
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc oxide nanoparticles embedded photo-crosslinkable PLA-block-PEG toward effective antibacterial coatings†
Nabasmita
Maity
 a,
Netta
Bruchiel-Spanier
b,
Orna
Sharabani-Yosef
c,
Daniel
Mandler
*b and
Noam
Eliaz
*a
a,
Netta
Bruchiel-Spanier
b,
Orna
Sharabani-Yosef
c,
Daniel
Mandler
*b and
Noam
Eliaz
*a
aDepartment of Materials Science and Engineering, Tel Aviv University, Tel Aviv 6997801, Israel. E-mail: neliaz@tau.ac.il
bInstitute of Chemistry, The Hebrew University of Jerusalem, Jerusalem 9190401, Israel. E-mail: daniel.mandler@mail.huji.ac.il
cDepartment of Biomedical Engineering, Tel Aviv University, Tel Aviv 6997801, Israel
First published on 28th June 2023
Abstract
Post-implantation infections are one of the major issues curtailing the clinical performance of numerous biomedical devices, leading to implant failure. With the advancement of modern surgery, improvement of implant surfaces is highly desirable to address biomaterial-associated biofilm formation. Herein, we report a photo-crosslinkable polymeric coating material composed of dimethacrylate end-terminated polylactic acid (PLA)/polyethylene glycol (PEG) diblock copolymers (PLEGDA) bequeathed with ZnO nanoparticles (NPs) for the surface modification of biomaterials via a facile photo-grafting method. Incorporation of 1 wt% ZnO NPs to the as-synthesized PLEGDA bestows the surface with potential contact-killing antibacterial properties against S. aureus and E. coli bacteria. The suitable hydrophobic/hydrophilic balance of PLA8600/PEG2000 segments in combination with ZnO NPs offers low fouling against both bacteria, with superior resistance against E. coli. Moreover, the homogeneous coating exhibits excellent biocompatibility as indicated by in vitro cell viability and cell adhesion behavior. Therefore, ZnO NPs-embedded biocompatible polymer-based, low-fouling, UV-assisted coating has great potential in the surface modification of different biomaterials for combatting implant-associated infections while preventing multiple drug resistance.
1. Introduction
Biomedical implants have been extensively used nowadays to improve the life quality of numerous patients. However, frequent microbial contamination of these implants still remains a major challenge1,2 that can cause infection, inflammation, and even implant failure, leading to serious health and economic issues for patients after surgery.3,4 Therefore, the development of new antibacterial surface coatings is of paramount importance to minimize implant-associated bacterial colonization and biofilm formation. For this purpose, a variety of methods have been developed. Examples include introducing various antibacterial agents such as antibiotics, quaternary ammonium compounds, antibacterial peptides, silver nanoparticles (NPs), and metal oxides on the surface of the implants.5–11 Recently, efforts have also been taken to integrate of those antibacterial agents into different forms of biomaterials or surgical dressings, for example, hydrogels, electrospun scaffolds, sponge and foams for the development of controlled antibacterial and anti-infection performance.12–15 Although these methods are found efficient for combatting bacterial infections, several drawbacks limit their applications, such as limited operational time, apparent cytotoxicity, narrow antibacterial spectrum, and establishment of transmitting multidrug resistance.16–18 Additionally, most of these monofunctional coatings are insufficient to resist fouling by proteins, platelets, and dead bacterial cell debris.19,20 To overcome these limitations, proper surface modifications to generate multifunctional surfaces with both antibacterial and antifouling activity are highly desirable. Recently, efforts have been made to develop contact-active antibacterial and antifouling dual-functional coatings.21–25 However, based on market availability, a multifunctional coating, which is highly efficient, is still rare, and further research is needed to optimize both materials and methods.Among the degradable polymers, poly(lactic acid) (PLA) has shown great potential as a biocompatible and bioresorbable Food and Drug Administration (FDA) approved green synthetic polymer. It has been successfully deployed in tissue engineering and scaffold fabrication for years.26,27 At the same time, poly(ethylene glycol) (PEG) has attracted much attention in the development of antifouling coatings due to its hydrophilicity, biocompatibility, and nontoxicity.28,29 PEG is also widely utilized as FDA approved nanocarriers in the biomedical industry,30,31 and hence, it allows loading antibacterial NPs, which form homogeneous polymer-NPs composite materials.
Compared with other metal-oxide NPs, ZnO NPs are gaining much interest as a preferred antibacterial agent due to their relatively low cost and toxicity, and renowned use in healthcare products.32 Furthermore, ZnO has been listed as “generally recognized as safe (GRAS)” by the FDA. Recently, few studies have reported the effective antibacterial activity of ZnO NPs against both Gram-positive and Gram-negative bacteria.33,34 Interestingly, different sizes and morphologies of the NPs have been found to significantly affect their antibacterial efficacy.35
To confer the desired antifouling and antibacterial properties to the implant surfaces, we envision herein that the use of biocompatible polymer-NPs composite coatings would provide a facile strategy to minimize the vulnerability to bacterial infections while incorporating the integrated material properties into the implants. Hence, in this study, we report an efficient thin-film composite coating for surface modification of biomaterials based on methacrylate end-functionalized rationally balanced block-copolymers combining synthetic biopolymer PLA and antifouling PEG (PLEGDA) embedded with antibacterial ZnO NPs (1 wt%). Ti–6Al–4V alloy is used as the substrate as it is the workhorse titanium alloy for surgical implantation due to its good biocompatibility, osseointegration, mechanical properties, and corrosion resistance.36 The PLEGDA/ZnO NPs composite coatings were prepared by mixing ZnO NPs suspension with PLEGDA, followed by dip-coating and photo-crosslinking. Exploiting the photo-grafting approach, both anchoring and crosslinking of the coatings were achieved on the silanized implant at the same time. The PLEGDA/ZnO NPs coatings were found to have good surface coverage with homogeneous distribution. Furthermore, in vitro studies demonstrated efficient contact-killing antibacterial properties and some fouling-resistance of the coatings against S. aureus and E. coli, while exhibiting non-cytotoxic behavior toward hFOB osteoprogenitor human cells. Tailoring these multifunctional polymer-nanomaterial coatings holds a significant potential toward substrate-independent UV-assisted surface modification of biomaterials to minimize implant-associated infections.
2. Results and discussion
2.1. Synthesis of ZnO NPs
Initially, a series of ZnO NPs were synthesized from zinc nitrate by a chemical precipitation method,37,38 followed by annealing at 150, 200, 350, and 450 °C for 2 hours. Further studies were conducted on ZnO NPs annealed at 350 °C (Fig. 1) as it was found preferable in terms of chemical purity and average particle size. The size of ZnO NPs greatly contributes to their bactericidal properties; the smaller their size, the higher is the antibacterial activity.35,38 The X-ray diffraction (XRD) pattern (Fig. 1a) reveals the presence of a highly crystallized ZnO phase (Zincite, JCPDS 5-0664), with no unwanted peaks related to impurities, including the precursor (Fig. S1, ESI†). The morphology and the size of the NPs were studied using transmission electron microscopy (TEM). Fig. 1b reveals the polygonal shape of ZnO NPs, with an average diameter of 55 ± 15 nm. This crystallite size was further supported by scanning electron microscopy (SEM, inset of Fig. 1b) and XRD measurements. To determine the surface charge, the variation of the zeta potential of ZnO NPs dispersion (Fig. 1c and Fig. S2, ESI†) as a function of the pH of the medium was studied. Fig. 1c shows that the NPs exhibit a characteristic positive surface charge/zeta potential value of +24.7 mV at pH 7.4, which decreases or reverses to negative values as the pH of the medium decreases or increases, respectively. This strong positive surface charge of ZnO NPs at physiological pH provides high cytotoxicity against negatively charged cell walls and organisms. | ||
| Fig. 1 (a) XRD pattern, (b) TEM image (inset: SEM image) of as-synthesized ZnO NPs after annealing at 350 °C, and (c) change in zeta potential values at different pH values of the solutions. | ||
2.2. Synthesis of dimethacrylate of PLA-block-PEG (PLEGDA) copolymer
Scheme 1a shows the synthetic route of dimethacrylate end-functionalized PLA-block-PEG (PLEGDA). Briefly, PLA-diol was prepared using ring-opening polymerization of L-(–)-lactide (LA) initiated by 1,4-butanediol. The molecular structure and weight were determined by 1H NMR and MALDI-TOF-MS spectra (Fig. S3, ESI†). The molecular weight (Mn ∼ 8600) deduced from matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) MS spectra was found to be quite similar to the molecular weight deduced from 1H NMR. In addition, a separation of m/z 144.1 between the peaks was measured, representing the molecular weight of LA and confirming that PLA-diol consists of LA repeating units. In the next step, PEG (Mn ∼ 2000) was coupled to the as-synthesized PLA-diol via the coupling reaction with hexamethylene diisocyanate (HDI) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. One of the terminal hydroxyl groups of the PEG chains undergoes a reaction with one of the terminal isocyanate groups of HDI, forming PEG-HDI pre-polymer. In the next step, the second isocyanate group is available as a reactive attaching site for the hydroxyl groups of PLA chains and produces PLA-block-PEG copolymers. In the final step of the synthesis, end-capping of the as-synthesized block copolymer was carried out by its reaction with 2-isocyanatoethyl methacrylate (IEMA). The final product, dimethacrylate end-terminated PLA-block-PEG (PLEGDA), was characterized by 1H NMR (Fig. S4, ESI†) and the molecular weight was determined using GPC (Fig. S5, ESI†). Two other UV-curable PLA-block-PEG copolymers, PDA1 [PLA(Mn ∼ 3600)/PEG(Mn ∼ 2000)] and PDA2 [PLA(Mn ∼ 17000)/PEG(Mn ∼ 2000)], were synthesized in a similar approach in order to examine the effect of the hydrophobic-hydrophilic balance on the production of an effective surface coating.
:1 molar ratio. One of the terminal hydroxyl groups of the PEG chains undergoes a reaction with one of the terminal isocyanate groups of HDI, forming PEG-HDI pre-polymer. In the next step, the second isocyanate group is available as a reactive attaching site for the hydroxyl groups of PLA chains and produces PLA-block-PEG copolymers. In the final step of the synthesis, end-capping of the as-synthesized block copolymer was carried out by its reaction with 2-isocyanatoethyl methacrylate (IEMA). The final product, dimethacrylate end-terminated PLA-block-PEG (PLEGDA), was characterized by 1H NMR (Fig. S4, ESI†) and the molecular weight was determined using GPC (Fig. S5, ESI†). Two other UV-curable PLA-block-PEG copolymers, PDA1 [PLA(Mn ∼ 3600)/PEG(Mn ∼ 2000)] and PDA2 [PLA(Mn ∼ 17000)/PEG(Mn ∼ 2000)], were synthesized in a similar approach in order to examine the effect of the hydrophobic-hydrophilic balance on the production of an effective surface coating.
 | ||
| Scheme 1 (a) Synthetic route of PLA-b-PEG dimethacylate (PLEGDA). (b) Schematic illustration of the post-modification of titanium substrate to produce PLEGDA/ZnO NPs-based antibacterial surface coating. | ||
2.3. Surface coating and characterization
Scheme 1b represents the schematic illustration of the photo-grafting process where Ti–6Al–4V substrates were used as model biomedical surfaces due to their multifunctional applications, high biocompatibility, osseointegration and corrosion resistance. In a typical process, ZnO NPs-embedded PLEGDA solutions were prepared by mixing 1 wt% ZnO NPs with 60 wt% PLEGDA in chloroform, followed by the addition of 0.5 wt% of TPO-L as a photoinitiator producing the final formulation. To facilitate surface grafting, the titanium substrates were subjected to pre-treatment which enabled the silanization of the surfaces by the reaction with 3-(trimethyoxysilyl)-propyl methacrylate (TMSPMA). Dip coating of PLEGDA/ZnO NPs nanocomposites on these silanized titanium substrates led to smooth coatings that were obtained after crosslinking by UV-irradiation.To assess and characterize successful modification during the stepwise coating process, X-ray photoelectron spectroscopy (XPS) analyses of all the surfaces were conducted before and after modification, see Fig. 2a–e. As shown in Fig. 2a, the relative intensity of the Ti 2p peak reduced after silanization, and the peak completely disappeared after photo-grafting of PLEGDA and PLEGDA/ZnO NPs (1 wt%) composites. These results indicate that the surface has been completely covered by the composite material. After silanization by TMSPMA, a peak of Si 2p (Fig. 2b) was observed, suggesting the presence of immobilized silane groups on the titanium surface. As expected, the peak showed a decrease in the intensity after application of the surface coating. Additionally, the appearance of Zn 2p peak after dip coating of PLEGDA/ZnO NPs (1 wt%) composite confirms the presence of ZnO NPs on the coated surface. Comparing the C 1s spectra of PLEGDA coatings before and after ZnO NPs addition, (Fig. 2d and e) it is noticed that the relative intensity of the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak increases and the –COO peak decreases compared to the –C–O peak after ZnO NPs mixing, which results from the interaction between the carbonyl groups of polymer chains and ZnO NPs. Furthermore, the TEM studies reveal that the block polymer PLEGDA aggregates in a vesicular arrangement (inset, Fig. 2d) that produces a stable polymer-NPs dispersion (inset, Fig. 2e).
O peak increases and the –COO peak decreases compared to the –C–O peak after ZnO NPs mixing, which results from the interaction between the carbonyl groups of polymer chains and ZnO NPs. Furthermore, the TEM studies reveal that the block polymer PLEGDA aggregates in a vesicular arrangement (inset, Fig. 2d) that produces a stable polymer-NPs dispersion (inset, Fig. 2e).
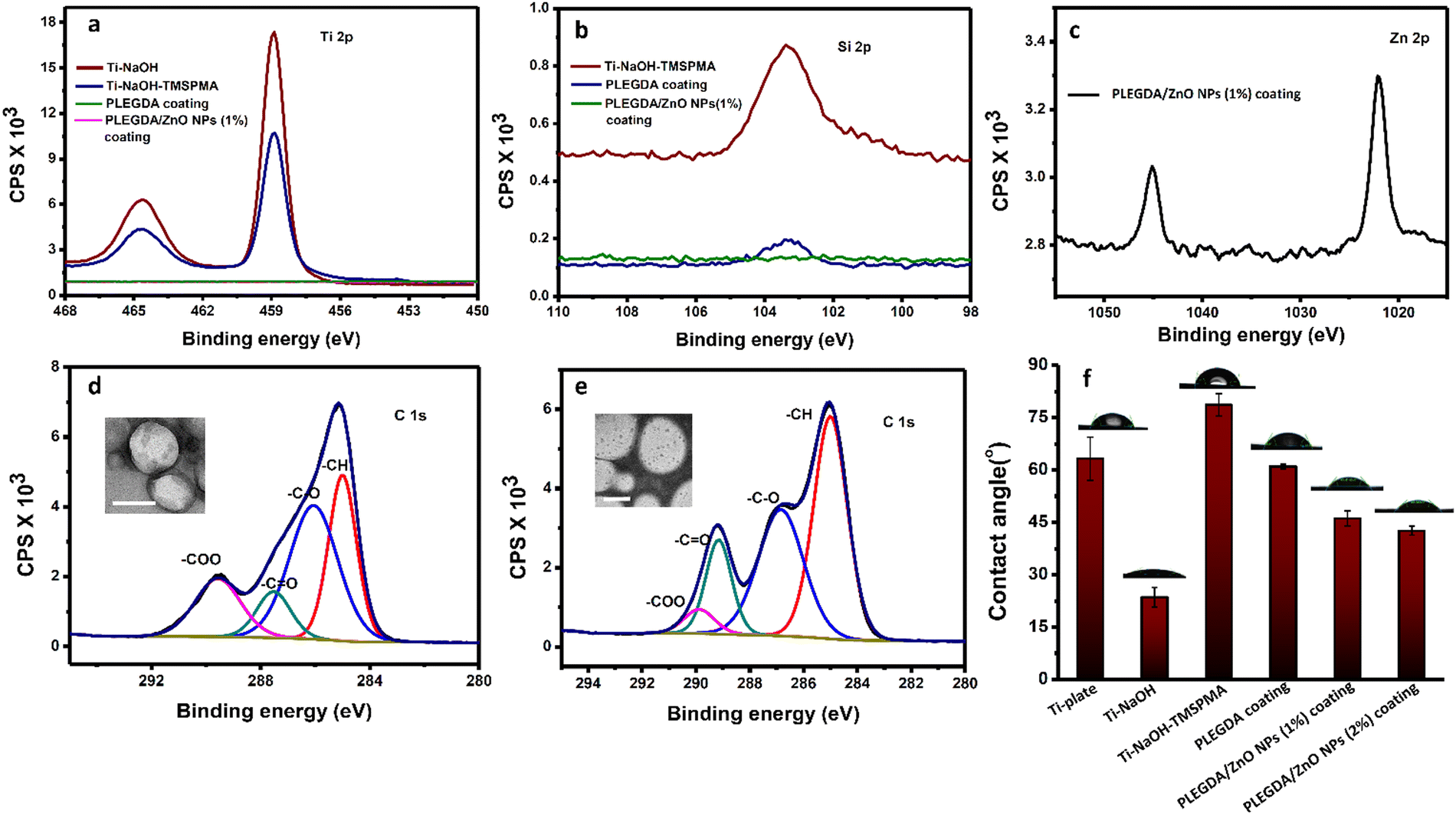 | ||
| Fig. 2 Surface characterization of the stepwise coating by (a) Ti 2p, (b) Si 2p, (c) Zn 2p XPS spectra; and C1s XPS spectra of (d) PLEGDA (inset: TEM image, scale bar: 500 nm) and (e) PLEGDA/ZnO NPs (1 wt%) coatings (inset: TEM image, scale bar: 500 nm). (f) Contact angle values of bare Ti and different coatings during step-by-step modifications. | ||
Further investigation of the surfaces before and after the application of coatings was performed using contact angle measurements of a sessile water drop. The results in Fig. 2f show a reduction of the contact angle after NaOH treatment due to an increase in the hydrophilicity aroused from newly generated hydroxyl groups on the titanium surfaces. With the introduction of TMSPMA, the titanium surfaces became relatively hydrophobic, and the contact angle increased to 79 ± 3°. The surfaces after photo-grafting with PLEGDA, PLEGDA/ZnO NPs (1 wt%) and PLEGDA/ZnO NPs (2 wt%) coatings demonstrate a distinct change in surface wettability, with contact angles of ca. 61°, 46° and 42°, respectively. The presence of PEG chains in the block copolymers as well as ZnO NPs in the composite has made the surface relatively hydrophilic. Furthermore, the hydrophilicity of the surfaces increases as the concentration of ZnO NPs increases. These results are in good agreement with the XPS data, illustrating the stepwise surface modification of titanium substrates.
To visualize the surface morphology of PLEGDA/ZnO NPs (1 wt%) coatings on pre-treated titanium substrates, SEM studies were performed. Fig. 3a(i and ii) indicates the morphological characteristics of a typical polymer film that covers the entire surface. The coating is uniform and homogeneous. The cross-sectional SEM image (inset, Fig. 3a(ii)) demonstrates the efficacy of coating formation using photo-grafting in preparing NPs-embedded polymer layers of ca. 2 μm thickness. The thickness can be altered by varying the concentration of the polymer. Further energy dispersive X-ray spectroscopy (EDS) analysis of the coating, Fig. 3b(i–v), reveals the presence of a large amount of carbon and oxygen, which indicates a uniform coverage of a coating containing PLEGDA polymer as the major constituent. In addition, the results confirm that the coating is composed of Zn, which is well distributed throughout the coating layer and originates from the ZnO NPs. In order to evaluate the pertinence of this process toward the coating of biomaterials, a commercial Ti–6Al–4V dental implant was also used as a substrate. Fig. 3c(i and ii) shows SEM images of the coated implant. The results clearly reveal that the geometry of the implants imposes no detrimental effect on the coating formation and that the coating has high surface coverage.
 | ||
| Fig. 3 (a) SEM (inset of aii: cross-sectional image, scale bar: 1 μm), and (b) X-ray maps (scale bar: 2 μm) and EDS spectrum of the PLEGDA/ZnO NPs (1 wt%) coating on pre-treated Ti substrate. (c) SEM images of the PLEGDA/ZnO NPs (1 wt%) coating on commercial dental implant. | ||
As a coating for implants, it is essential to investigate the stability of PLEGDA/ZnO NPs (1 wt%) coatings in a physiological environment. To evaluate their stability, the coated surfaces were immersed in phosphate-buffered saline (PBS, pH 7.4) at 37 °C and their weight losses were measured up to 15 days. Up to 1% of the coating was lost after 3 days of incubation, while a weight loss of 2.5% was observed after incubation for 15 days (Fig. 4a). These results indicate a low weight loss for the coating, and therefore, it is expected to possess stability suitable for implant applications. Following this study, another experiment was conducted to evaluate the leakage ZnO NPs. 50 mg of PLEGDA/ZnO NPs (1 wt%) samples (4 replicates) were placed in 5 mL of PBS solution. 50 μL of the solutions were taken after 3 days and added to 3 mL of 1 wt% of nitric acid. As the antibacterial tests on the samples were designed to be performed after 24 hour of immersion, we evaluated the leakage after 3 days. The zinc ion concentration of the samples was determined using IC-PMS and the total leakage percentage of zinc oxide was calculated from it. The result shows that (4.2 ± 0.3)% of the ZnO were depleted within 3 days. However, prolonged study indicates higher amount of leakage (17–35%) (Fig. 4b).
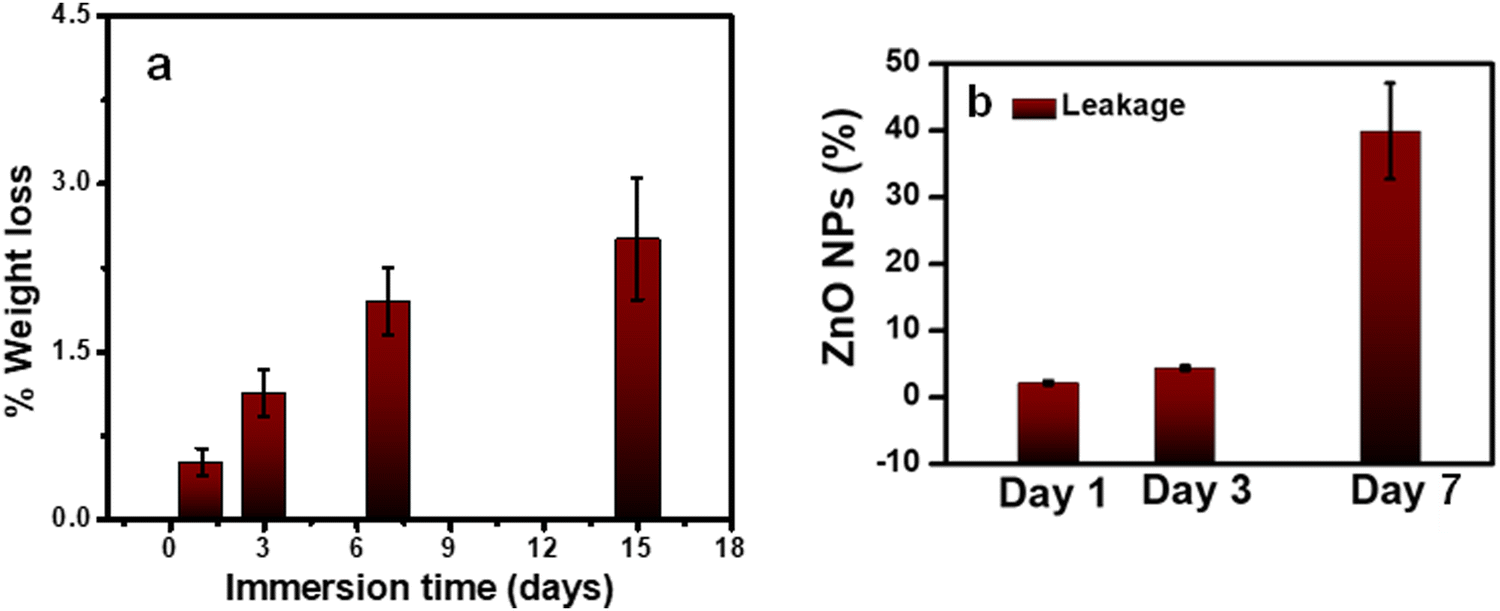 | ||
| Fig. 4 (a) Degradation behavior of PLEGDA/ZnO NPs (1%) coatings in PBS (pH 7.4) at 37 °C for up to 15 days of immersion; (b) % leakage of ZnO from PLEGDA/ZnO NPs (1%) samples in PBS (pH 7.4) at 37 °C for 7 days of immersion. | ||
2.4. Antibacterial activity of the PLEGDA/ZnO coatings
To assess the antibacterial activity of the ZnO NPs-loaded PLEGDA coating, both Gram-positive S. aureus and Gram-negative E. coli were tested in vitro as model bacteria. These bacteria account for the majority of implant-associated infections. ZnO NPs are known for their antibacterial properties in terms of contact killing.33,39 Hence, bacterial suspension of known concentration was added on uncoated and differently coated surfaces. After incubation for 4 hours, the bacterial growth medium was added to the samples and they were incubated for additional 20 hours to allow the bacterial growth. To examine the viability of the bacteria, the resulting bacterial suspensions were then analyzed for colony counting and the surfaces were investigated by live/dead bacterial cell staining. Throughout the study, non-mutagenic bacteria were used. After each experiment, the bacteria were immersed in 5% of sodium hypochlorite. As evidenced from Fig. 5a and b, the growth of both bacteria was completely inhibited in contact with PLEGDA/ZnO NPs (1 wt%) surfaces, while the bacteria continued to proliferate when exposed to the surfaces without embedded ZnO NPs. In addition, Fig. 5c–j demonstrates that the colony counting results were in agreement with the results obtained from the live/dead bacterial cell staining of the surfaces. It is worth noticing from Fig. 5e and i that fouling-resistant PEG-containing PLEGDA surfaces inhibited the initial attachment of bacteria and delayed the bacterial growth on the surfaces, showing the highest repelling ability against Gram-negative E. coli. As expected, PLEGDA did not kill the bacteria as it contained no antibacterial activity (Fig. S6, ESI†). Following the trend, it was found that PLEGDA did not completely inhibit the growth of Gram-positive S. aureus, which were able to attach to the surfaces. Fig. 5f and j reveal that after the incorporation of ZnO NPs, antibacterial activity was added to the antifouling property of PLEGDA, leading to the complete eradication of bacteria in contact with the surfaces. These results clearly demonstrate that apart from the low-fouling properties, an efficient bacterial inhibition was ensured by the direct contact with ZnO NPs during the first 4 hours of incubation. The result was again supported by the the bacteriostatic circle test (Fig. S7, ESI†). In the experiment, crosslinked films of PLADA (methacrylate of PLA), PLEGDA (methacrylate of PLA-block-PEG copolymer) and PLEGDA/ZnO NPs (1%) were added on agur plates containing the desired bacteria. The plates were then incubated for 24 hours at 37 °C. PLEGDA/ZnO NPs (1%) exhibited antibacterial activity against both E. coli and S. aureus, as evidenced by the inhibition of bacterial growth. | ||
| Fig. 5 (a and b) CFU count in the solution, and (c–j) confocal scanning laser microscope images of S. aureus and E. coli, respectively, after 4 hours bacterial culture in contact with bare Ti and different coatings, followed by 18 hours incubation in growth medium. The graph is representative of three replicates. Patterned circles indicate no colony observed. The green and red regions are representative of live (Syto9 stain) and dead (Propidium Iodide stain) bacteria, respectively. Scale bar: 20 μm. | ||
2.5. Cytotoxicity of the PLEGDA/ZnO coatings
Assessing the optimum balance between efficient antibacterial activity and good cytocompatibility is challenging, as a higher amount of nanomaterials might result in higher antibacterial effects as well as increased cytotoxicity. In light of the antibacterial studies, it was essential to investigate the effect of ZnO NPs on the cytocompatibility of the coating. Therefore, the cell viability (Fig. 6a–f) of PLEGDA coated surfaces without and with ZnO NPs loading was tested in vitro by XTT analysis on hFOB osteoprogenitor human cells (Fig. S8, ESI†). The surfaces were incubated in 1:1 DMEM/F12 growth medium for 24, 48, 72 hours, and 7 days. The extracted growth media were used to culture hFOB cells, and the viability tests were performed after 24 hours of incubation. In Fig. 6a and b, the XTT results reveal excellent cell viability both for the PLEGDA and PLEGDA/ZnO NPs (1 wt%) surfaces, with no significant difference between them, illustrating that ZnO NPs (1 wt%) embedded PLEGDA coatings are highly cytocompatible. Viability tests of coatings containing 1.5 and 2 wt% NPs were found to exhibit ∼85% and ∼70% cell viability (not shown), respectively. Thus, coatings with 1 wt% ZnO NPs were found ideal both for efficient antibacterial activity and cell viability. To visualize the cell viability more intensely, we further conducted a live-dead cell staining assay on hFOB cells cultured for 24 hours in the extracted growth media (incubation of 24 hours and 7 days with the coatings), see Fig. 6c–f. Staining with fluorescein diacetate shows cell membrane of live cells as green, while propidium iodide stains the DNA of dead cells as red.40 The results are consistent with the XTT assay, the coatings being found nontoxic as evidenced from the high population of live cells. Next, hFOB cells were seeded on the PLEGDA/ZnO NPs (1 wt%) coating and tested after 4 days of cell culture using staining with 4′,6-diamidino-2-phenylindole (DAPI) that indicates cell nuclei as blue. Interestingly, Fig. 6g and h shows that the PLEGDA/ZnO NPs (1 wt%) coating supports cell adherence and cell viability, and hence, it may be used as a coating for implants.
 | ||
| Fig. 6 Cytocompatibility of coatings by XTT studies of (a) PLEGDA, and (b) PLEGDA/ZnO NPs (1 wt%) coatings. (c and d) Live-dead viability test on hFOB cells treated for 24 hours with extracted media from PLEGDA coatings after (c) 24 hours and (d) 7 days, respectively. (e and f) Live-dead viability test on hFOB cells treated for 24 hours with extracted media from PLEGDA/ZnO NPs (1 wt%) coatings after (e) 24 hours and (f) 7 days, respectively. Scale bar: 500 μm. Green staining represents live cells; red staining represents dead cells. (g and h) Fluorescence micrographs of hFOB cells cultured on PLEGDA/ZnO NPs (1 wt%) coatings for 4 days, stained with DAPI (blue). | ||
It is important to mention here that the optimal hydrophobic–hydrophilic balance of the coating has a crucial role in determining its performance. Therefore, PLEGDA was used after screening using PDA1 and PDA2 coatings (not shown). As expected, PDA2 with the lowest PEG2000/PLA17000 ratio (∼0.11) did not show intense fouling resistance against bacteria. On the other hand, PDA1 with the higher PEG2000/PLA3600 ratio (∼0.55) exhibited better antifouling activity as it contains more hydrophilic PEG over PLA compared to PLEGDA. Nevertheless, PLEGDA with a moderate PEG2000/PLA8600 ratio (∼0.23) contains a suitable hydrophilic–hydrophobic balance. It affects the fouling property of the coating in an effective way, showing fouling resistance against bacteria while promoting cell adhesion. Moreover, it is apparent from the results that the incorporation of NPs into PLEGDA offers a platform for implants coating with an optimum hydrophobic–hydrophilic balance, which preserves the cytocompatibility, and in turn, influences the cell adhesion while preventing biofilm formation.
3. Conclusions
A facile, straightforward method of developing methacrylate-based PLA-block-PEG (PLEGDA) co-polymeric coating embedded with ZnO NPs using photo-crosslinking is reported. The combination of PLA and PEG offers a general strategy to provide an intrinsically biocompatible and fouling resistant polymer architecture on the biomaterial surface. The good surface coverage and uniform distribution of the coating material on titanium alloy with different geometries implies the applicability of the coating for medical implants. The optimal balance of hydrophobic–hydrophilic segments of PLEGDA with an appropriate amount of ZnO NPs (1 wt%) collectively imparted to potential contact-killing antibacterial surfaces that can repel bacteria to some extent, while exhibiting excellent cell viability in vitro. In addition, the ZnO NPs-embedded surfaces were found to improve cell adhesion. Here leakage of ZnO needs to be optimized and reduced, especially in case of prolonged applications. Our team is working on the development of a modified version of the coating to improve it's stability and leakage.Exploiting strategic synthetic design to tailor multifunctional polymer-nanomaterial coatings, this approach can be extended as a promising strategy toward material-independent UV-assisted surface grafting of biomaterials to minimize implant-associated infections.
4. Experimental section
Synthesis of PLA-diol
PLA-diol was synthesized via ring-opening polymerization (ROP) of L-(–)-lactide (Carbosynth Limited, UK) in the presence of 1,4-butanediol (Aldrich) as initiator and stannous (II) 2-ethylhexanoate [Sn(Oct)2] (Sigma-Aldrich) as a catalyst. In brief, 0.34 mol L-(–)-lactide was mixed with 0.45 × 10−2 mol 1,4-butanediol and 0.34 × 10−2 mol Sn(Oct)2 in a three-neck flask under nitrogen and stirred at 110 °C for 3 hours. The product was dissolved in chloroform, precipitated in a large excess of cold diethyl ether (Bio-Lab, Israel) and repeatedly washed with cold methanol (Honeywell, USA). Finally, PLA-diol was obtained after drying in a fume hood. 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.53–1.71 (m, 12H), 1.71–1.77 (m, 6H), 1.92–2.08 (m, 4H), 2.68 (s, 2H), 4.21 (m, 4H), 4.32–4.41 (m, 2H), 5.21 (m, 2H). Mn, NMR: 9200 g mol−1, Mn, MALDI-TOF: 8600 g mol−1 (Fig. S3, ESI†). Mn, GPC: 8943 g mol−1 and PDI: 1.72 (Fig. S5, ESI†).Synthesis of dimethacrylate of PLA-block-PEG copolymer (PLEGDA)
Poly(ethylene glycol) (PEG 2K) (Sigma-Aldrich) was dried in a three-necked flask under vacuum at 120 °C. To ensure the formation of PEG-HDI pre-polymer, hexamethylene diisocyanate (HDI) (Alfa-Aesar, Belgium) was added dropwise to it in a mole ratio of 1:1 in the presence of Sn(Oct)2 catalyst at 80 °C under nitrogen. In the next step, PLA-b-PEG was synthesized via coupling of PEG-HDI pre-polymer and PLA-diol at 1:1 mole ratio of isocyanate content of PEG-HDI to diol content of PLA. One of the terminal hydroxyl groups of the PEG chains underwent a reaction with one of the terminal isocyanate groups of HDI, while the second isocyanate group of HDI acts as the anchoring site for the PLA chains. The reaction was conducted for 4 hours under nitrogen at 70 °C. Then, dimethacrylation was done by adding 2-isocyanatoethyl methacrylate (IEMA) (Aldrich,) and catalytic amount of Sn(Oct)2 to the as synthesized block copolymer in dry dioxane (Bio-Lab, Israel), which was dried prior to use over molecular sieves (Sigma-Aldrich). The reaction continued for 2 hours at 80 °C under nitrogen. The reaction mixture was then dissolved in chloroform and precipitated in cold diethyl ether. The process was repeated for three times, and the final product (PLA-b-PEG dimethacrylate (PLEGDA)) was obtained after drying in a fume hood. 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.61 (s, 3H), 3.68 (m, 4H), 5.15-5.27 (m, 1H), 5.62 (d, 2H), 6.14 (d, 2H) (Fig. S4, ESI†). Mn, GPC: 11270 g mol−1 and PDI: 1.55 (Fig. S5, ESI†).
Synthesis of zinc oxide nanoparticles (ZnO NPs)
ZnO NPs were synthesized via a facile chemical precipitation method using sodium hydroxide.37 First, 0.1 M sodium hydroxide (Alfa Aesar) solution was added dropwise to 0.1 M zinc nitrate hexahydrate (Alfa Aesar) solution at room temperature under vigorous stirring. The precipitate was then filtered, washed with distilled water, followed by methanol, and dried at 80 °C for 5 hours. Finally, the ZnO NPs were obtained from the precursor after annealing at 150, 200, 350 and 450 °C for 2 hours.Preparation of ZnO NPs-embedded UV-curable PLA-block-PEG copolymer solution (PLEGDA/ZnO NPs)
ZnO NPs-embedded dimethacrylate end-functionalized PLA-b-PEG copolymer solutions were prepared by mixing 60 wt% solution of UV-curable PLEGDA in chloroform with 1, 1.5 and 2 wt% of ZnO NPs (annealed at 350 °C). The as-synthesized ZnO NPs annealed at 350 °C were used for further studies as they were found preferable in terms of purity and particle size distribution. Prior to use, 0.5 wt% of photoinitiator, TPO-L (Alfa Aesar) was added to the mixture to produce the final formulation.Photografting of ZnO NPs-embedded PLEGDA coatings
To develop photo-grafted surface coatings, Ti–6Al–4V alloy rods (Bramil LTD) were subjected to pre-treatment.41 First, the rods were machined, ground manually on silicon carbide papers (grit 600 and grit 4000, Microcut, Buehler, USA), washed in distilled water and acetone, acid etched in HF(40%)/HNO3(65%) solution (2 and 20 vol%, respectively) for 2 min, and grit blasted with high-purity alumina powder (59–68 μm) (Calbex Mineral Trading, Inc., China). Following cleaning and air oxidation for 1 h, the substrates were soaked in 5 M NaOH at 60 °C for 24 hours, cleaned and dried in cool air. The samples were then subjected to reaction with 3-(trimethyoxysilyl)-propyl methacrylate (TMSPMA, Sigma-Aldrich) (2% v/v) in dry toluene at 55 °C for 24 hours. After that, the samples were cleaned ultrasonically in toluene, ethanol, and distilled water, consecutively. Following silanization, the samples were dip-coated with PLEGDA/ZnO NPs and crosslinked using ultraviolet (UV) irradiation with a wavelength of 365 nm for 20 min to ensure complete photo-crosslinking. Also, commercial dental implants from SGS Dental Implants (Schaan, Liechtenstein) were tested after soaking in NaOH, followed by silanization and coating formation. All samples were stored in dark place before further use.Characterization
All 1H NMR spectra were recorded in CDCl3 with TMS internal standard using Bruker 400 MHz spectrometer. MALDI-TOF mass spectral measurements were performed with a MALDI-SYNAPT QTOF mass spectrometer (Waters, USA) combined with UPLC (Acquity, Waters) using 2,5-dihydroxybenzoic acid (DHB) as a matrix. Gel permeation chromatograph (Differential Separations Module Waters 2690 having Refractometer Detector Waters 410 and Millenium Chromatography Manager) was used to determine the molecular weight of as synthesized polymers using polystyrene standards. Powder X-ray diffraction (XRD) patterns of as-synthesized ZnO NPs were collected using a D8 ADVANCE DIFFRACTOMETER (Bruker AXS, Madison, WI, USA) equipped with Goebels mirrors and LYNXEYE-XE linear position sensitive device (PSD) detector. The average crystallite size (D) was calculated from the main peak using Scherrer's equation:42D = 0.9λ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Cosθ Cosθ |
Zeta (ζ) potential measurements of dispersed ZnO NPs at varying pH were carried out in a Zetasizer Malvern ZS system. For the morphological characterization, sample solutions/dispersions were drop-casted onto 400-mesh copper grids coated with thin carbon film (SPI, West Chester, PA, USA) and monitored after drying by using a JEM-1400Plus Transmission Electron Microscope (JEM) operating at 80 kV. Also, the powder NPs and coated surfaces were viewed using a scanning electron microscope (SEM, Quanta 200 FEG, FEI, Waltham, MA, USA) equipped with an energy-dispersive X-ray spectrometer (EDS, INCA detector, Oxford Instruments, Abington, UK). Surface wettability was checked via contact angle measurements using FTÅ 125 Dynamic, Contact Angle Analyzer (Crelab Instruments). X-ray photoelectron spectroscopy (XPS) analysis of the samples after surface treatments was performed in Kratos Axis ultrahigh-resolution XPS instrument (Kratos Analytical Ltd, UK).
Antibacterial property analysis
The preparation of live/dead assay: After incubation, the test samples were removed from the culture medium and washed three times with PBS to remove both the unbound bacteria and bacteria from the medium, followed by incubation in the staining solution containing propidium iodide and Syto9 (L13152 LIVE/DEAD® BacLight™ Bacterial Viability Kit, Molecular Probes, OR, USA) for 15 minutes. Then, confocal microscopy was performed using FV-1200 (Olympus, Japan).
:1 Dulbecco's Modified Eagle's Medium: Ham's F12 (DMEM/F12, Biological Industries, Israel) supplemented with 10 wt% Fetal Bovine Serum (FBS, Biological Industries), 0.3 mg mL−1 G418 (Apollo Scientific), and 2.5 mM L-glutamine (Biological Industries). The cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2. For the cytotoxicity analysis using an elusion test, the sterile samples were first incubated with the GM for 24, 48, 72 hours, and 7 days. The extracted GM was then used to culture hFOB cells, and the viability was determined after a 24 hour incubation period. Cell viability was evaluated using the XTT cell proliferation assay kit (Biological Industries) according to the manufacturer's instructions. In brief, 100 μL of the activation reagent was added to 5 mL of the XTT reagent. Then, 100 μL of the activated XTT solution was added to each well. After 2.5 hour incubation at 37 °C, the intensity of color was measured using an enzyme-linked immunosorbent assay (ELISA) microplate reader at 450 and 630 nm. Results are presented herein as mean ± standard deviation (n = 3). The fluorescent live/dead staining assay (Sigma Aldrich) containing 6.6 μg mL−1 fluorescein diacetate and 5 μg mL−1 propidium iodide was used to visualize the proportion of viable and non-viable cells that were cultured with the extracted GM for 24 hours. The labeled cells were viewed immediately afterwards using a Nikon Eclipse Tifluorescent microscope and images were captured by a Zylasc MOS camera using Nikon Intensilight C-HGFI fluorescent lamp.
000 cells in 0.2 mL of fresh GM were directly seeded on the samples and were cultured in GM. After 4 days of culture, the cells were stained with DAPI (Abcam, USA), as per the company protocol. They were subsequently imaged using a Nikon Eclipse Tifluorescent microscope. Images were captured by a Zylasc MOS camera using Nikon Intensilight C-HGFI fluorescent lamp.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project is supported by the Israel Ministry of Science, Technology and Space (Grant 3-15634).References
- D. W. Grainger, H. C. V.-D. Mei, P. C. Jutte, J. J. A. M. V.-D. Dungen, M. J. Schultz, B. F. A. M. V.-D. Laan, S. A. J. Zaat and H. J. Busscher, Biomaterials, 2013, 34, 9237–9243 CrossRef CAS PubMed.
- S. S. Magill, J. R. Edwards, W. Bamberg, Z. G. Beldavs, G. Dumyati, M. A. Kainer, R. Lynfield, M. Maloney, L. McAllister-Hollod, J. Nadle, S. M. Ray, D. L. Thompson, L. E. Wilson and S. K. Fridkin, N. Engl. J. Med., 2014, 370, 1198–1208 CrossRef CAS PubMed.
- C. Otto-Lambertz, A. Yagdiran, F. Wallscheid, P. Eysel and N. Jung, Dtsch Arztebl Int., 2017, 114, 347–354 Search PubMed.
- B. Allegranzi, S. B. Nejad, C. Combescure, W. Graafmans, H. Attar, L. Donaldson and D. Pittet, Lancet, 2011, 377, 228–241 CrossRef PubMed.
- M. Bagheri, M. Beyermann and M. Dathe, Antimicrob. Agents Chemother., 2009, 53, 1132–1141 CrossRef CAS PubMed.
- R. Tan, J. Yoo and Y. Jang, in Racing for the Surface, ed. B. Li, T. Moriarty, T. Webster and M. Xing, Springer, Cham, Switzerland, 2020, pp. 313–340 Search PubMed.
- X. Ding, S. Duan, X. Ding, R. Liu and F.-J. Xu, Adv. Funct. Mater., 2018, 28, 1802140 CrossRef.
- A. Taglietti, C. R. Arciola, A. D’Agostino, G. Dacarro, L. Montanaro, D. Campoccia, L. Cucca, M. Vercellino, A. Poggi, P. Pallavicini and L. Visai, Biomaterials, 2014, 35, 1779–1788 CrossRef CAS PubMed.
- A. Dong, Y.-J. Wang, Y. Gao, T. Gao and G. Gao, Chem. Rev., 2017, 117, 4806–4862 CrossRef CAS PubMed.
- O. Geuli, N. Metoki, N. Eliaz and D. Mandler, Adv. Funct. Mater., 2016, 26, 8003–8010 CrossRef CAS.
- Y. K. Jo, B. H. Choi, C. S. Kim and H. J. Cha, Adv. Mater., 2017, 29, 1704906 CrossRef PubMed.
- Y. Liang, J. He and B. Guo, ACS Nano, 2021, 15, 12687–12722 CrossRef CAS PubMed.
- G. Liu, Z. Bao and J. Wu, Chin. Chem. Lett., 2020, 31, 1817–1821 CrossRef CAS.
- A. Dodero, S. Scarfi, M. Pozzolini, S. Vicini, M. Alloisio and M. Castellano, ACS Appl. Mater. Interfaces, 2020, 12(3), 3371–3381 CrossRef CAS PubMed.
- Y. Liang, Y. Liang, H. Zhang and B. Guo, Asian J. Pharm. Sci., 2022, 17, 353–384 CrossRef PubMed.
- D. M. Mitrano, E. Rimmele, A. Wichser, R. Erni, M. Height and B. Nowack, ACS Nano, 2014, 8, 7208–7219 CrossRef CAS PubMed.
- L. Qi, H. Li, C. Zhang, B. Liang, J. Li, L. Wang, X. Du, X. Liu, S. Qiu and H. Song, Front. Microbiol., 2016, 7, 483 Search PubMed.
- D. Campoccia, L. Montanaro, P. Speziale and C. R. Arciola, Biomaterials, 2010, 31, 6363–6377 CrossRef CAS PubMed.
- A. M. C. Maan, A. H. Hofman, W. M. de. Vos and M. Kamperman, Adv. Funct. Mater., 2020, 30, 2000936 CrossRef CAS.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- Q. Gao, P. Li, H. Zhao, Y. Chen, L. Jiang and P. X. Ma, Polym. Chem., 2017, 8, 6386–6397 RSC.
- P. Yuan, X. Qiu, X. Wang, R. Tian, L. Wang, Y. Bai, S. Liu and X. Chen, Adv. Healthcare Mater., 2019, 8, 1801423 CrossRef PubMed.
- B. Wang, Q. Xu, Z. Ye, H. Liu, Q. Lin, K. Nan, Y. Li, Y. Wang, L. Qi and H. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 27207–27217 CrossRef CAS PubMed.
- H. Yuan, B. Yu, L.-H. Fan, M. Wang, Y. Zhu, X. Ding and F. J. Xu, Polym. Chem., 2016, 7, 5709–5718 RSC.
- X. Ding, C. Yang, T. P. Lim, L. Y. Hsu, A. C. Engler, J. L. Hedrick and Y.-Y. Yang, Biomaterials, 2012, 33, 6593–6603 CrossRef CAS PubMed.
- P. Mainil-Varlet, R. Rahm and S. Gogolewski, Biomaterials, 1997, 18, 257–266 CrossRef CAS PubMed.
- M. Santoro, S. R. Shahb, J. L. Walker and A. G. Mikos, Adv. Drug Delivery Rev., 2016, 107, 206–212 CrossRef CAS PubMed.
- J. Blümmel, N. Perschmann, D. Aydin, J. Drinjakovic, T. Surrey, M. Lopez-Garcia, H. Kessler and J. P. Spatz, Biomaterials, 2007, 28, 4739 CrossRef PubMed.
- N. Maity, N. Mansour, P. Chakraborty, D. Bychenko, E. Gazit and D. Cohn, Adv. Funct. Mater., 2021, 31, 2108436 CrossRef CAS.
- K. R. Gajbhiyea, A. Pawara, K. R. Mahadika and V. Gajbhiye, Colloids Surf., B, 2020, 187, 110770 CrossRef PubMed.
- A. Harguindey, D. W. Domaille, B. D. Fairbanks, J. Wagner, C. N. Bowman and J. N. Cha, Adv. Mater., 2017, 29, 1700743 CrossRef PubMed.
- A. Krol, P. Pomastowski, K. Rafinska, V. Railean-Plugaru and B. Buszewski, Adv. Colloid Interface Sci., 2017, 249, 37–52 CrossRef CAS PubMed.
- O. Geuli, I. Lewinstein and D. Mandler, ACS Appl. Nano Mater., 2019, 2, 2946–2957 CrossRef CAS.
- V. L. Prasanna and R. Vijayaraghavan, Langmuir, 2015, 31, 9155–9162 CrossRef PubMed.
- K. R. Raghupathi, R. T. Koodali and A. C. Manna, Langmuir, 2011, 27, 4020–4028 CrossRef CAS PubMed.
- N. Eliaz, Materials, 2019, 12, 407 CrossRef CAS PubMed.
- C. Chen, P. Liu and C. Lu, Chem. Eng. J., 2008, 144, 509–513 CrossRef CAS.
- O. Yamamoto, Int. J. Inorg. Mater., 2001, 3, 643–646 CrossRef CAS.
- A. Singh and A. K. Dubey, ACS Appl. Bio Mater., 2018, 1, 3–20 CrossRef CAS.
- C. Diaferia, M. Ghosh, T. Sibillano, E. Gallo, M. Stornaiuolo, C. Giannini, G. Morelli, L. Adler-Abramovich and A. Accardo, Soft Matter, 2019, 15, 487–496 RSC.
- O. Geuli, N. Metoki, T. Zada, M. Reches, N. Eliaz and D. Mandler, J. Mater. Chem. B, 2017, 5, 7819–7830 RSC.
- Y. T. Chung, E. Mahmoudi, A. W. Mohammad, A. Benamor, D. Johnson and N. Hilal, Desalination, 2017, 402, 123–132 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00169e |
| This journal is © The Royal Society of Chemistry 2023 |