
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biological activity and structure–activity relationship of dehydrodieugenol B analogues against visceral leishmaniasis†
Maiara
Amaral‡
ab,
Hannah
Asiki‡
 c,
Claire E.
Sear
c,
Snigdha
Singh
c,
Pauline
Pieper
c,
Marius M.
Haugland
c,
Edward A.
Anderson
*c and
Andre G.
Tempone
*b
c,
Claire E.
Sear
c,
Snigdha
Singh
c,
Pauline
Pieper
c,
Marius M.
Haugland
c,
Edward A.
Anderson
*c and
Andre G.
Tempone
*b
aInstituto de Medicina Tropical, Faculdade de Medicina, Universidade de Sao Paulo, Sao Paulo – 05403-000, Brazil
bCentre for Parasitology and Mycology, Instituto Adolfo Lutz, São Paulo – 01246-000, Brazil. E-mail: andre.tempone@ial.sp.gov.br; atempone@usp.br
cChemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: edward.anderson@chem.ox.ac.uk
First published on 26th May 2023
Abstract
Visceral leishmaniasis is a neglected protozoan disease with high mortality. Existing treatments exhibit a number of limitations, resulting in a significant challenge for public health, especially in developing countries in which the disease is endemic. With a limited pipeline of potential drugs in clinical trials, natural products could offer an attractive source of new pharmaceutical prototypes, not least due to their high chemodiversity. In the present work, a study of anti-L. (L.) infantum potential was carried out for a series of 39 synthetic compounds based on the core scaffold of the neolignan dehydrodieugenol B. Of these, 14 compounds exhibited activity against intracellular amastigotes, with 50% inhibitory concentration (IC50) values between 3.0 and 32.7 μM. A structure–activity relationship (SAR) analysis demonstrated a requirement for polar functionalities to improve activity. Lacking mammalian cytotoxicity and presenting the highest potency against the clinically relevant form of the parasite, compound 24 emerged as the most promising, fulfilling the hit criteria for visceral leishmaniasis defined by the Drugs for Neglected Diseases initiative (DNDi). This study emphasizes the potential of dehydrodieugenol B analogues as new candidates for the treatment of visceral leishmaniasis and suggests 24 to be a suitable compound for future optimization, including mechanism of action and pharmacokinetic studies.
Introduction
Leishmaniasis are neglected tropical diseases caused by parasites of the genus Leishmania which affect more than 12 million people worldwide, mainly in low-income populations. The disease is present in 98 countries, with approximately 1.3 million new cases per year and 350 million people at risk of infection.1,2 Human visceral leishmaniasis (VL) is a systemic disease which typically affects mononuclear phagocytic system cells. If treated, VL mortality rates can be as low as 10 to 20%; but if untreated, the disease is 100% fatal within two years.3 Although VL is the most severe form of the disease, it has received relatively little attention in terms of new treatments; a limited chemotherapeutic arsenal is available, with many current therapies eliciting adverse effects.4 As a result, the search for safe and accessible therapeutic alternatives is of exceptional importance.In the search for new drugs, natural products stand out as compounds that offer enormous pharmacological potential. Newman and Cragg (2020) showed that approximately 50% of all FDA-approved drugs feature natural product scaffolds as the basis of their pharmacophores.5 Our previous studies described the anti-Leishmania (L.) donovani and anti-Trypanosoma (T.) cruzi activity of a small family of neolignans isolated from branches and leaves of Nectandra leucanta (Lauraceae).6–8 Subsequently, using dehydrodieugenol B, four new semi-synthetic compounds were prepared and their antileishmanial activity reported.9
A total synthesis of dehydrodieugenol B was developed based on a copper-catalyzed Ullmann coupling to form the biaryl ether core (Fig. 1).10 This enabled site-specific modifications, and evaluation of a series of analogues against T. cruzi, resulting in the identification of several compounds that displayed enhanced bioactivity and selectivity against the trypomastigote form of the parasite, and meeting the Drugs for Neglected Diseases initiative (DNDi) criteria for hit compounds.11
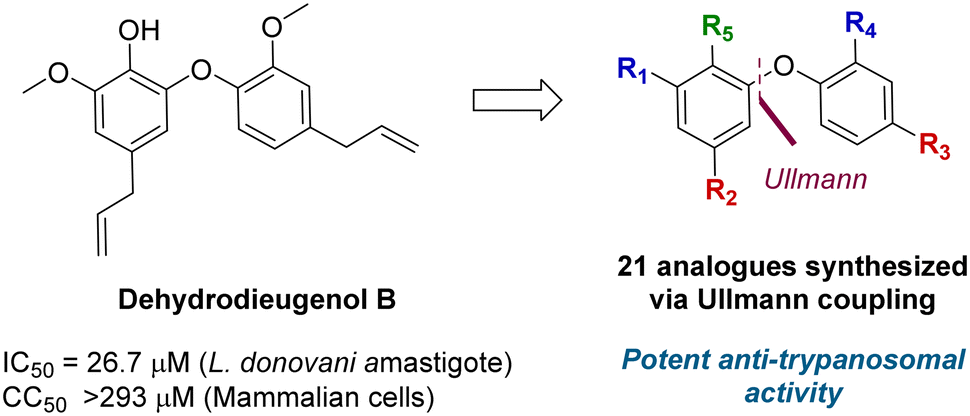 | ||
| Fig. 1 Previous work on dehydrodieugenol B synthesis and anti-parasitic activity of natural product analogues. | ||
In this work, the bioactivity of a series of 39 analogues of dehydrodieugenol B was studied against Leishmania (L.) infantum intracellular amastigotes. These compounds feature a variety of structural modifications which aim to improve biological activity over the natural product, and also to obtain information about the pharmacophore. Additionally, absorption, distribution, metabolism, excretion and toxicity (ADMET) parameters of the most promising compounds were evaluated using an in silico platform to investigate the drug-likeness profile.
Results and discussion
A series of 39 compounds was synthesized to explore the structure–activity relationships (SAR) of this family as antileishmanial compounds. Three different approaches were employed: I) substitution on the phenol group of the A-ring (S1); II) saturation, substitution or removal of the allyl side chains (S2-A and B), and III) removal of the methoxy groups (S3-A or B) (Fig. 2). These compounds were prepared by similar chemistry to our previous work,10 using an Ullmann coupling to form the C–O biaryl ether bond to the A ring from appropriately functionalized precursors (see the ESI† for synthesis details).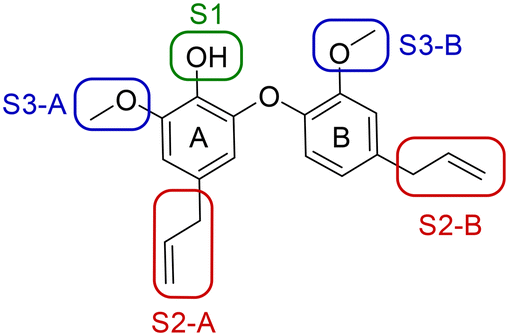 | ||
| Fig. 2 SAR strategy to explore antileishmanial activity of the dehydrodieugenol B scaffold. | ||
The synthesized compounds were evaluated for their anti-L. (L.) infantum activity on intracellular amastigotes following 96 h treatment, with standard drug miltefosine used as a control. The 50% inhibitory concentration (IC50) was determined by the infection index,12 and mammalian toxicity (CC50) for the series was evaluated against NCTC cells.
We first considered modification at the S1 (phenol) position of dehydrodieugenol B (Table 1). Although the p-methoxybenzyl ether of the natural product (1) had shown high activity against T. cruzi trypomastigotes in our previous work (4.0 ± 1.4 μM), this derivative proved inactive against L. (L.) infantum amastigotes. The introduction of a polar group on the sidechain seems to be necessary for activity, as other modifications at this position were inactive. Having a larger pKa is beneficial for these compounds as the more basic amines (4, 5) exhibited lower IC50 values than their corresponding amides (6, 7), although these four compounds also displayed moderate mammalian cytotoxicity.
| Comp. | S1 | IC50 (μM) ± SD | CC50 (μM) ± SD | SI |
|---|---|---|---|---|
| CC50: 50% cytotoxic concentration in NCTC cells; IC50: 50% inhibitory concentration in intracellular amastigotes; SI: selectivity index, given by the ratio between CC50 and IC50; SD: standard deviation; ND: not determined; NA: not active (>50 μM); Milt: miltefosine. | ||||
| 1 | OPMB | NA | >200 | ND |
| 2 | OMOM | NA | >200 | ND |
| 3 | OAllyl | NA | >200 | ND |
| 4 |
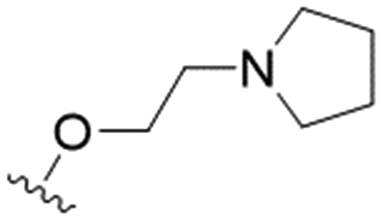
|
4.1 ± 1.1 | 24.9 ± 2.6 | 6 |
| 5 |
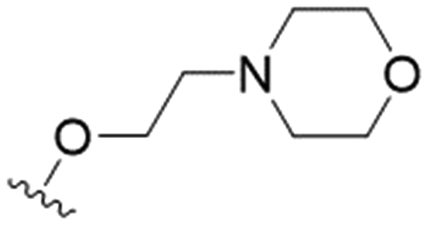
|
9.6 ± 3.6 | 63.7 ± 0.2 | 6.6 |
| 6 |
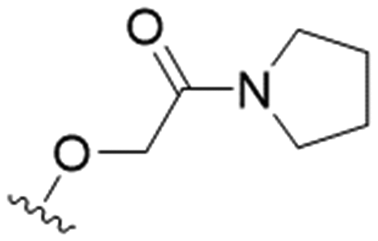
|
20.3 ± 5.9 | 52.9 ± 15.2 | 2.6 |
| 7 |
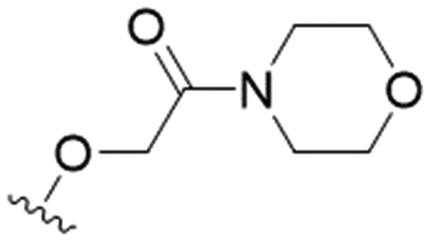
|
NA | 111.5 ± 8.2 | ND |
| 8 | H | NA | >200 | ND |
| Milt. | — | 6.5 ± 3.0 | 119.7 ± 4.2 | 18.4 |
Next, the effect of modifying the allyl sidechain functionalities on each of the A and B rings (positions S2) by substitution, saturation or deletion was considered (Table 2). Saturation or deletion of either of the allyl groups did not lead to antileishmanial activity in any of the analogues prepared, with the exception of moderate activity in 9 and 11. We noted a detrimental effect on the CC50 values in 15, 17, and 18, but this is more likely due to the presence of a free phenol in position S1 and not a result of the saturations or deletions (a result we had observed in our analogous work on T. cruzi); all other derivatives showed no toxicity to mammalian cells at the tested concentrations.
| Comp | S1 | S2A | S2B | IC50 (μM) ± SD | CC50 (μM) ± SD | SI |
|---|---|---|---|---|---|---|
| CC50: 50% cytotoxic concentration in NCTC cells; IC50: 50% inhibitory concentration in intracellular amastigotes; SI: selectivity index, given by the ratio between CC50 and IC50; SD: standard deviation; ND: not determined; NA: not Active (>50 μM); Milt: miltefosine. | ||||||
| 9 | OPMB | Allyl | H | 24.2 ± 18.2 | >200 | >8.3 |
| 10 | OPMB | H | Allyl | NA | >200 | ND |
| 11 | OAcyl | Allyl | H | 32.7 ± 5.0 | >200 | >6.1 |
| 12 | OAcyl | H | Allyl | NA | >200 | ND |
| 13 | OMe | Allyl | H | NA | >200 | ND |
| 14 | OMe | H | Allyl | NA | >200 | ND |
| 15 | OH | Allyl | H | NA | 31.6 ± 11.3 | ND |
| 16 | OH | H | Allyl | NA | >200 | ND |
| 17 | OH | Allyl | n-Pr | NA | 42.0 ± 3.8 | ND |
| 18 | OH | n-Pr | Allyl | NA | 14.2 ± 0.1 | ND |
| 19 | OMe | Allyl | n-Pr | NA | >200 | ND |
| 20 | OPMB | Allyl | n-Pr | NA | >200 | ND |
| 21 | OPMB | n-Pr | Allyl | NA | >200 | ND |
| 22 | OPMB | n-Pr | n-Pr | NA | >200 | ND |
| 23 | OPMB | Allyl |
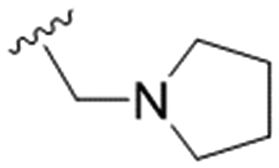
|
NA | 6.2 ± 1.2 | ND |
| 24 | OPMB | Allyl |
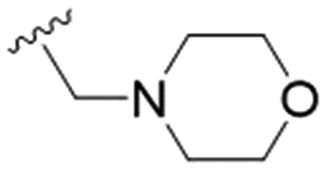
|
9.7 ± 2.0 | >200 | >20.6 |
| 25 | OPMB | n-Pr |
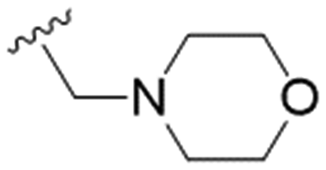
|
7.7 ± 0.8 | 74.4 ± 4.4 | 9.7 |
| 26 | OPMB | n-Pr |

|
3.7 ± 0.5 | 14.5 ± 0.2 | 3.9 |
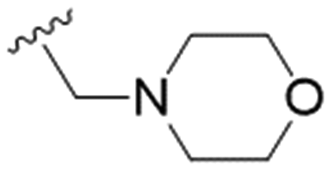
| 27 | OPMB |
|
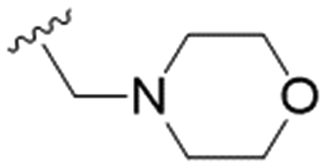
|
27.8 ± 7.4 | 74.4 ± 5.8 | 2.7 |
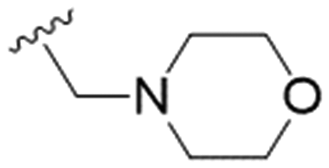
| 28 | OPMB |
|
n-Pr | 16.4 ± 4.3 | >200 | >12.2 |
| 29 |
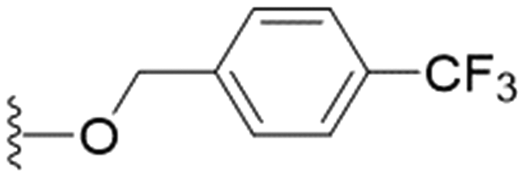
|
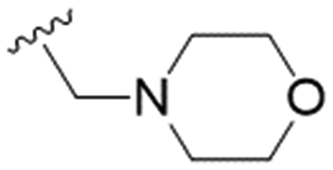
|
n-Pr | 13.2 ± 2.1 | >200 | >15.1 |
| 30 |
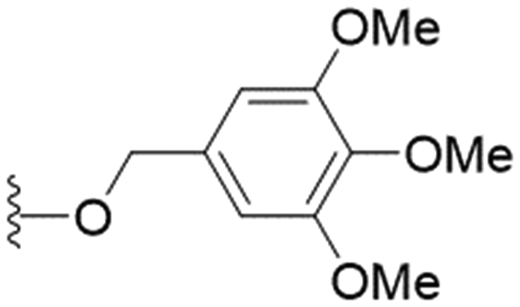
|
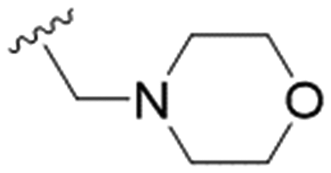
|
n-Pr | 26.3 ± 3.2 | >200 | >7.6 |
| 31 | OH | Allyl |
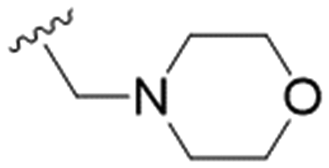
|
NA | 51.4 ± 4.7 | ND |
| 32 | OH | Allyl |
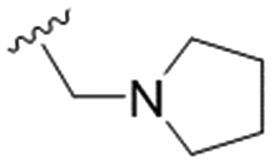
|
NA | 43.3 ± 5.3 | ND |
| Milt. | — | 6.5 ± 3.0 | 119.7 ± 4.2 | 18.4 | ||
The substitution of the allyl sidechains with polar morpholine or piperidine groups led to increased activity in all cases 24–30, although a pyrrolidine substitution (23) did not. Substitution at position S2-B tends to result in higher antileishmanial activity (IC50 ranging from 3.7 to 9.7 μM) than S2-A (IC50 ranging from 13.2 μM to inactive). An attempt to amplify the activity increase with a double substitution (27) did not have the desired effect, instead resulting in an IC50 value comparable to the less potent S2-A substitutions. Unlike position S1 there does not appear to be a consistent anti-parasitic IC50 trend corresponding to the different pKa values for the morpholine (least basic), piperidine and pyrrolidine (most basic) derivatives at position S2. The compounds broadly fit the trend of higher pKa corresponding to higher mammalian cytotoxicity with pyrrolidine containing compound 23 having the worst CC50 value for this series.
Our next step was to investigate ortho-methoxy groups at position S3 (Table 3). Deletion of the methoxy group at S3-B in compounds 33 and 34 was well-tolerated, with no effect on antileishmanial activity. The same is not true for S3-A: 35 was inactive against L. (L.) infantum upon deletion of this methoxy group compared to the corresponding highly active compound 25. Therefore, the methoxy group at S3-B is expendable, which could offer synthetic benefits, while that at S3-A is crucial for maintaining anti-parasitic activity.
| Comp | S1 | S2A | S2B | S3A | S3B | IC50 (μM) ± SD | CC50 (μM) ± SD | SI |
|---|---|---|---|---|---|---|---|---|
| CC50: 50% cytotoxic concentration in NCTC cells; IC50: 50% inhibitory concentration in intracellular amastigotes; SI: selectivity index, given by the ratio between CC50 and IC50; SD: standard deviation; ND: not determined; NA: not active (>50 μM); Milt: miltefosine. | ||||||||
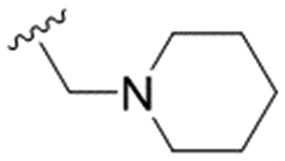
| 33 | OPMB |
|
n-Pr | OMe | H | 3.0 ± 1.1 | 22.7 ± 0.9 | 7.6 |
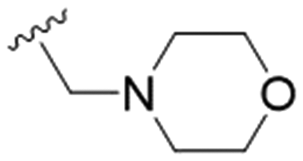
| 34 | OPMB |
|
n-Pr | OMe | H | 15.3 ± 1.9 | >200 | >13.1 |
| 35 | OPMB | n-Pr |
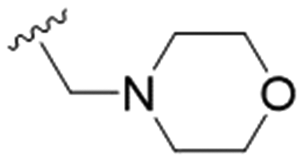
|
H | OMe | NA | 33.8 ± 4.9 | ND |
| 36 | OPMB | Allyl | Allyl | OMe | H | NA | >200 | ND |
| 37 | OH | Allyl | Allyl | OMe | H | NA | 123.4 ± 9.4 | ND |
| 38 | OPMB | Allyl | Allyl | H | OMe | NA | >200 | ND |
| 39 | OH | Allyl | Allyl | H | OMe | NA | 128.6 ± 5.2 | ND |
| Milt. | — | 6.5 ± 3.0 | 119.7 ± 4.2 | 18.4 | ||||
These results demonstrate that the presence of a polar functionality such as morpholine, pyrrolidine or piperidine is generally required for anti-Leishmania (L.) infantum activity, with 12 of the 14 active analogues containing one of these functional groups. 9 and 11 are the only exceptions but display a fairly low activity of 24.2 and 32.7 μM respectively. This polar group requirement could be linked to compound solubility and ability to permeate cell membranes; due to the nature of Leishmania (L.) infantum amastigotes residing in intracellular vacuoles the compound must pass through multiple cell membranes to act directly on the parasite. The presence of a free phenol at position S1 (as in the natural product itself) led to no anti-parasitic activity in every example tested, even when paired with a polar functionality as in 31 and 32, indicating the importance of a substituent in the S1 position. The series 28 to 30 attempted to investigate the effect of decreasing electron density at the benzylic S1 substituent. Electron withdrawing para-trifluoromethylbenzyl group showed a slightly lower IC50 value than strongly electron donating trimethoxybenzyl, with para-methoxybenzyl having an IC50 in between the two. However, these changes were of a small magnitude indicating electron density at this position is unlikely to play a major role in compound activity. Interestingly, the introduction of a piperidine group leads to lower IC50 values than morpholine substitution: compounds 26 and 33 have potent IC50 values of approximately 3 μM compared with the corresponding morpholine analogues 25 and 34 (7.7 and 15.3 μM respectively). However, this trend is also manifested in the mammalian cytotoxicity, with higher CC50 values for the morpholine analogues, which overall gives the morpholine analogues a better selectivity index.
Compound 24 emerged as the most favourable with the best selectivity index due to its high anti-parasitic activity (9.7 μM) coupled with no mammalian cytotoxicity (>200 μM). It was therefore selected for an in silico study of pharmacokinetic properties alongside closely related analogues 28 and 34. An analysis of pharmacokinetic properties and structural alerts for compounds 24, 28 and 34 was conducted using ADMETlab 2.0 (Table 4).13 The predictions were performed to explore the safety and drug-likeness profile of these analogues. The physicochemical property analysis of 24 predicts a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value of 5.18, a close to optimum logD value of 3.82 and water solubility (logS) of −5.27, which falls into the moderate solubility class – an important characteristic that relates to oral bioavailability which we show here to be high (≥30%). Polarity of 24 is also predicted to be favourable (0 < TPSA < 140 Å2). All these characteristics are improved in 24 compared to the related analogues 28 and 34, further justification for choosing 24 as the best compound from this series (see the ESI† for further ADMET analysis on 24).
P value of 5.18, a close to optimum logD value of 3.82 and water solubility (logS) of −5.27, which falls into the moderate solubility class – an important characteristic that relates to oral bioavailability which we show here to be high (≥30%). Polarity of 24 is also predicted to be favourable (0 < TPSA < 140 Å2). All these characteristics are improved in 24 compared to the related analogues 28 and 34, further justification for choosing 24 as the best compound from this series (see the ESI† for further ADMET analysis on 24).
| Properties | 24 | 28 | 34 |
|---|---|---|---|
| TPSA: topological polar surface area; P-gp: glycoprotein P; HIA: human intestinal absorption; BBB: blood brain barrier; CYP: cytochrome P450; DILI: drug induced liver injury. | |||
| Molecular weight | 505.25 | 507.26 | 477.25 |
| LogP |
5.18 | 5.65 | 5.69 |
| LogD |
3.82 | 4.12 | 4.17 |
| LogS |
−5.27 | −6.03 | −6.07 |
| TPSA | 58.62 | 58.62 | 49.39 |
| Caco-2 permeability | −5.03, optimal | −5.46, moderate | −5.30, moderate |
| Pgp inhibitor | Yes | Yes | Yes |
| Pgp substrate | No | No | No |
| HIA (≥30%) | Yes | Yes | Yes |
| Oral bioavailability | ≥30% F | ≥30% F | ≥30% F |
| BBB penetration | No | No | No |
| Distribution (VD) (L K−1) | 0.72, optimal | 0.72, optimal | 0.95, optimal |
| CYP1A2 inhibitor | No | No | No |
| CYP2C19 inhibitor | Yes | Yes | Yes |
| CYP2C9 inhibitor | Yes | No | Yes |
| CYP2D6 inhibitor | No | No | No |
| CYP3A4 inhibitor | Yes | Yes | Yes |
| Clearance (mg min−1 kg−1) | 11.6, moderate | 11.6, moderate | 11.7 moderate |
| hERG blockers | Yes | Yes | Yes |
| Human hepatotoxicity | No | No | No |
| AMES toxicity | No | No | No |
| DILI | Moderate | Moderate | Moderate |
| Lipinski rules | Fail | Fail | Pass |
| PAINs | 0 alerts | 0 alerts | 0 alerts |
The series is predicted to have a high Human Intestinal Absorption (HIA) being ≥30%, moderate (compounds 28, 34) to optimum (compound 24) Caco-2 permeability – another model of intestinal absorption, and pleasingly no permeability to the blood–brain barrier, which could lead to side-effects in the nervous system.14 The cytochrome P450 superfamily of enzymes are responsible for compound metabolism; inhibition of these enzymes can induce adverse effects and toxicity due to accumulation of drug metabolites.15 Of the five main isoforms (CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4), 24 is proposed to be an inhibitor of three with CYP2C9 and CYP3A4 having only a weak positive result.
Compound 24 exceeds the suggested limit of flexibility (number of rotatable bonds < 11) having 12 rotatable bonds, and lies close to the recommended boundary for size criteria (100 < MW < 600) and lipophilicity (0 < logP < 3.0), although values up to 5.0 are still considered reasonable by Lipinski's rules.16 Compounds 24 and 28 fail Lipinski's rule due to the high molecular weight (>500), although they are close to the boundary at 505 and 507 respectively. This may imply flexibility and size issues which should be addressed with future structural optimization. Otherwise, compound 24 demonstrated favourable predictions for toxicity, with no indication of human hepatotoxicity or AMES mutagenicity, and only moderate drug induced liver injury measurements. It is also important to note there were no structural similarities to pan-assay interference compounds (PAINs).
According to Drugs for Neglected Diseases initiative (DNDi), a new hit compound for Visceral Leishmaniasis, should: i) be synthesized in no more than 8 steps; ii) present IC50 lower than 10 μM in amastigotes; iii) have selectivity index higher than 10; iv) exhibit an appropriate in silico ADME profile and, v) show no structural alerts.11,17 Based on these criteria and the results obtained herein, compound 24 can be considered as a promising hit for Visceral Leishmaniasis.
Experimental
General experimental procedures
MTT was purchased from Molecular Probes® (Invitrogen™). Fetal bovine serum (FBS) was obtained from Gibco. All absorbance readings were performed using the FilterMax F5 Multi-Mode Microplate Reader spectrofluorimeter (Molecular Devices). Proton (1H) NMR spectra were recorded at 400 or 500 MHz and carbon (13C) NMR spectra at 101 or 126 MHz with 1H decoupling. Spectra were recorded on Bruker AVIIIHD 400 or Bruker AVIIIHD 500 spectrometers with CDCl3 as reference. High-resolution mass spectra (HRMS) were recorded by the Departmental Mass Spectrometry Service, University of Oxford on a Thermo Scientific Exactive Mass Spectrometer (using a Waters Equity autosampler and pump) for electrospray ionisation (ESI).Animals
Golden hamsters (Mesocricetus auratus) and BALB/c mice were obtained from the animal breeding facility of the Instituto Adolfo Lutz – SP and maintained in sterilized cages with controlled environment. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the Brazilian National Council of Animal Experimentation (COBEA) and had the approval of the Animal Ethics Committee (CEUA IMT-USP 000404A) from University of São Paulo and Instituto Adolfo Lutz – Secretary of Health of Sao Paulo State (CEUA 05/2018).Parasite and mammalian cell maintenance
Leishmania (L.) infantum (MHOM/BR/1972/LD) amastigotes were obtained by differential centrifugation from golden hamsters (Mesocricetus auratus) spleens with 60–70 days of infection and the number of parasites was determined by the Stauber method.18 Murine conjunctive fibroblasts NCTC clone 929 (ATCC) were maintained in RPMI-1640 medium supplemented with 10% FBS at pH 7.2 and 37 °C in a humidified incubator with 5% CO2. Peritoneal macrophages were by obtained by washing the peritoneal cavity of BALB/c mice with RPMI-1640 medium supplemented with 10% FBS, pH 7.2 and maintained at 37 °C in a humidified incubator with 5% CO2.Anti-L. (L.) infantum activity in vitro
Peritoneal macrophages (1 × 105/well) were added in 16-well slide chambers (NUNC) and incubated overnight at 37 °C with 5% CO2. Subsequently, the cells were infected with L. (L.) infantum amastigotes (10:1 amastigotes/macrophage) for 24 h, treated with compounds serially diluted (6.25 to 50 μM) and incubated for 96 h at 37 °C with 5% CO2. Miltefosine was used as standard drug and untreated cells as a negative control. Slides were fixed with methanol, stained with Giemsa and observed under a light microscope.19 The 50% inhibitory concentration (IC50) was determined by the infection index (number of infected macrophages × amastigotes/total macrophages).12
Mammalian toxicity in vitro
NCTC cells (6 × 104/well) were added to 96-well plates and maintained at 37 °C in a 5% CO2 humidified incubator for 96 h with compounds serially diluted (1.6 to 200 μM). The 50% cytotoxic concentration (CC50) was determined using the MTT colorimetric method and the selectivity index was calculated by the ratio: CC50 against NCTC cells/IC50 against amastigotes.20In silico analysis
Predictions were performed using the web server ADMETlab 2.0. This platform includes several models for physicochemical properties, solubility, pharmacokinetic/pharmacodynamic parameters, drug-likeness profile and structural alerts for pan-assay interference compounds (PAINS).13Statistical analysis
The determination of CC50 and IC50 values was performed by sigmoid dose–response curves using the Graph Pad Prism 5 software. The samples were tested in duplicate and the assays were reproduced at least twice.Conclusions
Among 39 tested analogues, 14 were active against the intracellular amastigote form of Leishmania (L.) infantum. SAR analysis suggests that polar functionalities are crucial for the activity of this family, with morpholine substituted analogues displaying the best SI. Derivative 24 was the most promising compound from this series, with excellent potency and selectivity, and can be considered a promising hit for VL. It is synthesized in 5 steps in the longest linear sequence (and 6 steps in total), displays promising bioactivity (9.7 μM), low mammalian toxicity (SI > 20.6), and meets the predicted ADME criteria.Author contributions
Conceptualization, AGT, EAA and MMH; methodology, MA, HA, CES, SS, PP; software, MA, HA; investigation, MA, HA, CES, SS, PP; resources, EEA and AGT; data curation, EAA and AGT; writing—original draft preparation, MA, HA, writing—review and editing, AGT, EAA, HA and MMH. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MA and AGT thank the São Paulo State Research Foundation for support (FAPESP 2021/04464-8, 2018/25128-3, 2017/50333-7). AGT thanks the Conselho Nacional de Pesquisa e Desenvolvimento scientific research award. HA thanks the Wellcome Trust for a studentship (Grant ID: 218514/Z/19/Z). CES thanks the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine for a studentship (EP/L015838/1), generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB, and Vertex. PP thanks the Swiss National Science Foundation for an SNSF Fellowship and the Marie Skłodowska-Curie actions for an Individual Fellowship (GA No. 832700). EAA and MMH thank the EPSRC for support (EP/S013172/1).Notes and references
- Integrating Neglected Tropical Diseases into Global Health and Development: Fourth WHO Report on Neglected Tropical Diseases, World Health Organisation, 2017 Search PubMed.
- S.-M. Lee, M.-S. Kim, F. Hayat and D. Shin, Molecules, 2019, 24(21), 3886 CrossRef CAS PubMed.
- T. Hailu, M. Yimer, W. Mulu and B. Abera, J. Vector Borne Dis., 2016, 53, 193–198 Search PubMed.
- V. K. Jain and K. Jain, Drug Discovery Today, 2018, 23(1), 161–170 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83(3), 770–803 CrossRef CAS PubMed.
- T. A. D. Costa-Silva, S. S. Grecco, F. S. de Sousa, J. H. G. Lago, E. G. A. Martins, C. A. Terrazas, S. Varikuti, K. L. Owens, S. M. Beverley, A. R. Satoskar and A. G. Tempone, J. Nat. Prod., 2015, 78, 653–657 CrossRef PubMed.
- S. S. Grecco, T. A. Costa-Silva, G. Jerz, F. S. de Sousa, G. A. A. Conserva, J. T. Mesquita, M. K. Galuppo, A. G. Tempone, B. J. Neves, C. H. Andrade, R. L. O. R. Cunha, M. Uemi, P. Sartorelli and J. H. G. Lago, Phytomedicine, 2017, 24, 62–67 CrossRef CAS PubMed.
- S. S. Grecco, T. A. Costa-Silva, G. Jerz, F. S. de Sousa, V. S. Londero, M. K. Galuppo, M. L. Lima, B. J. Neves, C. H. Andrade, A. G. Tempone and J. H. G. Lago, Chem.-Biol. Interact., 2017, 277, 55–61 CrossRef CAS PubMed.
- M. Amaral, F. S. de Sousa, T. A. C. Silva, A. J. G. Junior, N. N. Taniwaki, D. M. Johns, J. H. G. Lago, E. A. Anderson and A. G. Tempone, Sci. Rep., 2019, 9, 6114 CrossRef PubMed.
- C. E. Sear, P. Pieper, M. Amaral, M. M. Romanelli, T. A. Costa-Silva, M. M. Haugland, J. A. Tate, J. H. G. Lago, A. G. Tempone and E. A. Anderson, ACS Infect. Dis., 2020, 6, 2872–2878 CrossRef CAS PubMed.
- K. Katsuno, J. N. Burrows, K. Duncan, R. H. Van Huijsduijnen, T. Kaneko, K. Kita, C. E. Mowbray, D. Schmatz, P. Warner and B. T. Slingsby, Nat. Rev. Drug Discovery, 2015, 14, 751–758 CrossRef CAS PubMed.
- K. P. Chang, C. A. Nacy and R. D. Pearson, in Methods Enzymol., Academic Press, 1986, vol. 132, pp. 603–626 Search PubMed.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu, A. Lu, X. Chen, T. Hou and D. Cao, Nucleic Acids Res., 2021, 49, W5–W14 CrossRef CAS PubMed.
- W. Zheng, J. Toxicol., Clin. Toxicol., 2001, 39(7), 711–719 CrossRef CAS PubMed.
- J. Kirchmair, A. H. Göller, D. Lang, J. Kunze, B. Testa, I. D. Wilson, R. C. Glen and G. Schneider, Nat. Rev. Drug Discovery, 2015, 14, 387–404 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- R. O. B. Don and J.-R. Ioset, Parasitology, 2014, 141, 140–146 CrossRef PubMed.
- L. A. Stauber, E. M. Franchino and J. Grun, J. Protozool., 1958, 5, 269–273 CrossRef CAS.
- A. G. Tempone, M. de S. C. Melhem, F. O. Prado, G. Motoie, R. M. Hiramoto, M. M. Antoniazzi, C. F. B. Haddad and C. Jared, Lett. Drug Des. Discovery, 2007, 4, 67–73 CrossRef CAS.
- H. Tada, O. Shiho, K.-I. Kuroshima, M. Koyama and K. Tsukamoto, J. Immunol. Methods, 1986, 93, 157–165 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and copies of 1H NMR data for all novel compounds (PDF). In silico bioavailability predictions. See DOI: https://doi.org/10.1039/d3md00081h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |