
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiblock-copolymerisation-derived sulfonated-poly(p-phenylene)-based polymer electrolyte membranes with simultaneously enhanced proton conductivity and mechanical strength†
Miru
Yoshida-Hirahara
 ab,
Masahiro
Yoshizawa-Fujita
a,
Yuko
Takeoka
a and
Masahiro
Rikukawa
*a
ab,
Masahiro
Yoshizawa-Fujita
a,
Yuko
Takeoka
a and
Masahiro
Rikukawa
*a
aDepartment of Materials and Life Sciences, Sophia University, 7-1 Kioi-cho, Chiyoda-ku, Tokyo 102-8554, Japan. E-mail: m-rikuka@sophia.ac.jp; Fax: +81 3 3238 4198; Tel: +81 3 3238 4250
bAdvanced Technology Research Laboratories, Idemitsu Kosan Co., Ltd., 1280 Kami-izumi, Sodegaura, Chiba 299-0293, Japan
First published on 8th September 2023
Abstract
To achieve cooperative improvements in the proton conductivity and mechanical properties of sulfonated hydrocarbon-type polymer electrolyte membranes (PEMs) for fuel cell applications, a series of hydrophilic–hydrophobic multiblock copolymers with controlled unit ratio and ion-exchange capacity (IEC) was synthesised using a four-step technique: ([1] sulfonation of a monomer with a protecting group to prepare the hydrophilic segment, [2] synthesising dichloro-terminated oligomers with controlled chain length via the SNAr reaction to obtain the hydrophobic segment, [3] direct copolymerisation of the two products generated in the preceding steps by Ni(0)-catalysed coupling, and [4] cleavage of the protecting group of the copolymers). Six types of sulfonated poly(4-phenoxybenzoyl-1,4-phenylene)-b-poly(arylene ether ketone) multiblock copolymers, S-6X (n) x:y, were obtained by varying the sulfonated-monomer/hydrophobic-oligomer feed ratio. The IEC and weight-average molecular weight of the copolymers were 0.98–2.02 meq g−1 and 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300–239000 g mol−1, respectively. The proton conductivity of the S-6X specimens at 80 °C and 90% relative humidity (RH) was 2.2 × 10−2–1.3 × 10−1 S cm−1. The tensile strength and ultimate elongation of S-6X under similar conditions were 19.3–40.7 MPa and 21.6–210%, respectively. S-6X exhibited superior conductivity and mechanical properties with suppressed excess swelling compared to those of conventional hydrocarbon-based PEMs. Electrochemical atomic-force-microscopy images of S-6X revealed a phase-separated morphology with continuous hydrophilic domains. In fuel cell tests conducted at 80 °C and 100% RH, S-6X exhibited an optimal maximum-power-density of 611 mW cm−2 and a limiting current density of 1800 mA cm−2, which are comparable to those of perfluorosulfonic-acid-type PEMs.
300–239000 g mol−1, respectively. The proton conductivity of the S-6X specimens at 80 °C and 90% relative humidity (RH) was 2.2 × 10−2–1.3 × 10−1 S cm−1. The tensile strength and ultimate elongation of S-6X under similar conditions were 19.3–40.7 MPa and 21.6–210%, respectively. S-6X exhibited superior conductivity and mechanical properties with suppressed excess swelling compared to those of conventional hydrocarbon-based PEMs. Electrochemical atomic-force-microscopy images of S-6X revealed a phase-separated morphology with continuous hydrophilic domains. In fuel cell tests conducted at 80 °C and 100% RH, S-6X exhibited an optimal maximum-power-density of 611 mW cm−2 and a limiting current density of 1800 mA cm−2, which are comparable to those of perfluorosulfonic-acid-type PEMs.
1. Introduction
Sulfonated polymer electrolyte membranes have been studied as potential alternatives to perfluorosulfonic-acid-based specimens for utilisation in polymer electrolyte fuel cells (PEFCs).1 The main technical drawback of these hydrocarbon-type polymer electrolyte membranes (or proton-exchange membranes; PEMs) is achieving simultaneous improvements in proton conductivity and mechanical integrity under the wide-ranging operating conditions of PEFCs. In particular, rapid proton conduction in low-humidity, high-temperature environments is highly desired.2,3 However, realising improvements only by altering the primary structure of hydrocarbon polymers is difficult owing to the trade-off relationship between the two properties. Consequently, several approaches focussing on nanostructured polymer design that incorporates three-dimensional (3D) intermolecular interactions have been adopted to overcome these issues.4–7Block copolymers can have diverse long-range-ordered nanostructures that are generated by microphase separation between the constituent blocks. Because the blocks of a copolymer are interconnected by covalent bonds, the thermodynamic forces counterbalanced by the entropic forces from the covalent linkages drive phase separation at the scale of tens of nanometres.8–10 In recent studies on nanostructured PEMs, block copolymers with hydrophilic and hydrophobic segments have been extensively investigated. Thermodynamic incompatibility between the hydrophilic and hydrophobic building blocks induces microphase separation, thereby forming ion nanochannels.6,7,11 Continuous, ordered, and interconnected ion nanochannels can achieve rapid proton transport without deteriorating the mechanical integrity, primarily because of the mechanically robust hydrophobic domains surrounding the ion nanochannels.
Precise polymerisation methodologies must be implemented to develop the aforementioned nanostructured materials. To date, sulfonated aromatic copolymers with hydrophilic and hydrophobic segments have primarily been synthesised through nucleophilic-aromatic-substitution (SNAr)-type polycondensation reactions between two different telechelic oligomers.4,12 For example, McGrath et al. synthesised multiblock copolymers of sulfonated poly(arylene ether sulfone) (PAES) between fluorine- and phenoxide-terminated oligomers, and observed their highly phase-separated morphology using transmission electron microscopy and small-angle X-ray scattering (SAXS) profiles.13–15 Moreover, they found that the large-scale morphological order of the multiblock copolymers evolved with increasing block size, thereby considerably influencing their mechanical toughness, water uptake, and proton transport. Similarly, Yoo et al. synthesised sulfonated poly(arylene ether ketone) (PAEK) multiblock copolymers with a high local concentration of sulfonic acid groups.16 Wang et al. synthesised multiblock PAEKs with sulfoalkoxyl side chains through polycondensation of oligomers, and the resulting membranes achieved high power densities under low-relative-humidity (RH) conditions.17 Although SNAr polycondensation is a simple and powerful technique for synthesising poly(arylene ether)-type copolymers, it restricts the synthesis of aromatic polymers without heteroatom linkages. Moreover, limited monomer structures can be employed owing to the high reaction temperature (>180 °C), and the molecular weights of the polymers tend to be low. Therefore, new methodologies that permit the synthesis of sulfonated block copolymers for use in PEMs must be established.
Wholly aromatic poly(p-phenylene) derivatives with substantially high molecular weights have previously been synthesised by our group for practical PEM applications.18–20 Although the rigid poly(p-phenylene) structure is inconvenient because of its poor solubility and undesirable film-forming properties, sulfonated polyphenylenes with appropriate side-chain structures can yield durable and reliable membranes for PEFC applications.21–25 In particular, the benzoyl-type substituent is known to increase the solubility of wholly aromatic materials in certain common organic solvents, while preserving the outstanding thermal and mechanical properties of the conjugated backbones.26,27 Sulfonated poly(4-phenoxybenzoyl-1,4-phenylene) (S-PPBP) with a high molecular weight (up to 438000 g mol−1) was previously synthesised by our group via Ni(0)-catalysed coupling polymerisation, which is a useful synthesis method for creating carbon–carbon aryl bonds.18,28 S-PPBP exhibits particularly high proton conductivity and hydrolysis stability owing to its pendant chain structure. More recently, a series of S-PPBP-based random copolymers with controlled ion-exchange capacity (IEC) was prepared by our group through combined Ni(0) coupling polymerisation and a novel sulfonation procedure for precursor monomers.20 This sulfonation process, which employed neopentylsulfonate ester as a protective group, provided precise control of the IEC and structures with high molecular weights up to 465000 g mol−1. However, the products exhibited significantly decreased proton conductivity at IECs below 1.69 meq g−1. This was presumably due to the randomly distributed structure of the copolymers, which led to inferior connectivity between the hydrophilic domains. To achieve cooperative improvements in conductivity and mechanical integrity, block copolymerisation of S-PPBP and another hydrophobic segment could be implemented as a promising strategy. Therefore, a novel synthesis process for sulfonated block copolymers aimed at PEM applications can be devised by combining the aforementioned sulfonation procedure and other synthesis techniques.
In this study, SNAr-type oligomerisation and the Ni(0)-catalysed coupling polymerisation were combined with the aforementioned sulfonation to synthesise a series of hydrophilic–hydrophobic multiblock copolymers – S-PPBP-b-PAEK6X (or S-6X) – with precisely controlled IEC. Conventionally, multiblock copolymers are synthesised using technically complicated processes such as living polymerisation under limited conditions with multi-step procedures. In contrast, robust multiblock structures can be synthesised using our method through one-step direct copolymerisation of hydrophilic monomers and hydrophobic oligomers. Moreover, sulfonic acid groups can be introduced selectively and precisely onto the pendant chains of the hydrophilic blocks, enabling the formation of a phase-separated morphology and, consequently, interconnected ion nanochannels. Two types of PAEK structures were initially selected as the hydrophobic blocks for their ability to form flexible and tough film-forming materials. A dichlorobenzophenone was introduced into the oligomer structure as an end-capper to facilitate the Ni(0)-catalysed coupling copolymerisation with hydrophilic monomers. Six types of S-6X multiblock copolymers were synthesised with the goal of developing mechanically resilient PEMs without diminishing their conductivity. The water sorption properties, mechanical characteristics, and proton conductivity of S-6X were investigated to determine the effects of the hydrophilic–hydrophobic multiblock structure on the fundamental membrane properties. The morphologies of the S-6X membranes were examined by electrochemical atomic force microscopy (e-AFM) and SAXS analysis. Moreover, the superior performance of the synthesised multiblock copolymer for PEFC applications was demonstrated through single-fuel-cell tests.
2. Experimental
2.1. Materials
2,2-Bis(4-hydroxyphenyl)propane (bisphenol A; FUJIFILM Wako) was recrystallised from ethyl acetate/n-hexane (1/3, v/v) and dried in vacuo at 65 °C for 12 h. 4,4′-Dichlorobenzophenone (DCBP; Sigma-Aldrich Japan) and 2,2-bis(4-hydroxyphenyl)hexafluoropropane (bisphenol AF; FUJIFILM Wako) were recrystallised from toluene and dried in vacuo at 65 °C for 12 h. Potassium carbonate (K2CO3; FUJIFILM Wako) was dried in vacuo at 50 °C prior to use. 1,3-Dimethyl-2-imidazolidinone (DMI; FUJIFILM Wako) was dried with excess calcium hydride (CaH2; FUJIFILM Wako) for 24 h and purified by distillation under reduced pressure (1 mmHg, 83 °C). Bis(triphenylphosphine) nickel(II) dichloride (NiCl2(PPh3)2; Kanto Chemical) was dried in vacuo at 100 °C prior to use. Triphenylphosphine (PPh3; FUJIFILM Wako) was recrystallised from ethanol and dried in vacuo at 50 °C for 12 h. Sodium iodide (NaI; FUJIFILM Wako) was recrystallised from distilled water and dried in vacuo at 70 °C for 24 h. Zn powder (325 mesh; Merck Ltd) was washed three times with HCl (1.0 mol dm−3) in diethyl ether and dried in vacuo at 100 °C for 6 h and then at room temperature (RT) for 12 h. N-Methylpyrrolidone (NMP; Kanto Chemical) was dried with excess CaH2 for 24 h, purified by distillation under reduced pressure (1 mmHg, 54 °C), degassed by bubbling N2, and then dehydrated over molecular sieves dried at 200 °C (MS4A; FUJIFILM Wako) prior to use. All other solvents were purchased from FUJIFILM Wako or Kanto Chemical and purified appropriately where necessary.2.2. Synthesis of hydrophilic monomer
2,2-Dimethylpropyl-4-[4-(2,5-dichlorobenzoyl)phenoxy] benzenesulfonate (or neopentyl-protecting sulfonated DPBP; NS-DPBP) was synthesised according to our previously reported method.20 Briefly, chlorosulfuric acid was added to 2,5-dichloro-4′-benzophenone at RT for 3 h to achieve chlorosulfonation, and the resulting product was treated with neopentyl alcohol in pyridine at RT for 3 h to yield NS-DPBP.2.3. Synthesis of hydrophobic oligomers
SNAr polycondensation was performed to synthesise two types of dichloro-terminated PAEKs (PAEK-Cl).:10, v/v). The obtained product was washed by Soxhlet extraction with methanol for three days and dried in vacuo at 50 °C for 12 h. PAEK6H-Cl was obtained as a yellow powder in 67% yield (10.71 g).
1H NMR (300 MHz, CDCl3): δ 7.78 (d, J = 8.6 Hz), 7.72 (d, J = 8.6 Hz), 7.44 (d, J = 8.6 Hz), 7.26 (d, J = 8.6 Hz), 7.04–6.98 (m), 1.71 (s) ppm. IR (KBr): 3037, 2966, 1657, 1593, 1498, 1416, 1363, 1240, 1090, 1012, 874–682 cm−1. Elemental analysis: calcd for (CHN)n C, 81.87%; H, 5.36%. Found C, 81.52%; H, 5.25%. Molecular weight (1H NMR): Mn = 5940 g mol−1, n = 14.
1H NMR (300 MHz, CDCl3): δ 7.82 (d, J = 8.6 Hz), 7.73 (d, J = 8.6 Hz), 7.47 (d, J = 8.6 Hz), 7.43 (d, J = 8.6 Hz), 7.11 (t, J = 10.0 Hz) ppm. IR (KBr): 3047, 1660, 1595, 1500, 1246, 1205, 1091, 1016, 874–683 cm−1. Elemental analysis: calcd for (CHN)n C, 65.23%; H, 3.14%. Found C, 65.43%; H, 3.03%. Molecular weight (1H NMR): Mn = 5400 g mol−1, n = 10.
2.4. Synthesis of multiblock copolymers
Ni(0)-Mediated coupling was performed to synthesise two types of sulfonated poly(4-phenoxybenzoyl-1,4-phenylene)–poly(arylene ether ketone)s (S-PPBP-b-PAEK).:10, v/v; 550 mL). The crude product was purified by reprecipitation from a chloroform solution into HCl/methanol (1:10, v/v) at least twice to remove the residual catalyst, washed using a Soxhlet extractor with methanol for three days, and then dried in vacuo at 50 °C for 12 h. NS-PPBP-b-PAEK6H (NS-6H) was obtained as a white solid in 72–92% yields.
The neopentyl-protecting group of NS-6H was cleaved by acidolysis with (C2H5)2NH·HBr. NS-6H was placed in a three-neck round-bottom flask filled with nitrogen, which was equipped with a mechanical stirring shaft. NMP was added to the flask, and the mixture was heated to 80 °C and stirred. A solution of (C2H5)2NH·HBr in NMP was added, and the reaction mixture was heated to 120 °C and stirred for 24 h. After cooling to RT, the resulting mixture was poured into a solution of excess methanol and 10% HCl, and then stirred for 24 h. The precipitate was immersed in 1 mol dm−3 HCl (aq.) for 48 h to exchange the diethylammonium ions with protons. After conversion to the acidic form, the crude product was purified twice by reprecipitation from NMP into methanol and then dried in vacuo at 80 °C for 12 h. S-PPBP-b-PAEK6H (S-6H) was obtained as a reddish brown solid in 58–96% yields. The amounts of all reactants used in the synthesis and the characterisation data are provided in the ESI.†
2.5. Membrane preparation
Polymer membranes were prepared by solution casting S-6H or S-6F onto a flat glass plate with a 5 wt% dimethyl sulfoxide (DMSO) solution. Each polymer solution was cast onto a cleaned glass substrate (10 × 10 cm), heated at 80 °C for 12 h at ambient pressure, and then dried in vacuo at 80 °C for 3 h. The membranes were immersed in deionised water to help peel them from the substrates, and the detached membranes were subsequently immersed in 1 mol dm−3 HCl (aq.) at 80 °C for 2 h, followed by treatment with deionised water at 80 °C for 3 h, and drying in vacuo at 80 °C for 12 h.2.6. Characterisation
1H nuclear magnetic resonance (NMR; 300 or 500 MHz) and 13C NMR (125 MHz) measurements were performed using ECA-300 and ECA-500 spectrometers (JEOL). Chloroform-d1 (CDCl3; FUJIFILM Wako), dichloromethane-d2 (CD2Cl2; Sigma-Aldrich), and DMSO-d6 (Kanto Chemical) were used as solvents, and tetramethylsilane (TMS; FUJIFILM Wako) was used as an internal standard for the 1H and 13C NMR spectroscopy. Fourier transform infrared (FT-IR) spectra were recorded using a Nicolet 6700 spectrometer (Thermo Fisher Scientific) in the wavenumber range 650–4000 cm−1. Solid samples were analysed using the KBr or attenuated-total-reflection (ATR) methods. Elemental analysis was performed using a PE2400-II instrument (PerkinElmer) at 975 °C. Gel permeation chromatography (GPC) was performed using a LC-10AD chromatosystem (Shimadzu) equipped with a Shodex LF-804 column (Showa Denko K.K.). The GPC measurements were conducted at 50 °C using N,N-dimethylformamide (DMF; FUJIFILM Wako) containing 0.1% lithium bromide (LiBr; Wako) as an eluent at a flow rate of 1.0 mL min−1. Poly(ethylene oxide) standards (Sigma-Aldrich) were used for the GPC analysis.The experimental IEC values were determined using elemental analysis and back titration, as described henceforth. The dried membranes were immersed in 1.0 mol dm−3 HCl (aq.) at 80 °C for 2 h, and then treated with deionised water at 80 °C for 3 h. The IEC was then estimated by titration with 10 mmol dm−3 NaOH (aq.). Thermogravimetry/differential thermal analysis (TG-DTA) was performed using a TG-DTA7200 device (Seiko Instruments) at a heating rate of 10 °C min−1 from 25 to 500 °C under nitrogen flow.
Weight-based water uptake (WU) measurements were performed using an MSB-AD-V-FC isothermal-adsorption-measurement system (MicrotracBEL) equipped with a temperature- and humidity-controlling chamber. The measurements were performed at 80 °C with varying RH (25–85%). The WU was calculated as follows:
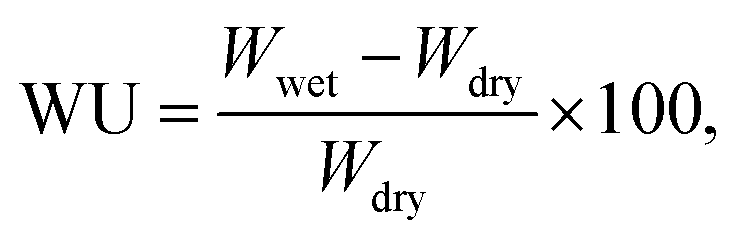
where 18.01 is the molecular weight of water.
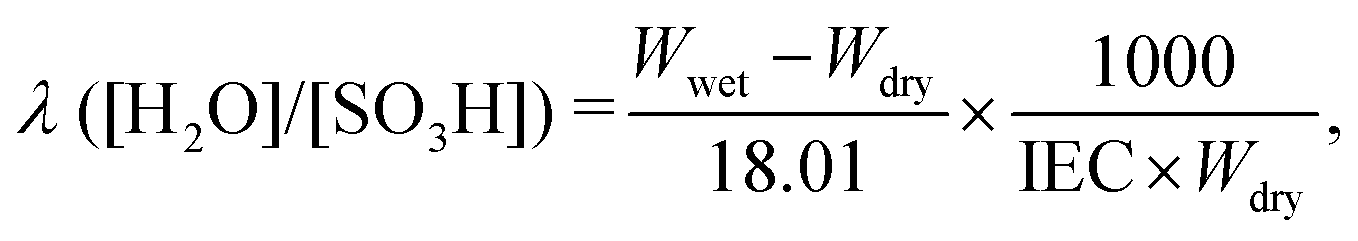
The in-plane proton conductivity σ (S cm−1) of the polymer membranes was measured using an SI1260 electrochemical impedance analyser (Solartron). Each sample was clamped between Pt electrodes and placed in a four-probe conductivity cell. The measurements were performed at 80 °C with varying RH (30%–90%), and σ was calculated as follows:
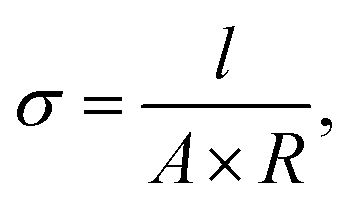
Tensile tests were performed using a Tensilon RTG-1210 instrument (A&D) equipped with a temperature- and humidity-controlling chamber under humid air flow. The tensile strength and ultimate elongation of membrane samples with areas of 10 mm × 20 mm were measured under ambient conditions (RT, no extra humidification) at an elevated temperature (80 °C) and humidity (90% RH).
e-AFM images were captured using a JSPM-5400 device (JEOL) equipped with a humidity-controlling unit. Pt-coated cantilevers – NSC05/Pt2.0 (NT-MDT Spectrum Instruments) and TAP-300E (BudgetSensors® Innovative Solutions Bulgaria) – with force constants of 12 and 40 N m−1 and resonance frequencies of 250 and 300 kHz, respectively, were used. The membrane sample was placed on a gold-plated conductive sample stage using Nafion® dispersion DE-2020 (DuPont) as an adhesive. Bias voltage (−1.5 to −3.0 V) was applied to the sample stage during the observations. Topography-, phase-, and current-mapping images based on proton conduction were obtained simultaneously. Prior to the e-AFM measurements, the samples were placed in a humidity-controlling unit for at least 1 h.
SAXS measurements were conducted using a NANO-Viewer system (Rigaku) equipped with a humidity-controlling unit. Ni-filtered CuKα radiation (λ = 0.15418 nm) was finely focussed using a confocal max-flux mirror and collimated into an airtight sample chamber (Rigaku) with Kapton windows using a three-slit system. SAXS analysis was performed at 40 kV using a 30 mA power beam, and patterns were recorded on a PILATUS detector sheet using the transmission method, with the sample-to-detector distance set to 75 cm. The X-ray scattering patterns for each sample were recorded for 1 h. The scattered intensity I(q) from the SAXS patterns was recorded in steps of the scattering vector, which was calculated as follows:
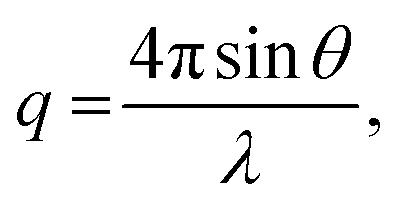
Membrane electrode assemblies (MEAs) were constructed as described henceforth. A catalyst ink was prepared by mixing a Pt/Vulcan-XC-72 system (amount of Pt = 47 wt%), purified water, ethanol, and a 5 wt% Nafion® dispersion. The obtained catalyst ink was coated on SIGRACET® GDL 35BC gas-diffusion layers (SGL Carbon Japan). The electrodes were then pressed onto the membrane samples (35 mm × 35 mm) at 6 MPa and 125 °C for 10 min. The active area and Pt catalyst loading were 5 cm2 and 1.0 mg cm−2, respectively. Each MEA was constructed into a single cell (ElectroChem) between graphite-based bipolar plates with serpentine flow channels. Single-fuel-cell tests with H2/air feed were conducted using an 890B fuel cell test system (Scribner Associates) at a cell temperature of 80 °C under humid conditions. Dry or humid H2 and air were continuously supplied at a flow rate of 500 and 1000 mL min−1, respectively, and the back pressure was maintained at 0.1 MPaG.
3. Results and discussion
3.1. Synthesis of a hydrophilic–hydrophobic multiblock structure with controlled IEC
The multiblock copolymers S-PPBP-b-PAEK6X (S-6X) were synthesised in four steps: (1) preparation of a sulfonated monomer with a protective group, (2) synthesis of a dichloro-terminated hydrophobic oligomer via SNAr polycondensation, (3) copolymerisation of the products from Steps 1 and 2 via Ni(0)-catalysed coupling, and (4) cleaving the protective group of the copolymer. The S-6X synthesis route and an illustration of the multiblock architecture are shown in Scheme 1 and Fig. 1, respectively. The sulfonated monomer NS-DPBP was synthesised according to our previously reported procedure.20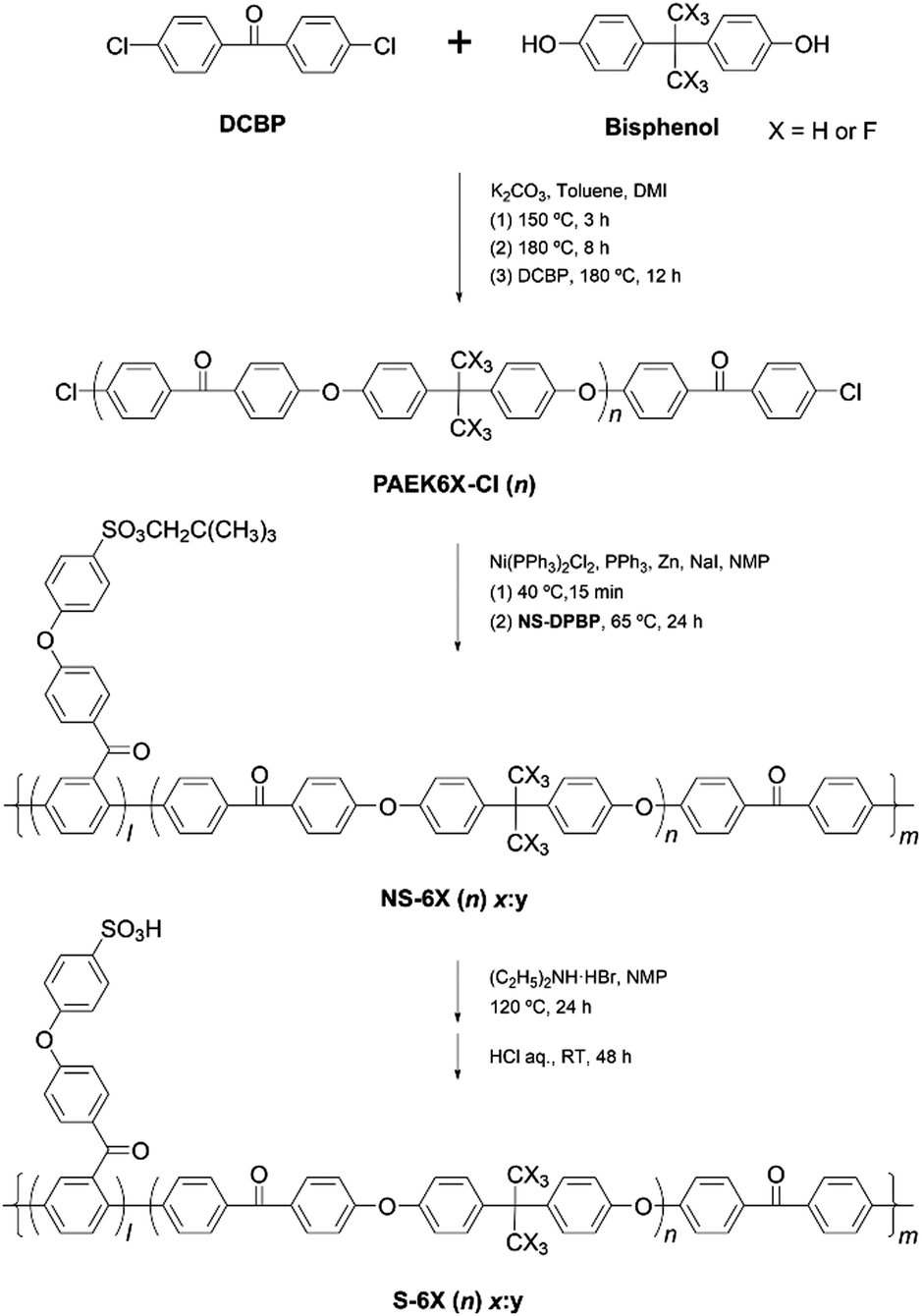 | ||
| Scheme 1 Synthesis of multiblock copolymers S-6X. | ||
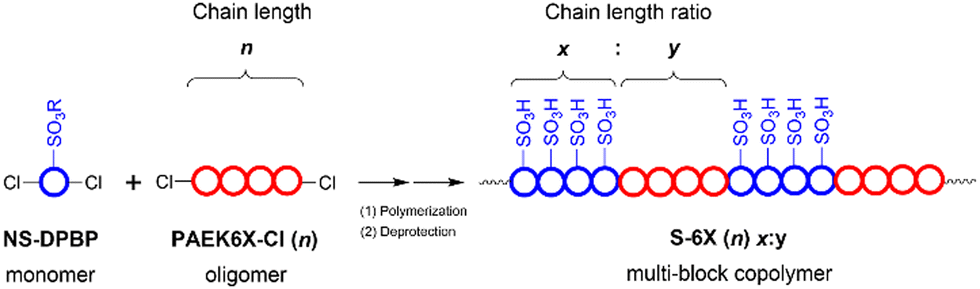 | ||
| Fig. 1 Schematic illustrating the polymer architecture of S-6X. | ||
The chemical structures of PAEK6H-Cl and PAEK6F-Cl were confirmed by FT-IR and 1H NMR analyses. The FT-IR spectra of the oligomers showed absorption bands at 1012 and 1240 cm−1, which corresponded to the C–O–C stretching vibration of the linking group, indicating progress of the polycondensation reaction (Fig. 2a and Fig. S1, S2, ESI†). Absorption bands associated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (1657 cm−1), C–H stretching (2966 cm−1; PAEK6H-Cl), C–F stretching (1205 cm−1; PAEK6F-Cl) also appeared. Small absorption bands derived from the C–Cl stretching vibration of the terminal group appeared at 1090 cm−1, validating the synthesis of the dichloro-terminated structure. The 1H NMR spectra of PAEK6H-Cl and PAEK6F-Cl showed verifiable signals (Fig. 3a and Fig. S3, S4, ESI†). Three large chemical shifts appeared at ∼6.9–7.9 ppm, which were assigned to the aromatic protons (a)–(d) on the repeat units. The distinct shift at ∼1.7 ppm was assigned to the methyl group protons (‘e’; PAEK6H-Cl). The two small shifts at ∼7.7 and 7.4 ppm were ascribed to the terminal aromatic protons (a′) and (b′), respectively, on the DCBP end-capping groups. The 1H NMR signals were employed to determine the number of repeat units (n) and the absolute number-average molecular weight (Mn) of the oligomers. This was accomplished by integration of the terminal aromatic protons (a′) and (b′) relative to the repeat-unit protons (a)–(d). The repeat units (n) of PAEK6H-Cl and PAEK6F-Cl were calculated to be ∼14 (Mn = 5940 g mol−1) and ∼10 (Mn = 5400 g mol−1), respectively. All oligomers were adequately soluble in conventional solvents such as chloroform, methylene chloride, tetrahydrofuran (THF), dimethylacetamide (DMAc), and NMP.
O stretching (1657 cm−1), C–H stretching (2966 cm−1; PAEK6H-Cl), C–F stretching (1205 cm−1; PAEK6F-Cl) also appeared. Small absorption bands derived from the C–Cl stretching vibration of the terminal group appeared at 1090 cm−1, validating the synthesis of the dichloro-terminated structure. The 1H NMR spectra of PAEK6H-Cl and PAEK6F-Cl showed verifiable signals (Fig. 3a and Fig. S3, S4, ESI†). Three large chemical shifts appeared at ∼6.9–7.9 ppm, which were assigned to the aromatic protons (a)–(d) on the repeat units. The distinct shift at ∼1.7 ppm was assigned to the methyl group protons (‘e’; PAEK6H-Cl). The two small shifts at ∼7.7 and 7.4 ppm were ascribed to the terminal aromatic protons (a′) and (b′), respectively, on the DCBP end-capping groups. The 1H NMR signals were employed to determine the number of repeat units (n) and the absolute number-average molecular weight (Mn) of the oligomers. This was accomplished by integration of the terminal aromatic protons (a′) and (b′) relative to the repeat-unit protons (a)–(d). The repeat units (n) of PAEK6H-Cl and PAEK6F-Cl were calculated to be ∼14 (Mn = 5940 g mol−1) and ∼10 (Mn = 5400 g mol−1), respectively. All oligomers were adequately soluble in conventional solvents such as chloroform, methylene chloride, tetrahydrofuran (THF), dimethylacetamide (DMAc), and NMP.
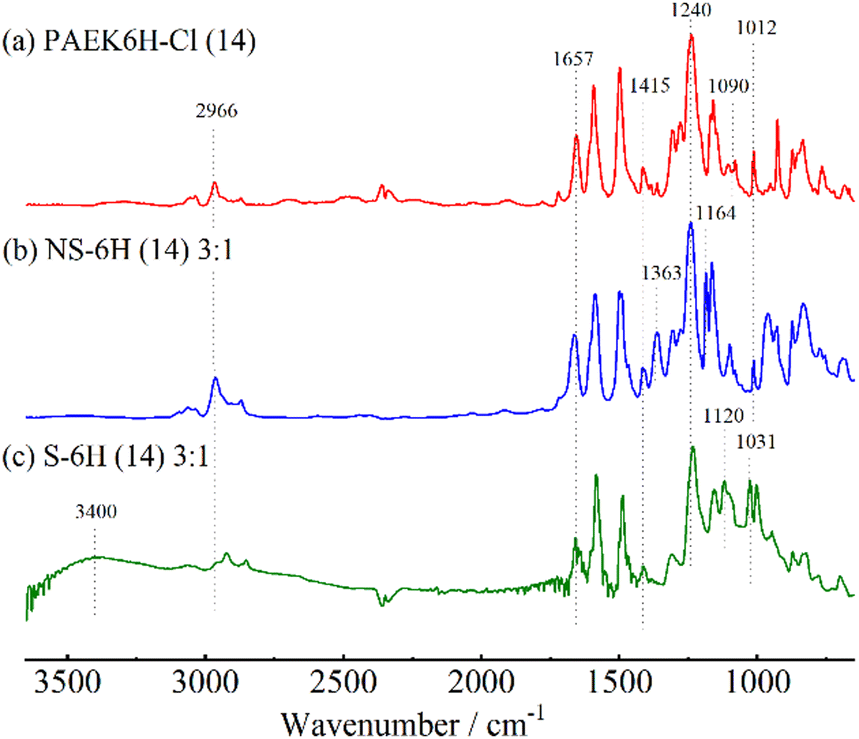 | ||
| Fig. 2 FT-IR spectra of (a) PAEK6H-Cl (14), (b) NS-6H (14) 3:1, and (c) S-6H (14) 3:1. | ||
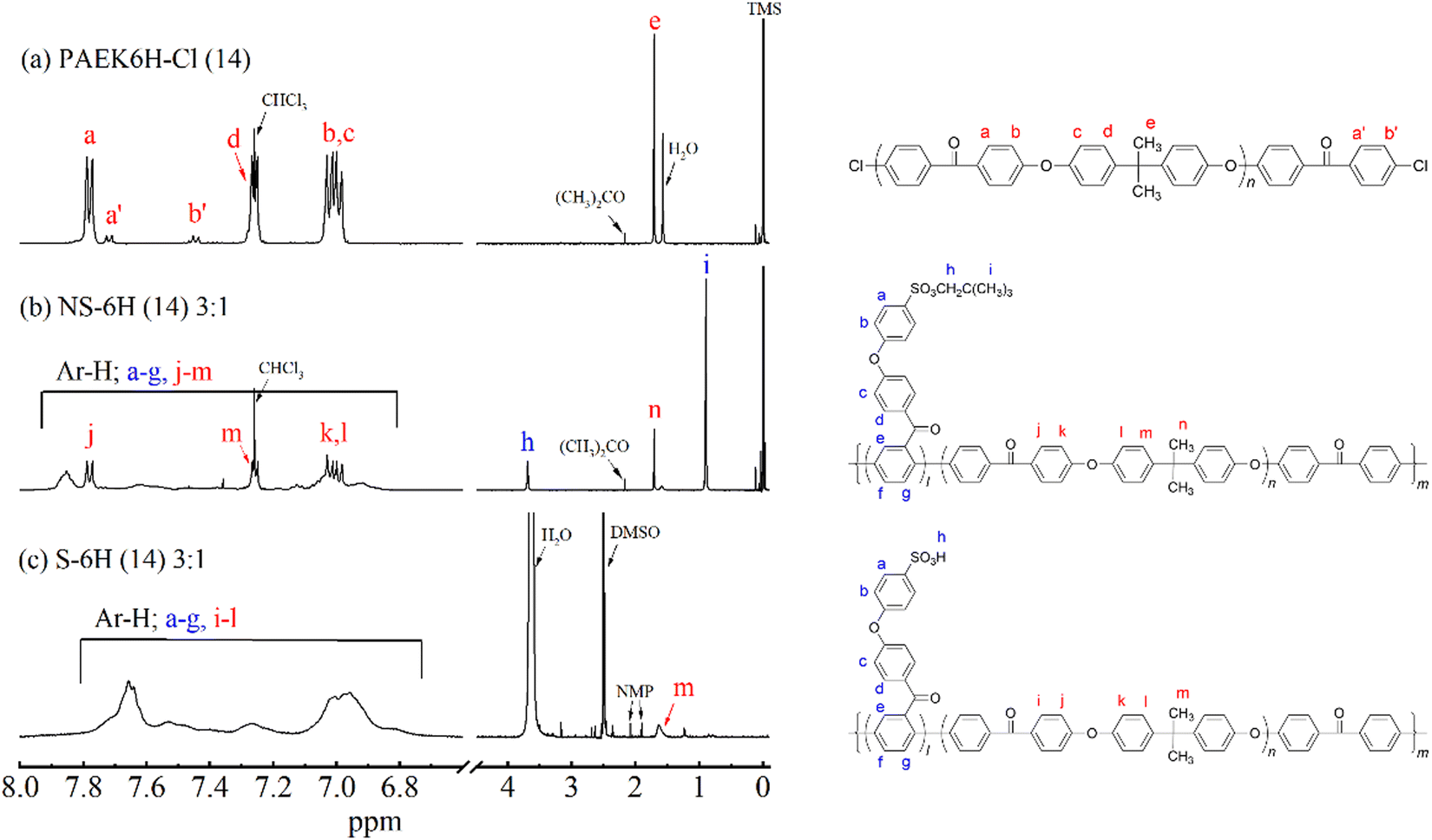 | ||
| Fig. 3
1H NMR spectra of (a) PAEK6H-Cl (14) in CDCl3, (b) NS-6H (14) 3:1 in CDCl3, and (c) S-6H (14) 3:1 in DMSO-d6. | ||
A multiblock structure with moderately periodic architectures was generated via direct copolymerisation of the hydrophilic monomer NS-DPBP and hydrophobic oligomer PAEK6X-Cl through one-pot Ni(0)-catalysed coupling reactions. A Ni(0)-complex catalyst solution was prepared through reduction of the Ni(II) species in Ni(PPh3)2Cl2 with activated zinc. Triphenylphosphine and sodium iodide were used as the ligand and co-catalyst, respectively. NS-DPBP and PAEK6X-Cl were copolymerised with the catalyst solution in purified NMP at 65 °C for 24 h. In contrast to our previously reported protocol for synthesising S-PPBP random copolymers, the reduction of Ni(II) to Ni(0) in this study was achieved in the presence of PAEK6X-Cl prior to adding NS-DPBP.20 This difference in catalyst preparation probably yielded PAEK–Ni(II) intermediates quickly by oxidative addition of PAEK6X-Cl to Ni(0), leading to efficient coupling of PAEK6X-Cl and NS-DPBP. The ratio of chain lengths of the NS-PPBP and PAEK6X segments in the copolymers (x:y) was adjusted by controlling the feed molar ratio of the corresponding monomers and oligomers. Six types of NS-6X (n) x:y specimens were successfully prepared in 72–92% yields by varying the NS-DPBP/PAEK6X-Cl feed ratio.
The structures of the NS-6X copolymers were confirmed by FT-IR and 1H NMR analyses. FT-IR spectra of NS-6X showed new absorption bands at 1363 and 1164 cm−1, which corresponded to the OSO asymmetric and symmetric stretching vibrations of the neopentylsulfonate ester group (Fig. 2b and Fig. S5, S6, ESI†). All investigated copolymers showed absorption bands associated with CO stretching (1658 cm−1), C–O–C stretching (1240 cm−1), C–H stretching (2966 cm−1), and CH3 bending vibration (1415 cm−1). The disappearance of the absorption band at 1090 cm−1, which corresponded to C–Cl stretching, indicated progress in the polymerisation. In the 1H NMR spectra of NS-6X (Fig. 3b and Fig. S7, S8, ESI†), the chemical shifts at ∼6.8–8.0 ppm were assigned to the aromatic protons, whereas those at 3.7 and 0.9 ppm were ascribed to the aliphatic protons (h) and (i), respectively, on the neopentylsulfonate ester group of the hydrophilic segments. In the 1H NMR spectrum of NS-6H, the distinct shift at ∼1.7 ppm was attributed to the methyl group protons (n) in the hydrophobic segment. Based on the 1H NMR signals, the simplified chain length ratios (x:y) for NS-6H were estimated to be 3.03:1, 2.04:1, and 1.03:1 by calculating the integration ratio of the neopentyl group protons (h) and (i) relative to the hydrophobic block protons (j)–(n). The obtained ratios are consistent with the theoretical ratios calculated using the monomer/oligomer feed ratios (Table 1). Similarly, the x:y values for NS-6F calculated from the 1H NMR spectra were close to the theoretical ratios. These results indicate that NS-6X copolymers with a moderately controlled polymer sequence were successfully synthesised through one-pot Ni(0)-catalysed coupling reactions. All copolymers exhibited decent solubility in common solvents, which was comparable with the high solubility of PAEK6X-Cl and the NS-PPBP homopolymer.20
| Sample | PAEK6X block na | S-PPBP: PAEK6X x:yb |
IEC/meq g−1 | GPC dataf | Water uptakeg | Swelling ratioh/% | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Targetc | Exp.d | Targetc | Exp.e | M n | M w | M w/Mn | WU/wt% | λ | Δt | Δl | Δt/l | ||
| a Block length of hydrophobic segment calculated from 1H NMR spectra of PAEK6X-Cl. b Simplified chain length ratio of hydrophilic and hydrophobic segments. c Calculated from the feed molar ratio of NS-DPBP and PAEK6X-Cl. d Determined by 1H NMR of NS-6X. e Determined by back titration. f Determined by GPC (eluent: DMF). g Water uptake (WU) and the number of water molecules per a sulfonic acid group (λ) of membranes. Each membrane was immersed in water at 30 °C for 2 h. h Swelling ratio of membranes in the through-plane (Δt) and the in-plane direction (Δl). Each membrane was immersed in water at 80 °C for 2 h. | |||||||||||||
| PAEK6H-Cl (14) | 14 (Mn = 5940) | — | — | — | — | — | — | — | — | — | — | — | — |
| PAEK6F-Cl (10) | 10 (Mn = 5400) | — | — | — | — | — | — | — | — | — | — | — | — |
| S-6H (14) 3:1 |
14 | 3:1 |
3.03:1 |
2.05 | 1.96 | 70600 |
176000 |
2.50 | 99 | 28 | 39 | 7.3 | 5.3 |
| S-6H (14) 2:1 |
14 | 2:1 |
2.04:1 |
1.80 | 1.80 | 58000 |
124000 |
2.13 | 82 | 25 | 27 | 6.2 | 4.4 |
| S-6H (14) 1:1 |
14 | 1:1 |
1.03:1 |
1.32 | 1.15 | 22000 |
69200 |
3.16 | 29 | 14 | 7.4 | 5.5 | 1.3 |
| S-6F (10) 3:1 |
10 | 3:1 |
3.24:1 |
1.91 | 2.02 | 50800 |
118000 |
2.31 | 68 | 19 | 40 | 8.3 | 4.8 |
| S-6F (10) 2:1 |
10 | 2:1 |
1.99:1 |
1.64 | 1.65 | 61400 |
239000 |
3.89 | 88 | 30 | 40 | 5.7 | 7.0 |
| S-6F (10) 1:1 |
10 | 1:1 |
0.91:1 |
1.15 | 0.98 | 29900 |
47300 |
1.58 | 29 | 17 | 20 | 4.8 | 4.2 |
| S-PPBP | — | — | — | 2.79 | 2.84 | 178000 |
359000 |
2.02 | 160 | 31 | 90 | 14 | 6.4 |
Using our previously reported protocol,20 acidolysis of NS-6X with neopentyl protective groups was performed using diethylamine hydrobromide to yield sulfonated copolymers. Six types of S-6X (n) x:y specimens were successfully obtained without any undesired side reaction such as gelation. FT-IR spectra of S-6X showed new absorption bands at ∼3400 cm−1, which corresponded to O–H stretching vibrations, indicating successful conversion of the sulfonic acid groups from the neopentylsulfonate ester groups (Fig. 2c and Fig. S9, S10, ESI†). The absorption band related to OSO stretching vibration also shifted to 1120 and 1031 cm−1, indicating formation of sulfonic acid groups. In the 1H NMR spectra of S-6H acquired using DMSO-d6 solvent, the disappearance of the chemical shifts at 3.7 and 0.90 ppm, corresponding to the neopentyl group, also indicated progress in the conversion of the neopentyl sulfonate ester groups into sulfonic acid groups (Fig. 3c and Fig. S11, ESI†). The weight-average molecular weight (Mw) values of S-6X, which were determined by GPC (Table 1), were in the range 47300–239000 g mol−1. The GPC results indicated that the molecular weights of the copolymers decreased with increasing content of the hydrophobic segment. This tendency was presumably due to the lower concentration of reactive terminated groups in PAEK6X-Cl than that of NS-DPBP during the copolymerisation. However, all copolymers exhibited moderate molecular weights, which was conducive to the production of self-supported films.
The solubility of the synthesised polymers was subsequently assessed (Table 2). The solubility of S-6X differed drastically from that of the NS-6X copolymers with protected groups. The S-6X specimens were soluble in polar aprotic solvents, such as NMP, DMF, DMAc, and DMSO, but insoluble in commonly used polar protic solvents and halogen solvents, such as methanol, chloroform, and methylene chloride. Moreover, none of the polymers were soluble in water, even after being subjected to 80 °C for 2 h. This solubility behaviour is similar to that of the S-PPBP homopolymer. Interestingly, no difference in solubility was observed with respect to the different composition ratios of the hydrophilic and hydrophobic segments. Moreover, no difference was observed based on the presence or absence of the fluorine substituents in the hydrophobic segment.
| Sample | CH3OH | CHCl3 | THF | NMP | DMF | DMSO | |
|---|---|---|---|---|---|---|---|
| a THF: tetrahydrofuran; NMP: 1-methyl-2-pyrrolidone; DMF: N,N-dimethylformamide; DMSO: dimethyl sulfoxide; (++): soluble at room temperature; (−): insoluble. b Homopolymer used as controls.20 | |||||||
| PAEK6H-Cl | − | ++ | ++ | ++ | ++ | − | |
| PAEK6F-Cl | − | ++ | ++ | ++ | ++ | − | |
| NS-6H | − | ++ | ++ | ++ | ++ | − | |
| NS-6F | − | ++ | ++ | ++ | ++ | − | |
| S-6H | − | − | − | ++ | ++ | ++ | |
| S-6F | − | − | − | ++ | ++ | ++ | |
| NS-PPBPb | − | ++ | ++ | ++ | ++ | − | |
| S-PPBPb | − | − | − | ++ | ++ | ++ | |
The experimental IECs of the obtained membranes were determined by back titration. The IEC data of the S-6X membranes (0.98–2.02 meq g−1) are consistent with the theoretical IECs calculated using the NS-DPBP/PAEK6X-Cl feed ratios. Membranes with the S-PPBP homopolymer (Mw = 359000, IEC = 2.84 meq g−1) and Nafion®112 were used as controls. The properties of all investigated polymer products are summarised in Table 1.
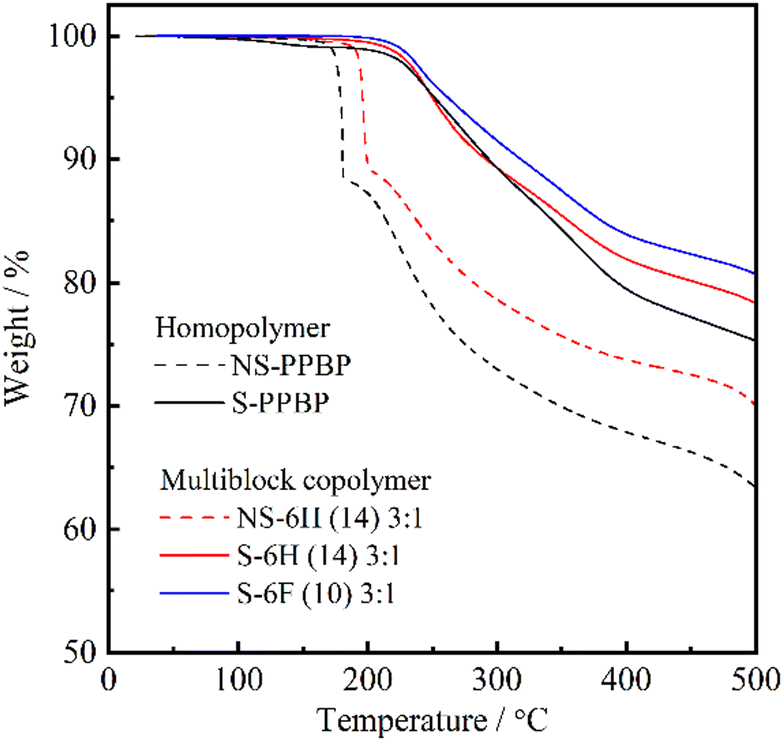 | ||
| Fig. 4 TGA curves of NS-6H (14) 3:1, S-6H (14) 3:1, S-6F (10) 3:1, NS-PPBP, and S-PPBP. | ||
The S-6X specimens obtained after deprotection exhibited two-step degradation. First, a continuous weight loss occurred at temperatures above 220–230 °C, which corresponded to the elimination of sulfonic acid groups from the polymer backbones. The second weight loss, which was observed at temperatures above 480 °C, was assigned to the degradation of the polymer backbones. Moreover, S-6F and S-6H showed similar thermal behaviour. Overall, these results indicate that all S-6X copolymers could exhibit moderate thermal stability in fuel cells operated at temperatures below 200 °C.
3.2. Cooperative improvements in conductivity and strength
This study was aimed at achieving cooperative improvements in proton conductivity and mechanical properties using the designed multiblock structures. Typically, sulfonated aromatic polymers require high IEC values to achieve high conductivity, because a large amount of water is essential for realising rapid proton transport as well as constructing interconnected ion channels. However, high water sorption can lead to excess swelling and deteriorate the membrane properties. Efficient proton conduction with moderate water contents is required to achieve further improvements in PEMs.Recently, various-typed poly(phenylene) (PP) and PAEK copolymers have been studied as both the PEM and the anion exchange membrane (AEM).29–34 In the recent publications, Miyatake et al. reported sulfonated PP based copolymers composed of m- and p-phenylene groups.29,30 Their membrane showed the phase separated structure with continuous water cluster, and thus high proton conductivity. As recent examples of AEMs, quaternized PP-PAE multiblock copolymers reported by Bae et al., and Friedel–Crafts-typed PES copolymers reported by Zhang et al., exhibited high conductivities about 100 mS cm−1 at fully hydrated conditions.31,32 These newly membranes which are currently being studied, tend to exhibit excellent ionic conductivity with suppressed water sorption. On the other hand, considering the durability when these materials are used in fuel cells, the mechanical strength needs further improvement. We anticipate that our multiblock strategy with pendant-typed PP and PAEK structure will show further superiority in terms of mechanical strength. Therefore, the water sorption properties, mechanical attributes, and proton conductivity of S-6X were analysed, as shown below, to determine the effects of the hydrophilic–hydrophobic multiblock structures on conductivity and mechanical properties.
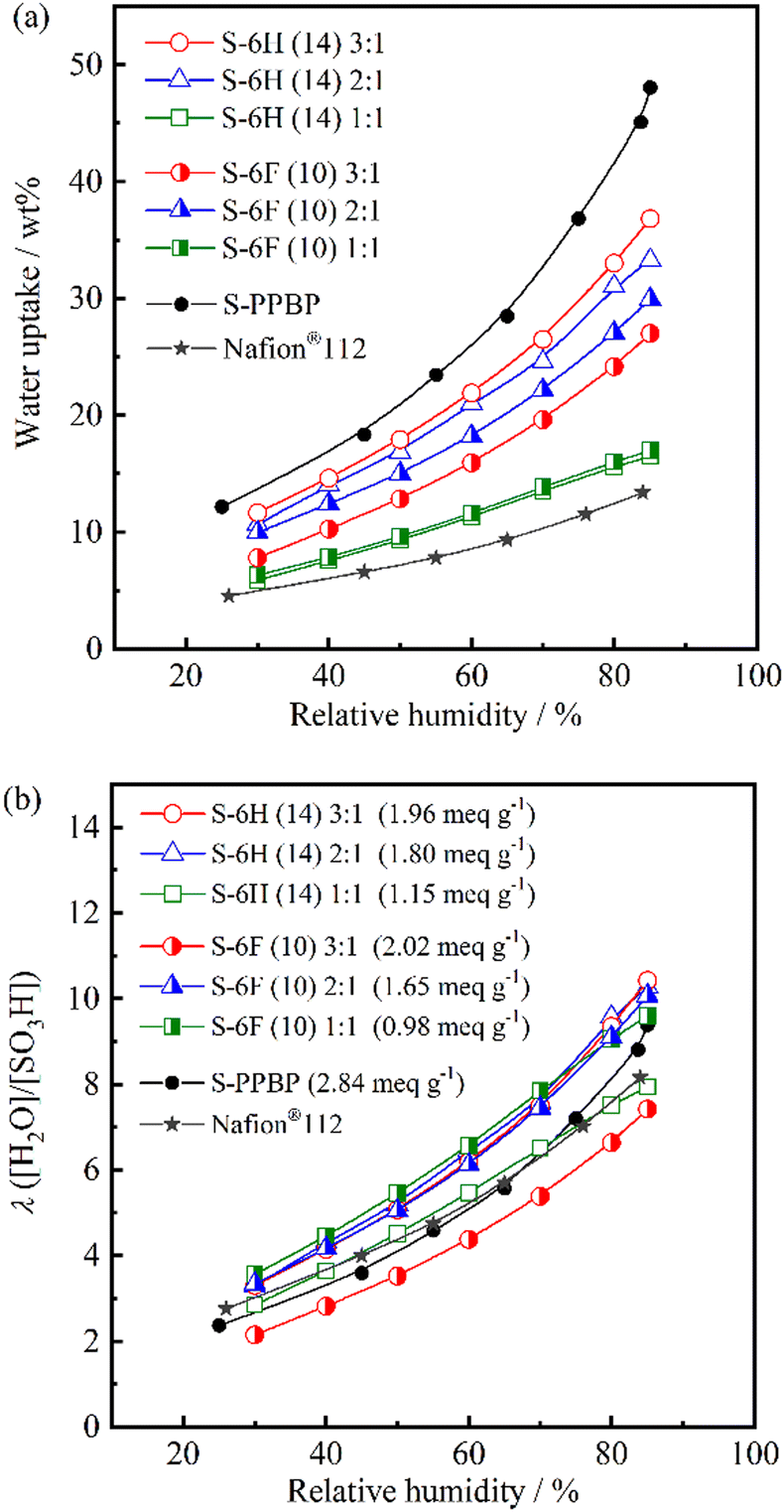 | ||
| Fig. 5 (a) Water uptake and (b) the number of adsorbed water molecules per sulfonic acid group (λ) for S-6H, S-6F, S-PPBP, and Nafion® 112 as a function of relative humidity at 80 °C. | ||
At 80 °C and 30–85% RH, the WU was found to be 6–37 wt% for S-6H and 6–30 wt% for S-6F. Generally, the WU of PEMs increases with increasing IEC because the amount of sulfonic acid groups directly affects the IEC value.7,16,35 As expected, the WU tended to increase with increasing IEC for S-6X. S-6F exhibited slightly lower WU values than those of S-6H, owing to the incorporation of the partially fluorinated polymer structure.14 S-6F (10) 2:1 showed slightly higher WU than 3:1, probably due to the morphology of water channel structure in the hydrophilic domain. Moreover, all S-6X membranes exhibited a lower WU than that of S-PPBP (12–48 wt%), presumably owing to their lower IEC and multiblock structures. After immersion in water, the WU of S-6X (29–99 wt%) was approximately two-to-five-times-lower than that of S-PPBP (160 wt%; Table 1). These results highlight the ability of the multiblock structures with PAEK hydrophobic blocks to prevent excessive water sorption.
The hydration number λ is another objective parameter for estimating the water-sorbing-ability of membranes. At 80 °C and 30–85% RH, λ was found to be 2.9–10.4 for S-6H and 2.2–10.1 for S-6F. While all the polymers exhibited WU values that increased with increasing sulfonation level, their λ values at a given RH appeared to be less dependent on their IEC. S-6H (14) 3:1, S-6H (14) 2:1, S-6F (10) 2:1, and S-6F (10) 1:1 exhibited comparable λ values (3.3–10.4), and displayed curves that were above that of S-PPBP (Fig. 5b). Moreover, the increasing tendency of these copolymers was similar to those of the S-PPBP and Nafion®112 membranes. In our previous study, S-PPBP random copolymers (0.86–2.40 meq g−1) exhibited lower λ values than those of the S-PPBP homopolymer and Nafion®112 at RH values above 65%.20 The low λ values and weak humidity dependence was believed to arise from the disordered morphology of the random copolymers. S-6X exhibited high-λ properties with suppressed WU, suggesting that the multiblock copolymers likely exhibited a well-defined morphology, which led to the developed hydrophilic domains facilitating local, dense water sorption. These results indicate that the multiblock structures developed effective water-sorption capabilities rather than the random copolymers. After immersion in water, the S-6X, S-PPBP, and Nafion®112 membranes exhibited λ values of 14–30, 31, and 14, respectively. The IEC dependence of λ intensified under fully hydrated conditions owing to the sorption of free water.
The swelling ratios in the through-plane (Δt) and in-plane (Δl) directions were evaluated by comparing the fully hydrated and dry membrane states (Table 1). S-6X exhibited Δt and Δl values of 7.4–40% and 5.5%–8.3%, respectively, at 80 °C. Similar to the WU values, the swelling ratios of S-6X were significantly lower than those of the S-PPBP homopolymer. S-6X exhibited anisotropic swelling behaviour (Δt/l = 1.3–7.0), indicating greater swelling in the through-plane direction. This tendency was similar to that of S-PPBP (Δt/l = 6.4). McGrath et al. reported that random copolymers with a disulfonated-PAES-based structure exhibited isotropic swelling behaviour, and that multiblock copolymers with a similar basic structure exhibited anisotropic swelling.36,37 Moreover, the through-plane swelling ratios of their multiblock copolymers increased with increasing block length, suggesting that this tendency is related to the development of an ordered morphology. Similarly, the anisotropic swelling behaviour of S-6X was presumably due to the phase-separated morphology created by both the multiblock structure and the pendant structure of the hydrophilic domain. The diminished in-plane swelling of S-6X can assist in improving dimensional stability during the wet–dry cycling of fuel cells.
:1 exhibited the highest conductivities (1.9 × 10−3–1.3 × 10−1 S cm−1), which were comparable to those of Nafion®112 and the S-PPBP homopolymer.
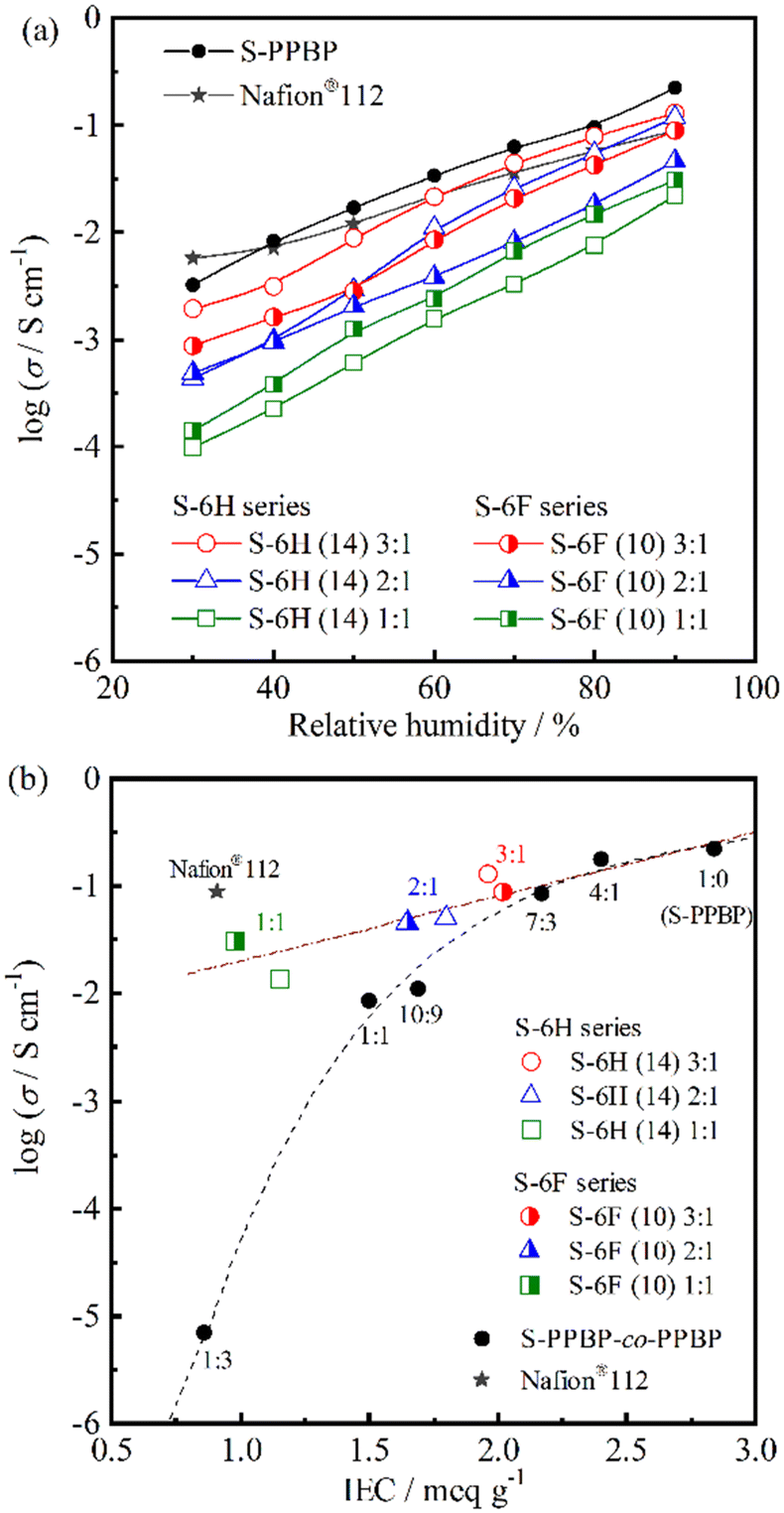 | ||
| Fig. 6 (a) Proton conductivity of S-6H, S-6F, and S-PPBP as a function of relative humidity at 80 °C. (b) Relationship between proton conductivity and IEC of S-6H, S-6F, and S-PPBP-co-PPBP at 80 °C and 90% RH. | ||
To gain further insight into the effects of the multiblock structures on proton-transporting properties, the proton conductivities of S-6X were plotted as a function of the IEC (Fig. 6b). An S-PPBP homopolymer and random copolymers (S-PPBP-co-PPBP x:y) with controlled IECs of 0.86–2.84 meq g−1 were used as controls.20 S-6X showed a linear increase in the proton conductivity with increasing IEC. However, S-PPBP-co-PPBP showed a remarkably different tendency. Our previous study indicated that the proton conductivity of random copolymers is strongly related to their IEC values. S-PPBP-co-PPBP with IEC values below 1.69 meq g−1 exhibited a rapid decrease in conductivity with decreasing IEC. The conductivity of S-6H (14) 2:1 and S-6F (10) 2:1 at 80 °C and 90% RH was 5.0 × 10−2 and 4.6 × 10−2 S cm−1, respectively; these values were four times higher than that of S-PPBP-co-PPBP 10:9 (1.1 × 10−2 S cm−1) with a comparable IEC. Furthermore, the conductivity of S-6H (14) 1:1 and S-6F (10) 1:1 was approximately three-orders-of-magnitude greater than that of S-PPBP-co-PPBP 1:3 (6.9 × 10−6 S cm−1). These results indicate that our multiblock structure helped prevent the drastic decrease in conductivity under low-IEC conditions.
Interestingly, the fits to the S-6H and S-6F conductivity data overlapped those of S-PPBP-co-PPBP 4:1 and the S-PPBP homopolymer (Fig. 6b). In contrast, the random copolymers showed significantly decreased conductivity with decreasing IEC. The major difference in these tendencies could be ascribed to morphological effects. The multiblock copolymers evidently formed continuous, interconnected ionic channels through micro-phase separation, which facilitated efficient proton conduction, in contrast to that in the fully sulfonated S-PPBP homopolymer. The low conductivity of the random copolymers was presumably due to morphological barriers such as poorly connected ionic channels and the isolated dead-end-channel pockets created by their ambiguous morphology. Several groups have reported higher proton conductivities of nanostructured polymer electrolytes than those of random copolymer counterparts.38–43 Our multiblock copolymers exhibited higher conductivities than those reported previously. These results indicate that the proton conductivity of the nanostructured PEMs was considerably affected by the local structures of the ionic channels.
000, IEC = 2.79 meq g−1) with a molecular weight close to those of the S-6X specimens, was used as a control. Under ambient conditions, S-6X exhibited σmax, εmax, and E values of 48.5–79.9 MPa, 5.5–71.6%, and 1504–2727 MPa, respectively. All S-6X membranes exhibited 3–70% higher strengths than that of S-PPBP. In the hydrated state (80 °C, 90% RH), the S-6X specimens exhibited σmax, εmax, and E values of 19.3–40.7 MPa, 21.6–210%, and 283–1004 MPa, respectively. All the S-6X and S-PPBP membranes exhibited lower strength, lower modulus, and higher elongation values than those of the counterparts in the ambient state.
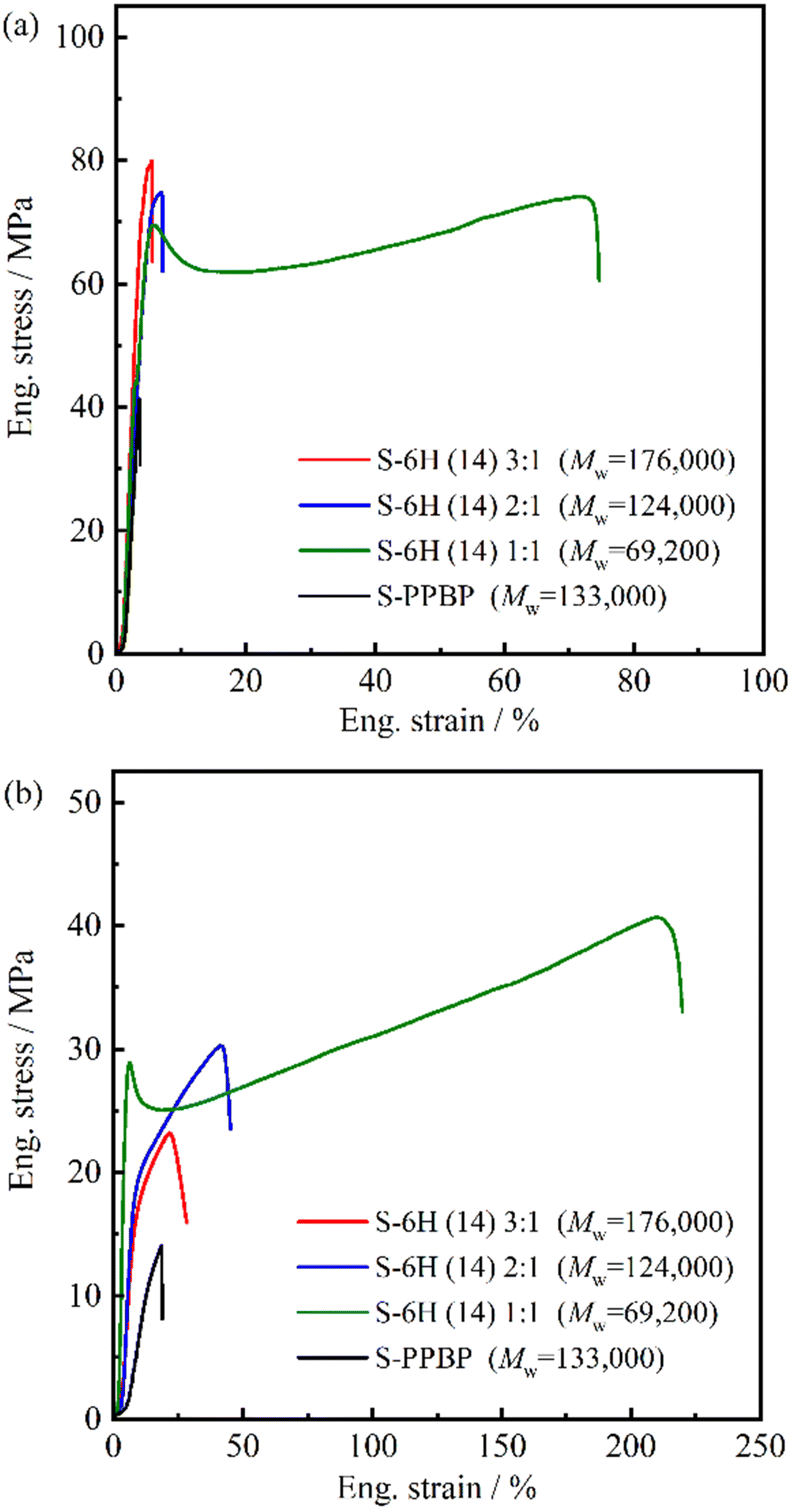 | ||
| Fig. 7 Stress–strain curves of S-6H and S-PPBP acquired under (a) ambient conditions (RT, no extra humidification) and (b) at an elevated temperature and humidity (80 °C, 90% RH). | ||
| Sample | Ambient atmosphere | 80 °C, 90% RH | ||||
|---|---|---|---|---|---|---|
| σ max/MPa | ε max/% | E/MPa | σ max/MPa | ε max/% | E/MPa | |
| a Tensile strength (σmax), ultimate elongation (εmax), and Young's modulus (E) determined from mechanical tensile tests under ambient atmosphere condition (r.t., non extra-humidified) and high temperature with humidification condition (80 °C, 90%RH). | ||||||
| S-6H (14) 3:1 |
79.9 | 5.5 | 2727 | 23.2 | 21.6 | 336 |
| S-6H (14) 2:1 |
74.8 | 6.9 | 2074 | 30.3 | 41.4 | 466 |
| S-6H (14) 1:1 |
67.9 | 71.6 | 2499 | 40.7 | 210.0 | 1004 |
| S-6F (10) 3:1 |
57.6 | 11.7 | 2189 | 19.3 | 28.6 | 283 |
| S-6F (10) 2:1 |
55.6 | 26.0 | 1504 | 33.4 | 68.0 | 649 |
| S-6F (10) 1:1 |
48.5 | 57.0 | 1857 | 23.5 | 95.9 | 589 |
| S-PPBP | 41.4 | 3.6 | 1816 | 14.1 | 18.7 | 130 |
| Nafion®112 | 31.0 | 200.2 | 200 | 11.0 | 333.7 | 47 |
Generally, hydrated PEMs with tethered acid groups tend to exhibit higher elongation and considerably lower mechanical strength than those of the dry equivalents, which is attributed to the plasticisation of the polymer matrix. In weakly hydrated Nafion membranes, strong electrostatic interactions between the sulfonic acid sites and water are believed to act as physical crosslinks, thereby improving the membrane stiffness.44–47 However, in the highly hydrated state, the high fraction of water domains weakens the ionic interactions, and the plasticisation effect dominates the mechanical response.45–51 In particular, the drastic decrease in mechanical strength under highly hydrated conditions has hindered the development of hydrocarbon-based PEMs. The tensile tests suggested that the S-6X samples maintained a mechanical strength of ∼20 MPa even under highly hydrated conditions, which is superior to that of conventional hydrocarbon-based PEMs. This was presumably due to the cooperative interplay between the diminished hydration and multiblock architectures of the designed copolymers. Recently, several groups have reported multiblock copolymers exhibiting unique physical properties.52–60 Diblock copolymers are susceptible to brittle fracture because their bonds are established only through van der Waals interactions and chain entanglements. Spontak et al. reported that symmetric lamellar (AB)n multiblock copolymers are capable of sustaining an applied load to considerably higher strain levels before failing compared to those of diblock copolymers, owing to an increase in the number of bridged middle blocks.52 Bates et al. found that when the number of blocks in a lamellar-structured copolymer increases, the bridging and entangled (knotted) looping configurations formed by the middle blocks effectively prevent the domain fracture caused by chain pull-out, thereby increasing the toughness and maximum strain.53–58 Based on the results from the two preceding studies, the higher tensile strength of the S-6X membranes than that of conventional PEMs under both ambient and humid conditions can mainly be attributed to the domain bridging and looping of the middle blocks in multiblock configurations. A comparison of S-6X and the S-PPBP homopolymer with similar molecular weights corroborates this explanation.
Interestingly, the elongation properties of S-6X were highly dependent on the chain length ratio of the S-PPBP and PAEK blocks. In particular, S-6H (14) 1:1 and S-6F (10) 1:1 exhibited yielding and ductility, whereas their 2:1 and 3:1 counterparts were brittle. The 1:1 membrane exhibited stretching behaviour after the yield point, followed by strain hardening prior to rupture. This ductile deformation behaviour is similar to that of thermoplastic elastomers, which comprise hard segments with constrained molecules, which prevent plastic deformation, and soft segments with rubber elasticity. As mentioned above, the stretching behaviour of S-6X after the yield point likely depended on two important relaxation features: the relaxation of rapid crack propagation owing to the plasticising effect of water in the hydrophilic domains, and the relaxation of entanglements and reorientations induced by the entangled loop structure formed in the flexible hydrophobic domains. Only the 1:1 membranes with the lowest water content among the specimens exhibited elastomeric behaviour, suggesting that the hydrophobic block conformation dominated the elongation properties of S-6X. In terms of the mechanical properties of PEMs, Diet et al. established the early fibrillar model for Nafion membranes, and Liu et al. developed the 3D bundle-cluster model for both Nafion and sulfonated PAES copolymers.44,60,61 The authors suggested that the primary relaxation mechanism in PEMs beyond yielding may involve continuous unchaining in the bundle of polymer backbone aggregates that primarily comprise the hydrophobic phase of the copolymer. Based on these models, the hydrophobic morphology was considered to significantly influence the mechanical properties of our S-6X multiblock-based PEMs; additional investigations focussing on the morphological effects are currently underway.
3.3. Morphology of S-6X membranes
The surface morphologies of the S-6X membranes were examined through e-AFM observations under ambient (RT, 30–50% RH) and humid conditions (RT, 85% RH). Representative phase-mapping images of the S-6H membranes are shown in Fig. 8, and all the phase-, topography-, and current-mapping images are shown in Fig. S14 and S15 (ESI†). The dark domains in the phase images were derived from the soft-polymer moieties and likely corresponded to the hydrophilic domains in the PEMs created by the plasticisation effect of adsorbed water. Certain S-6X membranes with fewer hydrophilic components showed the opposite correspondence under ambient conditions, presumably owing to their inferior water-sorption properties. Each domain was identified by comparing the current- and phase-mapping images. | ||
| Fig. 8 e-AFM observations of S-6H membranes. Phase images of (a) S-6H (14) 3:1, (b) S-6H (14) 2:1, and (c) S-6H (14) 1:1 acquired under humid conditions (RT, 85% RH); scan size, 1000 × 1000 nm2. (d) Phase (yellow coloured), (e) current-mapping (blue coloured), and (f) stacked images of S-6H (14) 3:1 obtained under ambient conditions; scan size, 500 × 500 nm2. | ||
The phase images of the S-6X membranes revealed a distinct microphase separation between the hydrophilic and hydrophobic domains. The membranes had a domain size of ∼16–36 nm, with the S-6X having highly hydrophilic components with a larger domain size. Interestingly, no significant volume changes were observed with respect to the domain size in all the S-6X samples at 85% RH compared to those under ambient conditions. This result is consistent with the swelling rate tendency, suggesting that the hydrophobic domain that formed the microphase-separated structure suppressed significant WU and structural failure. The current-mapping images helped investigate the relationship between the surface morphology and proton conduction.62,63 The bright domains in the current-mapping images were derived from the proton-conducting regions, that is, the hydrophilic domains. Continuous hydrophilic domains were observed in all the S-6X membranes. In particular, a morphology with continuous hydrophilic domains surrounding spherical hydrophobic zones was observed in the S-6X specimens with highly hydrophilic components (2:1 and 3:1), as illustrated in Fig. 8. The high proton conductivities of S-6X were ostensibly induced by the formation of hydrophilic domains with a continuous microphase-separated structure. Therefore, multiblock copolymerisation was visually demonstrated to be effective for forming continuous proton-conducting paths as well as maintaining the hydrated structure.
The internal morphologies of S-6X were examined by SAXS analysis. Scattering profiles of fully hydrated S-6H and S-6F membranes were acquired (Fig. 9). The SAXS profiles of S-6H and S-PPBP showed a scattering peak in the wide-angle region (q = 1.61–1.92 nm−1). This peak – also known as the ionomer peak – is the hallmark feature of nanophase-separated ionic aggregates (that is, mutiplets of ion pairs) that generate interparticle interferences that yield the characteristic scattering maximum.38,64 The mean separation distances between ionic domains in S-6H (14) 3:1, 2:1, 1:1, and S-PPBP, which were calculated using the relative position of the SAXS maxima with Bragg's law (d = 2π/q), were 3.90, 3.72, 3.27, and 3.72 nm, respectively. The SAXS profiles of S-6H (14) 2:1 and 1:1 showed an additional low-angle scattering intensity at q = 0.220 and 0.361 nm−1, which can be ascribed to structural correlations between the hydrophilic–hydrophobic block domains.38 The interdomain distance in the S-6H (14) 2:1 and 1:1 specimens, estimated using Bragg's law, was 28.6 and 17.4 nm, respectively. These results correlate decently with those obtained by e-AFM (Fig. 8). The wide-angle scattering peak did not appear in the S-6H (14) 3:1 profile, probably because its domain size was larger than that in the 2:1 and 1:1 specimens and exceeded the measurable q region. Similar to the S-6H profiles, the SAXS spectra of the S-6F membranes displayed two scattering peaks: an ionomer peak located at q = 1.73–2.03 nm−1 (d = 3.10–3.63 nm), and a hydrophilic–hydrophobic domain peak located at q = 0.238–0.365 nm−1 (d = 17.2–26.4 nm). These results indicate that the remarkable structural organisation achieved via self-assembly of the hydrophilic–hydrophobic blocks, which formed amplified large-scale-ordered domains, was expressed on both the surface and interior of the S-6X membranes. Additionally, the phase-separated structure of S-6X was evidently more affected by the block ratio than the substituent in the hydrophobic segments.
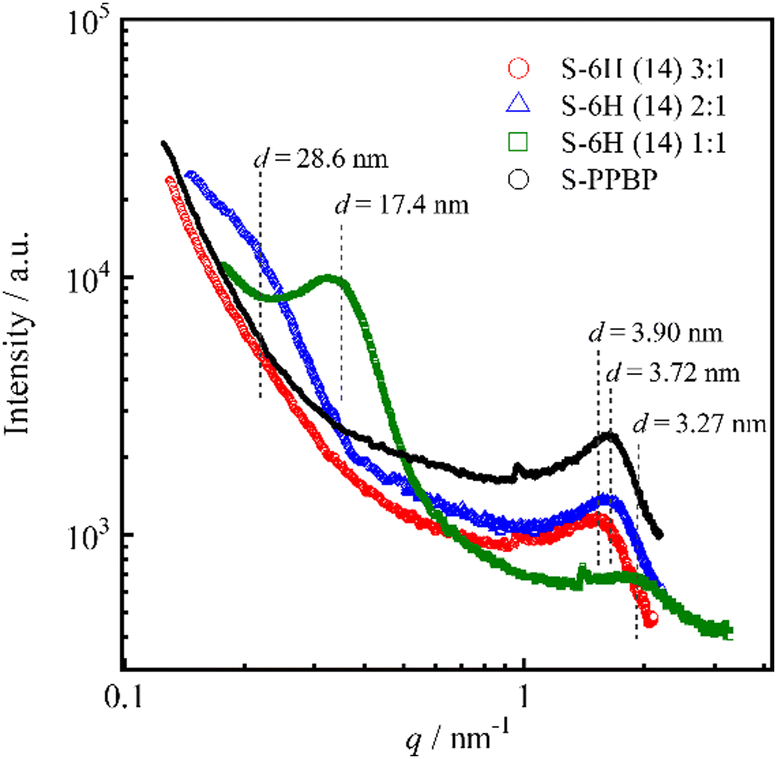 | ||
| Fig. 9 SAXS profiles of S-6H and S-PPBP membranes. Dotted lines indicate the interdomain spacing calculated using Bragg's law. | ||
3.4. Fuel cell performance
Single-cell tests were conducted using the S-6H, S-6F, and S-PPBP membranes at a cell temperature of 80 °C under fully hydrated conditions (100% RH; gas temperature, 80 °C) for both electrodes. Fuel-cell polarisation curves of the membranes were collected (Fig. 10). The IR-free IV curves evaluated from the high frequency resistance (HFR), which is equal to the ohmic resistance including membrane resistance and contact resistance, are replotted in Fig. 11.65 The HFR were found to be 12–31 mΩ cm2 for S-6H and 15–38 mΩ cm2 for S-6F. All the HFR values of S-6X were similar to those expected from the proton conductivity and membrane thickness (about 50 μm), suggesting that the contact resistance with the catalyst layer is low. The open-circuit voltage (OCV) of the S-6H (14) 3:1, 2:1, 1:1, and S-PPBP membranes was 0.98, 1.00, 1.00, and 0.99 V, respectively, at 100% RH. Similarly, the OCV of the S-6F membranes ranged from 0.97 to 1.00 V. All S-6X membranes exhibited a high OCV (>0.95 V), indicating the low hydrogen-gas permeability of the system from the anode to the cathode through the membrane.
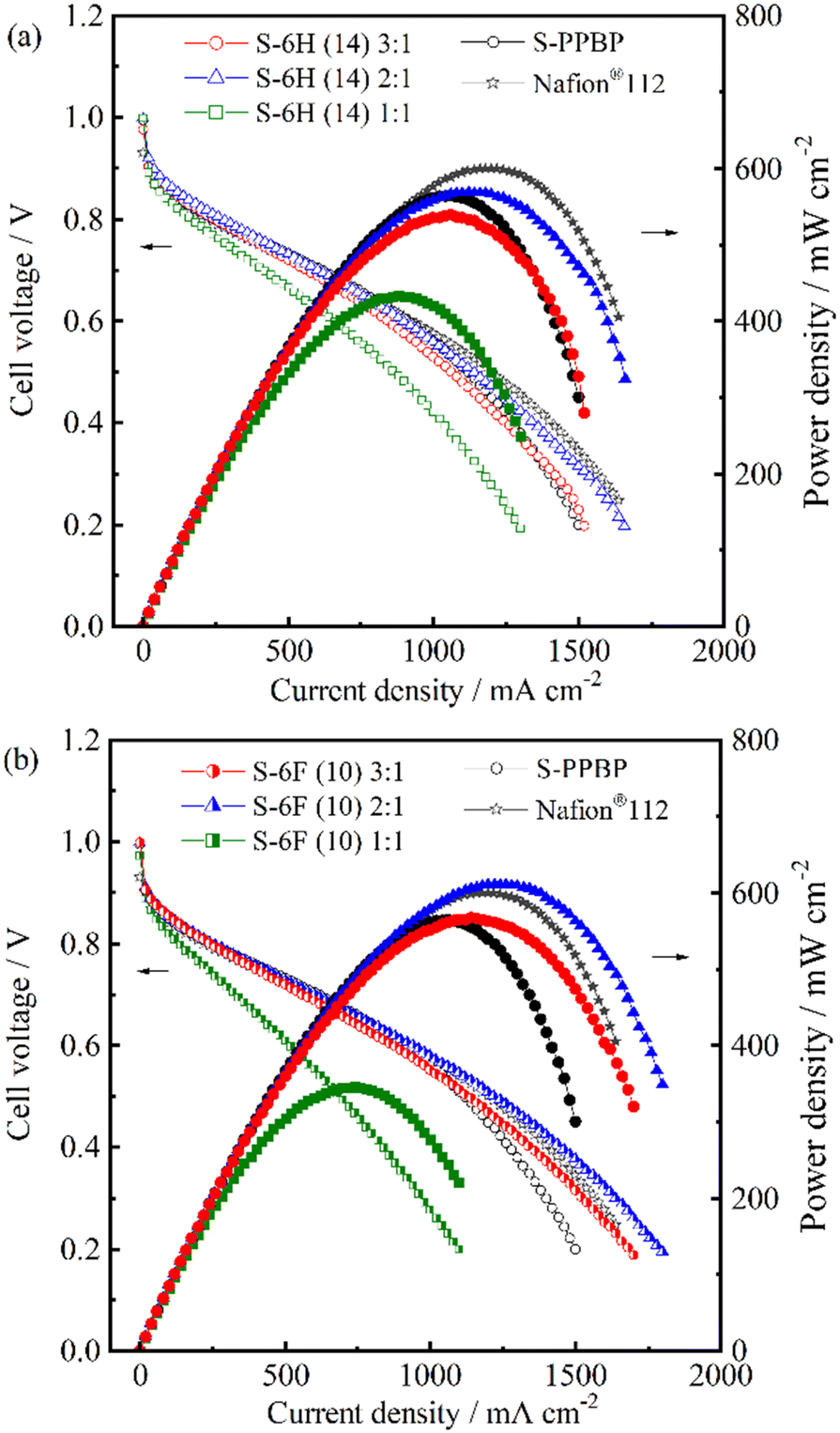 | ||
| Fig. 10 Cell voltage and power density as a function of current density for a 5 cm2-sized PEFC with H2 (500 mL min−1, 0.1 MPaG) and air (1000 mL min−1, 0.1 MPaG) feeds for (a) S-6H and (b) S-6F membranes at 80 °C and 100% RH. | ||
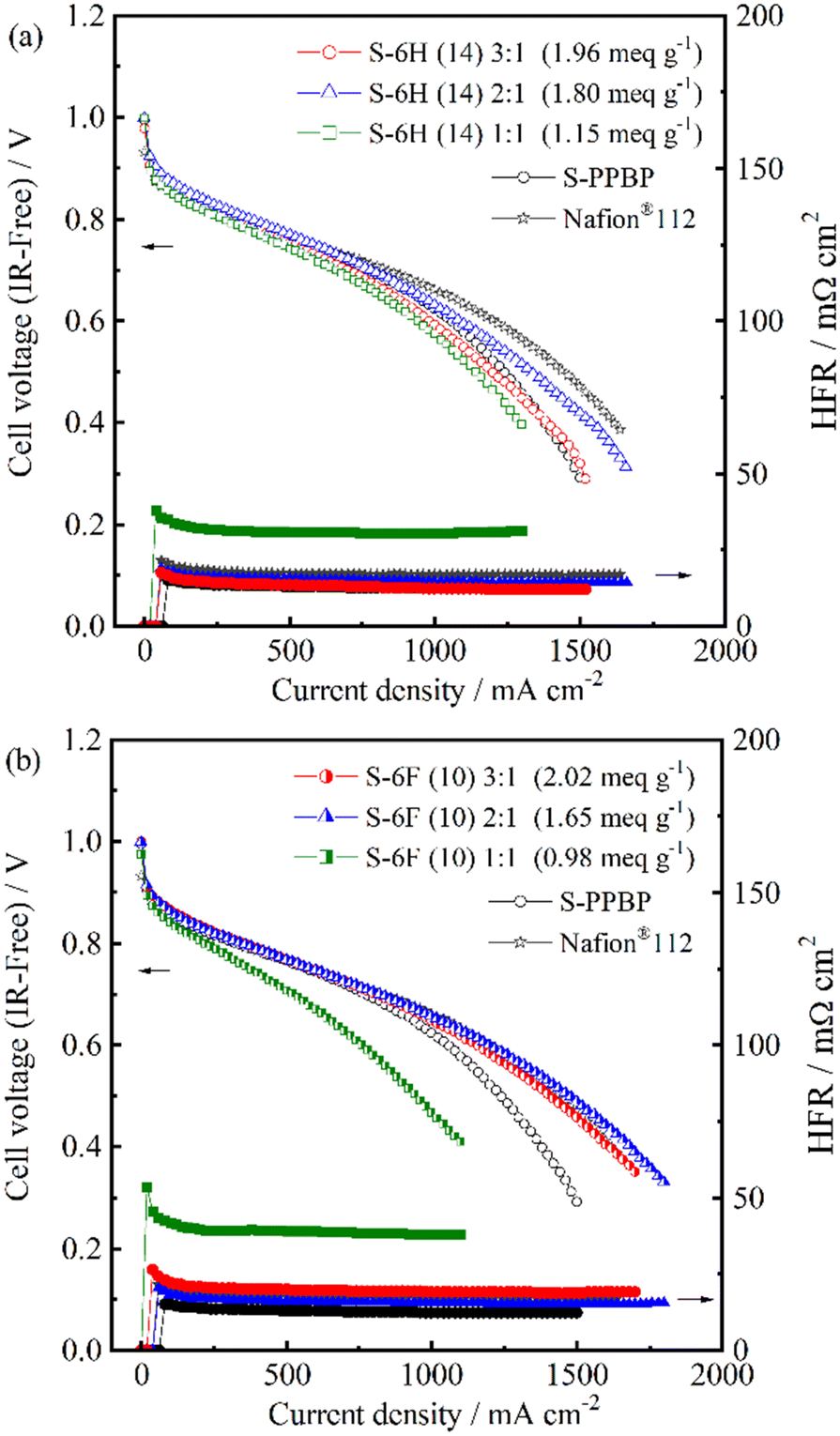 | ||
| Fig. 11 IR-free cell voltage and HFR as a function of current density for a 5 cm2-sized PEFC with H2 (500 mL min−1, 0.1 MPaG) and air (1000 mL min−1, 0.1 MPaG) feeds for (a) S-6H and (b) S-6F membranes at 80 °C and 100% RH. | ||
The maximum power density and limiting current density of S-6H (14) 2:1 were 569 mW cm−2 and 1660 mA cm−2, respectively, which were higher than those of the S-PPBP homopolymer (564 mW cm−2; 1500 mA cm−2). Interestingly, S-6H (14) 2:1 exhibited the highest fuel-cell performance among the S-6H membranes. This tendency was also displayed by the S-6F membranes, with S-6F (14) 2:1 showing the best performance (611 mW cm−2; 1800 mA cm−2) among all the membranes, including Nafion®112 (600 mW cm−2; 1640 mA cm−2). Compared to conventional hydrocarbon-based PEMs, the S-6X membranes exhibited superior fuel-cell performance, despite their relatively low IEC, and did not exhibit a simple dependence on the IEC. Because the fuel cell tests were performed using MEAs, factors other than the proton conductivity of the membranes probably acted as an overpotential. The morphology of the PEMs was likely a crucial factor that influenced the cell performance, with the effects being particularly evident in the 2:1 membranes. In conclusion, S-6X membranes with hydrophilic components exhibited outstanding fuel-cell performance under hydrated conditions.
To develop advanced materials with block architectures for PEFC applications, the structure–property relationships of PEM materials with nanoscale morphologies must be elucidated. Careful examination of the multiblock structure through cutting-edge nanometre-scale microstructural analysis are under progress.
4. Conclusions
As an alternative synthesis methodology to SNAr polycondensation, a novel Ni(0)-coupling-based procedure was implemented in this study to synthesise nanostructured PEM materials. A series of hydrophilic–hydrophobic multiblock copolymers (S-6X) with simple control of the IEC and unit ratios was prepared using a four-step procedure. A multiblock structure with a moderately periodic architecture was generated via direct one-pot copolymerisation by leveraging the presence of a hydrophobic oligomer with a controllable block length.The S-6X membranes achieved cooperative improvements in proton conductivity and mechanical properties owing to their multiblock structure. In contrast to previously reported hydrocarbon-based PEMs, which show a rapid nonlinear decrease in conductivity with decreasing IEC, the S-6X specimens showed a linear increase in conductivity with increasing IEC, indicating the potency of our multiblock structure in maintaining high conductivity under low-IEC conditions. In particular, the conductivity of S-6H (14) 2:1 and S-6F (10) 2:1 was approximately four times higher than that of S-PPBP random copolymers, and these specimens exhibited a tensile strength exceeding 30 MPa under hydrated conditions, presumably because of their multiblock architectures. SAXS profiles, which were acquired for morphological analysis, indicated that the S-6X membranes formed 10–30 nm-sized phase-separated hydrophilic–hydrophobic domains and 3–4 nm-sized nanophase-separated ionic aggregates. Moreover, direct observations of the proton-conducting regions via e-AFM revealed that the hydrophilic domains in S-6X formed a continuous phase-separated structure, indicating effective proton conduction in the absence of excess sorbed water.
Single-cell tests indicated that S-6X showed excellent fuel-cell performance under fully hydrated conditions. S-6H (14) 2:1 and S-6F (10) 2:1 exhibited optimal maximum-power-density values of 569–611 mW cm−2 and limiting current densities of 1660–1800 mA cm−2, which are comparable to those of Nafion membranes, which emphasises the potential of the S-6X PEMs for PEFC applications. Morphology was suspected to considerably influence the PEM properties; clarifying the structure–property relationships will assist in further improving PEM materials.
Finally, the novelty of this work is the establishment of a simple synthetic method for synthesizing multiblock PEMs with both high conductivity and mechanical strength. Our synthetic method has a wide selectivity of applicable structures. Further improvement in properties can be expected by tuning the backbone structure and the side chain structure, and by changing the polar group, it can be applied to an anion exchange type electrolyte membrane. We are confident that the results of our work will contribute to the further development of materials research in a wide range of applications, such as electrolytes for electrolytic reactions in addition to fuel cells. Also, systematic measurement data of multiblock PEMs were obtained in this study. In the future, more detailed structural analysis is expected to lead to direct elucidation of the proton conduction mechanism in PEM, which has not been achieved so far. Further structural analysis experiments are now under progress.
Author contributions
M. Y.-H. made substantially contributed to the design of this work, the data analysis, and original manuscript drafting. M. Y.-F. and Y. T. contributed to the interpretation of the results. M. R. substantially contributed to the study conceptualization and supervised the conduct of this study. All authors have approved the submitted version of the manuscript and agreed to be accountable for any part of the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the New Energy and Industrial Technology Development Organisation (NEDO) and a JSPS KAKENHI grant (21K05193).References
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587–4612 CrossRef CAS PubMed.
- C. H. Park, C. H. Lee, M. D. Guiver and Y. M. Lee, Prog. Polym. Sci., 2011, 36, 1443–1498 CrossRef CAS.
- Y. A. Elabd and M. A. Hickner, Macromolecules, 2011, 44, 1–11 CrossRef CAS.
- N. Li and M. D. Guiver, Macromolecules, 2014, 47, 2175–2198 CrossRef CAS.
- D. J. Kim, M. J. Jo and S. Y. Nam, J. Ind. Eng. Chem., 2015, 21, 36–52 CrossRef CAS.
- G. He, Z. Li, J. Zhao, S. Wang, H. Wu, M. D. Guiver and Z. Jiang, Adv. Mater., 2015, 27, 5280–5295 CrossRef CAS PubMed.
- D. W. Shin, M. D. Guiver and Y. M. Lee, Chem. Rev., 2017, 117, 4759–4805 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- J. K. Kim, S. Y. Yang, Y. Lee and Y. Kim, Prog. Polym. Sci., 2010, 35, 1325–1349 CrossRef CAS.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- G. Titvinidze, K.-D. Kreuer, M. Schuster, C. C. de Araujo, J. P. Melchior and W. H. Meyer, Adv. Funct. Mater., 2012, 22, 4456–4470 CrossRef CAS.
- M. G. Dhara and S. Banerjee, Prog. Polym. Sci., 2010, 35, 1022–1077 CrossRef CAS.
- J. R. Rowlett, Y. Chen, A. T. Shaver, O. Lane, C. Mittelsteadt, H. Xu, M. Zhang, R. B. Moore, S. Mecham and J. E. McGrath, Polymer, 2013, 53, 6305–6313 CrossRef.
- Q. Li, Y. Chen, J. R. Rowlett, J. E. McGrath, N. H. Mack and Y. S. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 5779–5788 CrossRef CAS PubMed.
- J. R. Rowlett, Y. Chen, A. T. Shaver, G. B. Fahs, B. J. Sundell, Q. Li, Y. S. Kim, P. Zelenay, R. B. Moore, S. Mecham and J. E. McGrath, J. Electrochem. Soc., 2014, 161, F535–F543 CrossRef CAS.
- K. H. Lee, J. Y. Chu, A. R. Kim and D. J. Yoo, ACS Appl. Mater. Interfaces, 2018, 10, 20835–20844 CrossRef CAS PubMed.
- T. Dong, J. Hu, M. Ueda, Y. Wu, X. Zhang and L. Wang, J. Mater. Chem. A, 2016, 4, 2321–2331 RSC.
- I. Tonozuka, M. Yoshida, K. Kaneko, Y. Takeoka and M. Rikukawa, Polymer, 2011, 52, 6020–6028 CrossRef CAS.
- T. Oshima, M. Yoshizawa-Fujita, Y. Takeoka and M. Rikukawa, ACS Omega, 2016, 1, 939–942 CrossRef CAS PubMed.
- M. Yoshida-Hirahara, S. Takahashi, M. Yoshizawa-Fujita, Y. Takeoka and M. Rikukawa, RSC Adv., 2020, 10, 12810–12822 RSC.
- H. Ghassemi and J. E. McGrath, Polymer, 2004, 45, 5847–5854 CrossRef CAS.
- S. Seesukphronrarak and A. Ohira, Chem. Commun., 2009, 4744–4746 RSC.
- S. G. Jo, T.-H. Kim, S. J. Yoon, S.-G. Oh, M. S. Cha, H. Y. Shin, J. M. Ahn, J. Y. Lee and Y. T. Hong, J. Membr. Sci., 2016, 510, 326–337 CrossRef CAS.
- H. Y. Shin, M. S. Cha, S. H. Hong, T.-H. Kim, D.-S. Yang, S.-G. Oh, J. Y. Lee and Y. T. Hong, J. Mater. Chem. A, 2017, 5, 12285–12296 RSC.
- S. Chandra Sutradhar, M. M. Rahman, F. Ahmed, T. Ryu, S. Yoon, S. Lee, J. Kim, Y. Lee, Y. Jin and W. Kim, Int. J. Hydrogen Energy, 2019, 44, 11311–11320 CrossRef CAS.
- E. C. Hagberg, D. A. Olson and V. V. Sheares, Macromolecules, 2004, 37, 4748–4754 CrossRef CAS.
- W. Xu, Y. Ding, T. Yang, Y. Yu, R. Huang, Z. Zhu, F. Hao and H. Hou, Macromolecules, 2017, 50, 9760–9772 CrossRef CAS.
- I. Colon and D. R. Kelsey, J. Org. Chem., 1986, 51, 2627–2637 CrossRef CAS.
- K. Shiino, T. Otomo, T. Yamada, H. Arima, K. Hiroi, S. Takata, J. Miyake and K. Miyatake, ACS Appl. Polym. Mater., 2020, 2, 5558–5565 CrossRef CAS.
- J. Miyake, T. Watanabe, H. Shintani, Y. Sugawara, M. Uchida and K. Miyatake, ACS Mater. Au, 2021, 1, 81–88 CrossRef CAS PubMed.
- A. F. Nugraha, S. Kim, S.-H. Shin, H. Lee, D. Shin and B. Bae, Macromolecules, 2020, 53, 10538–10547 CrossRef CAS.
- Z. Li, J. Guo, J. Zheng, T. A. Sherazi, S. Li and S. Zhang, Macromolecules, 2020, 53, 10998–11008 CrossRef CAS.
- F. Wijaya, S. Woo, H. Lee, A. F. Nugraha, D. Shin and B. Bae, J. Membr. Sci., 2022, 645, 120203 CrossRef CAS.
- J. Y. Lee, D. M. Yu, T.-H. Kim, S. J. Yoon and Y. T. Hong, J. Membr. Sci., 2015, 492, 209–219 CrossRef CAS.
- B. Date, J. Han, S. Park, E. J. Park, D. Shin, C. Y. Ryu and C. Bae, Macromolecules, 2018, 51, 1020–1030 CrossRef CAS.
- H.-S. Lee, A. Roy, O. Lane, S. Dunn and J. E. McGrath, Polymer, 2008, 49, 715–723 CrossRef CAS.
- Y. Chen, J. R. Rowlett, C. H. Lee, O. R. Lane, D. J. VanHouten, M. Zhang, R. B. Moore and J. E. McGrath, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2301–2310 CrossRef CAS.
- L. Assumma, H.-D. Nguyen, C. Iojoiu, S. Lyonnard, R. Mercier and E. Espuche, ACS Appl. Mater. Interfaces, 2015, 7, 13808–13820 CrossRef CAS PubMed.
- J. Aboki, B. Jing, S. Luo, Y. Zhu, L. Zhu and R. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 1173–1186 CrossRef CAS PubMed.
- X. Zhang, T. Dong, Y. Pu, T. Higashihara, M. Ueda and L. Wang, J. Phys. Chem. C, 2015, 119, 19596–19606 CrossRef CAS.
- A. Sannigrahi, S. Takamuku and P. Jannasch, Int. J. Hydrogen Energy, 2014, 39, 15718–15727 CrossRef CAS.
- H. Hu, T. Dong, Y. Sui, N. Li, M. Ueda, L. Wang and X. Zhang, J. Mater. Chem. A, 2018, 6, 3560–3570 RSC.
- J. R. Rowlett, V. Lilavivat, A. T. Shaver, Y. Chen, A. Daryaei, H. Xu, C. Mittelsteadt, S. Shimpalee, J. S. Riffle and J. E. McGrath, Polymer, 2017, 122, 296–302 CrossRef CAS.
- D. Liu, S. Kyriakides, S. W. Case, J. J. Lesko, Y. Li and J. E. McGrath, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 1453–1465 CrossRef CAS.
- F. Bauer, S. Denneler and M. Willert-Porada, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 786–795 CrossRef CAS.
- M. B. Satterfield and J. B. Benziger, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 11–24 CrossRef CAS.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- J. Han, L. Zhu, J. Pan, T. J. Zimudzi, Y. Wang, Y. Peng, M. A. Hickner and L. Zhuang, Macromolecules, 2017, 50, 3323–3332 CrossRef CAS.
- J. Pan, L. Zhu, J. Han and M. A. Hickner, Chem. Mater., 2015, 27, 6689–6698 CrossRef CAS.
- A. Ghorai, S. Roy, S. Das, H. Komber, M. M. Ghangrekar, B. Voit and S. Banerjee, ACS Appl. Polym. Mater., 2020, 2, 2967–2979 CrossRef CAS.
- A. K. Mohanty, E. A. Mistri, S. Banerjee, H. Komber and B. Voit, Ind. Eng. Chem. Res., 2013, 52, 2772–2783 CrossRef CAS.
- R. J. Spontak and S. D. Smith, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 947–955 CrossRef CAS.
- T. J. Hermel, S. F. Hahn, K. A. Chaffin, W. W. Gerberich and F. S. Bates, Macromolecules, 2003, 36, 2190–2193 CrossRef CAS.
- Y. Mori, L. S. Lim and F. S. Bates, Macromolecules, 2003, 36, 9879–9888 CrossRef CAS.
- L. S. Lim, T. Harada, M. A. Hillmyer and F. S. Bates, Macromolecules, 2004, 37, 5847–5850 CrossRef CAS.
- I. Lee, T. R. Panthani and F. S. Bates, Macromolecules, 2013, 46, 7387–7398 CrossRef CAS.
- T. R. Panthani and F. S. Bates, Macromolecules, 2015, 48, 4529–4540 CrossRef CAS.
- E. Grune, M. Appold, A. H. E. Mueller, M. Gallei and H. Frey, ACS Macro Lett., 2018, 7, 807–810 CrossRef CAS PubMed.
- Y. Matsumiya, H. Watanabe, A. Takano and Y. Takahashi, Macromolecules, 2013, 46, 2681–2695 CrossRef CAS.
- P. C. van der Heijden, L. Rubatat and O. Diat, Macromolecules, 2004, 37, 5327–5336 CrossRef CAS.
- D. Liu, M. A. Hickner, S. W. Case and J. J. Lesko, J. Eng. Mater. Technol., 2006, 128, 503–508 CrossRef CAS.
- N. Takimoto, S. Takamuku, M. Abe, A. Ohira, H.-S. Lee and J. E. McGrath, J. Power Sources, 2009, 194, 662–667 CrossRef CAS.
- A. Ohira, S. Kuroda, H. F. M. Mohamed and B. Tavernier, Phys. Chem. Chem. Phys., 2013, 15, 11494–11500 RSC.
- L. J. Anderson, X. Yuan, G. B. Fahs and R. B. Moore, Macromolecules, 2018, 51, 6226–6237 CrossRef CAS.
- C. Immerz, B. Bensmann, P. Trinke, M. Suermann and R. Hanke-Rauschenbach, J. Electrochem. Soc., 2018, 165, F1292–F1299 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00244f |
| This journal is © The Royal Society of Chemistry 2023 |