
Synthesis and structure–activity relationship (SAR) studies of 1,2,3-triazole, amide, and ester-based benzothiazole derivatives as potential molecular probes for tau protein†
Hendris
Wongso
 abc,
Maiko
Ono
d,
Tomoteru
Yamasaki
e,
Katsushi
Kumata
e,
Makoto
Higuchi
d,
Ming-Rong
Zhang
e,
Michael J.
Fulham
f,
Andrew
Katsifis
*f and
Paul A.
Keller
*a
abc,
Maiko
Ono
d,
Tomoteru
Yamasaki
e,
Katsushi
Kumata
e,
Makoto
Higuchi
d,
Ming-Rong
Zhang
e,
Michael J.
Fulham
f,
Andrew
Katsifis
*f and
Paul A.
Keller
*a
aSchool of Chemistry and Molecular Bioscience, Molecular Horizons, University of Wollongong, Wollongong, NSW 2522, Australia. E-mail: keller@uow.edu.au
bResearch Center for Radioisotope, Radiopharmaceutical, and Biodosimetry Technology, National Research and Innovation Agency, Puspiptek, Banten 15314, Indonesia
cResearch Collaboration Center for Theranostic Radiopharmaceuticals, National Research and Innovation Agency, Sumedang 45363, Indonesia
dDepartment of Functional Brain Imaging, National Institutes for Quantum Science and Technology, Chiba 263-8555, Japan
eDepartment of Advanced Nuclear Medicine Sciences, National Institutes for Quantum Science and Technology, Chiba 263-8555, Japan
fDepartment of PET and Nuclear Medicine, Royal Prince Alfred Hospital, Camperdown, NSW 2050, Australia
First published on 27th January 2023
Abstract
The pyridinyl-butadienyl-benzothiazole (PBB3 15) scaffold was used to develop tau ligands with improved in vitro and in vivo properties for imaging applications to provide insights into the etiology and characteristics of Alzheimer's disease. The photoisomerisable trans-butadiene bridge of PBB3 was replaced with 1,2,3-triazole, amide, and ester moieties and in vitro fluorescence staining studies revealed that triazole derivatives showed good visualisation of Aβ plaques, but failed to detect the neurofibrillary tangles (NFTs) in human brain sections. However, NFTs could be observed using the amide 110 and ester 129. Furthermore, the ligands showed low to high affinities (Ki = >1.5 mM–0.46 nM) at the shared binding site(s) with PBB3.
Introduction
Recent data indicate that Alzheimer's disease (AD) and other types of dementia are affecting approximately 35 million people worldwide.1–3 AD is associated with cognitive decline, including spatial disorientation, memory loss,4 language impairment, mood swings, and behavioural issues.5 Although the underlying cause of AD is still unknown,6 accumulating evidence suggests multifactorial factors instead of a single cause.Typical pathological hallmarks found in the brains of individuals with AD include the formation of extracellular amyloid plaques and phosphorylated tau fibrils. Overproduction and accumulation of amyloid in the brain has been considered as a major contributor to the pathogenic processes that can lead to early-onset AD. Alternatively, an imbalance between Aβ production and clearance may contribute to late-onset sporadic AD.7 Recent studies suggest that dysfunction in Aβ clearance in the central nervous system due to aging promotes the accumulation, aggregation, and deposition of Aβ1-40 and Aβ1-42, indicating that amyloid clearance may be clinically important in the development of AD.8
Hyperphosphorylated tau protein, or neurofibrillary tangle (NFT), is also a hallmark of AD pathology. The basic role of tau proteins is believed to be stabilisation of the microtubule cell cytoskeleton presenting as six different isoforms each comprising either 3-repeat units (3R) or 4-repeat units (4R) with different forms found in different tauopathies. In pathogenic conditions, amyloid plaques along with neuroinflammation lead to hyperphosphorylation of tau proteins, resulting in damage to the microtubules, affecting cell function and viability.9 It has been recognised that the density of amyloid plaques does not correlate with disease progression or cognitive impairment in AD. In contrast to the amyloid pathology, the density and neocortical spread of NFTs correlate well with progressive neuronal degeneration and cognitive decline in AD patients, thus making the non-invasive detection and measurement of NFTs using external imaging a desirable biomarker for AD.4 NFTs or paired helical filaments of tau protein are found within neurons whereas aggregates of amyloid-b protein (Ab) which are the constituents of plaques are distributed in the extracellular space.
During the last decade, several positron emission tomography (PET) probes have been developed for the in vivo detection of β-amyloid pathology, including [11C]-Pittsburgh compound B ([11C]PiB) 1, [11C]SB13 2, [18F]BAY94-9172 (florbetaben or florpyramine) 3, [18F]AV-45 (florbetapir) 4, [18F]flutemetamol 5, [18F]AZD4694 6, and [11C]AZD2184 7 (Fig. 1).10
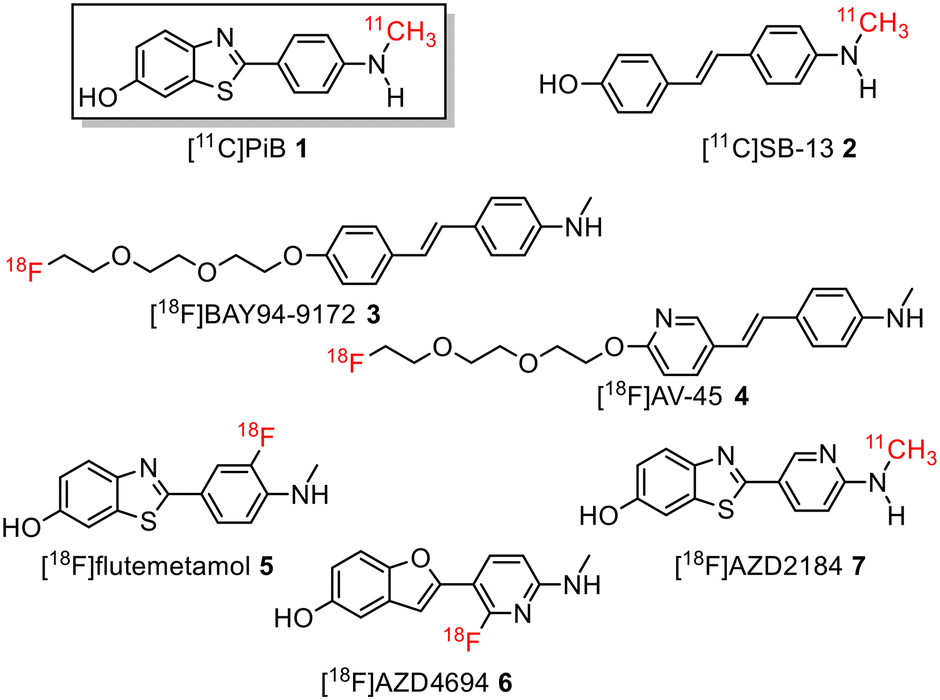 | ||
| Fig. 1 Structure of 11C-labelled and 18F-labelled radiotracers for imaging Aβ-plaques in Alzheimer's disease patients. | ||
The development of tau tracers with high specificity has lagged behind its amyloid counterparts. This is because tau is homologous in structure with β-amyloid, but is present in lower concentrations, thus requiring tau ligands to have selectivity (20–50 fold affinity) for tau over β-amyloid.11–13 Consequently, discovery efforts have translated into the development of small molecule PET imaging radiotracers with high binding affinity to NFTs and selectivity over β-amyloid.12 A number of PET tracers targeting aggregated forms of tau have been identified including first-generation tracers: [18F]FDDNP 8,14 [18F]THK5117 9, [18F]THK5351 10,15 [18F]-T807 (flortaucipir) (AV-1451) 11, [18F]-T808 (AV-680) 12 (Fig. 2)10 and PBB1–PBB5 13–17 (Fig. 3);16 and second-generation tracers: [18F]RO-948 18,17 [18F]MK6240 19,12 and [18F]JNJ311 20,18 and [18F]PM-PBB3 (aka APN-1607) 21 (Fig. 4).19
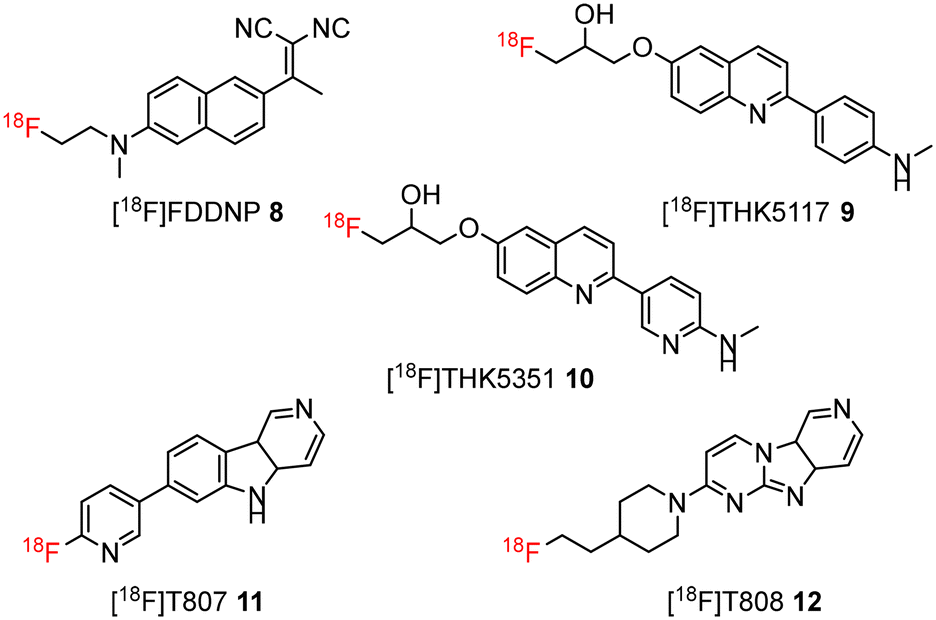 | ||
| Fig. 2 Structure of first-generation tau radiotracers. | ||
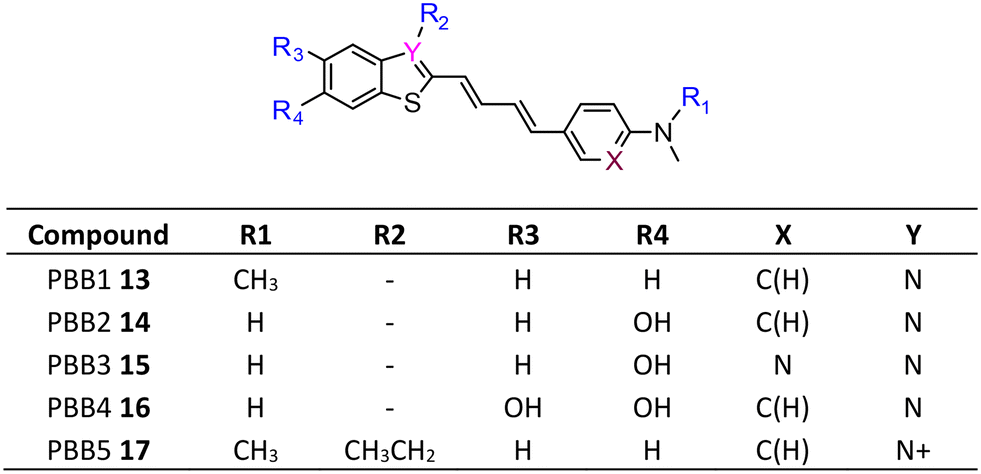 | ||
| Fig. 3 Structures and design of PBB1–PBB5 for labelling with carbon-11. | ||
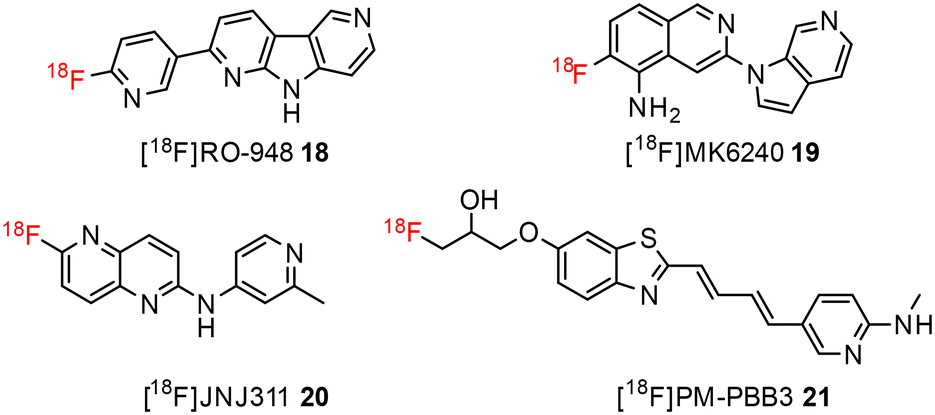 | ||
| Fig. 4 Structure of second-generation tau radiotracers. | ||
Recently a number of novel tau PET tracers based on different scaffolds have been reported.20 Many of these have limitations, including off-target binding, poor specificity, and low stability which hinder their potential in the clinical settings.21,22
[11C]PBB3 15 is used clinically for the in vivo detection of tau deposits in AD and non-AD tauopathies.23 Recent clinical studies showed that it differentiated tau in AD brains from healthy control brains, and specific binding was observed in the CA1 and subiculum areas in the hippocampus where a high accumulation of fibrillar tau protein exists.17
Despite the favourable in vivo properties, PBB3 15 incorporates a photoisomerisable trans-butadiene between the aminopyridine and the benzothiazole moieties. It was also found that only the (E,E)-isomers was active in binding to tau.22 Although, photoisomerisation can be entirely blocked using a UV-free LED light in the radiosynthesis and administration to subjects,22 the trans butadiene moiety of PBB3 15 provided an excellent structural platform for further tau ligand development. In this project, we focused on generating a new series of tau ligands by replacing the trans-butadiene bridge of PBB3 15 with 1,2,3-triazole, amide, and ester moieties (Fig. 5), with the aim of finding molecules with better stability whilst retaining its favourable in vivo properties. It was envisaged that a triazole would result in an inflexible linker whereas an amide and ester linker would result in a partially restricted linkage, thus generating a range of conformationally restricted examples. Furthermore, the latter amide and ester linkages attached to the benzothiazole could resemble the sulfoxide oxygen characteristic of the tau ligand [18F]N-methyllansoprazole.
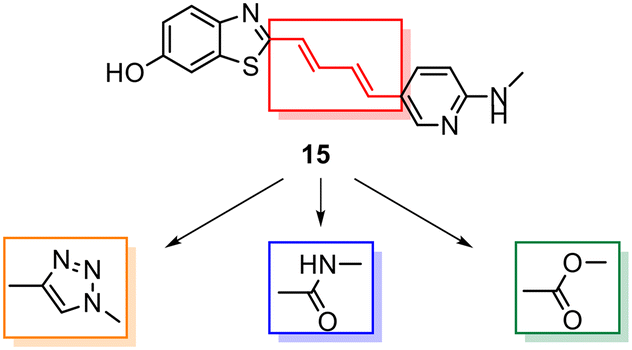 | ||
| Fig. 5 Strategy for the development of novel tau ligands through modification of the trans-butadiene bridge on PBB3 15. | ||
We report the preliminary data on the synthesis, structure–activity relationship (SAR) studies and biological evaluation (binding properties and fluorescence study) of 1,2,3-triazole, amide, and ester-based benzothiazole derivatives as potential ligands for detection of tau protein.
Results and discussion
Chemistry
The core structure of PBB3 15 consists of two building blocks; a benzothiazole and a pyridine connected by a trans-butadiene bridge. Replacing this trans-butadiene bridge with other bridging moieties provided scope for structural modifications between the benzothiazole region, and pyridine while offering greater photostability, through a variety of substitution patterns. The key component in our synthetic strategy of the target molecules was the incorporation of 1,4-disubstituted 1,2,3-triazole derivatives prepared by a Cu(I)-catalyzed azide–alkyne cycloaddition (click reaction) using appropriately substituted substrates.The synthesis of alkyne 25 for the first target was accomplished using a reported procedure24 with minor modifications (Scheme 1). Diazotization of the commercially available 6-methoxybenzo[d]thiazol-2-amine 22, followed by Sandmeyer iodination gave the iodobenzothiazole 23 in 70% yield, which was subjected to a Sonogashira cross-coupling reaction in the presence of 2.0 equivalents of ethynyltrimethylsilane to generate the protected alkyne 24 in 66% yield. Deprotection of the protected alkyne 24 with KF in MeOH at rt for 4 h to give the free alkyne 25 in 98% yield.
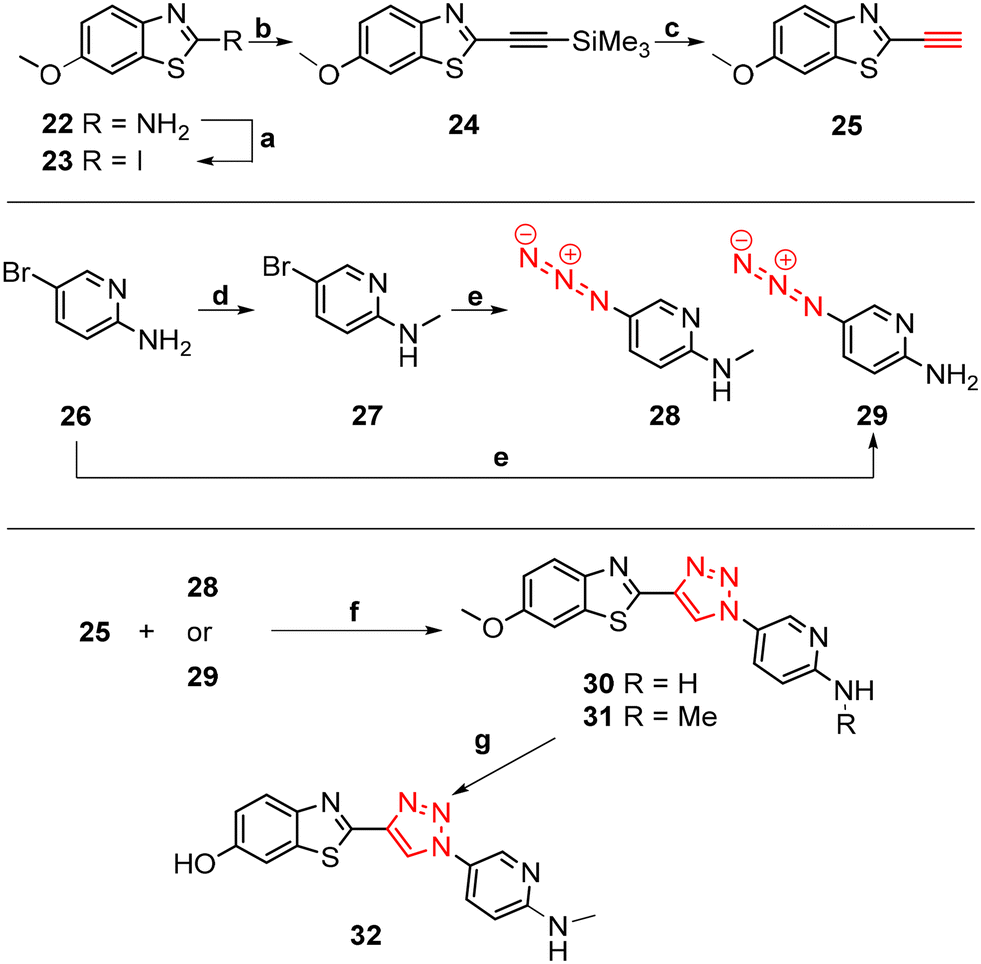 | ||
Scheme 1 Synthesis of triazole derivatives 30 and 31. Reagents and conditions: (a) p-TsOH·H2O, NaNO2, KI, MeCN/H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 0 °C – rt, 18 h, 70%. (b) PdCl2(PPh3)2, CuI, ethynyltrimethylsilane, DMF/TEA (2:1), rt – 65 °C, 8 h, 66%. (c) KF, MeOH, rt, 4 h, 98%. (d) i. NaOMe, paraformaldehyde, MeOH, 65 °C, 6 h; ii. NaBH4, 0–65 °C, 2 h, 70%. (e) Na ascorbate, N,N-dimethylethylenediamine, NaN3, EtOH/H2O (7:3), rt – 65 °C, 8 h, 23–45%. (f) Na ascorbate, CuSO4·5H2O, t-BuOH/H2O (1:1), rt, 24 h, N2, 59–69%. (g) 48% HBr in AcOH, 110 °C, 96 h, no conversion. :1), 0 °C – rt, 18 h, 70%. (b) PdCl2(PPh3)2, CuI, ethynyltrimethylsilane, DMF/TEA (2:1), rt – 65 °C, 8 h, 66%. (c) KF, MeOH, rt, 4 h, 98%. (d) i. NaOMe, paraformaldehyde, MeOH, 65 °C, 6 h; ii. NaBH4, 0–65 °C, 2 h, 70%. (e) Na ascorbate, N,N-dimethylethylenediamine, NaN3, EtOH/H2O (7:3), rt – 65 °C, 8 h, 23–45%. (f) Na ascorbate, CuSO4·5H2O, t-BuOH/H2O (1:1), rt, 24 h, N2, 59–69%. (g) 48% HBr in AcOH, 110 °C, 96 h, no conversion. | ||
The aryl azide 28 was prepared by N-methylation of aryl bromide 26via reductive amination to give 27 in 70% yield. Azidation of 27 in the presence of NaN3 and the stabilising ligand, N,N′-dimethylethylenediamine (DMEDA)25 gave the azide 28 in 23% yield. The low yield was attributed to the incomplete conversion of 27, and therefore product azide 28 was obtained as a mixture along with 27 in approximately 1:1 ratio as indicated by analysis of the 1H NMR spectrum. Attempts to separate the mixture using column chromatography and preparative TLC proved unsuccessful. Therefore, the azide 28 was used in the next reaction step as a mixture along with 27.
Aryl azide 29 was synthesised to that for azide 28. The reaction was optimised through careful choice of solvent mixture and temperature (Table 1) to give the aryl azide in 45% yield (entry 4), however, incomplete consumption of the starting bromide 26 was observed. Generally, a lower reaction temperature gave an incomplete conversion of aryl bromide 26 to azide 29 (entry 1–4), whereas at 70 °C the azide 29 eventually decomposed with no sign of the starting bromide 26 (entry 5). A compromise temperature of 65 °C allowed sufficient reactivity of the bromide 26 in the nucleophilic substitution to azide 29, while maintaining the stability of the in situ formed azide 29.
| Entry | Solvent | Temp (°C) | Time (h) | Yield (%) | Unreacted aryl bromidea (%) |
|---|---|---|---|---|---|
|
a Unreacted aryl bromides were obtained as the percent recovery (%) after flash chromatography over SiO2 gel (CH2Cl2/MeOH – 90:10).
|
|||||
| 1 | DMSO/H2O (1:1) |
65 | 8 | 25 | 51 |
| 2 | DMSO/H2O (7:3) |
65 | 8 | 22 | 50 |
| 3 | EtOH/H2O (1:1) |
65 | 8 | 40 | 46 |
| 4 | EtOH/H2O (7:3) |
65 | 8 | 45 | 40 |
| 5 | EtOH/H2O (7:3) |
70 | 8 | 11 | — |
The synthesis of triazoles 30 and 31 was achieved via CuAAC reactions of azide 28 (2.0 eq.) and 29 (2.0 eq.), and alkyne 25 (1.0 eq.) in the presence of Cu(II)/ascorbate in t-BuOH/H2O at rt for 24 h. Attempts to synthesise triazole 32 using excess 48% HBr in AcOH were unsuccessful, likely due to the low solubility of the starting triazole 31 in AcOH; therefore a new synthetic pathway via alkyne 39 was developed (Scheme 2).
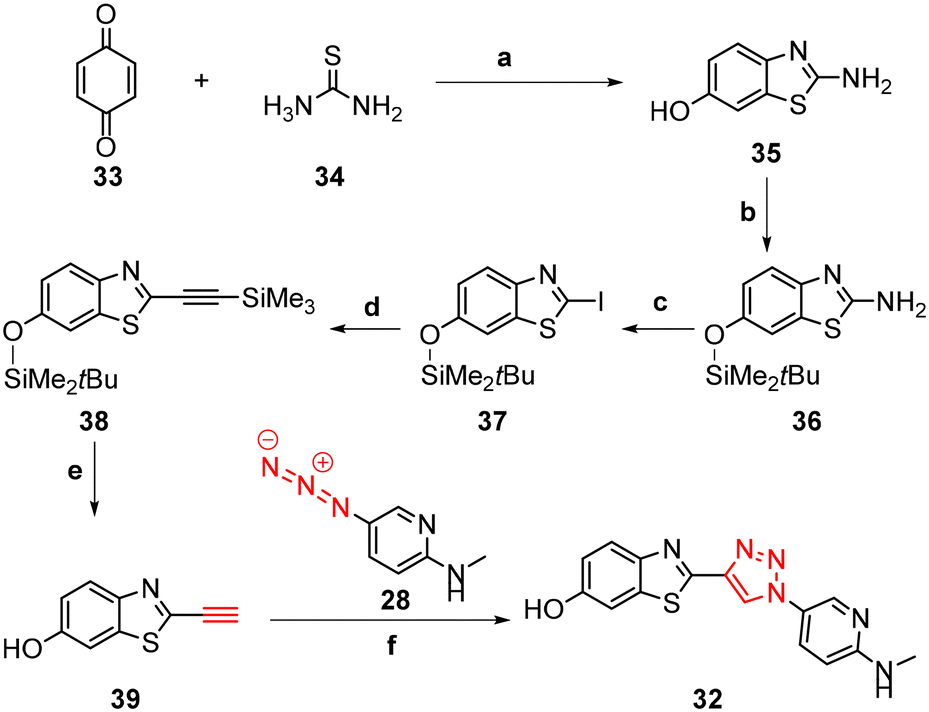 | ||
| Scheme 2 Synthesis of triazole 32. Reagents and conditions: (a) HCl, EtOH, rt, 48 h, 72%. (b) TBDMS-Cl, imidazole, DMF, rt, 24 h, 41%. (c) p-TsOH·H2O, NaNO2, KI, MeCN/H2O (28:1), 0 °C – rt, 18 h, 63%. (d) PdCl2(PPh3)2, CuI, ethynyltrimethylsilane, DMF/TEA (4:1), rt – 65 °C, 6 h, 74%. (e) KF, MeOH, rt, 4 h, 86%. (f) Na ascorbate, CuSO4·5H2O, t-BuOH/H2O (1:1), rt, 24 h, N2, 53%. | ||
The benzothiazole core 35 was synthesised in 72% yield by an acid-catalysed condensation reaction of benzoquinone 33 with thiourea 34 in EtOH at rt for 48 h, followed by protection of the phenol using t-butyldimethylsilyl (TBDMS) in the presence of imidazole in DMF to give 36 in 41% yield. Sandmeyer iodination of 36 in the presence of p-TsOH·H2O, NaNO2, and KI gave iodobenzothiazole 37 as a grey solid in 63% yield which was subjected to a Sonogashira cross-coupling reaction to afford the protected alkyne 38 in 74% yield. The synthesis of free alkyne 39 was achieved in 86% yield via fluoride desilylation using an excess of potassium fluoride (KF) and a solution of 38 in MeOH. Finally, the CuAAC reaction of free alkyne 39 with aryl azide 28 gave 32 in 53% yield (Scheme 2).
The triazole derivatives 30–32 were found to possess low solubility in most solvents, being only slightly soluble in DMSO and DMF. Therefore, in the search of analogues with improved solubility, additional derivatives were designed and synthesised incorporating different substitutions on the benzothiazole moiety (R) and phenyl ring (R1 and R2), and changes to the carbon linker (n) connecting the triazole unit and phenyl functionality (Fig. 6). To this end, free alkyne 43 was successfully synthesised using the same strategy as for alkyne 25 (Scheme 3).
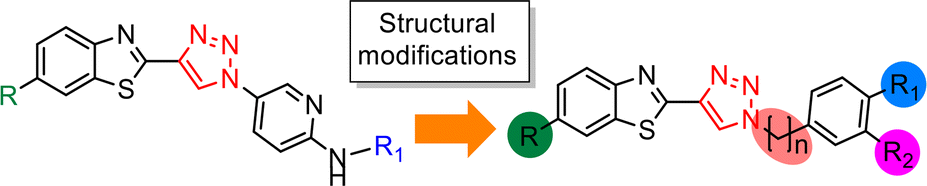 | ||
| Fig. 6 Rational design of triazole derivatives to target improved solubility. | ||
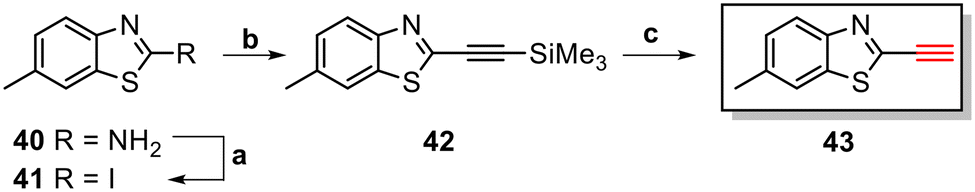 | ||
| Scheme 3 Synthesis of free alkyne 43. Reagents and conditions: (a) p-TsOH·H2O, NaNO2, KI, MeCN/H2O (9:1), 0 °C – rt, 18 h, 71%. (b) PdCl2(PPh3)2, CuI, ethynyltrimethylsilane, DMF/TEA (2:1), rt – 65 °C, 6 h, 76%. (c) KF, MeOH, rt, 2 h, 96%. | ||
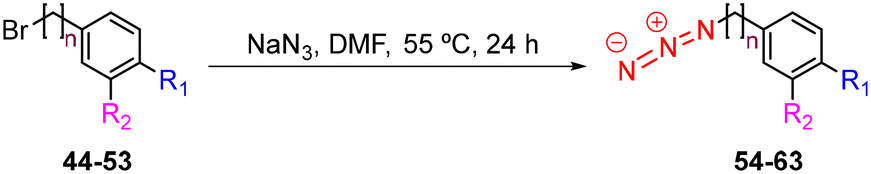
The synthesis of azides 54–63 was achieved in high to excellent yields (74–90%) by reacting the appropriate benzyl and phenylethyl bromides 44–53 with NaN3 in DMF at 55 °C for 24 h (Table 2).
|
|
||||
|---|---|---|---|---|
| Azide | R1 | R2 | n | Yield (%) |
| 54 | F | H | 1 | 83 |
| 55 | H | F | 1 | 75 |
| 56 | CF3 | H | 1 | 87 |
| 57 | H | H | 1 | 90 |
| 58 | NO2 | H | 1 | 77 |
| 59 | CN | H | 1 | 77 |
| 60 | F | H | 2 | 74 |
| 61 | CF3 | H | 2 | 83 |
| 62 | OH | H | 2 | 78 |
| 63 | NO2 | H | 2 | 78 |
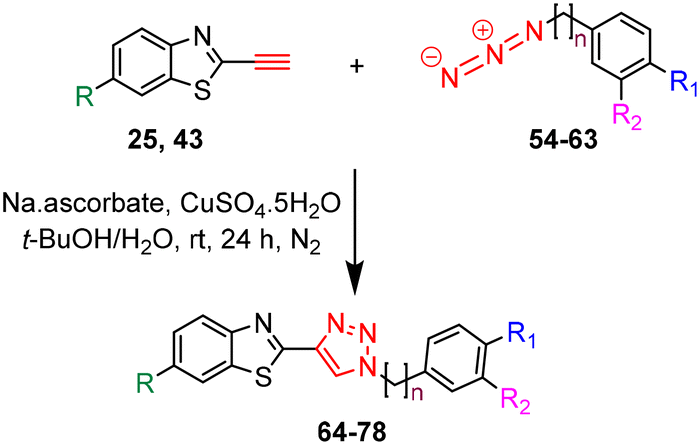
The target triazole derivatives were realised in moderate to high yields via CuAAC catalytic click reactions between the free alkyne 25 or 43 and the azides 54–63 (Table 3).
|
|
|||||
|---|---|---|---|---|---|
| Compound | R | R1 | R2 | n | Yield (%) |
| 64 | OCH3 | F | H | 1 | 77 |
| 65 | OCH3 | F | H | 2 | 69 |
| 66 | OCH3 | H | F | 1 | 84 |
| 67 | OCH3 | CF3 | H | 1 | 74 |
| 68 | OCH3 | CF3 | H | 2 | 71 |
| 69 | OCH3 | H | H | 1 | 86 |
| 70 | OCH3 | OH | H | 2 | 70 |
| 71 | OCH3 | NO2 | H | 1 | 79 |
| 72 | OCH3 | NO2 | H | 2 | 75 |
| 73 | OCH3 | CN | H | 1 | 73 |
| 74 | CH3 | F | H | 1 | 78 |
| 75 | CH3 | F | H | 2 | 69 |
| 76 | CH3 | CF3 | H | 1 | 73 |
| 77 | CH3 | OH | H | 2 | 81 |
| 78 | CH3 | NO2 | H | 1 | 75 |
Furthermore, to investigate the role of alkyl alcohol substituents for tau binding, triazole derivatives 89–93 were synthesised. The synthesis started with the azidation of bromo alkyl alcohols 79–83 using NaN3 in DMF at 55 °C for 24 h, followed by CuAAC reactions with free alkyne 25 (Scheme 4).
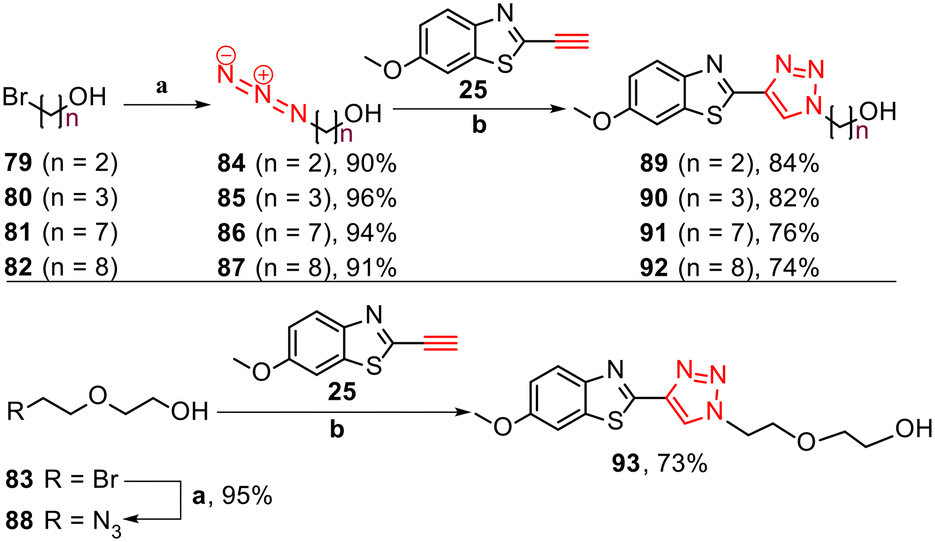 | ||
| Scheme 4 Synthesis of triazole derivatives 89–93. Reagents and conditions: (a) NaN3, DMF, 55 °C, 24 h, 90–96%. (b) Na ascorbate, CuSO4·5H2O, t-BuOH/H2O (1:1), rt, 24 h, N2, 73–84%. | ||
Standard amide coupling was used for the synthesis of amide derivatives from the benzothiazole carboxylic acid and commercially available amines. The synthesis of acid 95 was achieved through the addition of 1.0 mol L−1 NaOH (1.1 eq.) to a solution of the commercially available benzothiazole nitrile 94 in EtOH and heating at reflux for 18 h. Acidification of reaction mixture to pH 3 with a solution of HCl (18%) gave the acid 95 in 92% yield (Scheme 5).
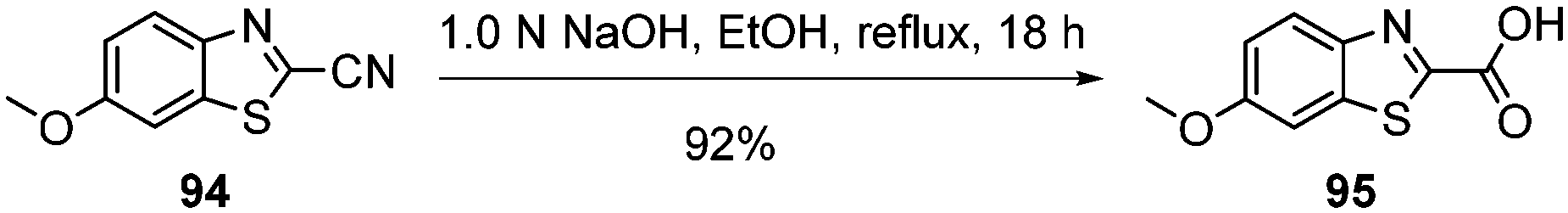 | ||
| Scheme 5 Synthesis of benzothiazole carboxylic acid 95. | ||
The synthesis of amide derivatives 109–121 was realised in high yields (70–77%) via amide coupling under EDCl and HOBT conditions in DMF at rt for 24 h (Table 4).
|
|
|||||
|---|---|---|---|---|---|
| Compound |
|
Yield (%) | Compound |
|
Yield (%) |
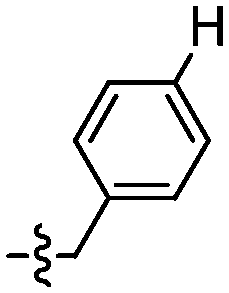
| 109 |
|
70 | 116 |
|
82 |
| 110 |
|
77 | 117 |
|
85 |
| 111 |
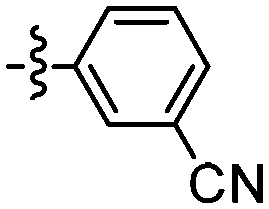
|
75 | 118 |
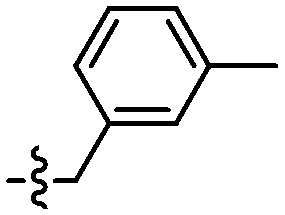
|
83 |
| 112 |
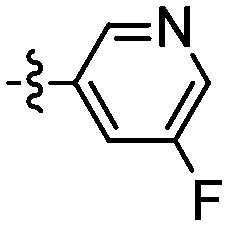
|
76 | 119 |
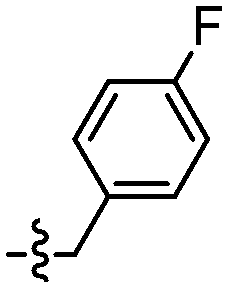
|
76 |
| 113 |
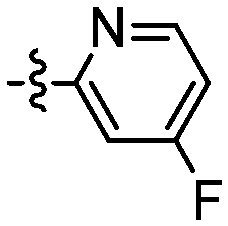
|
73 | 120 |
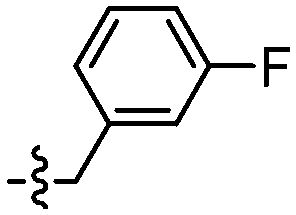
|
68 |
| 114 |
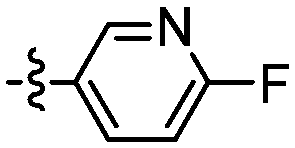
|
77 | 121 |
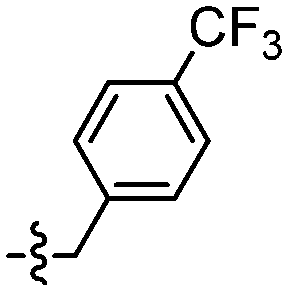
|
73 |
| 115 |
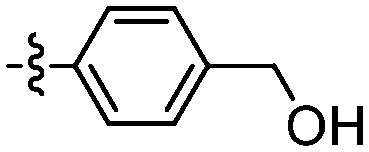
|
71 | |||
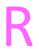
As isosteres of the amide linker, a series of ester analogues were made by the conversion of acid 95 to the corresponding sodium salt 122 in 98% yield. Reaction of benzothiazole salt 122 with the commercially available alkyl bromides 123–126 in DMF at rt for 24 h gave the ester products 127–130 in high to excellent yields (75–88%) (Table 5).
|
|
||
|---|---|---|
| Compound |
|
Yield (%) |
| 127 |
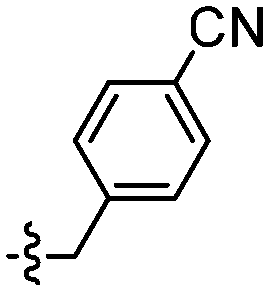
|
85 |
| 128 |
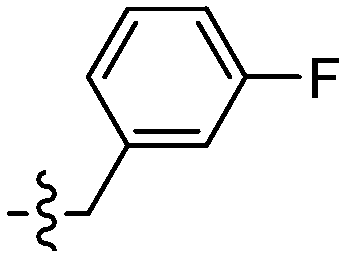
|
88 |
| 129 |
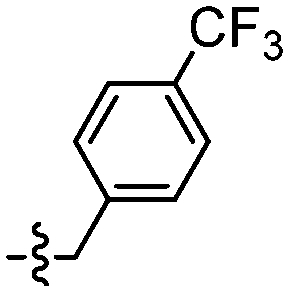
|
78 |
| 130 |
|
75 |

Biological evaluation
| Shared binding site with PBB3 (% inhibition) | Affinity at shared binding site(s) with PBB3 (Ki) | |
|---|---|---|
| Full | 95–100% | >PBB3 (high affinity, Ki < 4 nM) |
| Middle | 50–94% | |
| Low | 25–49% | =PBB3 (similar to PBB3, 4 nM < Ki < 10 nM) |
| Very little | 1–25% | |
| None | 0%, Ki not determinable | <PBB3 (low affinity, Ki > 10 nM) |
The results of the heterologous binding assays for selected tau ligands are shown in Table 7.
| Ligand | Inhibitory constant (Ki) | Inhibition |
|---|---|---|
| R 2: R-square for the binding curve of each compound fitted with Prism 6J. ND: not determinable: the inability to obtain the IC50 (Ki) values with certainty when the nonlinear regression using values for each concentration of test compound added did not converge for either 1- or 2-site curve fitting. | ||
| 30 | =PBB3 (6.3 nM, R2 = 0.8825) | Very little (19.9%) |
| 31 | <PBB3 (39.4 nM, R2 = 0.9620) | Low (32.3%) |
| 32 | =PBB3 (8.1 nM, R2 = 0.5741) | Very little (14.3%) |
| 64 | >PBB3 (3.8 nM, R2 = 0.2613) | Very little (10.1%) |
| 67 | <PBB3 (84.8 nM, R2 = 0.0659) | Very little (1.5%) |
| 69 | <PBB3 (341.4 nM, R2 = 0.3130) | Very little (5.7%) |
| 70 | <PBB3 (171.3 nM, R2 = 0.9215) | Low (27.2%) |
| 71 | <PBB3 (>1.5 mM, R2 = 0.5021) | Very little (13.5%) |
| 72 | >PBB3 (28.1 nM, R2 = 0.4872) | Very little (0.2%) |
| 73 | >PBB3 (0.46 nM, R2 = 0.1674) | Very little (7.7%) |
| 75 | ND | ND |
| 89 | <PBB3 (59.4 nM, R2 = 0.4957) | Very little (8.9%) |
| 90 | <PBB3 (28.5 nM, R2 = 0.7500) | Very little (8.0%) |
| 91 | <PBB3 (45.5 nM, R2 = 0.4785) | Very little (13.6%) |
| 92 | <PBB3 (241.7 nM, R2 = 0.4666) | Very little (7.9%) |
| 93 | <PBB3 (13.6 nM, R2 = 0.0798) | Very little (3.3%) |
| 110 | <PBB3 (761.0 nM, R2 = 0.8849) | Low (36.8%) |
| 111 | <PBB3 (>1.5 mM, R2 = 0.4912) | Very little (16.3%) |
| 115 | <PBB3 (215.0 nM, R2 = 0.9685) | Low (34.7%) |
| 127 | ND | ND |
| 128 | <PBB3 (13.2 nM, R2 = 0.3642) | Very little (7.9%) |
| 129 | >PBB3 (2.4 nM, R2 = 0.2924) | ND |
Analysis of the binding affinity (Ki) revealed that ligands 30 and 31 exhibited nanomolar affinity at the shared binding site(s) with [11C]PBB3 15 with Ki values of 6.3 and 39.4 nM, respectively. Substituting the methoxy unit on 31 with a hydroxy group (32) improved the affinity approximately 5-fold.
The replacement of the pyridine unit with fluorobenzene and the introduction of a one carbon linker between the triazole and phenyl moiety improved the affinity, as can be observed on ligand 64 (Ki = 3.8 nM). Furthermore, the substitution of the fluorine atom on 64 with a hydrogen atom (69) and a nitro group (71) decreased the binding affinity, suggesting that the high electronegative property of fluorine may be important for binding. The replacement of the fluorine atom with the polar nitrile group (73) led to improvement in affinity (Ki = 0.46 nM).
Ligands 89–93 showed nanomolar to submicromolar affinities at the shared binding site(s) with [11C]PBB3 15 (Ki = 13.6–241.7 nM), and interestingly, ligand 93 showed the highest affinity (Ki = 13.6 nM) compared to other ligands in this series. The superior affinity of ligand 93 suggests that the additional oxygen on the molecule should favourably interact with the site of the protein.
Furthermore, amide derivatives 110, 111, and 115 exhibited low affinity at the shared binding site(s) with [11C]PBB3 15 (Ki > 10 nM). By contrast, ester derivatives generally demonstrated better affinity than amide derivatives. Although ligand 129 seemed to display a high Ki value of 2.4 nM, this was associated with a high uncertainty and its shared binding status with PBB3 was not determinable.
Overall, the ligands showed no to low% inhibition (0.0–36.8%) at the shared binding site with [11C]PBB3 15. The ligands with a pyridine moiety (30–32) demonstrated very little to low% inhibition. Among the examples of this subset, the ligand with the mono N-methyl group (31) exhibited better inhibition (32.3%) than the ligand with a free amine group (30) (19.9%), suggesting that methyl substituent at pyridine unit may play an important role for the binding. However, the substitution of the methoxy group on 31 with a hydroxy unit (32) led to a decrease in inhibition value (14.3%).
The substitution of the pyridine unit with a phenyl moiety resulted in lower inhibition of [11C]PBB3 15 (0.2–13.5%). However, the hydroxy group attached to the phenyl ring in the para-position may be essential for achieving good inhibition, as exemplified by ligand 70 with an inhibition value of 27.2%.
Furthermore, the substitution of the pyridine unit with small alkyl alcohol (89–93) led to very little inhibitions of [11C]PBB3 15 (3.3–13.6%). The ligand 91 with seven alkyl carbons was more effective among these derivatives, and lower inhibitions were observed by either increasing or decreasing the alkyl carbon length. Interestingly, the presence of an oxygen atom in the middle of the carbon chain led to a significant decrease in inhibition of [11C]PBB3 15, as observed in ligand 93 (% inhibition = 3.3%).
In addition, three amide derivatives (110, 111, and 115) displaced 16.3–36.8% of [11C]PBB3 15 binding in the AD tissue. Among these derivatives, generally the 1-pyridine (110) and 4-hydoxymethyl (115) units were associated with better activity than the 3-cyano unit (111). Testing of ester derivatives (127–129) indicated that these ligands minimally blocked the [11C]PBB3 15.
Furthermore, the synthesised derivatives were subjected to calculated physiochemical properties to test the suitability of incorporation of the new linkers for future studies. The majority of the ligand derivatives showed good physicochemical properties according to Lipinski parameters, in relation to lipophilicity (clogP), number of hydrogen bond donor (HBD), number of hydrogen bond acceptor (HBA), polar surface area (PSA), and the ratio of the steady-state concentrations of a compound between the brain and the blood (logBB) (calculated by Molinspiration online services, ESI† S126–S128). In addition, the new derivatives are photo-stable, with some of them demonstrated moderate to high binding affinity against [11C]-PBB3 15 in in vitro heterologous blocking assay. Hence, further studies, including in vivo imaging using their corresponding radiolabelled derivatives, especially for ligands 110 and 129, which showed the capability to detect NFTs, are needed.
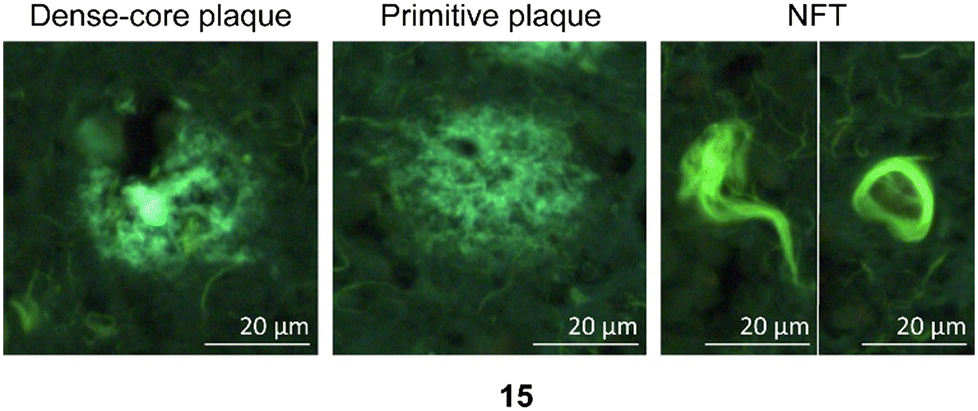 | ||
| Fig. 7 Representative staining of dense-core plaque, primitive plaque, and NFTs labeled with PBB3 (15), which are used as a positive control in these experiments.26 | ||
The in vitro staining assay of selected triazole derivatives revealed that ligands with pyridine units (30–32) poorly stained Aβ plaques and NFTs. The observation that the Aβ plaques and NFTs were not detectable may be due to the poor solubility properties of these probes in 50% ethanol in water, which limits tissue penetration, leading to insufficient imaging. However, this assumption needs to be confirmed by improving the bioavailability of the molecules. The substitution of a pyridine unit with phenyl moieties containing different substituents, and the introduction of the carbon linker between triazole and phenyl moieties improved the histology results. For example, ligands 67 and 75 which contain fluorine atoms displayed good staining of both dense-core plaque and primitive plaque (Aβ plaques) in human brain sections. This also suggests that the substitution of a methoxy group with a methyl group in the benzothiazole moiety improved the imaging of Aβ plaques as can be observed on ligand 75. Furthermore, the introduction of either a nitro (72), nitrile (73), or hydroxy (77) substituent on the phenyl ring led to ligands that showed good visualisation of Aβ plaques. It was also found that unsubstituted phenyl (69) intensely stained Aβ plaques (Fig. 8). Unfortunately, all triazole derivatives failed to detect NFTs.
 | ||
| Fig. 8 Representative in vitro histochemical staining of postmortem AD brain tissues with selected ligands. White arrow: dense-core plaque, yellow arrow: primitive plaque, red arrow: NFT. | ||
Unexpectedly, triazole ligands containing alkyl alcohols (89–92) and an alkoxy alcohol (93) failed to detect Aβ plaques and NFTs by fluorescence microscopy. In addition, most of the amide derivatives have failed to visualise Aβ plaques and NFTs. However, ligand 110 with a nitrogen atom at the 1-position of the phenyl ring selectively detected NFTs (Fig. 8). Furthermore, the ester derivative 129 bearing a trifluoro substituent on the para-position of the phenyl ring and one carbon linker between the ester group and the phenyl moiety exhibited specific staining to NFTs. However, ligands with a nitrile (127) and fluorine (128) substituent at the phenyl ring were unsuccessful in detecting Aβ and NFTs.
Experimental methods
Representative synthesis of triazole 32
:40): Rf = 0.85. 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 9.2 Hz, 1H), 7.28 (d, J = 2.8 Hz, 1H), 7.03 (dd, J = 9.2, 2.8 Hz, 1H), 3.86 (s, 3H). 13C NMR (400 MHz, CDCl3) δ 158.1, 149.0, 140.4, 123.0, 115.6, 103.1, 101.5, 55.8. MS (ESI +ve) m/z 292 ([M + H]+, 100%).
:40) to afford the protected alkyne 24 (1.10 g, 66%) as a light yellow oil. The product was used in the next reaction step immediately or stored in the freezer for a limited time due to stability issues. The spectroscopic data was in agreement with that previously reported. TLC (EtOAc/hexanes – 60:40): Rf = 0.86. 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 9.2 Hz, 1H), 7.25 (d, J = 2.8 Hz, 1H), 7.09 (dd, J = 9.2, 2.8 Hz, 1H), 3.85 (s, 3H), 0.30 (s, 9H). 13C NMR (400 MHz, CDCl3) δ 158.9, 147.3, 145.6, 136.8, 124.3, 116.6, 103.4, 102.3, 97.0, 55.8, 0.5. MS (ESI +ve) m/z 262 ([M + H]+, 100%).
:40) to afford the free alkyne 25 (0.71 g, 98%) as a pale yellow solid. The spectroscopic data was in agreement with that previously reported. TLC (EtOAc/hexanes – 60:40): Rf = 0.84. 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 9.2 Hz, 1H), 7.27 (d, J = 2.8 Hz, 1H), 7.12 (dd, J = 9.2, 2.8 Hz, 1H), 3.88 (s, 3H), 3.55 (s, 1H). 13C NMR (400 MHz, CDCl3) δ 159.1, 147.3, 144.8, 136.9, 124.6, 116.8, 103.5, 83.4, 77.0, 55.9. MS (ESI +ve) m/z 190 ([M + H]+, 100%).
:10) to afford the mono-N-methyl 27 (1.90 g, 70%) as a pale yellow solid. TLC (CH2Cl2/MeOH – 90:10): Rf = 0.49. 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 2.0 Hz, 1H), 7.49 (dd, J = 8.8, 2.0 Hz, 1H) 6.30 (d, J = 8.8 Hz, 1H), 4.59 (br s, 1H), 2.89 (d, J = 4.8 Hz, 3H). 13C NMR (400 MHz, CDCl3) δ 158.2, 148.8, 139.9, 107.8, 107.0, 29.3. MS (ESI +ve) m/z 187 (79Br [M + H]+, 100%), 189 (81Br [M + H]+, 100%).
:1, 22.5 mL) was stirred at rt for 30 min. NaN3 (0.38 g, 5.85 mmol) was added, and the reaction mixture was stirred at 65 °C for 8 h. The solution was cooled to rt, filtered through a layer of celite, concentrated, and extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with H2O (150 mL), brine (150 mL), dried (Na2SO4), filtered, and concentrated. The residue was subjected to flash chromatography over SiO2 gel (CH2Cl2/MeOH – 90:10) to afford the mixture of the azide 28 (100 mg, 23%) and starting material 5-bromo-N-methylpyridin-2-amine 27 (102 mg) as a grey solid which were unseparable. This mixture was used directly in the subsequent step. TLC (CH2Cl2/MeOH – 90:10): Rf = 0.44. 1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 2.8 Hz, 1H), 7.15 (dd, J = 8.8, 2.8 Hz, 1H) 6.29 (d, J = 8.8 Hz, 1H), 4.63 (br s, 1H), 2.89 (d, J = 4.8 Hz, 3H). MS (ESI +ve) m/z 150 ([M + H]+, 100%). HRMS (ESI +ve TOF) calcd for C6H8N5+ 150.0774, found 150.0773 ([M + H]+).
:H2O (2:1, 45 mL) was stirred at rt for 30 min. NaN3 (0.76 g, 11.69 mmol) was added, and the reaction mixture was stirred at 65 °C for 8 h. The solution was cooled to rt, filtered through a layer of celite, concentrated, and extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with H2O (150 mL), brine (150 mL), dried (Na2SO4), filtered, and concentrated. The residue was subjected to flash chromatography over SiO2 gel (CH2Cl2/MeOH – 90:10) to afford the azide 29 as a grey solid (0.35 g, 45%) which was used directly in the subsequent step. The spectroscopic data was in agreement with that previously reported. TLC (CH2Cl2/MeOH – 90:10): Rf = 0.41. 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 2.8 Hz, 1H), 7.16 (dd, J = 8.8, 2.8 Hz, 1H) 6.53 (d, J = 8.8 Hz, 1H), 4.44 (br s, 2H). 13C NMR (400 MHz, CDCl3) δ 156.0, 139.1, 128.9, 127.7, 109.5. MS (ESI +ve) m/z 136 ([M + H]+, 100%). HRMS (ESI +ve TOF) calcd for C5H6N5+ 136.0618, found 136.0623 ([M + H]+).
Following general procedure A2 (see ESI†). Alkyne 25 (40 mg, 0.21 mmol), CuSO4·5H2O (5 mg, 0.02 mmol), sodium ascorbate (8 mg, 0.04 mmol), and azide 29 (57 mg, 0.42 mmol) were stirred in t-BuOH/H2O (1
:1, 4.2 mL) to afford the 1,4-disubtituted-1,2,3-triazole 30 (40 mg, 59%) as a pale yellow solid after sequential trituration with CH2Cl2, MeCN, and hot MeOH. TLC (CH2Cl2/MeOH – 90:10): Rf = 0.03. Mp 182 °C (decomp). 1H NMR (400 MHz, DMSO) δ 9.35 (s, 1H), 8.47 (d, J = 2.4 Hz, 1H), 7.93 (dd, J = 8.8, 2.4 Hz, 1H), 7.92 (d, J = 9.2 Hz, 1H), 7.76 (d, J = 2.4 Hz, 1H), 7.16 (dd, J = 9.0, 2.4 Hz, 1H), 6.61 (d, J = 8.8 Hz, 1H), 6.50 (br s, 2H), 3.87 (s, 3H). 13C NMR (400 MHz, DMSO) δ 160.3, 157.6, 156.1, 147.6, 142.6, 140.8, 135.5, 130.7, 123.4, 123.1, 121.4, 116.0, 107.8, 105.0, 55.8. IR (neat) νmax 3462 (m), 3331 (m), 3222 (m), 3051 (w), 1645 (s), 1608 (m), 1563 (s), 1516 (s), 1466 (m), 1430 (m), 1413 (m), 1324 (s), 1249 (m), 1225 (s), 1079 (m), 1018 (s), 942 (s), 887 (s) cm−1. MS (ESI +ve) m/z 325 ([M + H]+, 100%). HRMS (ESI +ve TOF) calcd for C15H13N6OS+ 325.0866, found 325.0872 ([M + H]+).
:1, 10 mL), and alkyne 25 (127 mg, 0.67 mmol), CuSO4·5H2O (18 mg, 0.07 mmol), and sodium ascorbate (67 mg, 0.34 mmol) were added, and the reaction was stirred at rt for 24 hours. The resulting precipitate was filtered, and triturated with hot MeCN (5 mL) to afford the 1,4-disubtituted-1,2,3-triazole 31 (156 mg, 69%) as a pale yellow solid. TLC (CH2Cl2/MeOH – 90:10): Rf = 0.05. Mp 189 °C (decomp). 1H NMR (400 MHz, DMSO) δ 9.35 (s, 1H), 8.55 (d, J = 2.4 Hz, 1H), 7.93 (dd, J = 8.8, 2.4 Hz, 1H), 7.92 (d, J = 9.2 Hz, 1H), 7.76 (d, J = 2.4 Hz, 1H), 7.15 (dd, J = 9.0, 2.4 Hz, 1H), 7.07 (q, J = 4.8 Hz, 1H), 6.63 (d, J = 8.8 Hz, 1H), 3.86 (s, 3H), 2.84 (d, J = 4.8 Hz, 3H). 13C NMR (400 MHz, DMSO) δ 159.6, 157.6, 156.2, 147.7, 142.6, 140.6, 135.5, 130.2, 124.2, 123.1, 121.3, 116.0, 107.8, 105.0, 55.8, 28.0. IR (neat) νmax 3317 (m), 3100 (m), 3051 (w), 1608 (s), 1537 (s), 1493 (m), 1453 (m), 1401 (m), 1313 (s), 1290 (m), 1255 (m), 1218 (s), 1182 (m), 1079 (m), 1058 (m), 945 (s), 871 (s) cm−1. MS (ESI +ve) m/z 339 ([M + H]+, 100%). HRMS (ESI +ve TOF) calcd for C16H15N6OS+ 339.1023, found 339.1028 ([M + H]+).
Method B: following general procedure A2 (see ESI†). Alkyne 39 (37 mg, 0.21 mmol), CuSO4·5H2O (5 mg, 0.02 mmol), sodium ascorbate (8 mg, 0.04 mmol), and azide 28 (63 mg, 0.42 mmol) were stirred in t-BuOH/H2O (1
:1, 4.2 mL) to afford the 1,4-disubtituted-1,2,3-triazole 32 (36 mg, 53%) as a brown solid after sequential trituration with CH2Cl2, MeCN, and hot MeOH. TLC (CH2Cl2/MeOH – 90:10): Rf = 0.05. Mp 170 °C (decomp). 1H NMR (500 MHz, DMSO, 75 °C) δ 9.99 (s, 1H), 9.36 (s, 1H), 8.58 (d, J = 2.4 Hz, 1H), 7.94 (dd, J = 8.8, 2.4 Hz, 1H), 7.84 (d, J = 9.2 Hz, 1H), 7.46 (d, J = 2.4 Hz, 1H), 7.12 (br s, 1H), 7.01 (dd, J = 9.0, 2.4 Hz, 1H), 6.65 (d, J = 8.8 Hz, 1H), 2.83 (s, 3H). 13C NMR (500 MHz, DMSO) δ 159.6, 156.0, 155.1, 146.8, 142.8, 140.6, 135.5, 130.2, 123.3, 121.2, 116.2, 108.2, 107.0, 28.1. IR (neat) νmax 3243 (m), 3096 (m), 1613 (s), 1555 (s), 1455 (m), 1403 (m), 1380 (m), 1260 (m), 1235 (m), 1168 (m), 1035 (m), 902 (s), 831 (m), 799 (m) cm−1. MS (ESI +ve) m/z 325 ([M + H]+, 100%). HRMS (ESI +ve TOF) calcd for C15H13N6OS+ 325.0866, found 325.0866 ([M + H]+).
Conclusions
A series of tau ligands containing 1,2,3-triazole, amide, and ester moieties were synthesised. The heterologous blocking assay revealed that several of these ligands exhibited low to high affinities (Ki = >1.5 mM–0.46 nM) at the shared binding site(s) with PBB3 15. Ligands 30 and 64 had a similar affinity with PBB3 15 with Ki values of 6.3 and 3.8 nM, respectively. Furthermore, the ligands showed none to low % inhibition (0.0–36.8%) against [11C]PBB3 15. Results from in vitro fluorescence staining showed that some triazole derivatives showed good visualisation of Aβ plaques, but failed to detect the NFTs in human brain sections. However, NFTs were visualised with ligands 110 and 129. All except one of the novel tau ligands synthesised and tested thus far possess a methoxy or methyl substituent at the 6-position of the benzothiazole ring. PiB 1, PBB3 15 and many related amyloid and tau ligands are prepared with a free hydroxy group. It is not clear if biological efficacy can be improved following demethylation, although one analogue 32 bearing the hydroxy group did not show improved binding. Ligands 110 and 129 represent a good starting point for further tau ligand development.Author contributions
Conceptualization: AK, PAK, MH. Formal analysis HW, MO, TY, KK, MH, M-RZ, MF, PAK, AK. Funding acquisition PAK, AK, MF, MH. Investigation HW, MO, TY, KK, MH, M-RZ. Methodology HW, AK, MO, MH, MF, PAK, AK. Project administration AK, PAK. Resources AK, PAK, MF, M-RZ, MH, MO. Supervision AK, PAK, MH, M-RZ. Validation HW, M-RZ, MO, PAK. Visualization HW, M-RZ, MO, PAK. Writing – original draft HW, AK, PAK. Writing – review & editing HW, AK, PAK, MO, M-RZ, MH, MF.Conflicts of interest
There are no conflicts to declare.Acknowledgements
HW thanks Ministry of Research and Technology/National Research and Innovation Agency (Indonesia) for PhD scholarship funding under Research and Innovation in Science and Technology Project (RISET-Pro) No. 8245-ID.References
- W. Montgomery, K. Ueda, M. Jorgensen, S. Stathis, Y. Cheng and T. Nakamura, Clinicoecon. Outcomes Res., 2018, 10, 13–28 CrossRef PubMed.
- F. A. E. Sosso, O. Nakamura and M. Nakamura, Journal of Neurology and Neuroscience, 2017, 8, 1–3 Search PubMed.
- H. Tong, K. Lou and W. Wang, Acta Pharm. Sin. B, 2015, 5, 25–33 CrossRef PubMed.
- M. Ariza, H. C. Kolb, D. Moechars, F. Rombouts and J. I. Andres, J. Med. Chem., 2015, 58, 4365–4382 CrossRef CAS PubMed.
- P. V. Fish, D. Steadman, E. D. Bayle and P. Whiting, Bioorg. Med. Chem. Lett., 2019, 29, 125–133 CrossRef CAS PubMed.
- J. L. Haines, J. Law Med. Ethics, 2018, 46, 694–698 CrossRef PubMed.
- S. Liu, H. Suzuki, H. Ito, T. Korenaga, H. Akatsu, K. Meno and K. Uchida, Alzheimers Dement., 2019, 11, 85–97 Search PubMed.
- K. G. Mawuenyega, W. Sigurdson, V. Ovod, L. Munsell, T. Kasten, J. C. Morris, K. E. Yarasheski and R. J. Bateman, Science, 2010, 330, 1774 CrossRef CAS PubMed.
- Y. Madav, S. Wairkar and B. Prabhakar, Brain Res. Bull., 2019, 146, 171–184 CrossRef CAS PubMed.
- J. P. Holland, S. H. Liang, B. H. Rotstein, T. L. Collier, N. A. Stephenson, I. Greguric and N. Vasdev, J. Labelled Compd. Radiopharm., 2014, 57, 323–331 CrossRef CAS PubMed.
- B. Hall, E. Mak, S. Cervenka, F. I. Aigbirhio, J. B. Rowe and J. T. O'Brien, Ageing Res. Rev., 2017, 36, 50–63 CrossRef CAS PubMed.
- A. M. Walji, E. D. Hostetler, H. Selnick, Z. Zeng, P. Miller, I. Bennacef, C. Salinas, B. Connolly, L. Gantert, M. Holahan, S. O’Malley, M. Purcell, K. Riffel, J. Li, J. Balsells, J. A. OBrien, S. Melquist, A. Soriano, X. Zhang, A. Ogawa, S. Xu, E. Joshi, J. Della Rocca, F. J. Hess, J. Schachter, D. Hesk, D. Schenk, A. Struyk, K. Babaoglu, T. G. Lohith, Y. Wang, K. Yang, J. Fu, J. L. Evelhoch and P. J. Coleman, J. Med. Chem., 2016, 59, 4778–4789 CrossRef CAS PubMed.
- F. J. Rombouts, J. I. Andres, M. Ariza, J. M. Alonso, N. Austin, A. Bottelbergs, L. Chen, V. Chupakhin, E. Cleiren, K. Fierens, A. Fontana, X. Langlois, J. E. Leenaerts, J. Marien, C. Martinez Lamenca, R. Salter, M. E. Schmidt, P. Te Riele, C. Wintmolders, A. A. Trabanco, W. Zhang, G. Macdonald and D. Moechars, J. Med. Chem., 2017, 60, 1272–1291 CrossRef CAS PubMed.
- M. Scholl, A. Maass, N. Mattsson, N. J. Ashton, K. Blennow, H. Zetterberg and W. Jagust, Mol. Cell. Neurosci., 2019, 97, 18–33 CrossRef PubMed.
- R. Harada, N. Okamura, S. Furumoto, K. Furukawa, A. Ishiki, N. Tomita, T. Tago, K. Hiraoka, S. Watanuki, M. Shidahara, M. Miyake, Y. Ishikawa, R. Matsuda, A. Inami, T. Yoshikawa, Y. Funaki, R. Iwata, M. Tashiro, K. Yanai, H. Arai and Y. Kudo, J. Nucl. Med., 2016, 57, 208–214 CrossRef CAS PubMed.
- H. Shimada, S. Kitamura, M. Ono, Y. Kimura, M. Ichise, K. Takahata, S. Moriguchi, M. Kubota, T. Ishii, Y. Takado, C. Seki, S. Hirano, H. Shinotoh, N. Sahara, P. Tempest, G. Tamagnan, J. Seibyl, O. Barret, D. Alagille, M.-R. Zhang, S. Kuwabara, M.-K. Jang, K. Marek, T. Suhara and M. Higuchi, Alzheimer’s Dementia, 2017, 13(7), P1104 Search PubMed.
- N. Okamura, R. Harada, A. Ishiki, A. Kikuchi, T. Nakamura and Y. Kudo, Clin. Transl. Imaging, 2018, 6, 305–316 CrossRef PubMed.
- L. Declercq, F. Rombouts, M. Koole, K. Fierens, J. Marien, X. Langlois, J. I. Andres, M. Schmidt, G. Macdonald, D. Moechars, W. Vanduffel, T. Tousseyn, R. Vandenberghe, K. Van Laere, A. Verbruggen and G. Bormans, J. Nucl. Med., 2017, 58, 975–981 CrossRef CAS PubMed.
- K. Tagai, M. Ono, M. Kubota, S. Kitamura, K. Takahata, C. Seki, Y. Takado, H. Shinotoh, Y. Sano, Y. Yamamoto, K. Matsuoka, H. Takuwa, M. Shimojo, M. Takahashi, K. Kawamura, T. Kikuchi, M. Okada, H. Akiyama, H. Suzuki, M. Onaya, T. Takeda, K. Arai, N. Arai, N. Araki, Y. Saito, J. Q. Trojanowski, V. M. Y. Lee, S. K. Mishra, Y. Yamaguchi, Y. Kimura, M. Ichise, Y. Tomita, M. R. Zhang, T. Suhara, M. Shigeta, N. Sahara, M. Higuchi and H. Shimada, Neuron, 2021, 109, 42–58 CrossRef CAS PubMed , e8.
- L. Saint-Aubert, L. Lemoine, K. Chiotis, A. Leuzy, E. Rodriguez-Vieitez and A. Nordberg, Mol. Neurodegener., 2017, 12, 1–21 CrossRef PubMed.
- A. Leuzy, K. Chiotis, L. Lemoine, P. G. Gillberg, O. Almkvist, E. Rodriguez-Vieitez and A. Nordberg, Mol. Psychiatry, 2019, 24, 1112–1134 CrossRef CAS PubMed.
- H. Hashimoto, K. Kawamura, N. Igarashi, M. Takei, T. Fujishiro, Y. Aihara, S. Shiomi, M. Muto, T. Ito, K. Furutsuka, T. Yamasaki, J. Yui, L. Xie, M. Ono, A. Hatori, K. Nemoto, T. Suhara, M. Higuchi and M. R. Zhang, J. Nucl. Med., 2014, 55, 1532–1538 CrossRef CAS PubMed.
- Y. T. Wang and P. Edison, Curr. Neurol. Neurosci. Rep., 2019, 19, 1–14 CrossRef PubMed.
- J. Qi, M. S. Han, Y. C. Chang and C. H. Tung, Bioconjugate Chem., 2011, 22, 1758–1762 CrossRef CAS PubMed.
- S. Potratz, A. Mishra and P. Bauerle, Beilstein J. Org. Chem., 2012, 8, 683–692 CrossRef CAS PubMed.
- M. Ono, N. Sahara, K. Kumata, B. Ji, R. Ni, S. Koga, D. W. Dickson, J. Q. Trojanowski, V. M. Lee, M. Yoshida, I. Hozumi, Y. Yoshiyama, J. C. van Swieten, A. Nordberg, T. Suhara, M. R. Zhang and M. Higuchi, Brain, 2017, 140, 764–780 Search PubMed.
- H. C. W. Kolb, J. C. Walsh, W. Xhang, U. B. Gangadarmath, D. Kasi, K. Chen, A. Sinha, G. Chen, V. P. Mocharla, P. J. Scoot, H. C. Padgett, Q. Liang, Z. Gao, T. Zhao and C. Xia, Imaging agents useful for identifying AD pathology. Siemens Medical Solutions USA, Inc., PCT/US2009/051748, WO2010, 2010.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00358a |
| This journal is © The Royal Society of Chemistry 2023 |