
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of charge carrier dynamics in a Ti3C2Tx MXene for ultrafast photonics applications†
Ankita
Rawat
a,
Nitesh K.
Chourasia
 a,
Saurabh K.
Saini
b,
Gaurav
Rajput
a,
Aditya
Yadav
b,
Ritesh Kumar
Chourasia
c,
Govind
Gupta
b and
P. K.
Kulriya
*a
a,
Saurabh K.
Saini
b,
Gaurav
Rajput
a,
Aditya
Yadav
b,
Ritesh Kumar
Chourasia
c,
Govind
Gupta
b and
P. K.
Kulriya
*a
aSchool of Physical Sciences, Jawaharlal Nehru University, New Delhi 110067, India. E-mail: pkkulriya@mail.jnu.ac.in
bCSIR-National Physical Laboratory, New Delhi 110012, India
cPost-Graduate Department of Physics, Samastipur College (A Constituent College of L.N.M.U.-Darbhanga-846004, Bihar, India), Samastipur-848134, Bihar, India
First published on 2nd November 2023
Abstract
The rapid advancement of nanomaterials has paved the way for various technological breakthroughs, and MXenes, in particular, have gained substantial attention due to their unique properties such as high conductivity, broad-spectrum absorption strength, and tunable band gaps. This article presents the impact of the process parameters on the structural and optical properties of a Ti3C2Tx MXene for application in ultrafast dynamics. The synthesis of a singular-phase MXene derived from the MAX phase Ti3AlC2 was validated through X-ray diffraction (XRD) and Raman spectroscopy analyses. The complete etching of Al and increase in the interplanar distance are also observed upon centrifugation at a very high speed. The influence of varying centrifugation speeds, expressed in rotations per minute (rpm), has been employed to understand the ultrafast spectrum and charge carrier dynamics of the MXene. The investigation revealed the carrier lifetime is critically influenced by rotations per minute (rpm), e.g. showed by a notably swifter decay lifetime at 10 000 rpm compared to 7000 rpm. The electronic relaxation probed using the time-resolved photoluminescence (TRPL) technique exhibits average decay times (τav) of 5.3 ns and 5.13 ns at 7k and 10k rpm, respectively, which confirms that the optical properties of the MXene are strongly affected by the centrifugation speed. The synthesized MXene at 10k rpm typically suggests that radiative processes are due to a longer decay lifetime and it experiences fewer non-radiative losses, resulting in its enhanced luminescence properties.
1. Introduction
Recently, two-dimensional materials have received significant attention due to their unique physical and chemical properties compared to their bulk forms.1 Ever since the isolation of single-layer graphene in 2004, it has become a reference for all 2D materials and has opened the door to finding even more materials.2,3 Recently, various types of new 2D materials, such as transition metal dichalcogenides (TMDs), hexagonal boron nitride (h-BN), clays, transition metal oxides, etc., have been discovered.4 The new phases of 2D transition metal carbides, carbonitrides, and nitrides known as MXenes possess hydrophilic surfaces with abundant active sites, layered morphology, excellent flexibility, surface termination group-controlled tunable metallic or semiconducting behaviour, and good solution processability, making them highly suitable for supercapacitors,5 sensors,6,7,40 energy storage devices,1 optoelectronics,8 and biomedical applications.9 An MXene has a general formula of Mn+1XnTx, where n = 1, 2, or 3, M stands for an early transition metal, X represents nitrides, carbonitrides, and carbides, and Tx represents various surface termination groups such as –OH, –F, and –O.10 MXene materials can be produced by the removal of A group atoms (especially group 13th and 14th elements like Al, Si, Ga, etc.) from their corresponding MAX phases (Mn+1AXn). The “M” atoms of MXenes are organized in a packed hexagonal structure with a P63/mmc symmetry, similar to the MAX phases.11 Ideally, the M atoms are engaged in bonding with the “X” atoms between the octahedral sites.12 The M–X bond is a combination of ionic, covalent, and metallic characteristics, while the M–A bond is entirely metallic in nature. In contrast, the bonds that hold the layers together in Mn+1AXn phases are much stronger than the van der Waals interactions observed in 2D materials like graphene and TMDs. Due to the robust nature of these bonds, it is not possible to mechanically exfoliate Mn+1AXn phase compounds into 2D layers.13 In the last decade, hundreds of MXenes and their precursor compositions have been suggested by computational studies and more than 30 MXene phases have been experimentally synthesized. However, there are only a few reports on the optimization of the process parameters for the synthesis of impurity free single phase MXenes. For example Zhang et al.14 synthesized a single-phase MXene by exfoliation at different temperatures ranging from 30 °C to 55 °C and delamination using sonication under Ar flow for one hour, followed by centrifugation at 3500 rpm for one hour. Lipatov et al.15 optimized the LiF–HCl etching of Ti3AlC2 for the production of large high-quality MXene flakes having low defect concentration for electronic property studies. Maleski et al.'s16 research provided insights into how an MXene's flake size can be controlled by different sonication times and centrifugation speeds and investigated the effect of the MXene flake size on the optical and electrochemical properties of the MXene. The comparative studies on the production of a Ti3C2Tx MXene by HF etching and LiF–HCl etching showed that the LiF–HCl etched MXene can be delaminated easily by sonication in DI water, whereas the HF etched MXene requires additional steps for intercalation with organic molecules to improve its delamination.17 Kumar et al.18 optimized the rotational speed and observed improvement in the energy storage capacity with the highest specific capacitance and the lowest internal resistance at 10k rpm due to the enhancement of the surface area, capacitance, and interfacial contact of the device. Thus, the properties of an MXene critically depend upon the synthesis parameters such as the centrifugation speed, sonication time, molarity of HCl, chemical composition of the MAX phase, flake size, and intercalated species. Therefore, in the present study, the effect of the centrifugation speed on the structural and optical properties of the Ti3C2Tx MXene is investigated. Apart from the optimization of the synthesis process of the MXene, the effect of process parameters on the optical properties of Ti3C2Tx is also investigated in detail. Jhon et al.19 reported MXene-based saturable absorbers for the generation of ultrafast pulses at 1.5 μm and 2.0 μm bands. The dynamic behavior and optical characteristics of the Ti3C2Tx MXene can be investigated through the ultrafast examination of the material at a high energy pump wavelength. Researchers can explore diverse processes and elicit quick responses by employing a high-energy pump, giving insight into the material's distinctive properties. This study offers a greater comprehension of the possible uses of the Ti3C2Tx MXene and lays the way for ground-breaking results in the fields of materials science and photonics. The photoexcited carrier dynamics in the Ti3C2Tx MXene investigated using ultrafast transient absorption spectroscopy (UTAS) showed a fast relaxation process followed by a slower decay process due to the trapping of photoexcited carriers by surface states.20 The observation of efficient charge separation and a long charge carrier lifetime demonstrated the potential of this MXene in photovoltaic and photocatalytic applications. Colin-Ulloa et al.21 investigated the dynamics of plasmons and free carriers generated by a laser pulse using time-resolved spectroscopy. Fu et al.22 probed the dynamics of the light-MXene interaction and flash thermal dissipation using ultrafast transient spectroscopy. To the best of our knowledge, the effect of synthesis parameters on the optical properties of Ti3C2Tx has not been investigated using ultrafast transient absorption spectroscopy. Zhang et al.23 reported the ultrafast transient spectra (450–1100 nm) of the Ti3C2Tx MXene in response to light excitations. They also reported photo-induced transient spectra at 0.5, 10, and 30 ps upon excitation at 325, 500, and 780 nm, respectively. Although they have recorded a wide spectrum (450–1100 nm), the article is more focused on the ultrafast transient spectra of the Ti3C2Tx MXene and its dynamics in the visible region. This manuscript is mainly focused on the ultrafast transient spectra and dynamics of the Ti3C2Tx MXene in visible, near-infrared and far-infrared (400 nm to 1400 nm) regions. To record this wide spectrum, we have used the pump wavelengths of 325 nm and 550 nm, respectively. Apart from this, we have also synthesized the Ti3C2Tx MXenes at different centrifugation speeds and compared their charge carrier dynamics.In this article, technologically important impurity-free titanium carbide (Ti3C2Tx) MXene is synthesized by LiF/HCl etching and controlled delamination, employing various sonication times and centrifugation speeds. The structural, microstructural, compositional, and optical properties of the as-prepared MXene have been investigated using XRD, micro-Raman spectroscopy, SEM, EDS, and photoluminescence and time-resolved photoluminescence (TRPL) spectroscopy. Furthermore, the excited state dynamics and electronic structures of the MXene were also probed using ultrafast transient absorption spectroscopy in the femtosecond to nano-second time scale. The ultrafast transient absorption spectra and carrier dynamics of samples S1 and S2, synthesised at 7000 rpm and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, respectively, are revealed at a 350 nm (3.54 eV) pump wavelength in the broad visible to near-infrared probe range (450–1500 nm), which is greater than the bandgap of the Ti3C2Tx MXene.23 We observed ground-state bleaching (GSB) and excited-state absorption (ESA) with a fast decay profile. Furthermore, sample S2 (10000 rpm) has a shorter charge carrier decay lifetime than sample S1 (7000 rpm), which is further assessed using TRPL spectroscopy.
000 rpm, respectively, are revealed at a 350 nm (3.54 eV) pump wavelength in the broad visible to near-infrared probe range (450–1500 nm), which is greater than the bandgap of the Ti3C2Tx MXene.23 We observed ground-state bleaching (GSB) and excited-state absorption (ESA) with a fast decay profile. Furthermore, sample S2 (10000 rpm) has a shorter charge carrier decay lifetime than sample S1 (7000 rpm), which is further assessed using TRPL spectroscopy.
2. Experimental details
2.1. Materials
Analytical grade reactants such as titanium aluminium carbide Ti3AlC2 (>90%, <40 μm), lithium fluoride powder (300 mesh), hydrochloric acid (HCl, conc. 35%) and deionized water (pH ∼ 7) procured from Sigma Aldrich (India), Thermo Fisher Scientific India Pvt. Ltd and Organo Biotech Laboratories Pvt. Ltd, respectively, were used throughout the experiments.2.2. Ti3AlC2 etched through LiF/HCl
A single-phase impurity free MXene has been synthesized using a wet chemical method. Fig. 1 provides an overview of the steps involved in the synthesis of the MXene. Firstly, Al atoms were removed from their precursor MAX phase Ti3AlC2 through selective etching with an LiF/HCl etchant. The etchant contained 0.8 g of LiF dissolved in 10 mL of an aqueous hydrochloric acid (9 mol L−1) solution, and after 30 minutes of stirring, 0.5 g of Ti3AlC2 MAX phase powder was gradually added to the reaction mixture within the course of 15 minutes. After that, a black dispersion was formed after being stirred at room temperature for 24 h. The raw product was cleaned many times with deionized water and centrifuged at 4500 rpm until the pH reached nearly six. The high-yield precipitated MXene, also known as the sediment, is then obtained after multiple washes at 4500 rpm centrifugation, as illustrated in Fig. 1.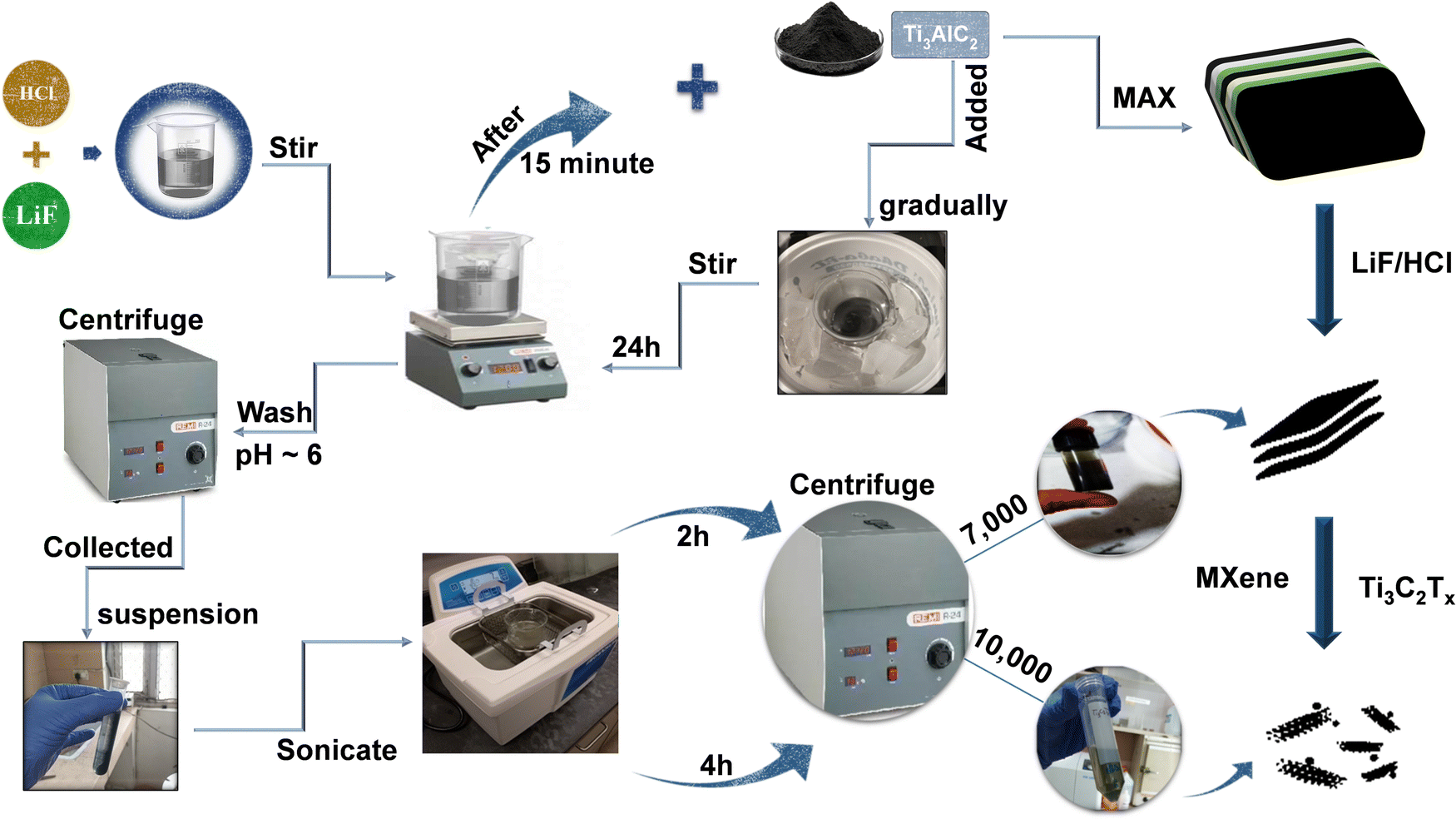 | ||
| Fig. 1 The process for the synthesis of an impurity free single phase Ti3C2Tx MXene. | ||
The recovered sediment is then mixed with DI water, sonicated for 2 h, and then centrifuged at 7000 (7k) rpm for 30 minutes. The obtained dark greenish supernatant is named S1. To further improve the quality of the synthesised MXene, the sonication time is increased to 2 h followed by centrifugation at a higher speed of 10000 (10k) rpm for 30 minutes. The obtained light greenish supernatant is named S2. This helps in the dispersion of the sheets into individual layers and the formation of an impurity free MXene. In order to obtain a completely exfoliated MXene, a series of repeated centrifugations at progressively higher speeds with higher sonication times were also performed. Finally, the Ti3C2Tx MXene sediment was dried in a hot vacuum oven and kept in a N2 environment. The S1 and S2 supernatants were used for further characterization.
2.3. Materials characterization
The diffraction pattern of precursor Ti3AlC2 was recorded using an X-ray diffractometer, whereas the grazing incidence X-ray diffraction (GIXRD) patterns of Ti3C2Tx MXene supernatant S1 and S2 were recorded by using a PANalyticalX’Pert PRO diffractometer equipped with a Cu Kα (λ = 0.15406 nm) X-ray source in the range of 5° to 60° with a step size of 0.02° and a scan time of 5 s at a grazing angle of 1°. Raman spectra were recorded on a Raman spectrometer (WITec alpha 300 RA), using 532.5 nm laser excitation. The surface morphology and elemental composition of the Ti3C2Tx MXene were characterized by using scanning electron microscopy (SEM, ZEISS EVO 40) and energy dispersive spectroscopy (EDS). The optical features were studied using a UV-Vis spectrophotometer (HITACHI U-3900), photoluminescence spectroscopy (FLS980 D2D2, Edinburgh, a double monochromatic system with a xenon lamp) and TRPL 266 nm laser spectroscopy. The ultrafast transient absorption spectroscopy (UFTAS) technique utilizes a Ti:Sapphire laser system, with a Coherent Micra (oscillator) and a Coherent Legend USP (amplifier). The oscillator produces mode-locked Gaussian-shaped pulses with a pulse width of 35–45 fs and a repetition rate of 80 MHz at 800 nm with a tuning capability from 780 to 810 nm. The amplifier provides a pulse width of 35–45 fs with a repetition rate of 1 kHz centered at 800 nm with an average output power of 3–4 mJ. The output of the amplifier beam was then split into two parts using beam splitters. One portion was directed to the operational parametric amplifier (OPA) to generate a pump, while the other portion was delayed and entered the spectrometer (Helios from Ultrafast Systems Inc.) after passing through a sapphire disk to produce a white light continuum (WLC) that serves as the probe beam,24 which is used to perform kinetics and carrier dynamics studies. The fitting process is carried out using the Surface Xplorer and Origin 9.1 software programs.3. Results and discussion
3.1. Structural and microstructural properties
X-ray diffraction (XRD) and Raman spectroscopy techniques are used to probe the structural properties of the Ti3C2Tx MXene and its supernatants. Fig. 2 shows the XRD patterns of (a) Ti3AlC2 powder, Ti3C2Tx MXene (before sonication), Ti3C2Tx MXene supernatants S1 and S2 (after sonication) and (b) the Raman spectra of Ti3C2Tx MXene supernatants S1 and S2. It can be seen that the diffraction peak corresponding to the MAX phase of Ti3AlC2 appeared at 2θ ≈ 9.8° and was assigned to the (002) plane and a new broad peak also appeared at a lower angle 2θ ≈ 6.5° after etching with the HCl + LiF solution for 24 h, followed by sonication and centrifugation at 7k rpm. In addition, a small sharp peak of Al (104) at 2θ ≈ 38.6° is also observed. The presence of these peaks is clear evidence that the MAX phase is not completely etched at 7k rpm. When the centrifugation speed is increased from 7k rpm to 10k rpm, the peaks corresponding to the Ti3AlC2 MAX phase, the (002) peak and the Al (104) peak, disappeared and two distinct peaks at 2θ ≈ 6.1° and an insignificant peak of Al at 38.6°, assigned to the (002) and (104) planes of the Ti3C2Tx MXene, respectively, are observed. The lower angle shifting of the MXene is direct evidence for the synthesis of the single phase Ti3C2Tx MXene, which is also consistent with previous studies on MXenes.18,25,26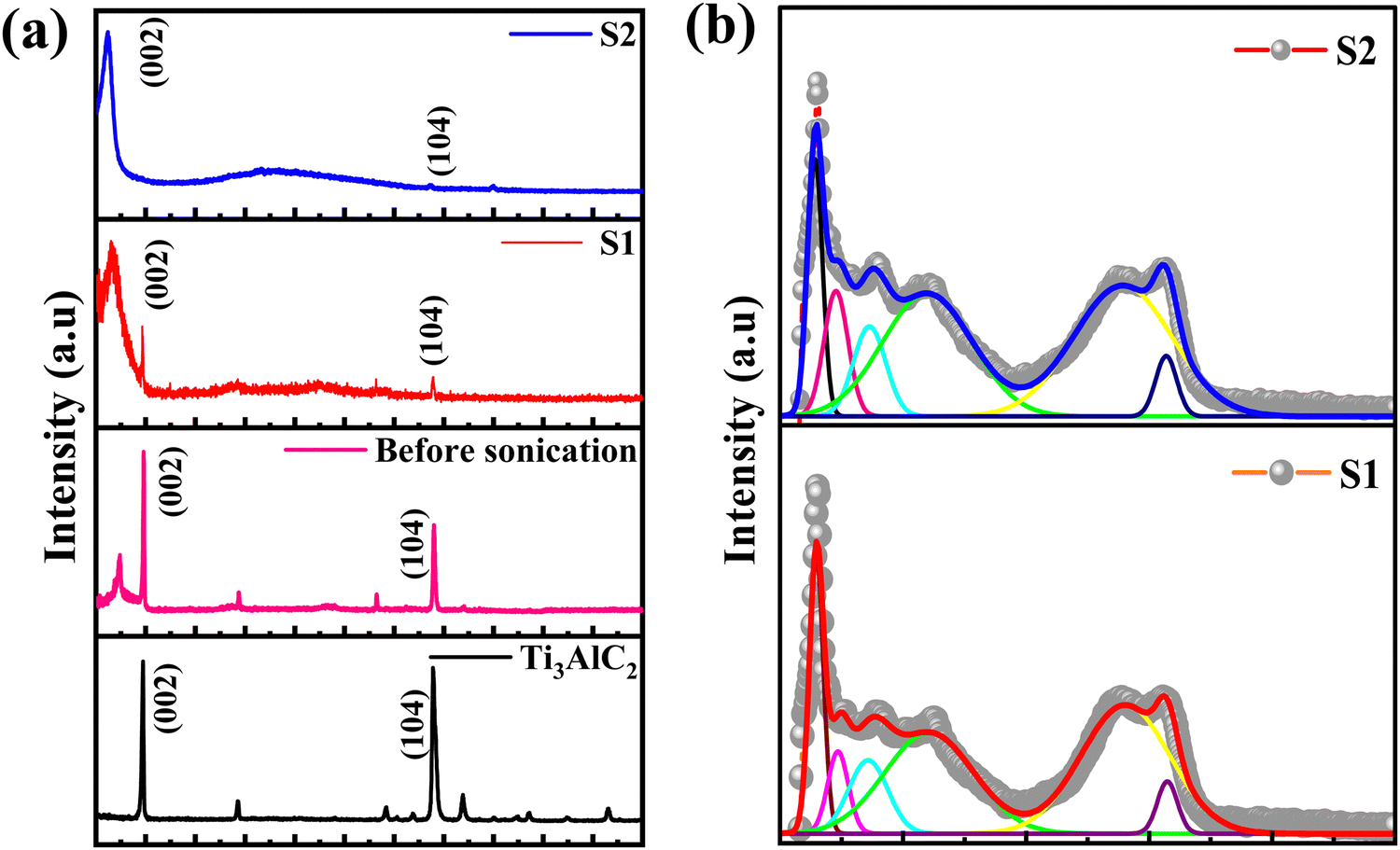 | ||
| Fig. 2 XRD patterns of (a) Ti3AlC2 powder, Ti3C2Tx MXene (before sonication), Ti3C2Tx MXene supernatants S1 and S2 (after sonication) and (b) Raman spectra of Ti3C2Tx MXene supernatants S1 and S2. | ||
It may be noted that the position of the (002) diffraction peak is shifted from 6.5° (for S1) to 6.1° (for S2) upon sonication and centrifugation of the Ti3C2Tx sample, indicating an increase in the interplanar distance from 9.12 Å (for Ti3AlC2) to 13.58 Å (for S1) and 14.47 Å (for S2), respectively. The calculated values of the lattice parameters are 18.03 Å, 27.17 Å and 28.95 Å for Ti3AlC2, S1 and S2 samples, respectively, which are consistent with earlier reported data on Ti3C2Tx crystal structures.27,28 The structural parameters of the as-synthesized MXenes are shown in Table 1.
| Sample | Plane | 2θ (°) | d-spacing (Å) | c (Å) |
|---|---|---|---|---|
| MAX (Ti3AlC2) | (002) | 9.8 | 9.12 | 18.03 |
| MXene (Before sonication) | (002) | 7.4 | 11.93 | 23.87 |
| MXene at 7000 rpm (S1) | (002) | 6.5 | 13.58 | 27.17 |
| MXene at 10000 rpm (S2) |
(002) | 6.1 | 14.47 | 28.95 |
Raman spectroscopy that provides information about the short-range structural ordering is also used for characterizing the structural and vibrational properties of the Ti3C2Tx MXene. The Raman spectra of the initial MAX phase Ti3AlC2 is shown in ESI† [Fig. S4]. The Raman peaks of Ti3AlC2 located around the wavenumbers of 198 cm−1, 268 cm−1 and 621 cm−1, respectively, are observed due to the shear and longitudinal oscillations of the Ti and the Al atoms.25 In particular, the peak located at 268 cm−1 is associated with the vibrations of Al as reported earlier.26 The absence of the Al peak in the Raman spectrum [Fig. 2(b)] is a direct outcome of the extensive etching of the Al atoms that leads to the formation of the MXene structure.27 The Raman spectrum of the Ti3C2Tx MXene shows several prominent peaks due to its different vibrational modes. The most intense peak at 145 cm−1 is attributed to the doubly degenerated Eg modes, which correspond to the vibration of the Ti atoms in the Ti3C2Tx layers.28 The other peaks are observed at 235 cm−1, 357 cm−1 and 606 cm−1 for S1 and at 227 cm−1, 363 cm−1 and 597 cm−1 for S2, respectively. The peaks that appeared at ∼230 cm−1 and ∼360 cm−1 are associated with the Ti–C vibrations and O atom vibrations, whereas the peak observed at ∼600 cm−1 is related to the Eg vibrations of the carbon in the Ti3C2Tx MXene that has terminal groups.18,29 Two additional bands appeared at 1392 cm−1 and 1565 cm−1 for S1 and at 1371 cm−1 and 1548 cm−1 for S2 and are assigned as D and G bands, respectively.28 Here, the D band represents the vacancies and other structural defects in the Ti3C2Tx MXene, whereas the G band represents the in-plane stretching vibration of the carbon in the MXene layers.30 The observation of the D and G bands between 1000 and 1800 cm−1 indicates that S1 and S2 have an almost similar structure.18,28 Furthermore, the observation of a lower intensity D band as compared to the G band is also in good agreement with the previous reported literature.31
The compositions and surface morphologies of the MAX phase and the as-synthesized MXenes were investigated using energy dispersive spectroscopy (EDS) and scanning electron microscopy (SEM), respectively. The microsheets of the Ti3AlC2 MAX phase exhibit a modest laminated pattern on their side surfaces [Fig. 3(a)].32 After LiF/HCl etching, the morphologies of the MXene powder and S1 and S2 MXene supernatants have completely developed into an accordion-like laminated structure with fabric-like layered features [Fig. 3(b)–(d)].33 This demonstrates the successful exfoliation of the MAX phase into a multilayer MXene. The elemental distributions of the Ti3AlC2 MAX phase and Ti3C2Tx MXenes (S1 and S2) determined using an EDX detector (see ESI,† Fig. S1–S3) show that the laminated Ti3C2 primarily consists of Ti and C, along with small amounts of O, F and Cl elements.34
 | ||
| Fig. 3 Surface morphologies of (a) the Ti3AlC2 MAX phase, (b) the Ti3C2Tx MXene sediment, and Ti3C2Tx MXene supernatants (c) S1 and (d) S2 after sonication. | ||
The existence of F and O may be related to the terminal groups, while the presence of Ti and C is predictable. However, the identification of low quantities of Al and Cl is most likely attributable to an incomplete etching process or a residue that was not washed away, which leads to impurities or the formation of a terminal group.31 The concentrations of different elements are shown in the ESI† (see Fig. S1–S3) for the Ti3AlC2 MAX phase, S1, and S2, respectively. It was found that the majority of the Al atoms are removed during the etching process, as S1 has 1.29 atomic percentage of Al and S2 has 0.20 atomic percentage, in comparison to the Ti3AlC2 MAX phase, which has 20.85 atomic percentage of Al. It is important to note that S2 contains very a low concentration of Al element and almost a single phase of the Ti3C2Tx MXene. EDX analysis confirmed that the major elements of Ti3C2Tx in both supernatants S1 and S2 are Ti, C, and O. In addition, the traces of F and Cl were also found as elements of the terminal groups in both MXene supernatants S1 and S2, respectively. During the wet-etching process, the surface of the MXene predominantly adsorbs hydroxyl (OH), fluorine (F), and oxygen (O) groups.35
3.2. Optical properties
The optical properties and electronic structure of the Ti3C2Tx MXene have been probed using photoluminescence spectroscopy, time-resolved photoluminescence (TRPL) spectroscopy and ultrafast transient absorption spectroscopy techniques. According to Zhang et al.,36 the band gap of Ti3C2Tx MXene powder is just about 0.1 eV, which is not large enough to emit light in the visible or near-infrared region. Xue et al.37 suggested that the luminescence properties of the Ti3C2Tx MXene depend on the size and surface defects, which lead to a strong quantum confinement effect due to its small lateral size. Thus, a reduction in the size of the MXene leads to an increase in its bandgap, enhancing its light emitting efficiency. Apart from that, the luminescence of the Ti3C2Tx MXene also arises from the surface defects, which create energy states within the bandgap of the MXene that assist in light emission when it is excited. The surface defects can be caused by various factors, such as oxidation, surface functionalization, or structural defects.38The imperfections in the supernatant of the Ti3C2Tx MXene provide sites where oxygen can be adsorbed onto its surface or solution, leading to a reaction with the Ti atoms and the subsequent formation of TiOx.36 Thus, the surface defects play important role in the determination of the optical properties of the Ti3C2Tx MXene. The photoluminescence spectroscopy measurement was performed in the wavelength range of 390 nm to 800 nm with an excitation wavelength of 374 nm at room temperature. The photoluminescence spectra of the MXene supernatants (S1 and S2) are shown in Fig. 4(a) and (b). The two prominent emission peaks of the MXene supernatants (S1 & S2) were positioned at 428 nm and 434 nm. The deconvolution and fitting of broad PL spectra unveiled three well-resolved shallow trap levels at the wavelengths of 460 nm, 490 nm, and 550 nm for supernatant S1. Additionally, supernatant S2 displayed two peaks at 408 nm and 460 nm. The peaks observed at high energies (428 nm for S1 and 434 nm for S2), attributed to band edge luminescence, are assigned to the Ti3C2Tx MXene supernatant. Conversely, the lower-energy peaks are attributed to the presence of oxygen vacancies and surface functional groups, arising from varying experimental conditions such as temperature, sonication, solvent, and aging time during synthesis.39 Moreover, the existence of oxygen vacancy states within the MXene supernatant can play a role in the emergence of visible light absorption bands.37 The absorption spectra of MXene supernatants S1 and S2 are shown in the ESI† [Fig. S5].
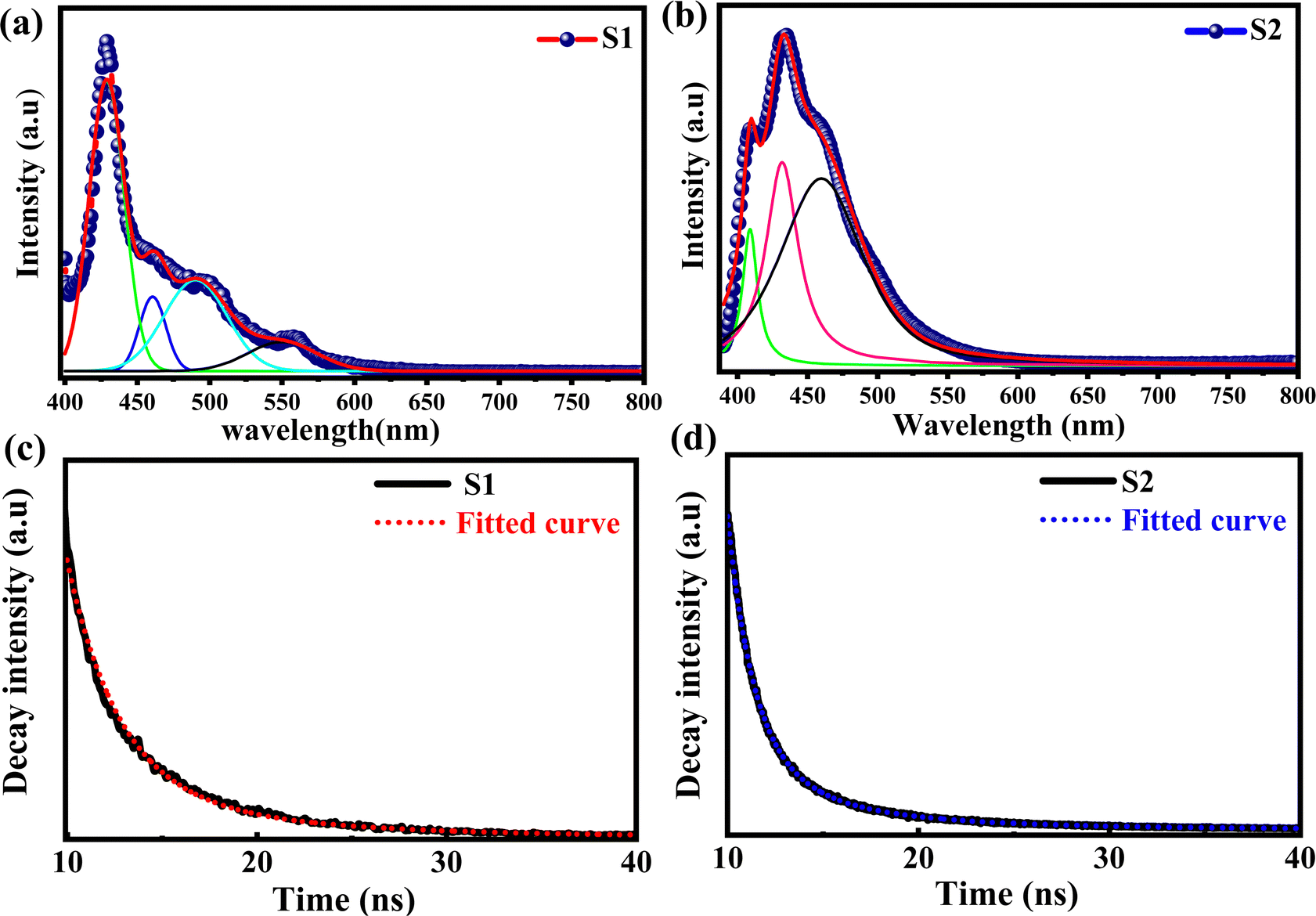 | ||
| Fig. 4 Emission spectra of Ti3C2Tx MXenes (a) S1 and (b) S2. Time-resolved photoluminescence (TRPL) spectra of Ti3C2Tx MXene supernatants, (c) S1 and (d) S2. | ||
To analyse dynamical processes such as excitation, relaxation, and recombination, time-resolved photoluminescence (TRPL) spectroscopy of the Ti3C2Tx MXene has been performed.
The TRPL spectra of Ti3C2Tx MXenes synthesized at 7k rpm and 10k rpm are shown in Fig. 4(c) and (d). The decay time (τ) of the photoexcited charge carrier was estimated through bi-exponential TRPL decay fitting by using eqn (1),
| I(t) = A + B1exp(−t/τ1) + B2exp(−t/τ2) + B3exp(−t/τ3) + … Biexp(−t/τi) | (1) |
| τav = B1τ12 + B2τ22 + …Biτn2/B1τ1 + B2τ2 + ⋯ + Biτn | (2) |
A slower rate of luminous signal decay can be inferred from the TRPL spectrum of supernatant S1 which exhibits a greater lifespan decay. This gives the impression that the luminous material stays in its excited state for a longer period of time before falling back to its ground state.
In ultrafast spectroscopy, a pump–probe setup uses a special kind of powerful laser to study the behavior of materials over very short time periods. Basically, this laser has two components: the “pump” and “probe”, which totally depend on the wavelength of the light source. Generally, the pump light is much stronger and brighter than the probe light. To get the exact information about the properties of the material, both the light must hit the same spot of the sample, which is a very crucial parameter. The wavelength corresponding to the pump of the ultrafast spectroscope is responsible for exciting the sample and results in the changes in its state properties. However, the wavelength corresponding to the probe is used to detect these perturbations. In the present article, we synthesized a 2D Ti3C2Tx MXene using an etching process with the LiF salt and subjected it to sonication processes for intercalation and delamination. Remarkably, when we exposed a 350 nm pump wavelength to the MXene supernatant and recorded its spectrum over a broad-spectrum range, especially in the visible to near-infrared (VIS-NIR) region, we observed worthy transient absorption responses. To investigate these responses, we employed transient absorption spectroscopy based on pump–probe techniques and examined the kinetics of Ti3C2Tx MXene's excited-state dynamics. Furthermore, we systematically investigated alternative low-energy injection profiles in the ultrafast transient absorption spectrum using a 550 nm pump wavelength, and extended our examination across a broad spectrum range encompassing visible to near-infrared (VIS-NIR). However, no discernible transient absorption responses were observed under these conditions. Consequently, we abstained from exploring transient absorption and kinetic profiles of MXene supernatant at a 550 nm pump wavelength with a broad spectrum probe (VIS-NIR).
Furthermore, the transient absorption study of MXene supernatant materials is carried out through ultrafast transient absorption spectroscopy. The ultrafast spectrum is obtained for MXene supernatants S1 and S2 synthesized at two different rpm values (7k and 10k). The MXene materials were subjected to excitation with a 350 nm pump wavelength at an average pump power of 1 mW. The spectrum of supernatant S1 was recorded across a wide visible-to-near-infrared probe range (450 nm to 1450 nm), as illustrated in Fig. 5(a) and (b). When the MXenes absorb photons from ultraviolet (UV) light, internal electronic excitations occur. This means that the energy from the absorbed UV photons is transferred to the electrons within the MXene material, causing them to move to higher energy states.
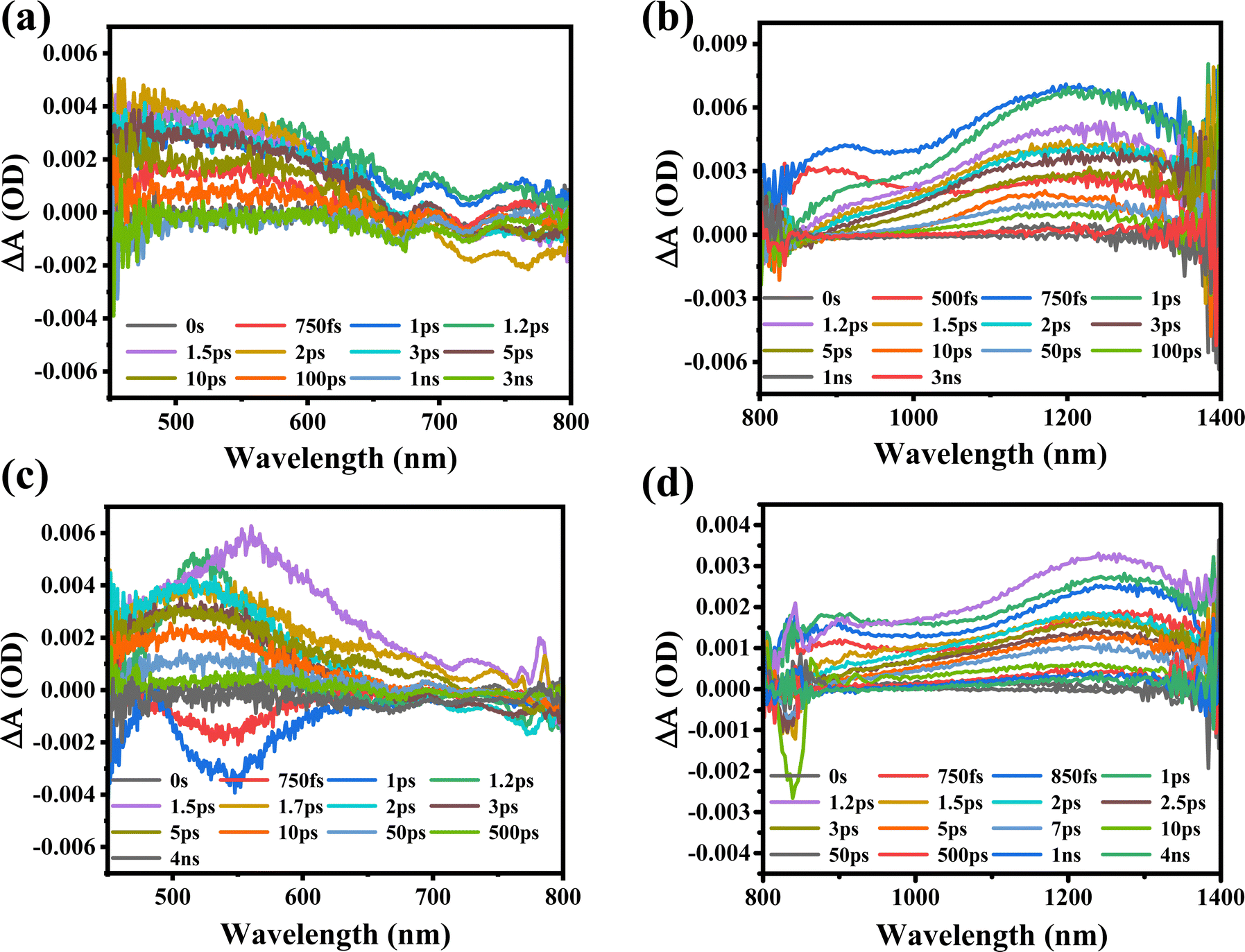 | ||
| Fig. 5 The transient absorption spectra for MXene supernatants S1 and S2 is excited at a 350 nm pump wavelength in both visible and NIR ranges, respectively. (a) supernatants S1 (7k) in the visible ranges (b) supernatants S1 (7k) in the NIR ranges (c) supernatants S2 (10k) in the visible ranges (d) supernatants S2 (10k) in the NIR ranges. | ||
The excitation of electrons can lead to two types of electronic absorption phenomena in the visible to near-infrared regions due to the specific Ti–C (titanium–carbon) structure within the MXenes. Photo-induced absorption (PIA) and ground state bleaching (GSB) are clearly observed in the visible probe range [Fig. 5(a)]. GSB occurs when the ground state of the material, which is the lowest energy state where electrons are typically found, loses some of its population due to electronic excitation, and this loss of population leads to a decrease in absorption in the visible range. This complex behavior could lead to the formation of trap-states due to bigger particle sizes, agglomeration, and incomplete etching of the MAX phase of the MXene at 7k rpm (S1). The trap states in the S1 sample are associated with bigger particle sizes, agglomeration, and incomplete etching of the MAX phase. The trap states are localized energy states that can capture charge carriers, slowing down their movement and recombination as shown in Table 2. However, the mention of “trap-states” suggests that some of these excited electrons may not return to their original state efficiently due to the presence of defects in the material. In the case of NIR probing, a broad PIA in the entire range of 800 nm to 1450 nm is also detected due to electron–phonon scattering [Fig. 5(b)]. When the probe wavelength is shifted to the NIR range, it is important to consider that the energy of the NIR photons is lower compared to that of visible light. In this context, the increase in the PIA signal with an increase in probe wavelengths indicates that the material's charge carrier density in the excited state is growing.
Furthermore, the MXene supernatant S2 is excited at the same pump wavelength, and spectra are also recorded in both visible and near-infrared probe ranges between 450 nm and 1450 nm, as shown in Fig. 5(c) and (d). The observation of a widespread photo-induced absorption (PIA) in the visible probe spectrum (450 nm to 800 nm) clearly indicates an electron–phonon interaction in the excited states. This indicates an increase in absorption due to the presence of hot carriers, showing that the material absorbs more light when excited. Owing to the migration of excited charge carriers from the valence to conduction bands, the ground state bleaching (GSB) observed between 750 fs and 1 ps probes a delayed periodicity, as depicted in Fig. 5(c). This observation implies a reduction in micro-strain and lattice relaxation, indicating more efficient charge carrier dynamics. The spectrum is also recorded in the broad NIR probe regime between 800 nm and 1450 nm, where PIA was observed across the probe range. At higher probe wavelengths, the transient absorption increases with the maximum absorption observed at 1230 nm [Fig. 5(d)]. The broad PIA is observed due to the electron–phonon scattering in the excited states of the conduction band. As the probe wavelength shifts to the NIR range, the lower energy of the probing light enables more charge carriers to transition to the excited state, as they now possess sufficient energy to bridge the energy gap.
Furthermore, the kinetic profiles of both probes are fitted through the model fitted by eqn (3) using the “Surface Xplorer” software and the fitting parameters such as t1, t2, and a1, a2 are calculated and are listed in Table 2. The times t1 and t2 represent the initial rise time and charge recombination time, respectively. These measurements show how quickly the charge carriers (such as electrons or holes) are generated and recombine or settle back to their original state. Based on the observations one can conclude that longer times may indicate less efficient charge carrier behavior. The following function enables fitting a kinetic trace with the sum of convoluted exponentials at a selected wavelength:
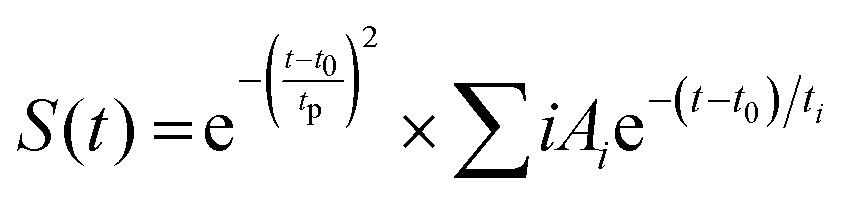 | (3) |
| Sample@pump wavelength (nm) | Wavelength (nm) | t 1 (ps) | a 1 (%) | t 2 (ps) | a 2 (%) |
|---|---|---|---|---|---|
| S1 (7k rpm) 350 nm, visible regime | 500 | 7.56 | 64.4 | 135 | 35.6 |
| 600 | 10.3 | 59.6 | 170 | 40.4 | |
| 755 | 1.78 | 93.2 | 3.15 | 6.8 | |
| S1 (7k rpm) 350 nm, NIR regime | 900 | 1.17 | 95.9 | 1.67 | 4.09 |
| 986 | 1.2 | 87.5 | 136 | 12.5 | |
| 1200 | 1.39 | 78.4 | 46.8 | 21.6 | |
| S2 (10k rpm) 350 nm, visible regime | 525 | 0.23 | 65.8 | 12 | 34.2 |
| 600 | 0.186 | 83.3 | 15.1 | 15.7 | |
| 728 | 0.187 | 85.1 | 1.93 | 14.9 | |
| S2 (10k rpm) 350 nm, NIR regime | 900 | 0.62 | 93.8 | 118 | 6.2 |
| 1154 | 1.18 | 63.2 | 36 | 36.7 | |
| 1253 | 1.12 | 74.2 | 33.3 | 25.8 |
Furthermore, the ultrafast carrier kinetic profiles of the MXene supernatant synthesized at 7k rpm (S1) in both visible and near-infrared probe regimes are shown in Fig. 6(a) and (b). The rise and recombination times of the S1 sample are getting higher due to agglomeration and the fact that the MAX phase in the MXene is not etched completely. This incomplete etching agglomeration leads to electron–photon coupling, which affects the behavior of the charge carriers [Table 2]. The corresponding kinetic profiles of the MXene supernatant synthesized at 10k rpm (S2) in both visible and near-infrared probe regimes are shown in Fig. 6(c) and (d). The kinetic profile in the visible range of MXene supernatant S2 shows the hot charge carrier interaction with phonons in the excited states as well as the relaxation of hot charge carriers from the highly excited states to quasi-thermal equilibrium states, which gives rise to the state filling of the MXene, as shown in [Fig. 6(c)].
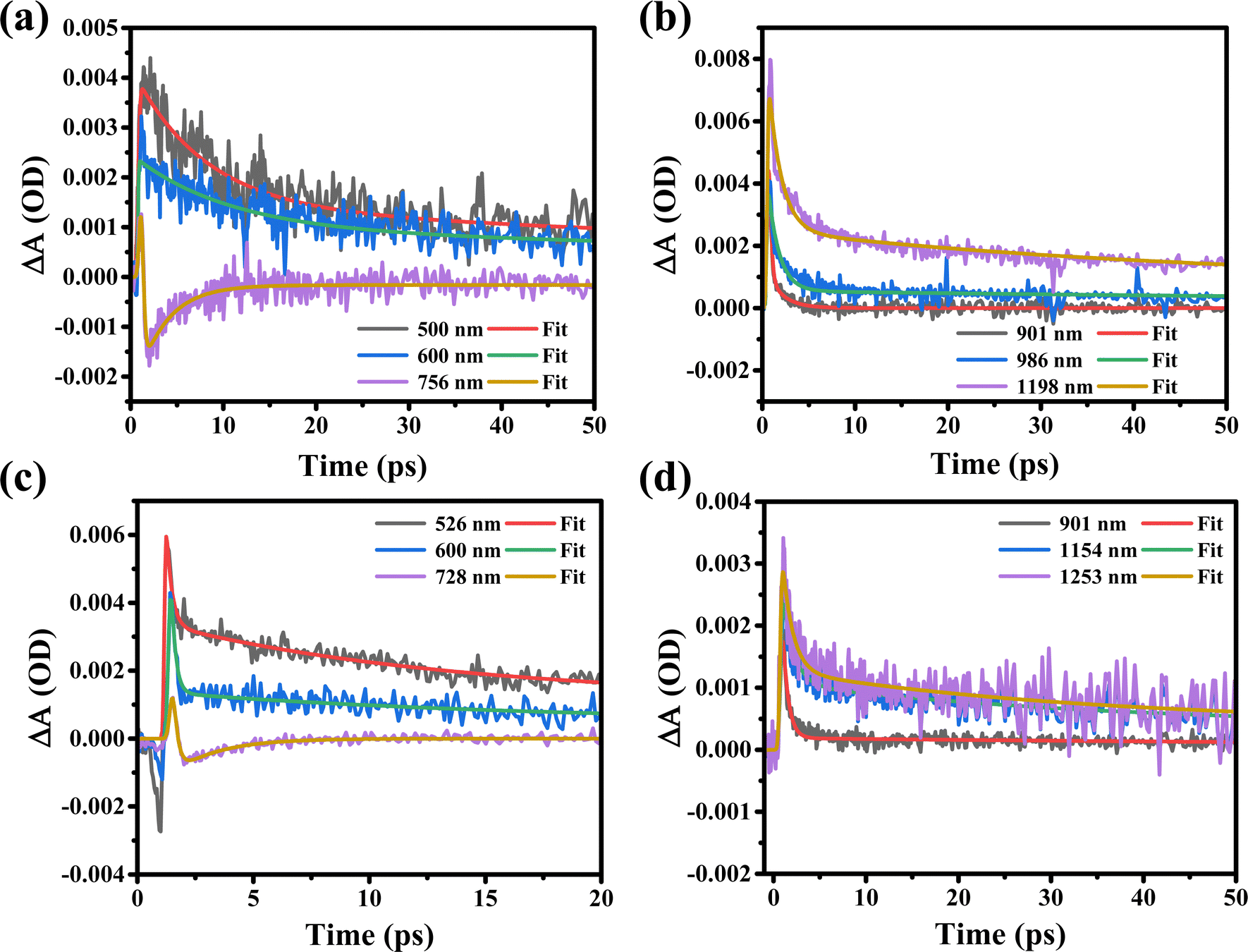 | ||
| Fig. 6 Transient absorption spectra of MXene supernatants S1 and S2 (a) and (b) and the kinetic profiles of the MXene supernatants synthesized at 7k rpm (S1) (c) and at 10k rpm (S2) (d) when excited at a 350 nm pump wavelength and in both the visible and NIR ranges. | ||
When a 350 nm pump wavelength laser excites the MXene, the electrons in the material absorb energy which leads to internal electronic excitations. There are different time scales in which the energy dissipation process ensues. The electrons interact with each other to realize electron thermalization (0.18 to 0.23 ps in the VIS region and 0.62 to 1.18 ps in the NIR region for sample-S2), which dampens the excitation energy. Notably, the degree of electron thermalization can be influenced by the characteristics of the excitation source, such as its wavelength and power. The highly energized electrons may engage with electrons near the Fermi level, leading to energy transfer and further thermalization of electrons. When the excitation is stronger, a large number of thermalized electrons take the lead, causing the relaxation process to slow down. In contrast, with lower-intensity excitation, fewer electrons become thermalized. In this situation, the key role shifts to the non-thermalized part, resulting in a quicker decay of energy relaxation. A 550 nm pump beam has a low photon energy and a low absorption efficiency as compared to a 350 nm pump wavelength; thus, the ultraviolet light could generate more thermalized electrons. Within the MXene, the thermalized electrons undergo a transformation into lattice vibrations, facilitated by the process of electron–phonon coupling (1.93 to 15.1 ps in the VIS region and 33.3 to 118 ps in the NIR region for sample-S2). In addition, the rising time of carriers is depicted in sub-femtoseconds due to intraband relaxation and electron–electron scattering in the excited states. The “rising time of carriers” refers to the time it takes for the population of hot carriers in the excited state after the initial excitation. Sub-femtosecond rise times suggest extremely fast processes, which could be associated with the efficient generation of hot carriers. As can be seen from Table 2, the rising time of sample-S2 is less as compared to that of sample-S1 which suggests that sample-S2 shows a fast charge carrier transfer in the excited state. Moreover, the kinetic profiles of sample-S2 in the VIS and NIR probe regions and sample-S1 in the NIR probe region show photo-induced absorption with a very short carrier rise time, as can be seen in [Fig. 6(b)–(d)] and Table 2. The charge recombination time (τ2) is increased due to electron–phonon coupling as shown in [Fig. 6(a), (b) and (d)] and Table 2. Electron–phonon coupling refers to the interaction between charge carriers (electrons) and lattice vibrations (phonons). During this interaction, some of the excess energy of the hot carriers is transferred to the phonons, causing the carriers to cool down and return to equilibrium. In this context, an increased recombination time means that the hot carriers take a longer time to return to their equilibrium state. This is because some of their excess energy is transferred to the phonons, slowing down the recombination process. The recombination time of sample-S2 is very less as compared to that of sample-S1 due to the shorter rise time of sample-S2 as can be seen in Table 2. In conclusion, based on the experimental observation we can say that after excitation in the UV range with VIS and NIR probe regions, the supernatant S2 rapidly generates a significant number of excited charge carriers. However, in the VIS to NIR region of S1, and the NIR region of S2, these carriers take a longer time to recombine and return to their equilibrium. In the visible spectrum of supernatant S2, at probe wavelengths of 525 nm, 600 nm, and 728 nm, rapid recombination times (τ2) are observed, with values of 12 ps, 15.1 ps, and 1.93 ps, respectively, as detailed in Table 2. These findings suggest variations in both the rise (τ1) and recombination time (τ2), signifying a potential influence on the material's properties.
In comparison, the kinetic profiles recorded in the VIS and NIR probe regimes for supernatants S1 and S2 suggest that supernatant S1 exhibits a more compact behavior in terms of the rise and recombination times of charge carriers due to agglomeration and the incomplete etching of the MAX phase in the MXene. Based on the above results we can say that the MAX phase in the S1 MXene material has not been completely removed, which could lead to variations in the behavior of the material. In the high-energy injection profile, the increase in the PIA signal at longer probe wavelengths (NIR range) indicates that more charge carriers are excited as the wavelength increases. This shows the enhancement of the charge carrier density in the excited state due to the excited charge carriers shifting from the valence to conduction bands and we observed worthy transient absorption responses. The transient absorption (TA) spectra of the S1 MXene are quite different from those of the S2 MXene due to the structural difference as can be seen from GIXRD data, where sample S1 has more Al impurities as compared to S2. Overall, the observed periodic GSB in the spectrum of the S2 sample indicates that it has favorable conditions for efficient charge carrier dynamics. In this case, it appears that the lattice relaxation in the S2 sample is efficient, allowing the charge carriers to move more freely, and exhibits a periodic GSB pattern as compared to the S1 sample. The kinetic profiles recorded in the VIS (visible) and NIR (near-infrared) probe regimes for supernatants S1 and S2 suggest that there are differences in the behavior of these two samples, particularly in terms of the rise and recombination times of charge carriers (as can be seen in Table 2). These differences may be due to the presence of agglomeration and the incomplete etching of the MAX phase in the MXene material.
4. Conclusion
In conclusion, the Ti3C2Tx MXene has been successfully synthesised by etching Al from the MAX phase by the LiF/HCl route. The effect of the centrifugation speed on the quality and the properties of the MXene reveals that the MXene synthesised at 10k rpm is better than that synthesised at 7k rpm from the material's quality and device perspectives. The MXene supernatant S2 at 10k rpm exhibits faster and more efficient charge carrier transfer as compared to 7k rpm, which is required for high-speed photonic devices. Using the time-resolved photoluminescence technique, the electronic relaxation (average decay time τav) was estimated at 5.3 ns and 5.13 ns for 7k and 10k rpm, respectively. This finding indicates that the optical properties of the MXene are strongly influenced by the process parameters used in the synthesis of the material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the “Science and Engineering Research Board,” India. Nitesh K. Chourasia acknowledges the “Science and Engineering Research Board’’ for the National Post-Doctoral Fellowship (PDF/2021/001490). Ankita Rawat thanks CSIR-UGC NET with file no. 09/263(1233)/2020-EMR-I for providing a Junior Research Fellowship (JRF). The authors are very thankful to Dr Mahesh Kumar for providing ultrafast facilities in CSIR-NPL, New Delhi, India.References
- J. Pang, R. G. Mendes, A. Bachmatiuk, L. Zhao, H. Q. Ta, T. Gemming, H. Liu, Z. Liu and M. H. Rummeli, Applications of 2D MXenes in Energy Conversion and Storage Systems, Chem. Soc. Rev., 2019, 48(1), 72–133, 10.1039/c8cs00324f.
- N. K. Chourasia, A. K. Singh, S. Rai, A. Sharma, P. Chakrabarti, A. Srivastava and B. N. Pal, A Lithography-Free Fabrication of Low-Operating Voltage-Driven, Very Large Channel Length Graphene Field-Effect Transistor with NH3 Sensing Application, IEEE Trans. Electron Devices, 2020, 67(10), 4385–4391, DOI:10.1109/TED.2020.3016606.
- N. K. Chourasia, V. K. Singh, A. Sharma, A. Srivastava and B. N. Pal, Lithography-Free Fabrication of Low Operating Voltage and Large Channel Length Graphene Transistor with Current Saturation by Utilizing Li+ of Ion-Conducting-Oxide Gate Dielectric, AIP Adv., 2020, 10(8), 085313, DOI:10.1063/5.0016466.
- X. Zhan, C. Si, J. Zhou and Z. Sun, MXene and MXene-Based Composites: Synthesis, Properties and Environment-Related Applications, Nanoscale Horiz., 2020, 5(2), 235–258, 10.1039/c9nh00571d.
- C. Zhao, Q. Wang, H. Zhang, S. Passerini and X. Qian, Two-Dimensional Titanium Carbide/RGO Composite for High-Performance Supercapacitors, ACS Appl. Mater. Interfaces, 2016, 8(24), 15661–15667, DOI:10.1021/acsami.6b04767.
- S. Nahirniak and B. Saruhan, MXene Heterostructures as Perspective Materials for Gas Sensing Applications, Sensors, 2022, 22(3), 972, DOI:10.3390/s22030972.
- N. K. Chourasia, A. Rawat, R. K. Chourasia, H. Singh, R. K. Kulriya, V. Singh and P. K. Kulriya, Unveiling the Potential of Ti3C2Tx MXene for Gas Sensing:Recent Developments and Future Perspectives, Mater. Adv., 2020, 1, 525–553, 10.1039/D3MA00631J.
- K. Hantanasirisakul and Y. Gogotsi, Electronic and Optical Properties of 2D Transition Metal Carbides and Nitrides (MXenes), Adv. Mater., 2018, 30(52), 1–30, DOI:10.1002/adma.201804779.
- B. Lu, Z. Zhu, B. Ma, W. Wang, R. Zhu and J. Zhang, 2D MXene Nanomaterials for Versatile Biomedical Applications: Current Trends and Future Prospects, Small, 2021, 17(46) DOI:10.1002/smll.202100946.
- M. W. Barsoum and M. Radovic, Elastic and Mechanical Properties of the MAX Phases, Annu. Rev. Mater. Res., 2011, 41, 195–227, DOI:10.1146/annurev-matsci-062910-100448.
- P. Eklund, M. Beckers, U. Jansson, H. Högberg and L. Hultman, The Mn+ 1AXn Phases: Materials Science and Thin-Film Processing, Thin Solid Films, 2010, 518(8), 1851–1878, DOI:10.1016/j.tsf.2009.07.184.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. C. Kent, Y. Gogotsi and M. W. Barsoum, Two-Dimensional, Ordered, Double Transition Metals Carbides (MXenes), ACS Nano, 2015, 9(10), 9507–9516, DOI:10.1021/acsnano.5b03591.
- Z. Sun, D. Music, R. Ahuja, S. Li and J. M. Schneider, Bonding and Classification of Nanolayered Ternary Carbides, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70(9), 1–3, DOI:10.1103/PhysRevB.70.092102.
- T. Zhang, L. Pan, H. Tang, F. Du, Y. Guo, T. Qiu and J. Yang, Synthesis of Two-Dimensional Ti3C2Tx MXene Using HCl + LiF Etchant: Enhanced Exfoliation and Delamination, J. Alloys Compd., 2017, 695, 818–826, DOI:10.1016/j.jallcom.2016.10.127.
- A. Lipatov, M. Alhabeb, M. R. Lukatskaya, A. Boson, Y. Gogotsi and A. Sinitskii, Effect of Synthesis on Quality, Electronic Properties and Environmental Stability of Individual Monolayer Ti3C2 MXene Flakes, Adv. Electron. Mater., 2016, 2(12) DOI:10.1002/aelm.201600255.
- K. Maleski, C. E. Ren, M. Q. Zhao, B. Anasori and Y. Gogotsi, Size-Dependent Physical and Electrochemical Properties of Two-Dimensional MXene Flakes, ACS Appl. Mater. Interfaces, 2018, 10(29), 24491–24498, DOI:10.1021/acsami.8b04662.
- M. Benchakar, L. Loupias, C. Garnero, T. Bilyk, C. Morais, C. Canaff, N. Guignard, S. Morisset, H. Pazniak, S. Hurand, P. Chartier, J. Pacaud, V. Mauchamp, M. W. Barsoum, A. Habrioux and S. Célérier, One MAX Phase, Different MXenes: A Guideline to Understand the Crucial Role of Etching Conditions on Ti3C2Tx Surface Chemistry, Appl. Surf. Sci., 2020, 530, 147209, DOI:10.1016/j.apsusc.2020.147209.
- S. Kumar, M. A. Rehman, S. Lee, M. Kim, H. Hong, J. Y. Park and Y. Seo, Supercapacitors Based on Ti3C2Tx MXene Extracted from Supernatant and Current Collectors Passivated by CVD-Graphene, Sci. Rep., 2021, 11(1), 1–9, DOI:10.1038/s41598-020-80799-9.
- Y. I. Jhon, J. Koo, B. Anasori, M. Seo, J. H. Lee, Y. Gogotsi and Y. M. Jhon, Metallic MXene Saturable Absorber for Femtosecond Mode-Locked Lasers, Adv. Mater., 2017, 29(40), 1–8, DOI:10.1002/adma.201702496.
- W. Song, Q. Zhang, H. Xu, J. Sun, K. Yuan, L.-H. Shao and J. Li, Ultrafast Dynamics and Energy Relaxation for Nanoporous Gold Materials: Lower Porosity and Faster Energy Exchange, J. Phys. Chem. C, 2020, 124(11), 6356–6363, DOI:10.1021/acs.jpcc.0c01435.
- E. Colin-Ulloa, A. Fitzgerald, K. Montazeri, J. Mann, V. Natu, K. Ngo, J. Uzarski, M. W. Barsoum and L. V. Titova, Ultrafast Spectroscopy of Plasmons and Free Carriers in 2D MXenes, Adv. Mater., 2022, 2208659, 1–10, DOI:10.1002/adma.202208659.
- B. Fu, J. Sun, C. Wang, C. Shang, L. Xu, J. Li and H. Zhang, MXenes: Synthesis, Optical Properties, and Applications in Ultrafast Photonics, Small, 2021, 17(11), 1–23, DOI:10.1002/smll.202006054.
- Q. Zhang, L. Yan, M. Yang, G. Wu, M. Hu, J. Li, K. Yuan and X. Yang, Ultrafast Transient Spectra and Dynamics of MXene (Ti3C2Tx) in Response to Light Excitations of Various Wavelengths, ACS Appl. Mater. Interfaces, 2020, 124(11), 6441–6447, DOI:10.1021/acs.jpcc.9b11652.
- S. K. Saini, N. K. Tailor, P. Sharma, L. Tyagi, N. Vashistha, R. Yadav, A. K. Chaudhary, S. Satapathi and M. Kumar, Revealing the Substrate Dependent Ultrafast Phonon Dynamics in Bi2Se3, Thin Films, 2023, 2201650, 1–11, DOI:10.1002/admi.202201650.
- A. Iqbal and N. M. Hamdan, Investigation and Optimization of Mxene Functionalized Mesoporous Titania Films as Efficient Photoelectrodes, Materials, 2021, 14(21), 6292, DOI:10.3390/ma14216292.
- A. Sarycheva and Y. Gogotsi, Raman Spectroscopy Analysis of the Structure and Surface Chemistry of Ti3C2T XMXene, Chem. Mater., 2020, 32(8), 3480–3488, DOI:10.1021/acs.chemmater.0c00359.
- R. M. Ronchi, J. T. Arantes and S. F. Santos, Synthesis, Structure, Properties and Applications of MXenes: Current Status and Perspectives, Ceram. Int., 2019, 45(15), 18167–18188, DOI:10.1016/j.ceramint.2019.06.114.
- M. Naguib, O. Mashtalir, M. R. Lukatskaya, B. Dyatkin, C. Zhang, V. Presser, Y. Gogotsi and M. W. Barsoum, One-Step Synthesis of Nanocrystalline Transition Metal Oxides on Thin Sheets of Disordered Graphitic Carbon by Oxidation of MXenes, Chem. Commun., 2014, 50(56), 7420–7423, 10.1039/c4cc01646g.
- T. Hu, J. Wang, H. Zhang, Z. Li, M. Hu and X. Wang, Vibrational Properties of Ti3C2 and Ti3C2T2 (T = O, F, OH) Monosheets by First-Principles Calculations: A Comparative Study, Phys. Chem. Chem. Phys., 2015, 17(15), 9997–10003, 10.1039/c4cp05666c.
- J. Zhu, Y. Tang, C. Yang, F. Wang and M. Cao, Composites of TiO 2 Nanoparticles Deposited on Ti3C2 MXene Nanosheets with Enhanced Electrochemical Performance, J. Electrochem. Soc., 2016, 163(5), A785–A791, DOI:10.1149/2.0981605jes.
- L. Lorencova, T. Bertok, E. Dosekova, A. Holazova, D. Paprckova, A. Vikartovska, V. Sasinkova, J. Filip, P. Kasak, M. Jerigova, D. Velic, K. A. Mahmoud and J. Tkac, Electrochemical Performance of Ti3C2Tx MXene in Aqueous Media: Towards Ultrasensitive H2O2 Sensing, Electrochim. Acta, 2017, 235, 471–479, DOI:10.1016/j.electacta.2017.03.073.
- C. E. Shuck, A. Sarycheva, M. Anayee, A. Levitt, Y. Zhu, S. Uzun, V. Balitskiy, V. Zahorodna, O. Gogotsi and Y. Gogotsi, Scalable Synthesis of Ti3C2Tx MXene, Adv. Eng. Mater., 2020, 22(31), 901241, DOI:10.1002/adem.201901241.
- T. H. Phuong Doan, W. G. Hong and J. S. Noh, Palladium Nanoparticle-Decorated Multi-Layer Ti3C2Tx dual-Functioning as a Highly Sensitive Hydrogen Gas Sensor and Hydrogen Storage, RSC Adv., 2021, 11(13), 7492–7501, 10.1039/d0ra10879k.
- M. A. Iqbal, A. Tariq, A. Zaheer, S. Gul, S. I. Ali, M. Z. Iqbal, D. Akinwande and S. Rizwan, Ti3C2-MXene/Bismuth Ferrite Nanohybrids for Efficient Degradation of Organic Dyes and Colorless Pollutants, ACS Omega, 2019, 4(24), 20530–20539, DOI:10.1021/acsomega.9b02359.
- Y. Gogotsi and M. W. Barsoum, Two-Dimensional Transition Metal Carbides, ACS Nano, 2012, 2, 1322–1331 Search PubMed.
- L. Zhang, W. Su, H. Shu, T. Lü, L. Fu, K. Song, X. Huang, J. Yu, C. T. Lin and Y. Tang, Tuning the Photoluminescence of Large Ti 3 C 2 T x MXene Flakes, Ceram. Int., 2019, 45(9), 11468–11474, DOI:10.1016/j.ceramint.2019.03.014.
- Q. Xue, H. Zhang, M. Zhu, Z. Pei, H. Li, Z. Wang, Y. Huang, Y. Huang, Q. Deng, J. Zhou, S. Du, Q. Huang and C. Zhi, Photoluminescent Ti3C2 MXene Quantum Dots for Multicolor Cellular Imaging, Adv. Mater., 2017, 29(15), 1–6, DOI:10.1002/adma.201604847.
- X. Sang, Y. Xie, M. W. Lin, M. Alhabeb, K. L. Van Aken, Y. Gogotsi, P. R. C. Kent, K. Xiao and R. R. Unocic, Atomic Defects in Monolayer Titanium Carbide (Ti3C2Tx) MXene, ACS Nano, 2016, 10(10), 9193–9200, DOI:10.1021/acsnano.6b05240.
- N. Xu, H. Li, Y. Gan, H. Chen, W. Li, F. Zhang and X. Jiang, Zero-Dimensional MXene-Based Optical Devices for Ultrafast and Ultranarrow Photonics Applications, Adv. Sci., 2020, 2002209, DOI:10.1002/advs.202002209.
- A. Rawat and P. K. Kulriya, 2D/3D Material for Gas Sensor, Smart Nanostructure Materials and Sensor Technology, Springer Nature, Singapore, 2022, pp. 161–178 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00429e |
| This journal is © The Royal Society of Chemistry 2023 |