
Mycobacterium tuberculosis CitA activity is modulated by cysteine oxidation and pyruvate binding†
Rasangi
Pathirage
 a,
Lorenza
Favrot‡
b,
Cecile
Petit§
b,
Melvin
Yamsek¶
b,
Sarbjit
Singh
c,
Jayapal Reddy
Mallareddy
c,
Sandeep
Rana
c,
Amarnath
Natarajan
acde and
Donald R.
Ronning
*a
a,
Lorenza
Favrot‡
b,
Cecile
Petit§
b,
Melvin
Yamsek¶
b,
Sarbjit
Singh
c,
Jayapal Reddy
Mallareddy
c,
Sandeep
Rana
c,
Amarnath
Natarajan
acde and
Donald R.
Ronning
*a
aDepartment of Pharmaceutical Sciences, University of Nebraska Medical Center, Omaha, NE 68198, USA. E-mail: don.ronning@unmc.edu
bDepartment of Chemistry and Biochemistry, University of Toledo, Toledo, OH 43606, USA
cEppley Institute for Cancer Research, University of Nebraska Medical Center, Omaha, NE 68198, USA
dDepartment of Genetics Cell Biology and Anatomy, University of Nebraska Medical Center, Omaha, NE 68198, USA
eFred & Pamela Buffett Cancer Center, University of Nebraska Medical Center, Omaha, NE, USA
First published on 5th April 2023
Abstract
As an adaptation for survival during infection, Mycobacterium tuberculosis becomes dormant, reducing its metabolism and growth. Two types of citrate synthases have been identified in Mycobacterium tuberculosis, GltA2 and CitA. Previous work shows that overexpression of CitA, the secondary citrate synthase, stimulates the growth of Mycobacterium tuberculosis under hypoxic conditions without showing accumulation of triacylglycerols and makes mycobacteria more sensitive to antibiotics, suggesting that CitA may play a role as a metabolic switch during infection and may be an interesting TB drug target. To assess the druggability and possible mechanisms of targeting CitA with small-molecule compounds, the CitA crystal structure was solved to 2.1 Å by X-ray crystallography. The solved structure shows that CitA lacks an NADH binding site that would afford allosteric regulation, which is atypical of most citrate synthases. However, a pyruvate molecule is observed within the analogous domain, suggesting pyruvate may instead be the allosteric regulator for CitA. The R149 and R153 residues forming the charged portion of the pyruvate binding pocket were mutated to glutamate and methionine, respectively, to assess the effect of mutations on activity. Protein thermal shift assay shows thermal stabilization of CitA in the presence of pyruvate compared to the two CitA variants designed to decrease pyruvate affinity. Solved crystal structures of both variants show no significant structural changes. However, the catalytic efficiency of the R153M variant increases by 2.6-fold. Additionally, we show that covalent modification of C143 of CitA by Ebselen completely arrests enzyme activity. Similar inhibition is observed using two spirocyclic Michael acceptor containing compounds, which inhibit CitA with ICapp50 values of 6.6 and 10.9 μM. A crystal structure of CitA modified by Ebselen was solved, but significant structural changes were lacking. Considering that covalent modification of C143 inactivates CitA and the proximity of C143 to the pyruvate binding site, this suggests that structural and/or chemical changes in this sub-domain are responsible for regulating CitA enzymatic activity.
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (M. tb), is the thirteenth leading cause of death around the world and the second leading infectious disease, the first one being COVID-19.1 TB is also the prime cause of death among HIV patients when co-infected. According to the estimation of WHO, approximately one-quarter of the world population is infected with TB and 5–15% of them have the active disease. The emergence of drug-resistant TB and co-infection with HIV emphasize the importance of identifying new antitubercular drugs.1,2Under environmental stresses such as acidic pH or low oxygen availability, many bacteria choose to adopt a latent state in which they stop their growth but maintain low metabolic activity.3,4M. tb reacts to environmental stresses in the same way and alters its metabolism.3,5,6 This phenomenon raises a challenge for treating TB as most of the TB drugs in clinical use target the replicating bacteria.3,7 In latency, the bacteria are tolerant to those antibiotics that target replicating bacteria making it more difficult to treat TB.3 Thus, it is critical to gain knowledge about M. tb primary metabolism and how the organism is able to alter its metabolism in order to enter a latent stage and resist antibiotic action.
The tricarboxylic acid (TCA) cycle is an essential part of this metabolism. The TCA cycle includes a sequence of chemical reactions that converts the acetyl moiety of acetyl-CoA (AcCoA) into carbon dioxide and in doing so makes nicotinamide adenine dinucleotide (NADH) that is later used in the electron transport chain to drive oxidative phosphorylation to make ATP. Electron transport chain has been identified as an interesting target for TB treatment over the years and one of the major examples for that is ATP synthase inhibition by cyclic peptides, aminoglycosides, diarylquinolines and bedaquiline.8–12 In addition to its essential role in providing cellular energy, the TCA cycle provides precursors for amino acids, lipids, and other essential macromolecules as well as other important metabolites. Many bacteria possess variations in the TCA cycle that respond to their specific needs and metabolic requirements.13 Some bacterial variants of the TCA cycle include oxidative and reductive pathways that favor the formation of biosynthetic precursors.
Citrate synthase is the first enzyme involved in the TCA cycle. This enzyme catalyzes the conversion of AcCoA and oxaloacetate (OAA) into citrate and free CoA. Numerous research articles have been published on the enzyme citrate synthase. Crystal structures of citrate synthase from various organisms have been solved over the years.14–25 The citrate synthase enzyme catalyzes an aldol–Claisen condensation.15,18 Kinetic studies are consistent with an ordered bi-substrate mechanism.26 Typically, citrate synthase binds oxaloacetate first and undergoes a conformational change that allows the creation of a binding pocket for the second substrate, AcCoA.14,27,28
Two different types of citrate synthase have been identified.22 Type I citrate synthases can be found in most organisms, such as eukaryotes and Gram-positive bacteria. The type I enzymes form a dimer and possess a shorter sequence compared to the type II enzymes and does not show any allosteric regulation. Type II citrate synthases are commonly found in Gram-negative bacteria and exhibits an extra N-terminal β-sheet domain.20,22,29 The type II enzymes usually form a hexameric structure. In some cases, the type II enzymes are allosterically inhibited by NADH, allowing regulation of the TCA cycle.30E. coli citrate synthase is an example of the type II group.
According to the in silico analyses, M. tb encodes three proteins that belong to the citrate synthase family including citrate synthase I encoded by gltA2 gene (Rv0896), citrate synthase II encoded by citA (Rv0889c) and methyl citrate synthase that is encoded by the prpC (Rv1131).31 Out of the two types of citrate synthases, the structure of the GltA2 has been solved to a resolution of 2.6 Å.32 The knockout of GltA2 shows growth defects whereas the CitA is known to be non-essential.33M. tb CitA is 40 kDa and bioinformatic analysis predicts that citrate synthase is a type II enzyme. Even though structural information is lacking for M. tb CitA thus far, the significance of the CitA enzyme has been discussed in two studies.
The first study, by Sassetti and co-workers, demonstrates that several growth-limiting stresses cause the bacteria to favor the carbon flux toward triglyceride synthesis (storage) instead of the TCA cycle.34 Additionally, overexpression of the citA gene showed no accumulation of triglycerides and stimulated the growth of M. tb in hypoxic conditions. Therefore, the sensitivity of M. tb to antibiotics was increased. This suggests that the CitA functions as a metabolic switch during the infection. Therefore, if it can be targeted by small-molecule compounds that maintain high levels of enzymatic activity, the carbon flux can be maintained to ensure bacteria remain in the active phase and retain susceptibility to antibiotics.
The second relevant study highlighting possible importance of CitA as a metabolic switch is by Antelmann et al. Glutathione is the major low molecular weight thiol in gram-negative bacteria and eukaryotes and is used to reversibly modify cysteine residues during oxidative stress. However, mycobacteria, streptomycetes and corynebacteria produce mycothiol instead of glutathione. Those low molecular weight thiols maintain redox homeostasis and neutralize ROS, toxins, heavy metals, antibiotics, and other xenobiotics. Antelmann et al. show mycothiolation of 58 proteins in total in Mycobacterium smegmatis when exposed to sodium hypochlorate stress. This group includes CitA being mycothiolated only at the conserved, solvent exposed cysteine residue, C143.35
Nonetheless, little is known about the activity and regulation of M. tb citrate synthase, CitA. The aim of this research was to gain more knowledge about the enzyme to assess druggability and the possible mechanisms of targeting CitA with small-molecule modulators of enzymatic activity. Here we report the first crystal structure of M. tb CitA solved to 2.1 Å resolution and a plausible mechanism of enzyme activity regulation by pyruvate, which has not been observed in other citrate synthases. We have also investigated the effect of C143 modification on the activity of the protein by chemical modification and mutagenesis to further understand the impact of reversible mycothiolation as a regulatory mechanism by targeting C143 with thiol-reactive compounds.
Results and discussion
Crystal structure of M. tb citrate synthase in complex with pyruvate
To gain more insight into M. tb citrate synthase, we crystallized the enzyme to solve its structure. Preliminary crystals of the enzyme were obtained with OAA and CoA. The rod shape crystals were analyzed at the APS and diffraction data were obtained to 2.9 Å resolution. The protein crystallized in a primitive hexagonal space group (unit cells: a = b = 150.3 Å, c = 200.1 Å, α = β = 90°, γ = 120°). Molecular replacement was attempted using various models of type II citrate synthases available in the RCSB Protein Data Bank at that time. Based on sequence alignment, M. tb citrate synthase shares approximately 30% sequence similarity with Thermus thermophilus (T. thermophilus) citrate synthase (PDB accession code 1IXE). The structure 1IXE was one of the main templates used for molecular replacement but no solution was obtained in any of the related space groups among the primitive hexagonal family. We attempted to index the diffraction data as a lower symmetry space group but once again, no molecular replacement solution was obtained. Thus, a selenomethionine-derivatized protein was prepared. M. tb citrate synthase possesses eleven methionine residues. Crystals of selenomethionine derivatized protein co-crystallized with CoA and OAA were optimized primarily by screening crystallization with various detergents. Most of the crystals analyzed diffracted poorly. However, one crystal of the complex grown in the presence of CYMAL-6 provided diffraction data to 2.1 Å. Data were collected at the peak of the selenium K-edge to support a SAD experiment for phasing (information about data collection and refinement statistics is displayed in Table S1† – CitA_SeMet* with PYR).The final refined crystal structure contains four molecules in the asymmetric unit forming an apparent tetramer, but size exclusion chromatography results suggest CitA forms a dimer in solution (ESI† Fig. S1). Each monomer possesses 15 α-helices and 4 β-sheets. The fold is similar to that of T. thermophilus citrate synthase. At the time, the available T. thermophilus citrate synthase structure exhibited the highest percentage of sequence identity to M. tb-CitA so was used as a model for molecular replacement. However, the molecular replacement attempts failed due to unexpected structural differences. Specifically, the interface between two molecules of T. thermophilus citrate synthase dimer involves 4 α-helices while only 3 α-helices are interacting between 2 molecules of M. tb citrate synthase (ESI† Fig. S2A and B). The fourth α-helix in the T. thermophilus citrate synthase structure forms a random coil in the M. tb citrate synthase that consists of several proline residues, phenylalanine residues or other hydrophobic residues. These four interfacial helices are found in all other citrate synthase structures from diverse organisms that are available in the RCSB Protein Data Bank, and the presence of this helix in the search model prevented appropriate packing in the M. tb CitA molecular replacement search.
Electron density in the active site representing OAA or CoA was lacking. However, unexpected electron density was observed in each monomer located near the center of the crystallographic CitA tetramer (Fig. S3 in ESI†). The pocket in which this electron density is capped by a helix–loop–helix motif formed from Q137 to Q155 residues (H5 loop H6 helices in Fig. 2) includes two arginine residues (R149 and R153) and a glutamine residue (Q137) that could stabilize a negatively charged molecule such as OAA or citrate. The crystallization condition does not contain any solute that could otherwise account for this density. As it is known that OAA undergoes spontaneous decarboxylation to pyruvate, we hypothesized that the unexpected density corresponds to pyruvate derived from OAA.36 We confirmed this hypothesis by co-crystallizing CitA with pyruvate and observing the same difference density (information regarding data collection and refinement statistics – CitA_PYR column of Table S1 in ESI†). Other minor secondary structural differences are shown in Fig. S4.†
Based on the structure, pyruvate binds in a pocket composed of mainly polar residues that complement the negative charge and hydrogen bond acceptors on pyruvate (Fig. 1). A water molecule binds at the small opening of the pyruvate binding site and interacts directly with the carboxylate group of pyruvate. Additionally, R153 and the backbone nitrogen of A68 stabilize the carboxylate moiety of pyruvate. The carbonyl group of pyruvate interacts through hydrogen bonding with the guanidinium group of R153 and polar interactions with S121. The methyl group of pyruvate is oriented towards a hydrophobic dead end of the binding pocket. Although binding of pyruvate is intriguing, superposition of the pyruvate bound CitA structure with the ligand free structure of CitA indicates a lack of any significant structural differences.
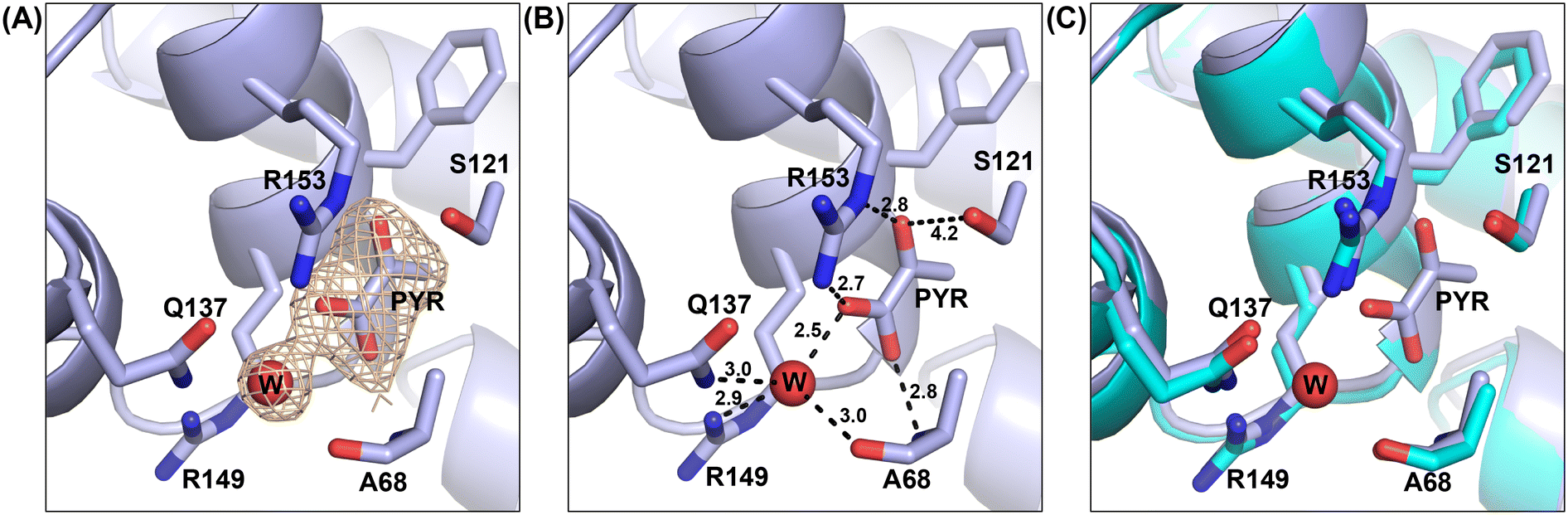 | ||
| Fig. 1 Structure of the pyruvate (PYR) binding site in M. tb citrate synthase. The red sphere represents the oxygen atom of the water molecule (W) interacting with pyruvate. (A) The electron density of a Fo − Fc omit map is shown contoured at 3σ (orange); PYR and W residues were omitted during map calculation. (B) The bonded lengths of interactions formed by pyruvate with the adjacent residues. (C) Alignment of the pyruvate bound CitA crystal structure (light blue) with the ligand free CitA crystal structure (cyan). | ||
To further assess this, we aligned the pyruvate-bound M. tb CitA crystal structure with the NADH-bound citrate synthase crystal structure of E. coli (PDB accession code 1OWB) and the OAA-bound structure of the primary M. tb citrate synthase, GltA2 (encoded by gene rv0896, PDB accession code 4TVM), for comparison. As shown in Fig. 2, The H2 and H3 helices at the NADH binding site of the E. coli citrate synthase structure are absent in the M. tb CitA structure and instead form a random coil. Furthermore, closer to the pyruvate binding site M. tb CitA possesses a short α-helix, labelled H5 in Fig. 2, whereas in E. coli citrate synthase and M. tb GltA2 it is a random coil instead. When comparing the residues involved in the NADH binding pocket in the E. coli citrate synthase with the corresponding residues from M. tb CitA, it is notable that only a few of the residues are conserved (Fig. S5 in ESI†). Those structural and sequence comparisons illustrate why M. tb CitA lacks NADH binding ability. To support this, we performed protein thermal shift assays to assess the binding of NADH to CitA. As expected, the melting temperature remained unchanged, further confirming the inability of CitA to bind with NADH (Fig. S7 in ESI†). Combined with the kinetic data showing that NADH does not affect CitA enzymatic activity, it can be conclusively stated that NADH concentration does not directly regulate CitA activity. Intriguingly, the pyruvate binding pocket resides roughly 7 Å from the NADH binding site in the E. coli homolog. Therefore, the absence of an NADH binding site, which is atypical for citrate synthases, and pyruvate being bound at a site only 7 Å away from the NADH binding site affords an argument that pyruvate may be the allosteric regulator of CitA instead of NADH.21,23,37
 | ||
| Fig. 2 Allosteric binding site differences between the NADH bound E. coli citrate synthase (light green) and M. tb GltA2 (brownish purple) with pyruvate (PYR) bound M. tb CitA (light blue). Helices of the E. coli citrate synthase are labelled in dark green and helices of M. tb CitA are labelled in purple. | ||
Another possibility is that M. tb CitA may require a conformational change to impart low affinity NADH binding. To assess this, we co-crystallized CitA in the presence of 6.5 mM NADH and solved the X-ray crystal structure. The enzyme crystallized in a P21 space group (unit cell: a = 75.33 Å, b = 121.033 Å, c = 99.584 Å, α = γ = 90°, β = 94.731°) and the crystal diffracted to a resolution of 2.4 Å (information about data collection and refinement statistics is displayed in Table S1 of the ESI†). Consistent with the negative results from the protein thermal shift and enzyme kinetic assays suggesting no effect of NADH on enzymatic activity, density corresponding to NADH was lacking in the solved crystal structure. However, an electron density was observed in the pyruvate binding pocket which accounts for an acrylate molecule, a component of the crystallization condition. Therefore, for further structural analysis we treated this structure as ligand free CitA. Since the structure does not show any evidence of bound NADH, nor does it display any conformational change with respect to the pyruvate bound form. This structure supports the hypothesis that NADH does not allosterically regulate M. tb CitA since the enzyme is incapable of binding NADH. To further investigate the plausible regulatory effect of pyruvate on CitA, studies were carried out to identify any impact that pyruvate binding may have on CitA thermal stability or enzyme function.
Assessment of pyruvate binding on the activity of the protein
To identify any changes in the pyruvate binding pocket that affect the CitA activity, the residues that are crucial for pyruvate binding, and how the binding of pyruvate affects for the activity of the protein, we created variants within the pyruvate binding site and used these to address those unknowns. As shown in Fig. 1, the R153 residue forms a bidentate interaction with the pyruvate molecule which is the main residue that stabilizes the pyruvate molecule via hydrogen bonding. Therefore, the R153 residue was mutated to methionine to disrupt the polar interactions that R153 forms with the pyruvate and weaken or prevent pyruvate binding. The R149 residue interacts with the water molecule (Fig. 1) that forms interactions with the pyruvate molecule. The R149 residue was mutated to glutamate with the expectation that the R149E could mimic the presence of the pyruvate. The R149E and R153M variants were obtained by site-directed mutagenesis and purified using the same procedure as wild-type enzyme.The crystal structure of the R149E variant was solved to a resolution of 2.6 Å in a P212121 space group (unit cell: a = 99.933 Å, b = 129.679 Å, c = 270.153 Å, α = β = γ = 90°). The structure of the R153M variant was solved in a P43 space group to a resolution of 2.96 Å (unit cell: a = 129.906 Å, b = 129.906 Å, c = 256.698 Å, α = β = γ = 90°). For the comparison of R149E and R153M crystal structures with the wild-type, the variant structures were superimposed with the wild-type CitA structure using PyMol. As shown in Fig. 3, the R149E residue occupies a similar space as the carboxylate group of the pyruvate molecule while the other residues in the pyruvate binding site remain the same. Due to the orientation of the R149E residue, the pyruvate binding pocket lacks sufficient volume to accommodate pyruvate as well as lacking appropriate charge. Therefore, the R149E variant is unlikely capable of binding pyruvate at this site. In the R153M variant structure, the hydrogen bonds that are formed with the pyruvate molecule by R153 residue in the wild-type are lacking due to the replacement of the R153 side chain by the methionine. Apart from the mutated residue R153M, other residues at the pyruvate binding site exhibit slight changes in position and orientation. Those slight changes and the mutation of R153 to disrupt the multiple interactions with pyruvate and likely ensures the inability of the R153M variant to bind pyruvate at this site. Again, no significant conformational changes were observed when comparing the wild-type structure and the variants.
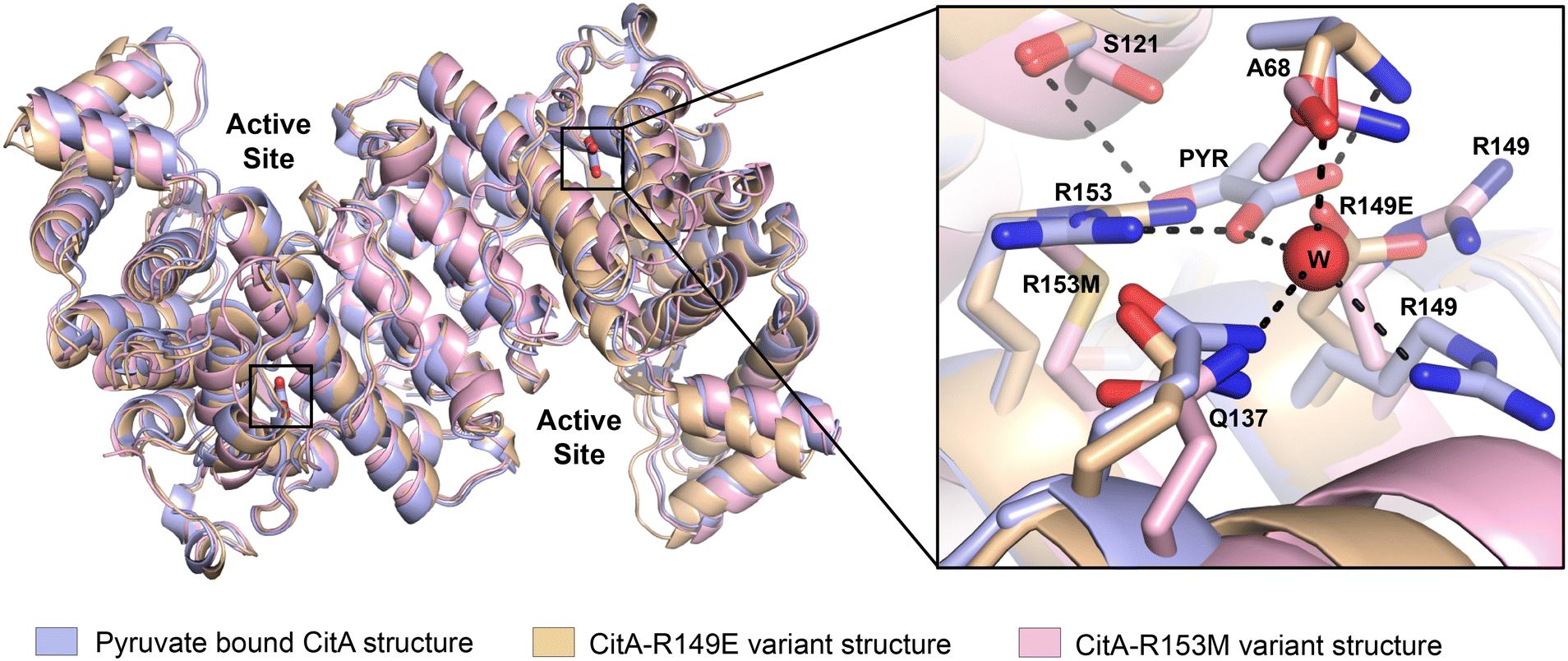 | ||
| Fig. 3 Superposition of CitA wild-type (light blue) with R149E (wheat) and R153M (light pink) variants shows only minor backbone variation and minor changes within the pyruvate binding pocket (inset). | ||
A protein thermal shift assay was conducted to assess the impact of pyruvate binding on the thermal stability of the protein and to assess the thermal stability of the variants with pyruvate present. As shown in Table 1, inclusion of pyruvate with wild-type CitA, increases the melting temperature (Tm) by 2.31 °C (derivative plots are included in ESI† Fig. S7). As expected, the two variants lack a significant change in the protein Tm when compared to the change in Tm of wild-type CitA when pyruvate is added. The slight increment of the Tm in the two CitA variants is likely due to pyruvate binding at the active site due to the structural and chemical similarity to OAA. Therefore, another set of protein thermal shift experiments was carried out in the presence of OAA as well as both OAA and pyruvate. Upon binding with OAA, wild-type CitA and the R149E variant exhibit a 11.16 and 10.84 °C shift in Tm, respectively. However, the R153M variant shows a larger thermal shift of 14.34 °C in the melting temperature. In the presence of both ligands, OAA and pyruvate, the Tm further increases for wild-type CitA whereas for the other 2 variants expected not to bind pyruvate exhibit no significant increment in the Tm. These results strongly suggest that the binding of pyruvate to the identified site on CitA is not a crystallographic artifact, that changes in the pyruvate binding site impact the interactions between CitA and OAA, and that pyruvate and OAA bind at different sites as indicated by the additive increase in thermal stability when both ligands are present.
| Ligand | ΔTmD mean (°C) | ||
|---|---|---|---|
| CitA WT | CitA-R149E | CitA-R153M | |
| Pyruvate | 2.31 ± 0.05 | 0.47 ± 0.05 | 0.51 ± 0.05 |
| OAA | 11.16 ± 0.00 | 10.84 ± 0.05 | 14.34 ± 0.05 |
| OAA + pyruvate | 12.32 ± 0.05 | 10.65 ± 0.05 | 14.29 ± 0.00 |
To further assess the significance of the pyruvate binding to CitA, protein thermal shift assays were also performed in the presence of other small metabolites containing carboxylic acids, as shown in Table 2 (derivative plots in ESI† Fig. S8). These results compare the WT CitA protein thermal shift results when using acetoacetate, glyoxylate and glycolate to the protein thermal shift observed with the analogous results when adding pyruvate. Inclusion of pyruvate shows a greater than 6-fold increment in the change in melting temperature versus the other carboxylates. Indeed, the magnitude of the Tm shift with the control carboxylates is similar to the Tm shift of the variants when adding pyruvate. These results strongly suggest that pyruvate forms more stabilizing interactions with CitA compared to other small carboxylate containing metabolites, and that pyruvate is likely the only biologically-relevant ligand for this site.
| Ligand | ΔTmD mean (°C) |
|---|---|
| Pyruvate | 2.31 ± 0.05 |
| Acetoacetate | 0.19 ± 0.05 |
| Glyoxylate | 0.10 ± 0.00 |
| Glycolate | 0.15 ± 0.05 |
Kinetic experiments were performed to further assess the effect of mutations on the activity of the protein in the presence of OAA, acetyl CoA and DTNB in the reaction mixture. As shown in Fig. 4(A), there was no difference in the catalytic efficiency between the wild-type and the R149E variant. However, the R153M variant shows 2.6-fold increased catalytic efficiency compared to the wild-type. The Km of the R153M variant is almost 4.5 times reduced compared to the wild-type suggesting that the R153M variant possesses higher affinity for OAA. This result is consistent with the protein thermal shift assay which showed a higher increment in the melting temperature for R153M compared to the wild-type. However, the catalytic rate of the R153M variant decreases by roughly 40% versus CitA WT with each showing kcat values of 198 min−1 and 340 min−1, respectively.
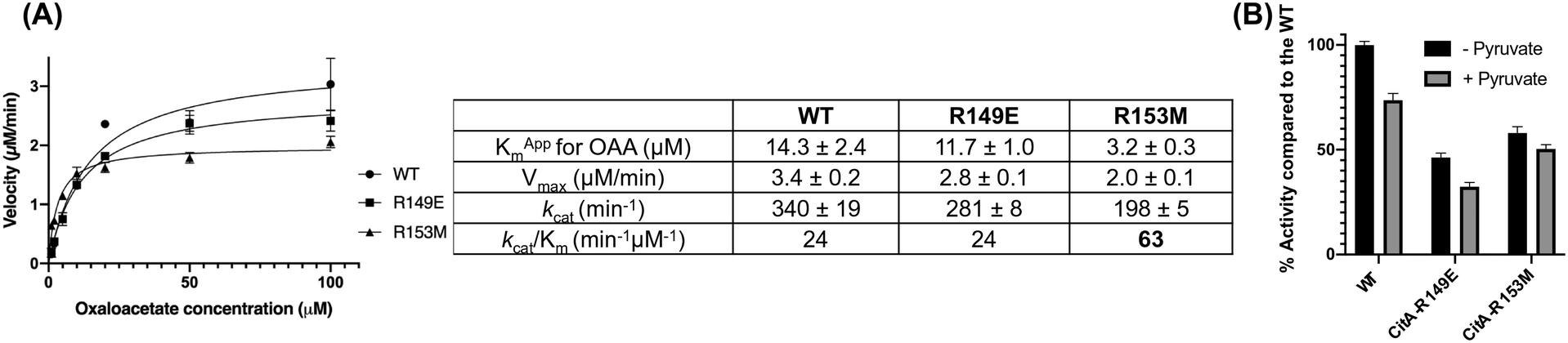 | ||
| Fig. 4 (A) Michaelis–Menten curves plotted for CitA wild-type (WT) and R149E, R153M variants with the table showing the data obtained from kinetic analysis using experimental results performed in triplicate. (B) The bar graph showing the percentage enzyme activity of CitA wild-type and the variants in the absence or presence of 500 μM pyruvate. | ||
The assay was also performed in the presence of 500 μM pyruvate in the reaction mixture along with OAA, acetyl CoA and DTNB, as shown in Fig. 4(B). Our hypothesis is that pyruvate, being the end product of glycolysis, would activate the CitA to enhance carbon flux through the TCA cycle. However, the observed result contradicts that hypothesis in that pyruvate showed concentration dependent inhibition of wild-type CitA. The activity of R149E and R153M variants were also tested in the presence of pyruvate and the results were similar to that of the wild-type with each variant showing a pyruvate-dependent reduction in enzymatic activity (Fig. 4(B)). Similar to hypothesis based on the protein thermal shift assay, pyruvate may bind to the active site of CitA due to the structural and chemical similarity to OAA and thereby functioning as a competitive inhibitor. That could be the reason for the reduced activity not only for the wild-type but also for the variants in which pyruvate cannot bind to the pyruvate binding site. Therefore, more thorough kinetic experiments are needed to fully assess the effect of pyruvate on the activity of the wild-type CitA.
Assessment of C143 modification on the activity of CitA
As previously mentioned, M. tb switches from active growth and division to a dormant state in response to oxidative stress. Previous research has identified various proteins undergoing mycothiolation of cysteine residues under conditions of oxidative stress, and CitA residue C143 was identified as one of these reversible mycothiolation sites.35 As CitA is recognized as a possible metabolic switch during infection, we hypothesized that mycothiolation of the solvent accessible C143 residue may regulate CitA function and thereby modulate M. tb metabolism.34 Interestingly, residue C143 is only 8 Å away from the identified pyruvate binding site and could represent an interesting target for regulation of M. tb CitA. Indeed, in the context of our current findings, it seemed likely that modification of C143 would arrest the activity of CitA.To understand the importance of the C143 residue and any changes around the C143 residue affecting enzyme activity, the C143 residue was subjected to mutagenesis to produce four variants C143A, C143S, C143W and C143K expected to exhibit varying impacts on CitA activity. The alanine variant was designed to eliminate the hydrogen bond between C143 and T152 and possibly enhance flexibility in the loop containing the C143 residue. The C143S mutation was expected to maintain the same hydrogen bond with the side chain of T152 residue but not be subject to oxidation like cysteine. The tryptophan mutation was introduced to disrupt all the side chain interactions that are formed by residue 143 and promote structural distortion of that region. The linear but charged lysine residue was introduced to mimic the modification of the cysteine residue by any compounds like mycothiol without potentially destabilizing this region by simply adding a bulky hydrophobic residue like methionine.
The enzymatic activity of each variant was tested in vitro and compared with wild-type CitA. The C143W and C143K variants exhibited 14% and 22% lower enzyme activity, respectively, when compared to the wild-type levels under Km conditions. The C143A and C143S variants were observed to be more active than the wild-type showing increases of 39% and 59% enzyme activity, respectively, compared to the wild-type (ESI† Fig. S9). To understand how the mutations affect the overall structure of the protein, crystallization experiments were performed and the structures of all four variants were solved (statistics of the structural refinements are provided in ESI† Table S2). However, based on the superposition of the variant structures with the wild-type enzyme, significant structural changes are lacking in the crystal structures for any of those variants compared to the wild-type CitA (ESI† Fig. S9). The C143W variant showed that the indole group is oriented in such a way that the N-atom forms a hydrogen bond with the neighbouring T152 residue just as C143 does in CitA WT. Therefore, there were no significant structural changes either around the C143 region or the overall structure. This is likely why there is no significant reduction of activity. The C143K residue is oriented towards the bulk solvent like a modified cysteine residue, however, this chemical change is not sufficient to stimulate any significant structural changes around residue 143 or the overall structure compared to the wild-type and, again, likely explains the lack of a significant reduction in enzyme activity. As expected, the C143S side chain forms a hydrogen bond with the neighboring T152 residue maintaining the same interaction as the C143 residue in the wild-type enzyme. However, the activity of CitA may have increased due to the fact that unlike C143 the C143S variant is not subject to oxidation. As the modification of C143 residue by mutagenesis was not sufficient to modulate CitA activity, likely due to side chain size limitations using only proteinogenic residues, we instead subjected C143 to chemical modification.
Regulation of CitA activity by chemical modification of the C143 residue
With the hypothesis that the activity of CitA could be altered by modifying the side chain of C143, we attempted to covalently modify the C143 residue using commercially available thiol-reactive compounds. First, we modified CitA by reacting it with Ebselen (Cayman Chemical), a fast thiol-reactive compound known to inhibit cysteine proteases and several other proteins that have an active site cysteine or a cysteine that allosterically regulates enzyme activity.38–41 For one set of experiments, CitA was purified in absence of any reductant and reacted with Ebselen in a 50 mM Tris pH 8.0 buffer. A control experiment was carried out without adding Ebselen to modify CitA. In a second set of experiments CitA was purified in the presence of a reductant during purification, and was incubated with Ebselen in the presence of tris(2-carboxyethyl)phosphine hydrochloride (TCEP) in 50 mM Tris pH 8.0 buffer, with a another control of unmodified CitA. The Ebselen modification reaction was carried out by overnight incubation followed by buffer exchange to remove the excess unreacted Ebselen and TCEP in the reaction buffers to prevent interference in the assay. As shown in Fig. 5A and B, the CitA enzymatic activity was arrested almost completely following reaction with Ebselen but in the absence of TCEP, due to the covalent modification of CitA. However, in the presence of TCEP, since the modification of thiols with Ebselen is reversible under mildly reductive conditions, the CitA activity is roughly equivalent to the unmodified CitA control. Indeed, the slightly lower enzymatic activity of the CitA sample purified without reductant may have slightly lower enzymatic activity due to oxidation of a subset of the CitA molecules in that sample. To strengthen the hypothesis that C143 is the residue modified by Ebselen and leading to enzyme inactivation, the CitA-C143S variant was incubated with Ebselen and the enzyme activity tested and compared to the enzyme activity of unmodified CitA-C143S. These data show that the enzymatic activity of the CitA-C143S variant incubated with Ebselen is unaffected by Ebselen addition since the activity is comparable to both wild-type CitA and wild-type CitA/Ebselen combination in reducing conditions (ESI† Fig. S10) Therefore, it is concluded that the activity of CitA is arrested by Ebselen modification of C143.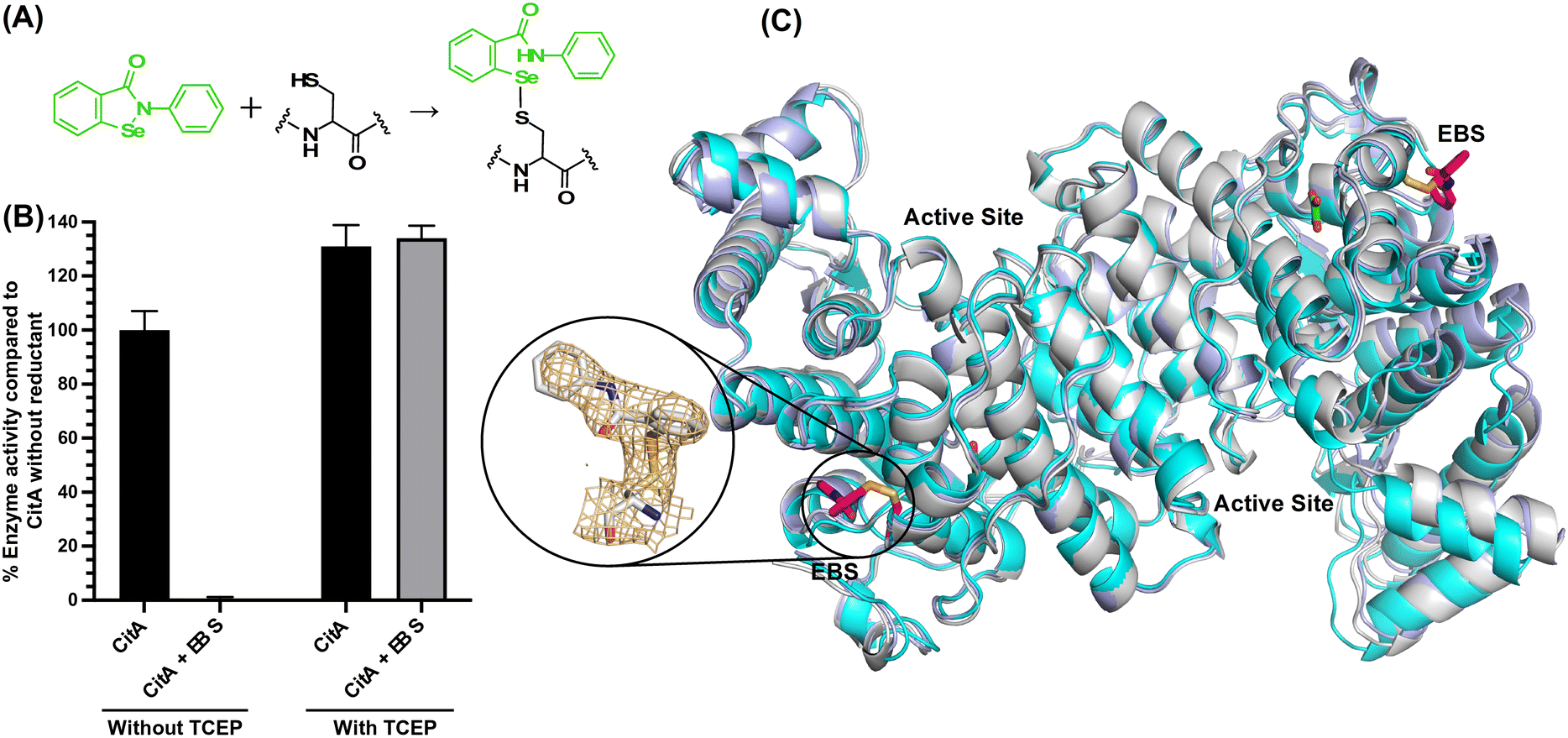 | ||
| Fig. 5 (A) The reaction between Ebselen and C143 residue (B) the bar graph showing the percentage activity difference between Ebselen modified CitA (CitA-EBS) versus unmodified wild-type CitA (CitA-WT) and recovery of CitA activity in presence of a reductant (C) the alignment of CitA unmodified ligand free structure (cyan), pyruvate bound CitA structure (light blue) and Ebselen modified CitA structure (gray) and the electron density of composite omit map for Ebselen modification of C143 residue. | ||
To further support this conclusion and possibly identify a molecular mechanism for the loss of activity, we solved the X-ray crystal structure of Ebselen-modified CitA to a resolution of 2.7 Å resolution in a P212121 space group (unit cell: a = 122.02 Å, b = 167.49 Å, c = 221 Å, α = β = γ = 90°). Clear difference density was observed for the modification of the C143 side chain that corresponds well with Ebselen. Of the three cysteine residues in CitA, only the solvent exposed C143 residue was observed to be modified with Ebselen. The buried cysteines C326 and C333 lack any additional difference density corresponding to modification by Ebselen. However, superposition of the Ebselen-modified CitA with the unmodified CitA shows no significant structural changes in the protein backbone or side chain rotomers due to the modification (Fig. 5).
Modification of the C143 residue by irreversible Michael acceptors
To determine if any thiol-reactive compound can inactivate CitA by modifying C143, an in-house library of Michael acceptor containing compounds was tested in the CitA enzymatic assay.42–47 These compounds were screened against 10 nM CitA at a concentration of 20 μM in the CitA enzymatic assay. Two of the 30 tested compounds were identified as hits by exhibiting a greater than 80% decrease in CitA activity. A follow up dose–response study with the two hit compounds (38-131 and 28-171) revealed dose-dependent inhibition of CitA activity. Fitting the data to the Hill equation yielded apparent IC50 values of 6.6 ± 0.2 μM and 10.9 ± 0.9 μM for 28-131 and 28-171, respectively (Fig. 6). Since the inhibition is the result of covalent modification, future studies will assess the time-dependent inhibition by these compounds. Additionally, a control experiment was carried out to ensure that the Michael acceptors fail to react with the thiol moiety of CoA produced by the CitA catalyzed reaction. These controls clearly show that during the timeframe of the assay used to determine the initial velocity, no measurable reaction occurs between the Michael acceptors and the CoA product. Therefore, the modification of C143 by 28-131, 28-171, or Ebselen as mentioned previously, accounts for the total reduction of CitA enzymatic activity.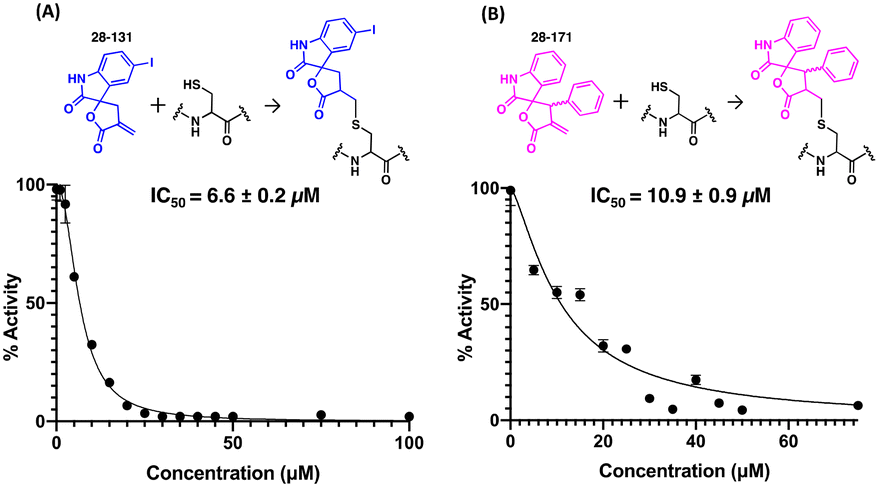 | ||
| Fig. 6 IC50 curves for Michael acceptor compounds. (A) The IC50 curve for the 28-131 and the reaction between 28-131 and C143. (B) The IC50 curve for the 28-171 and the reaction between 28-171 and C143. | ||
Experimental
Molecular cloning for WT CitA and mutants
The citA gene was amplified from H37Rv genomic DNA by PCR using the following set of primers (Integrated DNA Technology): 5′-GTCCCGGAGAATTTCGTCCCCGGCCTCGAC and 3′-TGGTGAGTACCCGCTCCCAGCCGTCGAC. The gene was amplified a second time using this PCR product obtained and a second pair of primers (5′-CACCTTCATATGACAGTGGTCCCGGAGAATTTCGTC and 3′-TTGGATCCTCAATGATGATGATGATGATGGGCGGTGGTGAGTACCCGCT). The second pair of primers includes the sequences corresponding to the restriction endonucleases recognition sites. The gene was then ligated into a pET32-based plasmid (EMD biosciences) between NdeI and BamHI restriction sites, including a non-cleavable C-terminal poly-histidine tag. Nucleotide sequencing was performed by Eurofins MWG Operon to confirm the gene sequence.Cloning of CitA variants including R149E, R153M, C143S, C143A, C143W and C143K was performed using the QuikChange Site-Directed Mutagenesis Kit (Aligent) with the designed primers (Integrated DNA Technology). The gene sequences were confirmed by sequencing the plasmids using Sanger sequencing at Genewiz.
Purification of M. tb CitA
The purified plasmid containing citA and the plasmids containing the genes encoding for variants of CitA were used to transform T7 Expression cells of E. coli (New England BioLabs) separately. Bacterial cells were cultured in Luria Broth (Research Products International) at 37 °C in presence of 100 mg L−1 of ampicillin (Gold Biotechnology). Once an OD600nm of 0.6 was reached, the cells were induced at 16 °C for 36 to 48 hours by the addition of 1 mM isopropyl b-D-1-thiogalactopyranoside (IPTG) (Gold Biotechnology). The cells were then harvested by centrifugation and resuspended in a buffer containing 20 mM Tris pH 8.0, 5 mM imidazole, 0.2 M sodium chloride and 5 mM β-mercaptoethanol (b-Me). The induced stock was stored at −80 °C until needed. The induced cells were thawed at room temperature. Then, the cells were lysed using 1 μM of lysozyme (Hampton Research) and 0.1 μM of DNaseI (Roche), as well as a sonication step (Sonicator 3000, Misonix). The crude lysate was pelleted by centrifugation at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (fixed-angle rotor, 5810-R Centrifuge, Eppendorf) for 30 minutes. The supernatant was filtered and then loaded onto a 5 mL HisTrap™ TALON crude column prepacked with TALON Superflow™ (GE Healthcare). The column was washed with 15 column volumes of resuspension buffer and was eluted using a linear gradient over 15 column volumes with a concentration of imidazole from 5 mM to 150 mM. Citrate synthase was concentrated using ammonium sulfate precipitation at a final concentration of 2.8 M. The pelleted protein was resuspended in a buffer containing 50 mM Tris pH 8.0 and 0.3 mM tris(2-carboxyethyl)phosphine (TCEP). The sample was further purified using size exclusion chromatography. The concentration of the citrate synthase was determined using absorbance spectroscopy at a wavelength of 280 nm using the theoretical extinction coefficient (50420 M−1 cm−1) determined by the ProtParam function from the ExPASY proteomics server.48
000 rpm (fixed-angle rotor, 5810-R Centrifuge, Eppendorf) for 30 minutes. The supernatant was filtered and then loaded onto a 5 mL HisTrap™ TALON crude column prepacked with TALON Superflow™ (GE Healthcare). The column was washed with 15 column volumes of resuspension buffer and was eluted using a linear gradient over 15 column volumes with a concentration of imidazole from 5 mM to 150 mM. Citrate synthase was concentrated using ammonium sulfate precipitation at a final concentration of 2.8 M. The pelleted protein was resuspended in a buffer containing 50 mM Tris pH 8.0 and 0.3 mM tris(2-carboxyethyl)phosphine (TCEP). The sample was further purified using size exclusion chromatography. The concentration of the citrate synthase was determined using absorbance spectroscopy at a wavelength of 280 nm using the theoretical extinction coefficient (50420 M−1 cm−1) determined by the ProtParam function from the ExPASY proteomics server.48
Preparation of selenomethionine M. tb CitA
The selenomethionine kit was purchased from Molecular Dimensions and the SelenoMet medium was prepared per the kit instructions. A 5 mL culture of BL21 E. coli cells containing the citA plasmid was grown overnight. The culture was harvested, and the cell pellet was resuspended with several milliliters of the SelenoMet medium. The resuspended cell pellet was used to inoculate the sterile SelenoMet medium. Bacterial cells were grown at 37 °C in the presence of 100 mg L−1 of ampicillin (Gold Biotechnology) to an OD600nm of 0.6. Selenomethionine solution (250 X) was added to the cells, which were then incubated for 15 min at 37 °C. The cells were then induced at 16 °C for 36 to 48 hours by addition of 1 mM IPTG (Gold Biotechnology). The cells were then harvested by centrifugation and resuspended into 20 mM Tris pH 8.0, 5 mM imidazole, and 5 mM β-Me. The protein was purified following the same protocol as the native enzyme. A cobalt affinity purification step, followed by size exclusion chromatography, was performed. The purity was analyzed using SDS-PAGE.Enzymatic activity assay
An absorbance-based assay was developed using 5,5′-dithio-bis-(2-nitrobenzoic acid), abbreviated as DTNB, which is commonly used to assess the amount of thiol groups in a sample.49 The absorbance-based assay utilizes oxaloacetate (OAA) and acetyl coenzyme A (AcCoA). In the presence of citrate synthase, the substrates are converted to citrate and CoA. The latter possesses a free thiol group, which acts as a nucleophile and attacks the DTNB compound, cleaving the disulfide bond present in DTNB and generating 3-thio-6-nitrobenzoate (TNB). The release of one CoA molecule corresponds to the formation of one TNB molecule. The absorbance can be monitored at 412 nm. Stock solutions of 5,5′-dithio-bis-(2-nitrobenzoic acid) (Sigma Aldrich), AcCoA (Roche) and oxaloacetic acid (Sigma Aldrich) were prepared in 50 mM Tris pH 8.0. AcCoA was regenerated prior to being utilized as described by Trievel and co-workers.50 Stock solutions of 5,5′-dithio-bis-(2-nitrobenzoic acid) (Sigma Aldrich), AcCoA (Roche) and oxaloacetic acid (Sigma Aldrich) were prepared in 50 mM Tris pH 8.0. AcCoA was regenerated prior to being utilized as described by Trievel and co-workers.50 The activity of M. tb citrate synthase was tested using 100 μM OAA and 200 μM AcCoA in the presence of 300 μM of DTNB and 10 nM enzyme. The reactions were carried out at 25 °C on a Synergy H4 Hybrid Reader (BioTek). The enzyme was dialyzed against 20 mM Tris pH 8.0 prior to use. Each of the reactions was performed in 50 mM Tris pH 8.0. A control reaction omitting the enzyme was performed in parallel. In the presence of citrate synthase, an increase of the absorbance at 412 nm is readily observed, indicating that the enzyme is active and converts the substrates to citrate and free CoA (Fig. 2). Additionally, the background for this assay is very low; no reaction is observed when only AcCoA and OAA are present. The Michaelis Menten curves were plotted using PRISM software (GraphPad).Modification of CitA with Ebselen and Michael acceptor
Modification of C143 residue of CitA with Ebselen (Cayman Chemicals) was performed by incubating the protein overnight with 5 μM of Ebselen in a 50 mM Tris pH 8.0 buffer without the presence of a reductant. The excess Ebselen was removed by buffer exchange prior to use for enzymatic reactions to prevent the reaction of Ebselen with DTNB in the reaction mixture. A control experiment was carried out for the C143S variant the same as the wild-type.For the assay with the Michael acceptor compounds, the compounds were directly added to the reaction mixture. However, a control experiment was carried out to test the rate at which the Michael acceptor compounds may react with the components of the reaction mixture.
Crystallization of M. tb CitA and variants
The selenomethionine derivatized citrate synthase (14.4 mg mL−1) was incubated with 5 mM oxaloacetate (Sigma Aldrich) and 5 mM CoA (Chem-Impex International Inc) for 30 min on ice. The complex crystallized against a well solution containing 0.1 M Tris pH 8.5, 0.5 M sodium chloride and 19% w/v polyethylene glycol (PEG) 3350 in the presence of 5.6 μM of a detergent, CYMAL-6 (6-cyclohexyl-b-D-maltoside). Additional PEG 3350 was added to the drop prior to looping the crystal for data collection.Native citrate synthase (30.8 mg mL−1) was also crystallized in presence of 6.5 mM NADH. The well solution contained 0.02 M magnesium chloride, 0.1 M HEPES pH 7.5, and 22% w/v poly(acrylic acid sodium salt) 5100 and the crystals were grown in presence of 0.1 M barium chloride (Hampton Research).
The CitA-R149E mutant was crystallized in 0.1 M Tris pH 8.5, 0.2 M lithium sulfate, and 25% PEG 3350. Glycerol was added to the drop to 25% immediately prior to flash-cooling in liquid nitrogen. The CitA-R153M variant (10 mg mL−1 in 0.4 M sodium citrate pH 5.5 buffer) was crystallized in 0.2 M magnesium chloride, 20 w/v% PEG 3350. The crystals were flash-cooled in liquid nitrogen right after the addition of 35% w/v PEG 3350 directly to the drop.
The Ebselen modified CitA crystals were obtained by soaking experiments. The apo crystals obtained from condition 0.2 M lithium sulfate, 0.1 M HEPES pH 7.5 and 25% w/v PEG 3350, were soaked with Ebselen for more than one day.
The C143S variant was crystallized by mixing an equal volume of 10 mg mL−1 CitA in 0.4 M sodium citrate pH 5.5 buffer and well solution containing 0.1 M sodium acetate pH 4.5, 3 M NaCl. The C143W variant was crystallized in the same condition but improved the crystals with the additive 5% w/v D-sorbitol in the drop. Both were cryoprotected with 33% v/v PEG 400 before flash cooling. The C143K variant was crystallized by mixing the well solution of 0.2 M ammonium sulfate, 20% PEG 3350 and 10 mg mL−1 of CitA in a 1:1 ratio. The C143A variant was crystallized using the well solution of 0.2 M sodium tartrate dibasic dihydrate, 20% w/v polyethylene glycol 3350. Both were cryoprotected with 35% w/v PEG 3350 before flash cooling. All crystals were flash-cooled in liquid nitrogen prior to data collection. Diffraction data were collected at the LS-CAT beamline at the Advance Photon Source Argonne National Laboratory (APS-ANL, IL).
Protein thermal shift assay for M. tb CitA and variants
The protein thermal shift assay was performed with a protein concentration of 25 μM in 50 mM Tris pH 8.0 buffer in the presence of 2 mM of sodium pyruvate, OAA, lithium acetoacetate, sodium glycolate or sodium glyoxylate. Protein Thermal Shift™ dye from ThermoFisher 1000X solution was diluted to have a final concentration of 2X in the reaction mixture. The temperature was ramped up 0.05 degrees per minute using the QuantStudio Real-Time PCR system from ThermoFisher to get the melting curves. The Tm was analyzed using generated derivative plots generated from the Applied Biosystems™ Protein Thermal Shift™ software v1.4.Structure determination of M. tb CitA and variants
A single diffraction data set was collected at a wavelength of 0.97856 Å for a crystal of selenomethionine derivatized citrate synthase crystallized in presence of oxaloacetate and CoA. HKL2000 was utilized to index, integrate and scaled as an anomalous data.51 The Autosol and Autobuild tools from PHENIX were used to build a model for the citrate synthase.52 Forty-four selenium sites were identified, leading to the determination of 4 copies of citrate synthase in one asymmetric unit since M. tb CitA possesses eleven methionine residues. The solution obtained provided a figure of merit of 0.347; the model correlation coefficient was 0.87. The PHENIX refine tool was used to carry out rigid body refinement, simulated annealing, positional and B-factor refinements52 while manual corrections of the model were performed using COOT.53 Structural alignment of CitA wild-type to the variants was performed using the PyMOL 2.4.0.Conclusions
The crystal structure of M. tb CitA was solved in an unliganded form and in a complex with pyruvate. In contrast to many other citrate synthases, M. tb CitA does not possess an NADH binding site. Two helices typically important for the formation of NADH binding pocket in NADH-regulated citrate synthases are absent in the M. tb crystal structure and are replaced by loops forming a portion of the M. tb CitA pyruvate binding site. The presence of the pyruvate binding site in what is typically an NADH-regulated allosteric sub-domain suggests that pyruvate may function as a feed-forward allosteric regulator of the M. tb citrate synthase.The protein thermal shift assays show a 2 degree increase in the protein melting temperature upon binding of pyruvate to wild-type CitA suggesting that pyruvate binding increases the thermal stability of the protein, compared to the two CitA variants that lack evidence for pyruvate binding. Compared to the other structurally similar carboxylate-containing metabolites, pyruvate shows a significant increase in the melting temperature of CitA upon binding strengthening the suggestion that pyruvate alone plays a regulatory role.
The enzyme kinetic results illustrate the activity of CitA changes upon modifications of the residues that are crucial for the binding of pyruvate. The R153M variant, in which the arginine residue that forms bidentate interaction with pyruvate is mutated to a methionine, exhibits an approximately 2.6-fold increment in the catalytic efficiency. Indeed, the proximity between C143 and the pyruvate binding site, a distance of only 8 Å, is unlikely a coincidence. While this subdomain is roughly 20 Å away from the active site, an analogous NADH-regulated domain is known in numerous citrate synthase enzymes to regulate OAA binding and subsequent enzyme activity. Further, residues now shown to be important for pyruvate binding and oxidation-dependent enzyme regulation, C143, R153 and R149, are in the helix–loop–helix that caps the pyruvate binding pocket (Fig. 7).
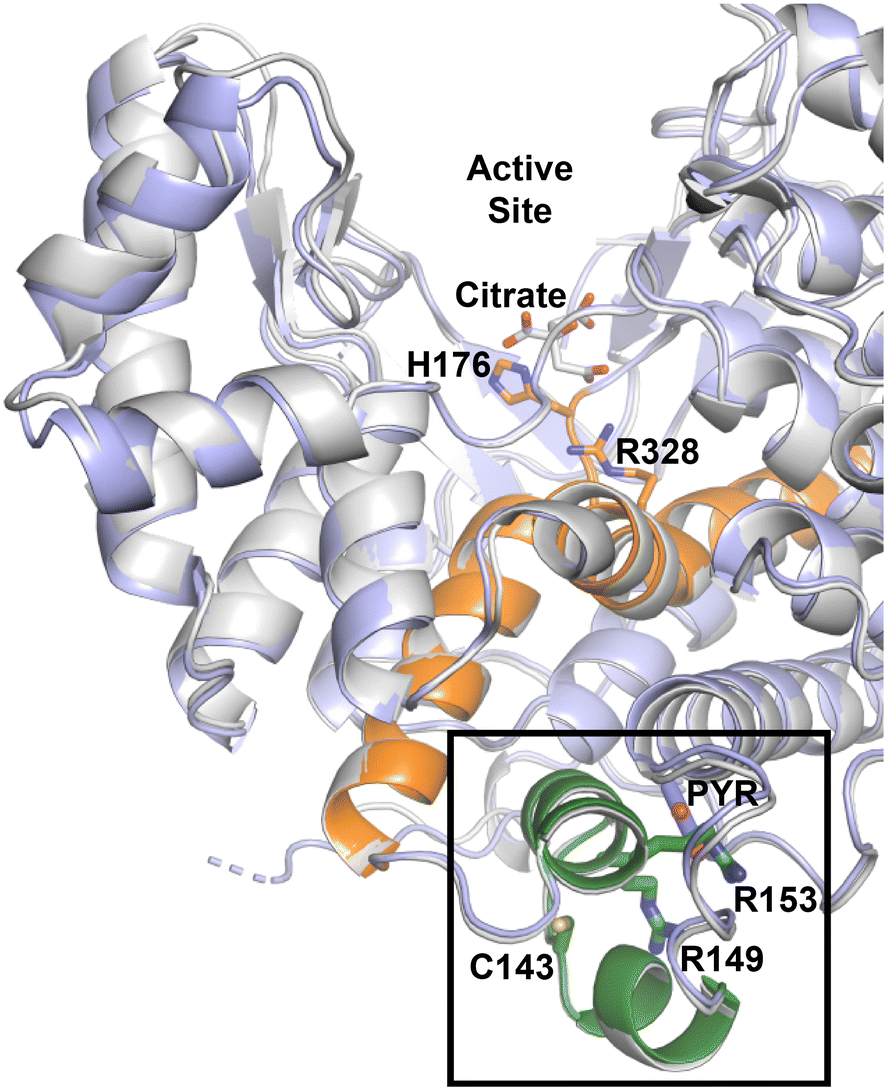 | ||
| Fig. 7 The alignment of citrate bound Ebselen modified M. tb CitA structure (grey) and pyruvate bound M. tb CitA structure highlighting the regulatory domain (green) and helices that connect the regulator domain to the active site (orange). | ||
Inspired by the results of Antelmann et al. showing that the C143 residue is modified by mycothiol under oxidative stress, we show that modification of the C143 residue by Ebselen arrests the activity of CitA. The thiol reactive Michael acceptor compounds 28-131 and 28-171 also show inhibition of enzyme activity with IC50 of 6.6 ± 0.2 μM IC50 of 10.9 ± 0.9 μM, respectively. Considering that the mutations of R153 and R149 as well as chemical modification of C143 affecting the activity of the protein suggests that this helix–loop–helix is acting as a regulatory domain for CitA. This regulatory domain is directly connected to one end of a helix that extends toward the active site as well as abutting one of the helices in the active site (Fig. 6). Therefore, the binding of small molecules at the pocket capped by this regulatory domain or chemical modifications that occur in this regulatory domain affect the activity of the protein. Therefore, this regulatory domain is a potential drug target to maintain high levels of CitA activity to make M. tb more susceptible to current TB drugs. Finally, this information can be further leveraged to re-evaluate regulation of other seemingly redundant enzymes of the TCA cycle.
Author contributions
D. R. R. and A. N. conceptualized the study. D. R. R., and A. N. provided resources, supervision, and validation. R. P., L. F., C. P., M. Y., S. S., J. R. M., and S. R. performed the experiments and formal analysis.Conflicts of interest
There are no conflicts to declare. The research presented here was not supported by nor represents experiments performed by BioNTech.Acknowledgements
Research was supported in part by NIH grant CA260749 to A. N. and NIH grant AI105084 to D. R. R. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).References
- WHO, Global Tuberculosis report, 2021 Search PubMed.
- S. Loewenberg, India reports cases of totally drug-resistant tuberculosis, Lancet, 2012, 379(9812), 205 CrossRef PubMed.
- M. Gengenbacher and S. H. Kaufmann, Mycobacterium tuberculosis: success through dormancy, FEMS Microbiol. Rev., 2012, 36(3), 514–532 CrossRef CAS PubMed.
- C. S. Hayes and D. A. Low, Signals of growth regulation in bacteria, Curr. Opin. Microbiol., 2009, 12(6), 667–673 CrossRef CAS PubMed.
- W. P. Gill, et al., A replication clock for Mycobacterium tuberculosis, Nat. Med., 2009, 15(2), 211–214 CrossRef CAS PubMed.
- D. A. Mitchison and A. R. Coates, Predictive in vitro models of the sterilizing activity of anti-tuberculosis drugs, Curr. Pharm. Des., 2004, 10(26), 3285–3295 CrossRef CAS PubMed.
- J. E. Gomez and J. D. McKinney, M. tuberculosis persistence, latency, and drug tolerance, Tuberculosis, 2004, 84(1–2), 29–44 CrossRef PubMed.
- P. Anand and Y. Akhter, A review on enzyme complexes of electron transport chain from Mycobacterium tuberculosis as promising drug targets, Int. J. Biol. Macromol., 2022, 212, 474–494 CrossRef CAS PubMed.
- Q. Li and X. Lu, New antituberculosis drugs targeting the respiratory chain, Chin. Chem. Lett., 2020, 31(6), 1357–1365 CrossRef CAS.
- B. S. Lee, et al., Dual inhibition of the terminal oxidases eradicates antibiotic-tolerant Mycobacterium tuberculosis, EMBO Mol. Med., 2021, 13(1), e13207 CAS.
- A. M. Thompson and W. A. Denny, Inhibitors of enzymes in the electron transport chain of Mycobacterium tuberculosis, in Annual reports in medicinal chemistry, Elsevier, 2019, pp. 97–130 Search PubMed.
- B. J. Berube and T. Parish, Combinations of respiratory chain inhibitors have enhanced bactericidal activity against Mycobacterium tuberculosis, Antimicrob. Agents Chemother., 2018, 62(1), e01677–e01717 CrossRef PubMed.
- J. R. Guest, The Leeuwenhoek Lecture, 1995, Adaptation to life without oxygen, Philos. Trans. R. Soc., B, 1995, 350(1332), 189–202 CrossRef CAS PubMed.
- S. Remington, G. Wiegand and R. Huber, Crystallographic refinement and atomic models of two different forms of citrate synthase at 2.7 and 1.7 A resolution, J. Mol. Biol., 1982, 158(1), 111–152 CrossRef CAS PubMed.
- M. Karpusas, B. Branchaud and S. J. Remington, Proposed Mechanism for the Condensation Reaction of Citrate Synthase - 1.9-a Structure of the Ternary Complex with Oxaloacetate and Carboxymethyl Coenzyme-A, Biochemistry, 1990, 29(9), 2213–2219 CrossRef CAS PubMed.
- M. Karpusas, D. Holland and S. J. Remington, 1.9-a Structures of Ternary Complexes of Citrate Synthase with D-Malate and L-Malate - Mechanistic Implications, Biochemistry, 1991, 30(24), 6024–6031 CrossRef CAS PubMed.
- D. I. Liao, M. Karpusas and S. J. Remington, Crystal-Structure of an Open Conformation of Citrate Synthase from Chicken Heart at 2.8-a Resolution, Biochemistry, 1991, 30(24), 6031–6036 CrossRef CAS PubMed.
- S. J. Remington, Structure and mechanism of citrate synthase, Curr. Top. Cell. Regul., 1992, 33, 209–229 CAS.
- R. J. Russell, et al., The crystal structure of citrate synthase from the hyperthermophilic archaeon pyrococcus furiosus at 1.9 A resolution, Biochemistry, 1997, 36(33), 9983–9994 CrossRef CAS PubMed.
- J. A. Francois, et al., Structure of a NADH-insensitive hexameric citrate synthase that resists acid inactivation, Biochemistry, 2006, 45(45), 13487–13499 CrossRef CAS PubMed.
- R. Maurus, et al., Insights into the evolution of allosteric properties. The NADH binding site of hexameric type II citrate synthases, Biochemistry, 2003, 42(19), 5555–5565 CrossRef CAS PubMed.
- N. T. Nguyen, et al., Comparative analysis of folding and substrate binding sites between regulated hexameric type II citrate synthases and unregulated dimeric type I enzymes, Biochemistry, 2001, 40(44), 13177–13187 CrossRef CAS PubMed.
- D. J. Stokell, et al., Probing the roles of key residues in the unique regulatory NADH binding site of type II citrate synthase of Escherichia coli, J. Biol. Chem., 2003, 278(37), 35435–35443 CrossRef CAS PubMed.
- D. R. Boutz, et al., Discovery of a thermophilic protein complex stabilized by topologically interlinked chains, J. Mol. Biol., 2007, 368(5), 1332–1344 CrossRef CAS PubMed.
- S.-H. Park, et al., Structural basis of the cooperative activation of type II citrate synthase (HyCS) from Hymenobacter sp. PAMC 26554, Int. J. Biol. Macromol., 2021, 183, 213–221 CrossRef CAS PubMed.
- D. A. Beard, K. C. Vinnakota and F. Wu, Detailed enzyme kinetics in terms of biochemical species: study of citrate synthase, PLoS One, 2008, 3(3), e1825 CrossRef PubMed.
- G. Wiegand, et al., Crystal structure analysis and molecular model of a complex of citrate synthase with oxaloacetate and S-acetonyl-coenzyme A, J. Mol. Biol., 1984, 174(1), 205–219 CrossRef CAS PubMed.
- G. Wiegand and S. J. Remington, Citrate synthase: structure, control, and mechanism, Annu. Rev. Biophys. Biophys. Chem., 1986, 15, 97–117 CrossRef CAS PubMed.
- P. D. Weitzman, Regulation of citrate synthase activity in escherichia coli, Biochim. Biophys. Acta, 1966, 128(1), 213–215 CrossRef CAS PubMed.
- P. D. Weitzman, Reduced nicotinamide-adenine dinucleotide as an allosteric effector of citrate-synthase activity in Escherichia coli, Biochem. J., 1966, 101(3), 44C–45C CrossRef CAS PubMed.
- S. Okay, In silico characterization of the citrate synthase family in Mycobacterium tuberculosis/Mycobacterium tuberculosis' te sitrat sentaz ailesinin in silico karakterizasyonu, Turk Biyokim. Derg., 2016, 41(2), 118–126 Search PubMed.
- D. M. Ferraris, et al., Structures of citrate synthase and malate dehydrogenase of M ycobacterium tuberculosis, Proteins: Struct., Funct., Bioinf., 2015, 83(2), 389–394 CrossRef CAS PubMed.
- A. Jinich, et al., Mycobacterium tuberculosis transposon sequencing database (MtbTnDB): a large-scale guide to genetic conditional essentiality, bioRxiv, 2021, preprint, 2021.03.05.434127, DOI:10.1101/2021.03.05.434127.
- S.-H. Baek, A. H. Li and C. M. Sassetti, Metabolic regulation of mycobacterial growth and antibiotic sensitivity, PLoS Biol., 2011, 9(5), e1001065 CrossRef CAS PubMed.
- M. Hillion, et al., Monitoring global protein thiol-oxidation and protein S-mycothiolation in Mycobacterium smegmatis under hypochlorite stress, Sci. Rep., 2017, 7(1), 1–20 CrossRef PubMed.
- C. Tsai, Spontaneous decarboxylation of oxalacetic acid, Can. J. Chem., 1967, 45(8), 873–880 CrossRef CAS.
- H. W. Duckworth, et al., Enzyme-substrate complexes of allosteric citrate synthase: evidence for a novel intermediate in substrate binding, Biochim. Biophys. Acta, 2013, 1834(12), 2546–2553 CrossRef CAS PubMed.
- M. J. Parnham and H. Sies, The early research and development of ebselen, Biochem. Pharmacol., 2013, 86(9), 1248–1253 CrossRef CAS PubMed.
- K. Amporndanai, et al., Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives, Nat. Commun., 2021, 12(1), 1–7 CrossRef PubMed.
- H. Sies and M. J. Parnham, Potential therapeutic use of ebselen for COVID-19 and other respiratory viral infections, Free Radical Biol. Med., 2020, 156, 107–112 CrossRef CAS PubMed.
- L. Favrot, et al., Mechanism of inhibition of Mycobacterium tuberculosis antigen 85 by ebselen, Nat. Commun., 2013, 4(1), 1–10 Search PubMed.
- S. Kour, et al., Stapling proteins in the RELA complex inhibits TNFalpha-induced nuclear translocation of RELA, RSC Chem. Biol., 2022, 3(1), 32–36 RSC.
- S. Kour, et al., Spirocyclic dimer SpiD7 activates the unfolded protein response to selectively inhibit growth and induce apoptosis of cancer cells, J. Biol. Chem., 2022, 298(5), 101890 CrossRef CAS PubMed.
- A. Natarajan and S. Rana, Dimers of covalent NFkappaB inhibitors, U.S. Pat., 11104684, U.S. Patent and Trademark Office, 2021 Search PubMed.
- S. Rana, et al., Isatin Derived Spirocyclic Analogues with alpha-Methylene-gamma-butyrolactone as Anticancer Agents: A Structure-Activity Relationship Study, J. Med. Chem., 2016, 59(10), 5121–5127 CrossRef CAS PubMed.
- S. Rana, et al., Dimers of isatin derived alpha-methylene-gamma-butyrolactone as potent anti-cancer agents, Bioorg. Med. Chem. Lett., 2022, 65, 128713 CrossRef CAS PubMed.
- S. Rana and A. Natarajan, Face selective reduction of the exocyclic double bond in isatin derived spirocyclic lactones, Org. Biomol. Chem., 2013, 11(2), 244–247 RSC.
- E. Gasteiger, C. Hoogland, A. Gattiker, S. Duvaud, M. R. Wilkins, R. D. Appel and A. Bairoch, Protein identification and analysis tools on the ExPASy server, in The proteomics protocols handbook, ed. J. M. Walker, Humana Press Inc, Totowa, NJ, 2005, pp. 571–607 Search PubMed.
- G. L. Ellman, Tissue sulfhydryl groups, Arch. Biochem. Biophys., 1959, 82(1), 70–77 CrossRef CAS PubMed.
- R. C. Trievel, F. Y. Li and R. Marmorstein, Application of a fluorescent histone acetyltransferase assay to probe the substrate specificity of the human p300/CBP-associated factor, Anal. Biochem., 2000, 287(2), 319–328 CrossRef CAS PubMed.
- Z. Otwinowski and W. Minor, Processing of X-ray diffraction data collected in oscillation mode, Methods Enzymol., 1997, 27, 307–326 Search PubMed.
- P. D. Adams, et al., PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(Pt 2), 213–221 CrossRef CAS PubMed.
- P. Emsley, et al., Features and development of Coot, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(Pt 4), 486–501 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00058c |
| ‡ Current address: Irving Institute for Cancer Dynamics, Columbia University, New York, NY, USA. |
| § Current address: BioNTech, 55131Mainz, Germany. |
| ¶ Current address: Department of Biochemistry and Molecular Biology, The University of Chicago, Chicago, IL, USA. |
| This journal is © The Royal Society of Chemistry 2023 |