. Yang, M. Grätzel and H. Han, A hole-conductor–free, fully printable mesoscopic perovskite solar cell with high stability, Science, 2014, 345, 295 CrossRef CAS PubMed.
- X. Xiao, Y. Chu, C. Zhang, Z. Zhang, Z. Qiu, C. Qiu, H. Wang, A. Mei, Y. Rong, G. Xu, Y. Hu and H. Han, Enhanced perovskite electronic properties via A-site cation engineering, Fundam. Res., 2021, 1(4), 385–392 CrossRef CAS.
- J. He, T. Ding and W. Wu, Surface Lattice Perturbation of Electron Transport Layer Reducing Oxygen Vacancies for Positive Photovoltaic Effect, Sol. RRL, 2022, 6(10), 2200226 CrossRef CAS.
- Y. Chen, D. Ma, Z. Wang, J. He, X. Gong and W. Wu, Tautomeric Dual-Site Passivation for Carbon-Based Printable Mesoscopic Perovskite Solar Cells, Adv. Mater. Interfaces, 2022, 9(17), 2200326 CrossRef CAS.
- L. Zhang, T. Liu, L. Liu, M. Hu, Y. Yang, A. Mei and H. Han, The effect of carbon counter electrodes on fully printable mesoscopic perovskite solar cells, J. Mater. Chem. A, 2015, 3(17), 9165–9170 RSC.
- X. Chen, L. Lu, D. Gu, X. Zhang, H. Yu, F. Chen, Y. Rui, J. Hou and Y. Yang, Chlorine management of a carbon counter electrode for high performance printable perovskite solar cells, J. Mater. Chem. C, 2021, 9(27), 8615–8622 RSC.
- M. Duan, Y. Rong, A. Mei, Y. Hu, Y. Sheng, Y. Guan and H. Han, Efficient hole-conductor-free, fully printable mesoscopic perovskite solar cells with carbon electrode based on ultrathin graphite, Carbon, 2017, 120, 71–76 CrossRef CAS.
- Y. Zhong, L. Xu, C. Li, B. Zhang and W. Wu, Needle coke: A predominant carbon black alternative for printable triple mesoscopic perovskite solar cells, Carbon, 2019, 153, 602–608 CrossRef CAS.
- C. Liu, C. Gao, W. Wang, X. Wang, Y. Wang, W. Hu, Y. Rong, Y. Hu, L. Guo, A. Mei and H. Han, Cellulose-Based Oxygen-Rich Activated Carbon for Printable Mesoscopic Perovskite Solar Cells, Sol. RRL, 2021, 5(9), 2100333 CrossRef CAS.
- W. Stöber, A. Fink and E. J. Bohn, Extension of the Stöber method to the preparation of monodisperse resorcinol-formaldehyde resin polymer and carbon spheres, J. Colloid Interface Sci., 1968, 26(1), 62–69 CrossRef.
- J. Liu, S. Qiao, H. Liu, J. Chen, A. Orpe, D. Zhao and G. Lu, Angew. Chem., Int. Ed., 2011, 50(26), 5947–5951 CrossRef CAS PubMed.
- J. Du, S. Zong, Y. Zhang, S. Hou and A. Chen, Co-assembly strategy for uniform and tunable hollow carbon spheres with supercapacitor application, J. Colloid Interface Sci., 2020, 565, 245–253 CrossRef CAS PubMed.
- J. Hou, X. Tu, X. Wu, M. Shen, X. Wang, C. Wang, C. Cao, H. Peng and G. Wang, Remarkable cycling durability of lithium-sulfur batteries with interconnected mesoporous hollow carbon nanospheres as high sulfur content host, Chem. Eng. J., 2020, 401(1), 126141 CrossRef CAS.
- J. Hou, T. Cao, F. Idrees and C. Cao, A co-sol-emulsion-gel synthesis of tunable and uniform hollow carbon nanospheres with interconnected mesoporous shells, Nanoscale, 2016, 8(1), 451–457 RSC.
- J. Sheng, X. Xu and W. Wu, Manganese doping: A novel approach to enhancing surface wettability of carbon electrodes, Carbon Trends, 2022, 9, 100217 CrossRef CAS.
- W. Sun, R. Mu, X. Lua and C. W. Miao, Effect of chloride salt, freeze-thaw cycling and externally applied load on the performance of the concrete, Cem. Concr. Res., 2002, 32(12), 1859–1864 CrossRef CAS.
- A. R. Campanelli and A. Domenicano, Structural variation, π-charge transfer, and transmission of electronic substituent effects through the carbon-carbon triple bond in β-substituted phenylacetylenes: a quantum chemical study, and a comparison with (E)-β-substituted styrenes, Struct. Chem., 2019, 30, 9–21 CrossRef CAS.
- C. Macias, G. Rasines, P. Lavela, M. C. Zafra, J. L. Tirado and C. O. Ania, Mn-Containing N-Doped Monolithic Carbon Aerogels with Enhanced Macroporosity as Electrodes for Capacitive Deionization, ACS Sustainable Chem. Eng., 2016, 4(5), 2487–2494 CrossRef CAS.
- Sutarsis, J. Patra, C. Y. Su, J. Li, D. Bresser, S. Passerini and J. K. Chang, Manipulation of Nitrogen-Heteroatom Configuration for Enhanced Charge-Storage Performance and Reliability of Nanoporous Carbon Electrodes, ACS Appl. Mater. Interfaces, 2020, 12(29), 32797–32805 CrossRef CAS PubMed.
- J. Zhao, H. Lai, Z. Lyu, Y. Jiang, K. Xie, X. Wang, Q. Wu, L. Yang, Z. Jin, Y. Ma, J. Liu and Z. Hu, Hydrophilic Hierarchical Nitrogen-Doped Carbon Nanocages for Ultrahigh Supercapacitive Performance, Adv. Mater., 2015, 27(23), 3541–3545 CrossRef CAS PubMed.
- L. Feng, K. Wang, X. Zhang, X. Sun, C. Li, X. Ge and Y. Ma, Flexible Solid-State Supercapacitors with Enhanced Performance from Hierarchically Graphene Nanocomposite Electrodes and Ionic Liquid Incorporated Gel Polymer Electrolyte, Adv. Funct. Mater., 2018, 28(4), 1704463 CrossRef.
- Y. Chen and J. Shi, Chemistry of Mesoporous Organosilica in Nanotechnology: Molecularly Organic–Inorganic Hybridization into Frameworks, Adv. Mater., 2016, 28(17), 3235–3272 CrossRef CAS PubMed.
- Y. Zhong, X. Xia, S. Deng, J. Zhan, R. Fang, Y. Xia, X. Wang, Q. Zhang and J. Tu, Popcorn Inspired Porous Macrocellular Carbon: Rapid Puffing Fabrication from Rice and Its Applications in Lithium-Sulfur Batteries, Adv. Energy Mater., 2018, 8(1), 1701110 CrossRef.
- L. Lin, F. Pei, J. Peng, A. Fu, J. Cui, X. Fang and N. Zheng, Fiber network composed of interconnected yolk-shell carbon nanospheres for high-performance lithium-sulfur batteries, Nano Energy, 2018, 54, 50–58 CrossRef CAS.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Poschl, Raman micro spectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information, Carbon, 2005, 43(8), 1731–1742 CrossRef CAS.
- Q. Yue, Y. Zhang, Y. Jiang, J. Li, H. Zhang, C. Yu, A. A. Elzatahry, A. Alghamdi, Y. Deng and D. Zhao, Nanoengineering of Core-Shell Magnetic Mesoporous Microspheres with Tunable Surface Roughness, J. Am. Chem. Soc., 2017, 139(13), 4954–4961 CrossRef CAS PubMed.
- K. S. W. Sing, Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity, Pure Appl. Chem., 1985, 57(4), 603–619 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms, J. Am. Chem. Soc., 1951, 73(1), 373–380 CrossRef CAS.
- S. Liu, D. Zhang, Y. Sheng, W. Zhang, Z. Qin, M. Qin, S. Li, Y. Wang, C. Gao, Q. Wang, Y. Ming, C. Liu, K. Yang, Q. Huang, J. Qi, Q. Gao, K. Chen, Y. Hu, Y. Rong, X. Lu, A. Mei and H. Han, Highly oriented MAPbI3 crystals for efficient hole-conductor-free printable mesoscopic perovskite solar cells, Fundam. Res., 2022, 2(2), 283–292 Search PubMed.
- H. Huang, P. Cui, Y. Chen, L. Yan, X. Yue, S. Qu, X. Wang, S. Du, B. Liu, Q. Zhang, Z. Lan, Y. Yang, J. Ji, X. Zhao, Y. Li, X. Wang, X. Ding and M. Li, 24.8%-efficient planar perovskite solar cells via ligand-engineered TiO2 deposition, Joule, 2022, 6(9), 2186–2202 CrossRef CAS.
- Q. Han, S. H. Bae, P. Sun, Y. T. Hsieh, Y. Yang, Y. S. Rim, H. Zhao, Q. Chen, W. Shi, G. Li and Y. Yang, Single Crystal Formamidinium Lead Iodide (FAPbI(3)): Insight into the Structural, Optical, and Electrical Properties, Adv. Mater., 2016, 28(11), 2253–2258 CrossRef CAS PubMed.
- Q. Guo, F. Yuan, B. Zhang, S. Zhou, J. Zhang, Y. Bai, L. Fan, T. Hayat, A. Alsaedi and Z. A. Tan, Passivation of the grain boundaries of CH3NH3PbI3 using carbon quantum dots for highly efficient perovskite solar cells with excellent environmental stability, Nanoscale, 2019, 11(1), 115–124 RSC.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Electron-hole diffusion lengths >175 μm in solution-grown CH3NH3PbI3 single crystals, Science, 2015, 347(6225), 967–970 CrossRef CAS PubMed.
- A. A. Zhumekenov, M. I. Saidaminov, M. A. Haque, E. Alarousu, S. P. Sarmah, B. Murali, I. Dursun, X. H. Miao, A. L. Abdelhady, T. Wu, O. F. Mohammed and O. M. Bakr, Formamidinium Lead Halide Perovskite Crystals with Unprecedented Long Carrier Dynamics and Diffusion Length, ACS Energy Lett., 2016, 1(1), 32–37 CrossRef CAS.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Origin and elimination of photocurrent hysteresis by fullerene passivation in CH3NH3PbI3 planar heterojunction solar cells, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- B. Jin, Y. Ming, Z. Wu, J. Cao, Y. Liu, Y. Zhu, S. Wang, Z. Liang and C. Wu, Silk fibroin induced homeotropic alignment of perovskite crystals toward high efficiency and stability, Nano Energy, 2022, 94, 106936 CrossRef CAS.
- X. Guo, J. Du, Z. Lin, J. Su, L. Feng, J. Zhang, Y. Hao and J. Chang, Enhanced efficiency and stability of planar perovskite solar cells using SnO2:InCl3 electron transport layer through synergetic doping and passivation approaches, Chem. Eng. J., 2021, 407, 127997 CrossRef CAS.
- W. Tress, M. Yavari, K. Domanski, P. Yadav, B. Niesen, J. P. C. Baena, A. Hagfeldt and M. Graetzel, Interpretation and evolution of open-circuit voltage, recombination, ideality factor and subgap defect states during reversible light-soaking and irreversible degradation of perovskite solar cells, Energy Environ. Sci., 2018, 11(1), 151–165 RSC.
- C. Tian, A. Mei, S. Zhang, H. Tian, S. Liu, F. Qin, Y. Xiong, Y. Rong, Y. Hu, Y. Zhou, S. Xie and H. Han, Oxygen management in carbon electrode for high-performance printable perovskite solar cells, Nano Energy, 2018, 53, 160–167 CrossRef CAS.
- L. Y. Yi, K. J. Dong, R. P. Zou and A. B. Yu, Coordination Number of the Packing of Ternary Mixtures of Spheres: DEM Simulations versus Measurements, Ind. Eng. Chem. Res., 2011, 50(14), 8773–8785 CrossRef CAS.
- J. You, Y. Yang, Z. Hong, T. B. Song, L. Meng, Y. Liu, C. Jiang, H. Zhou, W.-H. Chang, G. Li and Y. Yang, Moisture assisted perovskite film growth for high performance solar cells, Appl. Phys. Lett., 2014, 105(18), 183902 CrossRef.
- L. Zhuang, Synergy of Ammonium Chloride and Moisture on Perovskite Crystallization for Efficient Printable Mesoscopic Solar Cells, Acta Phys.-Chim. Sin., 2017, 33(4), 647–648 CAS.
- X. Chen, Y. Xia, Z. Zheng, X. Xiao, C. Ling, M. Xia, Y. Hu, A. Mei, R. Cheacharoen, Y. Rong and H. Han, In Situ Formation of δ-FAPbI3 at the Perovskite/Carbon Interface for Enhanced Photovoltage of Printable Mesoscopic Perovskite Solar Cells, Chem. Mater., 2022, 34(2), 728–735 CrossRef CAS.
- J. L. Urai, C. J. Spiers, H. J. Zwart and G. S. Lister, Weakening of rock salt by H2O during long-term creep, Nature, 1986, 324(6097), 554–557 CrossRef PubMed.
- P. J. Anderson and P. L. Morgan, Effects of H2O vapour on sintering of MgO, Trans. Faraday Soc., 1964, 60, 930–937 RSC.
- D. Vinh Quang, G. S. Han, T. Tran Quang, D. Le Thai, Y. U. Jin, B. U. Hwang, H. S. Jung and N. E. Lee, Methylammonium lead iodide perovskite-graphene hybrid channels in flexible broadband phototransistors, Carbon, 2016, 105, 353–361 CrossRef.
- X. Feng, Y. Fan, N. Nomura, K. Kikuchi, L. Wang, W. Jiang and A. Kawasaki, Graphene promoted oxygen vacancies in perovskite for enhanced thermoelectric properties, Carbon, 2017, 112, 169–176 CrossRef CAS.
- P. Jiang, T. W. Jones, N. W. Duffy, K. F. Anderson, R. Bennett, M. Grigore, P. Marvig, Y. Xiong, T. Liu, Y. Sheng, L. Hong, X. Hou, M. Duan, Y. Hu, Y. Rong, G. J. Wilson and H. Han, Fully printable perovskite solar cells with highly-conductive, low-temperature, perovskite-compatible carbon electrode, Carbon, 2018, 129, 830–836 CrossRef CAS.
- H. Zhang, K. Song, L. Zhu and Q. Meng, Back-interface regulation for carbon-based perovskite solar cells, Carbon, 2020, 168, 372–391 CrossRef CAS.
- D. Bogachuk, R. Tsuji, D. Martineau, S. Narbey, J. P. Herterich, L. Wagner, K. Suginuma, S. Ito and A. Hinsch, Comparison of highly conductive natural and synthetic graphites for electrodes in perovskite solar cells, Carbon, 2021, 178, 10–18 CrossRef CAS.
- R. Ishikawa, S. Yamazaki, S. Watanabe and N. Tsuboi, Layer dependency of graphene layers in perovskite/graphene solar cells, Carbon, 2021, 172, 597–601 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma01090a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |