
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt oxide decked with inorganic-sulfur containing vanadium oxide for chromium(VI) reduction and UV-light-assisted methyl orange degradation†
Sayanika
Saikia
a,
Manoshi
Saikia
a,
Salma A.
Khanam
a,
Seonghwan
Lee
b,
Young-Bin
Park
b,
Lakshi
Saikia
 c,
Gautam
Gogoi
a and
Kusum K.
Bania
*a
c,
Gautam
Gogoi
a and
Kusum K.
Bania
*a
aDepartment of Chemical Sciences, Tezpur University, Assam, 784028, India. E-mail: kusum@tezu.ernet.in; bania.kusum8@gmail.com
bDepartment of Mechanical Engineering, Ulsan National Institute of Science and Technology, UNIST-gil 50, Ulju-gun, Ulsan 44919, Republic of Korea
cMaterials Science and Technology Division (MSTD), CSIR-North East Institute of Science and Technology, Jorhat 785006, Assam, India
First published on 19th October 2023
Abstract
Cobalt oxide (CoOx) derived from a zeolitic imidazolate framework (ZIF) was modified with sulfur-doped vanadium oxide (VOx) using an impregnation method. The presence of inorganic sulfur in the mixed metal oxide matrix was confirmed through various physicochemical and spectroscopic techniques. The hybrid material designated as CoOx–S–VOx was applied as a catalyst for rapid reduction of toxic chromium, Cr(VI), in the presence of sodium borohydride, NaBH4. CoOx–S–VOx + NaBH4 (1 mmol) reduced Cr(VI) to Cr(III) within three minutes (3 min). The same catalyst possessing a band gap of ∼3.14 eV served as an effective photocatalyst for the degradation of methyl orange (MO) dye on irradiating with UV-light. The optimization of different parameters indicated that the degradation of photocatalytic dye proceeded very efficiently within 15 min with a total mineralization of 80.06%. The dye degradation experiments carried out under both light and dark phase conditions truly signified the impact of light irradiation on MO degradation. Mott–Schottky analysis and ultraviolet photoelectron spectroscopy (UPS) were performed to calculate the band edge positions of the material. Both the aforementioned analyses predicted that the hybrid catalyst possessed a perfect band structure that could provide high activity to the material. The mechanistic study of the dye degradation process suggested that the MO decomposition was mostly caused by free radicals. The presence of sulfur was considered to play an active role in enhancing the dye degradation process by reducing the electron–hole recombination rate. The catalyst was recyclable for ten consecutive cycles without any significant loss in catalytic activity.
1. Introduction
The gradual increase of industrial areas and the worldwide population boom along with the release of toxic metals or organic pollutants from various industries are causing an adverse impact on the aquatic life cycle.1,2 The contamination of water bodies with toxic metals and coloured organic molecules is posing a threat to human beings.3 Out of the many toxic metals, chromium (Cr) mostly used in dye and painting industries is considered to be highly toxic, when present in Cr(VI) form.4 Depending on the pH of ground water the hexavalent Cr remains as chromate (CrO42−) at pH > 6.5 and as HCrO4− when pH < 6.5.5–7 Studies on the toxicity of Cr(VI) have revealed that it can possibly become a health hazard by affecting the respiratory systems of living organisms. Moreover, its severity can affect other human organs such as kidneys and the liver and it also has the potential to cause major genetic disorder.8 Considering the life threatening impact of Cr(VI), stringent regulations have been imposed by the environmental protection agency (EPA). The EPA has further restricted the standard limit of Cr(VI) at 0.1 ppm.9 In contrast to Cr(VI), its low valent form i.e. Cr(III) is less toxic and is necessary for diabetic regulation and is therefore considered as an essential element. Cr(III) can remain as Cr(OH)3 or Cr2O3 in water, so they are measured to be less carcinogenic apart from causing some skin rashes when used in excess.10 Since Cr(III) doesn’t have any considerable negative impact on human health and the aquatic life, a substantial amount of previously published research work has been dedicated towards the reduction of poisonous Cr(VI) to non-poisonous Cr(III). Different chemical and photochemical processes have been developed for the reduction of Cr(VI) and accordingly different types of materials and catalysts have been synthesized.11–17 The reduction processes employ reducing agents like sodium borohydride (NaBH4) and ascorbic acid which are considered to be effective in the presence of suitable materials for the conversion of Cr(VI) to Cr(III).18–21 As far as the materials are concerned, sulfur-containing transition metals or the metal chalcogenides have shown some promising activity in the removal of Cr(VI).22–26 Our group has also reported a silver–sulfur–oxido–vanadium cluster for such a reduction process within a very short period of time with NaBH4 as a reducing agent.27 It is pertinent to mention that sulfur is a familiar insulator, but very recently, the viability and applicability of sulfur in the field of catalysis and photocatalysis have been explored. Compared to many non-metals, sulfur is non-toxic and inexpensive, and hence can be easily handled. However, the scopes of application of sulfur as a single element are lacking and narrow in the field of catalysis. But the advancement of new techniques such as surface modification, nanoscaling, heterojunction creation, doping etc. paved a new direction in the utilization of elemental sulfur in the synthesis of effective photoctalysts. It has been reported that the introduction of sulfur or modification of metal oxide with sulfur can bring significant changes by creating some defect sites.28 The introduction of sulfur in many photocatalysts is found to alter the band gap and enhance the photocatalytic activity of such materials. In this regard the earlier works of Liu et al. demonstrated the ability of sulfur crystals of cyclooctasulfur (S8) to act as a visible-light-active elemental photocatalyst.29 Furthermore, Jiang et al. developed a sulfur-doped g-C3N4/g-C3N4 heterostructure for the photocatalytic hydrogen evolution reaction with greater stability and reactivity.30 Similarly, another study conducted by Lu et al. synthesized a heterostructure of elemental sulfur supported on ultrathin titanic acid nanosheets in order to improve light and CO2 adsorption.31 So, finding the benefits of heterojunctions researchers are driven towards the design of newer photocatalysts or materials for sustainable development.32–34Other than Cr(VI), the coloured dye molecules that are mostly released from textile industries are also harmful for the aquatic life and passively for human beings as well.35,36 The most challenging part is the decomposition of such coloured dye molecules to CO2 and H2O. There are many approaches or chemical treatment techniques by which such toxic dye molecules can be removed from water.37–42 The photocatalytic decomposition of such dye molecules absorbing the solar energy is considered to be a green and clean process.43–45 But finding a suitable photocatalyst that can decompose dyes like methyl orange (MO) completely to CO2 and H2O is still under study and hence researchers are trying to develop a different low cost photocatalyst that can harvest the solar light and can perform the photodecomposition of dye molecules.
While considering the photocatalyst, the band gap as well as the pH under examination is considered as vital factors in improvising its photocatalytic performance.46–48 The literature report suggested that by doping metal oxides with sulfur with lower electronegativity than oxygen, polarized d-orbitals with a lone pair of electrons can modulate or alter the photoactivity of metal oxide by changing its band gap as well as the pH.28 Therefore, looking into the negative impact of Cr(VI) ions and MO dye on our environment, mostly on water and indirectly on human health, herein we have developed a new material by conjugating CoOx with sulfur containing VOx that showed excellent activity for the conversion of Cr(VI) to Cr(III). The same material also appeared as a suitable photocatalyst for the photodecomposition of MO. So far, the maximum reported results are mostly concerned with the preparation of one material for one electron transfer process, but the development of one material for two different chemical/photochemical processes will be always advantageous from both economic and synthesis aspects.
2. Experimental section
2.1. Materials
Cobalt(II) chloride hexahydrate (CoCl2·6H2O), ammonium metavanadate (NH4VO3) and methyl orange (MO) dye were purchased from Sisco Research Laboratory (SRL). Sodium sulphide flakes nonahydrate (Na2S·9H2O), glacial acetic acid (CH3COOH), hydrochloric acid (HCl), sulphuric acid (H2SO4) (95.0–98%), and nitric acid (HNO3) were received from E-Merck. Mohr's salt [Fe(NH4)2(SO4)2·6H2O], potassium nitrate (KNO3), and potassium dichromate (K2Cr2O7) were procured from HiMedia. Sodium hydroxide (NaOH), orthophosphoric acid (H3PO4) potassium iodide (KI), imidazole (C3H4N2), silver nitrate (AgNO3), isopropanol and ascorbic acid were received from E-Merck. All the HPLC grade solvents were received from Sigma-Aldrich.2.2. Synthesis of sulfur containing vanadium oxide, VOx (S–VOx)
The synthesis of sulfur-containing vanadium oxide designated as S–VOx is depicted schematically in Scheme 1a. For the synthesis of sulfur-containing vanadium oxide (S–VOx), a 0.1 M solution of NH4VO3 was prepared dissolving 1.17 g of NH4VO3 in 100 mL of distilled water. The solution was made acidic by adding a few drops of glacial acetic acid and stirred for about 30 min at room temperature. To this solution, 0.1 M (2.40 g in 100 mL of distilled water) of sodium sulfide (Na2S.9H2O) solution was given slowly and constantly stirred for 2 h at 60 °C. The black material produced during the course was repeatedly rinsed with water and finally dried in an oven at 60 °C.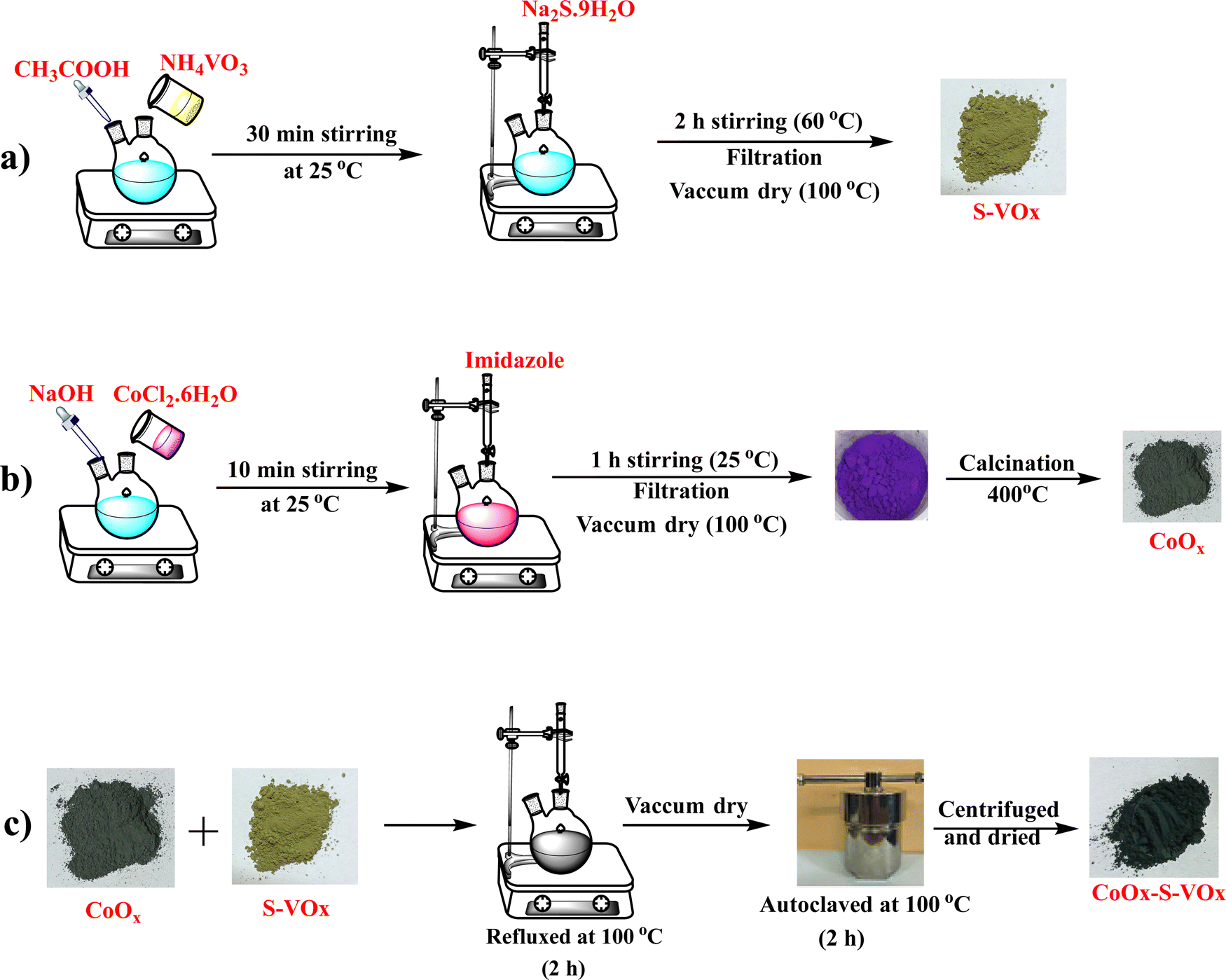 | ||
| Scheme 1 Schematic representation of the preparation of the CoOx–S–VOx catalyst. | ||
2.3. Synthesis of cobalt oxide (CoOx)
A solution of NaOH (0.1 M) was added to a round bottom flask containing 25 mL of CoCl2.6H2O solution (0.1 M). The solution was stirred while maintaining a pH of ∼12. To this solution, about 25 mL of 1 M imidazole was added under constant stirring for about 1 h at 60 °C. The violet precipitate obtained was filtered and washed repeatedly with water and ethanol. The oven-dried sample was calcined at 400 °C for about 4 h. The synthesis is depicted in Scheme 1b.2.4. Preparation of CoOx–S–VOx
CoOx–S–VOx was synthesized using a hydrothermal method by combining S–VOx and CoOx as shown in Scheme 1c. For this, an equal amount (50 mg each) of S–VOx and CoOx was mixed in a round bottom flask in water and refluxed for 2 h at 100 °C. The material was further autoclaved for 2 h at 100 °C. After centrifugation, a black material was obtained, which was subsequently dried to get CoOx–S–VOx. The overall synthesis process is shown in Scheme 1.2.5. Preparation of Co3O4–V2O5
The Co3O4–V2O5 material prepared previously by our group was used for the comparative study.492.6. Procedure for Cr(VI) reduction
A 50 mL prepared solution of K2Cr2O7 (50 ppm) was taken in a beaker. 10 mg of the CoOx–S–VOx catalyst and 1 mmol NaBH4 were added to the above dichromate solution under vigorous stirring conditions. A specific volume of an aliquot was taken out at a specific time interval, centrifuged, and the supernatant was analyzed using UV-Vis spectroscopy. Two UV visible peaks at around 259 and 357 nm were observed for the normal K2Cr2O7 solution. However, the peaks were slightly shifted to 271 and 372 nm upon the addition of NaBH4. The disappearance of the peak positions at 271 and 372 nm and the change in solution colour from yellow to colourless were thoroughly examined. It was found that the two UV peaks disappeared within 3 min after adding 10 mg of catalyst followed by 1 mmol NaBH4. The following equation was used to compute the percentage of reduction of Cr(VI).50,51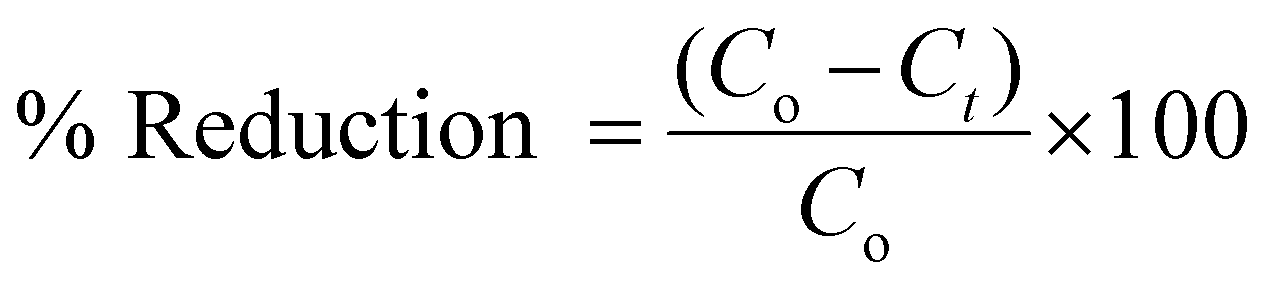 | (1) |
2.7. Dye degradation study
The decomposition of MO dye was studied under three different conditions: (i) in a normal laboratory environment without any external light stimuli, (ii) under the influence of artificial UV-light (λ = 254 nm) using a medium pressure mercury lamp (450 W) and (iii) under dark conditions. For all the cases, 15 mg of our synthesized catalyst CoOx–S–VOx in 20 mL of 10−6 M MO dye solution in water was taken and the reaction was allowed to proceed for 15 min. The artificial photocatalytic reaction was performed in a photoreactor as mentioned in our previous study.493. Results and discussion
The X-ray diffraction analysis (XRD) patterns of CoOx (blue), S–VOx (red) and CoOx–S–VOx(black) are shown in Fig. 1a. The presence of crystalline peak characteristics of Co3O4 (JCPDS-00-009-0418), CoO(JCPDS-00-001-1227), V2O5(JCPDS-00-003-0207), V3O7(JCPDS-00-020-1378) and inorganic sulfur(JCPDS-00-002-0324) in the material confirmed the formation of CoOx–S–VOx. FTIR spectra of the materials, S–VOx(blue), CoOx(black), and CoOx–S–VOx (red) are shown in Fig. 1b. FTIR analysis also confirmed the formation of CoOx–S–VOx species. The two bands at 577.4 cm−1 and 665.6 cm−1 resulting from Co+2–O vibration and Co+3–O vibration of tetrahedral and octahedral sites, respectively, of the Co3O4 lattice established the formation of Co3O4.52 The peaks at 1021.4 cm−1 and 975.8 cm−1 associated with the V = O band and the peak at 826.6 cm−1 associated with the V–(O)3 vibrational band of VOx.53 The Raman spectra of the materials S–VOx (blue), CoOx (black) and CoOx–S–VOx (red) are shown in Fig. 1c. The Raman spectra of S–VOx showed a vibrational band at ∼216 cm−1 corresponding to inorganic sulfur (S8). The breathing mode of the V2O4 cage was found at ∼353 cm−1, while the Ag mode of β-V2O5 appeared at ∼434 cm−1 and ∼467 cm−1. The peak at ∼152 cm−1 was for α-V2O5.54–56 The peaks at ∼550 cm−1 and ∼812 cm−1 represented the S–S stretching vibration and terminal V–O bonds, respectively (Fig. 1c (black)).56,57 Similarly, in the CoOx material, Raman shifts were noticed at ∼187 cm−1, ∼466 cm−1, ∼510cm−1, ∼608 cm−1,and ∼665 cm−1, confirming the formation of Co3O4 (Fig. 1c (red)). Out of which peaks at 187, 510 and 608 cm−1 were for F2g symmetry, the peak at 466 cm−1 was assigned to Eg symmetry and the peak at 665 cm−1 was for A1g symmetry.58 In the CoOx–S–VOx material, the Raman bands at ∼216 cm−1 were for S8 and the signals at ∼152 cm−1, ∼434 cm−1, and ∼468 cm−1 were for V2O5 and bands at ∼685 were for Co3O4 (Fig. 1c (blue)). The slight shifting of the signals indicated the well mixing of S–VOx with CoOx. The presence of the signals for VOx and CoOx in the composite material CoOx–S–VOx clearly indicated the formation of S8-containing mixed metal oxide of vanadium and cobalt. The diffused reflectance spectrum (DRS) of CoOx–S–VOx as depicted in Fig. 1d showed three broad signals at 340 nm (a charge transfer band of ligand-metal cation V+5), 450 nm (due to O2− → Co2+ charge transfer transition) and 751 nm (due to O2− → Co3+ charge transfer transition).59,60The material's band gap was determined using Tauc plot and was found to be ∼3.14 eV as shown in Fig. 1d. The occurrence of a band gap smaller than 4 eV signified that our synthesized material could absorb the UV light of the solar spectrum. The DRS plot (Fig. S1a and b, ESI†) and band gap (Tauc plot) (Fig. S1c and d, ESI†) of the other two materials CoOx and S–VOx, respectively, are depicted in Fig. S1 of the ESI.†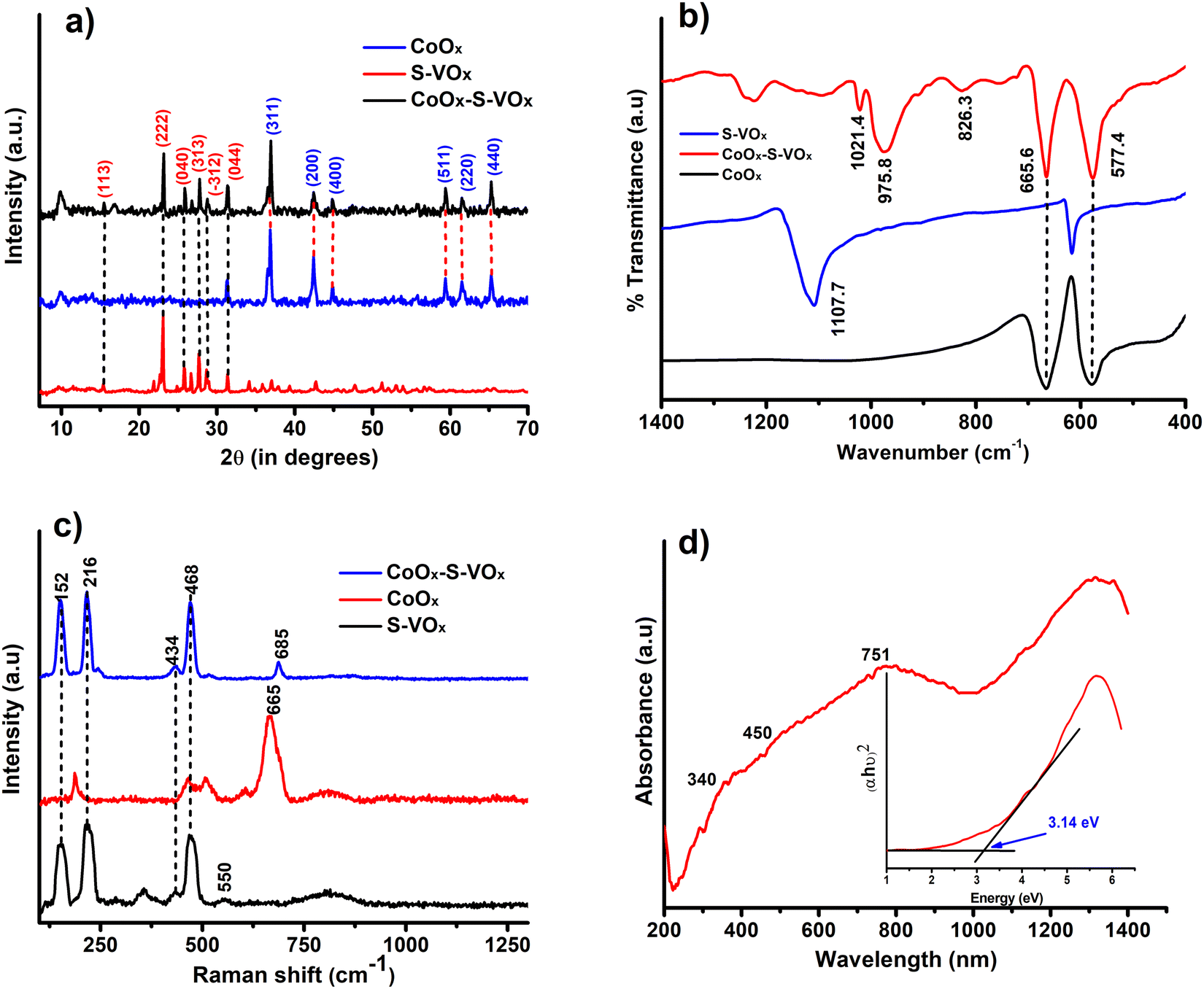 | ||
| Fig. 1 (a) XRD pattern of CoOx (blue), S–VOx (red) and CoOx–S–VOx(black). (b) FTIR spectra of CoOx (black), S–VOx (blue) and CoOx–S–VOx (red). (c) Raman spectra of CoOx (red), S–VOx (black) and CoOx–S–VOx (blue) and (d) DRS spectra of CoOx–S–VOx, with the inset showing Tauc plot. | ||
The X-ray photoelectron spectroscopy (XPS) study was performed for CoOx–S–VOx and the respective spectra are shown in Fig. 2a–d. The XPS spectrum of V (2p) depicted in Fig. 2a exhibited the V (2p) signals for V (2p1/2) and V (2p3/2) with binding energy (B.E) values of 522.6 eV and 515.4 eV, respectively, characteristic of vanadium(IV) species. Two peaks at 528.3 eV and 529.3eV originated from the O (1s) peak of vanadium oxide species.61 The B.E. values of Co (2p3/2) and Co (2p1/2) obtained at 780.3 eV and 796 eV, respectively, were assigned to Co+2 species while those at 778.5 eV and 793.5 eV were for Co+3 species. Along with these, the two satellite peaks at 786.2 eV and 802.5 eV resembled the spinel Co3O4 (Fig. 2b).46,62 In the S(2p) spectrum as shown in Fig. 2c, a doublet was obtained at 161.8 eV and 163.1 eV that indicated the formation of S(0) species and another doublet at 165.6 eV and 166.7 eV corresponded to the oxidize form of sulfur.63 The O (1s) binding energies were observed at 528.2 eV and 529.2 eV, signifying the formation of metal oxides (Fig. 2d). The XPS peaks were fitted with respect to C (1s) binding energy values as provided in Fig. S2 of the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 64
64
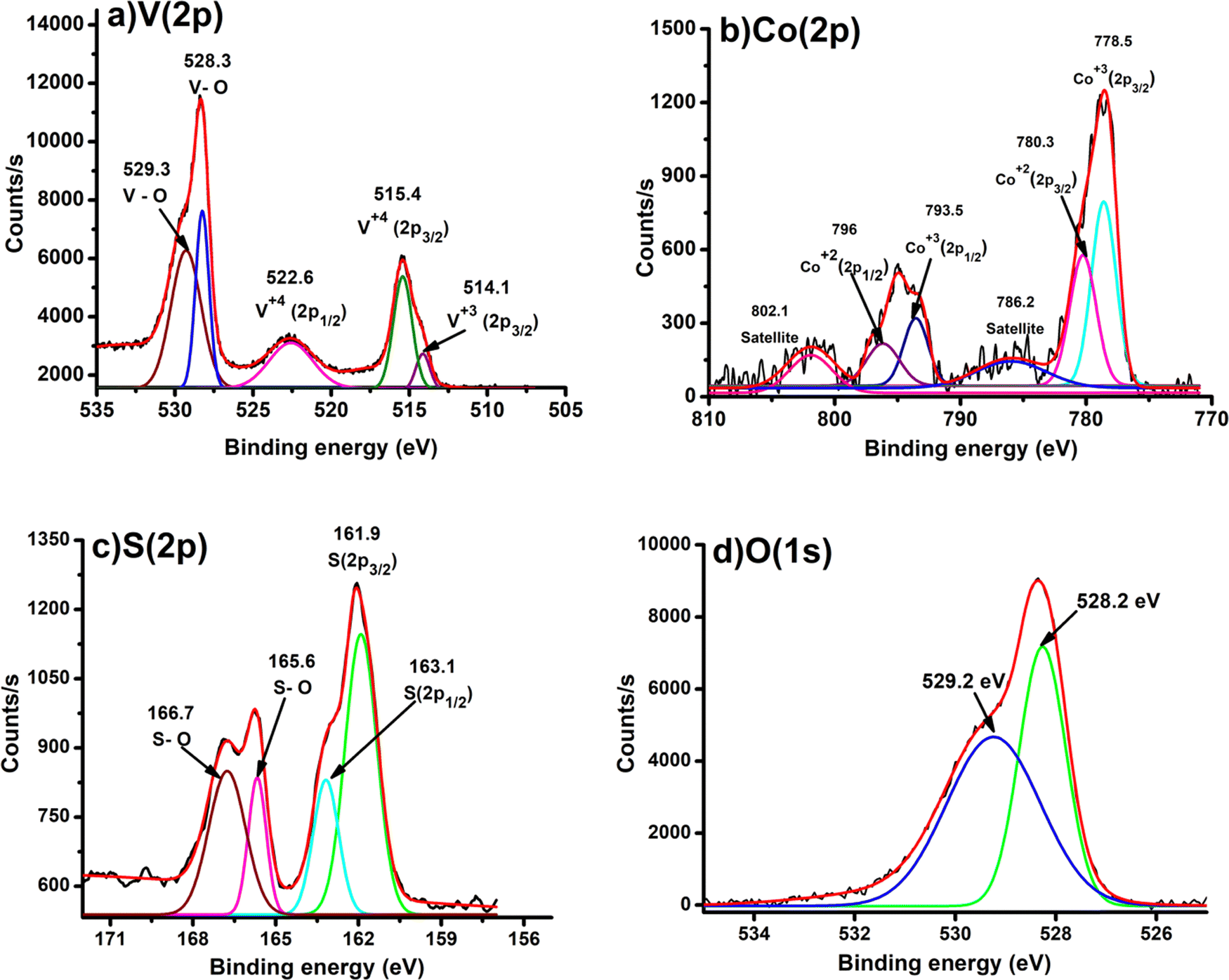 | ||
| Fig. 2 XPS spectra of (a) V(2p), (b) Co(2p), (c) S(2p), and (d) O(1s) present in CoOx–S–VOx. | ||
The formation of vanadium(IV) species was again confirmed by the electron paramagnetic resonance (EPR) study. The presence of a hyperfine structure for vanadium (I = 7/2) clearly indicated the occurrence of vanadium(IV) oxide with the CoOx matrix (Fig. S3a, ESI†).65 The thermal stability of the synthesized materials was determined using thermo gravimetric analysis (TGA). The TGA of the synthesized CoOx–S–VOx (red) and S–VOx (black) is shown in Fig. S3b (ESI†). The weight loss of 17.45% and 95.1% was observed from 150 °C to 250 °C in CoOx–S–VOx (red) and S–VOx, respectively. This weight loss was attributed to the sublimation of inorganic sulfur, leading to the formation of residual metal oxides.66 Also from the analysis, it was found that the thermal stability of S–VOx was enhanced on mixing CoOx.
From the scanning electron microscopy (SEM) analysis, we could see the formation of zeolitic-like crystals of the CoOx–S–VOx material (Fig. 3a–c). This was expected as we followed a zeolitic-imidazolate framework (ZIF) template approach during our synthesis.57,67 The ZIF-like structure was also seen from transmission electron microscopy (TEM) images (Fig. 3d and e). The SEM image (Fig. 3b) and the TEM image (Fig. 3e) represented an icosahedral-like morphology of the material.68 The d-spacing values were calculated for the regions marked as 1–4 in Fig. 3f. The enlarged version of these regions is shown individually in Fig. 3g–j. The d-spacing values of 0.385 nm and 0.159 nm correspond to the (131) and (511) plane of sulfur (S8) and Co3O4, respectively (Fig. 3g). The d-spacing values of 0.125 nm and 0.147 nm were assigned to the (620) and (440) planes of Co3O4 (Fig. 3h–j). The same with a d-spacing value of 0.318 nm was for the (−312) plane of V3O7 (Fig. 3i) and that with a d-value of 0.179 nm was for the (535) plane of S8 (Fig. 3j). The planes were assigned with reference to the JCPDS card numbers used in the XRD analysis. From the particle size distribution (PSD) analysis (Fig. 3k), the size of the nanoparticle was found to be 20–35 nm. From the EDX analysis, the average weight percentage of the constituent atoms was found to be 27.43% (Co), 12.54% (V), 46.10% (S), and 13.91% (O), as shown in Fig. 3l. The atomic percentage ratio of Co and V was found to be 1.8:1. The scanning transmission electron microscopy high-angle annular dark-field (STEM-HAADF) images clearly indicated that the icosahedral was mostly comprised of cobalt oxide with vanadium and sulfur (Fig. 4a–f). The elemental analysis and EDX mapping were also performed in different regions of the nanocrystal and are provided in the ESI† (Fig. S4).
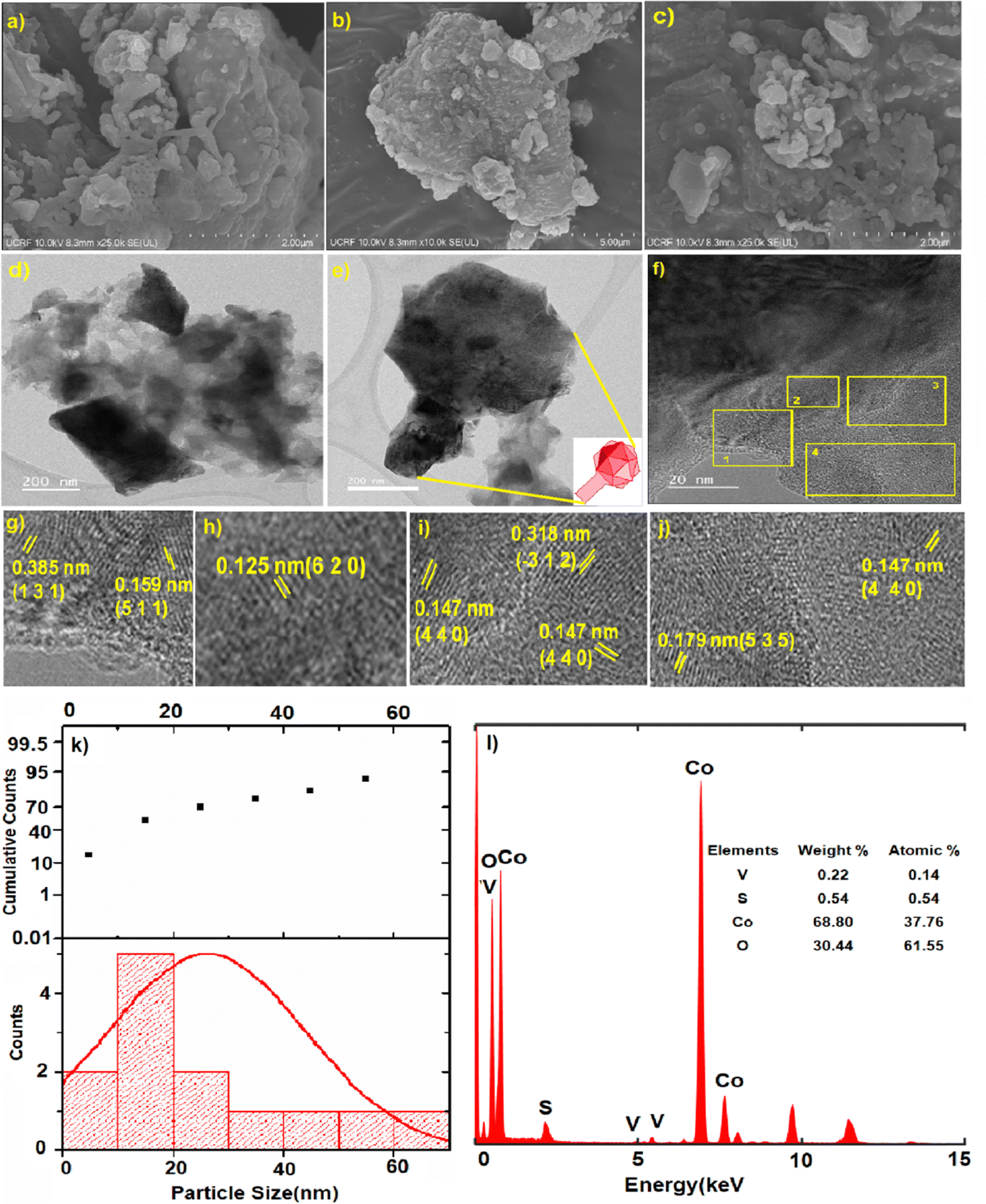 | ||
| Fig. 3 (a)–(c) SEM images of the CoOx–S–VOx material, (d)–(f) TEM images, (g)–(j) are enlarged images marked as 1–4 in f, (k) particle size distribution curve, (l) the EDX spectra showing the elemental distribution in the catalyst. | ||
 | ||
| Fig. 4 (a) Region selected for STEM-HAADF-EDX mapping of the CoOx–S–VOxmaterial showing the presence of all the elements, (b) Co, (c) V, (d) O, (e) S, (f) mixing of all elements. | ||
4. Reduction of Cr(VI)
The synthesized catalysts (CoOx–S–VOx, S–VOx and CoOx) were employed for the elimination of Cr(VI) ions from water. For this, a potassium dichromate (K2Cr2O7) solution with a concentration of 50 ppm was prepared. The initial assessment of the three catalysts for Cr(VI) removal without the external reducing agent was found to be negative. Our previous studies and various other literature reports predicted that NaBH4 served as a suitable reducing agent with an active catalyst for Cr(VI) reduction.18,19,27 Therefore, considering 0.1 mmol of NaBH4, 10 mL of Cr(VI) solution and 2 mg of CoOx–S–VOx catalyst, essential reaction parameters (time, NaBH4 amount, and catalyst amount) were optimized for Cr(VI) reduction. From the graph shown in Fig. 5a, three minutes (3 min) of reaction time was found to be optimum and from the graphical representation shown in Fig. 5b, maximum of 49.14% of Cr(VI) got reduced. The percentage of Cr(VI) reduction was calculated by using eqn (1). As the Cr(VI) reduction percentage was comparatively lower than most of the reported ones, the same experiment was again analyzed by varying the concentration of NaBH4. From the graphs (Fig. 5c and d), it was ascertained that 2 mg of the catalyst and 3 min of reaction time and 1 mmol of NaBH4 were required to improve the Cr(VI) reduction percentage from 49.14 to 60.34%. Further enhancement of NaBH4 concentration up to 2 mmol did not influence the Cr(VI) reduction process.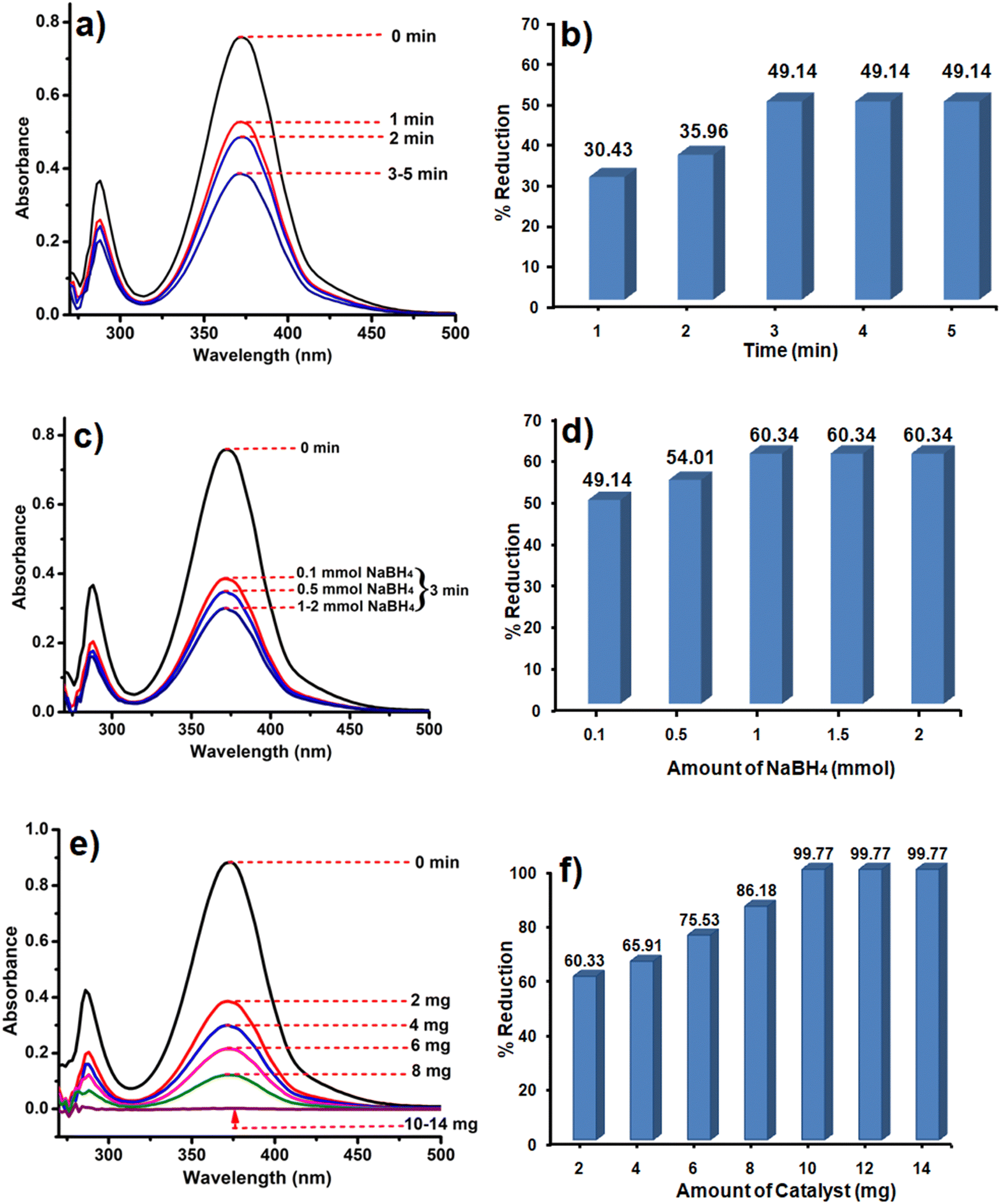 | ||
| Fig. 5 UV spectra and bar diagram showing the dependency of Cr(VI) reduction on time (a) and (b), NaBH4concentration (c) and (d) and catalyst amount (e) and (f). | ||
After getting the optimum time period and NaBH4 concentration, the amount of catalyst was varied as it played a crucial role in the Cr(VI) reduction process as the Cr(VI) reduction process was negative only with NaBH4. The catalyst amount was varied from 2 to 14 mg and the results are shown in Fig. 5e and f. It was observed that 10 mg of the catalyst resulted in 99.77% conversion of Cr(VI) to Cr (III). As a result of the aforementioned optimization, it was found that almost 100% of the Cr(VI) solution got reduced to Cr(III) within 3 min using 10 mg of the catalyst and 1 mmol NaBH4. The reduction of Cr(VI) was mostly confirmed with the consecutive diminishing and disappearance of UV peaks at 271 nm and 372 nm. This was further validated by the colour change of the Cr(VI) solution from light yellowish to colourless. In order to study the reaction kinetics of Cr(VI) reduction, UV spectra were recorded every 30 sec. The rate constant determined using the first order rate-law was k (rate constant) = 0.0334 min−1 (Fig. S5a and b, ESI†).69
Under the optimized conditions, the catalyst was further tested by varying the Cr(VI) concentration. The study predicted that the catalyst was capable of reducing Cr(VI) with same competency up to 50 ppm. However, on further increasing the strength of Cr(VI) to 250 ppm, its efficiency decreased to 56.28% (Fig. S6a and b, ESI†). As suggested by the environmental protection agency's (EPA) study, the toxicity limit of Cr(VI) is 0.1 ppm, so it can be said that our synthesized catalyst CoOx–S–VOx can serve as a suitable material for Cr(VI) reduction up to a very high concentration of 50 ppm with almost 100% efficiency and within a very short period of time (3 min). It was interesting to observe that CoOx–S–VOx was only able to reduce Cr(VI); however the other catalysts, CoOx, S–VOx and Co3O4–V2O5 were found to be ineffective under the optimized condition (Fig. S7a and b, ESI†). It was attributed to the fact that the single metal catalysts S–VOx and CoOx as well Co3O4–V2O5 could not provide the sufficient number of electrons, which was required for the reduction of Cr(VI) to Cr(III).
5. Mechanism of Cr(VI) reduction
When potassium dichromate (K2Cr2O7) is dissolved in water, an equilibrium is created between the dichromate (Cr2O72−) and chromate (CrO42−) ions as follows.70| Cr2O72− + H 2O ⇄ 2CrO42− + 2H+ | (2) |
Such types of equilibrium were found to be dependent on pH of the solution.71 In the presence of our catalyst, the pH of the solution was 7.05, while it was 6.17 for aqueous K2Cr2O7 solution. At this pH, the equilibrium equation becomes,
| CrO42− + 2H+ ⇄ HCrO4− | (3) |
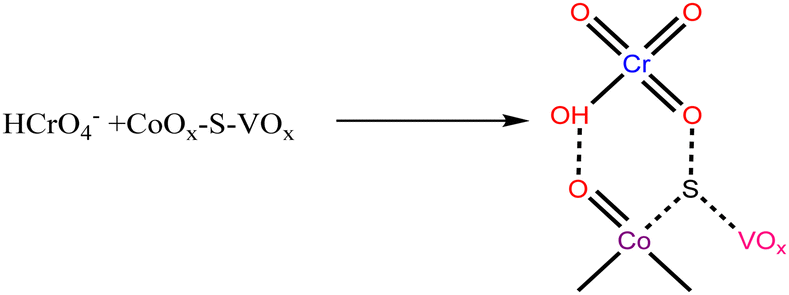 | (4) |
The HCrO4− species was expected to interact with the catalyst as shown in eqn (4). This HCrO4− species is reported to have a redox potential of +1.35 V.72 Therefore, it is considered to be highly reducible in the presence of the species that can behave as a Lewis base or an electron donor. In this case, the low valent V+4, Co+2 and S atoms can serve as an electron donor center and the required H+ ions will be provided by NaBH4. So, the reduction of Cr(VI) can be expressed by the following reactions.73
Hydrolysis of NaBH4:
| NaBH4 + 2H2O → NaBO2 + 4H2 | (5) |
| BO2− + 2H2O → H3BO3 + OH− | (6) |
| H3BO3 + H2O → B(OH)4− + H+ | (7) |
Reduction of Cr(VI):
| HCrO4− + 7H+ + 3e− → Cr+3 + 4H2O | (8) |
The reduction process was evaluated through the cyclic voltametric study (Fig. S8, ESI†). The cyclic voltamogram (CV) of the K2Cr2O7 solution represented a sharp cathodic peak at −0.24 V, and the potential value matches the reported one for CrO42−.74 This indicated that in an aqueous medium, Cr2O72−ionizes to CrO42−. On addition of the catalyst, almost a reversible couple with a sharp anodic peak at +0.83 V vs. Ag/AgCl (+1.03 V vs. NHE) was observed. This anodic peak matched the reported redox potential value for HCrO4− species.75 After addition of NaBH4, the anodic peak for HCrO4− got diminished and the voltamogram represented the hydrogen evolution reaction due to the reduction of H+ ions. The CV analysis was in accordance with our proposed mechanism, thereby proving the efficacy of our catalyst for Cr(VI) reduction.
6. Degradation of methyl orange (MO)
Like Cr(VI), there are many other organic dye components which are released from textile industries to nearby water bodies.76 One of such examples is the contamination of Yamuna river located in New Delhi, India.77 Therefore, the removal of dye contaminants for waste water treatment especially in the industrial urban areas is the need of the present environmental situation. After going through the literature, it is realized that the different catalysts or photocatalysts are designed, which are either effective for Cr(VI) reduction or for different dye degradation processes. But reports on single catalysts that have the potential to reduce Cr(VI) to Cr(III) as well as degrade dyes like MO are very less. MO is an anionic dye which is more frequently used in textile industries. They are toxic in nature if present in high concentration, so they need to be decomposed to CO2 and H2O.Initially, the reaction was studied using 5 mg of the CoOx–S–VOx catalyst and the dye degradation process was monitored through UV-visible spectroscopy. It was found that with 5 mg of the catalyst, only 42.83% of the dye got degraded in 15 min (Fig. 6a and b). Increasing the light irradiation time did not improve the percentage of degradation. Therefore, considering 15 min to be optimized light irradiation time, the catalyst amount was changed from 5 to 15 mg and found that the degradation process completed with 99.40% (Fig. 6c and d). Furthermore, increase in the catalyst amount did not improve the percentage of degradation. So, 15 mg of the catalyst and 15 min of light irradiation time were considered optimum reaction parameters. To fix the degradation kinetics, UV spectra were recorded after an interval of 3 min (Fig. 7a). The plot of ln(Co/Ct) vs. time generated a linear line plot, suggesting a 1st order kinetics with the rate constant (k) of 0.46238 min−1 for the MO dye degradation process (Fig. 7b).78
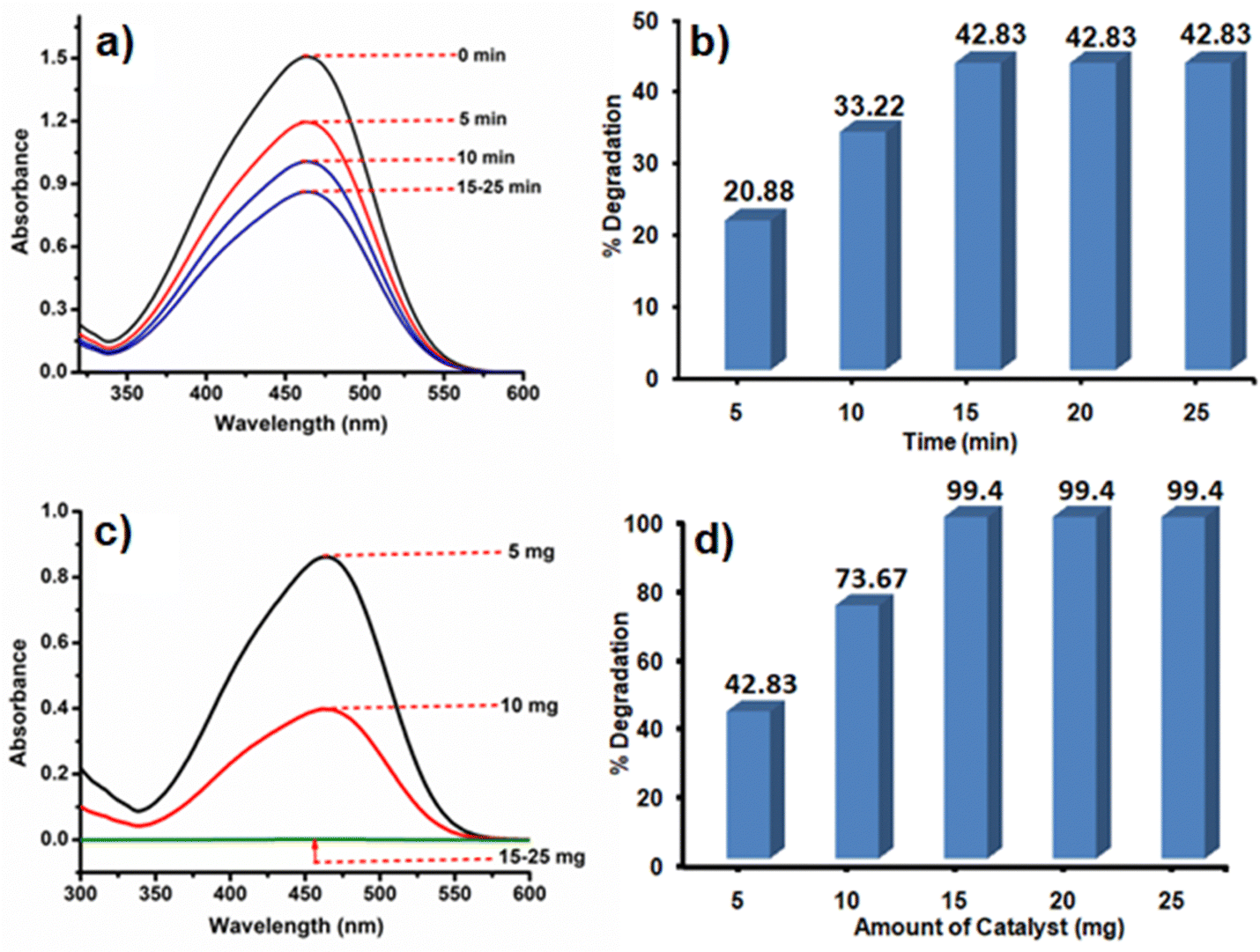 | ||
| Fig. 6 UV spectra and bar diagram showing the dependence of MO degradation on time (a) and (b), and catalyst amount (c) and (d). | ||
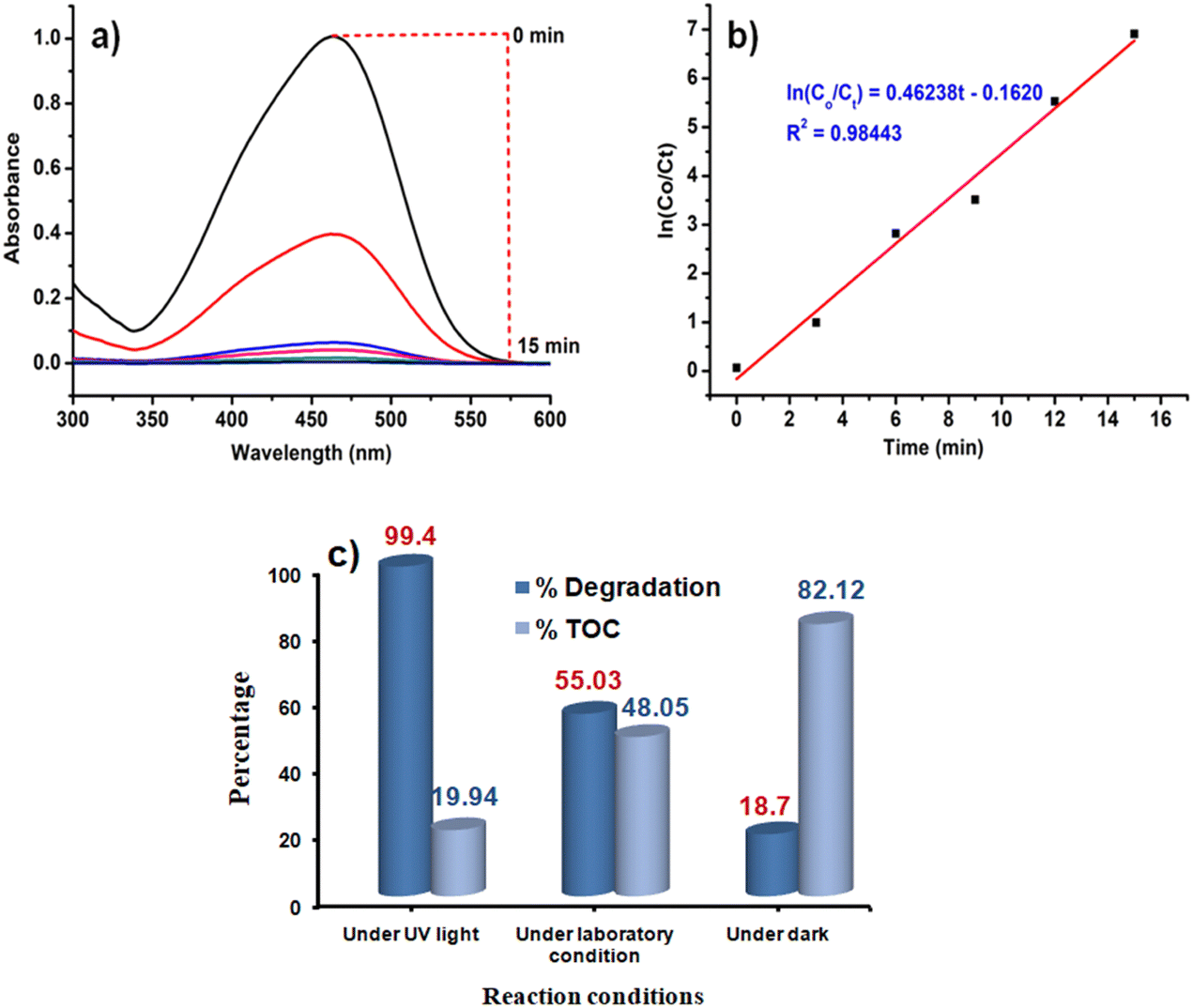 | ||
| Fig. 7 (a) UV spectra recorded over a 5 min time interval under optimized reaction conditions, (b) ln(Co/Ct) vs. time graph depicting a kinetic mechanism for MO degradation, and (c) comparative bar diagram of TOC and percentage degradation under various conditions. | ||
In the usual process, the decolourization of the dye and change in the absorbance value in the UV-visible spectrum do not signify the decomposition of dye into H2O and CO2. This is one of the challenging issues in the dye degradation process. Therefore, to validate that the synthesized catalyst decomposes the dye, the total organic content (TOC) over different time intervals under optimized conditions (15 mg of the catalyst CoOx–S–VOx) was calculated. The TOC was determined using the Walkley and Black method as previously stated by our group.79 The TOC was found to be 19.94% after the completion of the reaction under UV-light irradiation and suggested that percentage of degradation and decomposition was not the same (Fig. 7c). This reveals the fact that the catalyst was able to mineralize 80.06% of the total dye content to CO2 and H2O under the photocatalytic condition.
In order to confirm that dye degradation was occurring in the presence of light, the same reaction was performed under the optimized condition without UV-light irradiation. For this, the reaction mixture was stirred for 15 min under normal laboratory surrounding and dark conditions (Fig. S9a and b, ESI†). It was found that under this condition, the percentage degradation and TOC, respectively, were found to be 55.03% and 48.05% under normal laboratory conditions and 18.7% and 82.12% under dark conditions (Fig. 7c). It is pertinent to mention that when the reaction was carried out under normal conditions under stirring for 40 min, degradation was improved to 72.27%. Similarly, under dark conditions, degradation was found to be 33.92% after 40 min (Fig. S9c, ESI†). However, no further improvement was observed on performing the reaction for longer period. This implied that UV-light irradiation significantly facilitated the degradation of dye and its decomposition.
After finding the CoOx–S–VOx catalyst to be an effective catalyst for MO degradation, the other catalysts named Co3O4–V2O5, S–VOx and CoOx were also tested for MO degradation with 15 mg of the catalyst (Fig. S10a and b, ESI†). Co3O4–V2O5, S–VOx and CoOx could degrade only 22.72%, 39.39% and 51.55% MO dye, respectively, upon irradiation for 40 min. There may be two reasons for such ineffectiveness, one Co3O4–V2O5 and S–VOx exhibited a high band gap of 3.38 eV and 4.8 eV, respectively, while CoOx had a band gap of 3.32 eV but probably could not separate the electron–hole (e−–h+) pair effectively, leading to its low activity.49 Since a greater amount of MO is released into the water system, a study was performed to determine the effectiveness of our catalyst at higher molar concentrations of MO. Under the optimized condition, on increasing the dye concentration from 10−6 to 10−3 M, the degradation percentage got lowered to 33.22% (Fig. S11a and b, ESI†).
7. Point of zero charge (PZC) and Effect of pH on the degradation of MO
7.1. Determination of PZC of CoOx–S–VOx
Point of zero charge (PZC) of our synthesized catalyst was evaluated using the reported batch equilibrium method.80 For this, a solution of sodium chloride (0.1 M) was prepared and 20 mL of NaCl solution was taken in each of six beakers. Using 0.1 M H2SO4 or 0.1 M KOH, the pH of the solutions was changed to different pH values. 5 mg of the CoOx–S–VOx catalyst was added to each beaker, which was then stirred for 24 h, and then filtered and final pH values of all solutions were recorded after 24 h. By plotting the difference in the pH value against the initial pH value, the PZC of the catalyst was found to be 4 (Fig. S12a, ESI†).7.2. Effect of pH on the degradation of MO
To evaluate the effect of pH, six different solutions of 10−6 M MO were taken, and pH of each solution was calibrated to 2, 4, 6, 8, 10 and 12 using 0.1 M KOH and 0.1 M H2SO4. Under the different pH conditions, photocatalytic degradation was carried out with the optimized reaction parameters and the recorded results are given in Fig. S12b (ESI†). Degradation efficiency was found to decrease from 99.77% to 65% with an increase in pH from 2 to 12. This can be explained from the PZC value of the synthesized catalyst. When pH < PZC, the material acquired a net positive charge on its surface. As a result, the negatively charged MO dye molecules were more attracted to the positively charged catalyst surface, thereby enhancing the rate of degradation. Because of this reason, when pH > PZC, the rate of MO dye degradation got retarded at higher pH.808. Mechanistic study of dye degradation
The mechanism of dye degradation was believed to proceed via the excitation of electrons from the conduction band (CB) to the valence band (VB) of CoOx–S–VOx as shown in Fig. 8a. As the catalyst possessed an appropriate band gap of 3.14 eV, this electron transfer process creating the positive holes at the CB was expected to occur prominently. In general, in spite of having a suitable band gap, a catalyst may not perform well during photocatalytic dye degradation if the rate of electron and hole recombination was very fast and this can be a probable reason for the observed behavior of our synthesized material CoOx. Devi et al. have reported that the presence of a sulfur atom can help in separating the electrons and holes by oxidizing itself to SO42− ions and serving as an electron trapping agent, as shown in Fig. 8a.81 As the present S–VOx catalyst possessed an inorganic sulfur atom, from the XPS analysis, it was observed that under an oxidizing environment, it can oxidize to SO42−. Therefore, it was believed that in the present case, the sulfur atom oxidizes to SO42− ions and trapped the electron in the CB and thereby favored the photodegradation process keeping the holes (h+) free from negatively charged electrons. It was also reported that the SO42− ion can draw electrons from Lewis acidic sites through the inductive effect of S = O.81 As a result, Lewis acidic sites are transformed to Brønsted acidic sites (BAS). The BAS then abstracted a proton from the water and created more hydroxyl (OH−) ions which then combined with the holes to produce more hydroxyl radicals (OH˙). So probably in our catalyst having the V+4 and Co+3 sites may transform to BAS through the inductive effect of in situ-created SO42− ions and increased the generation of OH˙. The high valent Lewis acidic sites on the catalyst surface may also help in electron trapping as the Co+3 ion can fluctuate in between Co+2 and Co+3. Therefore, it can be said that the presence of a S atom with the conjugate metal oxide (VOx–CoOx) not only manifested a suitable band gap in the photocatalyst but also helped in electron–hole separation by converting itself to an electron scavenging agent.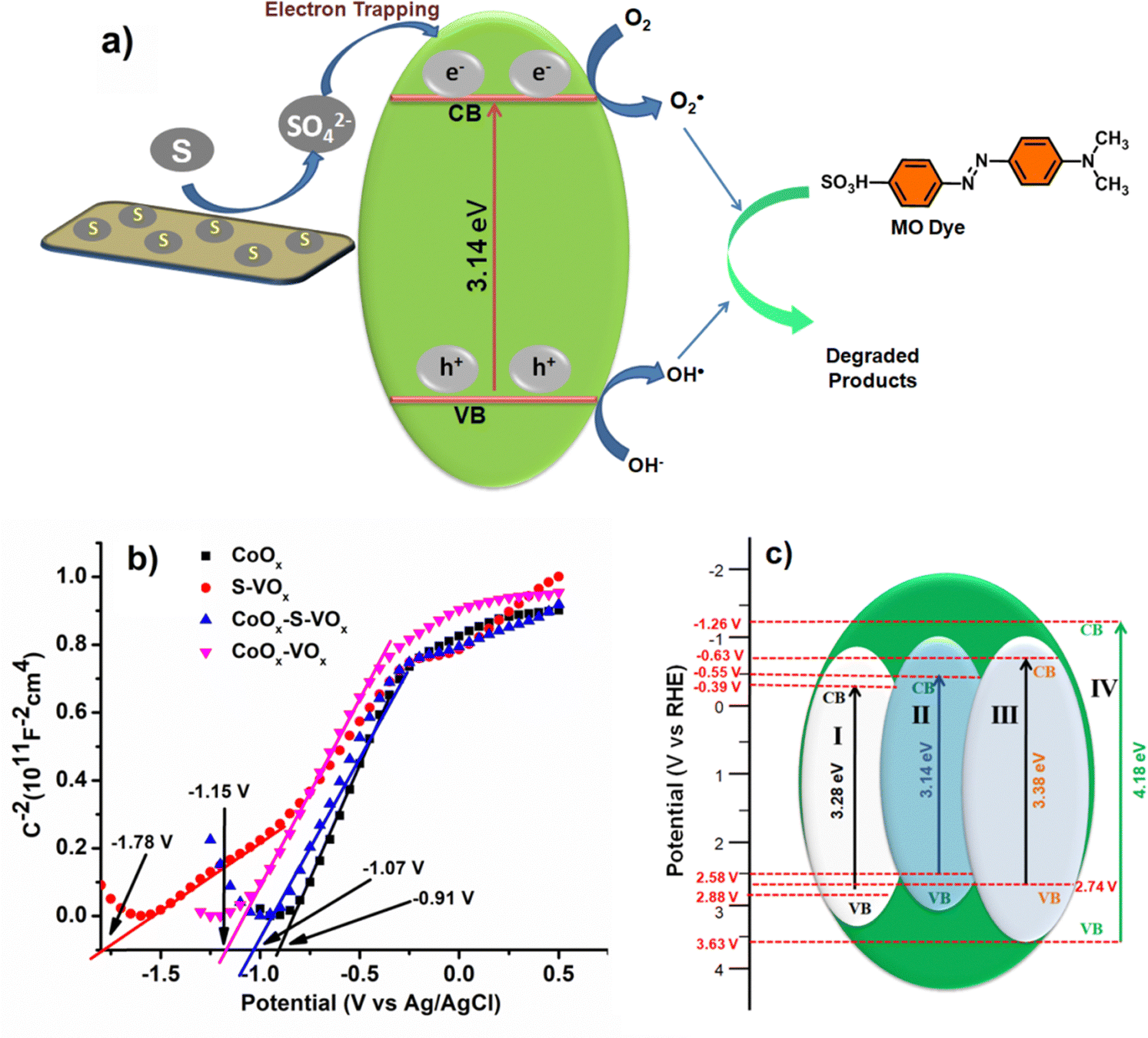 | ||
| Fig. 8 (a) Plausible charge transfer routes for the CoOx–S–VOx material, (b) Mott–Schottky (flat band potential) and (c) band edge structure plot of all the catalysts, (I) CoOx (II) CoOx–S–VOx, (III) Co3O4–V2O5, (IV) S–VOx. [Flat band potential (EFB) = Conduction band potential (ECB) + 0.1 V, Valance band potential (EVB) = Band gap + ECB].83 | ||
The photoluminescence (PL) spectroscopy analysis was performed with an excitation radiation of 450 nm and 740 nm (Fig. S13a and b, ESI†). When excited at 450 nm, CoOx–S–VOx showed emission bands at 687 nm and 1030 nm and S–VOx showed emission bands at 700 nm and 1057 nm, and CoOx did not show any emission band at this excitation wavelength. Similarly, when excited at 740 nm, both CoOx–S–VOx and CoOx showed emission signals at 905 nm, and S–VOx did not show any emission band upon excitation at 740 nm. The difference in the photoluminescence behavior was attributed to the difference in the absorption band as obtained from DRS analysis (Fig. 1d and Fig. S1a, b, ESI†). The appearance of the near infrared (NIR) emission band was attributed to the presence of electronic transitions at higher wavelength as observed from the DRS spectra (Fig. S1a and b, ESI†). It was observed from the PL spectra analysis that the intensity of the emission signal for CoOx–S–VOx appearing at 905 nm had low intensity compared to CoOx. Similarly, the emission band above 1000 nm also showed lowering of intensity in comparison with S–VOx. This indicated that the electron–hole recombination rate was suppressed in the case of CoOx–S–VOx in comparison with CoOx and S–VOx. This implied that CoOx–S–VOx can serve as a better photocatalyst in MO dye degradation by relaxing the electron–hole recombination rate.82
To further have a proper understanding of the mechanism, we also calculated the valence band position and conduction band position using the Mott–Schottky (MS) plot (Fig. 8b). MS plots were obtained by performing electrochemical impedance spectroscopy (EIS) over a range of applied potential values. The flat band potential (Vfb) values of the materials CoOx, S–VOx, Co3O4–V2O5 and CoOx–S–VOx were estimated by extrapolating the linear region of the MS plot. The obtained Vfb values of CoOx, S–VOx, Co3O4–V2O5 and CoOx–S–VOx were −0.91 V, −1.78 V, −1.15 V and −1.07 V, respectively vs. Ag/AgCl electrode. The 1/C2vs. V plots for all the materials exhibited positive slopes, indicating n-type semiconductor properties of the materials. Therefore, CB band position and VB position of the materials were calculated by combining the band gap (Eg) values obtained from DRS analysis and Vfb values obtained from MS plots, as shown in Fig. 8c. The CB position along with the VB position for all the synthesized catalysts are shown in Fig. 8c.83,84 From the diagram, it was observed that the CoOx–S–VOx catalyst had a CB potential that was slightly more negative than the O2/O2−˙ potential vs. NHE (−0.46 V). Similarly, the VB was approximately in the range of oxidation potential of H2O/OH˙. This further implied that our synthesized catalyst can effectively produce hydroxyl radicals (OH˙) or O2−˙ and can degrade the dye effectively. In contrast, the band edge potential for the other two catalysts differs significantly, especially in the case of S–VOx making it unsuitable for the photodegradation process. The same applies to Co3O4–V2O5 indicating the role of sulfur in modulating the band gap potential and thereby making the CoOx–S–VOx catalyst most suitable for the photodecomposition of MO. Although CoOx possessed CB position within the limit of water splitting reaction, probably as mentioned earlier due to non-availability of any electron scavenging agent, it did not participate well in the photooxidation process. The VB of the most effective catalyst (CoOx–S–VOx) was also calculated using UPS analysis shown in Fig. S14a and b (ESI†). It was found to be 2.8 eV approximately equivalent to that calculated from the MS plot. The work function was found to be 5.5 eV and was in the range of work function for cobalt oxide and vanadium oxide.85
Based on the proposed mechanism, it was understood that the photodegradation process probably proceeded either through the creation of hydroxyl radicals (OH˙) or superoxide radicals (O2−˙). So, in order to verify the involvement of such radical species, the photodegradation process was again studied by using some scavenging agents. Potassium iodide (KI) was used as a hole scavenging agent, silver nitrate (AgNO3) as an electron scavenging agent, isopropanol and ascorbic acid were used as OH˙ and O2−˙ radical scavenging agents, respectively.82 From the graph shown in Fig. S15 (ESI†), it was found that the efficacy in the presence of holes and electron scavengers remained almost the same, while it got retarded in the presence of radical scavengers. This implied that the radicals either O2−˙ or OH˙ were the active species for the decomposition of anionic MO dye.
9. Recyclability test
In order to check recyclability, the CoOx–S–VOx material was considered as the test catalyst and performed both Cr(VI) reduction and MO dye degradation reaction. Both the reactions were carried out up to 10th cycle. After 2nd cycle, 95% and 93% efficiency was observed in the case of Cr(VI) reduction and MO degradation reaction, respectively. Further decrease in efficiency was observed in the consecutive cycles as shown in Fig. S16 (ESI†). However, the decreasing rate was not significantly higher and ranged from 90 to 75% until the 10th cycle. These investigations revealed that the synthesized CoOx–S–VOx material was stable and could be recycled ten times without causing any substantial loss in efficiency. After 10th cycle, the remaining material was collected and then PXRD analysis (Fig. S17, ESI†), SEM and TEM analysis (Fig. S18, ESI†) were performed. The PXRD analysis of the recycled catalyst indicated the loss in the crystallinity of the original catalyst and probably influenced the catalytic activity. Similarly, from the SEM and TEM analysis, a change in the morphology of CoOx–S–VOx and a decrease in the presence of fine nanoparticles were observed leading to loss of activity. Thus, it can be said that the change in the catalyst structure and morphology resulted in the decrease of degradation activity for Cr(VI) reduction and MO dye degradation reaction.10. Conclusion
In summary, it can be said that the synthesized mixed metal oxide catalyst of cobalt and vanadium doped with inorganic sulfur served as a catalyst for Cr(VI) reduction as well as for MO dye degradation. The reduction of Cr(VI) to Cr(III) was found to be very fast in comparison to many other literature reports. The synthesized catalyst also appeared as a photocatalyst for the decomposition of MO dye harvesting the UV-light. The study predicted that the photocatalytic dye decomposition process was dependent on various parameters like time, catalyst amount, pH, and reaction environment. The dye degradation process was found to be more effective at low pH. The comparison of the activity of the bimetallic CoOx–S–VOx catalyst with its monometallic counterpart viz CoOx and S–VOx indicated that the mixed metal oxide was highly effective in comparison to the single metal oxides. The band structure of the catalysts was correlated with the mechanism of the photocatalytic process through various analyses. The mechanistic understanding revealed the dominant role of radicals in the photocatalytic process. The catalyst reduced Cr(VI) to Cr(III) even at a higher concentration level in very less time (3 min) and very effectively decomposed the toxic industrial waste dye with high mineralization percentage. Therefore, the present study can offer a new direction and perspective for sustainable development and the elimination of environmental pollutants, including toxic metals like hexavalent chromium.Author contributions
Sayanika Saikia: methodology, validation, formal analysis, investigation, writing – review & editing; Manoshi Saikia: formal analysis, writing – review & editing; Salma A. Khanam: formal analysis, investigation; Seonghwan Lee: resources, formal analysis; Young-Bin Park: resources, formal analysis; Lakshi Saikia: resources, formal analysis; Gautam Gogoi: validation, formal analysis; Kusum K. Bania: conceptualization, writing – original draft, writing – review & editing visualization, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KKB acknowledges SERB for the research grant (CRG/2019/000962) and CSIR (NO. 80(0094)/20/EMR-II). S. Saikia acknowledges University Grants Commission (UGC) for the Junior Research Fellowship. The authors also acknowledge Tezpur University for offering the analytical and infrastructural facilities required for this research work.References
- S. Rojas and P. Horcajada, Chem. Rev., 2020, 120, 8378–8415 CrossRef CAS PubMed.
- C. O. Okoye, C. I. Addey, O. Oderinde, J. O. Okoro, J. Y. Uwamungu, C. K. Ikechukwu, E. S. Okeke, O. Ejeromedoghene and E. C. Odii, Chem. Eng. J. Adv., 2022, 11, 100310 CrossRef CAS.
- M. Sajid, M. K. Nazal, N. Baig and A. M. Osman, Sep. Purif. Technol., 2018, 191, 400–423 CrossRef CAS.
- A. D. Bokare and W. Choi, Environ. Sci. Technol., 2010, 44, 7232–7237 CrossRef CAS PubMed.
- J. A. Jacobs and S. M. Testa, Overview of chromium(VI) in the environment: background and history, CRC Press, 2005, pp. 1–21 Search PubMed.
- M. Owlad, M. K. Aroua, W. A. W. Daud and S. Baroutian, Water, Air, Soil Pollut., 2009, 200, 59–77 CrossRef CAS.
- L. S. McNeill, J. E. McLean, J. L. Parks and M. A. Edwards, J. Am. Water Works Assoc., 2012, 104, E395–E405 CrossRef.
- P. Veerakumar, P. Thanasekaran, K. C. Lin and S. B. Liu, ACS Sustainable Chem. Eng., 2017, 5, 5302–5312 CrossRef CAS.
- R. M. Sedman, J. A. Y. Beaumont, T. A. McDonald, S. Reynolds, G. Krowech and R. Howd, J. Environment. Sci. Health C, 2006, 24, 155–182 CrossRef CAS PubMed.
- S. W. Hu, L. W. Yang, Y. Tian, X. L. Wei, J. W. Ding, J. X. Zhong and P. K. Chu, Appl. Catal. B: Environ., 2015, 163, 611–622 CrossRef CAS.
- Y. Ying, Y. Liu, X. Wang, Y. Mao, W. Cao, P. Hu and X. Peng, ACS Appl. Mater. Interfaces, 2015, 7, 1795–1803 CrossRef CAS PubMed.
- Y. Cheng, F. Yan, F. Huang, W. Chu, D. Pan, Z. Chen, J. Zheng, M. Yu, Z. Lin and Z. Wu, Environ. Sci. Technol., 2010, 44, 6357–6363 CrossRef CAS PubMed.
- H. L. Ma, Y. Zhang, Q. H. Hu, D. Yan, Z. Z. Yu and M. Zhai, J. Mater. Chem., 2012, 22, 5914–5916 RSC.
- P. K. Boruah, P. Borthakur, G. Darabdhara, C. K. Kamaja, I. Karbhal, M. V. Shelke, P. Phukan, D. Saikia and M. R. Das, RSC Adv, 2016, 6, 11049–11063 RSC.
- B. Xie, C. Shan, Z. Xu, X. Li, X. Zhang, J. Chen and B. Pan, Chem. Eng. J., 2017, 308, 791–797 CrossRef CAS.
- J. Nan, S. Guo, D. Alhashmialameer, Q. He, Y. Meng, R. Ge, S. M. El-Bahy, N. Naik, V. Murugadoss, M. Huang and B. B. Xu, ACS Appl. Nano Mater., 2022, 5, 8755–8766 CrossRef CAS.
- B. Gopal and A. Gupta, ACS Omega, 2019, 4, 20443–20449 CrossRef CAS PubMed.
- O. Njoya, S. Zhao, Y. Qu, J. Shen, B. Wang, H. Shi and Z. Chen, J. Environ. Manage., 2020, 275, 111165 CrossRef CAS PubMed.
- O. Njoya, S. Zhao, J. Shen, X. Kong, Y. Gong, B. Wang, J. Kang and Z. Chen, Sep. Purif. Technol., 2022, 301, 122051 CrossRef CAS.
- M. U. Rahman and S. A. Kazmi, J. Chem. Soc. Pak, 2011, 19, 201 Search PubMed.
- X. R. Xu, H. B. Li, X. Y. Li and J. D. Gu, Chemosphere, 2004, 57, 609–613 CrossRef CAS PubMed.
- Y. C. Zhang, J. Li, M. Zhang and D. D. Dionysiou, Environ. Sci. Technol., 2011, 45, 9324–9331 CrossRef CAS PubMed.
- W. Yang, L. Zhang, Y. Hu, Y. Zhong, H. B. Wu and X. W. Lou, Angew. Chem., 2012, 124, 11669–11672 CrossRef.
- H. Abdullah and D. H. Kuo, ACS Appl. Mater. Interfaces, 2015, 7, 26941–26951 CrossRef CAS PubMed.
- M. A. Pandit, D. S. H. Kumar, M. Ramadoss, Y. Chen and K. Muralidharan, RSC Adv., 2022, 12, 7762–7772 RSC.
- M. Madkour, Y. Abdelmonem, U. Y. Qazi, R. Javaid and S. Vadivel, RSC Adv., 2021, 11, 29433–29440 RSC.
- B. Das, M. Sharma, J. C. Sarmah and K. K. Bania, J. Environ. Chem. Eng., 2017, 5, 4212–4219 CrossRef CAS.
- R. Kamaraj and S. Vasudevan, ChemistrySelect, 2019, 4, 2428–2435 CrossRef CAS.
- G. Liu, P. Niu, L. Yin and H. M. Cheng, J. Am. Chem. Soc., 2012, 134, 9070–9073 CrossRef CAS PubMed.
- J. Jiang, Z. Xiong, H. Wang, G. Liao, S. Bai, J. Zou, P. Wu, P. Zhangand and X. Li, J. Mater. Sci. Technol., 2022, 118, 15–24 CrossRef CAS.
- S. Lu, W. Liao, W. Chen, M. Q. Yang, S. Zhuand and S. Liang, Appl. Surf. Sci., 2023, 614, 156224 CrossRef CAS.
- T. Zhu, J. Pan, Z. An, R. Zhe, Q. Ou and H. E. Wang, J. Mater. Chem. A, 2022, 10, 20375–20385 RSC.
- S. Y. Xiao, Y. Liu, X. F. Wu, L. T. Gan, H. Y. Lin, L. R. Zheng, S. Dai, P. F. Liu and H. G. Yang, J. Mater. Chem. A, 2021, 9, 14786–14792 RSC.
- Y. F. Ren, Z. L. He, H. Z. Zhao and T. Zhu, Rare Metals, 2022, 41, 830–835 CrossRef CAS.
- Y. Vasseghian, F. Almomani, M. Moradi and E. N. Dragoi, J. Hazard. Mater., 2022, 423, 127016 CrossRef CAS PubMed.
- Y. Vasseghian, E. N. Dragoi and F. Almomani, Chemosphere, 2022, 287, 132387 CrossRef CAS PubMed.
- S. Moosavi, C. W. Lai, S. Gan, G. Zamiri, O. Akbarzadeh Pivehzhani and M. R. Johan, ACS omega, 2020, 5, 20684–20697 CrossRef CAS PubMed.
- E. Routoula and S. V. Patwardhan, Environ. Sci. Technol., 2020, 54, 647–664 CrossRef CAS PubMed.
- R. Guo, T. Jiao, R. Li, Y. Chen, W. Guo, L. Zhang, J. Zhou, Q. Zhang and Q. Peng, ACS Sustainable Chem. Eng., 2018, 6, 1279–1288 CrossRef CAS.
- S. Rafaqat, N. Ali, C. Torres and B. Rittmann, RSC Adv., 2022, 12, 17104–17137 RSC.
- A. Ajmal, I. Majeed, R. N. Malik, H. Idriss and M. A. Nadeem, RSC Adv., 2014, 4, 37003–37026 RSC.
- Y. Wang, C. Liang, C. Fan, J. Chen, Z. Zhang and H. Liu, Environ. Sci.: Nano, 2023, 10, 891–901 RSC.
- C. Liu, X. Dong, Y. Hao, X. Wang, H. Ma and X. Zhang, RSC Adv., 2017, 7, 22415–22423 RSC.
- A. Pirkarami and M. E. Olya, J. Saudi Chem. Soc., 2017, 21, S179–S186 CrossRef CAS.
- M. Nazim, J. H. Kim, H. Y. Lee and S. K. Cho, Langmuir, 2021, 37, 12929–12939 CrossRef CAS PubMed.
- S. He, Y. Chen, X. Li, L. Zeng and M. Zhu, ACS ES&T Eng, 2022, 2, 527–546 Search PubMed.
- J. Luo, S. Zhang, M. Sun, L. Yang, S. Luo and J. C. Crittenden, ACS nano, 2019, 13, 9811–9840 CrossRef CAS PubMed.
- S. Gautam, H. Agrawal, M. Thakur, A. Akbari, H. Sharda, R. Kaur and M. Amini, J. Environ. Chem. Eng., 2020, 8, 103726 CrossRef CAS.
- M. J. Baruah, T. J. Bora, G. Gogoi, N. Hoque, N. K. Gour, S. K. Bhargava, A. K. Guha, J. K. Nath, B. Das and K. K. Bania, J. Colloid Interface Sci., 2022, 608, 1526–1542 CrossRef CAS PubMed.
- S. A. Khan, Z. Arshad, S. Shahid, I. Arshad, K. Rizwan, M. Sher and U. Fatima, Compos. B: Eng., 2019, 175, 107120 CrossRef CAS.
- M. A. Bhatti, A. A. Shah, K. F. Almani, A. Tahira, S. E. Chalangar, A. Dad Chandio, O. Nur, M. Willander and Z. H. Ibupoto, Ceramics International, 2019, 45, 23289–23297 CrossRef CAS.
- D. Khalafallah, O. Y. Alothman, H. Fouad and K. A. Khalil, Int. J. Hydrogen Energy, 2018, 43, 2742–2753 CrossRef CAS.
- M. M. Sajid, N. A. Shad, Y. Javed, S. B. Khan, Z. Zhang, N. Amin and H. Zhai, Surf. Interfaces, 2020, 19, 100502 CrossRef CAS.
- R. Baddour-Hadjean, M. B. Smirnov, K. S. Smirnov, V. Y. Kazimirov, J. M. Gallardo-Amores, U. Amador, M. E. Arroyo-de Dompablo and J. P. Pereira-Ramos, Inorg. Chem., 2012, 51, 3194–3201 CrossRef CAS PubMed.
- Z. Wang, X. Li, W. Guo and Y. Fu, Adv. Funct. Mater., 2021, 31, 2009875 CrossRef CAS.
- M. S. Weimer, R. F. McCarthy, J. D. Emery, M. J. Bedzyk, F. G. Sen, A. Kinaci, M. K. Chan, A. S. Hock and A. B. Martinson, Chem. Mater., 2017, 29, 2864–2873 CrossRef CAS.
- X. Wu, J. Wang, S. Liu, X. Wu and S. Li, Electrochim. Acta, 2011, 56, 10197–10203 CrossRef CAS.
- S. A. Khanam, N. Hoque, S. Lee, Y. B. Park, G. Gogoi and K. K. Bania, ACS Appl. Energy Mater., 2022, 5, 12651–12662 CrossRef CAS.
- T. V. Larina, S. V. Cherepanova, N. A. Rudina, B. A. Kolesov and A. N. Zagoruiko, Catal. Sustainable Energy, 2016, 2, 87–95 Search PubMed.
- S. Ambika, S. Gopinath, K. Saravanan, K. Sivakumar, C. Ragupathi and T. A. Sukantha, Energy Rep., 2019, 5, 305–309 CrossRef.
- T. Jenkins, J. A. Alarco, B. Cowie and I. D. Mackinnon, ACS Appl. Mater. Interfaces, 2021, 13, 45505–45520 CrossRef CAS PubMed.
- C. Alex, S. C. Sarma, S. C. Peter and N. S. John, ACS Appl. Mater. Interfaces, 2020, 3, 5439–5447 CAS.
- Y. Wang, C. Chen and D. Zhang, J. Mater. Chem. C, 2021, 9, 15445–15451 RSC.
- Y. Piao, V. N. Tondare, C. S. Davis, J. M. Gorham, E. J. Petersen, J. W. Gilman, K. Scott, A. E. Vladár and A. R. H. Walker, Compos. Sci. Technol., 2021, 208, 108753 CrossRef CAS.
- H. Kanso, R. M. Clarke, A. Kochem, H. Arora, C. Philouze, O. Jarjayes, T. Storr and F. Thomas, Inorg. Chem., 2020, 59, 5133–5148 CrossRef CAS PubMed.
- V. S. Wadi, K. K. Jena, K. Halique and S. M. Alhassan, ACS Appl. Polym. Mater., 2019, 2, 198–208 CrossRef.
- S. Zhou, Z. Ye, S. Hu, C. Hao, X. Wang, C. Huang and F. Wu, Nanoscale, 2018, 10, 15771–15781 RSC.
- S. E. Skrabalak, J. Chen, Y. Sun, X. Lu, L. Au, C. M. Cobley and Y. Xia, Acc. Chem. Res., 2008, 41, 1587–1595 CrossRef CAS PubMed.
- T. Xian, H. Li, X. Sun, Y. Huo, L. Di, C. Sun and H. Yang, ACS Appl. Nano Mater., 2022, 5, 15260–15271 CrossRef CAS.
- J. J. Bohning, J. Chem. Educ., 1960, 37, A443 CrossRef.
- R. Li, Q. D. An, B. Q. Mao, Z. Y. Xiao, S. R. Zhai and Z. Shi, J. Taiwan Inst. Chem. Eng., 2017, 80, 596–606 CrossRef CAS.
- P. C. Bandara, J. Peña-Bahamonde and D. F. Rodrigues, Sci. Rep., 2020, 10, 9237 CrossRef CAS PubMed.
- Z. Wu, X. Mao, Q. Zi, R. Zhang, T. Dou and A. C. Yip, J. Power Sources, 2014, 268, 596–603 CrossRef CAS.
- D. R. Lindsay, K. J. Farley and R. F. Carbonaro, J. Environ. Monit., 2012, 14, 1789–1797 RSC.
- J. W. Ball and D. K. Nordstrom, J. Chem. Eng. Data, 1998, 43, 895–918 CrossRef CAS.
- G. Sharma and B. Kandasubramanian, J. Chem. Eng. Data, 2020, 65, 396–418 CrossRef CAS.
- P. S. Minhas, J. K. Saha, M. L. Dotaniya, A. Sarkar and M. Saha, Sci. Total Environ., 2022, 808, 152001 CrossRef CAS PubMed.
- K. Wang, S. Zhan, D. Zhang, H. Sun, X. Jin and J. Wang, Colloids Surf. A: Physicochem Eng., 2021, 618, 126362 CrossRef CAS.
- G. Gogoi, P. Saikia, M. J. Baruah, S. Lee, Y. B. Park, R. Dutta and K. K. Bania, Microporous Mesoporous Mater., 2021, 326, 111392 CrossRef CAS.
- A. Abdelhaleem and W. Chu, J. Hazard. Mater., 2017, 338, 491–501 CrossRef CAS PubMed.
- L. G. Devi, P. M. Nithya and R. Kavitha, Mater. Today Commun., 2018, 17, 391–401 CrossRef.
- Z. Huang, S. Jia, J. Wei and Z. Shao, RSC Adv., 2021, 11, 16747–16754 RSC.
- Y. Xiao, Y. Jiang, X. Liu, W. Zhang, Z. Zhu, Y. Gao, H. Xu, J. Zhang, Z. Liu and L. Ni, J. Mater. Sci., 2020, 55, 14211–14228 CrossRef CAS.
- W. Yan, Y. Yu, H. Zou, X. Wang, P. Li, W. Gao, J. Wang, S. Wu and K. Ding, Solar RRL, 2018, 2, 1800058 CrossRef.
- N. R. Chodankar, H. D. Pham, A. K. Nanjundan, J. F. Fernando, K. Jayaramulu, D. Golberg, Y. K. Han and D. P. Dubal, Small, 2020, 16, 2002806 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The supporting information includes data related to physical measurements, DRS and Tauc's plot for S–VOx and CoOx, XPS analysis of C(1s) for CoOx–S–VOx, EPR, TGA spectra, STEM-HAADF-EDX mapping of the CoOx–S–VOx material in different regions, UV spectra and bar diagrams illustrating Cr(VI) reduction efficiency for different Cr(VI) solution concentrations using CoOx–S–VOx as well as Cr(VI) reduction efficiency with S–VOx and CoOx materials. The information also includes data related to CV plots for Cr(VI) reduction, the efficiency of the catalyst for MO degradation under different conditions, its efficiency in comparison with S–VOx and CoOx materials, UV spectra and bar diagrams showing MO degradation for different MO solution concentrations, point of zero charge (PZC) determination and effect of pH on MO degradation using CoOx–S–VOx, PL spectra at different excitation wavelengths, UPS spectra, photocatalytic degradation mechanism using an electron scavenger (AgNO3), a hole scavenger (KI), an OH˙ scavenger (isopropanol) and an O2−˙ scavenger (ascorbic acid), and bar diagram, PXRD spectra, SEM and TEM images for the recyclability test of CoOx–S–VOx. See DOI: https://doi.org/10.1039/d3ma00625e |
| This journal is © The Royal Society of Chemistry 2023 |