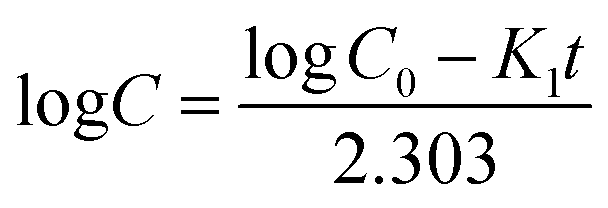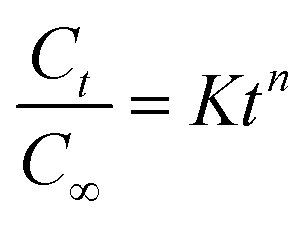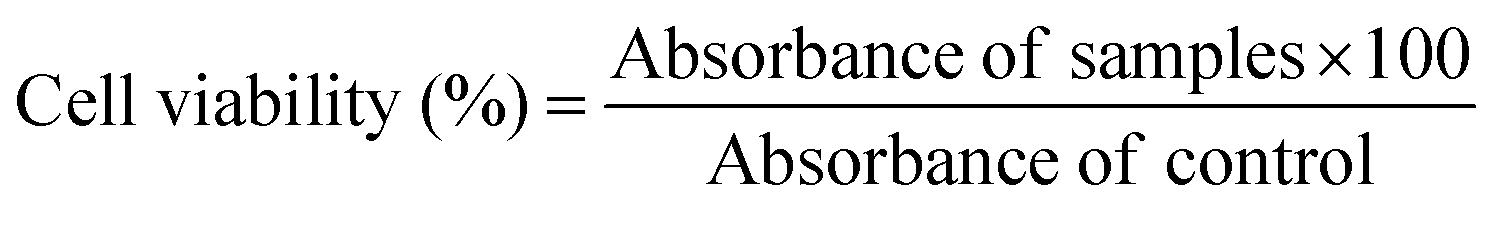DOI:
10.1039/D3MA00328K
(Paper)
Mater. Adv., 2023,
4, 5224-5237
Epidermal growth factor receptor targeted doxorubicin and vitexin loaded niosomes for enhanced breast cancer therapy†
Received
26th June 2023
, Accepted 18th September 2023
First published on 20th September 2023
Abstract
Breast cancer is one of the most frequently diagnosed cancers in women, and its current treatment modalities are limited by their non-specificity and adverse side effects. We evaluated the efficacy of cetuximab (C) conjugated doxorubicin (D) and vitexin (V) loaded niosomes (NIODVC) as a targeted delivery system for breast cancer. The combined release of doxorubicin and vitexin by antibody conjugated niosomes followed a biphasic release resulting in enhanced cytotoxicity (63%) and increased therapeutic efficiency compared to combined drug formulations (53%) without antibody. NIODVC demonstrated efficient cellular uptake in vitro, and its efficiency in treating breast cancer in BALB/c mice by local or systemic delivery was revealed by its potential anti-proliferation and anti-inflammatory activity. The localized delivery of NIODVC downregulated the genes that are over-expressed in breast cancer. These findings suggest that the combined delivery of doxorubicin and vitexin conjugated with cetuximab antibody provides a promising targeted therapy for breast cancer.
1. Introduction
Breast cancer is one of the most frequently diagnosed cancers in women globally with 2.26 million incident cases reported in 2020.1 Triple-negative breast cancer has poor prognosis due to its ability to spread quickly and develop resistance to multiple drugs. The primary drugs used to treat advanced breast cancer conditions include doxorubicin and paclitaxel, which cause adverse effects as they target both cancerous and normal tissues, which could partly be attributed to the systemic delivery of these drugs.2 In order to enhance the therapeutic efficiency of these drugs in targeting cancer cells and to reduce systemic toxicity, it is important to develop an alternative drug delivery system. Developing a targeted drug delivery system has significant advantages over conventional chemotherapy in breast cancer treatment, as it enables precise and effective delivery of therapeutic agents specifically to the tumor site.
The aim of the present study is to develop a targeted drug delivery system for breast cancer treatment. In this study, we aim to develop an endothelial growth factor receptor (EGFR) targeting niosome system to deliver doxorubicin and vitexin. Cetuximab was conjugated with the niosomes using gold nanoparticles as a linker. EGFR is highly expressed in most breast cancer which makes it an active target for delivery of drugs to breast cancer cells. Doxorubicin (D) is the most effective chemotherapeutic drug against triple-negative breast cancer (TNBC). The use of doxorubicin in treating TNBC is often limited due to several adverse reactions including fatigue, alopecia, nausea and vomiting, oral sores, bone marrow suppression and cardio toxicity. Doxorubicin induces oxidative stress, down regulates cardiac genes, apoptosis of cardiomyocyte and irreversible cardiomyopathy. The cardiotoxic mechanism is different from the anti-tumor mechanism of doxorubicin. This highlights the need to develop alternative ways to deliver doxorubicin and reducing its systemic toxicity.3,4 Several studies have suggested that combining doxorubicin with flavonoids enhances the cytotoxic effect in tumor cells while protecting normal cells.5–8 Vitexin (V), a flavone found in several medicinal plants, has numerous pharmacological effects, including anti-tumor, anti-oxidant, and anti-inflammatory effects.9 It has been found that vitexin protects myocardial cells against the effects of hypoxia, specifically from reoxygenation injury,10 and increases the bioavailability of drugs that are poorly soluble in water.
Niosomes (NIO) were selected in the present study as an alternative to the liposomal based drug delivery system.11 Although liposomes have the capacity to incorporate both hydrophilic and lipophilic substances with low toxicity, the unique advantages of niosomes, including their ability to penetrate the stratum corneum and stability, make them a potential candidate for targeted drug delivery. Niosomes could encapsulate both hydrophilic and hydrophobic drugs within the aqueous core or between the membrane bilayers.12,13 Niosomes are non-toxic,14 non-ionic surfactant vesicles with good chemical stability at different storage temperatures and in different biological environments. They penetrate the stratum corneum by disrupting its structure and fluidic properties, resulting in an improved delivery of drugs.15,16
To achieve targeted delivery, niosomes were conjugated with cetuximab (C) monoclonal antibody. Cetuximab which is primarily an EGFR inhibitor is used to treat colorectal and head and neck cancer. It binds to the external domain of epidermal growth factor receptor (EGFR) and inhibits signal transduction by blocking ligand binding.17–19 Gold nanoparticles were used to conjugate the antibody due to their excellent stability in solution, biocompatibility, and non-toxicity.20–24 Electrostatic adsorption was used for antibody conjugation, utilizing the interaction between the positively charged amino acid residues on the antibody surface and the negatively charged gold nanoparticles.
2. Experimental
2.1. Materials
Span 80, cholesterol, cetuximab, doxorubicin, vitexin, gold(III) chloride trihydrate (HAuCl4·3H2O), dialysis membranes (MWCO 1000 Da) and trisodium citrate were purchased from Sigma Aldrich and used without further purification. Potassium carbonate, phosphate buffer saline (PBS), and phospho-tungstic acid were purchased from Sisco Research Laboratories Pvt. Ltd. Chloroform and methanol were purchased from Merck, India. The human triple-negative breast cancer cell line (MDA-MB-231) was purchased from the National Centre for Cell Science, Pune, India. Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and 100× antimycotic solution were purchased from Sigma-Aldrich, USA. Dimethyl sulfoxide (DMSO) cell-culture grade, dimethyl thiazolyl tetrazolium bromide (MTT), fluorescein diacetate (FDA), and propidium iodide (PI) were purchased from HiMedia Laboratories, Mumbai, India.
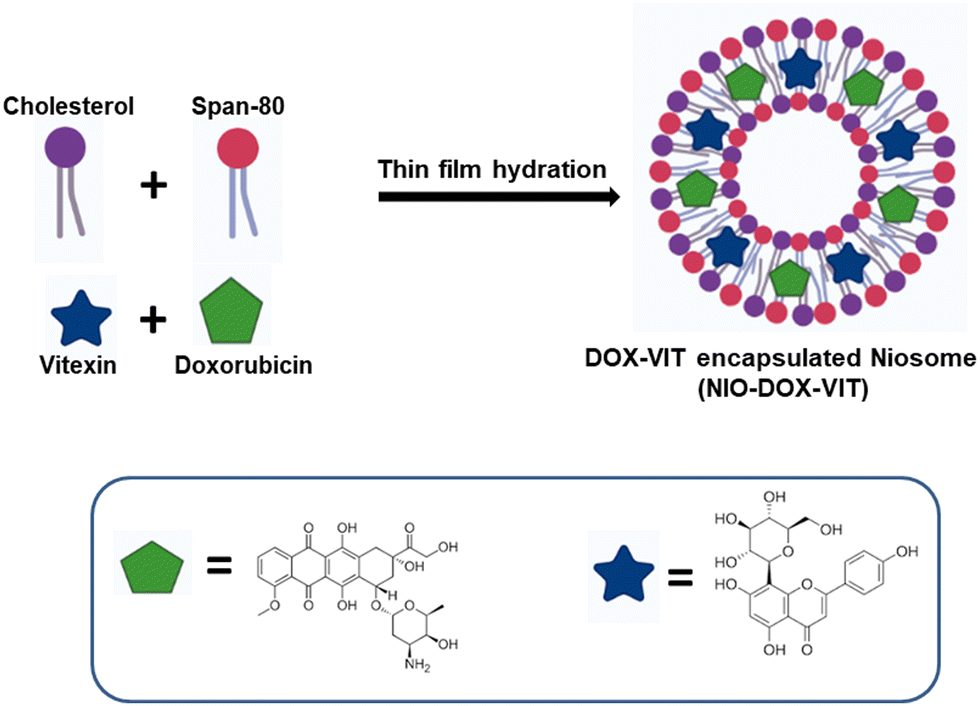
2.2. Preparation of virgin and drug loaded niosomes
The thin film hydration method was used to prepare niosomes.25 First, the non-ionic surfactant (Span 80) and cholesterol were dissolved in an equimolar ratio (0.1 M) in a chloroform and methanol mixture (6 mL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mL). The solvent was evaporated under vacuum using a rotary evaporator to form a thin film, which was further dried for 24 h in a vacuum desiccator. The film was then hydrated by resuspending in Milli-Q water (5 mL) and sonicated (M1800 ultrasonic bath) for 30 min to obtain niosomes. To prepare doxorubicin-loaded niosomes (NIOD), 100 μM doxorubicin was dissolved in 1 mL of PBS and added during the hydration step in the above mentioned procedure, followed by sonication to obtain doxorubicin-loaded niosomes. For vitexin-loaded niosomes (NIOV), 100 μM vitexin was dissolved in 1 mL of chloroform and added before the evaporation step. For doxorubicin- and vitexin-loaded niosomes (NIODV), the above procedures were carried out in the same suspensions (Scheme 1). Drug-loaded and virgin niosomes were stored at 4 °C until further use.
:3 mL). The solvent was evaporated under vacuum using a rotary evaporator to form a thin film, which was further dried for 24 h in a vacuum desiccator. The film was then hydrated by resuspending in Milli-Q water (5 mL) and sonicated (M1800 ultrasonic bath) for 30 min to obtain niosomes. To prepare doxorubicin-loaded niosomes (NIOD), 100 μM doxorubicin was dissolved in 1 mL of PBS and added during the hydration step in the above mentioned procedure, followed by sonication to obtain doxorubicin-loaded niosomes. For vitexin-loaded niosomes (NIOV), 100 μM vitexin was dissolved in 1 mL of chloroform and added before the evaporation step. For doxorubicin- and vitexin-loaded niosomes (NIODV), the above procedures were carried out in the same suspensions (Scheme 1). Drug-loaded and virgin niosomes were stored at 4 °C until further use.
 |
| | Scheme 1 Schematic representation of synthesis of drug loaded niosomes. | |
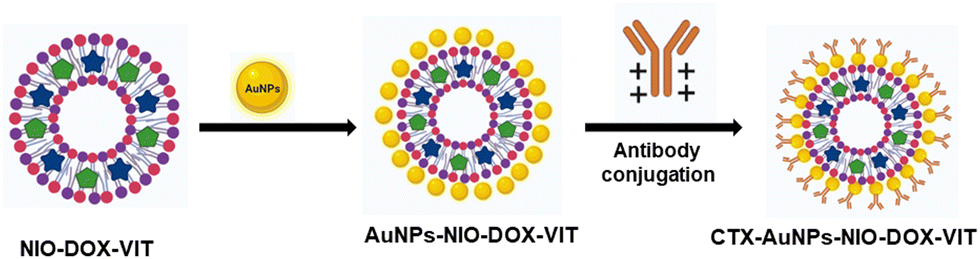
2.3. Synthesis of cetuximab conjugated niosomes
2.3.1. Synthesis of gold nanoparticles.
Gold nanoparticles were synthesised by boiling 50 mL of 0.2 mM Au3+ (gold chloride trihydrate) and adding 1 mL of trisodium citrate (1%) under vigorous stirring.26 The solution color changed from bluish-pink to ruby red after 10 min and the solution was continuously stirred at room temperature for 2 h. Nanoparticles were examined by ultraviolet-visible spectroscopy (UV-VIS) and transmission electron microscopy (TEM).
2.3.2. Synthesis of cetuximab conjugated niosomes.
The first step in the synthesis of cetuximab-conjugated niosomes is to prepare gold nanoparticle bound niosomes (NION). Gold nanoparticles (10−4 M, 500 μL) were added to virgin and drug-encapsulated niosomes separately under continuous vigorous stirring for 30 min. The resulting mixture was centrifuged for 10 min, and the pellet was washed with deionized water three times to remove any unbound gold nanoparticles. The NION pellet was resuspended in deionized water, and the pH was adjusted to 8.0 using potassium carbonate (1%) to reach the isoelectric point of cetuximab. To facilitate antibody conjugation, a solution containing 100 μg of cetuximab in 50 μL was added to the mixture, and then stirred at 4 °C for 1 h.27 The solution was then centrifuged at 15000 rpm (4 °C) for 15 min, and the resulting pellet was washed with deionized water to eliminate any unconjugated antibodies. The same procedure was followed to prepare cetuximab-conjugated doxorubicin niosomes (NIODC), vitexin niosomes (NIOVC), and both doxorubicin and vitexin loaded niosomes (NIODVC), and the pellets were stored at −20 °C until further use (Scheme 2).
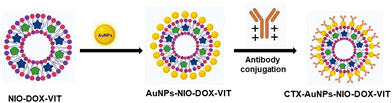 |
| | Scheme 2 Schematic representation of conjugation of antibody to drug-loaded niosomes. | |
2.4. Characterization of niosome formulations
Cetuximab conjugated virgin and drug-loaded niosomes were characterized using a FTIR/ATR spectrometer (FTIR-JASCO-6300v, JASCO International Co. Ltd, Tokyo, Japan). The size and shape of gold nanoparticles were determined using a Talos F200S HRTEM (Thermo Fisher Scientific Inc, Waltham, MA, USA). The surface morphology was analyzed using a FEI Quanta FEG 200F HR-SEM. Dynamic light scattering (DLS) with a Zeta sizer Nanoseries (DLS-Malvern Zetasizer Nano-ZS) was used to measure the size and zeta potential. The encapsulation efficiency and the amount of drug released were quantified by UV-Visible spectroscopy (JASCO V-730 International Co. Ltd, Tokyo, Japan).28
2.5. Encapsulation efficiency of niosome formulations
To determine the encapsulation efficiency, the niosomes were solubilized in methanol. The amount of drug loaded was quantified using a UV-Visible spectrophotometer at 482 and 267 nm for doxorubicin and vitexin, respectively. All the experiments were performed in triplicate, and the encapsulation efficiency was determined by the following formula:
where EE is the encapsulation efficiency, De is the amount of drug encapsulated and Dt is the total amount of drug.
2.6.
In vitro drug release profile
The drug release kinetics of doxorubicin and vitexin from three different formulations NIOD, NIOV, and NIODV was determined by a dialysis method.29 In brief, 100 mg of the sample was placed in a cellulose dialysis tubing membrane bag (MWCO: 1000 Da) and immersed in 100 mL of phosphate buffer saline (pH 7.4) at 37 °C under continuous stirring at 120 rpm. At different time intervals, 2 mL of released medium was collected. The amount of drug released was determined by measuring absorbance at 482 and 267 nm, and the cumulative percentage of drug released from each niosome formulation was calculated.
2.7. Mathematical model of drug delivery
Different kinetic models were used to fit the drug release from different niosome formulations:
(1) Zero order release kinetics
where
C is the amount of drug released,
K0 is the zero-order constant,
t is the time and
C0 is the initial amount of drug in solution.
(2) First order release kinetics
where
C0 is the initial amount of drug,
C is the percentage of drug released at time
t and
K1 is the first-order constant.
(3) Higuchi kinetic model
where
Q is the percentage of cumulative amount of drug released at time
t and kH is the Higuchi constant.
(4) Korsmeyer–Peppas kinetic model
where
Ct/
C∞ is the fraction of drug released at time
t,
K is the release rate constant and
n is the release exponent.
(5) Hixson–Crowell model
where
Q0 is the initial weight of solid at time
t = 0,
Qt is the remaining weight of solid at time
t and kHC is the dissolution rate constant.
2.8.
In vitro cytotoxicity of niosomes
2.8.1. Cell culture.
MDA-MB-231, a human triple-negative breast cancer cell line, was cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum at 37 °C. The cells were incubated in a humidified CO2 incubator with 5% CO2. Cells were trypsinized and cultured at a seeding density of 2 × 104 cells for 24 h before each experiment.
2.8.2. Cytotoxicity assay.
The MTT assay was used to determine the cytotoxicity of all niosome formulations against the MDA-MB-231 cell line. The niosomes were filter sterilized using a 0.22 μm syringe filter and UV sterilized (1 h in laminar hood). 3 × 103 cells per well were seeded in a 96-well plate and incubated for 24 h. Cells were then treated with different concentrations (0–50 μg mL−1) of niosomes and incubated for 24 h. Virgin niosomes without drugs were used as controls. The experiments involved the use of controls, including PBS (negative control), niosome (vehicle control), and Doxorubicin (positive control). After 24 h, 100 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide solution (2 mg mL−1) was added to each well and incubated for 4 h. The culture medium was then removed carefully, and the formazan crystals formed were resuspended in 100 μL of DMSO. The absorbance was measured at 570 nm using the Epoch 2 microplate reader, and the percentage of viable cells was calculated:
2.8.3. Live/dead cell assay.
Live/dead cell assay was performed to visualize the morphological changes associated with the treatment of niosome formulations. MDA-MB-231 cells were seeded at a density of 3 × 103 cells per well in a 96 well plate and cultured for 24 h. Cells were treated with all niosome formulations individually at a concentration of 1 μg mL−1 and incubated for another 24 h. Later the medium was removed and the cells were washed with PBS (1×) for 5 min. Cells were then incubated with fluorescein diacetate (400 nM) and propidium iodide (1 μM) for 2 min. Stained cells were visualized under a fluorescence microscope (DMi8, Leica Microscopy, Germany).
2.8.4. Cellular uptake of drug loaded niosomes.
MDA-MB-231 cells were seeded at a density of 3 × 103 cells per well in a 48 well plate and cultured for 24 h. Later, the cells were treated individually with all niosome formulations at a concentration of 1 μg mL−1 for 24 h. The cells were washed twice with PBS, trypsinized and resuspended in 400 μL of PBS, and the cellular uptake of niosomes was determined by the fluorescence intensity of doxorubicin using a fluorescence microscope.
2.9.
In vivo efficiency of cetuximab conjugated drug loaded niosomes
Female BALB/c mice aged 8 to 10 weeks were used. The animal use and all procedures were approved by the Institutional Animal Ethics Committee (IAEC) (205/GO/ReBi-S/Re-1/2000/CPCSEA) of the Central Animal House Facility, Dr ALM PG-IBMS, University of Madras, Taramani Campus, Tamilnadu, India. A total of 30 mice were used in the present study; the animals were provided with unrestricted access to food and monitored regularly in a pathogen-free and controlled environment. Breast cancer was induced in BALB/c mice using 4T1 breast cancer cells.30 Mice were anaesthetized under general anaesthesia (ketamine at 100 mg kg−1 body weight). The body was shaved from the fourth nipple to the midline, and 4T1 cells at a density of 5 × 105 cells were suspended in an equal volume of serum free media and matrigel (100 μL) and injected into the fourth inguinal mammary fat pad. The tumor development was monitored by measuring tumor volume regularly using the following formula:31
where Tv is the tumor volume, Ds is the shortest diameter of the tumor and Dl is the longest diameter of the tumor.
When the tumor volume reached a minimum volume (150 mm3), the animals were divided into six different groups randomly with each group consisting of 5 animals. Of the six different groups two groups were randomly selected for localized delivery and another two for systemic delivery. Group I: control, group II: tumor control, group III: NIODV (local), group IV: NIODV (intravenous), group V: NIODVC (local), and group VI: NIODVC (intravenous). A single dose of niosome formulation contained doxorubicin (4 mg kg−1), vitexin (20 mg kg−1), and cetuximab (4 mg kg−1). For localized delivery, niosomes were injected at the fourth inguinal mammary fat pad, and for systemic delivery, the niosomes were injected intravenously through the caudal vein. During the study, the animals were weighed regularly and the tumor dimensions were measured daily and recorded up to 20 days (Fig. 1). The mice were euthanized on day 20, and the tumor was excised and weighed. The vital organs (liver, spleen, kidney, heart, and lungs) were removed from each animal, washed with phosphate buffered saline, and weighed individually and proceeded for histology and gene expression studies.
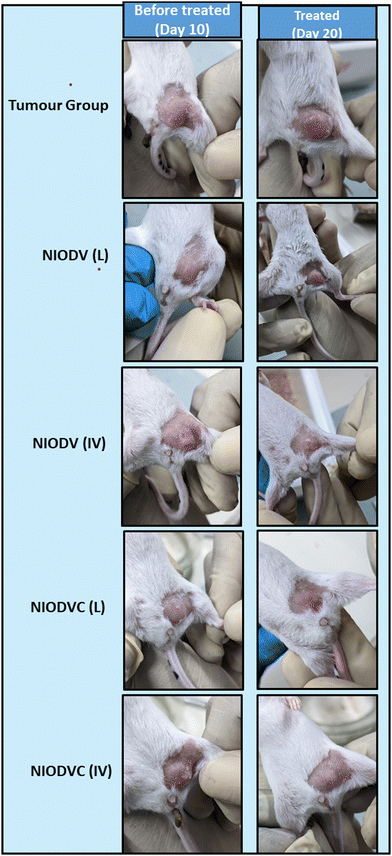 |
| | Fig. 1 Representative photographs of tumor induced mice before and after treatment. | |
2.9.1. Quantitative real-time PCR assay.
RNA was extracted from the mammary tissues using Trizol by a method reported earlier.32 The quality and quantity of RNA were analysed using a NanoDrop-spectrophotometer (Thermo 2000/2000c). RNA was reverse transcribed into cDNA using an Invitrogen SuperScript III First Strand synthesis kit following the manufacturer's protocol. The expression profile of selected genes, including BRCA1, EpCAM, C-MYC, NFKB, MMP9, miCAM, and iNOS, was determined by qPCR (CFX96 Touch Real-Time PCR Detection System, Bio-Rad) using SYBR green chemistry from Biorad (SsoAdvanced – Universal SYBR Green supermix). The amplification conditions employed were as follows: an initial denaturation step at 95 °C for 10 minutes, followed by 40 cycles of denaturation at 95 °C and annealing/extension at 55–60 °C for 15–30 seconds.33 β-Actin was used as the housekeeping gene. The primer sequence of all the genes tested is tabulated in Table 1.
Table 1 List of genes and primer sequences used in the real-time qPCR analysis
| Gene name |
Sequence of primers (5′-3′) |
| BRCA1 |
FP: CCACAGTGGGCTTACCTGTT |
| RP: GAGTCCATTCTCCCCGCTTC |
| C-MYC |
FP: CCTGCTTCTGGAGGGTGATG |
| RP: TGATGTGGTGTCTTGGAGAA |
| EpCAM |
FP: CATTTGCTCCAAACTGGCGT |
| RP: TTGTTCTGGATCGCCCCTTC |
| iNOS |
FP: GCACCGAGATTGGAGTTC |
| RP: GAGCACAGCCACATTGAT |
| miCAM |
FP: TGCTCAGGTATCCATCCATCC |
| RP: ACGGTGCCACAGTTCTCAA |
| MMP9 |
FP: GCCGACTTTTGTGGTCTTCC |
| RP: GGTACAAGTATGCCTCTGCCA |
| NFKB |
FP: CCTGCTTCTGGAGGGTGATG |
| RP: GCCGCTATATGCAGAGGTGT |
| β-Actin |
FP: TTCCAGCCTTCCTTCTTG |
| RP: GAGCCAGAGCAGTAATC |
2.9.2. Histological analysis.
Paraformaldehyde (4%) fixed mammary tissues were rehydrated and subsequently dehydrated using graded series of ethanol and embedded in paraffin wax. Sections of 4 μm thick were cut using a rotary microtome (Leica Rm2245) and the sections were stained with haematoxylin and eosin by the conventional method and visualized under a light microscope.34 Immunohistochemistry of Ki67 (Abcam ab15580) and PCNA (BioGenex AM252-5M) was carried out on de-paraffinized tissue sections following the guidelines provided by the respective manufacturers.
2.10. Statistical analysis
The results were presented as the mean value plus or minus standard error. Statistical analysis was carried out using Graph Pad Prism (version 9), and a P value <0.05 was considered as statistically significant.
3. Results and discussion
3.1. Characterization of niosome formulations
We examined the physicochemical properties of the niosomes, including size, morphology, surface charge, and their cytotoxicity against the triple-negative breast cancer MDMB-231 cell line. The average diameter of all the niosome formulations, polydispersity index (PDI), encapsulation efficiency (EE%), zeta potential, and the cumulative drug released at the end of 96 h are tabulated (Table 2). The average diameter of virgin niosomes was 380 ± 1.00 nm, but loading with doxorubicin, vitexin, and both doxorubicin and vitexin resulted in an increase in size to 410 nm ± 1.20, 389 nm ± 2.10, and 420 nm ± 1.00, respectively. The increase in size could be attributed to the rigidity of the bilayer in the liquid phase due to the addition of drugs.35 NIOD had a larger particle size than NIOV which is due to the higher molecular weight of doxorubicin. The average size of citrate stabilized gold nanoparticles was 30 nm. However, after the conjugation of the niosomes with cetuximab and gold nanoparticles, there was a significant increase in the size of the niosomes (425 nm ± 1.15). It has been reported that particles of size 425 nm are suitable for accumulation in solid tumor through the enhanced permeability and retention (EPR) effect or the passive targeting mechanism.36 For colloidal systems used in drug delivery, a polydispersity index (PDI) of less than 0.3 indicates a homogeneous population. The PDI values for all the niosome formulations ranged from 0.199 to 0.321, suggesting that the niosomes were relatively uniform in size.37 Zeta potential was measured to determine the stability of the formulations; values close to ±30 mV indicate a stable formulation.38 Zeta potential values of NIO, NIOD, NIOV, and NIODV were −28 ± 0.45, −29 ± 0.25, −34 ± 0.58, and −37 ± 0.56 mV, indicating that the niosome formulations without the antibody conjugation were having a negative charge.39 In the case of cetuximab-conjugated niosomes, citrate was used as a reducing agent in the synthesis of gold nanoparticles, which induced a negative charge on the surface of the nanoparticles, resulting in a Zeta potential of −22 ± 0.44 mV which prevents the nanoparticles from aggregation and precipitation over time (Table 2).40 Krasnici et al. found that cationic liposomes preferentially target the angiogenic endothelium of tumors.41 Similarly, Li et al. reported enhanced pulmonary insulin absorption compared to neutral liposomes.42 The encapsulation efficiencies for doxorubicin and vitexin were 72% and 81%, respectively. When both drugs were loaded together (NIODV), the encapsulation efficiencies for doxorubicin and vitexin were found to be 73% and 83%, respectively. Higher encapsulation efficiency for vitexin could be attributed to its lipophilicity; similar results were observed with apigenin liposomal formulations by Banerjee et al.43
Table 2 Physicochemical properties of niosome formulations
| Sample |
Average vesicle diameter (nm) |
PDI |
Zeta potential (mV) |
Encapsulation efficiency (%) |
Cumulative drug release (%) |
| NIO |
380 ± 1.00 |
0.321 ± 0.03 |
−28 ± 0.45 |
0 |
— |
| NIOD |
410 ± 1.20 |
0.28 ± 0.05 |
−29 ± 0.25 |
72.75 ± 0.96 |
71% |
| NIOV |
389 ± 2.10 |
0.199 ± 0.03 |
−34 ± 0.58 |
81.12 ± 0.12 |
53% |
| NIODV |
420 ± 1.00 |
0.201 ± 0.04 |
−37 ± 0.56 |
DOX:73.98 ± 0.56 |
85% |
| VIT:83.98 ± 0.75 |
67% |
| Gold nanoparticles (N) |
030 ± 0.005 |
— |
−22 ± 0.44 |
— |
— |
| NIODVN |
424 ± 1.25 |
0.271 ± 0.01 |
−34 ± 0.86 |
— |
— |
| NIODVC |
425 ± 1.15 |
0.291 ± 0.04 |
−35 ± 0.46 |
— |
— |
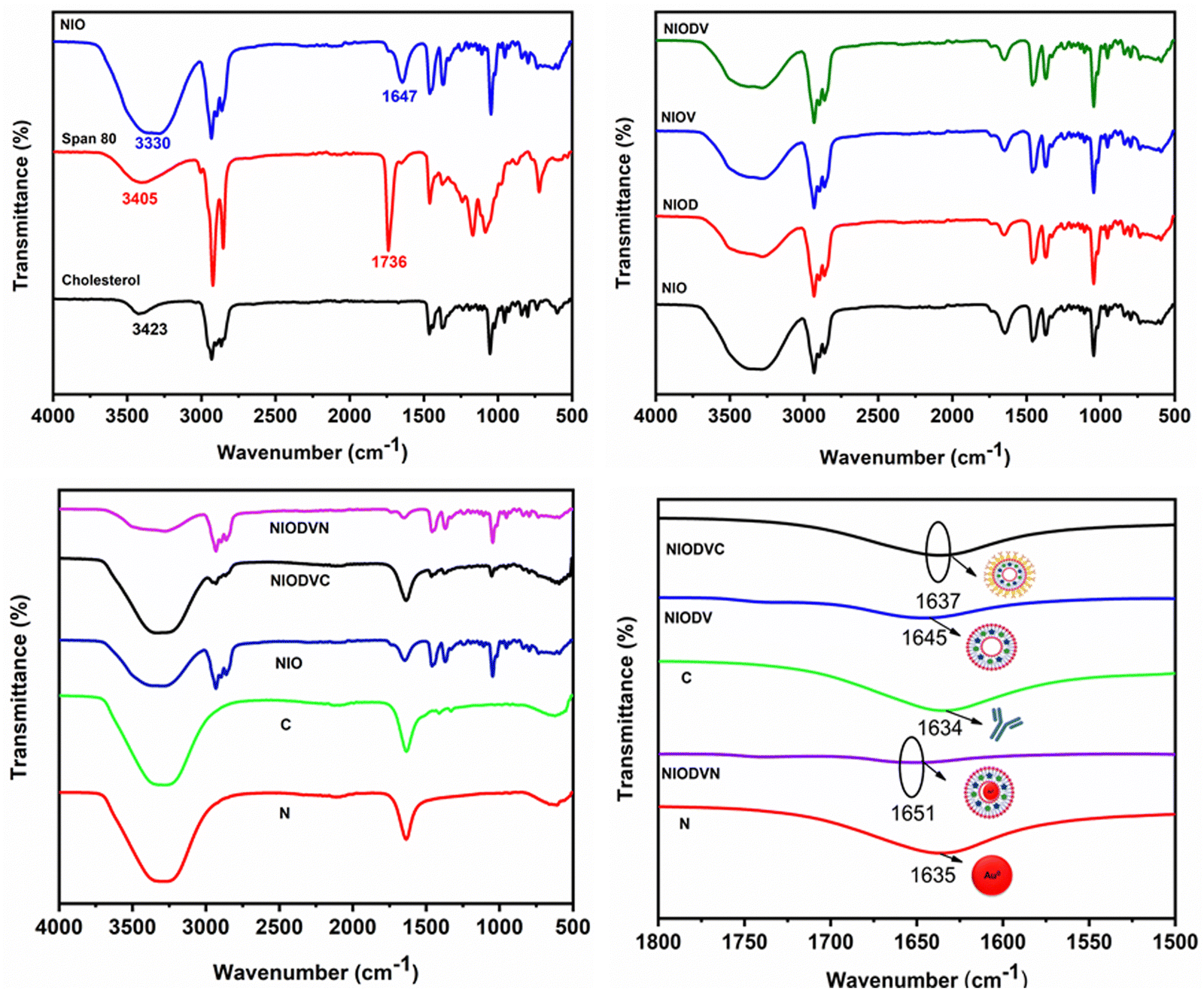
The FTIR spectrum of virgin niosomes (Fig. 2A) showed a characteristic ester peak at 1736 cm−1 that corresponds to Span 80 and a cholesterol hydroxyl peak at 3411 cm−1. The peak at 1647 cm−1 confirmed the presence of hydrogen bonding between the carbonyl and the hydroxyl group of span 80 and cholesterol.44 The structural integrity of the niosome is maintained by the presence of hydrophilic and hydrophobic groups that allow it to effectively encapsulate hydrophobic and hydrophilic drugs.45 The presence of additional hydroxyl (OH) groups from cholesterol could be responsible for the broad absorption band at 3330 cm−1. The distinct peaks at 2931 cm−1, 2895 cm−1, and 1464 cm−1 could be attributed to asymmetric stretching vibrations of the CH2 and CH3 groups of niosomes.46 The sharp peaks at 1168 cm−1 and 1047 cm−1 correspond to ring deformation in CH2 bending and in-plane C–H bending, respectively. The bands between 900 and 675 cm−1 represent the CH out-of-plane bending, and the band at 840 cm−1 indicates C–C–C stretching. FTIR spectra of doxorubicin, vitexin, and both drugs loaded niosomes (Fig. 2B) exhibited all the characteristic peaks of the niosomes, suggesting that no chemical reaction occurred between the drugs and niosomes. When gold nanoparticles were added to NIODV (Fig. 2C and D), the stretching frequency of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the citrate ligands shifted from 1635 cm−1 to 1651 cm−1, indicating an interaction between the carbonyl group and the hydrophilic surface of the niosomes. Conjugation of cetuximab showed its characteristic peak at 1637 cm−1, suggesting a π back bond between the gold nanoparticle and the cetuximab antibody resulting in the formation of a covalent bond between the two entities.47,48 The blue shift observed in the carbonyl stretching of gold nanoparticle conjugated NIODV compared to NIODVC confirmed the conjugation of cetuximab with the gold nanoparticles.
O of the citrate ligands shifted from 1635 cm−1 to 1651 cm−1, indicating an interaction between the carbonyl group and the hydrophilic surface of the niosomes. Conjugation of cetuximab showed its characteristic peak at 1637 cm−1, suggesting a π back bond between the gold nanoparticle and the cetuximab antibody resulting in the formation of a covalent bond between the two entities.47,48 The blue shift observed in the carbonyl stretching of gold nanoparticle conjugated NIODV compared to NIODVC confirmed the conjugation of cetuximab with the gold nanoparticles.
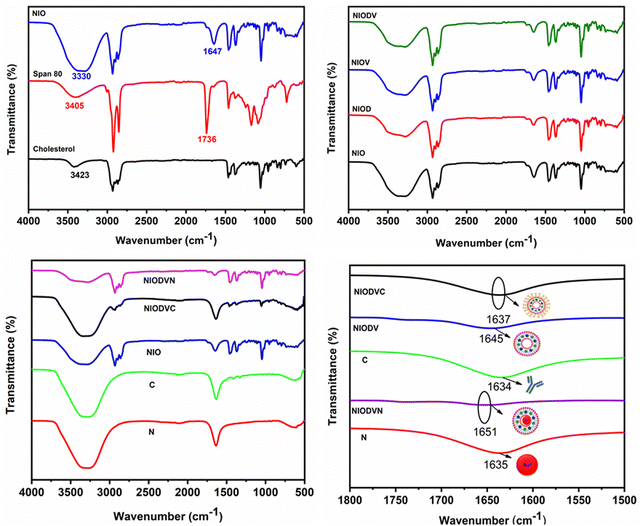 |
| | Fig. 2 FTIR spectra of blank niosomes (A), drug loaded niosomes (B), and gold nanoparticle and antibody-conjugated drug loaded niosome vesicles (C), and magnified view of the spectral region between 1800 and 1500 cm−1 (D). | |
3.2. High resolution electron microscopic analysis of virgin and drug loaded niosomes
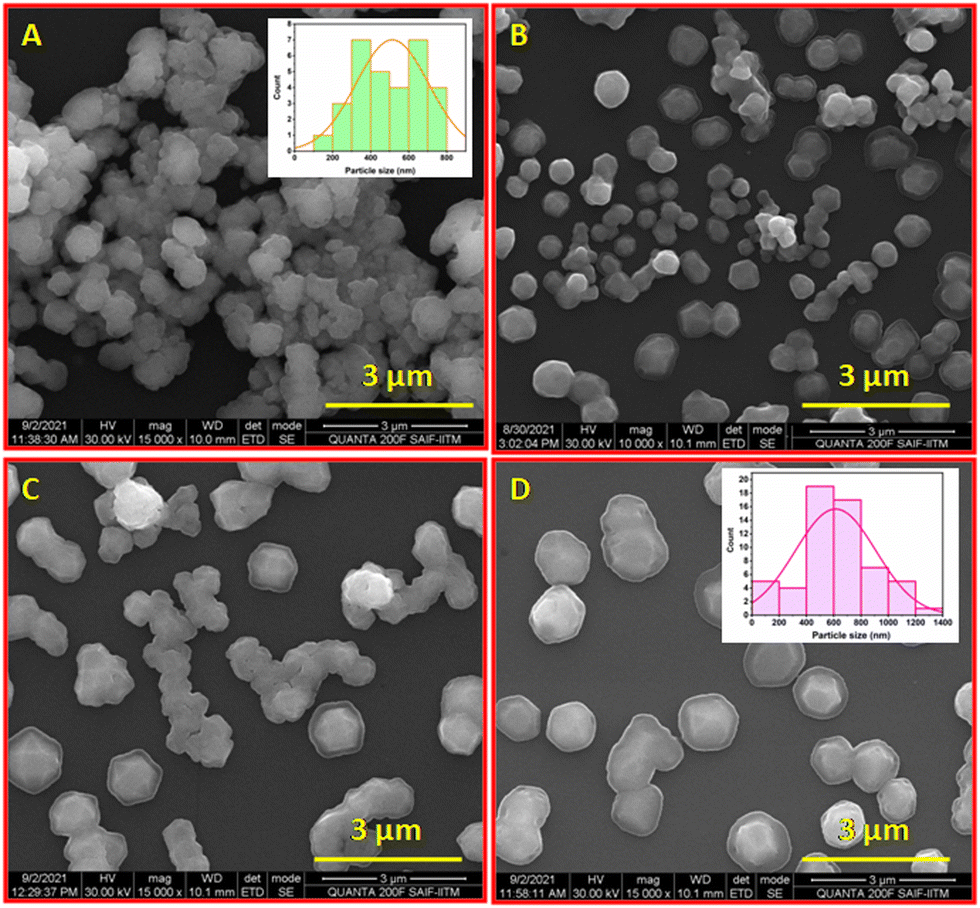
Size and surface morphology of all niosome formulations were evaluated by scanning electron microscopy (SEM) (Fig. 3) and DLS measurements respectively. The niosomes were stained with phospho-tungstic acid (1%) at room temperature and visualized under a high resolution scanning electron microscope. Niosomes were spherical, with a smooth outer surface, and contained a large internal aqueous area. The outer lipophilic domain appeared dark, while the inner hydrophilic domain was light. The average size of virgin niosomes was 513 nm (Fig. 3A), whereas niosomes with both doxorubicin and vitexin had a larger lipophilic domain, resulting in an average size of almost 614 nm (Fig. 3D).
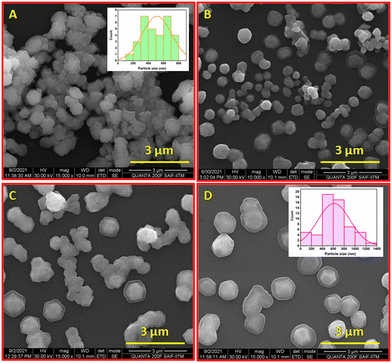 |
| | Fig. 3 High resolution scanning electron microscopic images of niosomes (A) (inset: particle size distribution), NIOD (B), NIOV (C), and NIODV (D) (inset: particle size distribution). | |
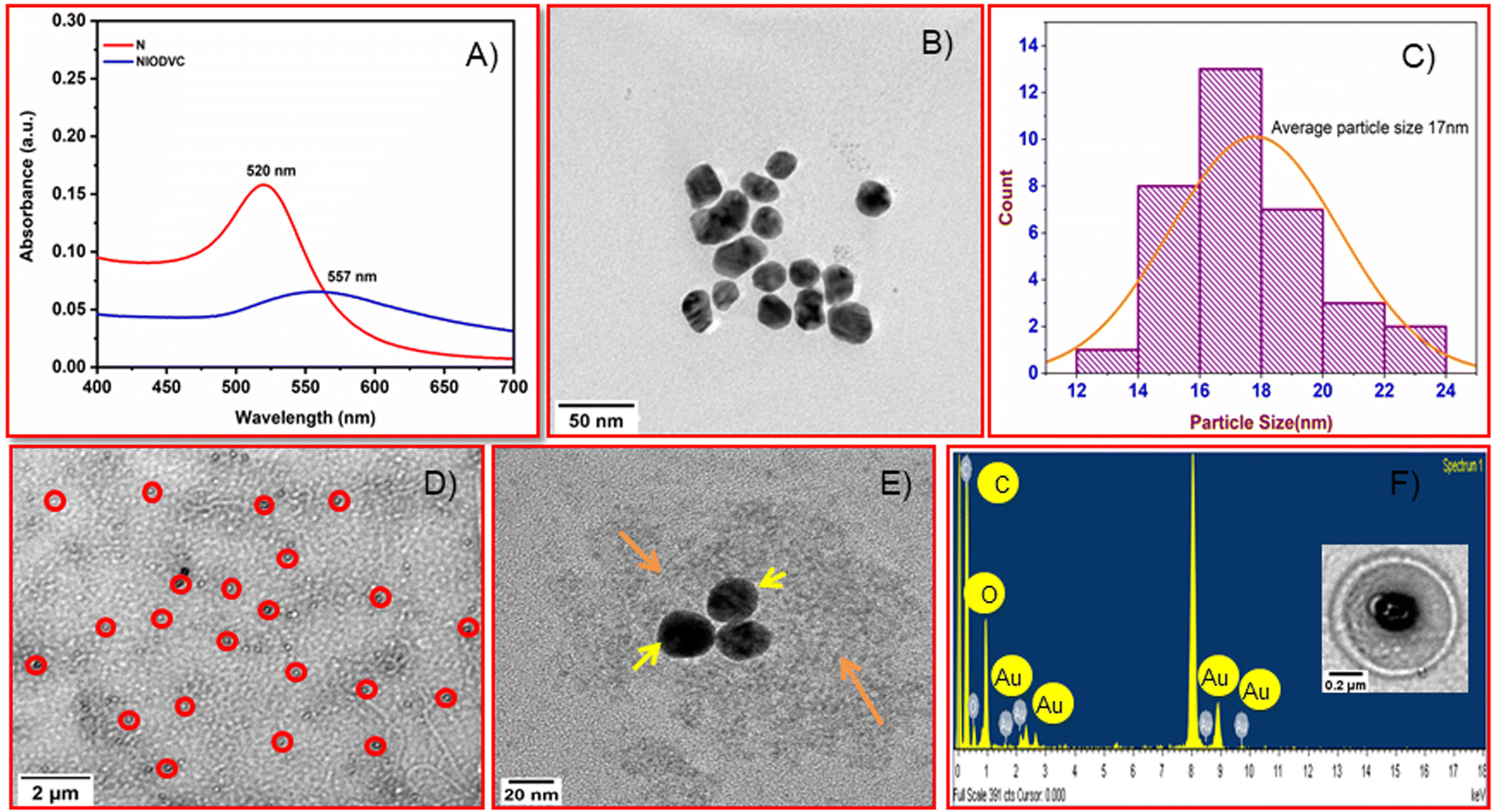
A high resolution transmission electron microscope (HRTEM) was used to characterize the antibody conjugated niosomes (Fig. 4). The Turkevich method used in the present study is a popular way to synthesize spherical gold nanoparticles because the citrate ions used are biocompatible with good crosslinking ability.26 Citrate acts as a stabilizer for the gold nanoparticles during the reaction and it also alters the pH of the system to around 6.7, which influences the size of gold nanoparticles, resulting in nearly monodisperse gold nanoparticles (20 to 40 nm). The absorption peak for gold nanoparticles was at 520 nm (Fig. 4A)49 but the maximum absorption of gold nanoparticles in NIODVC was at 557 nm. This shift could be attributed to the changes in the local refractive index because of protein adhesion.50 Gold nanoparticles were spherical (Fig. 4B), and the average particle size was around 17 nm (Fig. 4C). The niosomes had uniform, smooth bilayer morphology, and the size distribution was within the range of 0.34 to 0.4 μm. TEM images showed that the gold nanoparticles were found attached to the hydrophilic layer of the niosome (Fig. 4D and E) and the EDX analysis confirmed the same (Fig. 4F). EDX analysis confirmed the presence of carbon (K: 0.27 keV) and gold emission lines (M: 2.23 keV). Carbon and oxygen accounted for 88.2, and 5.86 wt% which could be attributed to both niosomes and antibody, while Au atoms on the surface of niosomes accounted for the remaining 5.86 wt%.
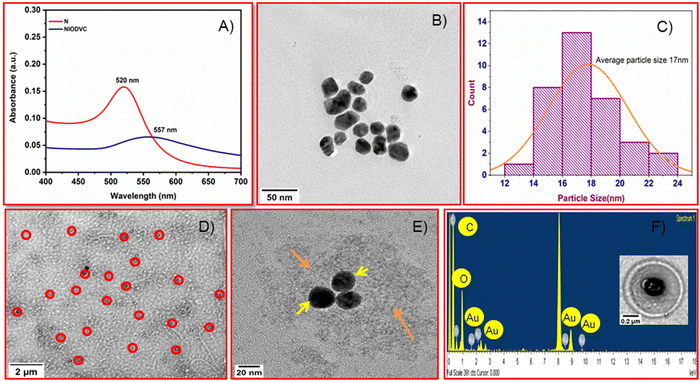 |
| | Fig. 4 Characterization of nanoparticles and niosomes: UV-visible spectrum (A), transmission electron microscopic images (B) and particle size distribution of gold nanoparticles (C). Low (D) and high (E) magnification transmission electron microscopic images and energy-dispersive X-ray spectroscopy of NIODVC. | |
3.3.
In vitro drug release kinetics
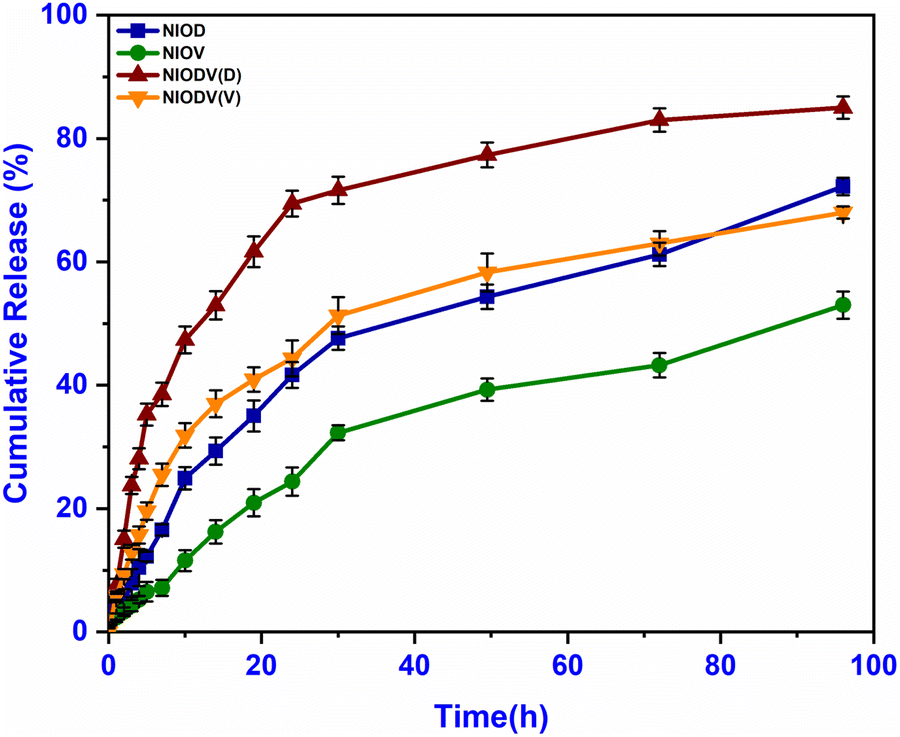
The release of doxorubicin, vitexin, and dual drugs from niosomes was studied in PBS (pH 7.4) at 37 °C. The amount of drug released was determined at regular time intervals for a period of 96 h and the cumulative release (mean ± standard deviation) against time was plotted (Fig. 5). A biphasic drug release pattern was found, with an initial burst release for 3–9 hours followed by a controlled release for 72 h. The burst release could be attributed to the release of the drug from the surface of the niosomes and smaller niosomes,51 while the controlled release could be attributed to drug diffusion through the vesicle bilayer. The highest percentage of drug was released from NIODV (85% for D and 63% for V), followed by NIOD (69%) and NIOV (49%). The differences in the release of same drugs in two different formulations could be attributed to the differences in size. The size of dual drug loaded niosomes was ∼614 nm. In general, the presence of high levels of cholesterol in niosome formulations enhances membrane stability and increases the hydrophobicity of the vesicles. As a result, drug release from the niosomes is slowed due to reduced permeability across the bilayer membrane.52 Previous studies have reported that niosomes loaded with timolol maleate for glaucoma treatment exhibited a sustained release pattern, and the entire drug was released from the vesicles in approximately 24 h but the free form of the drug was released completely within 4 h.53 Similarly, doxycycline, an ocular antibiotic, was fully released within the initial hour in its free form, while its niosome formulation sustained the release up to 20 h.54 In NIODVC the release of doxorubicin and vitexin was sustained up to 96 h.
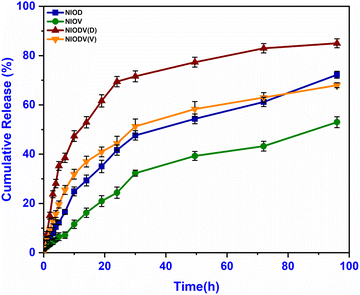 |
| | Fig. 5
In vitro drug release profiles of niosome formulations containing doxorubicin (NIOD), vitexin (NIOV), and both drugs (NIODV). | |
The correlation coefficient (R2) of various mathematical models used to fit the drug release profiles is tabulated in Table 3. Drug release from the niosomes followed different kinetics depending on the formulation. NIOD fits the Korsmeyer–Peppas model, suggesting a non-Fickian diffusion and erosion mechanism (Fig. S1, ESI†), whereas NIOV followed zero order kinetics with an initial burst release followed by continuous release (Fig. S2, ESI†). In the case of NIODV, both doxorubicin and vitexin followed Higuchi kinetics, indicating a diffusion-based drug release from the vesicles (Fig. S3, ESI†). These results were in line with meta-analysis findings of different release kinetics extrapolated from different drug delivery vehicles.55
Table 3 Release kinetics of niosome formulations
| Formulation code |
Correlation coefficient (R2) |
| Zero order |
First order |
Higuchi |
Hixson–Crowell |
Korsmeyer–Peppas |
| NIOD |
0.973 |
0.985 |
0.986 |
0.979 |
0.988 |
| NIOV |
0.993 |
0.992 |
0.951 |
0.946 |
0.976 |
| NIODV (DOX release) |
0.885 |
0.414 |
0.976 |
0.944 |
0.742 |
| NIODV (VIT release) |
0.925 |
0.786 |
0.988 |
0.952 |
0.989 |
3.4. Screening of niosomes efficiency against breast cancer cells in vitro
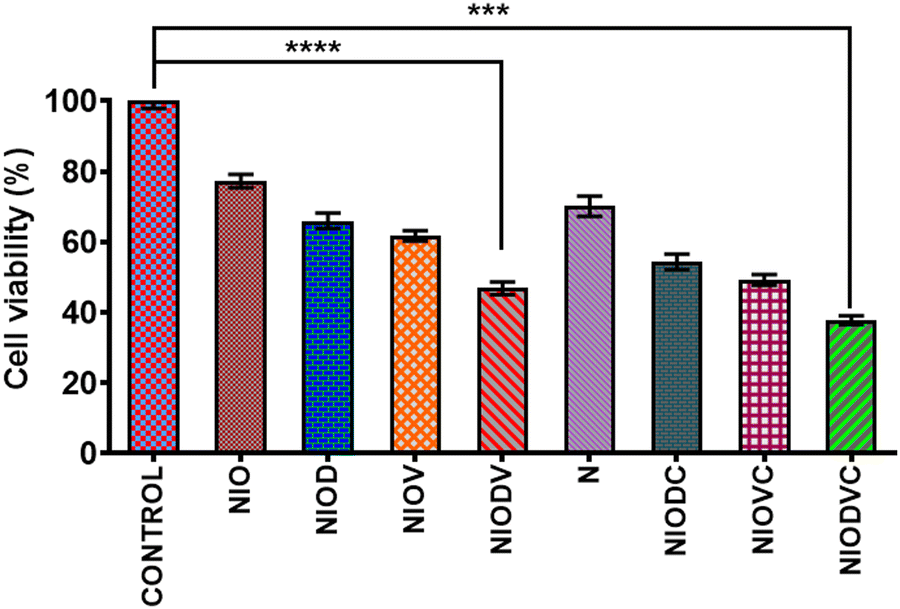
The cytotoxicity of virgin niosomes, drug loaded niosomes, and gold nanoparticles at a fixed concentration (10 μM) was tested against MDA-MB-231 cells (Fig. 6). MTT assay showed that virgin niosomes and gold nanoparticles had minimal cytotoxicity, while NIODVC was more cytotoxic (35% cell viability) than NIODV (45% cell viability). The delivery of doxorubicin and vitexin together was 1.5 times higher than individual drug delivery, revealing the synergistic effects. Combination of flavonoids and anticancer drugs helps to overcome the systemic toxicity of chemotherapeutic agents and increases the anticancer effect of the drug at lower concentrations.56 Similar results have also been reported with the co-delivery of doxorubicin and quercetin from PEGylated niosomes.57
 |
| | Fig. 6 Cytotoxicity of virgin and drug loaded niosomes against MDA-MB-231 cells after 24 hours of incubation. Results are presented as the mean ± standard deviation (SD) of three independent experiments. ***p < 0.001, ****p < 0.0001. | |
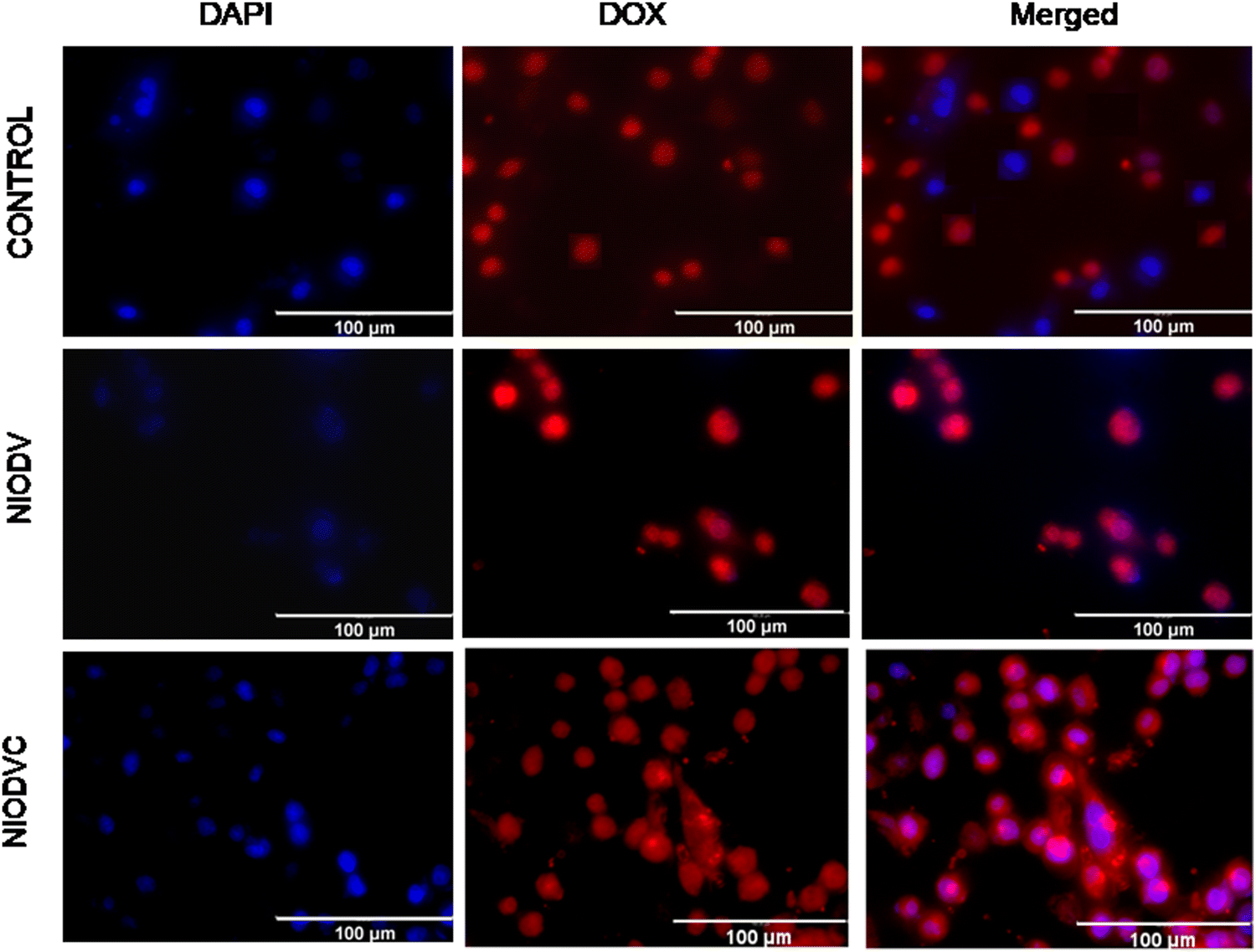
Fluorescence of doxorubicin was used to detect the cellular uptake in MDA-MB-231 cells; cells were counter stained with DAPI after niosome treatment to visualize the nucleus (Fig. 7). Cetuximab conjugated niosomes exhibited strong fluorescence intensity in the cytoplasm compared to the NIODV which could be attributed to the specificity and selective binding of the antibody to the epidermal growth factor receptor. This opens a new window for targeted cancer treatment.
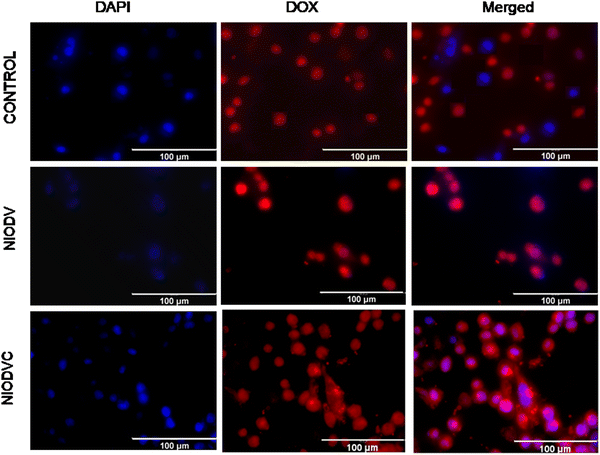 |
| | Fig. 7 MDA-MB-231 cellular uptake of niosome formulations counter stained with DAPI (blue) and doxorubicin (red). The merged image shows the co-localization of both DAPI and doxorubicin inside the cells. | |
The FDA/PI staining showed niosome induced apoptosis in MDA-MB-231 cells (Fig. S4, ESI†). FDA penetrates the live cells and emits green fluorescence, while PI stains the dead cell nuclei red. Cells treated with NIOD, NIOV, and NIODV displayed characteristics of early and late apoptotic cells. Doxorubicin induces apoptosis by inhibiting DNA replication and repair,58 while vitexin promotes apoptosis by inducing oxidative stress and inhibiting cell proliferation.59 The increased apoptosis with the combined delivery of doxorubicin and vitexin could be attributed to the synergistic effect of the combination of these drugs.
3.5.
In vivo efficiency of niosomes on a mouse tumor model: systemic vs. local delivery
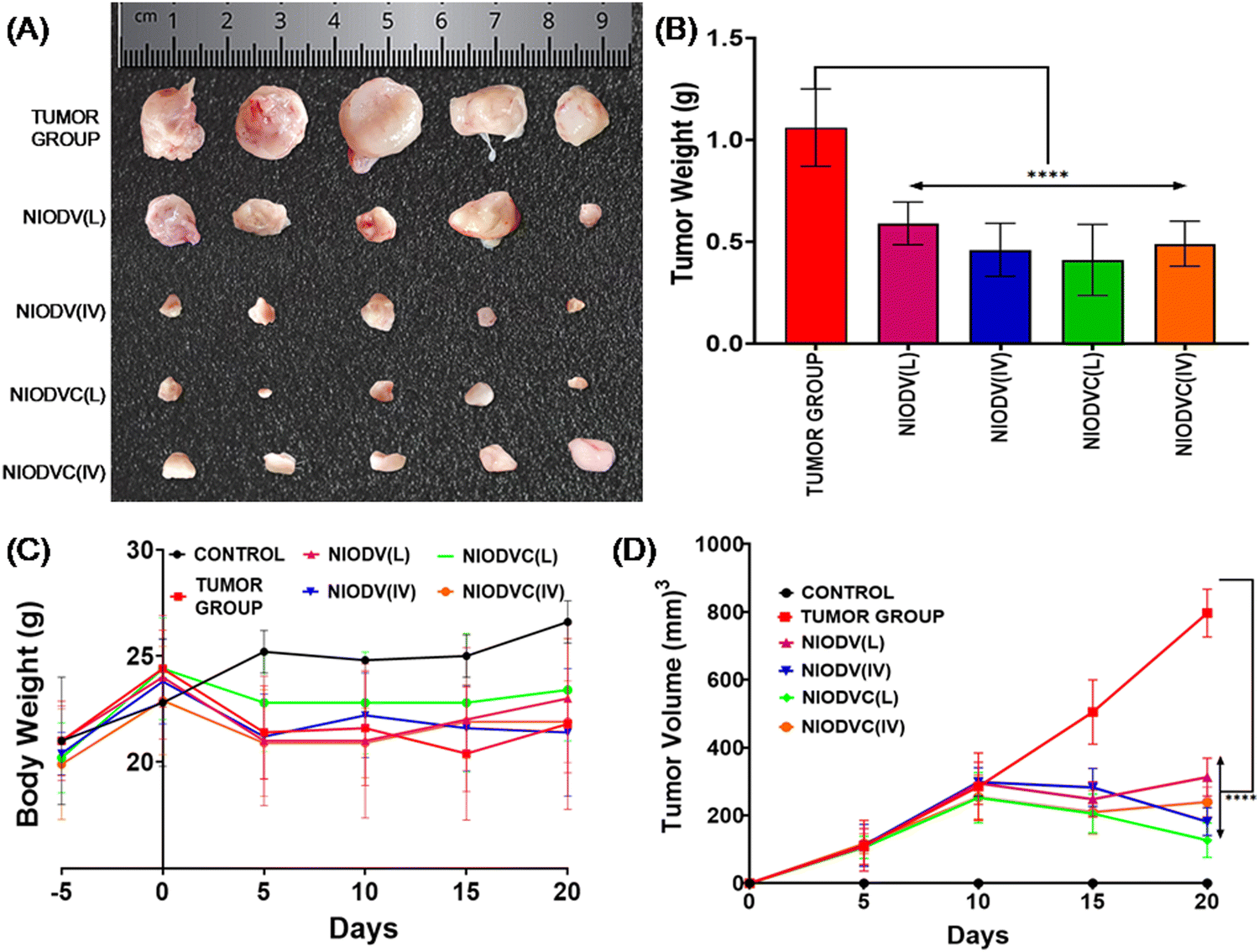
The in vivo efficacy of dual drug loaded niosomes, with and without antibody conjugation, was assessed on a 4T1 cell induced tumor model in BALB/c mice using two distinct delivery methods. For localized delivery, the niosomes were injected at the fourth mammary tumor tissue and to determine their efficiency in targeted delivery, the niosomes were injected intravenously through the caudal vein. Representative images of the dissected tumor after 20 days of treatment are represented in Fig. 8 showing the changes in tumor volume, tumor weight and body weight. The average tumor volume was 796.7 ± 285.3 mm3 in the tumor group (Group 2). In the case of NIODV when delivered locally it was 313 ± 56 mm3 (Group-3), and for systemic delivery it was 181.7 ± 41 mm3 (Group-4). In the case of NIODVC, when delivered locally it was 126.8 ± 50.7 mm3 (Group-5), and for intravenous it was 240 ± 43.8 mm3 (Group-6). All the niosome formulations exhibited significant anti-tumor activity compared to the tumor group. Tumor shrinkage was higher with NIODV when delivered systemically (2.54 fold increase than local injection). Interestingly, the tumor shrinkage was higher with NIODVC when delivered locally compared to systemic delivery (6.28 and 4.38 fold increase), which could be attributed to the efficiency of specific binding of cetuximab to the epidermal growth factor receptor (EGFR), which is overexpressed in many cancer cells.60 The difference in systemic delivery could be attributed to the bioavailability of NIODVC. Similar results were observed with liposomes conjugated with Fab fragments of cetuximab, which resulted in a 6 fold increase in drug accumulation in MDA-MB-468 human breast cancer xenografts.61
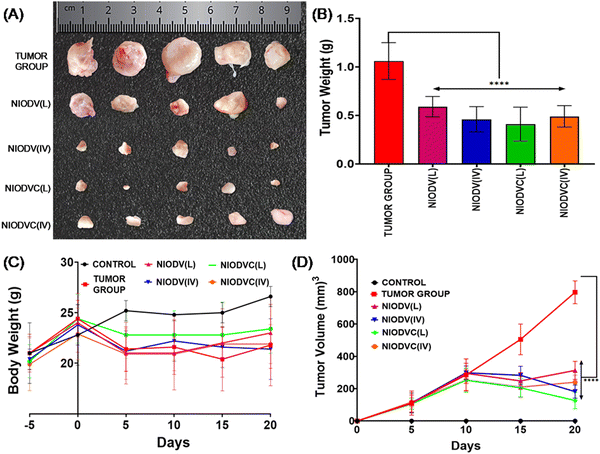 |
| | Fig. 8 Photographic images captured 20 days after niosome treatment, illustrate changes in the average tumor volume (A) and tumor weight (B), body weight (C) and tumor volume (D). Note: ***p < 0.001, indicating statistical significance. | |
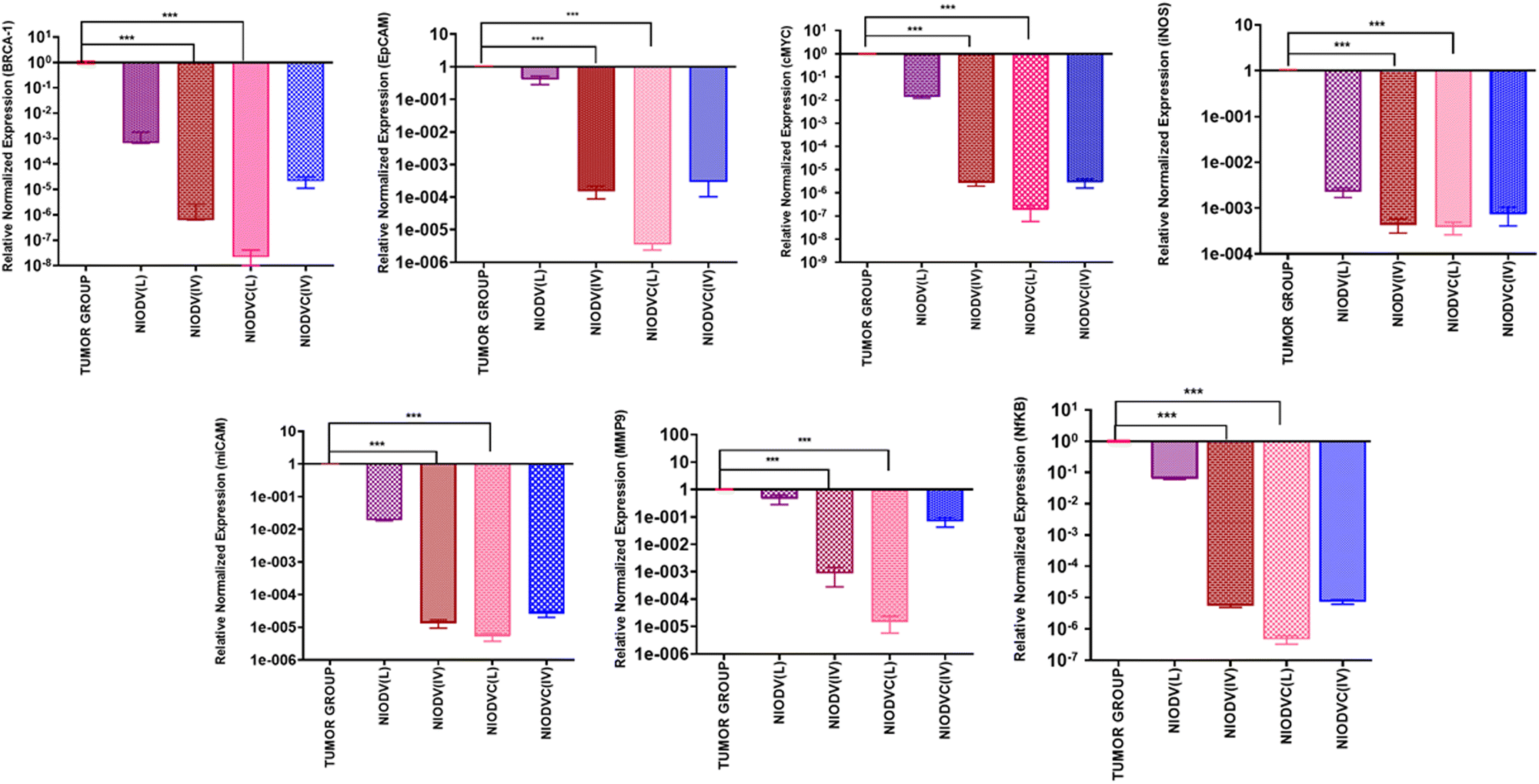
Fig. 9 shows quantitative mRNA expression profiles of cell proliferative genes such as BRCA1, cMYC, EpCAM, iCAM and MMP9 and inflammatory genes iNOX and NF-κB in mammary tissue. In the tumor group, there was an increase in the expression levels of genes related to proliferation and inflammation. However, the expression levels of these genes were significantly altered in the niosome treated mice. Previous reports have shown that cetuximab combined with chemotherapeutic drugs was effective in treating TNBC.62,63 Cetuximab binds to the epidermal growth factor receptor (EGFR), which is important in metastasis, tumorigenesis, drug resistance, and cell proliferation.62,63 Doxorubicin acts as a DNA inter-chelator and inhibits the topoisomerase II enzyme resulting in the inhibition of cancer cell growth; it also causes dsDNA damage and activates downstream apoptotic pathways.64 Similarly, vitexin promotes apoptosis and cell cycle arrest.65
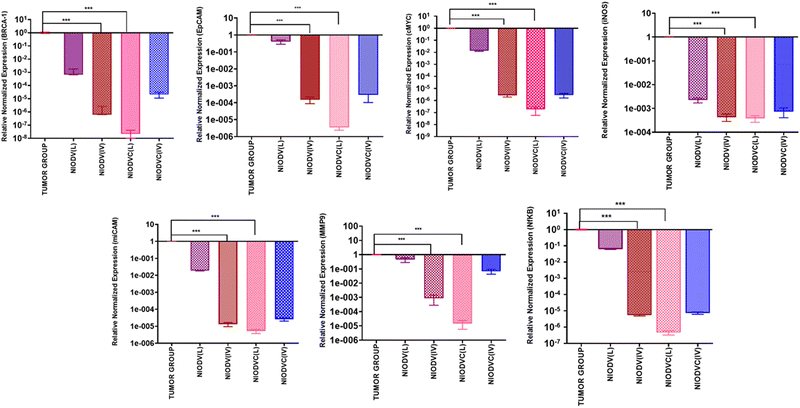 |
| | Fig. 9 Gene expression levels of BRCA1, EpCAM, cMYC, iNOS, miCAM, MMP9, and NFKB in 4T1 tumor cells after treatment with niosomes. The expression levels were normalized to the reference gene beta actin and presented as fold change relative to the control group. ***P < 0.01. | |
In the present study, elevated expression of BRCA1, cMYC, EpCAM, iCAM, MMP9, iNOS, and NFκB in the mammary tissues of the tumor group was observed. In contrast, mRNA expression of tumour suppressor gene BRCA1 was downregulated in the case of niosome treated groups. However, the local delivery of NIODVC resulted in decreased expression of proliferative, angiogenic, and inflammatory genes. It exerts inhibitory effects on cell migration and angiogenesis by downregulating cMYC, EpCAM, iCAM, and MMP9.66 Additionally, the down-regulation of NF-κB and iNOS indicates reduced inflammation and immune responses.67
Breast cancer susceptibility gene 1 (BRCA1) primarily a tumor suppressor gene plays a crucial role in DNA repair, replication fork protection, cell cycle regulation, and preservation of genome stability.68 Mutations or abnormalities in the BRCA1 gene make the cell more vulnerable to various cancers. Cellular myelocytomatosis (c-MYC) is a proto-oncogene that encodes a multifunctional transcription factor. It plays an important role in cell cycle progression from G1 to S phase and is overexpressed in various cancers, including breast cancer.69 The epithelial cell adhesion molecule (EpCAM) is a transmembrane glycoprotein that is overexpressed in many epithelial cancers and it is involved in gene regulation, cell proliferation, cancer stemness, and interactions with cell adhesion molecules.70 The intracellular adhesion molecule (iCAM) is a transmembrane glycoprotein receptor that plays a vital role in cell adhesion, cell signaling, and transendothelial migration. Overexpression of iCAM has been reported in various malignancies, including TNBC.71 Matrix metalloprotease (MMP)-9 is a zinc- and calcium-dependent protease that digests and remodels a wide range of extracellular matrix components. It is overexpressed in various cancer cells during invasion and plays a key role in tumorigenesis by inducing angiogenesis in the tumor microenvironment.72 Nuclear factor kappa B subunit 1 (NFκB) is a transcription factor that regulates the activation of various proinflammatory cytokine genes.73 Inducible nitric oxide synthase (iNOS) is a pro-inflammatory enzyme that is involved in various chronic inflammatory conditions, including breast cancer.74
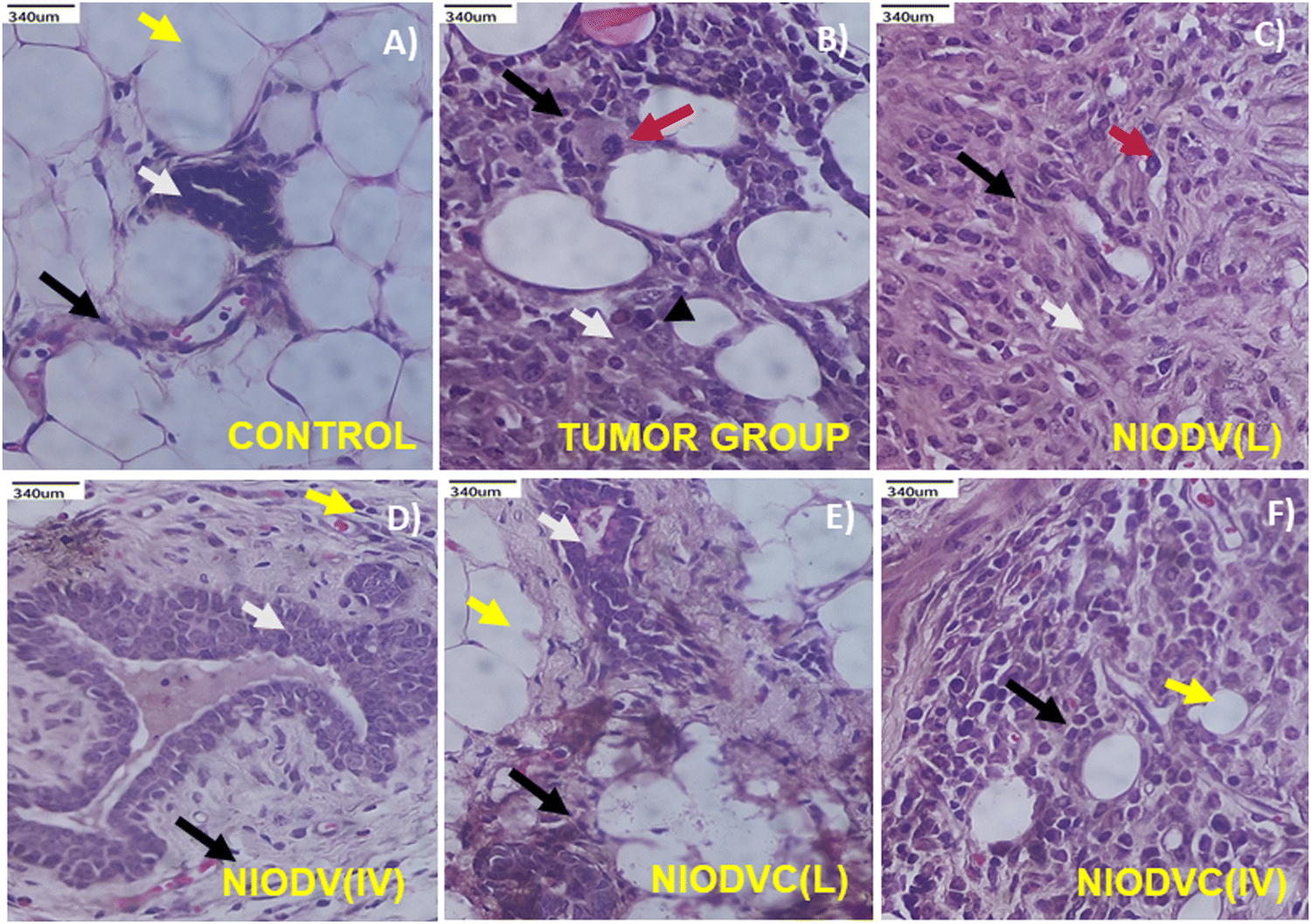
Fig. 10(A)–(F) shows the histopathological analysis of mammary tissue of control and experimental groups. The group I control animal showed normal histotype of ductal and stromal elements and normal mammary fat tissue (Fig. 10A). In contrast, the tumor group (Group II) showed severely infiltrated neoplastic ductal epitheloid cells with pleomorphic and hyperchromatic nuclei, and the average mitotic index was 5.4 per 10 high power field (HPF, 400×) (Fig. 10B). The niosome treated mice (Groups III to VI) exhibited residual tumours with usual ductal hyperplasia, which is a common mild-cancerous to noncancerous breast condition characterized by an overgrowth of cells in the ducts of the breast. In the case of NIODV delivered locally (Group III) there were mild infiltrating hyperplastic mammary epitheloid cells with moderate fibrosis, and the mitotic index reduced to 2.7 per 10 HPF (Fig. 10C); when it was delivered systemically (Group IV) there was usual ductal hyperplasia with mild fibrosis; and no mitotic figures were observed (Fig. 10D) in the case of NIODVC delivered locally (Group V). The tissue showed a normal architecture with mild fibrosis, and no mitotic figures (Fig. 10E) whereas when it was delivered systemically (Group VI) the tissues showed infiltrated hyperplastic mammary cell carcinoma with fibrosis, and an average mitotic index of 2.2 per 10 HPF (Fig. 10F). Histopathological analysis of the tumour group showed invasive ductal carcinoma (grade 2 to 3) with increased mitotic activity, whereas locally delivered NIODVC showed regression in tumour lesion with improved ductal architecture and reduced mitotic index.
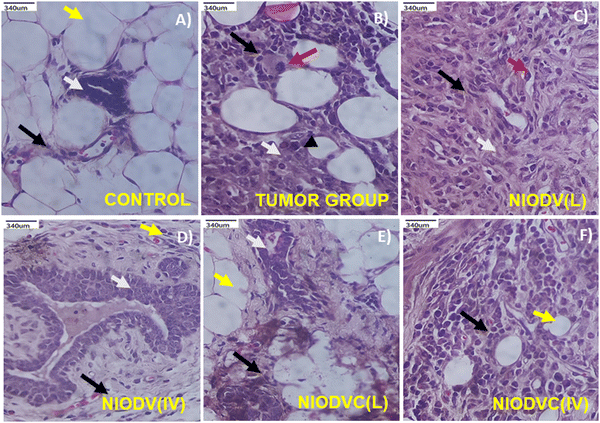 |
| | Fig. 10 Histopathological analysis of control (A) and tumor group (B) with infiltrating ductal adenocarcinoma of the breast – grade II. NIODV injected locally (C) and systemically (D) with residual infiltrating ductal adenocarcinoma of the breast – grade II and ductal hyperplasia and inflammatory infiltration. NIODVC injected locally (E) and systemically (F) with ductal epithelioid hyperplasia and inflammatory infiltration. Black arrow – mononuclear inflammatory infiltrates, white arrow – ductal elements, yellow arrow – fat component of breast tissue, red arrow – tumor giant cells. | |
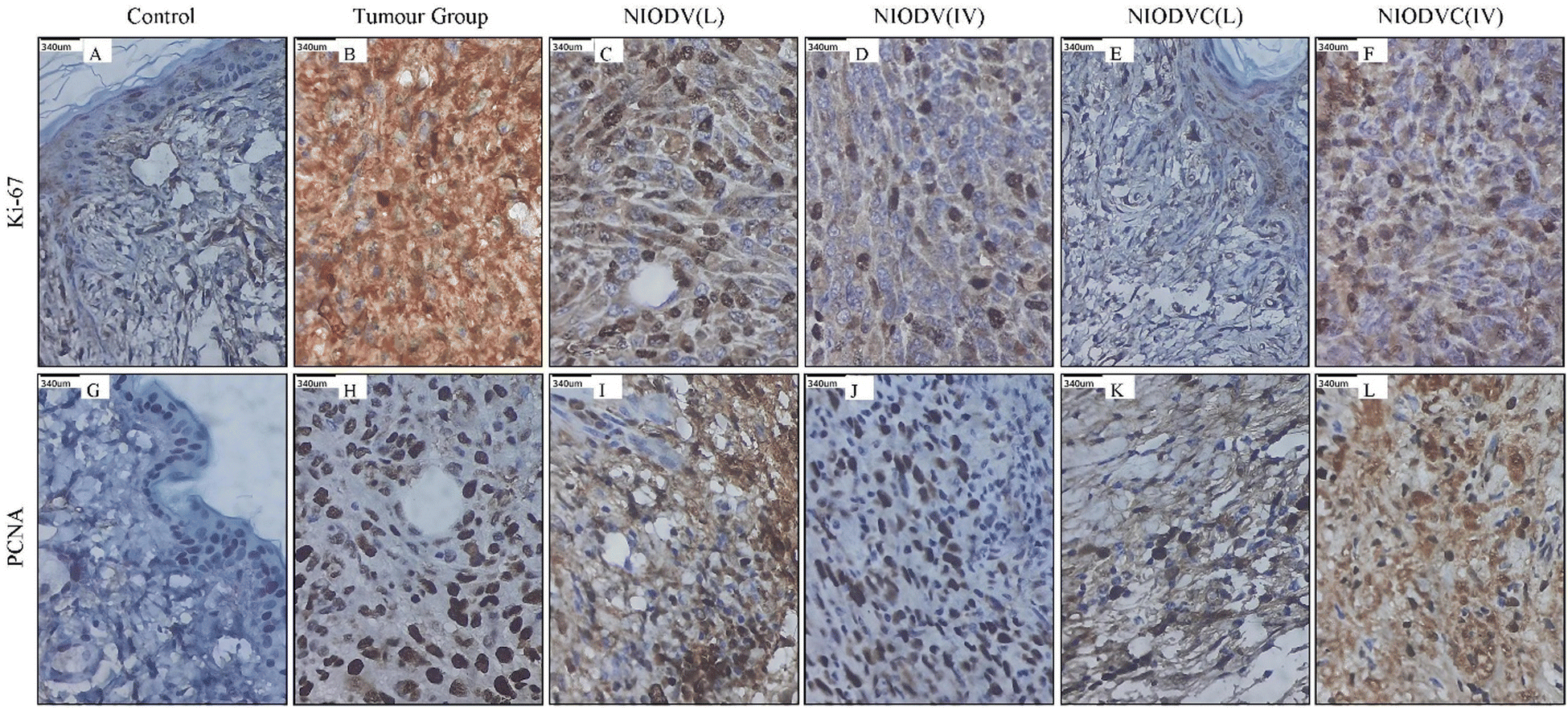
Fig. 11 shows the immunohistochemical staining of Ki67 and PCNA in control, tumor, and niosome treated groups respectively. There was a significant increase in the level of Ki67 and PCNA in the tumor group (Group-2), indicating active tumor cell proliferation. However, the level of these markers decreased significantly in niosome treated groups. In the present study, the local delivery of NIODVC (Group-5) resulted in the complete absence of Ki67 and PCNA expression in the ductal epithelial cells, which is in line with the tumor shrinkage volume, followed by NIODV (Group-4), NIODVC (Group-6), and NIODV (Group-3). Ki-67 is a 358 kDa nonhistone nuclear protein, which is overexpressed in actively dividing cells and it is widely used as a marker to assess the proliferative index in various cancers, including breast cancer.75 Proliferating cell nuclear antigen (PCNA) is a 36 kDa nonhistone nuclear cyclin protein, which is predominantly expressed in cells that undergo cell division and it is also used as a proliferative marker in several cancers.76 The present study revealed that conjugation of cetuximab helps to achieve the anti-proliferative effect of the treatment, as evidenced by the complete absence of Ki67 and PCNA expression in the ductal epithelial cells after local administration.
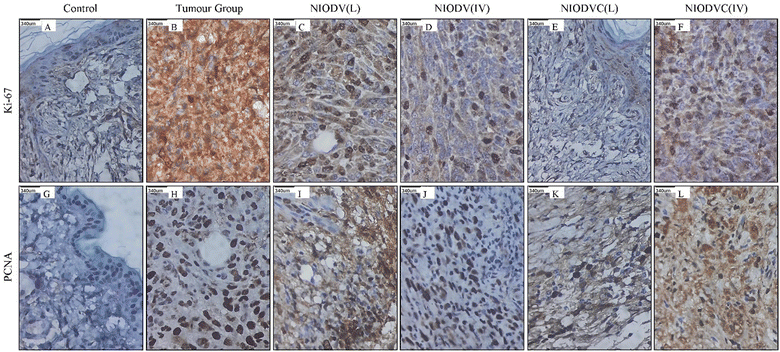 |
| | Fig. 11 Representative images of immunohistochemical staining for Ki-67 in control (A), tumor group (B), NIODV injected locally (C) and systemically (D), and NIODVC injected locally (E) and systemically (F) and for PCNA in control (G), tumor group (H), NIODV injected locally (I) and systemically (J), and NIODVC injected locally (K) and systemically (L). Brown staining indicates a positive expression of Ki-67 or PCNA. | |
4. Conclusion
In conclusion, the current study demonstrated the successful formulation of niosomes with high encapsulation efficiency (doxorubicin: 85% and vitexin: 67%) and stability. Cetuximab conjugated doxorubicin and vitexin loaded niosomes showed promising anti-tumor activity in 4T1 induced breast cancer mouse models. The release profile of doxorubicin, vitexin and the combination of both drugs exhibited a biphasic pattern, with an initial burst release followed by sustained slow release. The combined delivery of doxorubicin and vitexin from NIODV enhanced cellular uptake and cytotoxicity. NIODVC exhibited higher anti-tumor activity than other niosome formulations which are not conjugated with the antibody. Histopathological analysis revealed the potential anti-inflammatory effects of the niosome formulation on breast tissue, with downregulation of all genes tested.
Conflicts of interest
None.
Acknowledgements
Dr S. Malathi acknowledges UGC for the Dr D. S. Kothari Post-Doctoral scheme (No: F.4-2/2006 (BSR)/CH/18-19/0122). Dr V. Singaravel acknowledges RUSA 2.0 Research, Innovation and Quality Improvement, University of Madras. Ms V. Sisila acknowledges CSIR for financial support through the Senior Research Fellowship (CSIR-SRF-NET) (31/006(0432)/2017-EMR-I). Dr S. Narayana Kalkura acknowledges UGC for the UGC-BSR faculty fellowship (No: F.4-5(11)/2019(BSR). We extend our thanks to Dr Kaushik Biswas, Professor of Biological Sciences at the Unified Academic Campus of Bose Institute in Kolkata, India, for providing the 4T1 cell line. The authors thank Ms Divyarathna Bhuvarahamurthy for her help in reviewing the manuscript. The authors thank the Central Facility, CGC, Anna University, for electron microscopic analysis and the Department of Medical Biochemistry, PG-IBMS, University of Madras, for the animal facility.
References
- L. Wilkinson and T. Gathani, Br. J. Radiol., 2022, 95, 20211033 CrossRef PubMed.
- L. Minig, M. G. Patrono, N. Romero, J. F. Rodriguez Moreno and J. Garcia-Donas, World J. Clin. Oncol., 2014, 5, 86–92 CrossRef PubMed.
- S. Licata, A. Saponiero, A. Mordente and G. Minotti, Chem. Res. Toxicol., 2000, 13, 414–420 Search PubMed.
- P. S. Rawat, A. Jaiswal, A. Khurana, J. S. Bhatti and U. Navik, Biomed. Pharmacother., 2021, 139, 111708 CrossRef CAS PubMed.
- G. Du, H. Lin, M. Wang, S. Zhang, X. Wu, L. Lu, L. Ji and L. Yu, Cancer Chemother. Pharmacol., 2010, 65, 277–287 CrossRef CAS PubMed.
- M. Hamzeh-Mivehroud, S. Rahmani, M.-A. H. Feizi, S. Dastmalchi and M.-R. Rashidi, Arch. Pharm., 2014, 347, 738–747 CrossRef CAS PubMed.
- G. Scambia, F. O. Ranelletti, P. B. Panici, R. De Vincenzo, G. Bonanno, G. Ferrandina, M. Piantelli, S. Bussa, C. Rumi, M. Cianfriglia and S. Mancuso, Cancer Chemother. Pharmacol., 1994, 34, 459–464 CrossRef CAS PubMed.
- R. Václavíková, E. Kondrová, M. Ehrlichová, A. Boumendjel, J. Kovář, P. Stopka, P. Souček and I. Gut, Bioorg. Med. Chem., 2008, 16, 2034–2042 CrossRef PubMed.
- F. Babaei, A. Moafizad, Z. Darvishvand, M. Mirzababaei, H. Hosseinzadeh and M. Nassiri-Asl, Food Sci. Nutr., 2020, 8, 2569–2580 CrossRef CAS PubMed.
- X. Che, X. Wang, J. Zhang, C. Peng, Y. Zhen, X. Shao, G. Zhang and L. Dong, Am. J. Transl. Res., 2016, 8, 3319–3328 CAS.
- R. Bartelds, M. H. Nematollahi, T. Pols, M. C. A. Stuart, A. Pardakhty, G. Asadikaram and B. Poolman, PLoS One, 2018, 13, e0194179 CrossRef PubMed.
- A. Sankhyan and P. Pawar, J. Appl. Pharm. Sci., 2012, 2(6), 20–32 Search PubMed.
- D. Pando, M. Matos, G. Gutiérrez and C. Pazos, Colloids Surf., B, 2015, 128, 398–404 CrossRef CAS PubMed.
- C. A. Hunter, T. F. Dolan, G. H. Coombs and A. J. Baillie, J. Pharm. Pharmacol., 1988, 40, 161–165 CrossRef CAS PubMed.
- N. G. Kotla, B. Chandrasekar, P. Rooney, G. Sivaraman, A. Larrañaga, K. V. Krishna, A. Pandit and Y. Rochev, ACS Biomater. Sci. Eng., 2017, 3, 1262–1272 CrossRef CAS PubMed.
- R. Muzzalupo, L. Pérez, A. Pinazo and L. Tavano, Int. J. Pharm., 2017, 529, 245–252 CrossRef CAS PubMed.
- S. Khambata-Ford, C. T. Harbison, L. L. Hart, M. Awad, L.-A. Xu, C. E. Horak, S. Dakhil, R. C. Hermann, T. J. Lynch and M. R. Weber, J. Clin. Oncol., 2010, 28, 918–927 CrossRef CAS PubMed.
- J. A. Khan, R. A. Kudgus, A. Szabolcs, S. Dutta, E. Wang, S. Cao, G. L. Curran, V. Shah, S. Curley, D. Mukhopadhyay, J. D. Robertson, R. Bhattacharya and P. Mukherjee, PLoS One, 2011, 6, e20347 CrossRef CAS PubMed.
- S.-H. Ryu, S.-w Lee, Y.-J. Yang, S. Y. Song, J. H. Kim, E. K. Choi and S. D. Ahn, Lung Cancer, 2012, 77, 482–487 CrossRef PubMed.
- M. Z. Ahmad, S. Akhter, Z. Rahman, S. Akhter, M. Anwar, N. Mallik and F. J. Ahmad, J. Pharm. Pharmacol., 2013, 65, 634–651 CrossRef CAS PubMed.
- W. H. De Jong and P. J. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS PubMed.
- M. M. Joseph, S. R. Aravind, S. Varghese, S. Mini and T. T. Sreelekha, Colloids Surf., B, 2013, 104, 32–39 CrossRef CAS PubMed.
- J. Sakamoto, A. Annapragada, P. Decuzzi and M. Ferrari, Expert Opin. Drug Delivery, 2007, 4, 359–369 CrossRef CAS PubMed.
- H. Xie, S. Gu, J. Zhang, Q. Hu, X. Yu and J. Kong, C. R. Chim, 2018, 21, 104–111 CrossRef CAS.
- I. F. Uchegbu and S. P. Vyas, Int. J. Pharm., 1998, 172, 33–70 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- Y. Qian, M. Qiu, Q. Wu, Y. Tian, Y. Zhang, N. Gu, S. Li, L. Xu and R. Yin, Sci. Rep., 2014, 4, 7490 CrossRef CAS PubMed.
- B. Sivakumar, R. G. Aswathy, Y. Nagaoka, S. Iwai, K. Venugopal, K. Kato, Y. Yoshida, T. Maekawa and D. N. Sakthi Kumar, RSC Adv., 2013, 3, 20579–20598 RSC.
- B. Kocatürk and H. H. Versteeg, J. Vis. Exp., 2015, 8, 51967 Search PubMed.
- A. Manigandan, V. Handi, N. S. Sundaramoorthy, R. Dhandapani, J. Radhakrishnan, S. Sethuraman and A. Subramanian, Bioconjugate Chem., 2018, 29, 275–286 CrossRef CAS PubMed.
- A. Rodallec, C. Vaghi, J. Ciccolini, R. Fanciullino and S. Benzekry, PLoS One, 2022, 17, e0274886 CrossRef CAS PubMed.
- A. A. Samadani, N. Nikbakhsh, S. Fattahi, R. Pourbagher, S. M. Aghajanpour Mir, N. Mousavi Kani, Z. Abedian and H. Akhavan-Niaki, Int. J. Mol. Cell. Med., 2015, 4, 54–59 Search PubMed.
- N. Nordin, S. K. Yeap, H. S. Rahman, N. R. Zamberi, N. E. Mohamad, N. Abu, M. J. Masarudin, R. Abdullah and N. B. Alitheen, Molecules, 2020, 25, 2670 CrossRef CAS PubMed.
-
J. D. Bancroft and C. Layton, in Bancroft's Theory and Practice of Histological Techniques, ed. S. K. Suvarna, C. Layton and J. D. Bancroft, Elsevier, 8th edn, 2019, pp. 1–557 Search PubMed.
- E. A. Essa, Asian J. Pharm., 2010, 4(4), 227–233 CrossRef CAS.
- W. B. Liechty and N. A. Peppas, Eur. J. Pharm. Biopharm., 2012, 80, 241–246 CrossRef CAS PubMed.
- M. Chen, X. Liu and A. Fahr, Int. J. Pharm., 2011, 408, 223–234 CrossRef CAS PubMed.
- C. Detroja, S. Chavhan and K. Sawant, Sci. Pharm., 2011, 79, 635–651 CrossRef CAS PubMed.
- M. Ghazwani, U. Hani, A. Alam and M. H. Alqarni, Gels, 2023, 9, 401 CrossRef CAS PubMed.
- F. Rezaie Amale, S. Ferdowsian, S. Hajrasouliha, R. Kazempoor, A. Mirzaie, M. Sedigh Dakkali, I. Akbarzadeh, S. Mohammadmahdi Meybodi and M. Mirghafouri, Adv. Powder Technol., 2021, 32, 4711–4722 CrossRef CAS.
- S. Krasnici, A. Werner, M. E. Eichhorn, M. Schmitt-Sody, S. A. Pahernik, B. Sauer, B. Schulze, M. Teifel, U. Michaelis, K. Naujoks and M. Dellian, Int. J. Cancer, 2003, 105, 561–567 CrossRef CAS PubMed.
- Y. Li and A. K. Mitra, Pharm. Res., 1996, 13, 76–79 CrossRef CAS PubMed.
- K. Banerjee, S. Banerjee, S. Das and M. Mandal, J. Colloid Interface Sci., 2015, 453, 48–59 CrossRef CAS PubMed.
- B. Nasseri, Int. J. Pharm., 2005, 300, 95–101 CrossRef CAS PubMed.
- V. Sharma, S. Anandhakumar and M. Sasidharan, Mater. Sci. Eng., C, 2015, 56, 393–400 CrossRef CAS PubMed.
- W. Ren, G. Tian, S. Jian, Z. Gu, L. Zhou, L. Yan, S. Jin, W. Yin and Y. Zhao, RSC Adv., 2012, 2, 7037–7041 RSC.
- L. García-Fernández, J. Garcia-Pardo, O. Tort, I. Prior, M. Brust, E. Casals, J. Lorenzo and V. F. Puntes, Nanoscale, 2017, 9, 6111–6121 RSC.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- K. Tripathi and J. D. Driskell, ACS Omega, 2018, 3, 8253–8259 CrossRef CAS PubMed.
- X. Liu, M. Atwater, J. Wang and Q. Huo, Colloids Surf., B, 2007, 58, 3–7 CrossRef CAS PubMed.
- L. Thomas and V. Viswanad, Sci. Pharm., 2012, 80, 731–748 CrossRef CAS PubMed.
- A. A. Ramadan, S. A. Eladawy, A. S. M. A. El-Enin and Z. M. Hussein, J. Pharm. Invest., 2020, 50, 59–70 CrossRef CAS.
- V. Gugleva, S. Titeva, S. Rangelov and D. Momekova, Int. J. Pharm., 2019, 567, 118431 CrossRef CAS PubMed.
- C. Mircioiu, V. Voicu, V. Anuta, A. Tudose, C. Celia, D. Paolino, M. Fresta, R. Sandulovici and I. Mircioiu, Pharmaceutics, 2019, 11, 140 CrossRef CAS PubMed.
- M. Siddiqui, B. Abdellatif, K. Zhai, A. Liskova, P. Kubatka and D. Büsselberg, Cancers, 2021, 13, 1576 CrossRef CAS PubMed.
- M. Hemati, F. Haghiralsadat, F. Yazdian, F. Jafari, A. Moradi and Z. Malekpour-Dehkordi, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 1295–1311 CrossRef CAS PubMed.
- M. Giorgio, M. Pierantonio, S. Emanuela, C. Gaetano and G. Luca, Pharmacol. Rev., 2004, 56, 185 CrossRef PubMed.
- Y. Zhou, Y. E. Liu, J. Cao, G. Zeng, C. Shen, Y. Li, M. Zhou, Y. Chen, W. Pu, L. Potters and Y. E. Shi, Clin. Cancer Res., 2009, 15, 5161–5169 CrossRef CAS PubMed.
- M. Frattini, P. Saletti, F. Molinari and S. De Dosso, Gastrointest. Cancer: Targets Ther., 2015, 21–38 Search PubMed.
- C. Mamot, D. C. Drummond, C. O. Noble, V. Kallab, Z. Guo, K. Hong, D. B. Kirpotin and J. W. Park, Cancer Res., 2005, 65, 11631–11638 CrossRef CAS PubMed.
- T. Tanei, D. S. Choi, A. A. Rodriguez, D. H. Liang, L. Dobrolecki, M. Ghosh, M. D. Landis and J. C. Chang, Breast Cancer Res., 2016, 18, 6 CrossRef PubMed.
- N. E. Mohamad, N. Abu, S. K. Yeap, K. L. Lim, M. F. Romli, S. A. Sharifuddin, K. Long and N. B. Alitheen, Nutr. Metab., 2019, 16, 49 CrossRef PubMed.
- H. A. Wahba and H. A. El-Hadaad, Cancer Biol. Med., 2015, 12, 106–116 CAS.
-
A. Kumar, J. White, R. James Christie, N. Dimasi and C. Gao, in Annual Reports in Medicinal Chemistry, ed. R. A. Goodnow, Academic Press, 2017, vol. 50, pp. 441–480 Search PubMed.
- Y. Chen, X. Yuan, C. Pei, Z. Deng, X. Du, J. Liang, L. He and S. Hou, J. Funct. Foods, 2022, 95, 105190 CrossRef CAS.
- S. H. Tsai, S. Y. Lin-Shiau and J. K. Lin, Br. J. Pharmacol., 1999, 126, 673–680 CrossRef CAS PubMed.
- M. Feng, C. Feng, Z. Yu, Q. Fu, Z. Ma, F. Wang, F. Wang and L. Yu, Int. J. Clin. Exp. Med., 2015, 8, 346–357 CAS.
- X. Fu, W. Tan, Q. Song, H. Pei and J. Li, Front. Cell Dev. Biol., 2022, 10, 813457 CrossRef PubMed.
- M. E. Menezes, S. K. Das, L. Emdad, J. J. Windle, X. Y. Wang, D. Sarkar and P. B. Fisher, Adv. Cancer Res., 2014, 121, 331–382 CrossRef PubMed.
- S. Z. Eslami, L. E. Cortés-Hernández and C. Alix-Panabières, Cells, 2020, 9, 1836 CrossRef PubMed.
- H. Wei, Z. Wang, Y. Kuang, Z. Wu, S. Zhao, Z. Zhang, H. Li, M. Zheng, N. Zhang, C. Long, W. Guo, C. Nie, H. Yang and A. Tong, Front. Immunol., 2020, 11, 573823 CrossRef CAS PubMed.
- H. Huang, Sensors, 2018, 18, 3249 CrossRef PubMed.
- T. Liu, L. Zhang, D. Joo and S.-C. Sun, Signal Transduction Targeted Ther., 2017, 2, 17023 CrossRef PubMed.
- M. A. Cinelli, H. T. Do, G. P. Miley and R. B. Silverman, Med. Res. Rev., 2020, 40, 158–189 CrossRef CAS PubMed.
-
A. Sahin and H. Zhang, in Pathobiology of Human Disease, ed. L. M. McManus and R. N. Mitchell, Academic Press, San Diego, 2014, pp. 934–951 DOI:10.1016/B978-0-12-386456-7.03204-4.
- L. H. Malkas, B. S. Herbert, W. Abdel-Aziz, L. E. Dobrolecki, Y. Liu, B. Agarwal, D. Hoelz, S. Badve, L. Schnaper, R. J. Arnold, Y. Mechref, M. V. Novotny, P. Loehrer, R. J. Goulet and R. J. Hickey, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19472–19477 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a