
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface and bulk defect formation during hydrothermal synthesis of LiCoPO4 crystals and their electrochemical implications†
Moohyun
Woo
 ,
Jinhyuk
Lee
and
George P.
Demopoulos
*
,
Jinhyuk
Lee
and
George P.
Demopoulos
*
Materials Engineering, McGill University, 3610 rue University, Montréal, QC H3A 0C5, Canada. E-mail: george.demopoulos@mcgill.ca
First published on 22nd September 2023
Abstract
Lithium cobalt phosphate (LiCoPO4, LCP) is a high-voltage cathode material with a lot of promise in delivering high energy density in comparison to the established LiFePO4 counterpart. However, the road to developing LCP is hampered not only by electrolyte interfacial reaction due to high-voltage but also by the lack of critical knowledge regarding material crystal properties linked to synthesis that limit the attainment of full discharge capacity. Herein, we study in-depth the synthesis of LCP by the hydrothermal method and its post-synthesis modifications by high-energy planetary milling and conductive carbon coating in order to shed light on the crystal chemistry affecting its electrochemical performance. Via adjusting the Li/Co molar ratio and pH of precursor solution, the supersaturation is controlled to achieve high-purity and well-crystalline LCP particles with sub-micron size. After carefully characterizing the hydrothermally synthesized LCP crystalline material, we discovered the presence of two types of defects, surface composition inhomogeneities and bulk cation mixing, which adversely affect the Li-ion intercalation kinetics and storage capacity. More specifically, we identified (i) the formation of undesired nano-scale Co(OH)2 passivation layer on LCP surface and (ii) abundant anti-site defects blocking one-dimensional (1-D) Li-ion diffusion channels. These crystal defects we show to impose critical limitations to hydrothermally produced LCP materials in delivering near theoretical discharge capacities; hence on the basis of these new insights, alternative crystal engineering approaches need to be developed in pursuit of high-performance LCP cathodes.
1. Introduction
Rechargeable Li-ion batteries bring a wide spectrum of applications in modern society from mobile electronic devices to large-scale energy storage for electromobility and the smart grid.1–5 With this trend, the ever-growing demand for state-of-the-art rechargeable Li-ion batteries with greater energy density has triggered intensive research on high-voltage cathode materials.6–11Lithium transition metal phosphates (LiMPO4: M = Fe, Mn, Co and Ni) represent a large class of Li-ion cathode materials. Out of all members of this family, LiFePO4 (LFP) has been the most successful cathode so far for its high safety, low cost, excellent cyclability, and high power capability.12–18 The LFP cathode has been heavily investigated and optimized but cannot provide high-energy density as demanded by the electromobility sector due to its low voltage. As a result, there has been a strong interest in developing the rest of LiMPO4 cathode materials. In particular, LiCoPO4 (LCP) has received much attention for its very high voltage of 4.8 V vs. Li/Li+ (as compared to 3.5 V vs. Li/Li+ for the commercially available LFP).7,10,19–22 Having the same specific capacity as LFP (∼167 mA h g−1), the LCP cathode with a much higher operating voltage than LFP has a significantly increased theoretical energy density of 800 W h kg−1vs. 590 W h kg−1 of LFP.23–25 Also similarly to LFP, the strong P–O bonds guarantee a robust host framework for Li-ion storage, resulting in excellent thermal safety.26–28
Hence, considerable work has been undertaken on developing LCP cathodes from various synthesis routes (e.g., solid-state, sol–gel, or hydrothermal and solvothermal synthesis)29–39 to crystal modification via doping23,24,31,33,40,41 and/or coating29,34,37,42,43 approaches. Among the synthesis methods, the hydrothermal process has been suggested as the most cost-effective and energy-efficient one, as it employs water as a green solvent rather than organic solvents and chemicals.44,45 However, it has been found the hydrothermally synthesized LCP (HT-LCP) is characterized by poor electrochemical performance, as evidenced by low reversible discharge capacity compared to LCP made with a different method such as the solid-state method (SS-LCP).34–37 Several arguments have been made in previous research to explain the poor electrochemical response of HT-LCP, ranging from electrolyte decomposition owing to the relatively higher potential of Co2+/Co3+ redox couple34,46,47 to cation exchange (anti-site defects) developed during Li-ion intercalation process.32,48,49 However, both issues, i.e., the electrolyte decomposition and the cycling-induced cation exchange,50 are not observed only with HT-LCP but also with other LCP materials but not to the same extent.22 Hence, there is a gap in understanding the origin of this poor electrochemical capacity problem exhibited by HT-LCP from a material chemistry standpoint.
Given the importance of the scalable and green process of hydrothermal (HT) synthesis in the development of functional high-voltage LCP cathodes, in this work, we have investigated the impact of key HT-synthesis parameters (Li molar ratio & pH value of precursor solutions) but also post-synthesis modifications (nanosizing by planetary milling (PM) and carbon coating (C-coating)) on LCP crystal structure/morphology/purity and electrochemical storage properties. Upon monitoring the evolution of surface and bulk structure during material synthesis, the formation of unreported defects was identified the nature of which was characterized and their impact on Li-ion intercalation responsible for the low discharge capacity was determined. During the study, particular attention was paid to differentiating the effect of electrolyte decomposition from the effects of LCP material defects that they are commonly wrapped together obstructing clear understanding. These findings open new avenues toward developing LCP cathodes with fully unlocked high-voltage capacity.
2. Experimental section
2.1 Material preparation
In typical experiments, solutions with different concentrations (0.4, 0.6, 0.8, 1.0, and 1.2 M) of LiOH·H2O along 0.4 M of CoSO4·7H2O, 0.4 M of H3PO4, and 0.0095 M of C6H8O6 were prepared in 200 mL of deoxygenated and deionized water to give a different molar ratio of Li to cobalt (Co) namely 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1.5:1, 2:1, 2.5:1, and 3:1. The pH of the precursor solutions was controlled at 8, 9, and 10 with NH4OH added dropwise. The pH variations before and after HT-synthesis is measured and summarized in Table S1 (ESI†). The prepared precursor solution was transferred afterward to a 450 mL stainless steel autoclave reactor (Parr Instruments, Moline, IL) equipped with a glass liner. The whole solution preparation procedure was carried out in a nitrogen (N2)-filled glovebox to prevent possible Co oxidation from Co2+ to Co3+. Preliminary HT-synthesis done at different temperatures (data shown in Fig. S1, ESI†) determined 220 °C for 6 hours and 300 rpm impeller agitation to achieve uniform mixing of reactants and produced particles and was used throughout this study. Additionally, the exact temperature and pressure (psi) were monitored and controlled with Parr 4848 reactor controller during the synthesis. After HT-reaction, the reactor vessel was quenched down to room temperature within 30 minutes through internal tube cooling water flowing. The synthesized solids were separated from the solution via centrifugation and then rinsed with deoxygenated and deionized water twice and with ethanol once, followed by evaporating the remained ethanol in a vacuum oven at 80 °C overnight after several times purging with N2 gas.
:1, 1.5:1, 2:1, 2.5:1, and 3:1. The pH of the precursor solutions was controlled at 8, 9, and 10 with NH4OH added dropwise. The pH variations before and after HT-synthesis is measured and summarized in Table S1 (ESI†). The prepared precursor solution was transferred afterward to a 450 mL stainless steel autoclave reactor (Parr Instruments, Moline, IL) equipped with a glass liner. The whole solution preparation procedure was carried out in a nitrogen (N2)-filled glovebox to prevent possible Co oxidation from Co2+ to Co3+. Preliminary HT-synthesis done at different temperatures (data shown in Fig. S1, ESI†) determined 220 °C for 6 hours and 300 rpm impeller agitation to achieve uniform mixing of reactants and produced particles and was used throughout this study. Additionally, the exact temperature and pressure (psi) were monitored and controlled with Parr 4848 reactor controller during the synthesis. After HT-reaction, the reactor vessel was quenched down to room temperature within 30 minutes through internal tube cooling water flowing. The synthesized solids were separated from the solution via centrifugation and then rinsed with deoxygenated and deionized water twice and with ethanol once, followed by evaporating the remained ethanol in a vacuum oven at 80 °C overnight after several times purging with N2 gas.
:solvent = 1:2 volume ratio) at 300 rpm for 6 hours and subsequently dried in an oven at 50 °C overnight. After chemical mixing and drying, two times of calcination processes with a ramping rate of 5 °C min−1 were accomplished at 400 °C for 10 hours in air first to remove the crystal water and at 800 °C for 10 hours in argon (Ar) atmosphere to obtain pure LCP phase.29,30
For the C-coating process, lactose anhydrous (C12H22O11, Sigma-Aldrich) as a carbon source was dissolved and stirred in deoxygenated and deionized water with a concentration of 22.4 g L−1 for 30 minutes.51 Then, 5 mL of the lactose solution was applied to 1 g of pulverized LCP powder in a graphite crucible. The slurry was dried in a vacuum oven at 80 °C and transferred to MTI mini tube furnace (model OTF-1200X) and heated consecutively at three different intervals (120 °C–1 h, 400 °C–1 h, and 700 °C–3 h) for lactose decomposition and carbonization on the surface of pulverized LCP particles.
2.2 Materials characterization
Laboratory XRD was employed for phase analysis with Bruker D8 Discovery X-ray diffractometer using Co K-alpha source with wavelength (λ) of 1.78892 Å from 2θ = 10 to 80°. The diffraction patterns were collected by using Gadd software. Defect concentration was fitted and estimated with XRD Rietveld refinement by using the TOPAS Academic V5 program. The morphology characterization was done with a Hitachi cold-field emission SU-8000 scanning electron microscope (CFE-SEM). Inductively coupled plasma optical emission spectroscopy (ICP-OES, Thermo Scientific iCAP 6500 ICP spectrometer) was applied to determine the concentration of remaining elements in solution after HT-synthesis but also to determine the exact elemental ratio of Co/Li in different LCP samples. Prior to ICP-OES measurements, LCP samples were digested by using 50 vol.% concentrated hydrochloric acid (HCl, Fisher Scientific) followed by dilution with 4 vol.% nitric acid (TraceMetal Grade, HNO3, Fisher Scientific). Brunauer–Emmett–Teller (BET) specific surface area measurements were made by using the TriStar 3000 analyzer (Micromeritics) in N2 (−196 °C). The size-distribution analysis of LCP particles was achieved with Zetasizer Nano ZS using the dynamic light scattering analysis. Raman Spectroscopy (Witec Alpha 3000) with 514 nm wavelength was applied to confirm the existence of carbon layers on the surface of pulverized LCP particles and its quality with a D/G ratio. Transmission electron microscope (TEM) and high-resolution TEM (HR-TEM) images were achieved through Thermo Scientific Talos F200X G2 STEM. X-ray Photoelectron Spectroscopy (XPS) measurements were carried out with Thermo-Scientific K-Alpha using aluminum (Al) K-alpha micro-focused monochromator. The applied X-ray spot size was 400 μm and an electron flood gun was used for the charge compensation. The spectrometer energy scale was calibrated using C 1s characteristic peak at 284.8 eV (C–C) as a reference. XPS data processing was performed with the Avantage data analysis system for peak fitting of C 1s, Li 1s, Co 2p, P 2p, and O 1s spectra with single or multiple peaks. Fourier Transform Infrared (FT-IR) spectroscopy (Perkin Elmer Spectrum II FT-IR Spectrometer) was employed in the wavenumber range of 4000 to 400 cm−1 with a resolution 0.5 cm−1.2.3 Electrochemistry
Free-standing working electrodes without Al current collector were prepared with dry-method as follows: 70 wt.% of LCP active material was gently mixed with 20 wt.% of acetylene black (Alfa Aesar) with an average particle size of 60 nm and an area of 75 m2 g−1via mild PM at 300 rpm for 1 h using 20 e.a. of 10 mm ZrO2 grinding balls. After mixing, the jar was transferred and disassembled in the glovebox filled with high-purity Ar gas. 10 wt.% of Polytetrafluoroethylene (PTFE, (C2F4)n, DuPont, Teflon 8 A) binder was added to the mixed powder and subsequently spread with sufficient pressure through the hand-pressing process using stainless steel bar until a film with 0.16 cm2 area and ∼ 2.5 mg weight was obtained. A polypropylene film (Celgard 2200) was used as the separator. A custom-made electrolyte with stabilizing additive for high-voltage cathode operation was produced with 1.2 M of Lithium hexafluorophosphate (99.99% Trace metals basis LiPF6, Sigma-Aldrich) dissolved in a 3:7 (wt.%) mixture of ethylene carbonate (EC, 99% (CH2O)2CO, Sigma-Aldrich) and ethyl methyl carbonate (EMC, 99% CH3CH2OCOOCH3, Sigma-Aldrich) plus 2 wt.% tris(trimethylsilyl) phosphite (95% [(CH3)3SiO]3P, Sigma-Aldrich).41 The galvanostatic charge–discharge data was collected on an Arbin cycler. The battery cycling was carried out at a C/20 rate (1C = 167 mA h g−1) in the voltage range of 3.5–5.2 V vs. Li/Li+ at room temperature. Cyclic voltammetry (CV) was performed by using an electrochemical workstation (Bio-Logic) at the specific conditions between 3.5–5.2 V with a scan rate of 0.1 mV s−1. Electrochemical Impedance Spectroscopy (EIS) measurements of the pristine working electrodes were made after 6 hours relaxation in the equilibrium state in the potentiostat mode between 1 MHz and 10 mHz at open circuit voltage (OCV). The charge-transfer resistance was determined by fitting the Nyquist plot with the equivalent electric circuit by the Z-fit method provided by ZView® 4.0 software.
3. Results and discussion
3.1 Hydrothermal synthesis parameter effects on crystal formation
To identify the conditions to synthesize high-purity HT-LCP at 220 °C, we first investigated the effects of Li molar ratio (0.4. 0.6, 0.8, 1, and 1.2 M) and pH (8, 9, and 10) on LCP properties. The Li molar ratio was found to influence the crystallization of LCP, leading to cube-like shape crystals when added in 50% Li excess (1.5:1:1) over the stoichiometry (1:1:1) (Fig. 1(a)–(d)). Cubic shape crystal formation was completed with 100% Li excess (2:1:1). It is postulated the cubic crystals grow via oriented attachment of the initially nucleating crystallites.52–56 Further crystal growth was not observed if the Li excess was raised to 150% excess (2.5:1:1), but instead, there was particle size reduction from 5 μm to less than 1 μm.
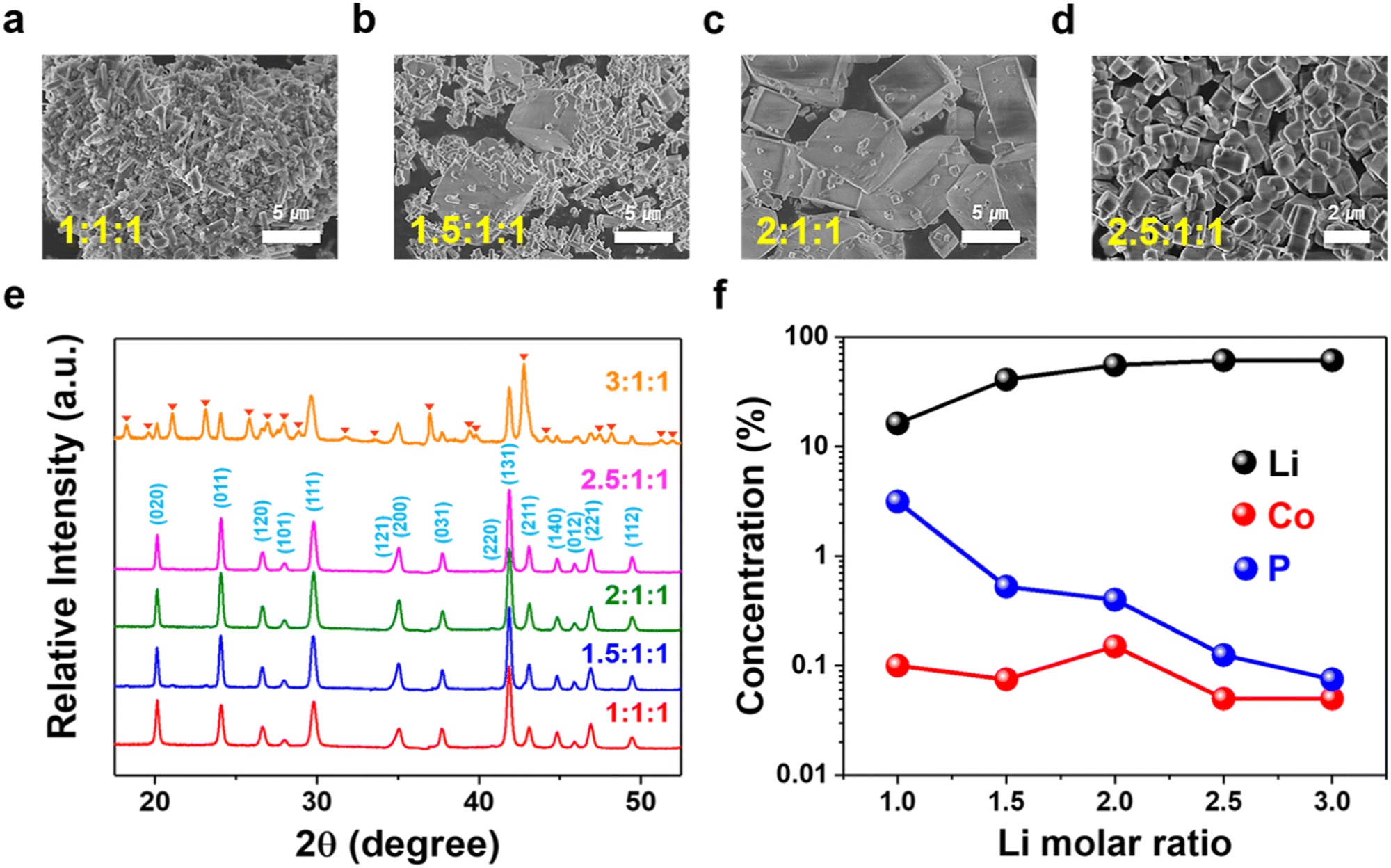 | ||
| Fig. 1 Morphology, phase, and residual solution composition of LCP materials obtained with different Li/Co/P molar ratios at pH 9 and 220 °C via (a)–(d) SEM, (e) XRD, and (f) ICP-OES. | ||
Fig. 1(e) shows XRD patterns of HT-LCP materials made with different Li molar ratios at 220 °C for 6 hours. The patterns of the HT-LCP materials prepared up to a 2.5:1:1 molar ratio can be clearly indexed to the orthorhombic phase with the space group, Pnma (JCPDS 89-6192). Further, from the sharpening of the XRD peaks, we can see the crystallinity of LCP improves as the over-stoichiometric Li ratio increases, consistent with the SEM image observations. However, if the Li ratio increases beyond 2.5:1:1, extra peaks appear, corresponding most likely to the formation of Li3PO4 (JCPDS 07-2815) and Co3O4 (JCPDS 78-1969). In addition, according to the residual element composition in the solution shown in Fig. 1(f), it can be seen the concentration of Co and P (PO4) decreases with increasing Li concentration, implying the excess Li drives the reaction to a higher yield.
Other than pH 9, additional tests were performed with various Li molar ratios at pH 8 and 10, the results of which are shown in Fig. S2 (ESI†). In this case, all samples produced at pH 8 were found to contain impurities. Nanosized and pure LCP phase was produced (at a lower yield of 83%), on the other hand, at pH 10, but this time, only when the precursor was solution prepared with the stoichiometric ratio of 1:1:1 (Fig. S3, ESI†). Interestingly, in this case (i.e. at pH 10), as the Li ratio was increased above 1, the XRD patterns (Fig. S2c, ESI†) revealed complex and not well developed patterns not matching that of well-crystallized LCP. Therefore, based on the above findings, we conclude that the optimal conditions for producing high-purity sub-micron size LCP crystals are with a 2.5:1:1 molar ratio and pH 9, denoted as “HT-LCP”.
The formation of high-crystalline sub-micron-sized LCP cube-shaped particles using a 2.5:1:1 molar ratio at pH 9 and 220 °C can be understood on the basis of governing nucleation & growth kinetics.57–59 The nucleation rate (J) as a function of supersaturation, S, is given by equation (1) described below,
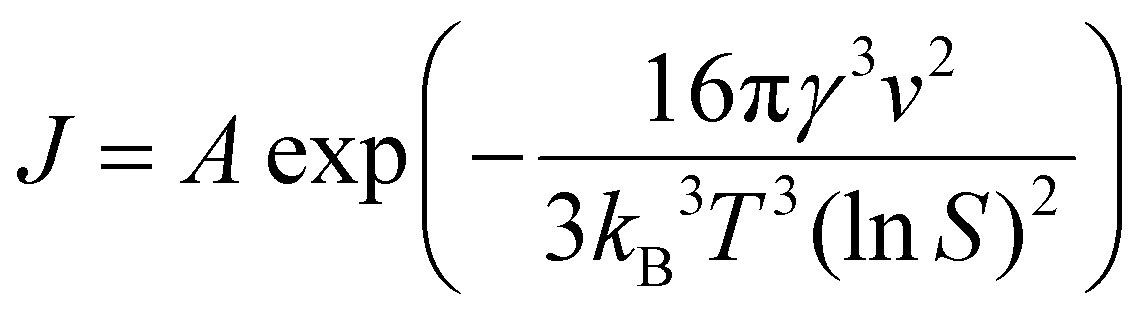 | (1) |
| S = α/αeq | (2) |
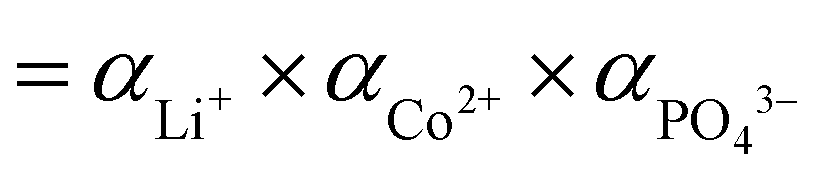 ), αeq is the activity of the solid compound in saturated solution i.e. at equilibrium state represented by the solubility product:
), αeq is the activity of the solid compound in saturated solution i.e. at equilibrium state represented by the solubility product: 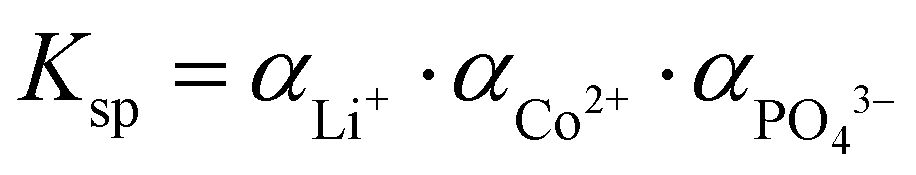 . The higher the supersaturation the lower the nuclei size as described in eqn (3).
. The higher the supersaturation the lower the nuclei size as described in eqn (3).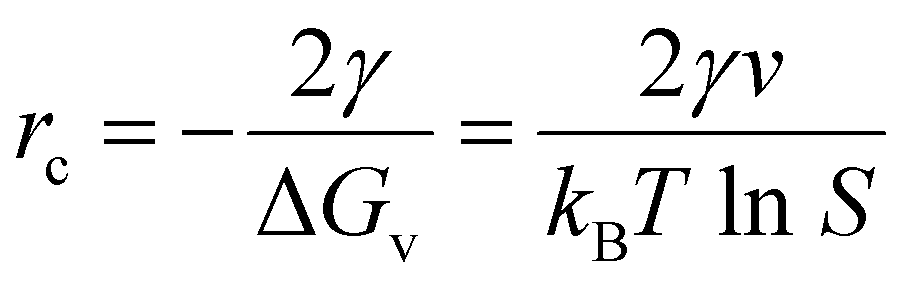 | (3) |
The activity of PO43− anions (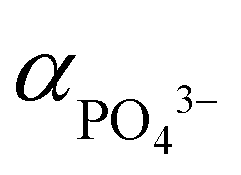 ) meanwhile depends on pH value and temperature:60–62
) meanwhile depends on pH value and temperature:60–62
| H3PO4 = H2PO4− + H+ K1 = 7.52 × 10−3 |
| H2PO4− = HPO42− + H+ K2 = 6.23 × 10−8 |
| HPO42− = PO43− + H+ K3 = 4.80 × 10−13 |
It is postulated that at 220 °C and pH 9, HPO42− to dominate and thus maintain low 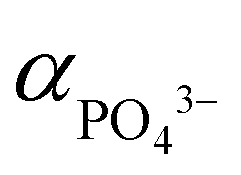 ,62 hence the necessity of excess αLi+ to modulate supersaturation which in turn controls the nucleation rate to yield smaller size crystals (<1 μm) as predicted by eqn (3). This is a delicate equilibrium state as at pH 8, the appearance of impurity formation implies
,62 hence the necessity of excess αLi+ to modulate supersaturation which in turn controls the nucleation rate to yield smaller size crystals (<1 μm) as predicted by eqn (3). This is a delicate equilibrium state as at pH 8, the appearance of impurity formation implies 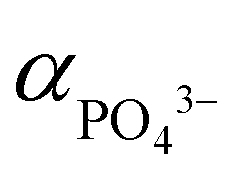 to be too low, leading to parasitic reactions involving dihydrogen phosphate, while at pH 10 is too high; hence only stoichiometric amount of Li produced LCP. Excess Li apparently was counter-productive in the case of pH 10 solution as the high supersaturation regime resulted in the formation of amorphous LCP nanoparticles.
to be too low, leading to parasitic reactions involving dihydrogen phosphate, while at pH 10 is too high; hence only stoichiometric amount of Li produced LCP. Excess Li apparently was counter-productive in the case of pH 10 solution as the high supersaturation regime resulted in the formation of amorphous LCP nanoparticles.
3.2 Post-synthesis LCP crystal modification
Since the intrinsic ionic and electronic conductivities of LCP are low, it was decided to consider nanosizing (to boost ion conductivity) and carbon coating (to boost electron conductivity). Nanosizing of HT-LCP was induced by high energy planetary milling (PM) at different times from 1 to 4 hours. The related size distribution, BET surface area, SEM, and XRD results are summarized in Fig. 2(a)–(c). According to size-distribution analysis (shown in Fig. 2(a)), their mean size was reduced after 2 h of PM to around 200 nm (from 800 nm), and the corresponding specific surface area (Fig. 2(b)) increased from 1.59 m2 g−1 to 28.29 m2 g−1. In addition, inset SEM images in Fig. 2(b) show the morphology of the obtained particles after milling.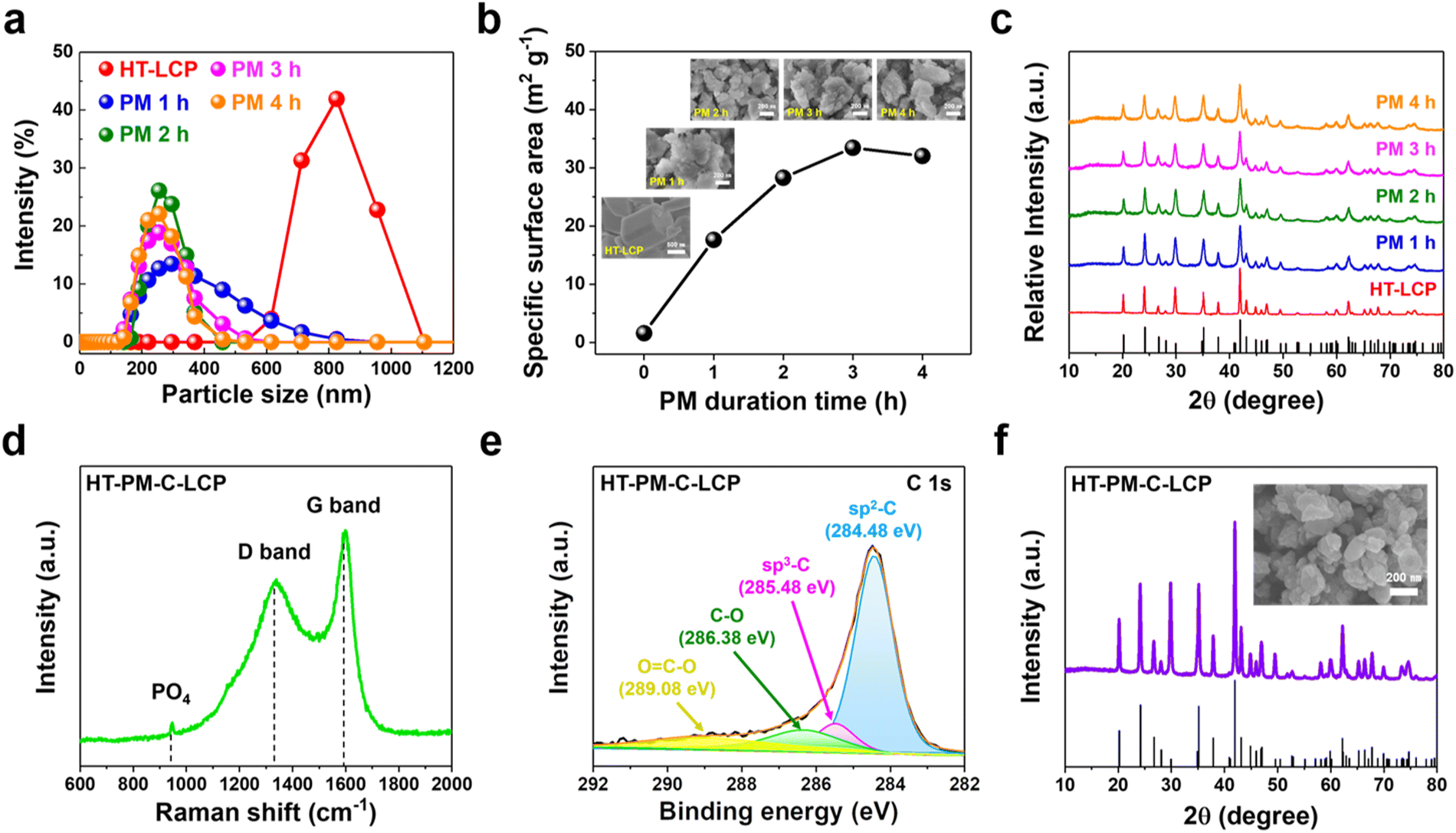 | ||
| Fig. 2 Characterization of HT-LCP crystals after post-synthesis modifications (high-energy PM and conductive C-coating) via (a) size-distribution measurement with the dynamic light scattering, (b) BET analysis, and (c) XRD with different milling duration time; (d) Raman, (e) XPS C 1s spectra, and (f) XRD. | ||
Meanwhile as per XRD patterns (Fig. 2(c)), nanosizing caused progressive crystal disordering with increasing milling time, as evidenced by the peak broadening, an observation that is in agreement with previous reports.63 Of the different nanosized LCP materials, the one obtained after 2 h of PM (hereafter referred to as “HT-PM-LCP”) was retained for subsequent electrochemical performance evaluation as it provided a short diffusion length (Fig. S4, ESI†) without major loss of crystallinity. Finally, the nanosized HT-PM-LCP was subjected to C-coating by mixing the powder with lactose solution and annealing under an inert Ar atmosphere.59 The planetary-milled/carbon-coated material was labelled “HT-PM-C-LCP”.
Fig. 2(d) shows the Raman spectra for HT-PM-C-LCP material recorded at the laser excitation wavelength of 514 nm. Three peaks with high intensity were clearly detected at 947.09, 1340.65, and 1590.10 cm−1. The first peak in the 600–1100 cm−1 region corresponds to the intramolecular stretching modes of the PO4 group in LCP structure;64 and the other two peaks indicate the presence of a carbon layer on the surface of HT-PM-LCP particles. These peaks are referred to as the “D” (1340 cm−1) and “G” (1590 cm−1) bands, respectively.20,65,66 Hence, the carbon layer was favourably developed on the surface of HT-PM-LCP particles. The intensity ratio (ID/IG) from the deposited carbon layer was 0.77, indicating that the carbon layer mainly consists of disordered carbon.67 In order to further verify the nature of carbon, we performed XPS measurement on HT-PM-C-LCP. The C 1s spectrum (Fig. 2(e)) was fitted with multiple peaks at different binding energies: sp2 C–C at 284.48 eV, sp3 C–C at 285.48 eV, C–O group at 286.38 eV, and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O group at 289.08 eV, respectively.68–70 By comparing the relative atomic ratios of sp2 (88.92%) and sp3 (11.02%), sp2 representing C–C bonding is found to be dominant in the carbon layer on HT-PM-C-LCP over sp3, which indicates C–H bonding. The XPS result is consistent with the ID/IG in Raman analysis.
C–O group at 289.08 eV, respectively.68–70 By comparing the relative atomic ratios of sp2 (88.92%) and sp3 (11.02%), sp2 representing C–C bonding is found to be dominant in the carbon layer on HT-PM-C-LCP over sp3, which indicates C–H bonding. The XPS result is consistent with the ID/IG in Raman analysis.
Moreover, as shown in Fig. 2(f), the phase and morphology of HT-PM-C-LCP were successfully preserved after C-coating without suffering agglomeration. In addition, the crystallinity was markedly restored due to the high-temperature treatment required for lactose decomposition and carbonization.71
In order to evaluate the impact of HT-synthesis on LCP crystal properties and electrochemistry, LCP material was also synthesized via conventional solid-state reaction.29,30 Again, three samples were prepared (summarized in Fig. S5, ESI†), a pristine one (SS-LCP) and two after post-synthesis treatments (SS-PM-LCP and SS-PM-C-LCP).
3.3 Electrochemistry
After the satisfactory synthesis and post-synthesis modifications, we investigated the electrochemical properties of the differently prepared LCP materials either via HT-method or solid-state reaction so to probe synthesis-impacted LCP functionality. Galvanostatic charge–discharge tests were carried out at a C/20 rate within the voltage range of 3.5–5.2 V vs. Li/Li+ at ambient (∼22 °C) temperature. The initial charge–discharge curves are summarized in Fig. 3(a) and (b). Despite the high-purity and well-developed crystallinity of HT-LCP material, it exhibited a poor discharge capacity of only 33 mA h g−1 (vis-a-vis the theoretical capacity of 167 mA h g−1). The 1st discharge capacity was marginally increased to 53 mA h g−1 after PM because of nanosizing (HT-PM-LCP), whereas the capacity was dropped to 25 mA h g−1 after the subsequent C-coating process (HT-PM-C-LCP). In comparison, even with the agglomerated and larger particle size (shown in Fig. S5a, ESI†), SS-LCP could deliver 53.3 mA h g−1, while after the PM treatment (SS-PM-LCP), its discharge capacity was improved to 76.2 mA h g−1 and after C-coating, registered a slightly dropped discharge capacity at 62 mA h g−1. The unexpected drop in discharge capacity after carbon coating of the planetary-milled LCP obtained by both synthesis methods (HT and SS) is discussed later.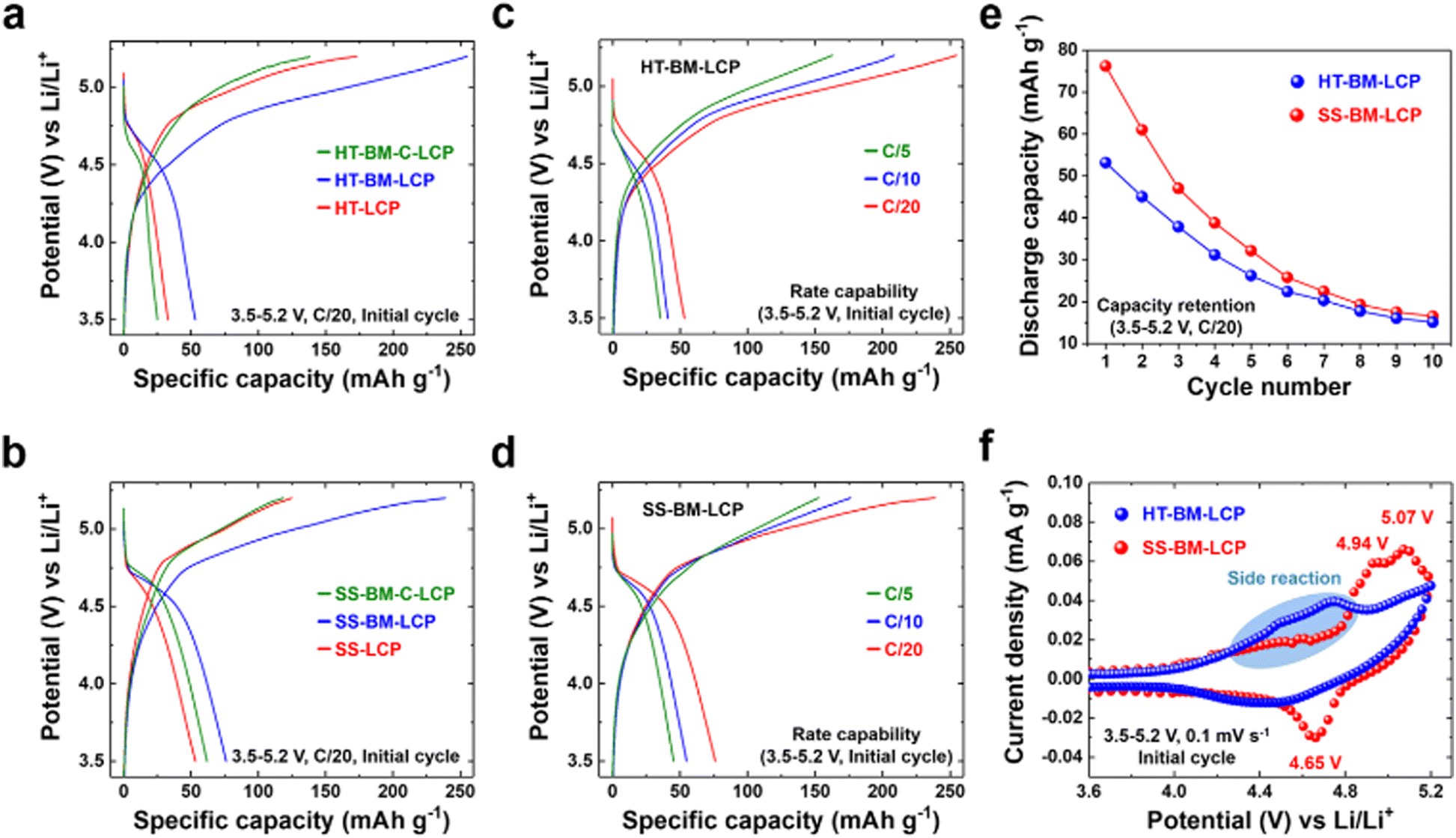 | ||
| Fig. 3 Comparison of the electrochemical properties of LCP materials obtained by hydrothermal (HT) and solid-state (SS) synthesis: (a) and (b) galvanostatic charge–discharge, (c) and (d) rate capability, (e) capacity retention, and (f) cyclic voltammetry (CV). | ||
Fig. 3(c) and (d) display the rate capability of HT-PM-LCP and SS-PM-LCP, which were found to deliver respectively the highest initial charge–discharge capacity among the different samples. As the C-rate is gradually increased, both milled LCP samples reveal significantly reduced initial discharge capacity of 53, 40.8, and 35.3 mA h g−1 at C/20, C/10, and C/5 for HT-PM-LCP and 76, 54.9, and 45.3 mA h g−1 for SS-PM-LCP. In addition, Fig. 3(e) shows the capacity retention of HT-PM-LCP and SS-PM-LCP over 10 cycles. There is a severe capacity loss after 10 cycles, which at least in part is due to accelerated electrolyte decomposition at high voltage on the exposed surface of the nanosized LCP particles, as also observed by other researchers.50,72–74
In order to probe further the origin of the observed poor discharge capacity and retention, cyclic voltammetry (Fig. 3(f)) was performed over the 3.5–5.2 V range at a scan rate of 0.1 mV s−1. During the anodic sweep, SS-PM-LCP exhibits two distinct oxidation peaks at 4.94 and 5.07 V vs. Li/Li+. This two-step delithiation process is consistent with the formation of the previously identified intermediate phase with the stoichiometry Li2/3(Co2+)2/3(Co3+)1/3PO4.25,75,76 However, during the cathodic sweep, only one reduction peak is observed (corresponding to Co2+/3+) at 4.65 V vs. Li/Li+ and a rather high polarization of 0.42 V. By comparison, the current linked to the redox couple of Co2+/3+ is substantially depressed for HT-PM-LCP. In addition, there is no clear peak during both oxidation and reduction processes. This broad curve feature obviously reflects a rather hampered Li-ion intercalation. Moreover, we noticed the magnitude of electrolyte decomposition to be more prominent in HT-PM-LCP (28.29 m2 g−1 in Fig. 2(b)) than in SS-PM-LCP (8.51 m2 g−1 in Fig. S6, ESI†) above 4.3 V vs. Li/Li+ (marked with blue-colored area in Fig. 3(f)) despite the use of stabilizing additive tris(trimethylsilyl) phosphite suggested by Allen et al.42 Although side reactions with electrolyte due to the LCP nanosizing are expected,40,72,77 the contrasting redox activity difference between the HT and SS samples points to other possible causes.
Hence, overall, we find poor cycling performance in the HT-synthesized LCP samples regardless of post-synthesis treatment, which is worse than that of the SS-synthesized LCP sample. Thus, elucidating the intrinsic material chemistry factors responsible for the poor charge-transfer kinetics and Li-ion storage of LCP is critical to unlocking its full theoretical capacity.
3.4 Nanoscale Co(OH)2 layer on the surface of HT-LCP
To investigate the factors that limit the performance of HT-LCP, we first performed TEM and HRTEM on a HT-LCP particle to see if there were any structural defects. As per Fig. S7a (ESI†), HT-LCP crystal d-spacing is 1.02 nm, which corresponds to the (100) plane of the LCP Pnma space group. In addition, the SAED pattern shown in Fig. S7b (ESI†) indicates that HT-LCP consists of a single-crystal motif.72 However, we discovered the existence of a nearly 3.8 nm thick surface layer covering the whole HT-LCP particle as shown in Fig. 4(a) and (b), which might be interfering with the electrochemical performance of HT-LCP by increasing charge-transfer resistance.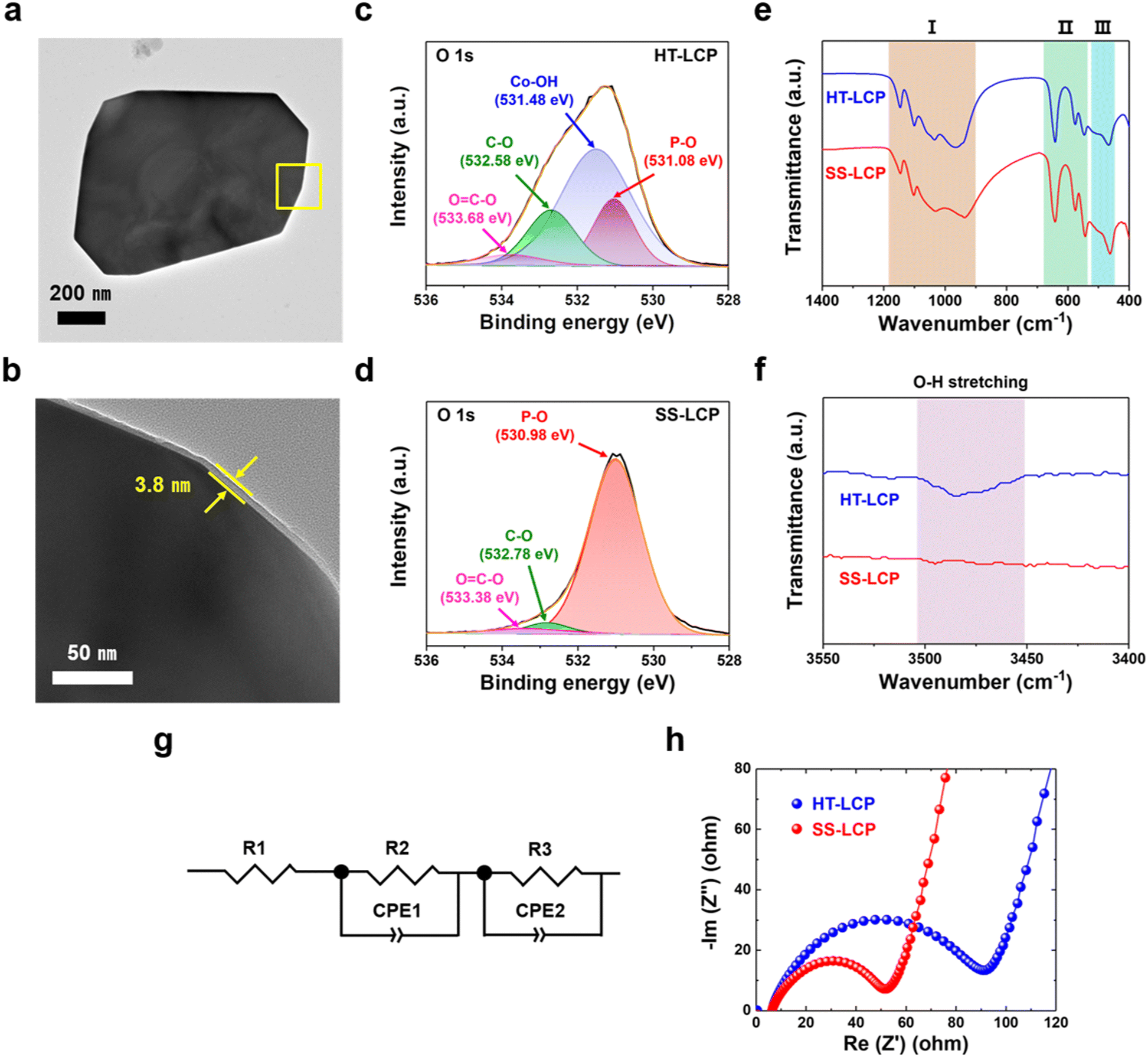 | ||
| Fig. 4 Characterization of nano-scale Co(OH)2 layer on the surface of HT-LCP. (a) and (b) TEM and HRTEM images of HT-LCP; (c) and (d) XPS O 1s and (e) and (f) FTIR spectra of HT-LCP and SS-LCP; (g) and (h) Nyquist plots of pristine HT-LCP and SS-LCP electrodes with equivalent circuit used to fit the EIS model. | ||
To understand the nature of the surface layer, we performed XPS on HT-LCP and SS-LCP (Fig. S8 and S9, ESI†). The chemical composition of the surface area is clearly comprising Li, Co, P, and oxygen (O) in both LCP samples, and the corresponding peaks can be associated with the binding energies of Li 1s, Co 2p, P 2p, O 1s, and C 1s. The high-resolution spectra of Co 2p, O 1s and C 1s were fitted with the Gaussian method. The Co 2p spectrum is resolved into Co 2p3/2 and Co 2p1/2 contributions centered at 781.18 and 797.38 eV for HT-LCP and 781.18 and 797.48 eV for SS-LCP, which can be ascribed to the Co2+ in LCP crystal structure.78–80 However, Fig. 4(c) and (d) reveals different chemical environments for O in the surface of HT-LCP and SS-LCP in regard to the high-resolution spectrum and curve fitting of O 1s. SS-LCP shows three deconvoluted O 1s peaks from the polyanion phosphate group (PO43−) group (530.98 eV), the C–O bonding (532.78 eV), and the OC–O bonding (533.38 eV) from surface contaminations. Yet, there is another O discernible peak in HT-LCP with binding energy of 531.48 eV indicative cobalt hydroxide (Co–OH) group.81–84 This result clearly thus identifies the thin surface layer on HT-LCP particles (as seen in TEM images) to be made up of Co(OH)2.
In addition, HT-LCP was subjected to XPS depth profile analysis with the integration of mild beam energy (200 eV) and Ar ion etching every 20 seconds, shown in Fig. S10 (ESI†). The profile result shows that the area under the fitting curve indicating nano-scale Co(OH)2 progressively decreases and entirely disappears after 80 second etching. We propose that the nano-scale Co(OH)2 layer forms via a side reaction after hydrothermal synthesis favoured by the alkaline solution pH.
We also performed FT-IR to verify the presence of nano-scale Co(OH)2 layer on HT-LCP particles. Fig. 4(e) and (f) show the FT-IR spectra of HT-LCP in comparison to those of SS-LCP. The spectra for both HT-LCP and SS-LCP samples in the lower wavenumber region are mainly dominated by the vibrations from the stretching and bending modes of PO43− guaranteeing the stable framework and the translational vibrations of Li-ion:20,85,86 (I) The broad band between 900–1100 cm−1 and 1050–1150 cm−1 corresponding to the symmetric and asymmetric stretching of PO43−, respectively. (II) The peaks around 500–700 cm−1 are attributed to the bending vibration mode of PO43−. (III) And the two peaks at 499 and 463 are derived from the translational vibrations of Li-ion adjacent to the oxygen atoms in the orthorhombic structure. However, we note that only HT-LCP shows the weakened O–H stretching band at 3484 cm−1 originating from the surface Co(OH)2 layer in the higher wavenumber region. These findings are further consistent with our XPS results (Fig. 4(c) and (d)), proving the existence of nano-scale Co(OH)2 layer on HT-LCP surface.
To study the impact of nano-scale Co(OH)2 layer on the electrochemical functionality of HT-LCP, as-made HT-LCP and SS-LCP electrodes were subjected to EIS analysis. A direct comparison between the two electrodes is made with the Nyquist plots in Fig. 4(g) and (h), where the EIS results are plotted together with the equivalent circuit model. Notably, HT-LCP displays a higher charge-transfer resistance (90.96 Ω) compared to SS-LCP (51.84 Ω), as can be inferred from the relative size of the semicircle in the high-frequency region.87 This result shows that the nano-scale Co(OH)2 layer on the HT-LCP acts as a passivation layer hampering charge transfer at the cathode/electrolyte interface and, as a consequence, resulting in poorer cycling performance compared to SS-LCP without the surface layer.
We note that the alkaline precursor solution with a high pH value above 8 creates an environment for Co(OH)2 formation, as indicated in the Pourbaix diagram of cobalt at 298.15 K (Fig. S11, ESI†)88,89 explaining why our HT-LCP has the Co(OH)2 surface layer. It is hypothesized residual Co2+ ions in hydrothermal solution adsorb on the surface of the precipitated LCP crystals and following surface hydrolysis due to high pH to form the in situ nano-scale Co(OH)2 layer. The Co(OH)2 layer would not have been a problem if Co(OH)2 were a fast Li-ion conductor. However, it is known that Co(OH)2 works as an anode material that goes through a sluggish conversion reaction at the lower voltage window (0.0–3.0 V vs. Li/Li+),90–92 which is below the operating potential of LCP. Therefore, this nano-scale Co(OH)2 layer would serve as an insulating layer during the cycling of LCP, consistent with the observed increased charge-transfer resistance for the HT-LCP compared to SS-LCP.
Meanwhile, since PM of the as-made HT-LCP particles should damage and at least partially remove the surface Co(OH)2 layer (Fig. S12, ESI†), it explains other than nanosizing, why HT-PM-LCP delivers a higher capacity. However, still the capacity of HT-PM-LCP is significantly below the theoretical one, implying that there must be other factors than the surface layer that further limit its cycling performance.
3.5 Anti-site defect evolution in HT-LCP
To probe other factors that may limit the cycling performance of HT-LCP, we tracked the defect concentrations in the bulk LCP crystal structure by applying XRD and ICP-OES techniques.32,48,49,93,94Fig. 5 shows the abundance of the anti-site defects (i.e., Co in Li site) and excess Co (i.e., excess amount of Co compared to the desired Li:Co = 1:1 ratio) in our LCP materials, extracted via XRD Rietveld refinement (also shown in Fig. S13, ESI†) and ICP-OES analysis, respectively. Noticeably, pristine HT-LCP (9.14%) has a high anti-site defect concentration nearly six times higher than that of pristine SS-LCP (1.73%). Such a significant amount of anti-site defects (9.14%) greatly limits 1-D Li-ion diffusion in the olivine structure, which constitutes another limiting factor explaining the small capacity in pristine HT-LCP samples.
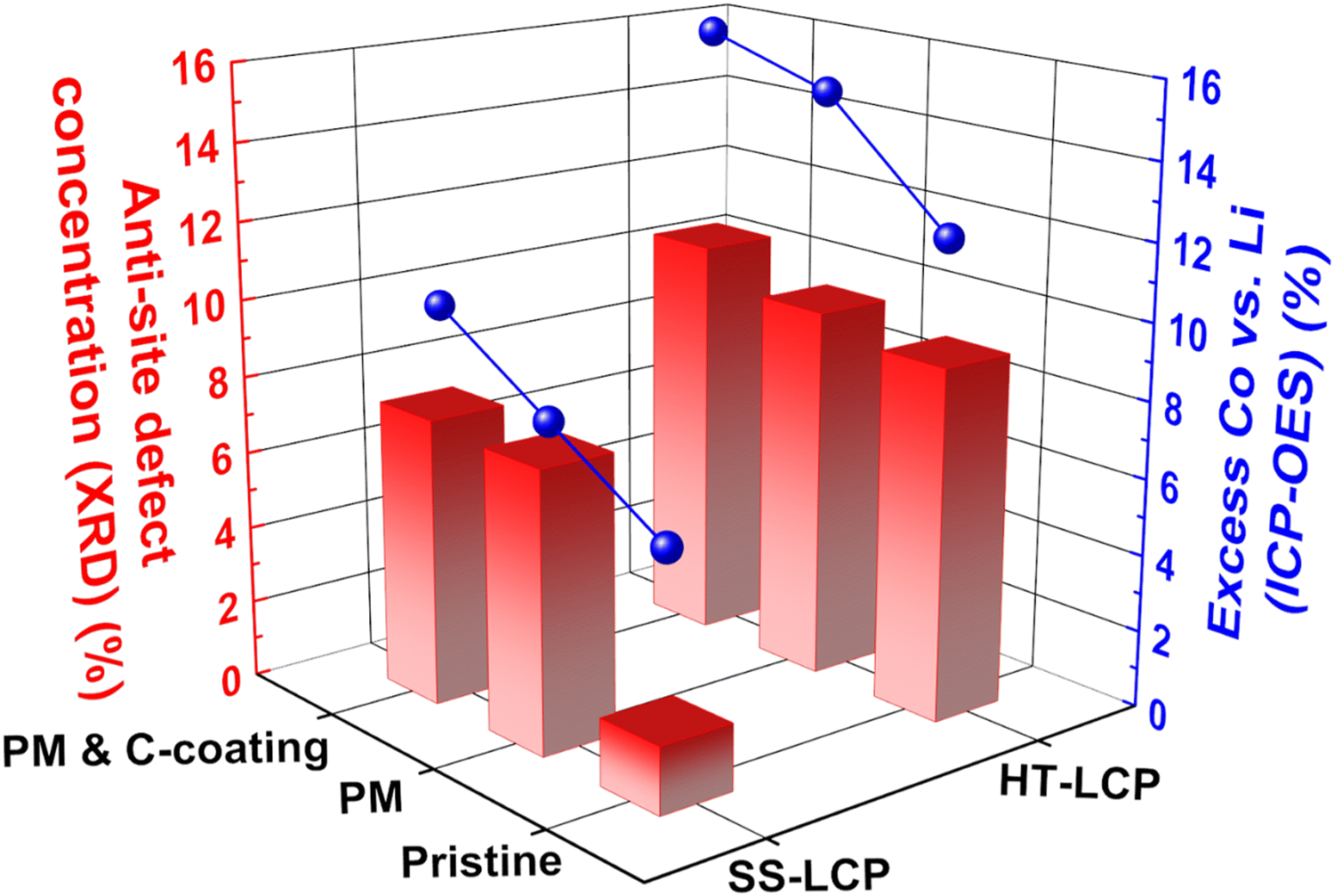 | ||
| Fig. 5 Anti-site defect concentration (red bar) and excess Co (blue sphere) calculated with XRD Rietveld refinement and ICP-OES analysis. | ||
In terms of anti-site defect evolution induced by PM and C-coating treatment steps, HT-LCP exhibits a modest increase from 9.63 to 10.52%. By comparison, anti-site defects in SS-LCP increased from 1.73% to 7.29 and 7.46% after the post-synthesis treatments. As mentioned earlier, PM causes a degree of disordering mobilizing the movement of ions hence introducing defects that seem to persist even after the C-coating at elevated temperature. Thus, it is reasonable to suspect that anti-site defects obtained during synthesis remained critical obstacles impeding Li-ion diffusion, and other than conventional annealing strategies need to be explored.
The ICP-OES results show that hydrothermal synthesis resulted in significant excess of Co (vs. Li) in HT-LCP composition (12.47%) compared to SS-LCP (6.4%). Since excess Co ions would occupy other cation sites than the original Co sites (thus Li sites), the greater excess Co found from HT-LCP than SS-LCP is consistent with the XRD Rietveld refinement results showing more anti-site defects (Co in Li sites) in HT-LCP. It must be noted that we used 150% excess Li (Li:Co:P = 2.5:1:1) in the precursor solution. Thus, it is surprising to observe Co excess instead of Li excess in HT-LCP, implying that this issue does not simply arise from the precursor ratio in the HT solution. Instead, this problem might be related to the nucleation and growth mechanism during the HT-synthesis, which limits full Li-ion incorporation to the Co–P–O intermediate species needed to form stoichiometric LCP. Finally, we note that the excess Co values increased after the post-synthesis treatments (HT-PM-LCP: 14.78%, HT-PM-C-LCP: 15.72% and SS-PM-LCP: 7.90%, SS-PM-C-LCP: 10.16%) following a similar trend with anti-site defect concentration. This increasing excess Co after post-synthesis treatments may be due to the loss of Li-ion to the isopropanol solvent used during PM or Li-ion evaporation upon the C-coating process at a high temperature.
Note that as LCP is a 1-D Li-ion diffusion material,26,95–97 keeping its stoichiometry would be critical to achieving high capacity because off-stoichiometry can either (i) block Li-ion diffusion in the crystal structure (found in Co-rich LCP such as Li0.8Co1.1PO4, excess Co in Li sites would block Li-ion diffusion) or (ii) limit the transition metal redox capacity (found in Li-rich LCP, e.g., Li1.1[Co2+0.8Co3+0.1]PO4, excess Li in Co sites which would increase the average Co oxidation state and decrease the Co-redox capacity). Our experiments suggest that off-stoichiometry from excess Co is another critical problem to overcome if HT-synthesis is to be used.
Overall, our experiments reveal two critical limitations of using the HT-synthesis of LCP. Although HT-synthesis can be green and has some important advantages (e.g., relatively easy control of particle size and morphology) over other synthesis methods, we reveal that a high pH value (pH 9) during the HT-synthesis (which is needed to form a high-purity and well crystalline LCP powder) also creates an environment that favors the nano-scale Co(OH)2 formation on the LCP particle surface introducing severe charge-transfer resistance. Moreover, even with the use of “excess Li” in the precursor solution, we reveal that HT-synthesis leads to the formation of LCP with “excess Co”, which introduces a large occurrence of anti-site defects in the bulk LCP crystal structure, significantly limiting the 1-D Li-ion diffusion. Avoidance of these stumbling blocks via modifications to the hydrothermal synthesis process or adoption of water-free synthesis routes should be pursued.
4. Conclusions
In conclusion, we successfully synthesized high-purity and single-crystal LiCoPO4 material (HT-LCP) with sub-micron particle size via HT-method by controlling the degree of supersaturation with over-stoichiometric Li contents (Li:Co:P = 2.5:1:1) and pH value of precursor solution 9 at 220 °C. Also, we applied nanosizing by planetary milling and C-coating on the HT-LCP to enhance its discharge capacity via shortening its diffusion length and increasing electron conductivity but with only partial success. Via a suite of surface and bulk crystal characterizations coupled with electrochemical analysis, we discovered hydrothermal synthesis to lead to (i) the formation of resistive nano-scale Co(OH)2 surface passivation layer and (ii) abundant anti-site defects arising from excess Co in HT-LCP samples compared to SS-LCP samples. These findings should allow for new routes to be explored so we ultimately design processes for achieving the full high-voltage capacity of LiCoPO4 and other types of polyanionic cathode materials.
Author contributions
M. W. performed the overall experiments and characterizations with the interpretation of the results. G. D. supervised all aspects of the research. All authors (M. W., J. L., and G. D.) contributed to the discussion of content. The manuscript was written by M. W. and revised by J. L. and G. D.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research project was funded by a NSERC Strategic Project grant to G. D. M. W. acknowledges support he received via the McGill Engineering Doctoral Award program.References
- G. E. Blomgren, J. Electrochem. Soc., 2016, 164, A5019–A5025 CrossRef.
- V. Sivaram, J. O. Dabiri and D. M. Hart, Joule, 2018, 2, 1639–1642 CrossRef.
- Y. Miao, P. Hynan, A. von Jouanne and A. Yokochi, Energies, 2019, 12, 1074 CrossRef CAS.
- C. Xu, Q. Dai, L. Gaines, M. Hu, A. Tukker and B. Steubing, Commun. Mater., 2020, 1, 99 CrossRef.
- A. Masias, J. Marcicki and W. A. Paxton, ACS Energy Lett., 2021, 6, 621–630 CrossRef CAS.
- D. Andre, S.-J. Kim, P. Lamp, S. F. Lux, F. Maglia, O. Paschos and B. Stiaszny, J. Mater. Chem. A, 2015, 3, 6709–6732 RSC.
- W. Li, B. Song and A. Manthiram, Chem. Soc. Rev., 2017, 46, 3006–3059 RSC.
- S. Zhang, J. Ma, Z. Hu, G. Cui and L. Chen, Chem. Mater., 2019, 31, 6033–6065 CrossRef CAS.
- E. M. Erickson, W. Li, A. Dolocan and A. Manthiram, ACS Appl. Mater. Interfaces, 2020, 12, 16451–16461 CrossRef CAS PubMed.
- J. Ling, C. Karuppiah, S. G. Krishnan, M. V. Reddy, I. I. Misnon, M. H. Ab Rahim, C.-C. Yang and R. Jose, Energy Fuels, 2021, 35, 10428–10450 CrossRef CAS.
- S. Zhao, Z. Guo, K. Yan, S. Wan, F. He, B. Sun and G. Wang, Energy Storage Mater., 2021, 34, 716–734 CrossRef.
- K. Striebel, J. Shim, A. Sierra, H. Yang, X. Song, R. Kostecki and K. McCarthy, J. Power Sources, 2005, 146, 33–38 CrossRef CAS.
- K. Zaghib, J. Dubé, A. Dallaire, K. Galoustov, A. Guerfi, M. Ramanathan, A. Benmayza, J. Prakash, A. Mauger and C. M. Julien, J. Power Sources, 2012, 219, 36–44 CrossRef CAS.
- C. Sun, S. Rajasekhara, J. B. Goodenough and F. Zhou, J. Am. Chem. Soc., 2011, 133, 2132–2135 CrossRef CAS PubMed.
- S. Wang, L. Lu and X. Liu, J. Power Sources, 2013, 244, 101–108 CrossRef CAS.
- H. Huang, S. C. Yin and L. F. Nazar, Electrochem. Solid-State Lett., 2001, 4, A170–A172 CrossRef CAS.
- C. S. Y. Sunhye Lim and J. Cho, Chem. Mater., 2008, 20, 4560–4564 CrossRef.
- K. Zaghib, A. Guerfi, P. Hovington, A. Vijh, M. Trudeau, A. Mauger, J. B. Goodenough and C. M. Julien, J. Power Sources, 2013, 232, 357–369 CrossRef CAS.
- G. Hautier, A. Jain, S. P. Ong, B. Kang, C. Moore, R. Doe and G. Ceder, Chem. Mater., 2011, 23, 3495–3508 CrossRef CAS.
- Q. D. Truong, M. K. Devaraju, Y. Ganbe, T. Tomai and I. Honma, Sci. Rep., 2014, 4, 3975 CrossRef PubMed.
- K. Amine, H. Yasuda and M. Yamachi, Electrochem. Solid-State Lett., 2000, 3, 178–179 CrossRef CAS.
- M. Zhang, N. Garcia-Araez and A. L. Hector, J. Mater. Chem. A, 2018, 6, 14483–14517 RSC.
- D. Liu, W. Zhu, C. Kim, M. Cho, A. Guerfi, S. A. Delp, J. L. Allen, T. R. Jow and K. Zaghib, J. Power Sources, 2018, 388, 52–56 CrossRef CAS.
- D.-W. Han, Y.-M. Kang, R.-Z. Yin, M.-S. Song and H.-S. Kwon, Electrochem. Commun., 2009, 11, 137–140 CrossRef CAS.
- F. C. Strobridge, R. J. Clement, M. Leskes, D. S. Middlemiss, O. J. Borkiewicz, K. M. Wiaderek, K. W. Chapman, P. J. Chupas and C. P. Grey, Chem. Mater., 2014, 26, 6193–6205 CrossRef CAS PubMed.
- C. A. J. Fisher, V. M. H. Prieto and M. S. Islam, Chem. Mater., 2008, 20, 5907–5915 CrossRef CAS.
- N. N. Bramnik, K. Nikolowski, D. M. Trots and H. Ehrenberg, Electrochem. Solid-State Lett., 2008, 11, A89–A93 CrossRef CAS.
- S. Theil, M. Fleischhammer, P. Axmann and M. Wohlfahrt-Mehrens, J. Power Sources, 2013, 222, 72–78 CrossRef CAS.
- I. C. Jang, H. H. Lim, S. B. Lee, K. Karthikeyan, V. Aravindan, K. S. Kang, W. S. Yoon, W. I. Cho and Y. S. Lee, J. Alloys Compd., 2010, 497, 321–324 CrossRef CAS.
- A. Freiberg, M. Metzger, D. Haering, S. Bretzke, S. Puravankara, T. Nilges, C. Stinner, C. Marino and H. A. Gasteiger, J. Electrochem. Soc., 2014, 161, A2255–A2261 CrossRef CAS.
- S. M. G. Yang, V. Aravindan, W. I. Cho, D. R. Chang, H. S. Kim and Y. S. Lee, J. Electrochem. Soc., 2012, 159, A1013–A1018 CrossRef CAS.
- Q. D. Truong, M. K. Devaraju, T. Tomai and I. Honma, ACS Appl. Mater. Interfaces, 2013, 5, 9926–9932 CrossRef CAS PubMed.
- R. Hanafusa, Y. Oka and T. Nakamura, J. Electrochem. Soc., 2015, 162, A3045–A3051 CrossRef CAS.
- A. V. Murugan, T. Muraliganth and A. Manthiram, J. Electrochem. Soc., 2009, 156, A79–A83 CrossRef.
- Y. Zhao, S. Wang, C. Zhao and D. Xia, Rare Met., 2009, 28, 117–121 CrossRef CAS.
- C. Neef, H.-P. Meyer and R. Klingeler, Solid State Sci., 2015, 48, 270–277 CrossRef CAS.
- Y. Maeyoshi, S. Miyamoto, Y. Noda, H. Munakata and K. Kanamura, J. Power Sources, 2017, 337, 92–99 CrossRef CAS.
- J. Ludwig, C. Marino, D. Haering, C. Stinner, D. Nordlund, M. M. Doeff, H. A. Gasteiger and T. Nilges, RSC Adv., 2016, 6, 82984–82994 RSC.
- X. Wu, M. Meledina, H. Tempel, H. Kungl, J. Mayer and R.-A. Eichel, J. Power Sources, 2020, 450, 227726 CrossRef CAS.
- D. Liu, C. Kim, A. Perea, D. Joel, W. Zhu, S. Collin-Martin, A. Forand, M. Dontigny, C. Gagnon, H. Demers, S. Delp, J. Allen, R. Jow and K. Zaghib, Materials, 2020, 13, 4450 CrossRef CAS PubMed.
- J. L. Allen, J. L. Allen, T. Thompson, S. A. Delp, J. Wolfenstine and T. R. Jow, J. Power Sources, 2016, 327, 229–234 CrossRef CAS.
- K. J. Kreder and A. Manthiram, ACS Energy Lett., 2016, 2, 64–69 CrossRef.
- Y. Wang, J. Chen, J. Qiu, Z. Yu, H. Ming, M. Li, S. Zhang and Y. Yang, J. Solid State Chem., 2018, 258, 32–41 CrossRef CAS.
- J. A. Darr, J. Zhang, N. M. Makwana and X. Weng, Chem. Rev., 2017, 117, 11125–11238 CrossRef CAS PubMed.
- M. K. Devaraju and I. Honma, Adv. Energy Mater., 2012, 2, 284–297 CrossRef CAS.
- K. N. Natalia, N. Bramnik, C. Baehtz, K. G. Bramnik and H. Ehrenberg, Chem. Mater., 2007, 19, 908–915 CrossRef.
- Y. Wang, H. Ming, J. Qiu, Z. Yu, M. Li, S. Zhang and Y. Yang, J. Electroanal. Chem., 2017, 802, 8–14 CrossRef CAS.
- M. K. Devaraju, Q. D. Truong, T. Tomai, H. Hyodo, Y. Sasaki and I. Honma, RSC Adv., 2014, 4, 52410–52414 RSC.
- A. Boulineau and T. Gutel, Chem. Mater., 2015, 27, 802–807 CrossRef CAS.
- X. Wu, M. Meledina, J. Barthel, Z. Liu, H. Tempel, H. Kungl, J. Mayer and R.-A. Eichel, Energy Storage Mater., 2019, 22, 138–146 CrossRef.
- M. Uceda, H. C. Chiu, J. Zhou, R. Gauvin, K. Zaghib and G. P. Demopoulos, Nanoscale, 2020, 12, 23092–23104 RSC.
- R. L. L. Stefania Ferrari, D. Capsoni, E. Quartarone, A. Magistris, P. Mustarelli and P. Canton, J. Phys. Chem. C, 2010, 114, 12598–12603 CrossRef.
- B. Ellis, W. H. Kan, W. R. M. Makahnouk and L. F. Nazar, J. Mater. Chem., 2007, 17, 3248–3254 RSC.
- K. Dokko, S. Koizumi, H. Nakano and K. Kanamura, J. Mater. Chem., 2007, 17, 4803–4810 RSC.
- X.-L. Pan, C.-Y. Xu and L. Zhen, CrystEngComm, 2012, 14, 6412–6418 RSC.
- Z. Li, K. Zhu, J. Li and X. Wang, CrystEngComm, 2014, 16, 10112–10122 RSC.
- J. Manuel García-Ruiz, J. Struct. Biol., 2003, 142, 22–31 CrossRef PubMed.
- G. P. Demopoulos, Hydrometallurgy, 2009, 96, 199–214 CrossRef CAS.
- L.-D. Shiau, Crystals, 2018, 8, 403 CrossRef.
- R. Zeitoun and A. Biswas, J. Electrochem. Soc., 2020, 167, 127507 CrossRef CAS.
- J. Conrad and P. R. Tremaine, Phys. Chem. Chem. Phys., 2021, 23, 10670–10685 RSC.
- D. R. Kester and R. M. Pytkowicx, Limnol. Oceanogr., 1967, 12, 243–252 CrossRef CAS.
- K. T. Lee and J. Cho, Nano Today, 2011, 6, 28–41 CrossRef CAS.
- K. J. Kreder, G. Assat and A. Manthiram, Chem. Mater., 2015, 27, 5543–5549 CrossRef CAS.
- E. Markevich, R. Sharabi, O. Haik, V. Borgel, G. Salitra, D. Aurbach, G. Semrau, M. A. Schmidt, N. Schall and C. Stinner, J. Power Sources, 2011, 196, 6433–6439 CrossRef CAS.
- R. Sharabi, E. Markevich, V. Borgel, G. Salitra, G. Gershinsky, D. Aurbach, G. Semrau, M. A. Schmidt, N. Schall and C. Stinner, J. Power Sources, 2012, 203, 109–114 CrossRef CAS.
- P. Puech, M. Kandara, G. Paredes, L. Moulin, E. Weiss-Hortaka, A. Kundu, N. Ratel-Ramond, J.-M. Plewa, R. Pellenq and M. Monthioux, C, 2019, 5, 69 CAS.
- D. Y. Kang, C. Kim, G. Park and J. H. Moon, Sci. Rep., 2015, 5, 18185 CrossRef CAS PubMed.
- E. Sviridova, A. Barras, A. Addad, E. Plotnikov, A. Di Martino, D. Deresmes, K. Nikiforova, M. Trusova, S. Szunerits, O. Guselnikova, P. Postnikov and R. Boukherroub, Biomater. Adv., 2022, 134, 112697 CrossRef PubMed.
- S. Sawalha, M. Assali, M. Raddad, T. Ghneem, T. Sawalhi, M. Almasri, A. Zarour, G. Misia, M. Prato and A. Silvestri, ACS Appl. Bio Mater., 2022, 5, 4860–4872 CrossRef CAS PubMed.
- M. Uceda, J. Zhou, J. Wang, R. Gauvin, K. Zaghib and G. P. Demopoulos, Electrochim. Acta, 2019, 299, 107–115 CrossRef CAS.
- M. Devaraju, Q. Truong, H. Hyodo, T. Tomai and I. Honma, Inorganics, 2014, 2, 233–247 CrossRef CAS.
- Y. Li and I. Taniguchi, Adv. Powder Technol., 2019, 30, 1434–1441 CrossRef CAS.
- S. Sreedeep, S. Natarajan and V. Aravindan, Curr. Opin. Electrochem., 2022, 31, 100868 CrossRef CAS.
- M. Kaus, I. Issac, R. Heinzmann, S. Doyle, S. Mangold, H. Hahn, V. S. K. Chakravadhanula, C. Kübel, H. Ehrenberg and S. Indris, J. Phys. Chem. C, 2014, 118, 17279–17290 CrossRef CAS.
- M. G. Palmer, J. T. Frith, A. L. Hector, A. W. Lodge, J. R. Owen, C. Nicklin and J. Rawle, Chem. Commun., 2016, 52, 14169–14172 RSC.
- T. Kozawa, K. Fukuyama, A. Kondo and M. Naito, Mater. Res. Bull., 2021, 135, 111149 CrossRef CAS.
- J. Su, B.-Q. Wei, J.-P. Rong, W.-Y. Yin, Z.-X. Ye, X.-Q. Tian, L. Ren, M.-H. Cao and C.-W. Hu, J. Solid State Chem., 2011, 184, 2909–2919 CrossRef CAS.
- B. Wu, H. Xu, D. Mu, L. Shi, B. Jiang, L. Gai, L. Wang, Q. Liu, L. Ben and F. Wu, J. Power Sources, 2016, 304, 181–188 CrossRef CAS.
- Y. Maeyoshi, S. Miyamoto, H. Munakata and K. Kanamura, J. Power Sources, 2018, 376, 18–25 CrossRef CAS.
- Y. Ge, K. Kan, Y. Yang, L. Zhou, L. Jing, P. Shen, L. Li and K. Shi, J. Mater. Chem. A, 2014, 2, 4961–4969 RSC.
- Y. Xu, Z. Liu, D. Chen, Y. Song and R. Wang, Prog. Nat. Sci.: Mater. Int., 2017, 27, 197–202 CrossRef CAS.
- H. Liu, D. Guo, W. Zhang and R. Cao, J. Mater. Res., 2017, 33, 568–580 CrossRef.
- A. Roy, H. S. Jadhav, G. M. Thorat and J. G. Seo, New J. Chem., 2017, 41, 9546–9553 RSC.
- A. Rajalakshmi, V. D. Nithya, K. Karthikeyan, C. Sanjeeviraja, Y. S. Lee and R. Kalai Selvan, J. Sol-Gel Sci. Technol., 2013, 65, 399–410 CrossRef CAS.
- W. Li, J. Hwang, W. Chang, H. Setiadi, K. Y. Chung and J. Kim, J. Supercrit. Fluids, 2016, 116, 164–171 CrossRef CAS.
- E. J. Kim, X. Yue, J. T. S. Irvine and A. R. Armstrong, J. Power Sources, 2018, 403, 11–19 CrossRef CAS.
- J. Chivot, L. Mendoza, C. Mansour, T. Pauporté and M. Cassir, Corros. Sci., 2008, 50, 62–69 CrossRef CAS.
- P. Meshram, U. Prakash, L. Bhagat, A. Abhilash, H. Zhao and E. D. van Hullebusch, Minerals, 2020, 10, 290 CrossRef CAS.
- Y.-S. He, D.-W. Bai, X. Yang, J. Chen, X.-Z. Liao and Z.-F. Ma, Electrochem. Commun., 2010, 12, 570–573 CrossRef CAS.
- J. Wu, W.-M. Lau and D.-S. Geng, Rare Met., 2017, 36, 307–320 CrossRef CAS.
- S. H. Lim, G. D. Park and Y. C. Kang, Chem. Eng. J., 2020, 401, 126121 CrossRef CAS.
- M. K. Devaraju, Q. D. Truong, H. Hyodo, Y. Sasaki and I. Honma, Sci. Rep., 2015, 5, 11041 CrossRef PubMed.
- A. Paolella, S. Turner, G. Bertoni, P. Hovington, R. Flacau, C. Boyer, Z. Feng, M. Colombo, S. Marras, M. Prato, L. Manna, A. Guerfi, G. P. Demopoulos, M. Armand and K. Zaghib, Nano Lett., 2016, 16, 2692–2697 CrossRef CAS PubMed.
- D. Morgan, A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, A30–A32 CrossRef CAS.
- D. J. D. M. Saiful Islam, C. A. J. Fisher and P. R. Slater, Chem. Mater., 2005, 17, 5085–5092 CrossRef.
- G. K. P. Dathar, D. Sheppard, K. J. Stevenson and G. Henkelman, Chem. Mater., 2011, 23, 4032–4037 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00455d |
| This journal is © The Royal Society of Chemistry 2023 |