
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile surface restructure by one-step sub-millisecond laser exposure promotes the CO2 methanation performance of cobalt oxide supported Pd nanoparticles with copper-oxide cluster decoration†
Dinesh
Bhalothia‡
 a,
Amisha
Beniwal‡
a,
Praveen Kumar
Saravanan
b,
Guo-Heng
Huang
a,
Mingxing
Cheng
ac,
Ming-Wei
Lin
ad,
Po-Chun
Chen
*b and
Tsan-Yao
Chen
*a
a,
Amisha
Beniwal‡
a,
Praveen Kumar
Saravanan
b,
Guo-Heng
Huang
a,
Mingxing
Cheng
ac,
Ming-Wei
Lin
ad,
Po-Chun
Chen
*b and
Tsan-Yao
Chen
*a
aDepartment of Engineering and System Science, National Tsing Hua University, Hsinchu 30013, Taiwan. E-mail: chencaeser@gmail.com; Tel: +886-3-5715131 ext. 34271
bDepartment of Materials and Mineral Resources Engineering, National Taipei University of Technology, Taipei 10608, Taiwan
cDepartment of Electrical Engineering and Electronics, University of Liverpool, Liverpool L69 3GJ, UK
dInstitute of Nuclear Engineering and Science, National Tsing Hua University, Hsinchu 30013, Taiwan
First published on 10th November 2023
Abstract
Carbon dioxide (CO2) methanation not only mitigates excessive CO2 emissions but also circumvents the difficulties associated with the storage and transportation of low-grade energies. However, the competitive reverse water gas shift (RWGS) reaction severely hampers the productivity of methane (CH4). In this context, a tri-metallic nanocatalyst (NC) comprising atomic CuOx cluster anchored Pd nanoparticles (NP)s on the cobalt-oxide support (hereafter denoted as CPCu) is developed. Furthermore, to optimize the CO2 methanation performance, the surface and sub-surface atomic arrangements of the as-prepared CPCu NCs were altered by a sub-millisecond pulsed laser irradiation with per pulse energies of 1 mJ and 10 mJ for a fixed duration of 10 s. For the optimum case (1 mJ per pulse energy input; denoted as CPCu-1), the CPCu-1 NC delivers an optimum CH4 productivity of ∼1346 mmol g−1 h−1 at 300 °C temperature, which is 13.6% enhanced as compared to the pristine conditions (∼1164 mmol g−1 h−1). On top of that, the CH4 selectivity is improved by 40% for CPCu-1 NCs as compared to the as-prepared conditions. The cross-referencing results of physical characterization along with electrochemical analysis indicate that such an improved activity and selectivity of CPCu-1 NCs originate from the significant surface restructure of CPCu-1 NCs, where the high density of surface exposed atomic CuOx species and neighbouring Pd sites, respectively, promotes CO2 activation and H2 dissociation steps during CO2 methanation. We believe that the obtained results will provide insight into designing high-performance catalytic materials for CO2 methanation by using sub-millisecond pulsed laser irradiation.
Introduction
It is widely acknowledged that the sequestration of carbon dioxide (CO2) is crucial for achieving global sustainability. However, the existing economic and technological challenges have compelled us to explore the recycling of CO2 into valuable fuels or chemicals. Among the various pathways under consideration, the selective hydrogenation of CO2 to methane (CH4), known as CO2 methanation, stands out as the most promising route for small to intermediate-scale industrial operations.1,2 Moreover, CH4 is a more manageable species and can leverage existing distribution networks, which addresses the main challenges associated with the safe storage and transportation of hydrogen.3 These aspects remain significant bottlenecks in utilizing hydrogen, particularly in areas distant from its production sites. Despite its numerous advantages, the competitive reverse water gas shift (RWGS) reaction, which produces carbon monoxide (CO) at ambient pressure, poses a significant obstacle to CO2 methanation, leading to a decrease in CH4 productivity.4,5 Consequently, there is an urgent need to develop highly efficient catalysts that can facilitate CO2 methanation at low temperatures, while maintaining high selectivity towards CH4 and minimizing CO production.Previous studies have highlighted that many group VIII metals exhibit high catalytic activity in CO2 methanation. While precious metal catalysts like Rh, Ru, and Pd have been extensively investigated,6–11 Ni-based catalysts have attracted attention due to their lower preparation cost and higher availability.12,13 However, Ni-based catalysts typically exhibit lower activity at low temperatures compared to their precious metal counterparts.14 Moreover, the metallic Ni active sites in these catalysts are prone to thermal sintering, especially in the presence of hotspots during CO2 methanation, leading to rapid catalyst deactivation.15 Therefore, considerable efforts have been devoted to developing precious metal-based catalysts with low metal loading and excellent low-temperature activity.
CO2 methanation is a multistep reaction and can proceed through various pathways such as CO and formate pathways.16 In the CO pathway, initially, CO is produced via the RWGS reaction (CO2 + H2 → CO + H2O(g)), which undergoes further hydrogenation (CO + 3H2 → CH4 + H2O) to yield CH4.17 Besides, intermediate species such as formate (HCOO−) are produced via direct hydrogenation of the adsorbed CO2, which again reacts with adsorbed hydrogen atoms (H*) to produce CH4.18 Taking into account the preceding discussion, it can be inferred that the CO2 methanation reaction is highly influenced by the choice of catalyst. To attain optimal CH4 selectivity and maximize productivity, the catalysts should possess two adjacent reaction sites, one for CO2 adsorption and another for H2 dissociation.19 The significance of having two adjacent reaction sites stems from the fact that catalytic materials with a single site for CO2 reduction are prone to rapid failure due to severe coke formation.20 On the other hand, catalysts with a single site for H2 dissociation exhibit inactivity towards CO2.21 Therefore, to achieve an optimal outcome, there is a need for synergistic collaboration between neighboring reaction sites for CO2 activation and H2 dissociation in sub-nanometer domains. Previously published literature frequently reported the importance of synergistic cooperation between adjacent reaction sites for CO2 methanation, where one reaction site promotes CO2 activation, while the other favours the H2 spillover.22 For instance, our recent work highlighted the potential synergy between oxygen vacancy enriched atomic scale CoOx clusters and adjacent Pd domains, where CO2 activation and H2 dissociation were achieved simultaneously on CoOx and adjacent Pd domains, respectively.19 Moreover, Zhang et al. demonstrated the synergistic effect between the Pd nanoparticles (NP)s and the UiO-66 support for enhanced CO2 methanation.23 These studies highlight the crucial role of the surface atomic configuration in catalytic activity and the importance of exploring next-generation techniques for manipulating the surface atomic arrangement. While thermal annealing has been commonly employed to control heteroatomic intermixing on the surface and subsurface domains of nanocatalysts, its limitations, such as longer operational times and high energy input, make it less suitable for large-scale commercial applications.24 In contrast, pulsed laser irradiation with controllable duration and high energy photon flux has emerged as a potential technique for precise atomic manipulation and designing materials with optimized functionality.25 Moreover, pulsed laser annealing allows for precise control over the energy and duration of the laser pulses. This level of control enables the manipulation of atomic structures with high precision, making it suitable for designing highly active and selective catalysts for CO2 methanation applications where exact atomic positioning is critical. On top of that, pulsed laser annealing is a non-contact and non-invasive technique. This means that it doesn't require physical contact with the sample, reducing the risk of contamination or damage to the material being manipulated. Moreover, the short, intense laser pulses can rapidly heat the material to very high temperatures and then quickly cool it down. This rapid heating and cooling cycle can be used to induce phase changes, crystallization, or amorphization of materials, allowing for the manipulation of atomic structures for various redox applications. Since the heating and cooling process occurs very quickly during pulsed laser annealing, it minimizes the thermal impact on the surrounding material. This is important for preserving the properties of the material and avoiding unwanted side effects. In our previous study, submillisecond laser annealing was utilized to form multiple metal-to-metal oxide heterogeneous interfaces in a trimetallic system (CuNiPd), resulting in enhanced CO2 reduction performance.26 This demonstrates the potential of pulsed laser irradiation in restructuring the nanocatalyst surface at the sub-nanometer scale, promoting strong heteroatomic intermixing, and enhancing catalytic activity.
In this study, we have tailored the physiochemical properties of a tri-metallic nanocatalyst (NC) comprising atomic scale CuOx clusters decorated Pd NPs on the cobalt-oxide support (hereafter denoted as CPCu) using a one-step sub-millisecond pulsed laser beam with per-pulse energies of 1 mJ and 10 mJ for enhanced CO2 methanation performance. In the ideal scenario, when the per pulse energy of a pulsed laser beam is 1 mJ, referred to as CPCu-1, the high density of CuOx atomic clusters are decorated in the defect sites of Pd NPs, which leads to the CPCu-1 NC exhibiting a CH4 productivity of ∼1346 mmol g−1 h−1 at 300 °C temperature, which is 13.6% enhanced as compared to the pristine CPCu catalyst (∼1164 mmol g−1 h−1). Of utmost importance, such surface atomic arrangement suppresses the competitive RWGS and thus the CH4 selectivity is improved by 40% as compared to pristine CPCu. The findings from various physical investigations and electrochemical analyses suggest that there is a potential synergistic effect between the surface CuOx species and the neighboring active sites of Pd. This synergism contributes to enhancing the CH4 productivity of the CPCu-1 mJ NC, where both the CuOx clusters and Pd reaction sites play a simultaneous role in promoting the activation of CO2 and H2 respectively. These combined effects contribute to the improved activity and selectivity observed in the CH4 production during the CO2 methanation reaction.
Experimental
Synthesis of tri-metallic CPCu NCs.
The CPCu NCs are first synthesized using a combination of wet chemical reduction and ultrasonication methods. Prior to the synthesis, the catalyst carrier, carbon black (UR-XC72, UniRegion Bio-Tech), is surface functionalized to enhance the metal–support interaction and achieve uniform dispersion. In the very first step, a total of 6 g of surface-functionalized carbon black (referred to as active carbon or AC) is dispersed in 3.06 g of an aqueous solution containing 0.1 M cobalt(III) chloride (CoCl3, Sigma-Aldrich Co.) and stirred at 600 rpm for 6 hours at 25 °C. This solution, denoted as “A”, consists of Co3+ ions adsorbed on the surface of AC (Co3+_ads-AC). The weight ratio of Co3+ ions to AC is 30 wt%, corresponding to 18 mg of Co3+ ions. Subsequently, 5 ml solution of sodium borohydride (NaBH4; 99%, Sigma-Aldrich Co.) in deionized (DI) water is rapidly added to solution A and stirred at 600 rpm for 10 seconds. This step leads to the formation of a mixed metallic and oxide phase of Co (Co/CoOx–AC) on the AC surface. To solution B (i.e., Co/CoOx–AC), 3.06 g of a 0.1 M Pd precursor solution is added to grow Pd nanoparticles over the CoOx support. The Pd precursor solution is prepared by dissolving palladium chloride (PdCl2, 99%, Sigma-Aldrich Co.) in 1.0 M hydrochloric acid (HCl(aq)). In this step, Pd2+ ions are reduced by the excess NaBH4 added in the previous step. The as-synthesized Co@Pd NPs are subjected to ultrasonication treatment at a frequency of 100 kHz. This treatment creates surface defects, acting as nano-tweezers for the surface-anchored atomic species. After the ultrasonication treatment, the surface anchoring of atomic CuOx species is achieved by the adsorption of Cu2+ ions (0.1 M solution of copper(II) sulfate pentahydrate; CuSO4·5H2O, 99%, Sigma Aldrich Co.) onto the defect sites of Co@Pd NPs. The adsorbed Co3+ ions on the Co@Pd NPs are subsequently reduced using NaBH4 to form CuOx clusters. The reduction occurs in the defect sites created during the ultrasonication treatment. The resulting product, denoted as CPCu, is washed sequentially with acetone, ethanol, and DI water. It is then centrifuged, dried at 120 °C for 12 hours, and exposed to an ambient environment. As prepared CPCu NCs are then subjected to sub-millisecond pulsed laser annealing with per pulse energies of 1 mJ and 10 mJ for fine-tuning the physiochemical properties of the material. Hereafter the CPCu NC irradiated with 1 mJ and 10 mJ per pulse energies are denoted as CPCu-1 and CPCu-10, respectively.Physical characterization of CPCu NCs
The physical properties of CPCu NCs and reference samples were examined through a combination of electron microscopy and X-ray spectroscopy techniques. At the National Tsing Hua University in Taiwan, high-resolution transmission electron microscopy (HRTEM) images were acquired to reveal the crystal structure and surface morphology of the prepared samples. X-Ray diffraction (XRD) spectra were obtained at the BL-01C2 beamline of the National Synchrotron Radiation Research Center (NSRRC) in Taiwan using an incident X-ray with a wavelength of 0.6888 Å (18.0 KeV). The experimental samples underwent X-ray absorption spectroscopy (XAS) at the BL-17C and 01C1 beamlines of NSRRC, Taiwan, and the data were normalized using ATHENA software. For extended X-ray absorption fine structure (EXAFS) analysis, the pre-edge and post-edge backgrounds were subtracted and normalized to the edge jump step from the XAS spectra. The resulting normalized spectra were transformed from energy to k-space and further weighted by k3 to differentiate backscattering interferences from various coordination shells. The backscattered amplitude and phase shift functions for specific atom pairs were theoretically estimated using the FEFF8.0 code. Surface characterization was performed using X-ray photoelectron spectroscopy (XPS) with a Thermo VG Scientific Sigma Probe operated at a voltage of 20 kV and a current of 30 mA. A monochromatic X-ray source (Al Kα) was utilized. XPS analysis aimed to investigate the oxidation states and surface compositions of the experimental samples. The surface compositions were determined by integrating each peak after subtracting the Shirley-type background and fitting the experimental curve using a combination of Lorentzian and Gaussian lines. Accurate binding energies were established by referencing the C 1s peak at 284.6 eV.Electrochemical characterization and gaseous product analysis
The electrochemical measurements were conducted at room temperature utilizing a CH Instruments Model 600B potentiostat (CHI 600B) equipped with a three-electrode system. To prepare the catalyst slurry, 5 mg of catalyst powder was dispersed in 1.0 ml of isopropanol (IPA) and 50 μl of Nafion-117 (99%, Sigma Aldrich Co.) using ultrasonic treatment. For the electrochemical measurements, 10.0 μl of the catalyst slurry was drop-cast and air-dried onto a glassy carbon rotating disk electrode (RDE) with a surface area of 0.196 cm2, serving as the working electrode. A Hg/HgCl2 electrode saturated in KCl aqueous solution was employed as the reference electrode, and a Pt wire was used as the counter electrode. Cyclic voltammetry (CV) curves were recorded with a voltage scan rate of 0.02 V s−1 within a potential range from 0.1 V to 1.3 V (vs. RHE) in a 0.1 M KOH electrolyte (pH 13) saturated with N2. Additionally, for CO-stripping analysis, an Ag/AgCl electrode was utilized as the reference electrode. The adsorption of CO onto the catalyst's surface was achieved by purging CO into 0.5 M H2SO4 at 0.05 V (vs. NHE) for 20 minutes. Subsequently, CO stripping voltammetry was performed in N2-saturated 0.5 M H2SO4 solution, scanning the potential between −0.10 and 1.20 V (vs. NHE) at a rate of 50 mV s−1. The catalytic performance of the experimental samples was evaluated using a previously established protocol.19,26 More details are given in the ESI† (note-1).Results and discussion
The physical structure inspection of CPCu NCs
The HRTEM technique was employed to investigate the crystal structure and surface atomic arrangements of pristine and laser-treated CPCu NCs. Fig. 1a presents the HRTEM image of the pristine CPCu NC, where very few Pd domains are exposed on the surface, denoted in the region (a-1), while the majority of Pd domains are covered by a thin layer of amorphous CuOx, as indicated in region (a-2). The veracity of these findings is substantiated by the application of forward Fourier transform (FFT) and inverse Fourier transform (IFT) techniques, which yielded corroborating patterns and corresponding line histograms. In particular, the presence of ring-like FFT patterns within the inner lattices (a-1) and fuzzy patterns pervading the outer space (a-2) provided compelling evidence for the co-existence of surface-exposed polycrystalline Pd NPs and a thin layer of amorphous CuOx.27,28 Line histograms were used to determine the interlayer spacing (d-spacing) of regions (a-1) and (a-2), which were found to be 0.225 nm and 0.248 nm, respectively. These values correspond to the Pd (111) plane and Cu2O (111) plane, respectively, consistent with the aforementioned observations.29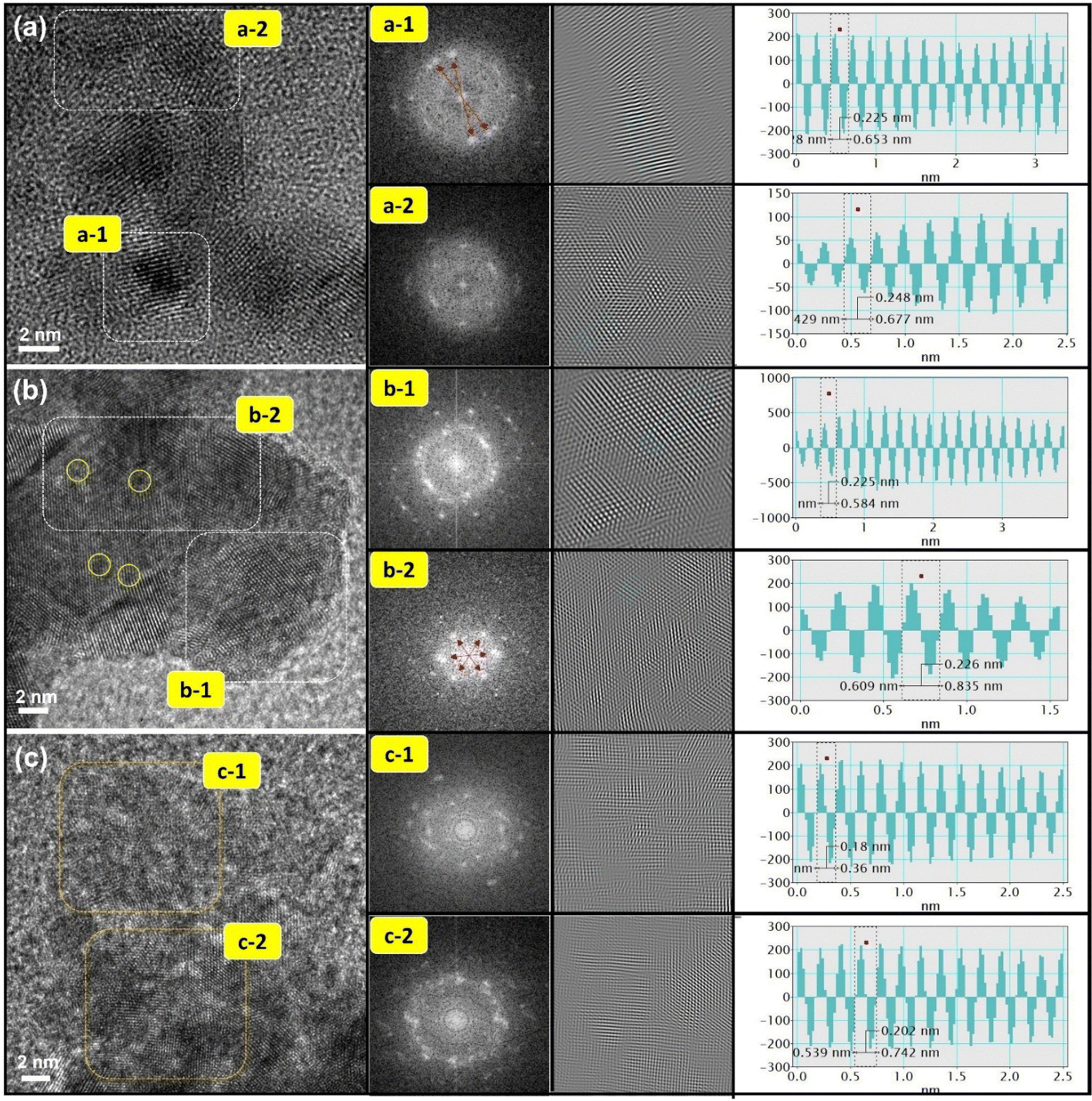 | ||
| Fig. 1 HRTEM images of (a) pristine CPCu, (b) CPCu-1 and (c) CPCu-10 NCs. The forward Fourier transform (FFT) patterns of the selected area in HRTEM images arex shown in the insets. The d-spacing values of experimental samples are calculated by using the inverse Fourier transform (IFT) technique and their corresponding line histograms (insets). | ||
Furthermore, upon exposure of the CPCu NCs to a sub-millisecond laser at an energy of 1 mJ per pulse (i.e., CPCu-1), a significant surface restructuring is observed. Fig. 1b reveals the presence of discrete local domains with clear lattice fringes oriented in different directions, suggesting that the majority of Pd domains are now exposed on the surface, which can be attributed to the removal of the surface amorphous CuOx layer due to laser exposure. More interestingly, it's worth noting that the atomic scale amorphous regions (denoted by yellow circles) are observed on the Pd surface and can be assigned to the atomic CuOx species in the defect sites of the Pd crystals. Such a scenario is obvious due to the less per-pulse energy of 1 mJ, where the atomic migration is limited only to the surface region.26 These findings are corroborated by FFT patterns, which exhibit symmetrically aligned hexagonal bright spots in inner lattices as well as outer space and suggest the formation of long-range ordered structures as a result of surface oxide removal. Furthermore, the CPCu-1 NC shares a nearly similar d-spacing to Pd (111) plane (0.225/0.226 nm) on the whole surface, underscoring the uniform distribution of the adorned CoOx species. Further raising the per-pulse energy to 10 mJ (i.e., CPCu-10) results in a completely different nanoarchitecture (Fig. 1c), where the higher per-pulse energy (10 mJ) induces substantial atomic migration in the sub-surface region as well, leading to the deposition of amorphous CoOx (from support) and CuOx on the surface of Pd NPs. Such an atomic rearrangement results in the formation of a thick unconformable layer of mixed CuOx and CoOx on the Pd NPs. These scenarios are cross-referenced by the fuzzy FFT patterns and severely reduced d-spacings (0.18 nm and 0.202 nm) across the surface. Herein the drastically reduced d-spacing can be attributed to the formation of CoPd and/or CuPd alloys or mixed oxide structures. For further clarification, the HRTEM images of control samples (Pd–AC and Co@Pd) are shown in Fig. S1 (ESI†). Moreover, the X-ray diffraction (XRD) patterns are shown in Fig. S2 (ESI†) to elaborate the crystal structure of CPCu NCs.
X-ray absorption near-edge spectroscopy (XANES) at Cu, Pd and Co K-edges was performed for a comprehensive understanding of the local atomic and electronic structure of the target atoms in the CPCu NCs. Fig. 2a shows the XANES spectra of experimental samples at the Cu K-edge, where the pre-edge “P” and absorption edge “A”, respectively, are assigned to the 1s-to-3d and 1s-to-4s/4p transitions.30 Given that the XANES spectra at the Cu K-edge display three prominent characteristics: the pre-edge “P”, the position of the inflection point (If), and the intensity of the absorption edge (HA). These features provide valuable insights into various aspects related to the target atom. Specifically, they shed light on the local geometry surrounding the target atom, the relative oxidation state or valence compared to a reference sample, and the extent of unoccupied states resulting from electron relocation to neighboring atoms associated with the target atom.31 Accordingly, the inflection point (If) position of CPCu NCs shifted to higher energies as compared to the Cu-foil, confirming that Cu is present in the form of oxide. These scenarios are further confirmed by the first deviation curve of XANES spectra (inset of Fig. 2a), where the peak X* is shifted to a higher energy level similar to the If in the XANES spectra. Moreover, the highest white line intensity (HA) of CPCu-1 NC refers to the highest extent of surface chemisorption on the surface of CPCu-1 NC,27,29,32 which is obvious due to the highest exposure of Cu atoms on the surface. Fig. 2b shows the Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra of CPCu NCs at the Cu K-edge and the corresponding fitting curves are presented in Fig. S3 (ESI†), while the fitting parameters are listed in Table 1. In Fig. 2b, the peaks labelled as “M” and “N”, correspond to Cu–O and Cu–Cu bond pairs, respectively. Additionally, Table 1 confirms the absence of coordination number (CN) for the Cu–Co bond pairs in pristine CPCu and CPCu-1 NCs, while observed in CPCu-10 NC. These findings provide complementary evidence that there is no heteroatomic intermixing between Cu and Co domains in pristine and CPCu-1 NCs, while the heteroatomic intermixing between Cu and Co domains is present in CPCu-10 NC due to the atomic migration triggered by the pulse energy. These results are consistent with former HRTEM observation. Moreover, with the same Cu content, a slightly reduced CN for the Cu–O bond pair refers to a certain extent of Cu oxide decomposition in the laser-treated CPCu-1 and CPCu-10 NCs.
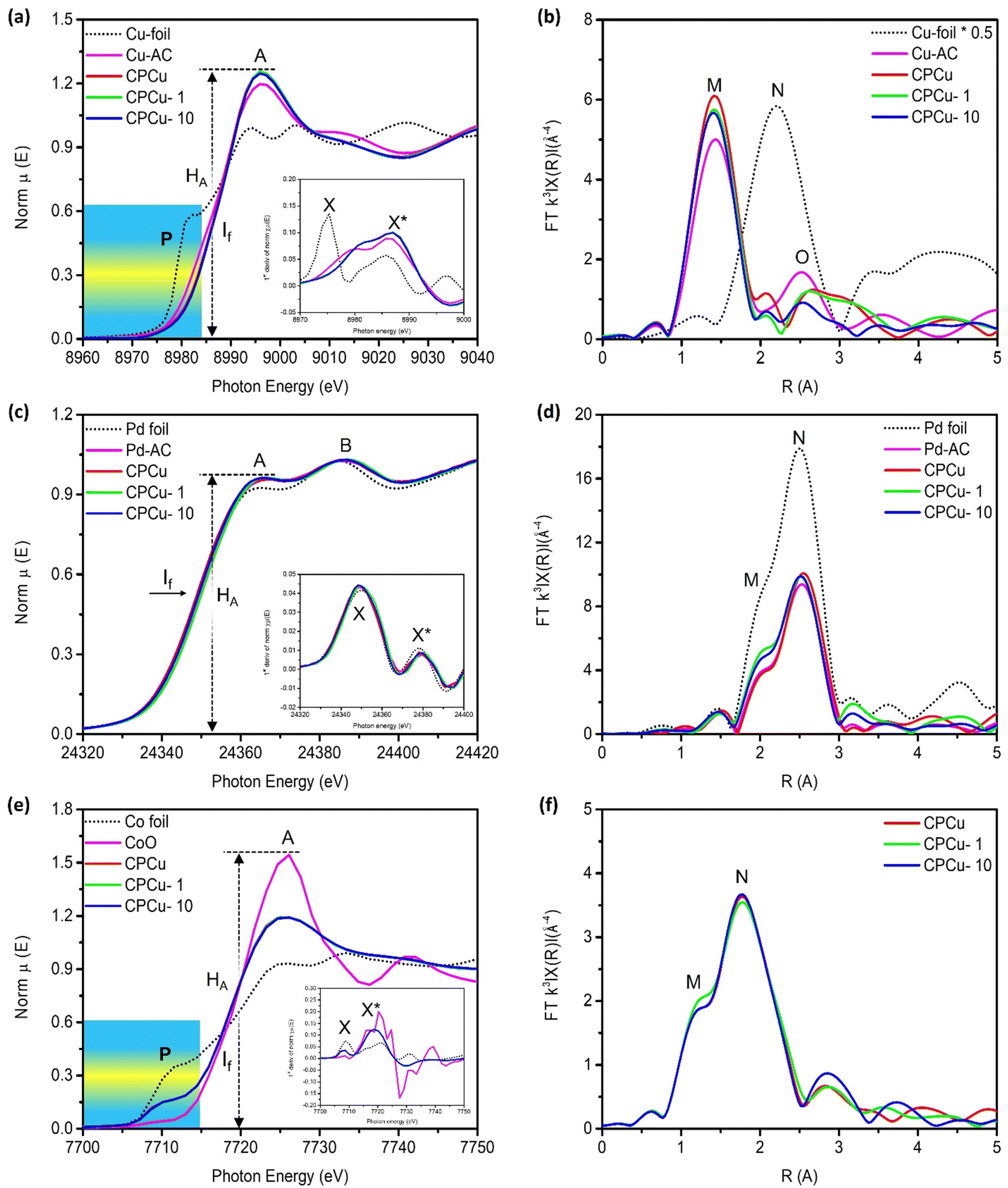 | ||
| Fig. 2 X-Ray absorption spectroscopy of the experimental NCs compared with those of reference samples. (a) XANES and (b) FT-EXAFS spectra of the experimental NCs at the Cu K-edge. (c) XANES and (d) FT-EXAFS spectra of the experimental NCs at the Pd K-edge. (e) XANES and (f) FT-EXAFS spectra of the experimental NCs at the Co K-edge. The first deviation curves of XANES spectra are presented in the insets. | ||
| Sample | Co K-edge | Pd K-edgeb | Cu K-edge | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Bond pair | CN | R | Bond pair | CN | R | Bond pair | CN | R | |
| CPCu | Co–O | 2.88 | 2.043 | Pd–O | 0.70 | 1.967 | Cu–O | 2.19 | 1.924 |
| Co–Cu | N/A | N/A | N/A | N/A | N/A | Cu–Cu | 0.70 | 3.149 | |
| Co–Co | 4.15 | 2.397 | Pd–Cu | 0.95 | 3.260 | Cu–Co | N/A | N/A | |
| Co–Pd | 1.29 | 3.152 | Pd–Pd | 5.06 | 2.739 | Cu–Pd | 0.62 | 3.339 | |
| CPCu-1 | Co–O | 1.92 | 2.044 | Pd–O | 0.62 | 2.052 | Cu–O | 2.13 | 1.923 |
| Co–Cu | 0.65 | 2.558 | N/A | N/A | N/A | Cu–Cu | 0.60 | 3.261 | |
| Co–Co | 4.38 | 2.398 | Pd–Cu | 0.90 | 3.292 | Cu–Co | N/A | N/A | |
| Co–Pd | 2.05 | 3.152 | Pd–Pd | 5.68 | 2.738 | Cu–Pd | 0.63 | 3.338 | |
| CPCu-10 | Co–O | 2.94 | 2.041 | Pd–O | 0.78 | 2.063 | Cu–O | 2.07 | 1.924 |
| Co–Cu | 1.80 | 2.542 | N/A | N/A | N/A | Cu–Cu | 0.61 | 3.192 | |
| Co–Co | 4.20 | 2.398 | Pd–Cu | 0.85 | 3.331 | Cu–Co | 1.2 | 2.899 | |
| Co–Pd | 1.82 | 3.229 | Pd–Pd | 5.58 | 2.739 | Cu–Pd | 0.64 | 3.058 | |
In Fig. 2c, the normalized XANES spectra of CPCu NCs at the Pd K-edge are presented. The absorption peaks labelled as “A” and “B” correspond to the probabilities of electron transition from the 1s orbital to the unoccupied 5p and 4f states, respectively.33 Notably, the inflection point (If) position of the CPCu NCs remains identical to that of the Pd-foil (consistent with peaks X and X* in the first deviation curve of XANES spectra (inset of Fig. 2c)), indicating the preservation of metallic states across all samples. These characteristics are further confirmed by the model simulated fitting results (Fig. S4 (ESI†) and Table 1) of the FT-EXAFS spectra of CPCu NCs at the Pd K-edge (Fig. 2d), where all the samples show higher CN for Pd–Pd bond pairs as compared to Pd–O bond pairs. Normalized Co K-edge XANES spectra of CPCu NCs are shown in Fig. 2e, along with those of standard samples (Co-foil and CoO) for comparison. The standard Co-foil exhibits two prominent features: the pre-edge peak “P” at around 7710 eV, which corresponds to the electric dipole-forbidden but quadrupole-allowed 1s-to-3d transition in a tetrahedral symmetry, and the absorption peak “A”, which is attributed to dipole-allowed 1s-to-4p transitions.34,35 In contrast, the pre-edge peak of standard CoO is much weaker, indicating a predominantly octahedral Co configuration.35 More interestingly, the pre-edge intensity of CPCu NCs lies in between Co-foil and CoO (consistent with peak X in the first deviation curve of XANES spectra (inset of Fig. 2e)), suggesting a distorted tetrahedral Co configuration. With a similar position of inflection point If (i.e. same valence/oxidation state) to CoO and the suppressed white line intensity (HA) could be attributed to the electron relocation or the shielding effects to Co by the neighboring atoms in the CPCu NCs. Fig. 2f illustrates the FT-EXAFS spectra at the Co K-edge. To provide clarity, Fig. S5 (ESI†) presents simulated fitting curves, and the corresponding structural parameters are summarized in Table 1. Accordingly, the presence of both Co–O and Co–Co bond pairs with nearly similar coordination numbers suggests that Co is present in mixed oxide and metallic phases in all CPCu NCs. Consistent with former HRTEM results, the pristine CPCu NC does not show CN for the Co–Cu bond pair, however, it was observed for laser-treated CPCu NCs, confirming the atomic migration.
The X-ray photoelectron spectroscopy (XPS) analysis at Pd-3d, Co-2p and Cu-2p orbitals was performed on experimental samples to investigate the surface chemical composition and binding energies of the constituent elements. Fig. 3a illustrates the comparative XPS spectra of pristine and laser-treated CPCu NCs at the Pd-3d orbitals. In the Pd-3d XPS spectrum, the doublet peaks observed at approximately ∼335.8 eV and ∼341.2 eV correspond to the photoelectron emission response from the Pd-3d5/2 and Pd-3d3/2 orbitals, respectively. It is evident from Fig. 3a that the laser-treated CPCu-1 and CPCu-10 NCs exhibit lower binding energy as compared to that of pristine CPCu NCs at Pd-3d orbitals, while the opposite behaviour is observed in the XPS spectra at the Co-2p orbital, where the laser-treated CPCu-1 and CPCu-10 NCs show higher binding energy as compared to that of pristine CPCu NC (Fig. 3b). These scenarios integrally confirm the severe electron relocation from Co atoms to Pd atoms (consistent with Fig. S6, ESI†). On top of that, the XPS spectra at Co-2p (Fig. 3b) and Cu-2p (Fig. 3c) orbitals confirm the degree of atomic migration and heteroatomic intermixing between Co–Pd–Cu domains in CPCu NCs under the exposure of laser with different per pulse energies. It is evident from Fig. 3b that the intensity of emission peaks in the XPS spectra of Co-2p orbitals increases with increasing per-pulse energies of the incident laser, while a reverse trend has been observed in the XPS spectra of Cu-2p orbitals (Fig. 3c). These scenarios can be attributed to the increasing atomic migration with increasing per-pulse energies and are in good agreement with the HRTEM and XAS analysis, where a higher amount of Cu in the form of amorphous CuOx is observed on the surface of pristine CPCu NCs (Fig. 1a), while the CN for Cu–Co bond pair is absent (Table 1) and therefore the pristine CPCu NC exhibits the highest intensity of emission peaks in the XPS spectra of Cu-2p orbitals and lowest for the Co-2p orbital. Furthermore, when the pristine CPCu NC is exposed to laser irradiation with 1 mJ per pulse energy, the surface CuOx layer is removed (consistently confirmed by the HRTEM and XAS analysis) and therefore the intensity for the emission peaks in the XPS spectra of Cu-2p orbitals for CPCu-1mJ NC is suppressed. Further raising the per pulse energy to 10 mJ leads to the higher order of atomic migration and therefore the Co atoms are observed on the surface (consistent with the CN analysis of EXAFS spectra at Cu K-edge, where CN for the Cu–Co bond pair is 1.2 for CPCu-10 NC), hence the intensity for the emission peaks in the XPS spectra of the Co-2p orbital is increased.
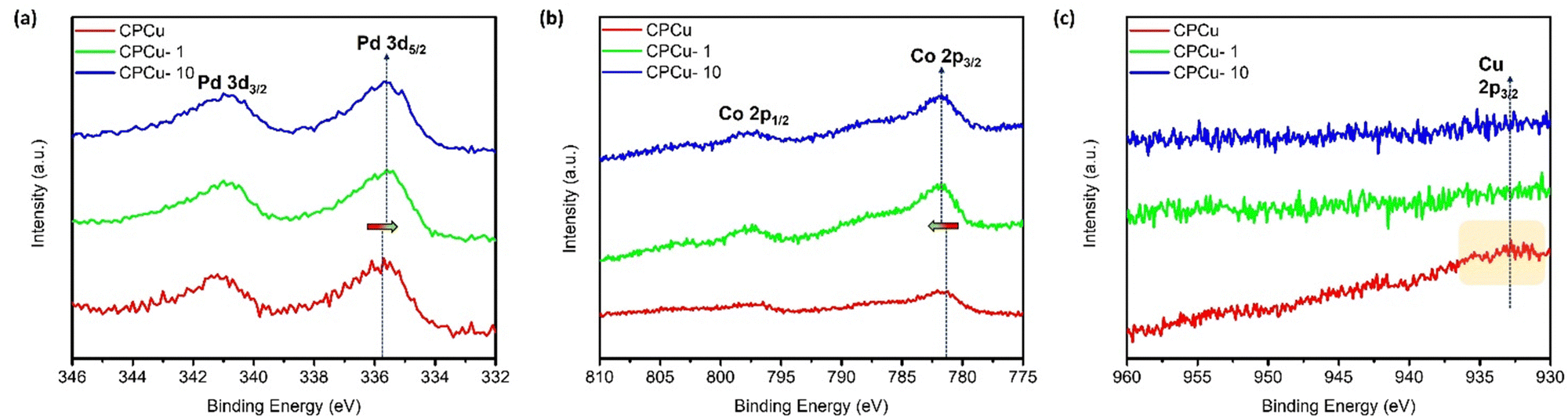 | ||
| Fig. 3 X-ray photoelectron spectroscopy of experimental NCs at (a) Pd-3d, (b) Co-2p and (c) Cu-2p orbitals of CPCu NCs in pristine and post-laser conditions. | ||
The surface chemical compositions of the experimental NCs were elucidated via CO-stripping and cyclic voltammetry (CV) analyses. Fig. 4a illustrates the CO-stripping curves of pristine CPCu NC in comparison to laser-treated ones and reference samples. The relative position of the CO oxidation peak within a CO-stripping curve generally serves as an indicator of the potential necessary for CO oxidation.36 Simultaneously, the extent of the area underneath the CO-oxidation peak corresponds to the density of active sites on the surface that are occupied by CO chemisorption.36 Accordingly, the current responses observed for bare Co-NPs (specifically, Co oxide) display a flattened behavior, indicating their inertness towards CO molecules. Moreover, the unmodified Pd NPs exhibit a distinct and pronounced CO-oxidation peak (A) at the highest potential, approximately 0.957 V vs. NHE, corresponding to the densely packed (111) facet. Notably, no CO-oxidation peak is observed at lower potentials. It is important to note that since all the experimental samples in this particular study were fabricated on the same carbon support, any electronic effects such as residual capacitance and interface contact can be disregarded as potential factors. Consequently, these collective observations strongly indicate that the surface of the Pd NPs possesses the highest energy barrier for CO oxidation and a lower surface selectivity between the open and closed facets of the Pd crystal. In the case of Co@Pd, a slight deviation of the primary CO oxidation peak by – 0.01 V, denoted as B (0.947 V vs. NHE), indicates a decreased energy barrier for CO oxidation compared to Pd NPs. This phenomenon can be attributed to electron localization from Co to Pd atoms due to their electronegativity difference and lattice mismatch. This observation aligns consistently with the XPS spectra of Co@Pd, as depicted in Fig. S6 (ESI†). Furthermore, the presence of an additional peak, labelled as B* (0.887 V vs. NHE.), signifies CO oxidation occurring at sites with lower energy barriers, such as the heterogeneous Co-Pd interface or CoPd alloys, resulting from limited intermixing between different atomic species. Simultaneously, the suppression of the CO-oxidation peak can be attributed to the coverage of Pd sites by a thin layer of amorphous CoOx due to the galvanic replacement reaction involving Co atoms and Pd2+ ions (Co0 + Pd2+ → Co3+ + Pd0) accompanied by re-deposition of remaining Pd2+ and Co3+ ions. This phenomenon is consistently supported by the flattened CO-stripping curve of Co NPs, indicating the inert behavior of CoOx towards CO molecules. Furthermore, a notable decrease in the intensity of the main CO-oxidation peak is observed for pristine CPCu NC and can be attributed to the surface coverage of Pd sites by a thin layer of amorphous CuOx, which is in good agreement with the HRTEM findings. Meanwhile, a slight shift of −0.002 V, indicates a lower energy barrier for CO oxidation on the CPCo-1 surface when compared to Co@Pd. Besides, the CPCu-1 NC exhibits a significantly intense main CO-oxidation (D) as compared to that of pristine CPCu NC, indicating the re-exposure of surface active Pd due to the removal of the surface oxide layer. On top of that, the lowest peak potential (0.927 V vs. NHE.) and highest peak area, respectively, suggest the lowest energy barrier for CO-oxidation and the highest density of active sites on the surface of CPCu-1 NC. These scenarios confirm that surface-decorated atomic CuOx species create additional active sites on the surface of CPCu-1 NC. Furthermore, when the per-pulse energy of the sub-millisecond laser beam is increased to 10 mJ, the main CO oxidation is shifted to a higher potential as compared to CPCu-1 NC, which is obvious and can be attributed to the additional surface coverage of Pd sites by CoOx species (from the support). Moreover, the presence of an additional peak (E) reveals the formation of CoPd or CuPd alloys due to the higher atomic migrations. These results are in good agreement with former HRTEM observations.
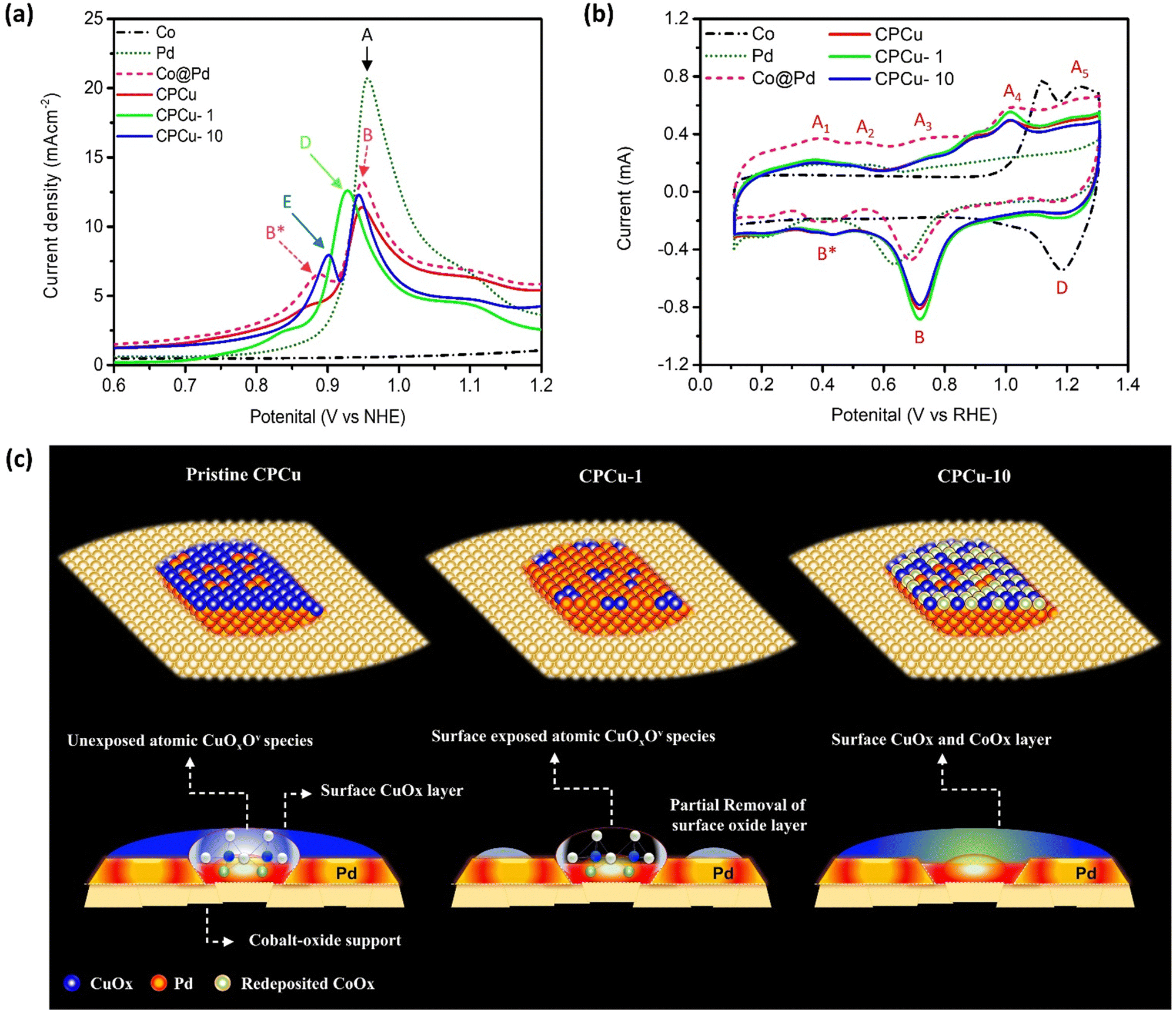 | ||
| Fig. 4 (a) CO-stripping and (b) cyclic voltammetry curves of the as-prepared CPCu, CPCu-1 and CPCu-10 NCs compared with reference samples (Co, Pd and Co@Pd). (c) The schematic representation for the atomic structures of pristine CPCu, CPCu-1 and CPCu-10 nanocatalysts. | ||
In accordance with Fig. 4b, the dashed black curve representing carbon-supported Co NPs exhibits a flattened profile during the under-potential deposition of hydrogen (HUPD) and in the double layer regions (ranging from 0.1 to 0.8 V vs. RHE). This behavior indicates the formation of a chemically inert Co oxide surface, rendering the Co NPs nearly inactive towards the intermediate species. On the other hand, the two peaks (A4 and A5) in the forward sweep and peak D in the reverse sweep correspond to the adsorption and desorption of oxygen species, such as OH−. Furthermore, the Pd NPs demonstrate a smeared peak profile in the HUPD region, ranging from 0.1 to 0.4 V vs. RHE, which is a typical characteristic of metallic Pd with a high affinity for proton (H+) adsorption. Additionally, a broad peak (B) at 0.637 V vs. RHE in the backward sweep, corresponds to the desorption of adsorbed oxygen (Oads) from metallic Pd. Regarding Co@Pd, the current peaks A1, A2, and A3 observed in the forward sweep are attributed to the formation of oxides on the surface involving different combinations of Co and Pd. In the backward sweep, a broad peak B* ranging from 0.53 to 0.3 V vs. RHE can be assigned to the desorption of Oads from these reaction sites. Additionally, a distinct peak “B” is evident in the backward sweep, corresponding to the reduction of oxide species on the Pd surface. Comparatively, when compared to Pd NPs, the significant offset of peak “B” indicates a weakening of Oads. Of utmost importance, considering the behavior of Co@Pd, it exhibits a similar peak profile to Pd NPs in the HUPD region and to Co NPs at higher potentials, characterized by the presence of current peaks A4 and A5 in the forward sweep. This suggests the formation of Pd NPs over the Co-oxide support. Furthermore, a particularly intriguing finding is that in comparison to Co@Pd, the CPCu NCs exhibit suppressed current peaks in the forward sweep, while the backward sweep exhibits higher onset potential for peaks B and B*, which is the highest for CPCu-1 NC. These observations serve to confirm an elevated affinity for oxygen species, indicating the presence of CuOx species with various oxidation states by the Co intermix on the surface of Pd NPs within the CPCu NCs. Consequently, these findings strongly imply an enhancement in the kinetics of CO2 dissociation, which holds promising implications for related processes. The proposed atomic structures of pristine CPCu, CPCu-1, and CPCu-10 NCs are presented in Fig. 4c, based on a comprehensive analysis that incorporates the results obtained from physical and electrochemical characterization techniques. By carefully cross-referencing these outcomes, we have arrived at a detailed understanding of the atomic configurations of the aforementioned catalysts. It is important to note that the following representations depict the structural arrangements derived from the experimental data and serve as a visual guide for further analysis and discussion.
The effect of laser treatment on the CO2 conversion performance of the CPCu NCs has been assessed in a flow reactor system operating at atmospheric pressure under a flowing H2/CO2 (3/1) atmosphere, across a temperature range from room temperature (RT) to 300 °C. Fig. 5a and b display the CO and CH4 productivity, respectively, for the CPCu NCs. It can be observed that the pristine CPCu NC exhibits higher CO productivity (Fig. 5a) as compared to that of CH4 (Fig. 5b) in the higher temperature range (250–300 °C), indicating that the RWGS reaction dominates CO2 methanation. However, the laser-treated CPCu-1 NC significantly suppresses the competitive RWGS reaction and thus the CO productivity decreases from ∼14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 120 mmol g−1 h−1 to ∼8912 mmol g−1 h−1 at 300 °C. Besides the CH4 productivity for CPCu-1 NC is improved from ∼1164 mmol g−1 h−1 to ∼1345 mmol g−1 h−1, which is 13.60% enhanced as compared to that of pristine CPCu NC. Such an improvement in CH4 productivity can be attributed to the potential synergism between adjacent reaction sites of Pd and atomic CuOx species, which, respectively, promote H2 splitting and CO2 dissociation. These results suggest that the surface atomic arrangement plays a crucial role in the CO2 methanation process. Meanwhile, the significantly suppressed CO2 conversion performance of CPCu-3 NC can be rationalized to the insufficient surface reaction sites for H2 dissociation to the presence of a thick oxide layer on the surface. Furthermore, the CO2RR results of control samples in pure CO2 ambient as well as in reaction gas are shown in Fig. S7 (ESI†) for reference. More importantly, the activation energy for CO (EaCO) and CH4 (EaCH4) production are summarized in Table S1 (ESI†).
120 mmol g−1 h−1 to ∼8912 mmol g−1 h−1 at 300 °C. Besides the CH4 productivity for CPCu-1 NC is improved from ∼1164 mmol g−1 h−1 to ∼1345 mmol g−1 h−1, which is 13.60% enhanced as compared to that of pristine CPCu NC. Such an improvement in CH4 productivity can be attributed to the potential synergism between adjacent reaction sites of Pd and atomic CuOx species, which, respectively, promote H2 splitting and CO2 dissociation. These results suggest that the surface atomic arrangement plays a crucial role in the CO2 methanation process. Meanwhile, the significantly suppressed CO2 conversion performance of CPCu-3 NC can be rationalized to the insufficient surface reaction sites for H2 dissociation to the presence of a thick oxide layer on the surface. Furthermore, the CO2RR results of control samples in pure CO2 ambient as well as in reaction gas are shown in Fig. S7 (ESI†) for reference. More importantly, the activation energy for CO (EaCO) and CH4 (EaCH4) production are summarized in Table S1 (ESI†).
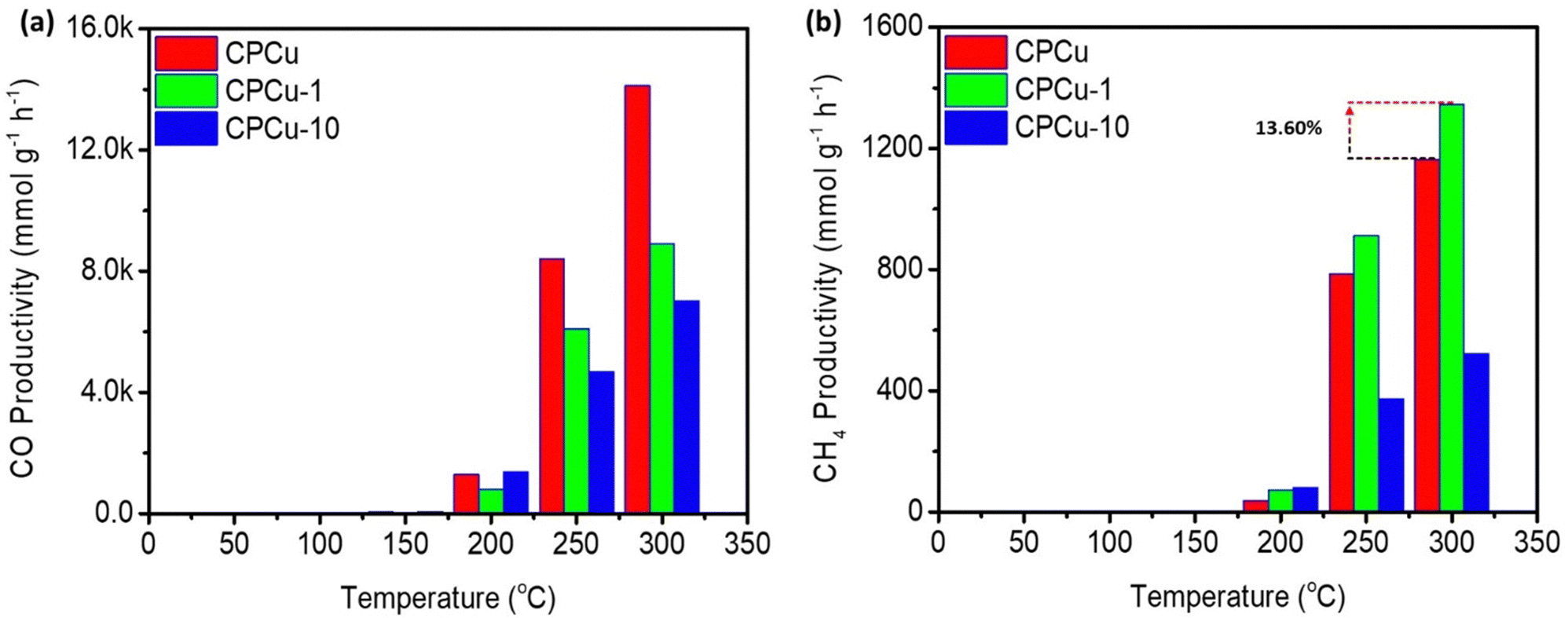 | ||
| Fig. 5 Gas chromatography (GC) determined CO2RR results for the CPCu NCs for (a) CO and (b) CH4 productivity in the reaction gas (CO2 + 3H2). | ||
Conclusions
We have demonstrated a novel approach for a facile surface restructuring of multimetallic nanocatalysts for various applications. To demonstrate such a concept, we have developed a tri-metallic nanocatalyst called CPCu, which consists of Co incorporated CuOx clusters in the CoOx supported Pd nanoparticles (NPs). To enhance the performance of CO2 methanation, we employed sub-millisecond pulsed laser irradiation at two different pulse energy levels (1 mJ and 10 mJ) for a fixed duration of 10 seconds to modify the surface and sub-surface atomic arrangements of the as-prepared CPCu NCs. Among the different conditions tested, we found that the optimum case, denoted as CPCu-1, achieved the highest CH4 productivity. At a temperature of 300 °C, CPCu-1 exhibited an optimal CH4 productivity of ∼1346 mmol g−1 h−1, which is a 13.6% improvement compared to the pristine condition (∼1164 mmol g−1 h−1). Additionally, the CH4 selectivity of CPCu-1 NC was enhanced by 40% compared to the as-prepared condition. The results obtained from various physical characterization studies and electrochemical analyses suggest that the improved activity and selectivity of CPCu-1 NC can be attributed to the significant surface restructuring caused by the sub-millisecond pulsed laser irradiation. Specifically, the high density of surface-exposed CuOx species and neighboring Pd sites on CPCu-1 NC promote the activation of CO2 and the dissociation of H2 during the CO2 methanation process. These findings provide valuable insights into the design of high-performance catalytic materials for CO2 methanation through the utilization of sub-millisecond pulsed laser irradiation. By harnessing the surface restructuring effects induced by this technique, we can enhance the efficiency and selectivity of catalysts, thereby contributing to the development of effective CO2 conversion processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to the staff of the National Synchrotron Radiation Research Center (NSRRC), Hsinchu, Taiwan for help with various synchrotron-based measurements. T.-Y. Chen acknowledges the funding support from the National Science and Technology Council, Taiwan (NSTC 109-2923-E-007-005, NSTC 109-3116-F-007-001, NSTC 109-2112-M-007-030-MY3 and NSTC 112-2112-M-007-026) and MA-Tek (2023-T-004) and Taiwan Space Agency (TASA-S-1120691). P.-C. Chen acknowledges the funding support from the National Science and Technology Council, Taiwan (NSTC 110-2221-E-027-022-MY3). In addition, the authors acknowledge the Precision Research and Analysis Centre at NTUT.References
- Z. Liu, X. Gao, B. Liu, Q. Ma, T.-S. Zhao and J. Zhang, Fuel, 2022, 321, 124115 CrossRef CAS.
- X. Gao, Z. Wang, Q. Huang, M. Jiang, S. Askari, N. Dewangan and S. Kawi, Catal. Today, 2022, 402, 88–103 CrossRef CAS.
- A. Tripodi, F. Conte and I. Rossetti, Energy Fuels, 2020, 34, 7242–7256 CrossRef CAS.
- R. Zhang, A. Wei, M. Zhu, X. Wu, H. Wang, X. Zhu and Q. Ge, J. CO2 Util., 2021, 52, 101678 CrossRef CAS.
- H. C. Wu, Y. C. Chang, J. H. Wu, J. H. Lin, I. K. Lin and C. S. Chen, Catal. Sci. Technol., 2015, 5, 4154–4163 RSC.
- E. Meloni, L. Cafiero, S. Renda, M. Martino, M. Pierro and V. Palma, Catalysts, 2023, 13, 488 CrossRef CAS.
- Y. Jiang, J. Lang, X. Wu and Y. H. Hu, Catal. Today, 2020, 356, 570–578 CrossRef CAS.
- A. Porta, L. Falbo, C. G. Visconti, L. Lietti, C. Bassano and P. Deiana, Catal. Today, 2020, 343, 38–47 CrossRef CAS.
- S. L. Rodríguez, A. Davó-Quiñonero, J. Juan-Juan, E. Bailón-García, D. Lozano-Castelló and A. Bueno-López, J. Phys. Chem. C, 2021, 125, 12038–12049 CrossRef PubMed.
- R. Chen, L. Shen, W. Zhang, Y.-F. Han, Z. Yang and M. Zhu, Greenhouse Gases: Sci. Technol., 2023, 13, 396–408 CrossRef CAS.
- C. Yan, C.-H. Wang, M. Lin, D. Bhalothia, S.-S. Yang, G.-J. Fan, J.-L. Wang, T.-S. Chan, Y.-L. Wang, X. Tu, S. Dai, K.-W. Wang, J.-H. He and T.-Y. Chen, J. Mater. Chem. A, 2020, 8, 12744–12756 RSC.
- F. Hu, R. Ye, Z.-H. Lu, R. Zhang and G. Feng, Energy Fuels, 2022, 36, 156–169 CrossRef CAS.
- M. Zhu, P. Tian, X. Cao, J. Chen, T. Pu, B. Shi, J. Xu, J. Moon, Z. Wu and Y.-F. Han, Appl. Catal., B, 2021, 282, 119561 CrossRef CAS.
- A. I. Tsiotsias, N. D. Charisiou, I. V. Yentekakis and M. A. Goula, Nanomaterials, 2021, 11, 28 CrossRef CAS PubMed.
- S. Abate, C. Mebrahtu, E. Giglio, F. Deorsola, S. Bensaid, S. Perathoner, R. Pirone and G. Centi, Ind. Eng. Chem. Res., 2016, 55, 4451–4460 CrossRef CAS.
- C. Yan, D. Bhalothia, S.-S. Yang, A. Beniwal, Y.-X. Chang, P.-C. Wang, Y.-C. Cheng, C.-L. Chen, S.-C. Wu and T.-Y. Chen, Catalysts, 2022, 12, 1127 CrossRef CAS.
- P. Panagiotopoulou, D. I. Kondarides and X. E. Verykios, J. Phys. Chem. C, 2011, 115, 1220–1230 CrossRef CAS.
- Q. Pan, J. Peng, T. Sun, S. Wang and S. Wang, Catal. Commun., 2014, 45, 74–78 CrossRef CAS.
- D. Bhalothia, S.-S. Yang, C. Yan, A. Beniwal, Y.-X. Chang, S.-C. Wu, P.-C. Chen, K.-W. Wang and T.-Y. Chen, Sustainable Energy Fuels, 2023, 7, 526–536 RSC.
- S. A. Al–Fatish, A. A. Ibrahim, A. H. Fakeeha, M. A. Soliman, M. R. H. Siddiqui and A. E. Abasaeed, Appl. Catal., A, 2009, 364, 150–155 CrossRef.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2094–2100 CrossRef CAS.
- P. K. Saravanan, D. Bhalothia, G.-H. Huang, A. Beniwal, M. Cheng, Y.-C. Chao, M.-W. Lin, P.-C. Chen and T.-Y. Chen, Nanomaterials, 2023, 13, 1801 CrossRef CAS PubMed.
- H. Jiang, Q. Gao, S. Wang, Y. Chen and M. Zhang, J. CO2 Util., 2019, 31, 167–172 CrossRef CAS.
- D. Bhalothia, C.-Y. Lin, C. Yan, Y.-T. Yang and T.-Y. Chen, ACS Omega, 2019, 4, 971–982 CrossRef CAS PubMed.
- R. C. Forsythe, C. P. Cox, M. K. Wilsey and A. M. Müller, Chem. Rev., 2021, 121, 7568–7637 CrossRef CAS PubMed.
- D. Bhalothia, W.-H. Hsiung, S.-S. Yang, C. Yan, P.-C. Chen, T.-H. Lin, S.-C. Wu, P.-C. Chen, K.-W. Wang, M.-W. Lin and T.-Y. Chen, ACS Appl. Energy Mater., 2021, 4, 14043–14058 CrossRef CAS.
- T. Yang, D. Bhalothia, H.-W. Chang, C. Yan, A. Beniwal, Y.-X. Chang, S.-C. Wu, P.-C. Chen, K.-W. Wang, S. Dai and T.-Y. Chen, Chem. Eng. J., 2023, 454, 140289 CrossRef CAS.
- D. Bhalothia, J.-P. Chou, C. Yan, A. Hu, Y.-T. Yang and T.-Y. Chen, ACS Omega, 2018, 3, 8733–8744 CrossRef CAS PubMed.
- D. Bhalothia, C. Yan, N. Hiraoka, H. Ishii, Y.-F. Liao, P.-C. Chen, K.-W. Wang, J.-P. Chou, S. Dai and T.-Y. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 16177–16188 CrossRef CAS PubMed.
- J. L. DuBois, P. Mukherjee, A. M. Collier, J. M. Mayer, E. I. Solomon, B. Hedman, T. D. P. Stack and K. O. Hodgson, J. Am. Chem. Soc., 1997, 119, 8578–8579 CrossRef CAS.
- D. Bhalothia, L. Shuan, Y.-J. Wu, C. Yan, K.-W. Wang and T.-Y. Chen, Sustainable Energy Fuels, 2020, 4, 2541–2550 RSC.
- S. Dai, J.-P. Chou, K.-W. Wang, Y.-Y. Hsu, A. Hu, X. Pan and T.-Y. Chen, Nat. Commun., 2019, 10, 440 CrossRef CAS PubMed.
- D. Bhalothia, D.-L. Tsai, S.-P. Wang, C. Yan, T.-S. Chan, K.-W. Wang, T.-Y. Chen and P.-C. Chen, J. Alloys Compd., 2020, 844, 156160 CrossRef CAS.
- C. Sarkar, P. Koley, I. Shown, J. Lee, Y.-F. Liao, K. An, J. Tardio, L. Nakka, K.-H. Chen and J. Mondal, ACS Sustainable Chem. Eng., 2019, 7, 10349–10362 CrossRef CAS.
- M. Wu, G. Zhang, H. Tong, X. Liu, L. Du, N. Chen, J. Wang, T. Sun, T. Regier and S. Sun, Nano Energy, 2021, 79, 105409 CrossRef CAS.
- K.-W. Wang, Z. Yu, A. Hu, Y.-Y. Hsu, T.-L. Chen, C.-Y. Lin, C.-W. Hu, Y.-T. Yang and T.-Y. Chen, RSC Adv., 2017, 7, 55110–55120 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00663h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |