
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments of imidazo[1,2-a]pyridine analogues as antituberculosis agents
Sauvik
Samanta
 a,
Sumit
Kumar
a,
Eswar K.
Aratikatla
a,
Sandeep R.
Ghorpade
a and
Vinayak
Singh
*ab
a,
Sumit
Kumar
a,
Eswar K.
Aratikatla
a,
Sandeep R.
Ghorpade
a and
Vinayak
Singh
*ab
aHolistic Drug Discovery and Development (H3D) Centre, University of Cape Town, Rondebosch 7701, South Africa. E-mail: vinayak.singh@uct.ac.za
bSouth African Medical Research Council Drug Discovery and Development Research Unit, Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Rondebosch 7701, South Africa
First published on 3rd March 2023
Abstract
Over the past 2000 years, tuberculosis (TB) has killed more people than any other infectious disease. In 2021, TB claimed 1.6 million lives worldwide, making it the second leading cause of death from an infectious disease after COVID-19. Unfortunately, TB drug discovery research was neglected in the last few decades of the twentieth century. Recently, the World Health Organization has taken the initiative to develop new TB drugs. Imidazopyridine, an important fused bicyclic 5,6 heterocycle has been recognized as a “drug prejudice” scaffold for its wide range of applications in medicinal chemistry. A few examples of imidazo[1,2-a]pyridine exhibit significant activity against multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB). Here, we critically review anti-TB compounds of the imidazo[1,2-a]pyridine class by discussing their development based on the structure–activity relationship, mode-of-action, and various scaffold hopping strategies over the last decade, which is identified as a renaissance era of TB drug discovery research.
 Sauvik Samanta | Dr. Sauvik Samanta obtained his Ph.D. in Synthetic Organic Chemistry from the Indian Institute of Technology Kanpur, India in 2015. He worked as an Erasmus Mundus postdoctoral fellow at the University of Warsaw, Poland from November 2014 for a year and thereafter joined as a Claude Leon Foundation postdoctoral fellow at the University of Cape Town in May 2016. Dr. Samanta joined H3D in July 2018 as a Medicinal Chemistry postdoctoral researcher and in February 2022 he was appointed as a Medicinal Chemistry Investigator at H3D. His research areas include medicinal chemistry and drug discovery research for infectious diseases. |
 Sumit Kumar | Sumit Kumar obtained his Ph.D. in 2019 in the field of Medicinal Chemistry from Guru Nanak Dev University, Amritsar in the research group of Prof. Vipan Kumar. He worked for one year at Sphaera Pharmaceutical Pvt. Ltd. in India as Research Associate and thereafter, moved to Seoul National University, South Korea for his first post-doctorate and gained experience in the field of nanomedicine. Sumit joined Dr. Sandeep at H3D Drug Discovery and Development as a Post-Doc in the year 2021 and is currently exploring the area of tuberculosis drug discovery. |
 Eswar K. Aratikatla | Dr. Eswar K. Aratikatla was born in Vizianagaram, Andhra Pradesh, India. He received his M.Sc. in Organic Chemistry (2010) from Andhra University, India, and subsequently worked as a Research Chemist (2010–2012) in the pharmaceutical industry. He received his Ph.D. in Organic and Natural Product Chemistry (2019) from CSIR-National Chemical Laboratory, Pune, India. He joined the Drug Discovery and Development Centre (H3D) at the University of Cape Town in 2022 as a postdoctoral research fellow. His current research at H3D aims at developing novel antituberculosis agents. His research interests include medicinal chemistry, natural product chemistry, and the synthesis of biologically active molecules. |
 Sandeep Ghorpade | Dr. Sandeep R. Ghorpade heads the medicinal chemistry section of the Holistic Drug Discovery and Development Centre (H3D) of the University of Cape Town and leads H3D's TB and AMR portfolios. Before joining H3D in 2014, he worked as a team leader and senior scientist at AstraZeneca India Pvt Ltd. Bangalore, India, for ten years. Dr. Ghorpade did a Master of Pharmaceutical Sciences from the University of Mumbai and completed a Ph.D. in organic chemistry at National Chemical Laboratory, India, followed by two years of post-doctoral studies in asymmetric catalysis at the University of North Dakota, USA. |
 Vinayak Singh | Vinayak Singh obtained his Ph.D. in Biochemistry from the University of Lucknow (India) in 2010, where he carried out his research at CSIR-Central Drug Research Institute, Lucknow. He then moved to the University of Cape Town (UCT) for his postdoctoral studies (2011–2016). Vinayak joined the Drug Discovery & Development Centre (H3D) at UCT in 2017; currently, he is leading tuberculosis and antimicrobial resistance biology research. Being an artist of molecular networks, genomics, and metabolomics, his main interest is to deconvolute the mechanism of action of potential compounds – to fulfil a broad and acute interest in the discovery of new innovative drugs. |
Introduction
Tuberculosis (TB) is an ancient airborne infectious disease caused by the acid-fast bacterium Mycobacterium tuberculosis (Mtb) that primarily affects the lungs and eventually can spread to other parts of the body. It is estimated that nearly 10.6 million people were affected by TB with 1.6 million deaths worldwide, in 2021.1 After the invention of the current 1st line drug regimen for the treatment of drug-sensitive TB in the 1960s, the TB drug discovery research was neglected for almost half a century, unfortunately. Recently, the World Health Organization (WHO) has taken several initiatives to reduce TB-related deaths by 95% and TB infection incidents by 90% by the end of 2035 to terminate the TB epidemic.1 WHO has set up a target of $2 billion annually1 for TB drug discovery research to develop new TB drugs.Drug resistance (DR) is one of the main challenges in TB treatment. Resistant Mtb strains of newly developed anti-TB drugs are generally found within 10 years of their first use.2,3 Almost 2 in 3 individuals who develop multi-drug resistant TB (MDR-TB) each year are not receiving appropriate treatment.2 Encouragingly, recent clinical uses of new and repurposed drugs have simplified and shortened the treatment regimens of MDR-TB.2–5 After almost 50 years of a drought of novel TB drugs, recently, new drugs bedaquiline (BDQ; Janssen Therapeutics, 2012)6 and pretomanid (TB Alliance, 2019)7 have been approved by the U.S. Food and Drug Administration (FDA), whereas delamanid (Otsuka Pharmaceutical Co., 2014)8 was approved by European Medicines Agency (EMA) for the treatment of DR-TB. There are a few new drug candidates in the clinical development pipeline.3,9 However, considering the reports of the emergence of DR against newly approved drugs and the high attrition rate of drugs in the pipeline, continuous discovery and development of new TB drugs are needed. At present, 17 drug candidates from 14 different chemical classes are being listed at various stages of clinical development (Table 1).3,9–11 With few exceptions, fused bicyclic rings are a commonly observed structural feature of the TB drugs in the clinical pipeline, which often leads to issues with physicochemical properties such as high lipophilicity and low aqueous solubility, etc. This review focuses on one such fused bicyclic ring system, imidazo[1,2-a]pyridine (IP), represented by telacebec (Q203) in this list, highlighting the efforts to overcome these challenges in identifying a compound with drug-like properties.
| Name | Chemical class | Structure | Phase |
|---|---|---|---|
| SQ109 | Ethylenediamine |

|
IIb/III |
| Delpazolid | Oxazolidinone |
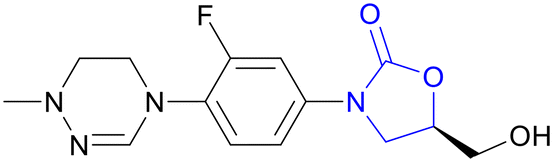
|
IIb |
| Sutezolid | Oxazolidinone |
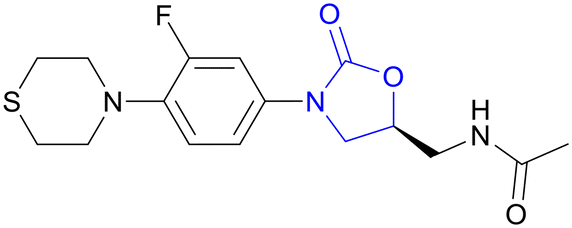
|
IIb |
| Sudapyridine (WX-081) | Triarylpyridine |
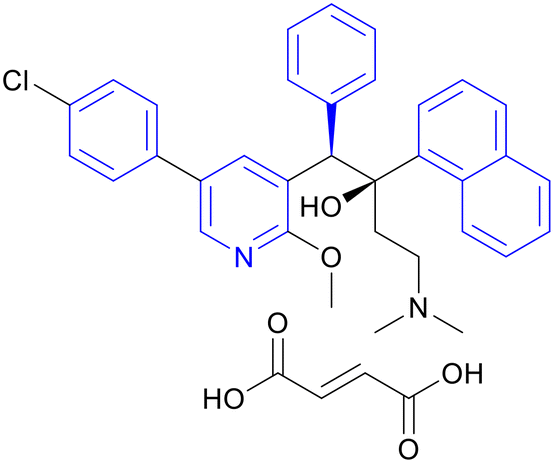
|
IIa/IIb |
| Telacebec (Q203) | Imidazo[1,2-a]pyridine |
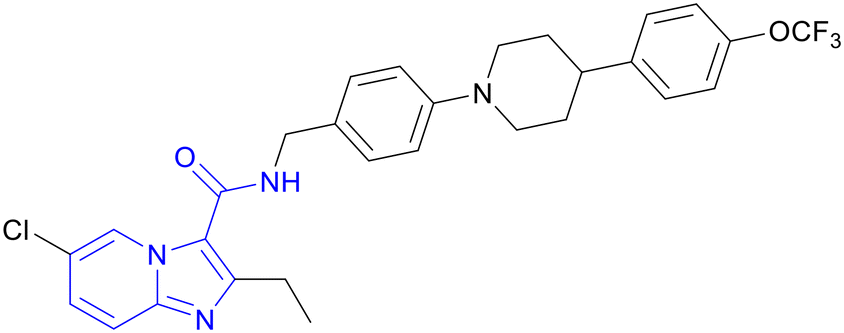
|
IIa |
| Macozinone (PBTZ169) | Benzothiazinone |
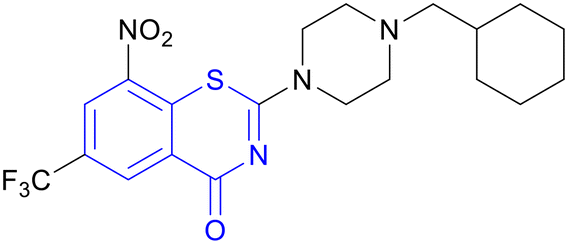
|
IIa |
| GSK3036656 | Benzoxaborole |
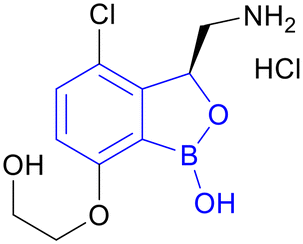
|
IIa |
| TBA-7371 | 1,4-Azaindole |
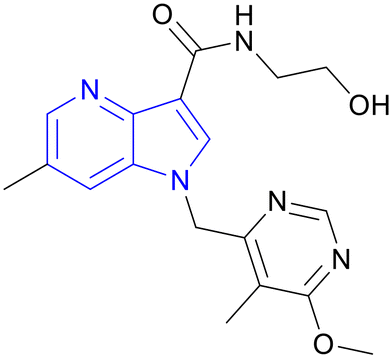
|
IIa |
| Pyrifazimine | Riminophenazine |
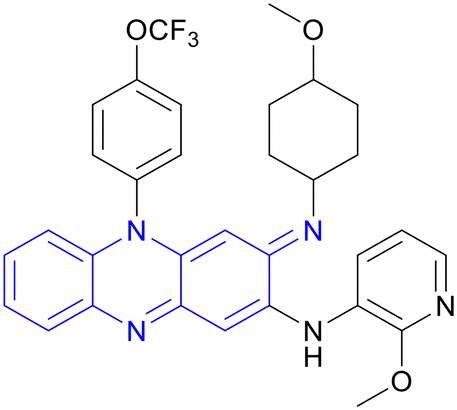
|
IIa |
| OPC-167832 | 3,4-Dihydrocarbostyril |
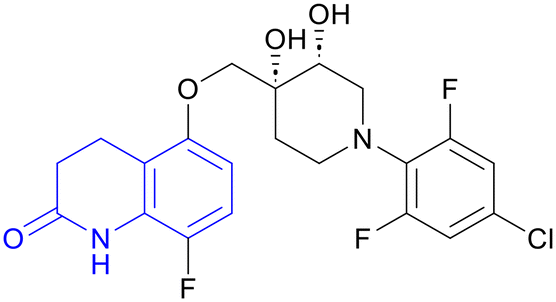
|
(I/IIa) IIb/IIc |
| BTZ043 | Benzothiazinone |
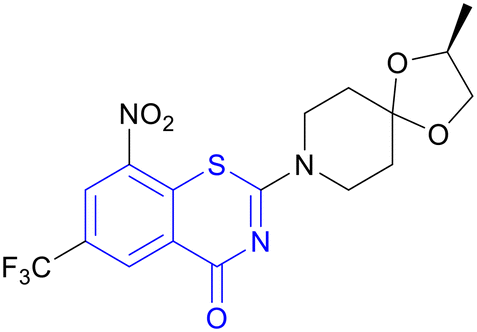
|
I/IIa |
| TBI-223 | Oxazolidinone |
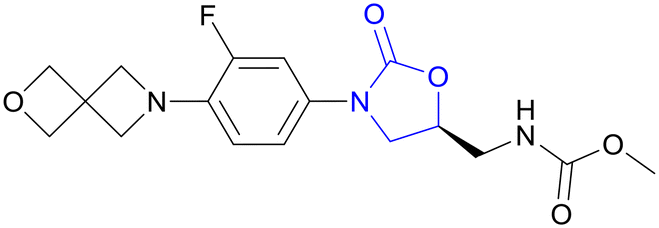
|
I |
| SPR720 | Benzimidazole urea |
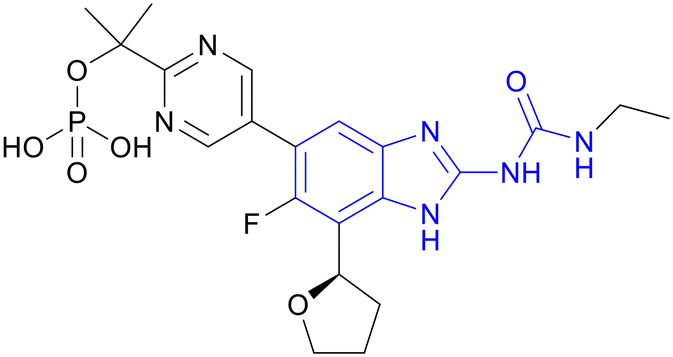
|
I |
| TBAJ-876 | Diarylquinoline |
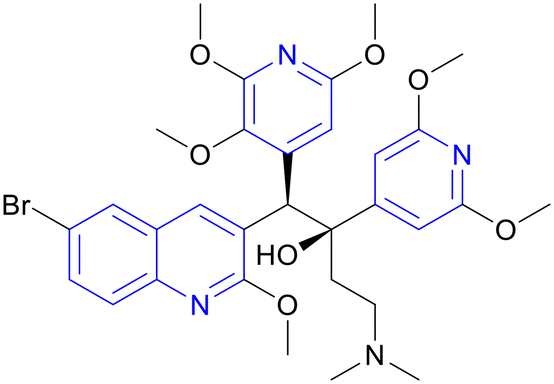
|
I |
| TBAJ-587 | Diarylquinoline |
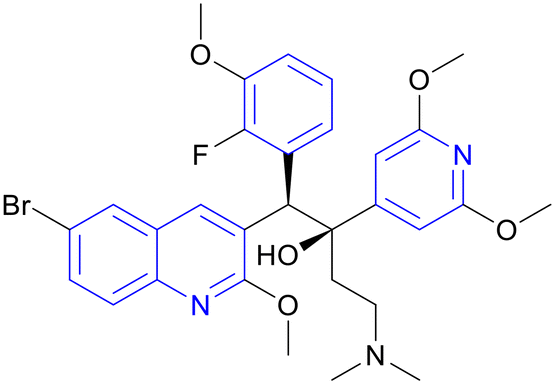
|
I |
| GSK2556286 | Pyrimidine-2,4-dione |
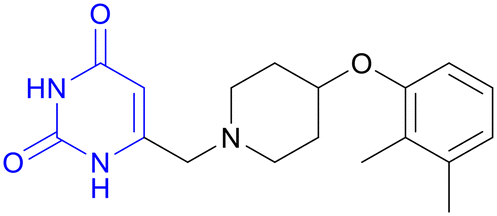
|
I |
| BVL-GSK098 | Amido-piperidine |
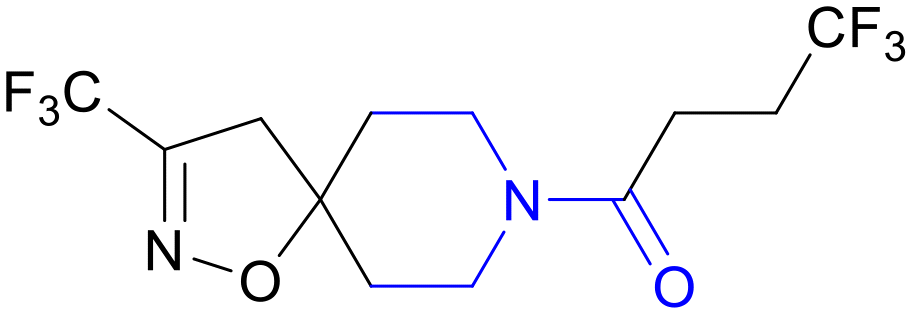
|
I |
IP, a 5,6-fused ring systems, is an important class of compounds having a range of medicinal chemistry applications. For example, zolimidine (1, gastroprotective drug),12,13 alpidem (2, anxiolytic drug),14,15 and zolpidem (3, insomnia drug)16 are in clinical use (Fig. 1). Along with anticancer, antileishmanial, anticonvulsant, antimicrobial, antiviral, antidiabetic, proton pump inhibition, and insecticidal activities, this class has also shown significant potency against Mtb.17 In this review, we discuss the recent developments of IP analogues as anti-TB agents.
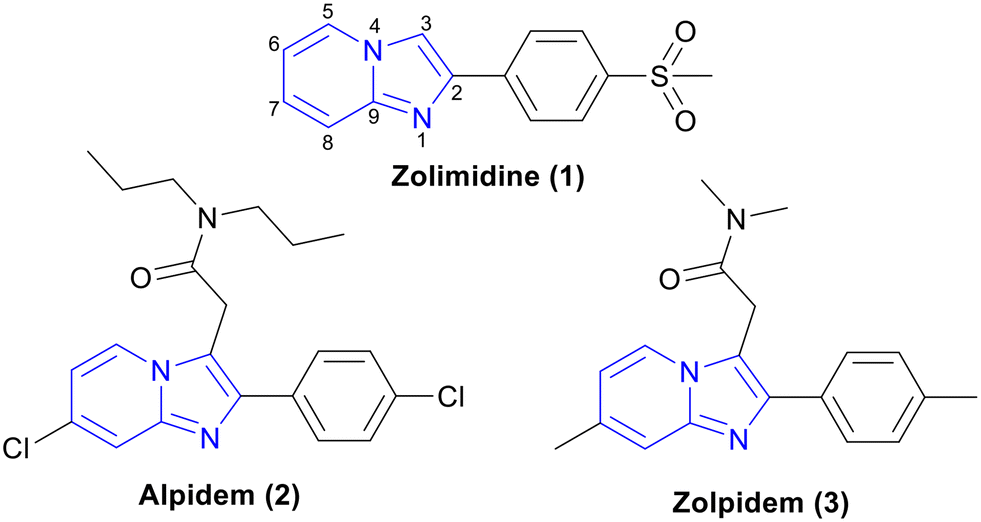 | ||
| Fig. 1 Imidazo[1,2-a]pyridine-based drugs available in the market. | ||
Imidazo[1,2-a]pyridine amides as QcrB inhibitors
Since the approval of BDQ in 2012, anti-TB drug discovery targeting energy generation and adenosine triphosphate (ATP) synthesis became an attractive approach.18–21 Oxidative phosphorylation (OxPhos) is the key factor for Mtb survival and growth.22 In this pathway, electrons are transferred from the oxidation of organic substrates to oxygen via a series of protein complexes. Simultaneously, proton transportation across the inner membrane creates a proton motive force that helps ATP synthase to produce ATP from ADP. Targeting the OxPhos pathway by interacting with a protein or by blocking any step in the pathway of OxPhos, the anti-TB agents can combat mycobacterial infections and may lead to the treatment-shortening.23,24 Complex III containing cytochrome bcc oxidase is one of the important protein complexes of Mtb, responsible for OxPhos pathway.25,26 The QcrB, a subunit of cytochrome bcc is a popular target for small molecule drug discovery, including telacebec (Q203) which is an imidazo[1,2-a]pyridine amide (IPA) class of anti-TB drug and currently in phase II clinical trials.25,26 Here we provide a brief overview of various reports on IPAs.Vast exploration of anti-TB activity of IP analogues was started at the beginning of the last decade. Abrahams et al. identified four hit compounds of the imidazo[1,2-a]pyridine class as potent inhibitors of Mtb and M. bovis BCG by performing a high throughput screening (HTS) approach.27 The minimum inhibitory concentration (MIC), resulting in an 80% reduction in bacterial growth (MIC80), was quite promising (0.03 to 5.0 μM) against the Mtb H37Rv strain.
Moraski and co-workers reported a set of nine 2,7-dimethylimidazo[1,2-a]pyridine-3-carboxamides showing in vitro anti-TB activity against replicating and non-replicating (MIC90, MIC resulting in a 90% reduction in bacterial growth, range: 0.4–1.9 μM), MDR (MIC90 range: 0.07–2.2 μM), and extensively drug-resistant (XDR, activity range: 0.07–0.14 μM) Mtb.28 These compounds were non-cytotoxic against the verda reno (VERO) cell line (IC50 >128 μM). They further explored the structure–activity relationship (SAR) of imidazo[1,2-a]pyridine-3-carboxamides and developed several compounds with improved activity against Mtb.29 Five compounds showed an impressive MIC90 of ≤0.006 μM against the Mtb. SAR investigation revealed that the bulky and more lipophilic biaryl ethers had a nanomolar range of potency. One of the front-runners 4 (Fig. 2) showed excellent activity against MDR and XDR Mtb strains (MIC90 range: ≤0.03–0.8 μM), which is better than one of the recently approved anti-TB drugs, pretomanid. Further, a promising in vivo mouse pharmacokinetic (PK) profile (AUC 3850 ng h mL−1 at 3 mg kg−1 PO, t1/2 13.2 h and bioavailability 31.1%) supported the series as an exciting class of anti-TB agents.
 | ||
| Fig. 2 First-generation highly potent IPAs. | ||
Zolpidem (3), an FDA-approved drug for the treatment of insomnia contains imidazo[1,2-a]pyridine core, which inspired the same investigators to re-purpose it for TB.30 Subsequently, 3 was modified to 3-carboxylates, 3-oxoacetamides, and 3-acetamides analogues, and the modified analogues were evaluated against replicating Mtb in various growth media. SAR studies revealed that 3-carboxylates series was more potent (MIC90 range: 0.003 to 0.05 μM) than 3-oxoacetamides and 3-acetamides. However, among all the synthesized series of compounds, benzyl amides containing analogues were the most active. Most potent compounds were profiled for the cytotoxicity against VERO, Hela (Henrietta Lacks' cells), PC-3 (prostate cancer), and MCF-7 (Michigan Cancer Foundation-7) cell lines. With a few exceptions, most of the compounds showed non-toxicity against the tested cell lines. These IPAs were also tested against non-tubercular mycobacteria and were found active. These compounds were also identified as QcrB inhibitors, and the most potent compound 6 showed an MIC90 of 0.004 μM against replicating Mtb and was nontoxic (Fig. 2). Compounds 4 and 6 in Fig. 2, which were synthetically accessible IPAs showing excellent potency against Mtb, can be considered as the 1st generation IPAs for the treatment of TB.
Japanese Global Health Innovative Technology (GHIT) Fund and Bill & Melinda Gates Foundation came together with the experts from the Medicines for Malaria Venture (MMV), the Drugs for Neglected Diseases initiative (DNDi), and the TB Alliance, to set disease-specific criteria for hits and leads for different infectious diseases including TB to achieve good quality starting point of the projects that can improve the likelihood of clinical success.31 The hit and lead compounds are shown in Fig. 2, which followed a set criteria (for detailed criteria of anti-TB hits and leads, please refer to the original report)31 in terms of activity, cytotoxicity, tractable SAR, etc. Compounds 4 and 6 were the ideal starting points to develop the IPA series for anti-TB drug discovery research.
Discovery of telacebec (Q203)
Kang and co-workers explored a novel series of imidazo[1,2-a]pyridine amides leading to a clinical candidate Q203 which was active against MDR- and XDR-TB.32 A library of novel IPAs was synthesized to obtain lead compounds with good drug-like properties against Mtb. IPA series was evaluated against both extracellular and intracellular Mtb. To prioritize the compounds for in vivo efficacy, metabolic studies were performed both in mouse and human liver microsomes and based on the data a good structure–property relationship (SPR) was established. Initial SAR studies were explored across 3-carboxamide. N-Benzylcarboxamide e.g., compound 7 showed potent activity (MIC80, 0.045 μM) while all other modifications like tertiary carboxamide, reversed amide, insertion of methylene between the IP ring, and amide functionality abolished the activity (Fig. 3a). Further exploration of substituents at C2 and C6 positions identified compound 8, having 2-ethyl-6-chloro imidazo[1,2-a]pyridine ring, with significantly improved potency against extracellular (MIC80, 0.0009 μM) as well as intracellular (MIC80, 0.00045 μM) Mtb with good microsomal stability (t1/2 – human 83 min and mouse 63 min). Unfortunately, the high lipophilic nature of this compound resulted in the precipitation and extended absorption in the gut resulting in suboptimal plasma exposure, hence requiring further lead optimization. Insertion of piperidine and piperazine (ring A) between two phenyl rings helped to improve microsomal stability (Fig. 3b). The second phenyl ring with C-4 substitution (ring B) was essential to maintain the potency. e.g., compound 9 with N-4-phenylpiperazine showed enhanced stability in both human and mouse liver microsomes with potent intracellular activity (MIC80, 0.0003 μM) (Fig. 3a). Additionally, the ionizable nature of the basic saturated ring viz. piperazine and piperidine increased the solubility under acidic conditions without losing overall activity. The metabolic stability was influenced by C4-substituent on ring B following an order of substituents viz. trifluoromethoxy > chlorine > fluorine, respectively. Further lead optimization afforded Q2O3 (10) showing MIC80 0.004 μM against extracellular and 0.0014 μM against intracellular Mtb with high microsomal stability and minimal cytochrome P450 inhibition (CYP450, IC50 >100 μM; Fig. 3a).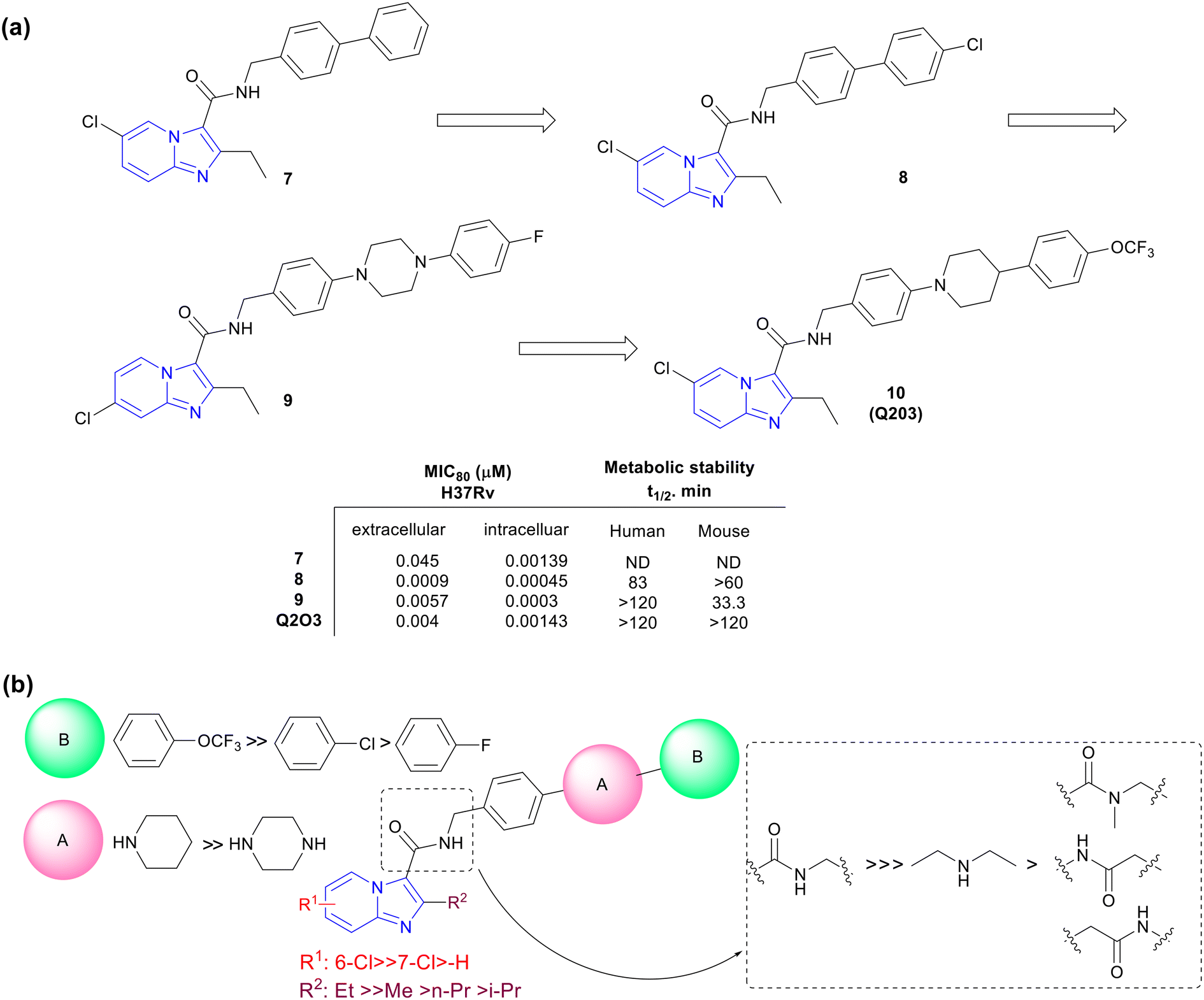 | ||
| Fig. 3 (a) Schematic representation of lead optimization towards the discovery of Q203. (b) Generic SAR for anti-Mtb potency of IPA series. | ||
Q2O3 showed a good PK profile (t1/2, 23.4 h; oral bioavailability, 90.7%; and IV-Cl, 4.0 mL min−1 kg−1) in mice and no cytotoxicity (IC50, >10 μM) against three human cell lines e.g., neuroblastoma cell line (SH-SY5Y), human embryonic kidney 293 (HEK293), and human liver cancer cell line (HepG2). The compound did not inhibit the human ether-à-go-go-related gene (hERG), suggesting a low risk of cardiotoxicity. The acute TB mouse model (BALB/c mice infected with Mtb H37Rv)33 indicated 90%, 99%, and 99.9% reduction of bacterial load when treated with 0.4, 2.0, and 10.0 mg kg−1 doses of Q203, respectively, after 4 weeks of treatment. Importantly, Q203 displayed pharmacokinetic and safety profiles compatible with once-daily dosing. Hence it was selected as a clinical candidate for further development.34
Side chain modification of Q203
Kang et al. focused on side chain modification of Q2O3 to understand the role of the right-hand side chain moiety on anti-tubercular activity.35 Two types of compounds were synthesized viz. type 1 (compound 11) and type 2 (compound 12), respectively (Fig. 4a). Type 1 compounds contained linkers like ether, methyl ether, amine, and methyl amines between two aryl moieties which made them conformationally more flexible than Q2O3. The introduction of 6-Br on the IP ring resulted in a decrease in activity by 16-fold while the Cl atom enhanced the potency, indicating a crucial role of a steric factor over an electronic factor in determining extracellular MIC80. Type 1 compounds with –OCH2, –O, and –NMe linkers in combination with trifluoromethoxy substituents on the terminal phenyl ring showed potent MICs whereas those with –NCH2, and –NH linkers showed significantly weaker MICs. Type 2 analogues contained cyclic amines like piperidine, pyrrolidine, and piperazine along with the extended phenyl ring. Regardless of the R2 substituent on the phenyl ring in type 2, analogues having ether linker next to the piperidine ring displayed excellent activity (MIC80, 0.003–0.026 μM). It was suggested that the lipophilicity is more important than the linearity of the side chain in retaining activity, and a flexible linker will help reorientation of the side chain for strong target binding. The in vivo pharmacokinetic properties study of the most potent analogues (11a, 11b, 12a, 12b; Fig. 4) showed low systemic clearance (2.23–5.77 mL min−1 kg−1) with good oral bioavailability (51.4–80.5%) indicating the potential to further evaluate theses analogues for their in vivo efficacy.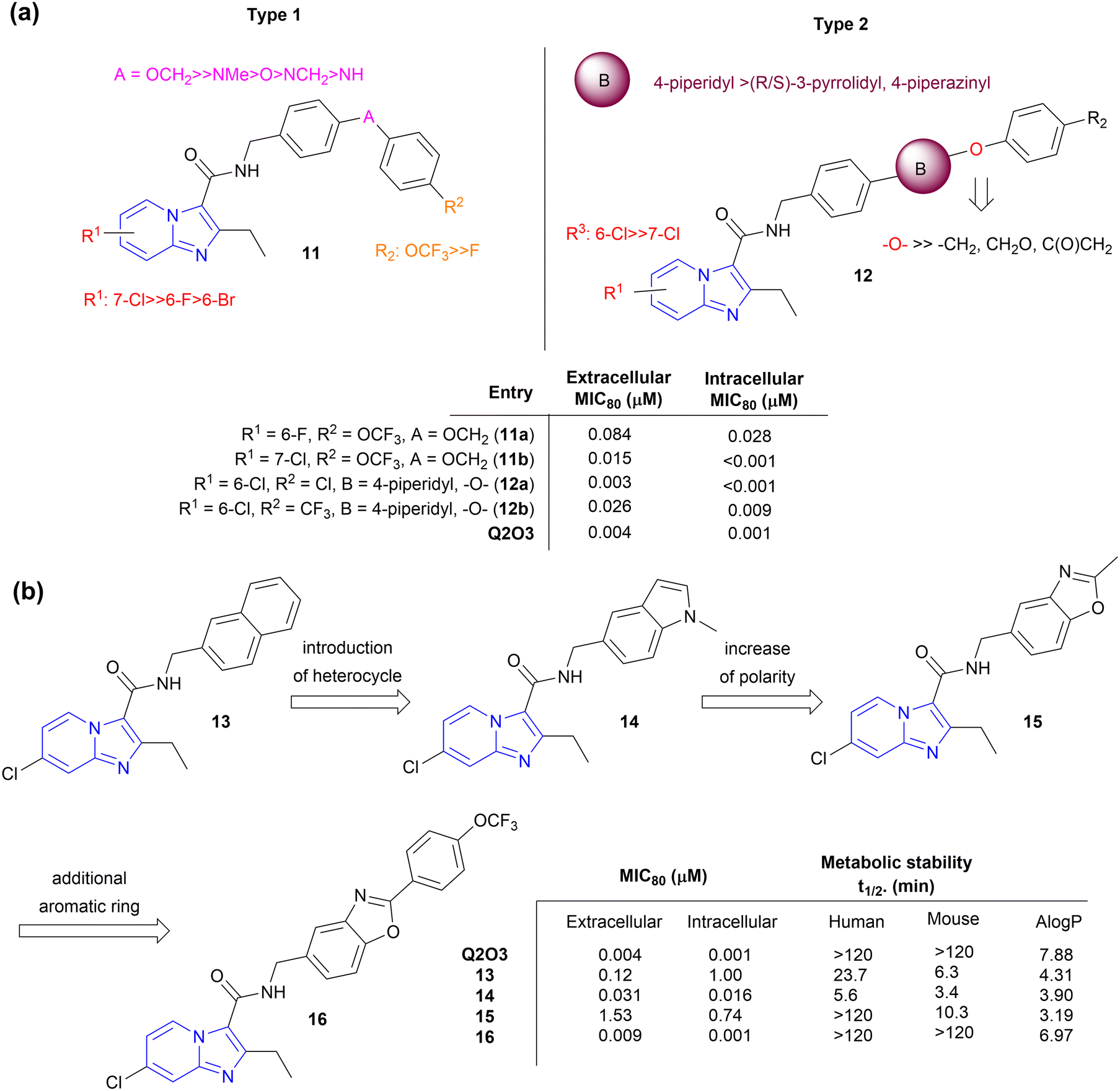 | ||
| Fig. 4 Side-chain modification of Q203. (a) Generic SAR of side-chain modification with linkers, (b) Lead optimization of side-chain modification with a fused ring system. | ||
Similarly, the introduction of fused bicyclic rings such as naphthyl, indole, and benzo[d]oxazole (compounds 12–15, Fig. 4b) maintained anti-TB activity with a reduction of lipophilicity compared to Q203 (AlogP, 4.3–3.2), but compounds were rapidly metabolized in human and mouse liver microsomes with exception of 15 that showed relatively better stability in human microsomes.36,37 Compound 16 with 4-F-phenyl group added at C2-position of benzo[d]oxazole ring showed >100-fold better anti-TB activity and high metabolic stability in both human and mouse microsomes. On oral administration at a dose of 10 mg kg−1, 16 showed low systemic clearance and high drug exposure levels (AUC0-inf, 116![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 h ng mL−1) with a long half-life of 36.3 h.
400 h ng mL−1) with a long half-life of 36.3 h.
Core modifications of Q203
The success of Q203 kindled interest in exploring scaffold hopping,38–42i.e., the replacement of the fused bicyclic imidazo[1,2-a]pyridine framework of IPAs with another fused bicyclic scaffolds, to identify molecules targeting the QcrB complex. In 2015, Tang and co-workers43 reported a scaffold hopping study on Ambien “anagram” (17)30 as an anti-TB agent, derived from the sleep aid, Ambien (zolimidine, 1). The investigators designed and synthesized a library of pyrazolo[1,5-a]pyridines-3-carboxamides (PPAs). One of the lead compounds 18 was encouragingly active against Mtb (Fig. 5a). Further SAR studies on 18 led to a highly potent anti-TB compound TB47 (19), a structurally matched pair of Q203 with a PPA core. TB47 was equipotent to Q203 (Fig. 5a) and was confirmed to be a QcrB inhibitor.44 The compound was not cytotoxic (CC50, >100 μM on Vero and HepG2 cells) or cardiotoxic (hERG IC50, >30 μM).43 The compound showed high plasma protein binding and poor CaCo-2 permeability but demonstrated an excellent rat PK profile (t1/2, 19 h; oral bioavailability, 94%). Furthermore, 19 was efficacious in the acute mouse model and showed synergy when combined with rifampicin (10 mg kg−1) or pyrazinamide (150 mg kg−1), but was less active in the chronic mouse model compared to Q203,45 but it demonstrated good in vivo synergy with other anti-TB agents such as clofazimine and linezolid in mouse relapse model of TB.46 It also showed good in vivo efficacy against M. ulcerans.47 The compound is undergoing preclinical safety studies.48 The backup candidate, analogue 20 with heterobiaryl side chain, was 3-fold more active against Mtb than 19 and did not show cytotoxicity against VERO cells (CC50, >110 μM, Fig. 5a).49 It displayed a moderate rat PK profile (t1/2, 5.1 h; oral bioavailability, 41%). Efficacy in an acute mouse model suggested that the compound has the potential to be a lead candidate for further anti-TB drug discovery.48,50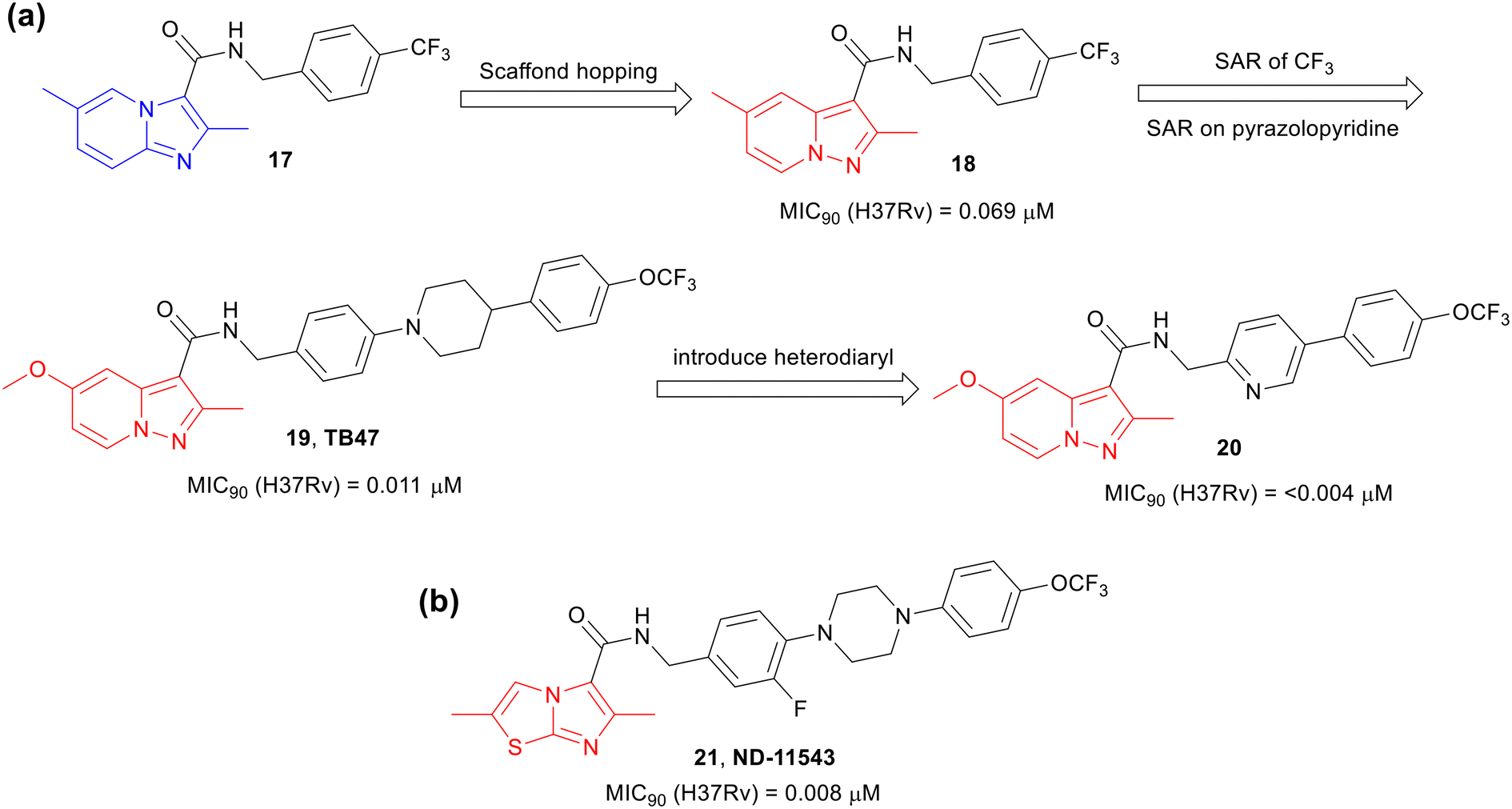 | ||
| Fig. 5 Scaffold hopping of Q203. (a) Development of TB47 and further optimization, (b) ND-11543. | ||
Moraski et al. performed scaffold hopping SAR study51 and developed imidazo[2,1-b]thiazole derivative ND-11543 (21) (Fig. 5b).52 Compound 21 exhibited excellent activities against both replicating (MIC90 0.008 μM) and nonreplicating (LORA, MIC90, 1.1 μM) Mtb and was shown to target QcrB. It was noncytotoxic against VERO cells (CC50, >100 μM), had reasonable solubility (50 μM), showed moderate human liver microsomal stability (t1/2, 28 min), but showed 100% plasma protein binding with low CaCo-2 permeability. The compound had reasonable total drug exposure that remained above the MIC for 24 h at 200 mg kg−1 oral dose in mice but showed only ∼0.3log CFU reduction in the chronic model most likely due to due to inadequate free drug exposure above MIC. Further optimization of the physicochemical and pharmacokinetic properties is warranted for progressing this class of compounds into preclinical studies.
Other IPs targeting QcrB
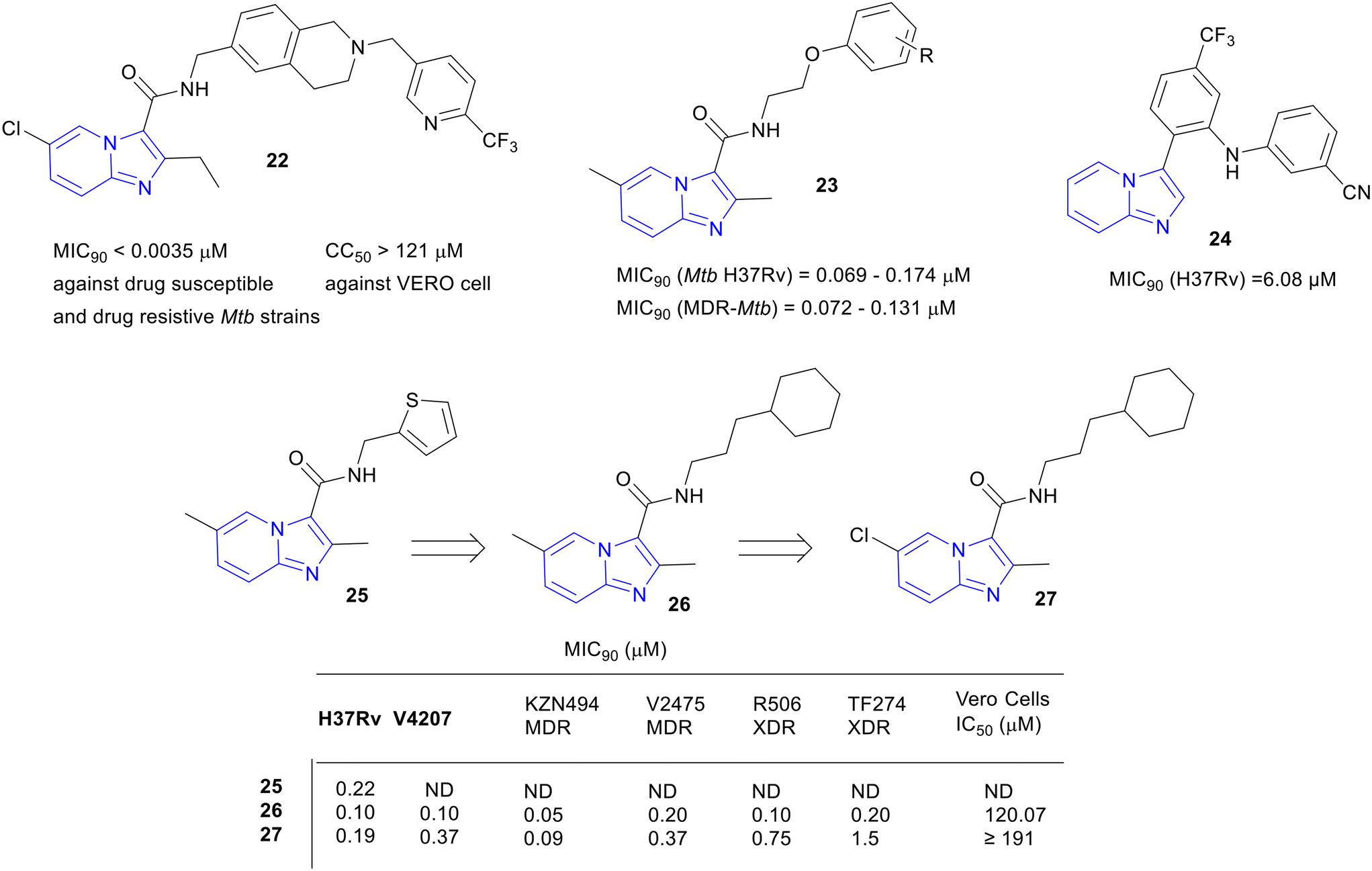
To reduce the lipophilicity of Q2O3, Wang and co-workers explored new series of IPA derivatives with basic heterocycles, tetrahydroisoquinolines, obtained by the fusion of benzene with piperidine or pyrrolidine rings.53 They optimized IPA 22 (Fig. 6) which was equipotent as Q203 with MIC90 <0.035 μM against both drug-susceptible (DS) and DR Mtb strains. It was non-cytotoxic against the VERO cell line (CC50, >121 μM) and showed better aqueous solubility compared to Q203 (2.10 mg mL−1vs. 0.079 mg mL−1) resulting in better drug levels in plasma at an oral dose of 25 mg kg−1 in mice (AUC0–∞ 54916 h ng mL−1vs. 46637 h ng mL−1).
 | ||
| Fig. 6 IPs as potent Mtb inhibitors, targeting QcrB. | ||
A series of N-(2-phenoxyethyl)imidazo[1,2-a]pyridine-3-carboxamides 23 was designed and synthesized as a novel anti-TB agent (Fig. 6).54 Seven most active compounds from the series showed excellent in vitro activity (MIC90, 0.069–0.174 μM) against DS Mtb. The compounds were also active against MDR-Mtb strains. The lead compound, where R = 4-Br, demonstrated an acceptable safety profile in the acute toxicity study and a good PK profile (AUC0–∞, 288.22 h ng mL−1; t1/2, 1.5 h) in rats.
With an intent to explore other IPs, Karale et al. reported a new potent anti-TB agent, 3-arylsubstituted IP, by replacing the extended amide side chain of Q203 with different substituted aryl or heteroaryl rings at the C-3 position.55,56 Initially they found a hit compound with a trifluromethylphenyl substitution at the C-3 position (MIC90, 21.16 μM). Further SAR exploration of the aromatic ring on C-3, led to amine-substituted 3-arylimidazo[1,2-a]pyridine analog with promising activity against Mtb. Despite good anti-Mtb activities, most of the compounds showed significant cytotoxicity against Chinese hamster ovary cells (CHO-K1). However, compound 24 (MIC90, 6.08 μM) indicated significant improvement in cytotoxicity (CHO-K1, IC50, 216.72 μM) (Fig. 6). The docking model of 24 in the active side of Mtb QcrB showed significant π–π interactions with amino acid residues, confirming 24 as a promising lead compound to proceed further (refer to Fig. 3 in the original article).55
Onajole et al. reported IPA analogues containing cyclic aliphatic rings at the amide end and evaluated against DS H37Rv, V4207, MDR KZN494, MDR V2475, XDR R506, and TF274 strains of Mtb.57 SAR studies revealed that the potency of analogues is dependent on the carbon spacer between amide nitrogen and the cyclic aliphatic ring. In general, a three-carbon spacer enhanced the overall activity. Most active compounds 26 and 27 were 1.5–3-fold more potent than the reference drug isoniazid (Fig. 6). Most potent compounds were tested against MDR and XDR clinical isolates, and results demonstrated their high efficiency to treat DR TB. The toxicity of the active compounds was screened against VERO cells and the compounds were non-cytotoxic with a good selectivity index (IC50 VERO/MIC90, ≥1024).
IPs with other MOAs
2-Trans-enoyl-acyl carrier protein reductase (InhA) inhibitors
Synthesis of various imidazo[1,2-a]pyridine-2-carboxamide derivatives and their profiling against Mtb and human cell line were done by Jose and co-workers.58 Synthesized series of IPAs (28) were screened against Mtb and the most potent derivatives of the series showed a MIC ranging from 17 to 30 μM (Fig. 7) with no cytotoxicity. The molecular docking study of the InhA domain with a few synthesized compounds showed good inhibition against the enzyme. The SAR study indicated that 2-carboxamides IPAs showed much weaker MICs than the IPAs with 3-carboxamides and a switch in the mode of action (MoA) could be a possible reason.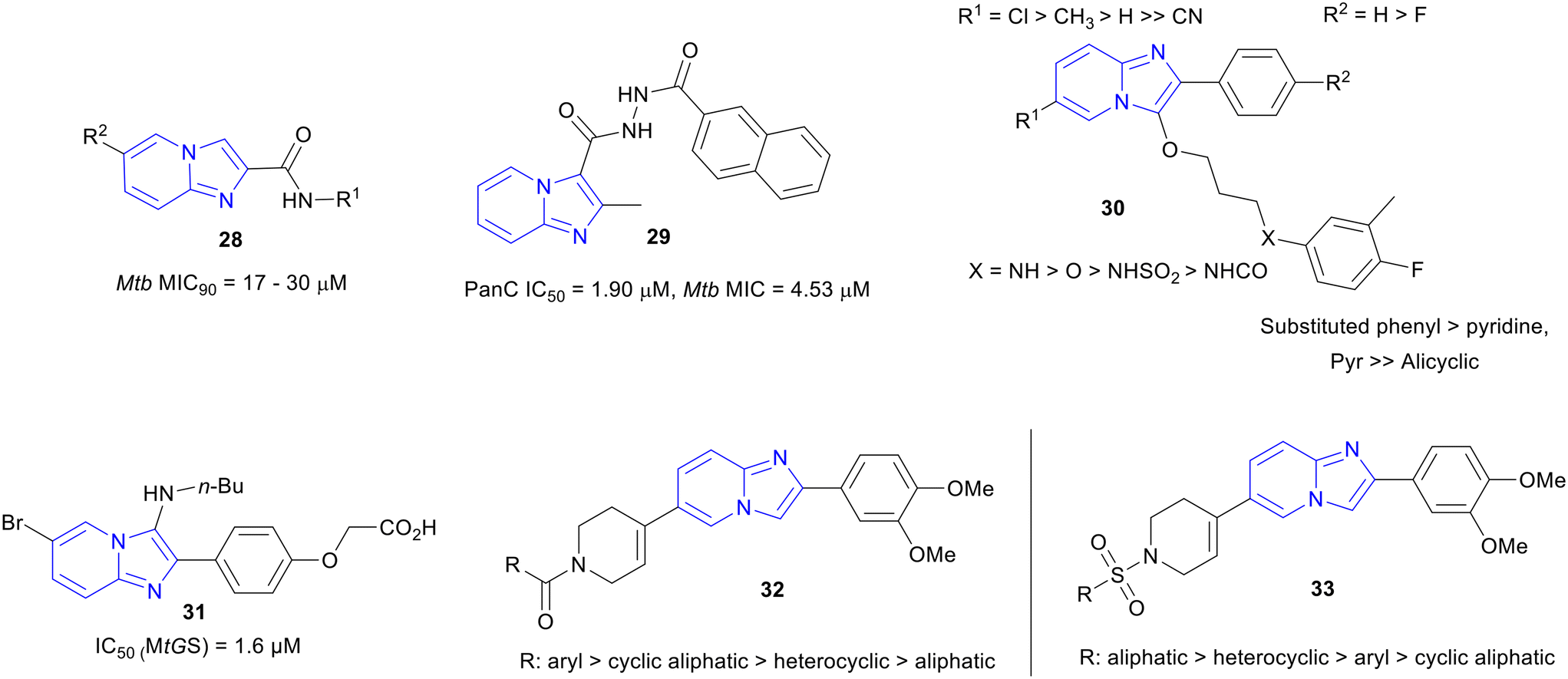 | ||
| Fig. 7 IPs as potent Mtb inhibitors with various MoAs. The “>” sign indicates better target and whole cell activity. | ||
Mtb pantothenate synthetase (PS) inhibitors
2-Methylimidazo[1,2-a]pyridine-3-carbohydrizides were identified as Mtb pantothenate synthetase (PS) inhibitors.59 Derivative 29 (Fig. 7) was the most active compound against Mtb (MIC90, 4.53 μM) from the library of synthesized hydrazides and it displayed no cytotoxicity against mouse macrophage cell line (RAW 264.7). The PS inhibition was confirmed by testing the compounds in a PS assay where adenosine monophosphate (AMP) was produced by the oxidation of NADH to NAD+, because of the condensation of β-alanine and pantoate through different kinases. Most of the compounds showed more than 50% inhibition against Mtb PS.ATP synthase inhibitors
Imidazo[1,2-a]pyridine ethers (IPE, 30) were discovered as selective and potent inhibitors of mycobacterial ATP synthesis (ATPS) and anti-Mtb activity (Fig. 7).60 The IPE hit was identified from the AstraZeneca library using HTS. During the hit to lead, a library of IPEs was developed based on both ATP synthesis inhibition61–64 and Mtb MIC. The most active compounds showed a potency of MIC80 <0.5 μM with ATPS IC50 <0.02 μM. The general SAR informed that the bridgehead nitrogen of IP and directly attached phenyl ring were essential for maintaining the biological activity (Fig. 7). Chloro/methyl substitutions at the R1 position were responsible for the best potency, and H got preference over F at the R2 position for improving potency. Interestingly, –NH– or –O– at the terminal aryl linkage yielded the best potency against both the target and the Mtb, among all the other linker functionalities. The detailed SAR trend showed that –NH– linkage acted slightly better than the –O– linker in terms of potency. The substituted phenyl was preferred over hetero aromatic rings at the terminal ring of the side chain for maintaining activity. However, the minimum bactericidal concentration (MBC) of many IPEs was >100 μM against Mtb. In addition, the compounds from this series faced the barrier of poor solubility and PK profile that decided to deprioritize the series for further development.Mtb glutamine synthetase (MtGS) inhibitors
3-Amino-imidazo[1,2-a]-pyridines as glutamine synthetase (MtGS) inhibitor, a potential target for the Mtb drug development, were developed.65,66 A library of 3-amino-imidazo[1,2-a]-pyridines was synthesized employing microwave-assisted one-pot Ugi-type multi-component reaction. The molecular docking model of one of the most active compounds 31 (MtGS IC50, 1.6 μM) on the MtGS complex was found to be a very good fit in the ATP-binding site and confirmed the target (Fig. 7).65IPs with unknown MOAs
Recently, two novel imidazopyridine series were reported: N-carbonyl substituted IP (C-IP) and N-sulphonyl substituted IP (S-IP).67 Libraries of C-IP (32) and S-IP (33) were synthesized and evaluated for in vitro anti-Mtb activity. A few compounds exhibited excellent activities (IPA MIC90, 0.11 μM; IPS MIC90, 0.98 μM). The SAR trends for both series were reported (Fig. 7). Aromatic and cyclic aliphatic amide groups were responsible for the improved potency of C-IP whereas, aliphatic and heterocyclic sulphonamides of S-IP were more active against Mtb. Active compounds from both series were found to be nontoxic against HEK cells (SI ≥66). Furthermore, predicted ADME (absorption–distribution–metabolism–excretion) values for the lead compounds were obtained within the range of the marketed drugs.Hybrid molecules
To expand SAR studies of potential anti-TB agents, a hybrid scaffold approach of combining two classes of known anti-TB compounds has been used. Majewski et al. reported the coupling of piperazino-1,3-benzothiazin-4-ones (PBTZs) with IP to explore the anti-TB potency of the synthesized conjugates.68 Benzothiazinones are anti-TB drug candidates that target arabinan synthesis.69–71 The conjugated compound 34 was synthesized by the coupling reaction between imidazopyridine carboxylic acid and TFA salt of PBTZ, obtained after Boc deprotection of PBTZ-Boc (Fig. 8). PBTZ-Boc showed an MIC90 of 0.28 μM against Mtb, whereas the conjugate 34 lost about 7.5-fold potency. They introduced a 4-aminomethylphenylcarboxyl spacer in between IP and PBTZ but the strategy (compound 36) was further detrimental to the activity. In summary, the conjugation of two potent anti-TB agents PBTZ and IPA did not yield improved potency.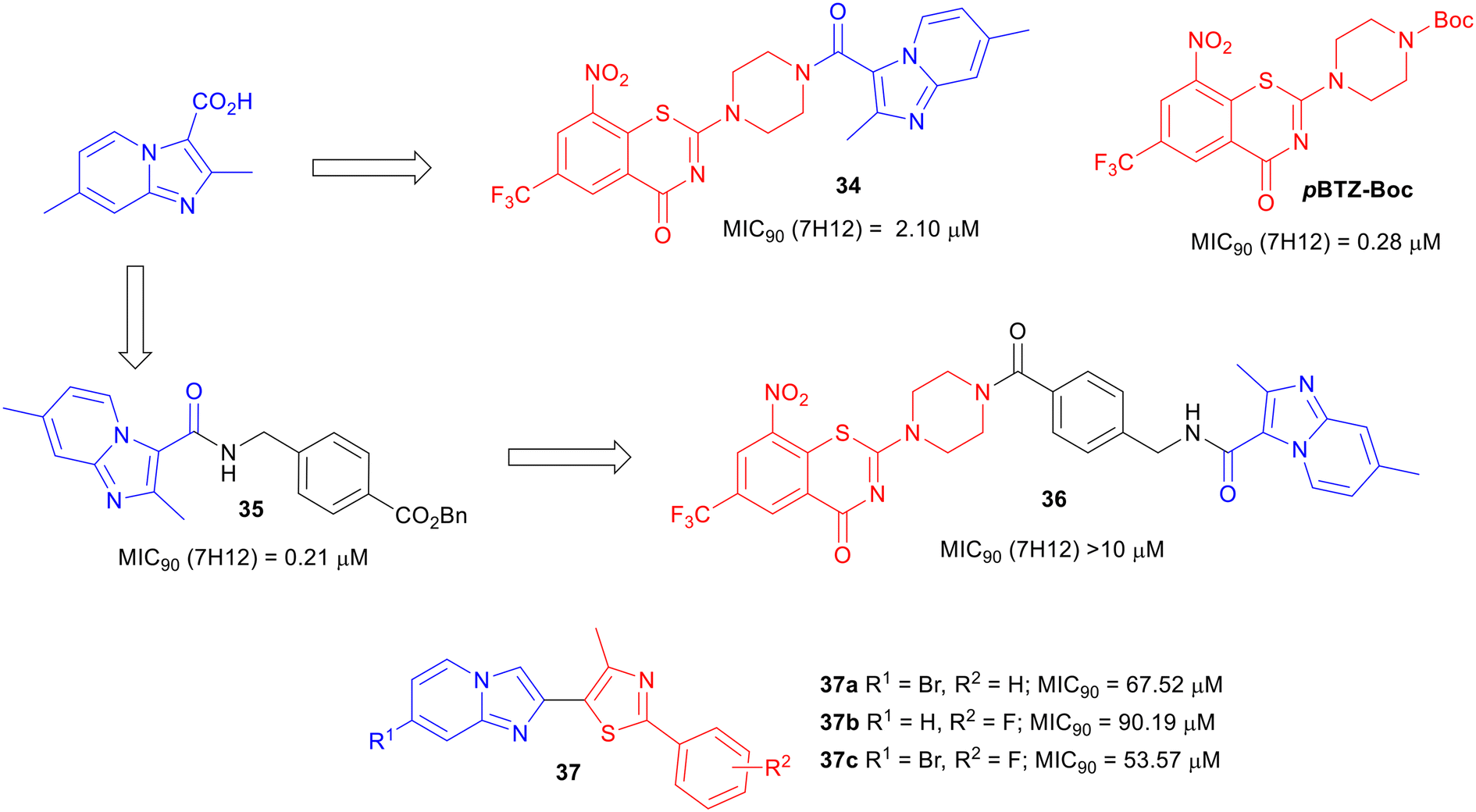 | ||
| Fig. 8 Hybrid molecules having IP scaffold showing activities against Mtb. | ||
Thiazoles are another important pharmacophore known for anti-TB activity.72–74
Abhale et al. synthesized a set of thiazole and IP conjugates 37.75 Only a few compounds (37a, 37b, and 37c) from their synthesized library showed a low potency against M. smegmatis (Fig. 8). Although a few conjugation strategies between two or more classes of pharmacophores have been able to yield anti-Mtb agents with good potency, the hybridization of IP with another class of compounds did not yield promising results in terms of TB drug discovery research.
Conclusions
Despite significant challenges, the recent advancements in TB drug discovery and development provide a positive outlook for the future. In this respect, the imidazopyridine class of compounds, recognized as the ‘drug prejudice’,76 have gained significant consideration in the recent past. Q203, the most advanced anti-TB drug candidate from this class, is in phase II clinical trial.77,78 The development of Q203 offered a new horizon to anti-TB drug discovery research targeting QcrB. Several synthetic modification strategies of imidazopyridine amides have been acquired around Q203, particularly the long-side chain modification to reduce lipophilicity and induce polarity. In vivo studies have been initiated with a few modified compounds. The case study of IPAs leading to the discovery of Q203 demonstrates that the introduction of polar functionalities, such as amides and higher sp3 character is essential to overcome issues of high lipophilicity and poor aqueous solubility of fused bicyclic systems, such as IPs, to deliver drug-like molecules. Further exploration in this area needs to incorporate these learnings. Scaffold hopping of Q203 contributed significant advancement to the anti-TB drug discovery research. TB47 is one of the promising scaffold-hopped candidates completing safety profiling at pre-clinical development. Other IP designs also showed promising initial results to take a few of them for further development. In summary, although TB drug discovery research of IP class looks promising, continuous discovery and development of anti-TB compounds are essential to fight against DR Mtb.Author contributions
S. S., S. K., E. K. A., S. R. G., and V. S. designed the content and flow of the paper. S. S., S. K., and E. K. A. collected the necessary information/data and prepared the first draft. S. S., S. K., E. K. A., S. R. G., and V. S. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Bill and Melinda Gates Foundation (Global Health Grant Number OPP1066878), South African Medical Research Council (SAMRC), Strategic Health Innovation Partnerships (SHIP) unit of the SAMRC, Technology Innovation Agency (TIA), South African Department of Science and Innovation, and South African National Research Foundation are gratefully acknowledged for support.References
- W. H. Organization, Global tuberculosis report 2022, 2022 Search PubMed.
- W. H. Organization, WHO operational handbook on tuberculosis: module 4: treatment: drug-susceptible tuberculosis treatment, Report 9240050760, 2022 Search PubMed.
- V. Singh and K. Chibale, Acc. Chem. Res., 2021, 54, 2361–2376 CrossRef CAS PubMed.
- B. B. Aldridge, D. Barros-Aguirre, C. E. Barry, R. H. Bates, S. J. Berthel, H. I. Boshoff, K. Chibale, X.-J. Chu, C. B. Cooper and V. Dartois, Nat. Med., 2021, 27, 1333–1337 CrossRef CAS PubMed.
- F. Conradie, A. H. Diacon, N. Ngubane, P. Howell, D. Everitt, A. M. Crook, C. M. Mendel, E. Egizi, J. Moreira and J. Timm, N. Engl. J. Med., 2020, 382, 893–902 CrossRef CAS PubMed.
- A. K. Kakkar and N. Dahiya, Tuberculosis, 2014, 94, 357–362 CrossRef CAS PubMed.
- S. J. Keam, Drugs, 2019, 79, 1797–1803 CrossRef PubMed.
- N. J. Ryan and J. H. Lo, Drugs, 2014, 74, 1041–1045 CrossRef CAS PubMed.
- G. F. Fernandes, A. M. Thompson, D. Castagnolo, W. A. Denny and J. L. Dos Santos, J. Med. Chem., 2022, 65, 7489–7531 CrossRef CAS PubMed.
- S. Huszár, K. Chibale and V. Singh, Drug Discovery Today, 2020, 25, 772–780 CrossRef PubMed.
- New TB Drugs Clinical Pipeline, https://www.newtbdrugs.org/pipeline/clinical Search PubMed.
- C. Parisio and F. Clementi, Lab. Invest., 1976, 35, 484–495 CAS.
- D. Belohlavek and P. Malfertheiner, Scand. J. Gastroenterol., Suppl., 1979, 54, 44–44 CAS.
- B. Zivkovic, E. Morel, D. Joly, G. Perrault, D. Sanger and K. Lloyd, Pharmacopsychiatry, 1990, 23, 108–113 CrossRef PubMed.
- D. Sanger and B. Zivkovic, Psychopharmacology, 1994, 113, 395–403 CrossRef CAS PubMed.
- J. Hoehns and P. Perry, Clin. Pharm., 1993, 12, 814–828 CAS.
- A. Deep, R. Kaur Bhatia, R. Kaur, S. Kumar, U. Kumar Jain, H. Singh, S. Batra, D. Kaushik and P. Kishore Deb, Curr. Top. Med. Chem., 2017, 17, 238–250 CrossRef CAS PubMed.
- A. M. Thompson and W. A. Denny, in Annual reports in medicinal chemistry, Elsevier, 2019, vol. 52, pp. 97–130 Search PubMed.
- C. S.-Y. Foo, K. Pethe and A. Lupien, Appl. Sci., 2020, 10, 2339 CrossRef CAS.
- I. K. Iqbal, S. Bajeli, A. K. Akela and A. Kumar, Pathogens, 2018, 7, 24 CrossRef PubMed.
- Q. Li and X. Lu, Chin. Chem. Lett., 2020, 31, 1357–1365 CrossRef CAS.
- D. Bald, C. Villellas, P. Lu and A. Koul, MBio, 2017, 8, e00272-17 CrossRef PubMed.
- G. M. Cook, K. Hards, C. Vilchèze, T. Hartman and M. Berney, Microbiol. Spectrum, 2014, 2, 2.3. 06 CrossRef PubMed.
- T. O’malley, T. Alling, J. V. Early, H. A. Wescott, A. Kumar, G. C. Moraski, M. J. Miller, T. Masquelin, P. A. Hipskind and T. Parish, Antimicrob. Agents Chemother., 2018, 62, e02439-17 CrossRef PubMed.
- H. Gong, J. Li, A. Xu, Y. Tang, W. Ji, R. Gao, S. Wang, L. Yu, C. Tian and J. Li, Science, 2018, 362, eaat8923 CrossRef PubMed.
- S. Safarian, H. K. Opel-Reading, D. Wu, A. R. Mehdipour, K. Hards, L. K. Harold, M. Radloff, I. Stewart, S. Welsch and G. Hummer, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- K. A. Abrahams, J. A. Cox, V. L. Spivey, N. J. Loman, M. J. Pallen, C. Constantinidou, R. Fernandez, C. Alemparte, M. J. Remuinan and D. Barros, PLoS One, 2012, 7, e52951 CrossRef CAS PubMed.
- G. C. Moraski, L. D. Markley, P. A. Hipskind, H. Boshoff, S. Cho, S. G. Franzblau and M. J. Miller, ACS Med. Chem. Lett., 2011, 2, 466–470 CrossRef CAS PubMed.
- G. C. Moraski, L. D. Markley, J. Cramer, P. A. Hipskind, H. Boshoff, M. A. Bailey, T. Alling, J. Ollinger, T. Parish and M. J. Miller, ACS Med. Chem. Lett., 2013, 4, 675–679 CrossRef CAS PubMed.
- G. C. Moraski, P. A. Miller, M. A. Bailey, J. Ollinger, T. Parish, H. I. Boshoff, S. Cho, J. R. Anderson, S. Mulugeta and S. G. Franzblau, ACS Infect. Dis., 2015, 1, 85–90 CrossRef CAS PubMed.
- K. Katsuno, J. N. Burrows, K. Duncan, R. H. Van Huijsduijnen, T. Kaneko, K. Kita, C. E. Mowbray, D. Schmatz, P. Warner and B. Slingsby, Nat. Rev. Drug Discovery, 2015, 14, 751–758 CrossRef CAS PubMed.
- S. Kang, R. Y. Kim, M. J. Seo, S. Lee, Y. M. Kim, M. Seo, J. J. Seo, Y. Ko, I. Choi and J. Jang, J. Med. Chem., 2014, 57, 5293–5305 CrossRef CAS PubMed.
- J. Rullas, J. I. García, M. Beltrán, P.-J. Cardona, N. Cáceres, J. F. García-Bustos and I. Angulo-Barturen, Antimicrob. Agents Chemother., 2010, 54, 2262–2264 CrossRef CAS PubMed.
- K. Pethe, P. Bifani, J. Jang, S. Kang, S. Park, S. Ahn, J. Jiricek, J. Jung, H. K. Jeon and J. Cechetto, Nat. Med., 2013, 19, 1157–1160 CrossRef CAS PubMed.
- S. Kang, Y. M. Kim, R. Y. Kim, M. J. Seo, Z. No, K. Nam, S. Kim and J. Kim, Eur. J. Med. Chem., 2017, 125, 807–815 CrossRef CAS PubMed.
- S. Kang, Y. M. Kim, H. Jeon, S. Park, M. J. Seo, S. Lee, D. Park, J. Nam, S. Lee and K. Nam, Eur. J. Med. Chem., 2017, 136, 420–427 CrossRef CAS PubMed.
- AlogP prediction was derived from Discovery Studio, BIOVIA, San Diego, CA, 2016 Search PubMed.
- H.-J. Böhm, A. Flohr and M. Stahl, Drug Discovery Today: Technol., 2004, 1, 217–224 CrossRef PubMed.
- H. Sun, G. Tawa and A. Wallqvist, Drug Discovery Today, 2012, 17, 310–324 CrossRef CAS PubMed.
- S. R. Langdon, P. Ertl and N. Brown, Mol. Inf., 2010, 29, 366–385 CrossRef CAS PubMed.
- Y. Hu, D. Stumpfe and J. R. Bajorath, J. Med. Chem., 2017, 60, 1238–1246 CrossRef CAS PubMed.
- P. Ertl, Identification of Bioisosteric Scaffolds using Scaffold Keys, ChemRxiv, 2021, preprint, DOI:10.26434/chemrxiv.13525457.
- J. Tang, B. Wang, T. Wu, J. Wan, Z. Tu, M. Njire, B. Wan, S. G. Franzblauc, T. Zhang and X. Lu, ACS Med. Chem. Lett., 2015, 6, 814–818 CrossRef CAS PubMed.
- S. Zhou, W. Wang, X. Zhou, Y. Zhang, Y. Lai, Y. Tang, J. Xu, D. Li, J. Lin and X. Yang, eLife, 2021, 10, e69418 CrossRef CAS PubMed.
- X. Lu, Z. Williams, K. Hards, J. Tang, C.-Y. Cheung, H. L. Aung, B. Wang, Z. Liu, X. Hu and A. Lenaerts, ACS Infect. Dis., 2018, 5, 239–249 CrossRef PubMed.
- W. Yu, B. Yusuf, S. Wang, X. Tian, H. A. Hameed, Z. Lu, G. Chiwala, M. S. Alam, G. M. Cook and D. A. Maslov, Antimicrob. Agents Chemother., 2021, 65, e00706–e00721 CrossRef CAS PubMed.
- Y. Liu, Y. Gao, J. Liu, Y. Tan, Z. Liu, C. Chhotaray, H. Jiang, Z. Lu, G. Chiwala and S. Wang, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- W. Yu, G. Chiwala, Y. Gao, Z. Liu, S. Sapkota, Z. Lu, L. Guo, S. A. Khan, N. Zhong and T. Zhang, Biomed. Pharmacother., 2020, 131, 110782 CrossRef CAS PubMed.
- X. Hu, B. Wan, Y. Liu, J. Shen, S. G. Franzblau, T. Zhang, K. Ding and X. Lu, ACS Med. Chem. Lett., 2019, 10, 295–299 CrossRef CAS PubMed.
- X. Lu, J. Tang, Z. Liu, M. Li, T. Zhang, X. Zhang and K. Ding, Bioorg. Med. Chem. Lett., 2016, 26, 5916–5919 CrossRef CAS PubMed.
- G. C. Moraski, N. Seeger, P. A. Miller, A. G. Oliver, H. I. Boshoff, S. Cho, S. Mulugeta, J. R. Anderson, S. G. Franzblau and M. J. Miller, ACS Infect. Dis., 2016, 2, 393–398 CrossRef CAS PubMed.
- G. C. Moraski, N. Deboosère, K. L. Marshall, H. A. Weaver, A. Vandeputte, C. Hastings, L. Woolhiser, A. J. Lenaerts, P. Brodin and M. J. Miller, PLoS One, 2020, 15, e0227224 CrossRef CAS PubMed.
- A. Wang, H. Wang, Y. Geng, L. Fu, J. Gu, B. Wang, K. Lv, M. Liu, Z. Tao and C. Ma, Bioorg. Med. Chem., 2019, 27, 813–821 CrossRef CAS PubMed.
- Z. Wu, Y. Lu, L. Li, R. Zhao, B. Wang, K. Lv, M. Liu and X. You, ACS Med. Chem. Lett., 2016, 7, 1130–1133 CrossRef CAS PubMed.
- U. B. Karale, A. U. Shinde, D. A. Babar, K. G. Sangu, S. K. Vagolu, V. K. Eruva, S. S. Jadav, S. Misra, S. Dharmarajan and H. B. Rode, Arch. Pharm., 2021, 354, 2000419 CrossRef CAS PubMed.
- S. Kalari, D. A. Babar, U. B. Karale, V. B. Makane and H. B. Rode, Tetrahedron Lett., 2017, 58, 2818–2821 CrossRef CAS.
- O. K. Onajole, S. Lun, Y. J. Yun, D. Y. Langue, M. Jaskula-Dybka, A. Flores, E. Frazier, A. C. Scurry, A. Zavala and K. R. Arreola, Chem. Biol. Drug Des., 2020, 96, 1362–1371 CrossRef CAS PubMed.
- G. Jose, T. S. Kumara, G. Nagendrappa, H. Sowmya, D. Sriram, P. Yogeeswari, J. P. Sridevi, T. N. G. Row, A. A. Hosamani and P. S. Ganapathy, Eur. J. Med. Chem., 2015, 89, 616–627 CrossRef CAS PubMed.
- G. Samala, R. Nallangi, P. B. Devi, S. Saxena, R. Yadav, J. P. Sridevi, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2014, 22, 4223–4232 CrossRef CAS PubMed.
- S. J. Tantry, S. D. Markad, V. Shinde, J. Bhat, G. Balakrishnan, A. K. Gupta, A. Ambady, A. Raichurkar, C. Kedari and S. Sharma, J. Med. Chem., 2017, 60, 1379–1399 CrossRef CAS PubMed.
- A. Matsuno-Yagi and Y. Hatefi, J. Biol. Chem., 1993, 268, 1539–1545 CrossRef CAS PubMed.
- J. Zheng and V. D. Ramirez, Br. J. Pharmacol., 2000, 130, 1115–1123 CrossRef CAS PubMed.
- A. Koul, N. Dendouga, K. Vergauwen, B. Molenberghs, L. Vranckx, R. Willebrords, Z. Ristic, H. Lill, I. Dorange and J. Guillemont, Nat. Chem. Biol., 2007, 3, 323–324 CrossRef CAS PubMed.
- T. Yano, S. Kassovska-Bratinova, J. S. Teh, J. Winkler, K. Sullivan, A. Isaacs, N. M. Schechter and H. Rubin, J. Biol. Chem., 2011, 286, 10276–10287 CrossRef CAS PubMed.
- A. Nordqvist, M. T. Nilsson, O. Lagerlund, D. Muthas, J. Gising, S. Yahiaoui, L. R. Odell, B. R. Srinivasa, M. Larhed and S. L. Mowbray, MedChemComm, 2012, 3, 620–626 RSC.
- L. R. Odell, M. T. Nilsson, J. Gising, O. Lagerlund, D. Muthas, A. Nordqvist, A. Karlén and M. Larhed, Bioorg. Med. Chem. Lett., 2009, 19, 4790–4793 CrossRef CAS PubMed.
- Y. M. Khetmalis, S. Chitti, A. U. Wunnava, B. K. Kumar, M. M. K. Kumar, S. Murugesan and K. V. G. C. Sekhar, RSC Med. Chem., 2022, 13, 327–342 RSC.
- M. W. Majewski, R. Tiwari, P. A. Miller, S. Cho, S. G. Franzblau and M. J. Miller, Bioorg. Med. Chem. Lett., 2016, 26, 2068–2071 CrossRef CAS PubMed.
- V. Makarov, G. Manina, K. Mikusova, U. Möllmann, O. Ryabova, B. Saint-Joanis, N. Dhar, M. R. Pasca, S. Buroni and A. P. Lucarelli, Science, 2009, 324, 801–804 CrossRef CAS PubMed.
- C. Trefzer, H. Škovierová, S. Buroni, A. Bobovská, S. Nenci, E. Molteni, F. Pojer, M. R. Pasca, V. Makarov and S. T. Cole, J. Am. Chem. Soc., 2012, 134, 912–915 CrossRef CAS PubMed.
- V. Makarov, B. Lechartier, M. Zhang, J. Neres, A. M. van der Sar, S. A. Raadsen, R. C. Hartkoorn, O. B. Ryabova, A. Vocat and L. A. Decosterd, EMBO Mol. Med., 2014, 6, 372–383 CrossRef CAS PubMed.
- M. R. Shiradkar, K. K. Murahari, H. R. Gangadasu, T. Suresh, C. A. Kalyan, D. Panchal, R. Kaur, P. Burange, J. Ghogare and V. Mokale, Bioorg. Med. Chem., 2007, 15, 3997–4008 CrossRef CAS PubMed.
- G. Turan-Zitouni, A. Özdemir, Z. A. Kaplancikli, K. Benkli, P. Chevallet and G. Akalin, Eur. J. Med. Chem., 2008, 43, 981–985 CrossRef CAS PubMed.
- G. S. Kumar, Y. Rajendraprasad, B. Mallikarjuna, S. Chandrashekar and C. Kistayya, Eur. J. Med. Chem., 2010, 45, 2063–2074 CrossRef PubMed.
- Y. K. Abhale, K. K. Deshmukh, A. V. Sasane, A. P. Chavan and P. C. Mhaske, J. Heterocyclic Chem., 2016, 53, 229–233 CrossRef CAS.
- A. K. Bagdi, S. Santra, K. Monir and A. Hajra, Chem. Commun., 2015, 51, 1555–1575 RSC.
- V. R. de Jager, R. Dawson, C. van Niekerk, J. Hutchings, J. Kim, N. Vanker, L. van der Merwe, J. Choi, K. Nam and A. H. Diacon, N. Engl. J. Med., 2020, 382, 1280–1281 CrossRef PubMed.
- I. Malík, J. Čižmárik, G. Kováč, M. Pecháčová and L. Hudecova, Ceska Slov. Farm., 2021, 70, 164–171 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |