
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAza-benzannulated-perylenebisimide-porphyrin dyad as an intensely absorbing donor in bulk-heterojunction organic solar cells†
Ayushi
Kaushik
 a,
Subhrajyoti
Bhandary
b,
Ganesh D.
Sharma
*c and
Jeyaraman
Sankar
*a
a,
Subhrajyoti
Bhandary
b,
Ganesh D.
Sharma
*c and
Jeyaraman
Sankar
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research, Bhopal (MP) 462066, India
bDepartment of Chemistry, Ghent University, 9000 Ghent, Belgium
cDepartment of Physics and Electronics and Communication Engineering, The LNM Institute of Information Technology, Jamdoli, Jaipur (Raj) 302031, India. E-mail: gdsharma273@gmail.com; gdsharma@lnmiit.ac.in
First published on 31st October 2023
Abstract
Porphyrins (PORPs) have demonstrated excellent photovoltaic performances in organic solar cells when applied in combination with suitable acceptors in molecular donor–acceptor (D–A) dyads. Perylenebisimides (PBIs) have emerged as important non-fullerene acceptors, and various derivatives of PBIs have been incorporated in D–A motifs as acceptors. In this work, we describe the design and photovoltaic performance of a novel N-benzannulated perylenebisimide-porphyrin dyad and its Zn(II) complex. Single-crystal X-ray diffraction (SCXRD) structures show a non-coplanar arrangement between the individual chromophores in the freebase dyad. The PBI and PORP chromophoric units show head-to-tail intermolecular π–π stacking interactions with a D–A distance of 3.4 Å. Solution-processed bulk heterojunction (BHJ) organic solar cells were fabricated using each of the two synthesized molecules as donors with PC71BM as the acceptor. The power conversion efficiencies (PCEs) for PDPP and Zn-PDPP were 7.25% and 9.10%, respectively, and they possess significant electron and hole mobilities.
Introduction
Organic solar cells (OSCs) based on a bulk heterojunction (BHJ) active layer are thriving as one of the promising technologies for renewable energy because of their potential low-cost fabrication, mechanical flexibility, lightweight, and color-tunable features.1–5 With the emergence of non-fullerene small molecule acceptors, the power conversion efficiency (PCE) of OSCs has exceeded 19% via optimizing the device structure and morphology of the BHJ active layer using polymers as a donor.6–9 However, the application of polymer donors is significantly limited by their inherent properties, such as dispersive molecular weight and limited reproducibility, thereby leaving several challenges for the commercialization of OSCs based on polymers as donors. Therefore, it is crucial to develop a successful replacement for polymer-based BHJ-OSCs that is effective, mechanically and electronically flexible, chemically durable, and economically viable, to allow the enhancement of the PCE along with better electron mobility. Solution-processed BHJ all small molecule-OSCs have attracted renewed interest owing to their well-defined chemical structure, easy purification, and easier energy level adjustment. They could overcome the shortcoming of polymers and have emerged as promising candidates for commercial production of OSCs.10–12 So far, the best PCEs of 11%13–15 and 16–17%,16–21 respectively, have been reported for all small molecule-OSCs based on fullerene and non-fullerene acceptors, which is comparable to those of polymers. Therefore, it is necessary to design simple and low-cost small molecule donors that will be compatible with both fullerene and non-fullerene acceptors for OSCs.Rylene diimides are an important class of compounds exhibiting a variety of applications in modern organic material-based devices. These molecules possess a rigid polyaromatic backbone, along with two imide groups positioned at the edge of their long axis. The imide substituents help in the solubility of these molecules and often do not influence the electronic properties due to the presence of nodes passing through the N–N axis.22 This class of molecules has also been extensively explored as a multi-redox system for modern optoelectronic applications.23 Perylenebisimides (PBIs) are one of the popular members of this family due to their chemical and photochemical stability. Their photophysical characteristics make them efficient materials suitable for organic photovoltaics and organic light-emitting devices.24 However, the narrow range of absorption of these materials needs to be improved to enhance their photovoltaic efficiency. Thus, developing newer derivatives of these compounds has been an active area of research in recent years.
The absorption characteristics of PBIs can be effectively tuned by functionalizing them at the available polyaromatic positions. Functionalization at the bay positions of PBIs has shown potential in influencing their electronic nature and thus dictating the photophysical properties effectively. The bay positions have been utilized for both π-extension and for π-expansion, depending on the nature of the application. The π-extension strategy has been widely exploited to achieve molecules with bathochromically shifted absorption, whereas π-expansion often results in a hypsochromic shift. However, neither method results in a panchromatic absorption ideal for applications such as artificial light harvesting. Thus, having only a PBI unit will not be sufficient to achieve this goal, but connecting a suitable accessory chromophore can augment the absorption profile. Also, the formation of optimal film morphologies for effective charge separation and transport in optoelectronics is severely hampered by the high tendency of large coplanar molecular structures for PBIs to form microscale aggregates, which significantly limits their photovoltaic performance.25–28 Different methods have been developed to reduce intermolecular interactions and excessive domain sizes in an effort to solve this issue.29,30 The most popular technique is to attach a bulky substituent at the bay position, which can thereby render the PBI derivatives non-planar. This aids in reducing excessive intermolecular contact and preventing the creation of large aggregates.
The π-conjugation extension strategy has been successfully used to achieve molecular platforms that have panchromatic absorption (Chart 1). One of the efficient accessory chromophores exploited for this is porphyrin, due to its complementing absorption profile with that of PBIs. Porphyrins have intense Soret bands (∼400 nm) and Q-bands (∼500–600 nm).31,32 Derivatives achieved via conjugation extension of PBIs with ethynyl porphyrins have wider absorption profiles and have been demonstrated to show impressive performance as active layers in organic solar cells.33,34 This is due to the electronic complementarity of the two chromophores and their absorption profiles. However, the concept of an accessory chromophore has not been explored with π-expanded PBIs for OSCs. This strategy may result in derivatives having entirely new electronic and photophysical characteristics, which will be interesting for our desired light-harvesting applications.
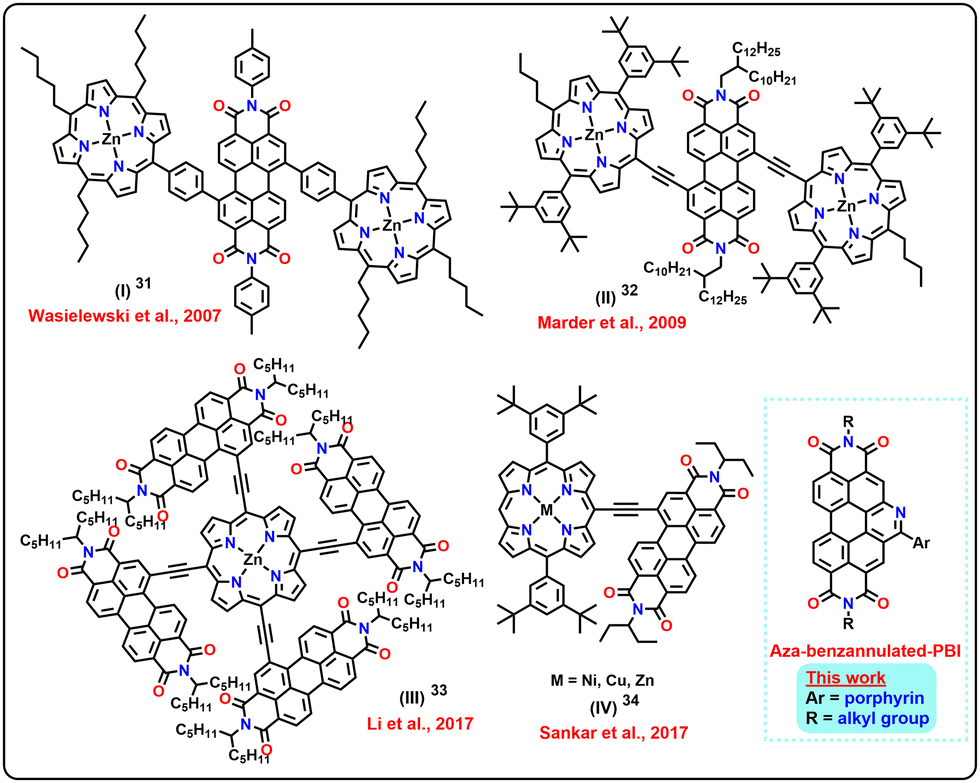 | ||
| Chart 1 Selected examples of PBI-porphyrin conjugates reported and the current work. | ||
Recently, there have been a few interesting reports on organic solar cells based on PBI and PORP with moderate PCEs; however, achieving higher efficiency is still challenging. Therefore, developing novel PORP-PBI-based small molecules with versatile structures and further obtaining insight into their potential photovoltaic application has aroused our interest. Inspired by the strategy for constructing low-bandgap materials with a D–A system, herein, the highly functionally flexible PORP platform was selected for developing D–A structural small molecule-based wide-range absorbing donors. In our previously reported work (molecule IV, Chart 1), even though the porphyrin and PBI were linked through an ethynyl bridge to incorporate electronic coupling, the performance was limited to 6% PCE. In this case, an effective charge-transfer between the donor and acceptor motifs widened the absorption range and thus provided a panchromatic absorption.34 Despite this, the PCE could not be further improved upon. This result hinted at screening different PBI derivatives as acceptors while retaining the porphyrin as a donor to check the effect on the PCE. In this direction, it was presumed that a core-expanded PBI would be an interesting candidate to investigate the effect of anchoring with porphyrin. Additionally, the presence of a heteroatom, if any, in the core would also perturb the electronic nature of the derivative. In this direction, it was decided to link an aza-benzannulated-PBI to the meso-position of both a freebase porphyrin and its zinc(II) complex.
Aza-benzannulated PBIs can be obtained by simple Pictet–Spengler reaction (PSR) of 1-amino-PBI with the respective aryl aldehydes in the presence of an acid, followed by aromatization. Even though PSR is known on simple PBIs35–38 and porphyrins individually39,40 there is no report on connecting the two chromophores using this strategy. We report the first successful synthesis of a porphyrin-appended aza-benzannulated-PBI using a one-pot, one-step PSR, along with its zinc metal complex, in high yields. This molecular platform is also unique due to two large polyaromatic units connected directly to each other; an N-doped semi-coronene diimide and a porphyrin. Most importantly, the resultant dyad is structurally characterized, and its photophysical properties are investigated. Light harvesting efficiency was investigated for the OSCs fabricated from these dyads as donors.
Results and discussion
The starting materials 1-amino-perylenebisimide (PBN) and 10,20-bis(3,5-ditertbutylphenyl)-porphyrin-5-carboxaldehyde (PCHO) were synthesized from PBI and PORP, respectively, using reported procedures.41,42 The desired molecular dyad PDPP was synthesized via a modified Pictet–Spengler condensation reaction of PBN and PCHO using trifluoromethanesulphonic acid (TFSA) as a catalyst in toluene (Scheme 1). Here, the selection of porphyrin aldehyde as one of the substrates and TFSA behaving as a reagent made this reaction happen in a single step. For the preparation of the dyad PDPP, two equiv. of the compound PCHO were taken with ten equiv. of TFSA with respect to one equivalent of PBN in toluene, and the reaction mixture was stirred and heated for 4 h at 110 °C under argon. No further dilution or exposure to visible light or addition of oxidant was required. The desired product was obtained with a good yield. The Zn(II)-complex was obtained by carrying out metalation of the freebase dyad using zinc acetate as shown in Scheme 1. All the PBI- and PORP-based precursors were characterized by NMR spectroscopy and mass spectrometry. Synthesis and characterization details are given in detail in the ESI† (Fig. S1–S37). The mechanistic pathway for the formation of PDPP in a single step is shown in Fig. S49 (ESI†).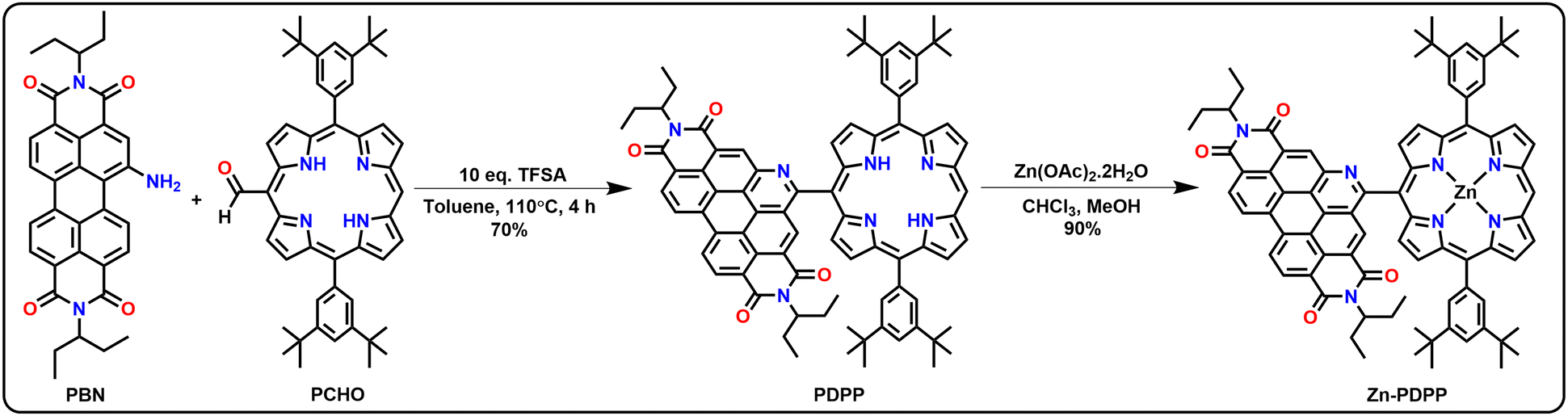 | ||
| Scheme 1 Synthesis of the aza-benzannulated-PBI-porphyrin dyad (PDPP) and its Zn-complex (Zn-PDPP). | ||
Crystallography
The single-crystal X-ray diffraction studies were carried out for PDPP and PDPP-triflate. Crystals suitable for diffraction were obtained by slow-diffusion of hexane into a toluene solution and chloroform solution for PDPP and PDPP-triflate, respectively. The PBI and PORP units in the dyad PDPP-triflate were found to be non-coplanar with a dihedral angle of 59.4°. The core of the porphyrin was found to be ruffled in the case of the triflate salt of PDPP (Fig. 1). The same is the case with the crystals obtained for free-base PDPP. The two chromophoric units are almost orthogonal to each other with π–π stacking between the donor and the acceptor in the case of PDPP, as shown in Fig. 1 and Fig. S47 (ESI†).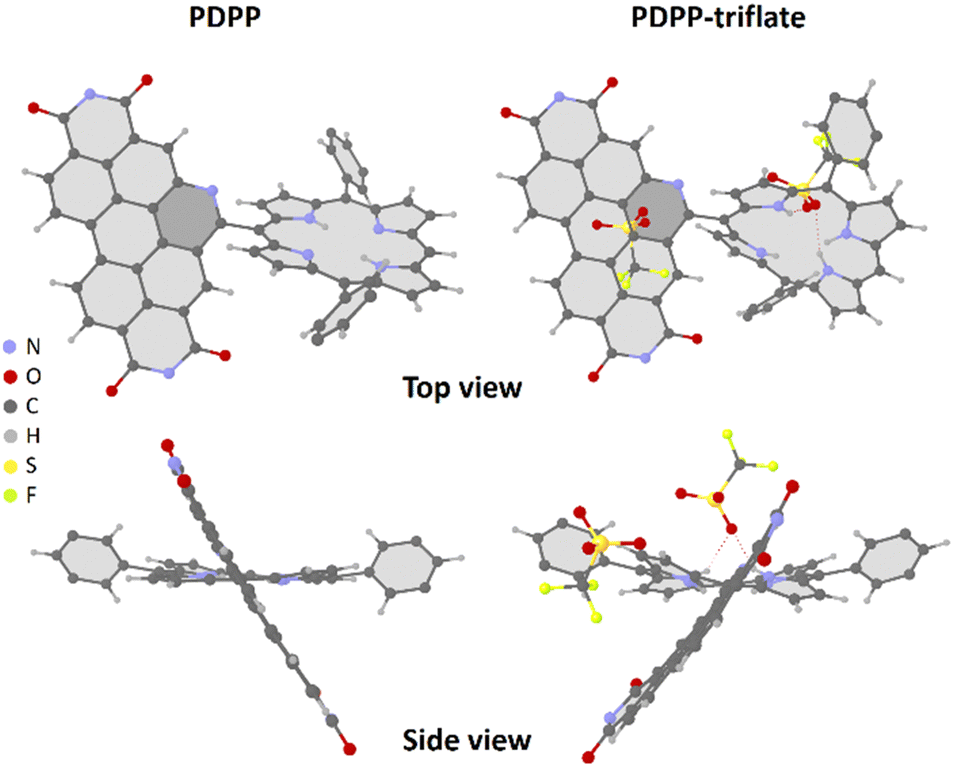 | ||
| Fig. 1 SCXRD structures of PDPP (CCDC 2255973†) and PDPP-triflate (CCDC 2255971†) salt (PDPP crystallized using triflic acid). Alkyl chains are omitted for clarity. | ||
Photophysical studies
Fig. 2a shows the absorption profiles for both the synthesized molecules in toluene. The dyad PDPP shows an absorption-maximum with the Soret band at 417 nm superimposing with absorption for N-benzannulated-PBI at around 467 nm, whereas its zinc(II) complex shows slightly red-shifted Soret and Q-bands. The emission spectra for these molecules were also recorded in toluene. It was found that the dyad PDPP showed a dual emission with PBI and porphyrin-based emission bands of reduced intensities (Fig. S38b, ESI†), whereas Zn-PDPP was non-emissive. Complete optical spectroscopic data are listed in Table 1. The absorption profiles confirm the limited electronic coupling in the ground state between the PBI and PORP units in both PDPP and Zn-PDPP. This results from the free rotation around the single bond that connects both and the non-coplanar arrangement of the two chromophores. Moreover, the PBI core in the dyads is expected to be comparatively less electron-deficient than the parent PBI (PBI) and thus have a reduced inductive effect on the PORP unit. The absence of any prominent CT bands in the bathochromic region further reiterates this inference. Interestingly, both the dyads possess similar molar extinction coefficients and thus hint at a marginal electronic effect by Zn(II) insertion.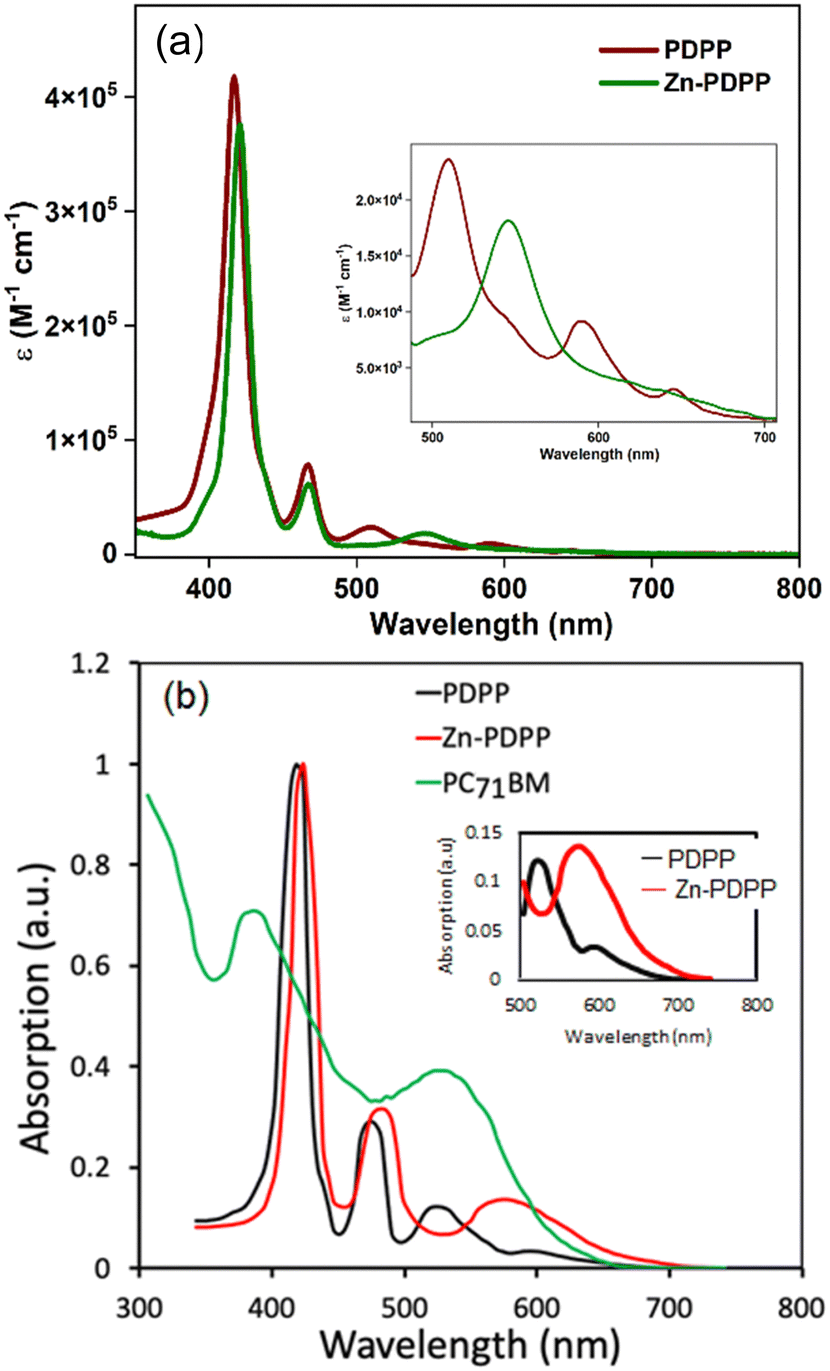 | ||
| Fig. 2 UV-visible absorption spectra of PDPP and Zn-PDPP (in toluene solvent, concentration = 1 μM) (a) and in a thin film cast from chloroform solution (b). The thin film absorption spectrum of PC71BM is also shown in (b). | ||
The thin film UV-visible absorption spectra of both the molecules along with the absorption spectra of PC71BM are shown in Fig. 2b. As compared to the absorption spectra in solution, the absorption spectra in the film get broadened and redshifted due to the intermolecular interactions present in the solid state. The redshift for Zn-PDPP is more as compared to that of PDPP, which is ascribed to a stronger intermolecular interaction due to the electron-rich Zn–porphyrin unit. The optical bandgaps estimated from the onset edge of these spectra are about 1.75 and 1.66 eV for PDPP and Zn-PDPP, respectively. The thin film absorption spectrum of PC71BM is complementary to both the synthesized molecules.
Electrochemical studies
The electrochemical properties of these new molecules were investigated by cyclic voltammetry (CV) in deaerated CH2Cl2 using Bu4NPF6 (TBAP) as a supporting electrolyte (Fig. 3). The dyad Zn-PDPP shows three reversible reductions and two reversible oxidations. In contrast, PDPP shows two irreversible oxidations along with three reversible reductions. Out of these, the first two reductions are expected to be PBI-centered, while the third one corresponds to a porphyrin-centered reduction. The two oxidations were expected to be of porphyrin type in each case. Further details on the reduction and oxidation potentials and electrochemical band gaps for these molecules are given in Table 1. The HOMO/LUMO energy levels obtained from electrochemical investigations are −5.44/−3.72 and −5.23/−3.66 eV, for PDPP and Zn-PDPP, respectively.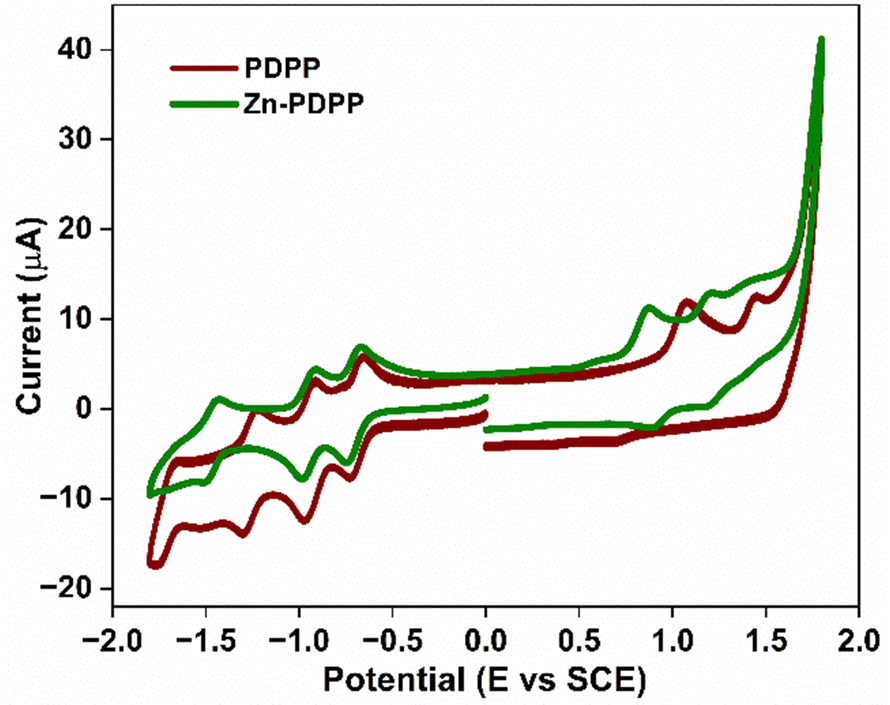 | ||
| Fig. 3 Cyclic voltammograms (CV) for PDPP, and Zn-PDPP in TBAP (0.1 M), and CH2Cl2 at 100 mV s−1. | ||
The LUMO offsets between PDPP or Zn-PDPP and PC71BM (LUMO = −4.12 eV) are about 0.30 or 0.38 eV, greater than the driving force needed for exciton dissociation and charge separation in the BHJ active layer.
Density functional theory (DFT) calculations
To visualize the electron delocalization, DFT calculations were carried out at the B3LYP/6-31g(d,p) level using the Gaussian 09 suite of programs for the dyads. The HOMOs and LUMOs for both molecules are shown in Fig. 4. The HOMO was found to be localized mainly on the porphyrin core while the LUMO was found to be localized over the PBI core along with its N-heteroring in each case. This is in line with the expectation for the individual chromophores.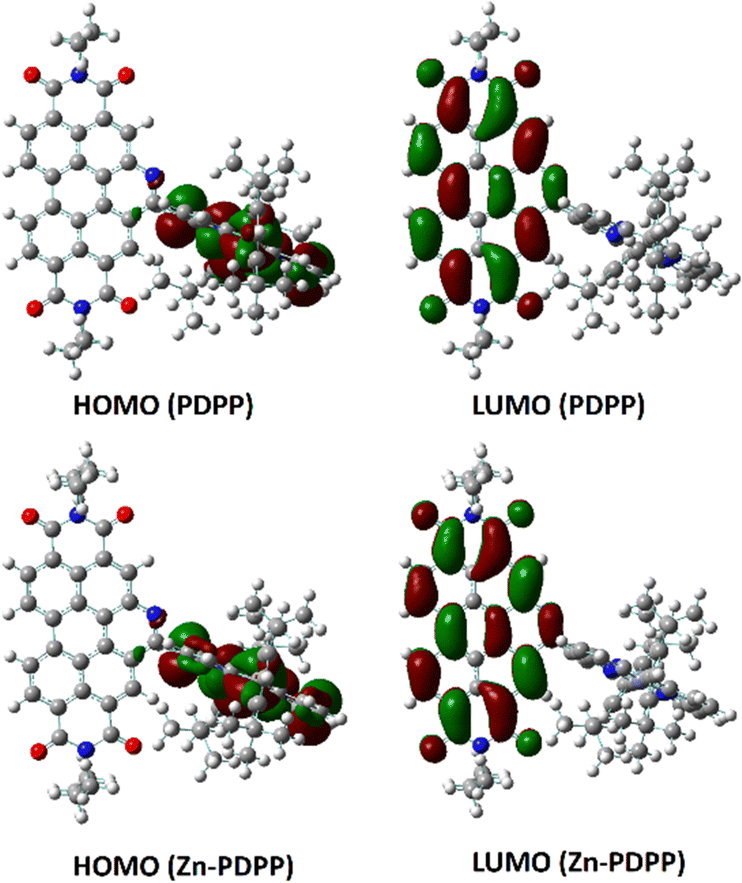 | ||
| Fig. 4 Frontier molecular orbitals of PDPP and Zn-PDPP obtained by DFT, calculated at the B3LYP/6-31G(d,p) level. | ||
Photovoltaic properties
The conventional BHJ-OSCs were fabricated with a structure of ITO/PEDOT:PSS/PDPP or Zn-PDPP:PC71BM/PFN/Al. The targeted BHJ active layer consisted of PDPP or Zn-PDPP as the donor and PC71BM as the acceptor prepared by spin coating the blend solution of the donor and acceptor with different D/A weight ratios in chloroform solution and it was found that the ratio with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.4 showed the best photovoltaic performance. To optimize the morphology of the BHJ active layer and to improve the photovoltaic performance, pyridine as the solvent additive was added to the chloroform solution (0.5 v%) prior to the spin coating process, it was found that the power conversion efficiency was improved after this modification. To improve the photovoltaic performance further, we have adopted solvent vapor treatment by exposing the optimized active layer with THF vapour for 40 s, before the deposition of the PFN layer. The current–voltage characteristics of the OSCs are presented in Fig. 5a and the corresponding photovoltaic parameters are summarized in Table 2. The optimized OSCs based on PDPP and Zn-PDPP showed overall PCEs of 7.25% and 9.10%, respectively. The OSC based on PDPP showed a higher value of VOC (0.94 V) than its Zn-PDPP counterpart owing to the deeper HOMO energy level of PDPP, the enhanced value of the PCE for Zn-PDPP-based OSCs is due to the improved values of both current density (JSC) and fill factor (FF). The higher value of JSC for the Zn-PDPP-based OSCs may be attributed to its lower optical bandgap as compared to PDPP, which is beneficial for high exciton generation in the former OSC than the latter.
:1.4 showed the best photovoltaic performance. To optimize the morphology of the BHJ active layer and to improve the photovoltaic performance, pyridine as the solvent additive was added to the chloroform solution (0.5 v%) prior to the spin coating process, it was found that the power conversion efficiency was improved after this modification. To improve the photovoltaic performance further, we have adopted solvent vapor treatment by exposing the optimized active layer with THF vapour for 40 s, before the deposition of the PFN layer. The current–voltage characteristics of the OSCs are presented in Fig. 5a and the corresponding photovoltaic parameters are summarized in Table 2. The optimized OSCs based on PDPP and Zn-PDPP showed overall PCEs of 7.25% and 9.10%, respectively. The OSC based on PDPP showed a higher value of VOC (0.94 V) than its Zn-PDPP counterpart owing to the deeper HOMO energy level of PDPP, the enhanced value of the PCE for Zn-PDPP-based OSCs is due to the improved values of both current density (JSC) and fill factor (FF). The higher value of JSC for the Zn-PDPP-based OSCs may be attributed to its lower optical bandgap as compared to PDPP, which is beneficial for high exciton generation in the former OSC than the latter.
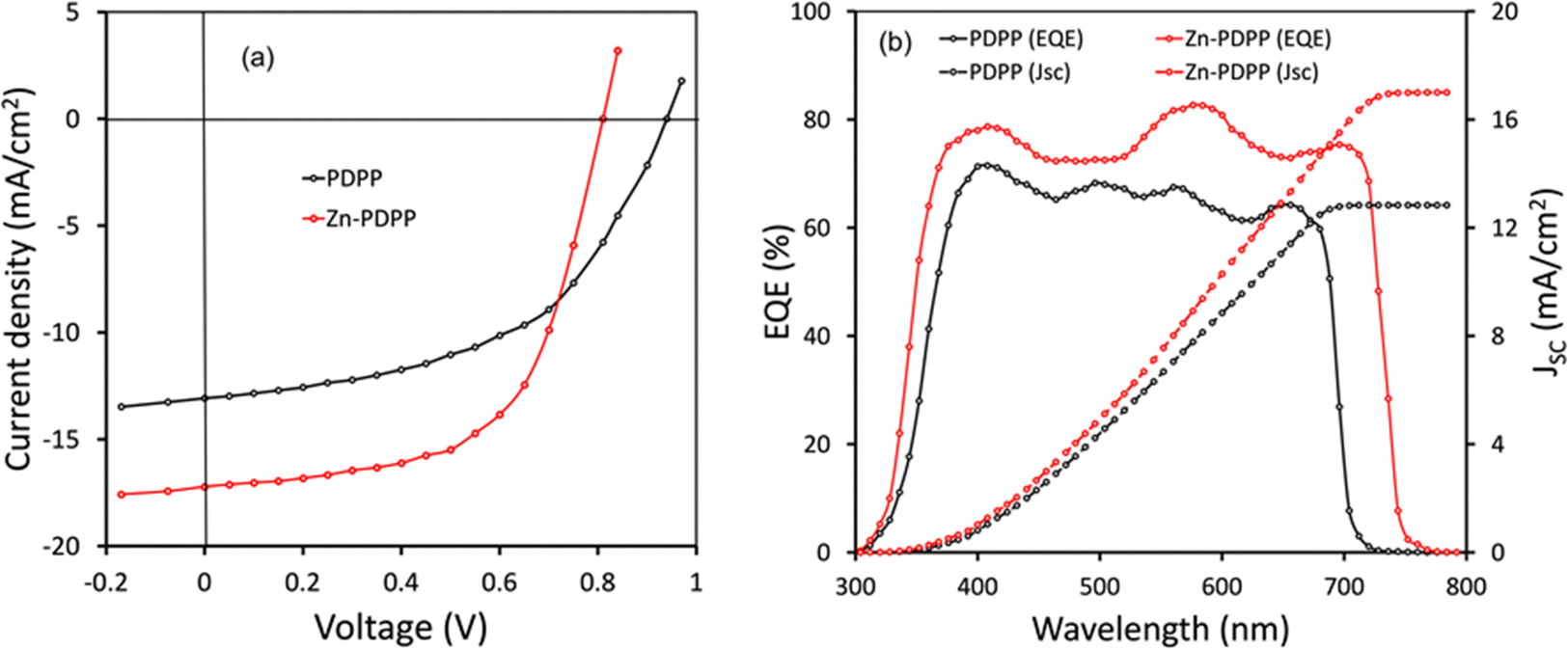 | ||
| Fig. 5 (a) J–V Characteristics under illumination (AM1.5 G. 100 mW cm−2) and EQE spectra of OSCs. Estimated values of JSC are also shown in (b). | ||
Furthermore, the external quantum efficiency (EQE) of the OSCs was measured as shown in Fig. 5b. As presented in Fig. 5b, the EQE spectra of the OSCs closely resemble the corresponding absorption spectra of the active layers (Fig. 2b). The EQE spectra for Zn-PDPP are extended up to 760 nm whereas for the PDPP counterpart it is limited up to 720 nm. These results suggest that the light-harvesting efficiency in the Zn-PDPP-based OSC is higher than that of the PDPP counterpart. The JSC values estimated from the integration of the EQE spectra are about 12.76 and 17.04 mA cm−2 for PDPP and Zn-PDPP-based OSCs, respectively, and this trend agrees with the values obtained from the J–V curves under illumination.
The exciton generation and their subsequent dissociation into free charge carriers, and the charge transport and recombination processes within the active layer, play a crucial role in the overall PCE. To get information about the exciton generation and their dissociation into free charge carriers, the variation of photocurrent density (Jph) with effective voltage (Veff)43,44 was investigated and shown in Fig. 6. The Jph is estimated as Jph = JL − JD, where JL and JD are the current densities under illumination and in the dark, respectively and Veff = Vo − Va, where Vo is the voltage when Jph is zero, and Va is the applied voltage.
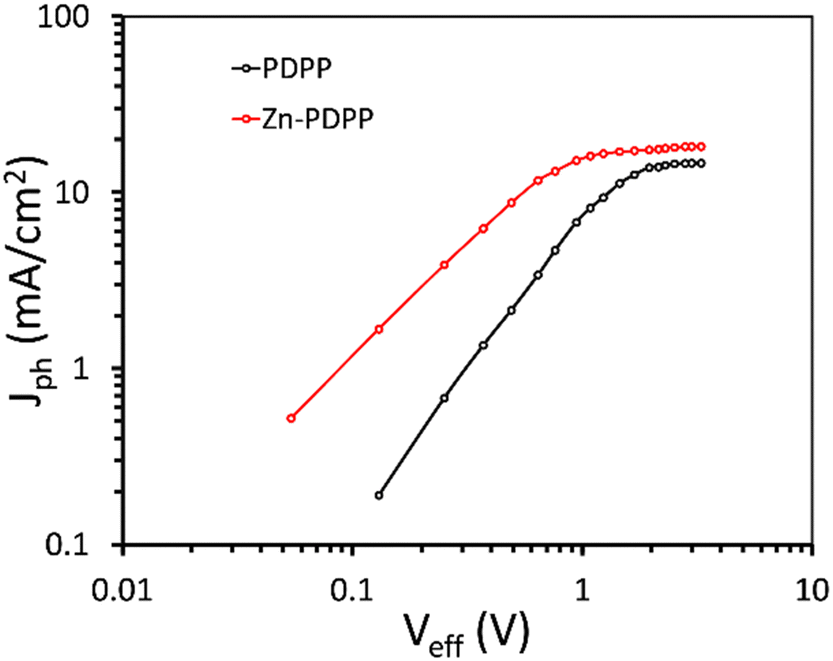 | ||
| Fig. 6 Variation of Jph with Veff for OSCs. | ||
In general, when Veff is increased to a sufficiently high value, the Jph is independent of Veff, and reached its saturation value (Jsat) and only depends upon the light harvesting efficiency of the active layer. The maximum exciton generation rate (Gmax) can be estimated as Gmax= Jsat/qL, where q is the elementary charge and L is the thickness of the active layer and is about 0.96 × 1028 m−3 s−1, and 1.15 × 1028 m−3 s−1 for PDPP:PC71BM and Zn-PDPP:PC71BM based OSCs, respectively, indicating that more excitons are generated in the latter device as compared to the former one. The values of exciton dissociation probability (Pdiss) and charge collection probability (Pcoll) were estimated as the ratio of Jph/Jsat under short circuit conditions and maximum power conditions, respectively. The values of Pdiss/Pcoll for the PDPP:PC71BM and Zn-PDPP:PC71BM are about 0.886/0.667 and 0.942/0.712, respectively. It can be seen from Fig. 6 that the Jph saturates at a higher value of Veff for PDPP:PC71BM than for Zn-PDPP:PC71BM, indicating that more effective internal electric field is needed to sweep the charge carriers towards the electrode for the former than the latter. These observations indicate that the Gmax, Pdiss and Pcoll are higher for Zn-PDPP:PC71BM than for PDPP:PC71BM, which may be one of the origins for the higher values of both JSC and FF for the former.
To get information about the charge transport, we have prepared hole only devices (ITO/PEDOT:PSS/active layer/Au) and electron only devices (ITO/Al/active layer/Al) and measured their dark J–V characteristics. We have fitted these curves with space-charge limited current (SCLC) to estimate the hole mobility (μh) and electron mobility (μe)45 (Fig. 7). The values of μe (3.12 × 10−4 cm2 V−1 s−1) and μh (2.34 × 10−4 cm2 V−1 s−1) for Zn-PDPP:PC71BM are higher with a μe/μh ratio of 1.33 than that for PDPP:PC71BM (μe = 3.06 × 10−4 cm2 V−1 s−1 and μh = 2.02 × 10−4 cm2 V−1 s−1 with μe/μh = 1.51), indicating that the charge transport in the former device is more efficient than the latter, resulting in high value of FF for Zn-PDPP:PC71BM-based OSCs. Here, V − Vbi (in volts) on the horizontal axis in Fig. 7 represents the difference in the applied voltage (V) and built-in potential (Vbi).
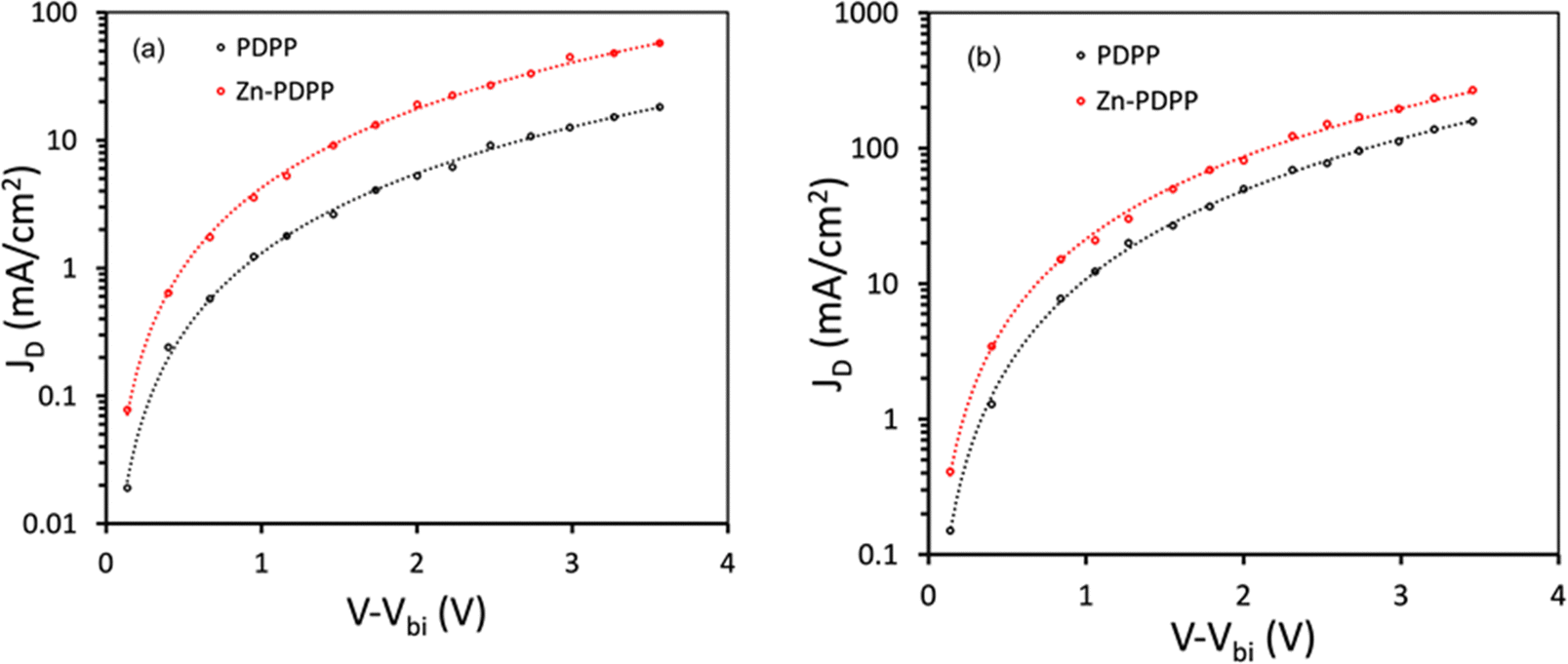 | ||
| Fig. 7 Dark J–V characteristics for (a) hole-only and (b) electron-only devices and SCLC fitting. | ||
We have examined the variation of JSC and VOC with illumination intensity (Pin) for these devices,46,47 as shown in Fig. 8. The variation of JSC with Pin follows the power law as JSC ∝ (Pin)α, where α is the exponent power factor and gives information about the degree of bimolecular recombination. When the value of α is unity, the bimolecular recombination is almost negligible, whereas if the value of α is less than unity, it indicates the occurrence of bimolecular recombination in the active layer. The value of α for the devices based on Zn-PDPP is higher (0.939) compared to those based on PDPP (0.881), indicating that the bimolecular recombination is suppressed for the former device as compared to the latter one. The variation of VOC with Pin follows the relationship as VOC = (nkT/q)ln(Pin), where k is Boltzmann's constant, T is the absolute temperature, q is the elementary charge, and n is the diode quality factor. The value of n estimated from the slope of the VOC–Pin curves is about 1.46 and 1.31 for PDPP and Zn-PDPP-based devices, indicating that the trap-assisted recombination is more suppressed for the Zn-PDPP-based OSCs, compared to the PDPP counterpart. The reduced bimolecular and trap-assisted recombination for Zn-PDPP-based-OSCs lead to higher values of both JSC and FF.
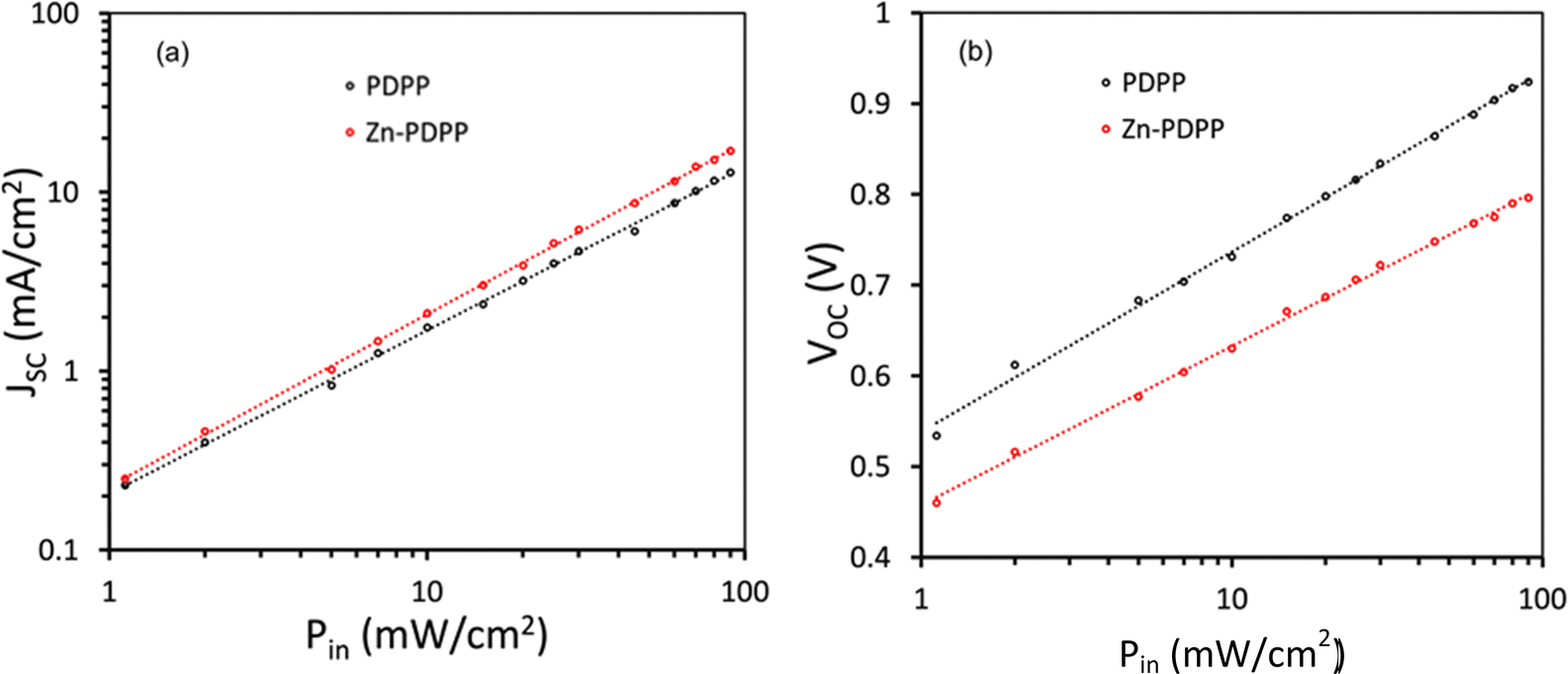 | ||
| Fig. 8 Variation of (a) JSC and (b) VOC for the OSCs. | ||
To gain further information about the charge carrier dynamics process in these OSCs, transient photocurrent (TPC) and transient photovoltage (TPV) decays were measured and shown in Fig. 9. The charge carrier lifetime and extraction time were derived from the TPV and TPC decay curves, respectively, using mono-exponential fits.48,49 The device based on Zn-PDPP showed shorter extraction time (1.41 s) than the PDPP counterpart (1.63 s) estimated from the TPC curves (Fig. 9a), indicating more efficient extraction of the charge carriers in the former device than the latter, which is in good agreement with the higher value of charge collection probability (Pcoll). As shown in Fig. 9b, the charge carrier lifetimes estimated from the TPV decay curves in the device based on PDPP and Zn-PDPP are 7.09 s and 8.36 s, respectively. The relatively longer charge carrier time in the Zn-PDPP-based device than the PDPP counterpart implies that significantly reduced charge recombination occurred in the former device than the latter, which is in good agreement with the higher value of FF.50
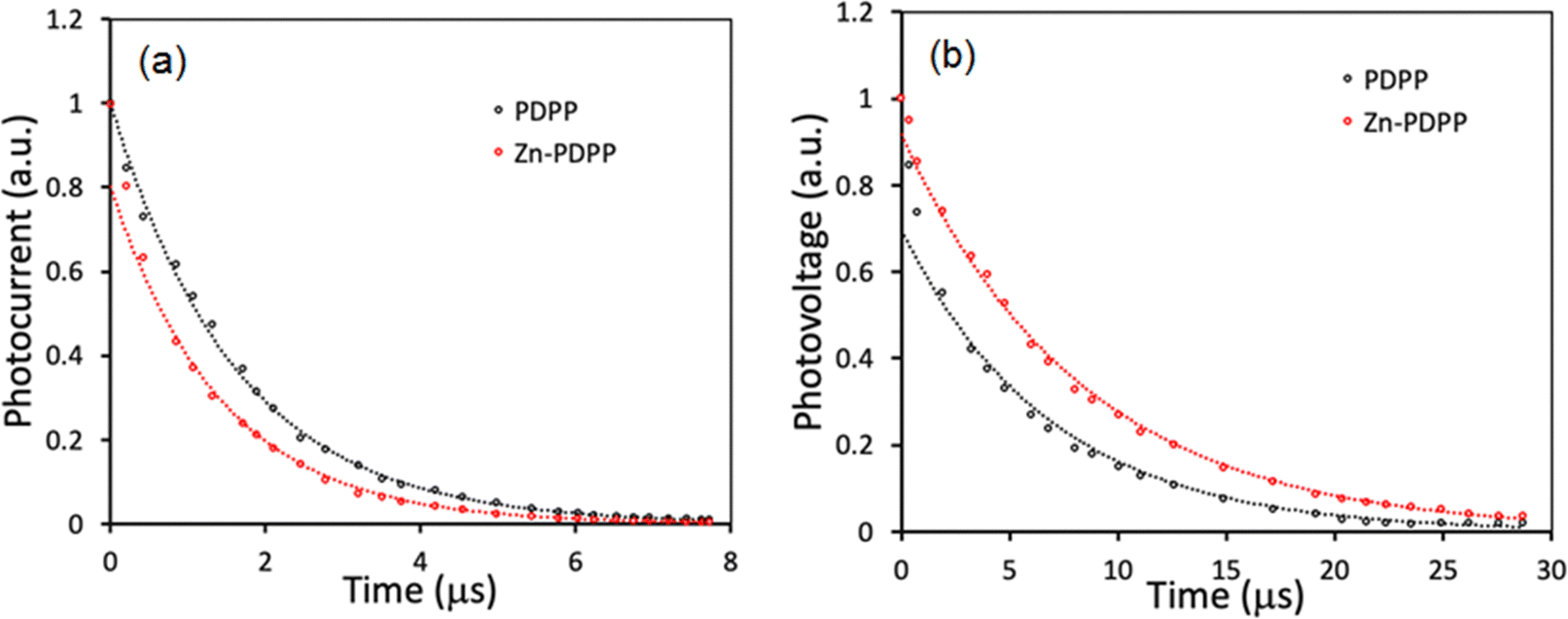 | ||
| Fig. 9 (a) Transient photocurrent and (b) transient photovoltage decay curves for OSCs. | ||
Conclusion
We present here a one-step synthesis for N-doped semicoronene diimides using a strong acid for its application as an intensely absorbing donor in organic photovoltaic devices. The current study confirms that the novel PBI-PORP dyad is a new and promising donor platform for BHJ-OSCs. In solution-processed small-molecule OSCs, the free-base and the zinc-metallated dyads demonstrated good photovoltaic performances when used as donors against PC71BM as the acceptor. The photovoltaic studies revealed an overall PCE of 7.25% and 9.10% for PDPP and Zn-PDPP, respectively, after optimizing the donor-to-acceptor weight ratio (1:1.4), which is much higher than that of the previously reported PBI-PORP dyads. Solvent vapor treatment is expected to improve the nanoscale morphology of the active layer, which is necessary for exciton formation and their dissociation into free charge carriers. The above findings imply that the novel PBI-PORP dyads are potential motifs in low-cost organic photovoltaic cells and can be employed to fabricate highly efficient solar cells.
Author contributions
AK synthesized and characterized all the molecules and analyzed the spectral data. SB solved the SCXRD data. GD and JS supervised and co-wrote the manuscript. All the authors have consented to the contents of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely acknowledge DST-SERB-EMR/2016/005768 and DST-SERB-CRG/2020/004632 for the financial support. The facilities and infrastructure provided by IISER Bhopal are highly acknowledged. AK is thankful to IISER Bhopal for a senior research fellowship.Notes and references
- W. Lowrie, R. J. E. Westbrook, J. Guo, H. I. Gonev, J. Marin-Beloqui and T. M. Clarke, J. Chem. Phys., 2023, 158, 110901 CrossRef CAS PubMed.
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H. J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K. Y. Jen and H. L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed.
- H. Yao and J. Hou, Angew. Chem., Int. Ed., 2022, 61, e202209021, DOI:10.1002/anie.202209021.
- B. Zhang, F. Yang and Y. Li, Small Sci., 2023, 3, 2300004, DOI:10.1002/smsc.202300004.
- Y. Li, G. Xu, C. Cui and Y. Li, Adv. Energy Mater., 2018, 8, 1–28 Search PubMed.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C. C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- D. Li, N. Deng, Y. Fu, C. Guo, B. Zhou, L. Wang, J. Zhou, D. Liu, W. Li, K. Wang, Y. Sun and T. Wang, Adv. Mater., 2023, 35, 1–9 Search PubMed.
- T. Gokulnath, J. Kim, H. Kim, J. Park, D. Song, H. Y. Park, R. Kumaresan, Y. Y. Kim, J. Yoon and S. H. Jin, ACS Appl. Mater. Interfaces, 2023, 15, 19307–19318, DOI:10.1021/acsami.3c01121.
- J. Fu, P. W. K. Fong, H. Liu, C. S. Huang, X. Lu, S. Lu, M. Abdelsamie, T. Kodalle, C. M. Sutter-Fella, Y. Yang and G. Li, Nat. Commun., 2023, 14, 1760 CrossRef CAS PubMed.
- S. D. Collins, N. A. Ran, M. C. Heiber and T. Q. Nguyen, Adv. Energy Mater., 2017, 7, 1602242, DOI:10.1002/aenm.201602242.
- H. Gao, Y. Sun, L. Meng, C. Han, X. Wan and Y. Chen, Small, 2023, 19, 2205594, DOI:10.1002/smll.202205594.
- B. Kan, Y. Kan, L. Zuo, X. Shi and K. Gao, InfoMat, 2021, 3, 175–200 CrossRef CAS.
- B. Kan, Q. Zhang, M. Li, X. Wan, W. Ni, G. Long, Y. Wang, X. Yang, H. Feng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 15529–15532 CrossRef CAS PubMed.
- D. Deng, Y. Zhang, J. Zhang, Z. Wang, L. Zhu, J. Fang, B. Xia, Z. Wang, K. Lu, W. Ma and Z. Wei, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- J. Wan, X. Xu, G. Zhang, Y. Li, K. Feng and Q. Peng, Energy Environ. Sci., 2017, 10, 1739–1745 RSC.
- J. Ge, L. Hong, H. Ma, Q. Ye, Y. Chen, L. Xie, W. Song, D. Li, Z. Chen, K. Yu, J. Zhang, Z. Wei, F. Huang and Z. Ge, Adv. Mater., 2022, 34, 1–9 Search PubMed.
- J. Qin, Z. Chen, P. Bi, Y. Yang, J. Zhang, Z. Huang, Z. Wei, C. An, H. Yao, X. Hao, T. Zhang, Y. Cui, L. Hong, C. Liu, Y. Zu, C. He and J. Hou, Energy Environ. Sci., 2021, 14, 5903–5910 RSC.
- Y. Sun, L. Nian, Y. Kan, Y. Ren, Z. Chen, L. Zhu, M. Zhang, H. Yin, H. Xu, J. Li, X. Hao, F. Liu, K. Gao and Y. Li, Joule, 2022, 6, 2835–2848 CrossRef CAS.
- Y. Gao, X. Yang, W. Wang, R. Sun, J. Cui, Y. Fu, K. Li, M. Zhang, C. Liu, H. Zhu, X. Lu and J. Min, Adv. Mater., 2023, 2300531, 1–11 Search PubMed.
- W. Gao, M. Jiang, Z. Wu, B. Fan, W. Jiang, N. Cai, H. Xie, F. R. Lin, J. Luo, Q. An, H. Y. Woo and A. K. Y. Jen, Angew. Chem., Int. Ed., 2022, 61, e202205168, DOI:10.1002/anie.202205168.
- X. Wang, Z. Li, X. Zheng, C. Xiao, T. Hu, Y. Liao and R. Yang, Adv. Funct. Mater., 2023, 2300323 CrossRef CAS.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- J. Shukla, V. P. Singh and P. Mukhopadhyay, ChemistryOpen, 2020, 9, 304–324 CrossRef CAS PubMed.
- A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC.
- H. Wang, M. Li, Y. Liu, J. Song, C. Li and Z. Bo, J. Mater. Chem. C, 2019, 7, 819–825 RSC.
- Y. Lin, Y. Wang, J. Wang, J. Hou, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2014, 26, 5137–5142 CrossRef CAS PubMed.
- S. Y. Liu, C. H. Wu, C. Z. Li, S. Q. Liu, K. H. Wei, H. Z. Chen and A. K. Y. Jen, Adv. Sci., 2015, 2, 1–7 Search PubMed.
- J. Zhao, Y. Li, J. Zhang, L. Zhang, J. Y. L. Lai, K. Jiang, C. Mu, Z. Li, C. L. C. Chan, A. Hunt, S. Mukherjee, H. Ade, X. Huang and H. Yan, J. Mater. Chem. A, 2015, 3, 20108–20112 RSC.
- J. Lee, R. Singh, D. H. Sin, H. G. Kim, K. C. Song and K. Cho, Adv. Mater., 2016, 28, 69–76 CrossRef CAS PubMed.
- Y. Liu, C. Mu, K. Jiang, J. Zhao, Y. Li, L. Zhang, Z. Li, J. Y. L. Lai, H. Hu, T. Ma, R. Hu, D. Yu, X. Huang, B. Z. Tang and H. Yan, Adv. Mater., 2015, 27, 1015–1020 CrossRef CAS PubMed.
- R. F. Kelley, W. S. Shin, B. Rybtchinski, L. E. Sinks and M. R. Wasielewski, J. Am. Chem. Soc., 2007, 129, 3173–3181 CrossRef CAS PubMed.
- S. A. Odom, R. F. Kelley, S. Ohira, T. R. Ensley, C. Huang, L. A. Padilha, S. Webster, V. Coropceanu, S. Barlow, D. J. Hagan, E. W. Van Stryland, J. L. Brédas, H. L. Anderson, M. R. Wasielewski and S. R. Marder, J. Phys. Chem. A, 2009, 113, 10826–10832 CrossRef CAS PubMed.
- A. Zhang, C. Li, F. Yang, J. Zhang, Z. Wang, Z. Wei and W. Li, Angew. Chem., Int. Ed., 2017, 56, 2694–2698 CrossRef CAS PubMed.
- R. Mishra, R. Regar, R. Singhal, P. Panini, G. D. Sharma and J. Sankar, J. Mater. Chem. A, 2017, 5, 15529–15533 RSC.
- L. Hao, W. Jiang and Z. Wang, Tetrahedron, 2012, 68, 9234–9239 CrossRef CAS.
- M. Schulze, M. Philipp, W. Waigel, D. Schmidt and F. Würthner, J. Org. Chem., 2016, 81, 8394–8405 CrossRef CAS PubMed.
- P. Hudhomme, R. El-Berjawi, L. Rocard, A. Goujon and T. Cauchy, J. Org. Chem., 2020, 85, 12252–12261 CrossRef PubMed.
- A. Goujon, L. Rocard, T. Cauchy and P. Hudhomme, J. Org. Chem., 2020, 85, 7218–7224 CrossRef CAS PubMed.
- K. Gao, N. Fukui, S. I. Jung, H. Yorimitsu, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 13038–13042 CrossRef CAS PubMed.
- D. K. Singh and M. Nath, Org. Biomol. Chem., 2015, 13, 1836–1845 RSC.
- H. Y. Tsai and K. Y. Chen, Dyes Pigm., 2013, 96, 319–327 CrossRef CAS.
- K. Susumu, T. Shimidzu, K. Tanaka and H. Segawa, Tetrahedron Lett., 1996, 37, 8399–8402 CrossRef CAS.
- P. W. M. Blom, V. D. Mihailetchi, L. J. A. Koster and D. E. Markov, Adv. Mater., 2007, 19, 1551–1566 CrossRef CAS.
- V. D. Mihailetchi, L. J. A. Koster, J. C. Hummelen and P. W. M. Blom, Phys. Rev. Lett., 2004, 93, 19–22 CrossRef PubMed.
- M. Sajedi Alvar, P. W. M. Blom and G. J. A. H. Wetzelaer, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- S. Zeiske, W. Li, P. Meredith, A. Armin and O. J. Sandberg, Cell Rep. Phys. Sci., 2022, 3, 101096 CrossRef CAS.
- A. K. K. Kyaw, D. H. Wang, V. Gupta, W. L. Leong, L. Ke, G. C. Bazan and A. J. Heeger, ACS Nano, 2013, 4569–4577 CrossRef CAS PubMed.
- S. Wood, J. C. Blakesley and F. A. Castro, Phys. Rev. Appl., 2018, 10, 1 Search PubMed.
- S. Wood, D. O’Connor, C. W. Jones, J. D. Claverley, J. C. Blakesley, C. Giusca and F. A. Castro, Sol. Energy Mater. Sol. Cells, 2017, 161, 89–95 CrossRef CAS.
- R. Yu, H. Yao, Y. Cui, L. Hong, C. He and J. Hou, Adv. Mater., 2019, 31, 1–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2255971 and 2255973. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ma00610g |
| This journal is © The Royal Society of Chemistry 2023 |