DOI:
10.1039/D3MD00113J
(Research Article)
RSC Med. Chem., 2023,
14, 947-956
Identification and analysis of small molecule inhibitors of FosB from Staphylococcus aureus†
Received
10th March 2023
, Accepted 19th April 2023
First published on 24th April 2023
Abstract
Antimicrobial resistance (AMR) poses a significant threat to human health around the world. Though bacterial pathogens can develop resistance through a variety of mechanisms, one of the most prevalent is the production of antibiotic-modifying enzymes like FosB, a Mn2+-dependent L-cysteine or bacillithiol (BSH) transferase that inactivates the antibiotic fosfomycin. FosB enzymes are found in pathogens such as Staphylococcus aureus, one of the leading pathogens in deaths associated with AMR. fosB gene knockout experiments establish FosB as an attractive drug target, showing that the minimum inhibitory concentration (MIC) of fosfomycin is greatly reduced upon removal of the enzyme. Herein, we have identified eight potential inhibitors of the FosB enzyme from S. aureus by applying high-throughput in silico screening of the ZINC15 database with structural similarity to phosphonoformate, a known FosB inhibitor. In addition, we have obtained crystal structures of FosB complexes to each compound. Furthermore, we have kinetically characterized the compounds with respect to inhibition of FosB. Finally, we have performed synergy assays to determine if any of the new compounds lower the MIC of fosfomycin in S. aureus. Our results will inform future studies on inhibitor design for the FosB enzymes.
Introduction
Antimicrobial resistance (AMR) poses a significant threat to human health around the world. In 2019, 1.27 million global deaths were directly attributable to bacterial AMR.1 Though bacterial pathogens can develop resistance through a variety of mechanisms, one of the most prevalent is the production of antibiotic-modifying enzymes,2 making them attractive targets for the development of new drugs that inhibit the antibiotic-modifying activity and restore potency of the original antibiotic. This combination approach has been proven effective, as exemplified by β-lactamase inhibitors being used in tandem with β-lactam antibiotics.3
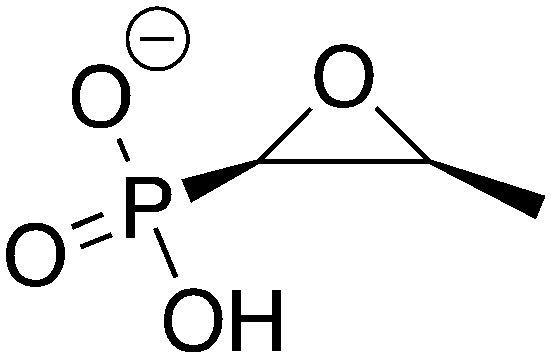
Fosfomycin, sold under the brand name Monurol®, is a broad-spectrum small molecule antibiotic that is effective against both Gram-negative and Gram-positive pathogens.4,5 It inhibits the enzyme UDP-N-acetylglucosamine-3-enolpyruvyltransferase (MurA), which catalyzes the first committed step in the cell wall biosynthesis pathway.6,7 Fosfomycin is naturally produced by certain strains of the genus Streptomyces8 and exhibits very low toxicity with few side effects in humans. Therefore, it is typically given as a single 3 g oral dose for urinary tract infections,9,10 and it is eliminated from the body almost entirely in its unmetabolized active form. However, fosfomycin-modifying enzymes (FosA, FosB, and FosX/M) have proven to be a significant complication for the extended usage of fosfomycin.
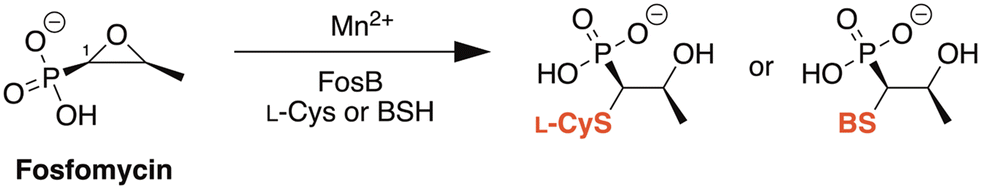
FosB, a fosfomycin-inactivating enzyme from Gram-positive species, is a Mn2+-dependent bacillithiol (BSH) or L-cysteine (L-Cys) transferase.11–13 It catalyzes the nucleophilic addition of the thiol to fosfomycin, which opens the epoxide ring and destroys the activity of the antibiotic (Fig. 1). FosB enzymes are found in pathogens such as Enterococcus faecium and Staphylococcus aureus,11,13 both of which are listed as high priority pathogens by the World Health Organization.14S. aureus is also one of the leading pathogens in deaths associated with AMR, due to methicillin-resistant S. aureus (MRSA) strains.1 We have previously established, through gene knockout experiments, that lack of the FosB enzyme restores fosfomycin activity, reducing the MIC from 64 μg mL−1 to 0.5 μg mL−1,15 thereby establishing FosB as an attractive drug target for a combination therapy approach.
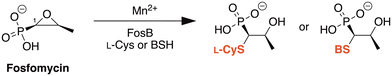 |
| | Fig. 1 Mechanism of fosfomycin inactivation by FosB, a Mn2+-dependent bacillithiol (BSH) or L-cysteine (L-Cys) transferase that catalyzes nucleophilic addition of the thiol to C1 of fosfomycin. | |
Herein, we have identified several lead compounds as potential inhibitors of FosB from S. aureus (FosBSa) using a high-throughput in silico screening approach. Next, we obtained crystal structures of FosB from Bacillus cereus (FosBBc) in the presence of each compound to identify protein–ligand interactions important for binding. We used FosBBc as a structural homolog for FosBSa in screening and crystallography experiments because the available structures of FosBSa are missing key active site residues necessary for identifying protein–ligand interactions. We then performed steady-state kinetic analyses to determine if any of the identified compounds effectively inhibit FosBSain vitro. The kinetic results suggest the compounds are competitive inhibitors for fosfomycin in FosBSa and provide the first evidence for potential cooperativity between two distinct FosBSa binding sites. All compounds have very low mammalian cytotoxicity. Unfortunately, even though our results provide valuable structural and mechanistic insights into inhibition of FosBSa, none of the identified compounds lowered the MIC of fosfomycin in cell-based synergy assays.
Experimental
General materials
Buffer salts were purchased from VWR. Magnesium chloride hexahydrate was purchased from Fisher Scientific, L-cysteine was purchased from Fisher Chemical, and fosfomycin disodium salt was obtained from MP Biomedicals, LLC.
Expression and purification of FosBBc and FosBSa
FosBBc and FosBSa were expressed and purified according to published protocols.11,12
High-throughput screening and compound identification
High-throughput screening and docking were performed according to a previously published procedure.15 The initial library of compounds used for screening was identified by searching the ZINC15 database for compounds with a 40% similarity cutoff to phosphonoformate (PPF).16–18
Purchasing and synthesis of compounds
Phosphonoacetate (PPA) was purchased from VWR. 2-Phosphonopropionic acid (2-PPP) was purchased from ABCR, and 2-phosphonoethylphosphonic acid (2-PEP) was purchased from Manchester Organics. 3-Phosphonopropionic acid (3-PPP) was purchased from TCI Chemicals, methylmalonate (MM) was purchased from AmBeed, and (1-hydroxypropan-2-yl)phosphonic acid (1H2Y) was purchased from ChemSpace. (1-Hydroxy-2-methylpropyl)phosphonic acid (1H2M) and 1-hydroxypropylphosphonic acid (1HPP) were custom synthesized as described below.
Preparation of compound 9.
To a mixture of dimethyl phosphite (2.50 mL, 27.3 mmol) and propanal (2.36 mL, 32.7 mmol) at 0 °C, triethylamine (1.14 mL, 8.19 mmol) was added dropwise. The reaction mixture was stirred at room temperature for 12 h and progress of the reaction was monitored by TLC (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19/MeOH:CH2Cl2, Rf 0.21). The organic layer was removed under reduced pressure and the residue was purified by flash column chromatography (SiO2, 1:19/MeOH:CH2Cl2) to afford compound 9 (1.68 g, 37%) as a colorless liquid: 1H NMR (500 MHz, (CD3)2SO, Fig. S1†) δ 5.46 (t, J = 6.8 Hz, 1H), 3.66 (d, J = 10.2 Hz, 3H), 3.64 (d, J = 10.2 Hz, 3H), 1.70–1.58 (m, 1H), 1.57–1.46 (m, 1H), 0.94 (td, J1 = 7.4 Hz, J2 = 0.9 Hz, 3H); 13C NMR (150 MHz, (CD3)2SO, Fig. S2†) δ 67.34 (d, J = 160.5 Hz), 52.84 (d, J = 6.5 Hz), 52.26 (d, J = 7.4 Hz), 24.59 (d, J = 3.2 Hz), 10.34 (d, J = 14.0 Hz).
:19/MeOH:CH2Cl2, Rf 0.21). The organic layer was removed under reduced pressure and the residue was purified by flash column chromatography (SiO2, 1:19/MeOH:CH2Cl2) to afford compound 9 (1.68 g, 37%) as a colorless liquid: 1H NMR (500 MHz, (CD3)2SO, Fig. S1†) δ 5.46 (t, J = 6.8 Hz, 1H), 3.66 (d, J = 10.2 Hz, 3H), 3.64 (d, J = 10.2 Hz, 3H), 1.70–1.58 (m, 1H), 1.57–1.46 (m, 1H), 0.94 (td, J1 = 7.4 Hz, J2 = 0.9 Hz, 3H); 13C NMR (150 MHz, (CD3)2SO, Fig. S2†) δ 67.34 (d, J = 160.5 Hz), 52.84 (d, J = 6.5 Hz), 52.26 (d, J = 7.4 Hz), 24.59 (d, J = 3.2 Hz), 10.34 (d, J = 14.0 Hz).
Preparation of compound 10.
To a mixture of dimethyl phosphite (1.67 mL, 18.2 mmol) and isobutyraldehyde (2.36 mL, 21.8 mmol) at 0 °C, triethylamine (0.76 mL, 5.46 mmol) was added dropwise. The reaction mixture was stirred at room temperature for 12 h and progress of the reaction was monitored by TLC (1:19/MeOH:CH2Cl2, Rf 0.24). The organic layer was removed under reduced pressure and the residue was purified by flash column chromatography (SiO2, 1:19/MeOH:CH2Cl2) to afford compound 10 (1.87 g, 56%) as a colorless liquid: 1H NMR (600 MHz, (CD3)2SO, Fig. S3†) δ 5.45 (dd, J1 = 8.2 Hz, J2 = 7.3 Hz, 1H), 3.68 (d, J = 10.1 Hz, 3H), 3.64 (d, J = 10.3 Hz, 3H), 3.58 (td, J1 = 7.4 Hz, J2 = 5.3 Hz, 1H), 1.95–1.87 (m, 1H), 0.96 (d, J = 6.8 Hz, 6H); 13C NMR (150 MHz, (CD3)2SO, Fig. S4†) δ 70.92 (d, J = 158.9 Hz), 52.89 (d, J = 7.5 Hz), 51.96 (d, J = 6.6 Hz), 29.88 (d, J = 4.4 Hz), 19.78 (d, J = 10.7 Hz), 17.73 (d, J = 6.6 Hz).
Preparation of compound 1 (1H2M).
A solution of compound 9 (1.00 g, 5.95 mmol) in 6 N HCl (5.0 mL) was refluxed overnight. The resulting solution was concentrated under reduced pressure and the residue obtained was dried overnight to afford compound 1 (712 mg, 93%) as a white solid: 1H NMR (600 MHz, D2O, Fig. S5†) δ 3.74 (ddd, J1 = 10.3 Hz, J2 = 6.7 Hz, J3 = 3.7 Hz, 1H), 1.88–1.79 (m, 1H), 1.69–1.59 (m, 1H), 1.05 (t, J = 7.4 Hz, 3H); 13C NMR (150 MHz, D2O, Fig. S6†) δ 68.92 (d, J = 158.1 Hz), 23.92 (d, J = 2.8 Hz), 9.70 (d, J = 14.0 Hz).
Preparation of compound 3 (1HPP).
A solution of compound 10 (1.00 g, 5.49 mmol) in 6 N HCl (5.0 mL) was refluxed overnight. The resulting solution was concentrated under reduced pressure and the residue obtained was dried overnight to afford compound 3 (798 mg, 94%) as a white solid: 1H NMR (600 MHz, D2O, Fig. S7†) δ 3.65 (dd, J1 = 8.3 Hz, J2 = 5.8 Hz, 1H), 2.10–2.02 (m, 1H), 1.05 (d, J = 7.0 Hz, 3H), 1.03 (d, J = 6.8 Hz, 3H); 13C NMR (150 MHz, D2O, Fig. S8†) δ 72.63 (d, J = 156.9 Hz), 29.59 (d, J = 4.0 Hz), 19.14 (d, J = 9.7 Hz), 17.16 (d, J = 7.7 Hz).
Absorbance-based kinetic assays
Kinetic assays using 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) were performed according to a published protocol.15 Kinetics were performed with different concentrations of each compound depending on their efficiency. This included 600 μM, 800 μM, and 1 mM PPA; 800 μM, 1.2 mM, and 1.6 mM 2-PPP; 3 mM 1HPP; 5 mM 1H2M; 6 mM 3-PPP; 6 mM 1H2Y; and 6 mM 2-PEP. Methylmalonate was tested with concentrations up to 10 mM with no observable inhibitory activity. The first concentration listed for each compound was the minimum amount required to observe inhibition. Kinetic data were fit using a global fitting approach (GraFit, Erithacus Software) with the following equations depending on the nature of inhibition:| | 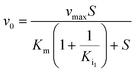 | (1) |
| | 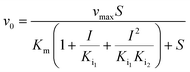 | (2) |
Eqn (1) represents simple, competitive inhibition with a single inhibition constant. Eqn (2) describes parabolic competitive inhibition19 where two molecules of the inhibitor bind to the enzyme. Here, Ki1 is the inhibition constant for the initial binding event and Ki2 is the inhibition constant for the second binding event.
Protein crystallization
Crystals of FosBBc with inhibitor compounds were grown using the vapor diffusion method in hanging drops containing a mixture of protein solution (10 mg mL−1 FosBBc in 20 mM HEPES buffer, pH 7.5 and 5 mM inhibitor) and reservoir solution and incubated against 2–3 μL of reservoir solution in a Hampton Research VDX plate at 298 K. The reservoir solutions that produced the best crystals differed based on the compound present in solution. Compound identities are presented in Table 1. The reservoir solutions were as follows: 125 mM Mg formate and 20% (w/v) poly(ethylene glycol) (PEG) 3350 (compound 1); 75 mM Mg formate and 14% (w/v) PEG 3350 (compound 2); 50 mM Mg formate and 16% (w/v) PEG 3350 (compound 3); 125 mM Mg formate and 16% (w/v) PEG 3350 (compounds 4, 5, and 6); 100 mM Mg formate and 18% (w/v) PEG 3350 (compound 7); 50 mM Mg formate and 14% (w/v) PEG 3350 (compound 8). Crystals formed in the presence of compounds 1, 3, 4, 5, and 6 were grown in drops containing 3 μL of each solution. Crystals formed in the presence of compounds 2 and 8 were grown in drops containing 2 μL of protein solution and 3 μL of reservoir solution, and crystals formed in the presence of compound 7 were grown in drops containing 3 μL of protein solution and 2 μL of reservoir solution. Each crystal was cryoprotected in its reservoir solution with 20% glycerol added prior to rapid freezing in liquid nitrogen.
Table 1 Structures, names, abbreviations, and Glide screening scores of the compounds in our resulting list
| Compound |
Structure and ZINC ID |
Chemical name |
Abbreviation |
Docking score |
Glide g-score |
| Control |
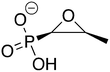 ZINC1530427 ZINC1530427 |
Fosfomycin |
Fos |
−10.734 |
−10.734 |
|
1
|
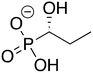 ZINC39255972 ZINC39255972 |
1-Hydroxypropylphosphonic acid |
1HPP |
−10.611 |
−10.611 |
|
2
|
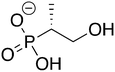 ZINC34127926 ZINC34127926 |
(1-Hydroxypropan-2-yl)phosphonic acid |
1H2Y |
−10.441 |
−10.441 |
|
3
|
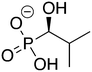 ZINC39275104 ZINC39275104 |
(1-Hydroxy-2-methylpropyl)phosphonic acid |
1H2M |
−10.420 |
−10.420 |
|
4
|
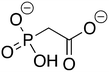 ZINC3869741 ZINC3869741 |
Phosphonoacetate |
PPA |
−10.113 |
−10.113 |
|
5
|
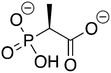 ZINC2008861 ZINC2008861 |
2-Phosphonopropionic acid |
2-PPP |
−10.113 |
−10.113 |
|
6
|
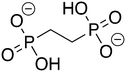 ZINC3953973 ZINC3953973 |
2-Phosphonoethylphosphonic acid |
2-PEP |
−9.305 |
−9.305 |
|
7
|
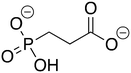 ZINC2008858 ZINC2008858 |
3-Phosphonopropionic acid |
3-PPP |
−8.025 |
−8.025 |
|
8
|
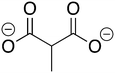 ZINC901289 ZINC901289 |
Methylmalonate |
MM |
−7.734 |
−7.734 |
Data collection, structure determination, and refinement
X-ray diffraction data were collected at 100 K at the 22-ID beamline at the Advanced Photon Source at the Argonne National Laboratory (Argonne, IL). The data were indexed, integrated, and scaled with HKL2000.37 The crystal structure determination was performed by molecular replacement with PHASER20 using PDB ID 4JH6 (ref. 12) as a search model. All metals and ligands were removed from PDB ID 4JH6 prior to phasing. All crystals belong to the P212121 space group, and all structures contain 270 amino acids and 2 metal ions in the asymmetric unit. Model building for each structure was performed using Coot model building software.21 Water molecules were placed with the Coot routine, Find Waters. The final models were obtained by iterative cycles of model building in Coot and structure refinement using Refmac5 (ref. 22) in the CCP4 suite of programs (Collaborative Computational Project, 1994). All protein figures were prepared with Chimera.23 A summary table consisting of the data collection and refinement statistics of FosB complexes is shown in Table S1.† All root-mean square deviation (RMSD) analyses were performed with respect to PDB ID 4JH6.
MIC assays
All compounds and fosfomycin were dissolved in water (final stock concentration of 10 mg mL−1). S. aureus JE2 was streaked on Mueller–Hinton (MH) agar and allowed to grow at 37 °C overnight. Enough colonies were scraped from the plate and suspended in MH broth with vortexing to reach an attenuance at 600 nm (OD600) of ∼0.5. The resulting culture was diluted 1:1000 and added to the prepared serially doubly diluted compound solutions in 96-well plates. For each row, one well was reserved for sterile control (which consisted of 200 μL of MH medium) and another well was reserved for growth control (which consisted of 100 μL of MH medium seeded with 100 μL of bacterial culture). Plates were incubated overnight at 37 °C and the MIC were visually determined to be the well with no growth for each compound (Table 3). For combination studies, fosfomycin was diluted horizontally across the plate, while the inhibitor was incubated vertically across the plate prior to addition of the seed culture.
Cytotoxicity analysis
The cytotoxicity against mammalian cells was measured based on a previously published protocol24 using human embryonic kidney (HEK-293) and epithelial-like morphology hepatocellular carcinoma (HepG2). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C with 5% CO2 until confluent. Cells at a concentration of 10000 cells per mL were plated in 96-well plate (100 μL) and grown overnight. The next day, the medium was replaced with 100 μL of fresh DMEM and 100 μL of DMEM containing test compounds with final concentrations of 64 to 4 μg mL−1. Media containing 1% Triton X-100™ was used for the positive control. The cells were incubated with tested compounds for another 24 h at 37 °C with 5% CO2. Cell survival was assessed by a resazurin assay, as follows. Each well was treated with resazurin (10 μL of a 2 mM solution) for 24 h. Live cells produced a pink color and the highly fluorescent dye, resorufin, that was detected by using a SpectraMax M5 plate reader to quantify the viability of the cells. These data are presented in Fig. 5 and S14.†
Results and discussion
Identification of FosB inhibitors
High-throughput virtual screening (HTVS) is widely used to identify inhibitors of target proteins.25–27 To identify inhibitors of FosBSa, we have used an HTVS method employing the program Glide28–30 in the Schrödinger program suite to dock ligands from the ZINC15 database,16–18 a free-to-use online library of small molecules that contains more than 997 million compounds separated based on molecular weight and logP values. We have previously used this screening method to identify and validate phosphonoformate (PPF) as a potent inhibitor of the FosBSa enzyme.15 To obtain new lead compounds, we searched the ZINC15 library for ligands with a 40% similarity cutoff to PPF using the pre-built search function in the database. The output was then screened by docking the molecules in the FosB active site using Glide as described in the methods. Our resulting list of hits, shown in Table 1, was initially selected based on the docking score with a cutoff value of −7.5. According to the Schrödinger website, an acceptable Glide score can vary from −8 to −12, depending on the system.31 A cutoff of −7.5 allows us to remove most false positives from our compound pool while still maintaining a diverse range of poses. Successful HTVS also requires a high-resolution crystal structure of the target protein. However, the crystal structure of FosBSa (PDB ID: 4NB2) is not suitable for the docking procedure due to disordered regions in the flexible loop in the fosfomycin binding site that spans from Ile90 to Asp100.11 As an alternative, we used the crystal structure of FosB from Bacillus cereus (FosBBc; PDB ID: 4JH6), which we have previously established as an acceptable structural and functional analogue of FosBSa.15
Purchasing and synthesis of compounds
Of the eight compounds found in our list, six of them were available to purchase from various vendors, as listed in the methods. The remaining two required custom synthesis. Compounds 1 (1H2M) and 3 (1HPP) were synthesized based on previously reported methods.32 The commercially available aldehydes (propanal and isobutyraldehyde) were reacted with dimethyl phosphite in the presence of a base to provide compounds 9 and 10. The acid hydrolysis of these intermediates resulted in the formation of compounds 1 (1H2M) and 3 (1HPP) (Scheme 1).
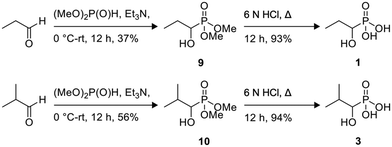 |
| | Scheme 1 Synthetic schemes for the preparation of compounds 1 (1H2M) and 3 (1HPP). | |
Crystal structures of FosBBc-inhibitor complexes
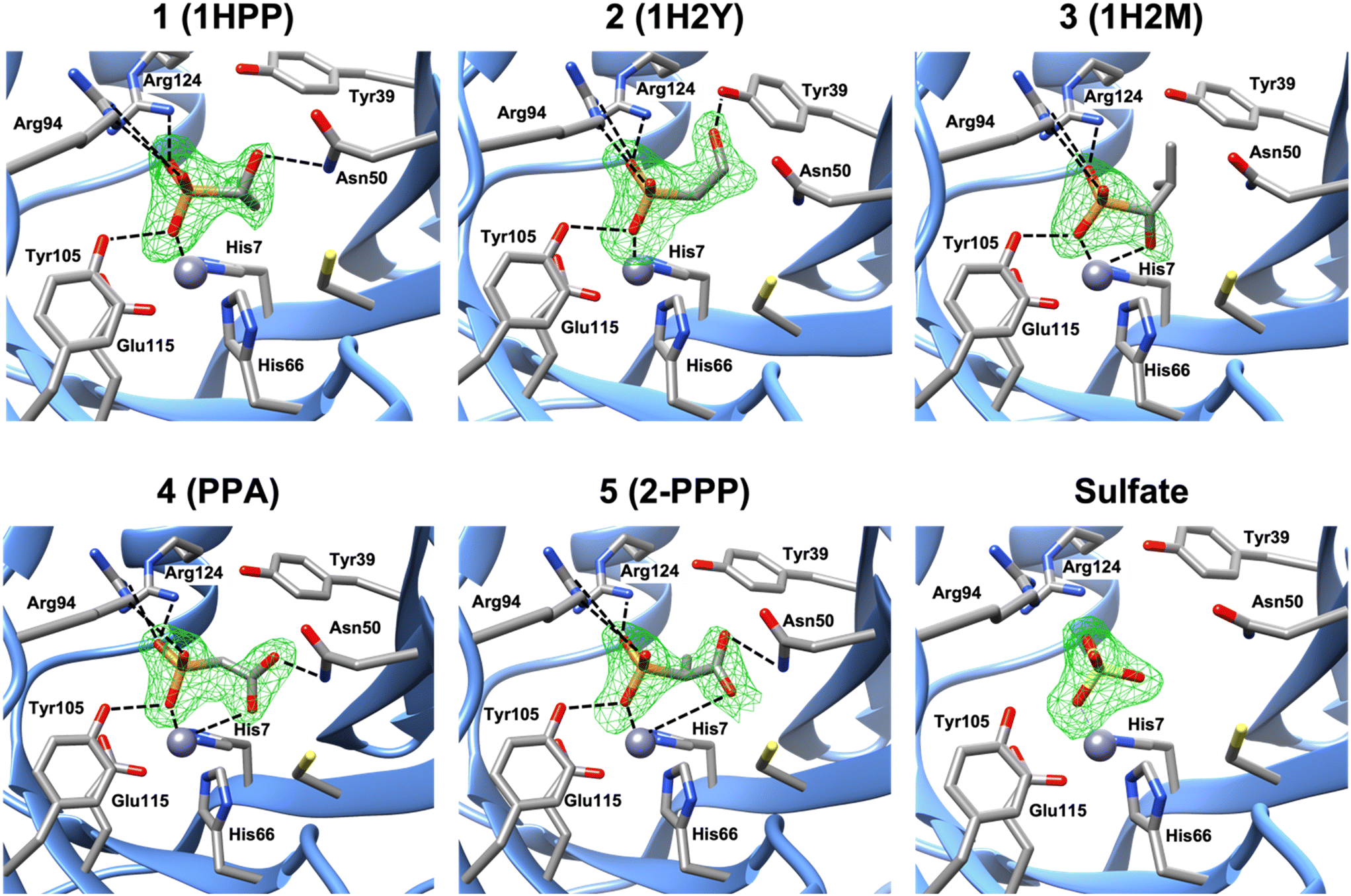
Molecular docking of each compound using Glide resulted in the predicted active site fit shown in Fig. S9.† Nevertheless, we obtained crystal structures of FosBBc in complex with five of the new compounds from our list, at resolutions between 1.8 and 2.2 Å, to investigate protein-ligand interactions. The structures were determined by molecular replacement using the structure of FosBBc with fosfomycin and Mn2+ bound (PDB ID 4JH6 (ref. 12)) as a search model. Although FosB is a Mn2+-dependent enzyme, it often purifies with Zn2+ in the divalent metal binding site due to the zinc-rich medium used in the purification process.12 Therefore, we have modeled Zn2+ into the metal sites in each structure, which is consistent with other structures of FosBBc (Fig. 2).11–13
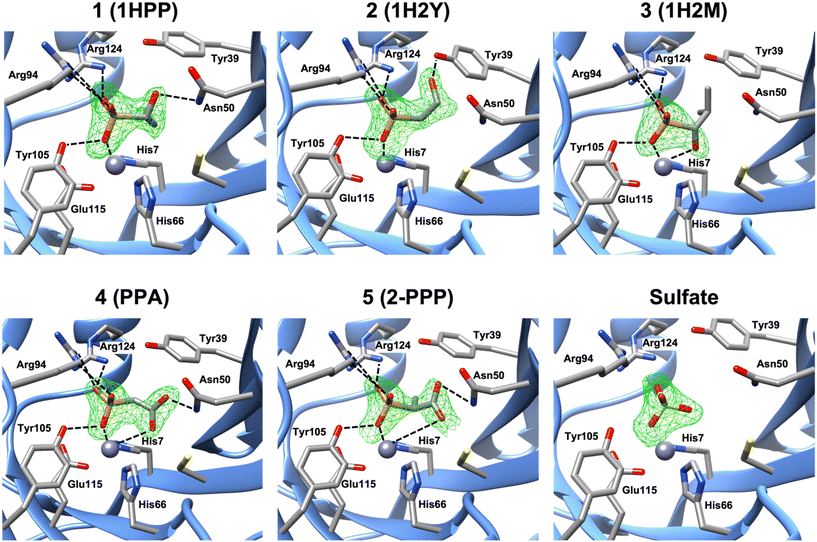 |
| | Fig. 2 A zoomed-in view of the active site of FosBBc bound to Zn2+ and compounds from our list. The omit electron density Fo–Fc difference map (mesh) was calculated before the modeling of the inhibitor and contoured at 3σ. Important interactions with the metal and active site residues (as listed in Table S2†) are shown as dashed lines. Interactions with Tyr64 have been omitted for visibility. | |
The overall structures of the new FosBBc-inhibitor complexes all conform to the same domain-swapped homodimeric βαβββ motif as all other currently characterized fosfomycin resistance enzymes.11–13,33–35 All ligands have been modeled into Fo–Fc electron difference density maps (Fig. 2) in the FosBBc active site. Table S2† summarizes the observed interactions of each compound with active site residues. Since we have modeled Zn2+ into each metal site, the crystal structure of FosBBc with Zn2+ and fosfomycin (PDB ID: 4JH3)12 has been used for the active site interaction comparisons to best compare all metal–ligand distances. Comparisons to our previous crystal structure with FosBBc with PPF (PDB ID: 8DTD)15 have also been included as an additional reference. Each compound containing a phosphonate group exhibits the same interactions as seen with the phosphonate group of fosfomycin, including coordination to the metal and hydrogen bonding interactions with the sidechains of Tyr64, Arg94, Tyr105, and Arg124.
Crystal structure of FosBBc·Zn2+·compound 1 complex (PDB ID: 8G7F)
The crystal structure of the FosBBc·Zn2+·compound 1 complex revealed that, in addition to the interactions observed with the phosphonate group, the hydroxyl group of compound 1 forms a hydrogen bond with the amine group of Asn50. This residue has been found to be responsible for stabilizing both the C1 of fosfomycin and the deprotonated thiol group of the incoming nucleophile in FosB.12 Interactions at this residue could therefore prove beneficial for a compound's inhibitory capability. This complex is, however, missing the bidentate metal chelation seen with both fosfomycin and PPF. The overall RMSD for all atoms between this structure and PDB ID 4JH6 is 0.166 Å.
Crystal structure of FosBBc·Zn2+·compound 2 complex (PDB ID: 8G7H)
The binding interactions observed in the active site of the FosBBc·Zn2+·compound 2 complex are very similar to those in the complex with compound 1. However, instead of forming a hydrogen bonding interaction with Asn50, the hydroxyl group of compound 2 interacts with Tyr39. Tyr39 has been implicated in facilitating the deprotonation of the incoming thiol nucleophile.12 As with Asn50, interactions with this residue may prove beneficial to the efficiency of an inhibitor in FosB. Compound 2 also does not exhibit bidentate metal coordination, and has an overall RMSD value of 0.135 Å.
Crystal structure of FosBBc·Zn2+·compound 3 complex (PDB ID: 8G7G)
In contrast to the complexes with compounds 1 and 2, the FosBBc·Zn2+·compound 3 structure does not show any interactions with Asn50 or Tyr39. Instead, the hydroxyl group is oriented 2.4 Å from the metal, comparable to the oxirane oxygen of fosfomycin (2.5 Å) and the carboxylate oxygen of PPF (2.2 Å). However, a hydroxyl group, with a pKa between 16 and 18, would be in its protonated state at physiological pH. Protonation of the oxirane oxygen has been proposed to trigger product release from the active site of FosB,36 suggesting compound 3 is likely not an ideal candidate for FosB inhibition. The overall RMSD of this structure when compared to PDB ID 4JH6 is 0.445 Å.
Crystal structure of FosBBc·compound 4 complex (PDB ID: 8E7R)
The crystal structure of FosBBc and compound 4 shows that, similarly to PPF, one carboxylate oxygen of compound 4 is oriented toward the metal at a distance of 3.4 Å. At this distance, the carboxylate may be engaging in an electrostatic interaction with the metal instead of the coordinate binding seen in fosfomycin and PPF. This suggests that compound 4 likely binds with lower affinity than fosfomycin or PPF, though it does retain some of the bidentate character. Moreover, the second carboxylate oxygen of compound 4 forms a hydrogen bond with the amine group of Asn50. The overall RMSD of PDB ID 8E7Q compared to the FosBBc·fosfomycin complex is 0.200 Å.
Crystal structure of FosBBc·compound 5 complex (PDB ID: 8E7Q)
Compound 5 binds in the FosBBc active site in a similar orientation to compound 4 and PPF, with a carboxylate oxygen facing the metal binding site. However, this carboxylate is even further from the metal than that of compound 4, sitting 4.4 Å away, though this is still within range to provide electrostatic interaction. The carboxylate also forms a hydrogen bond with the amine of Asn50, which likely helps stabilize this orientation in the active site. The overall RMSD of PDB ID 8E7R compared to PDB ID 4JH6 is 0.160 Å.
Crystal structure of FosBBc·sulfate complex (PDB ID: 8G7I)
Binding of compounds 6, 7, and 8 was not observed. Instead, only a sulfate ion was found in the active site. The sulfate ion remains bound to FosBBc following the ammonium sulfate precipitation step during purification. This has been observed in other crystal structures of FosB.11,12 Lack of binding of compound 6–8 in the FosBBc active site suggests that (1) the molecules have become too large, as may be the case for compounds 6 and 7, or (2) the phosphonate group, which compound 8 does not have, is likely important for binding affinity.
Mode of inhibition studies by steady-state kinetic assays
To elucidate the mechanism of inhibition of FosBSa by these compounds, and to determine the inhibitory efficiency of each, we performed Michaelis–Menten (MM) analyses of FosBSa in the presence of each compound using an absorbance-based DTNB assay (Table 2). This assay has already proven effective to determine the efficiency of the competitive FosB inhibitor, PPF.15 Consistent with previous experiments involving FosB enzymes, we have used L-cysteine as the nucleophilic substrate due to the high cost of BSH.
Table 2 Steady-state enzyme kinetics of FosBSa inhibition by compounds from our inhibitor library
| Inhibitor |
K
m (mM) |
k
cat (s−1) |
k
cat/Km (mM−1 s−1) |
K
i1 (mM) |
K
i2 (mM) |
|
Estimated Ki values from a single inhibitor concentration.
|
| None |
0.29 ± 0.03 |
2.95 |
10.33 |
— |
— |
|
1
|
0.25 ± 0.04 |
2.82 |
11.37 |
1.57 ± 0.37a |
— |
|
2
|
0.27 ± 0.03 |
2.91 |
10.65 |
5.29 ± 1.24a |
— |
|
3
|
0.25 ± 0.03 |
2.84 |
11.24 |
3.58 ± 0.78a |
— |
|
4
|
0.24 ± 0.02 |
2.83 |
11.81 |
0.90 ± 0.13 |
0.21 ± 0.05 |
|
5
|
0.25 ± 0.03 |
2.84 |
11.35 |
0.43 ± 0.06 |
— |
We began our steady-state kinetic analyses using compounds 4 and 5. These were selected from the set based on availability and similarity to PPF. All three compounds coordinate the active site metal ion in a bidentate manner through a phosphonate oxygen and a carboxylic acid oxygen. In addition, analysis of the series (PPF to 4 to 5) allowed us to investigate a structure activity relationship via growing the PPF molecule by one methylene carbon and then subsequently adding a methyl group to the methylene carbon. Kinetic assays for 4 and 5 were performed in the presence of multiple concentrations of inhibitor to determine the mechanism of inhibition. Initial velocities determined in the presence of 5 could be globally fit to eqn (1), consistent with simple competitive inhibition vs. fosfomycin with a Ki value of 0.43 mM (Fig. S10B†). Initial velocities in the presence of 4 gave a poor fit to eqn (1) suggesting a different mechanism of inhibition. Instead, inhibition data by 4 could be fit by eqn (2) (Fig. S10A†), which describes parabolic competitive inhibition.19 As predicted by the mechanism, replots of the individually-fit Lineweaver–Burk slopes for 4 yielded a parabolic curve (Fig. S11A†).
Parabolic competitive inhibition is seen when two molecules of the inhibitor can bind to the enzyme. In the case of 4, the first binding is weak but activates a second binding event with a much lower Ki value. Ki1 and Ki2 for 4 are 0.90 ± 0.13 and 0.21 ± 0.05 mM, respectively. This is interesting because the FosB enzymes are homodimers with two domain-swapped metal ion active sites per enzyme. Furthermore, each active site has two distinct substrate binding locations, one for fosfomycin and a second for the thiol co-substrate. Previous structural analyses have indicated that fosfomycin must bind first to FosB.37 Then, coordination of the phosphonate domain of fosfomycin by active site arginine residues forms and stabilizes the second thiol co-substrate binding site. Thus, there are four total binding sites on each enzyme and two possible explanations for the two inhibition events. The first is that binding of the inhibitor at one metal ion active site increases affinity at the other in an allosteric cooperative manner. If that is the case, this is the first evidence of cooperativity in the FosB enzymes. The second explanation is that a second molecule of compound 4 is binding in the thiol co-substrate binding site after it is formed. However, no evidence of 4 bound in the thiol binding site was observed in the crystal structures.
Compounds 1, 2, and 3 required significantly increased concentrations (as high as 6 mM) in order to observe any notable inhibition. Hence, they are poor FosBSa inhibitors. Therefore, we did not pursue a full inhibition kinetic analysis for these compounds. Nevertheless, to estimate kinetic parameters and Ki values for comparison of the remaining compounds, we completed kinetic assays using a single concentration of inhibitor and fit the data to a traditional MM equation for competitive inhibition (Fig. S13A–C†). We understand the limitations of only using one concentration, but we do not view these compounds as particularly valuable for future studies. Finally, compounds 6, 7, and 8 did not bind with high enough affinity to displace the sulfate ion in the active site of FosB from purification. Consequently, compounds 6, 7, and 8 did not effectively inhibit FosBSa; thus, we did not further pursue inhibition assays for these compounds.
Structure–activity relationship (SAR) of inhibitors
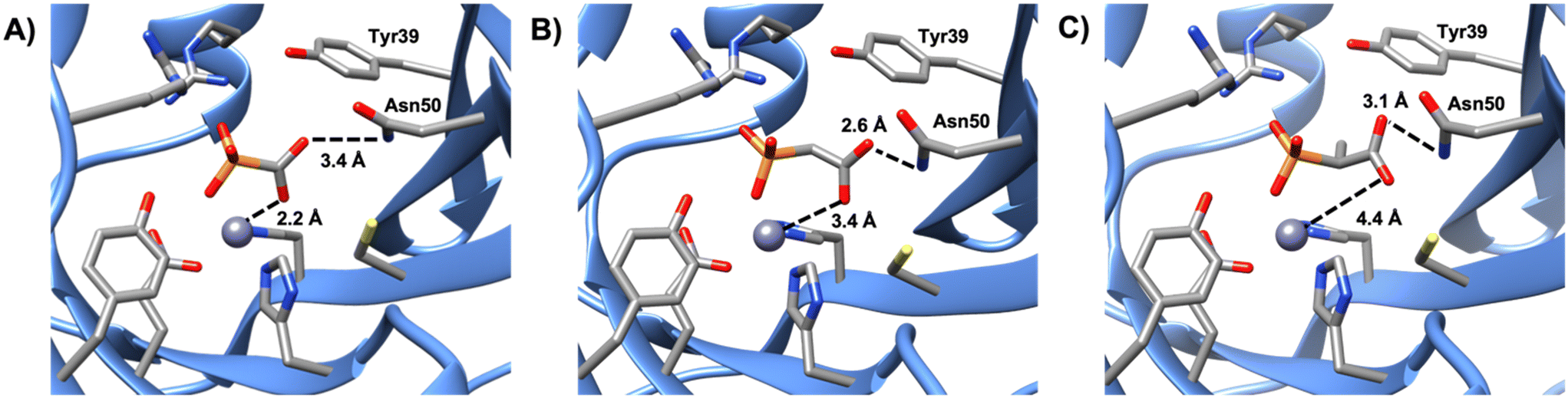
Of the phosphonate-containing compounds, the three that inhibited most effectively (PPF, compound 4, and compound 5) had bidentate coordination to the metal (Fig. 3). As the carboxylate oxygen of each compound moves further away, there is a strong correlation between the best Ki value and the carboxylate–oxygen metal distances: the carboxylate oxygen–metal distances for PPF, compound 4, and compound 5 are 2.2, 3.4, and 4.4 Å, respectively, which results in a Ki of 0.09 ± 0.01, 0.21 ± 0.05, and 0.43 ± 0.06 mM for each compound, respectively (Fig. S12†). Here we have used Ki2 for compound 4 because we cannot be certain if the crystal structure is a representation of the first or second binding event given that both active sites have the molecule bound in the same orientation. Nevertheless, all three compounds were significantly better inhibitors, according to either Ki value, than any of the compounds that formed only a single metal–ligand interaction between the metal and the phosphonate, indicating that a bidentate metal–ligand interaction should be pursued when identifying an effective inhibitor for FosB. In addition, functional groups that are similar in size and shape to the phosphonate, such as sulfates or sulfonates, might also be worth pursuing in the future.
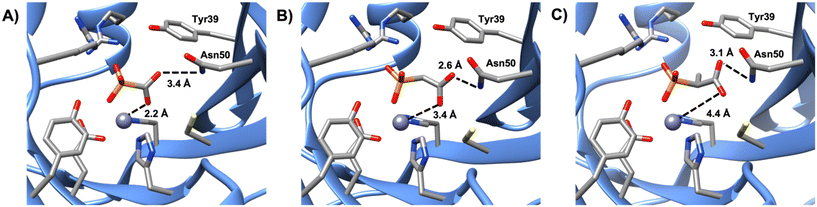 |
| | Fig. 3 Comparison of the metal-to-ligand distances of A) PPF, B) compound 4, and C) compound 5. | |
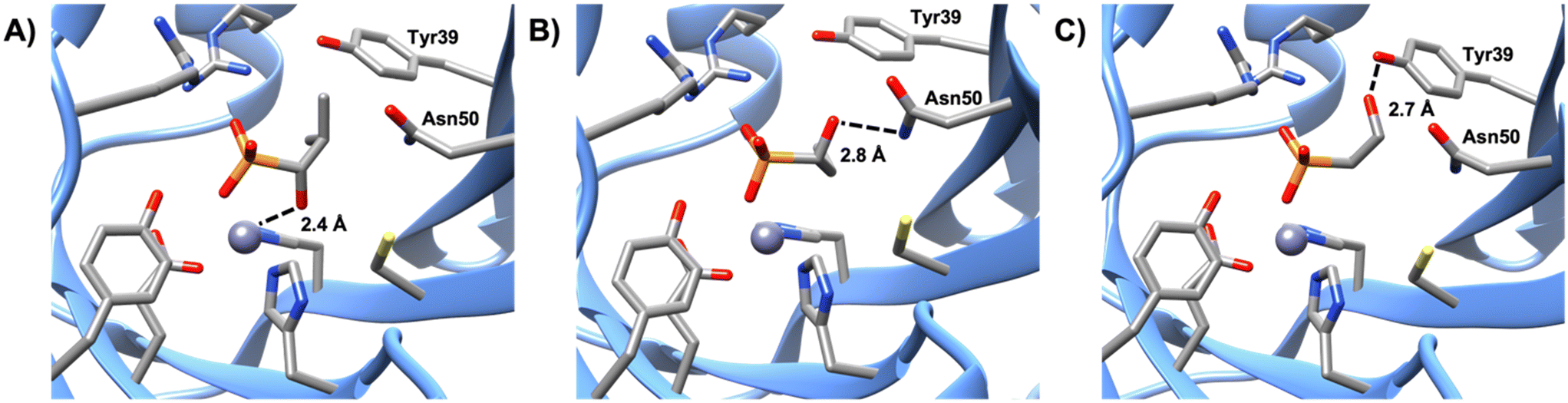
Like compounds 4 and 5, compound 3 also forms bidentate metal coordination, with the hydroxyl group positioned 2.4 Å from the metal. However, the estimated Ki is significantly higher. Protonation of the oxirane oxygen of fosfomycin to form the resulting hydroxyl product has been proposed to trigger product release from the active site of FosB.36 This may explain why compound 3 is a poor inhibitor even though it maintains the bidentate metal coordination. Furthermore, compound 3 is not stabilized by hydrogen bonding to active site amino acids Tyr39 or Asn50. Interaction with either Tyr39 or Asn50 may be advantageous for future inhibitor design. Of the evaluated compounds, the only one with no bonds to either residue was compound 3, which had an estimated Ki of 3.58 ± 0.78 mM. By comparison, compound 1 (estimated Ki value of 1.57 ± 0.37 mM), which lacks bidentate metal coordination, yet forms hydrogen bonding interactions with Asn50 (Fig. 4), is a more effective inhibitor than compound 3, indicating that while bidentate metal coordination is important, hydrogen bond interactions also contribute to inhibitor potency. Conversely, compound 2, which only forms hydrogen bonds with Tyr39, has an estimated Ki value of 5.29 ± 1.24 mM, making it the least effective of the tested compounds. This suggests that interactions with Tyr39 play less of a role in inhibition than those with Asn50. In future studies, compounds that exhibit bidentate metal binding could be functionalized to more closely investigate the effect interactions with Asn50 and Tyr39 have on the efficiency of FosB inhibitors.
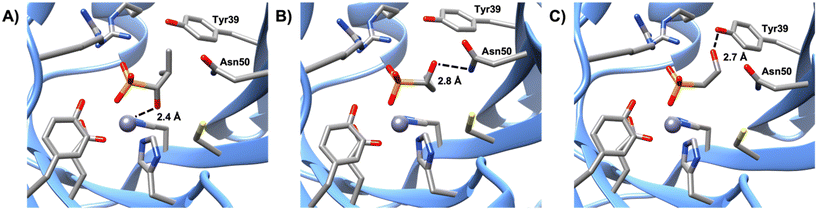 |
| | Fig. 4 Comparison of the hydrogen bonding distances of A) compound 3, B) compound 1 to the amine group of Asn50, and C) compound 2 to the hydroxyl group of Tyr39. | |
Synergy of inhibitors with fosfomycin
The double dilution method was used to determine the MIC of the FosB inhibitors (Table 3). A strain of S. aureus containing FosB, strain JE2, was used for the MIC experiments. While fosfomycin displayed an MIC value of 8 μg mL−1, all the inhibitory compounds displayed an MIC value of >128 μg mL−1. To understand if the inhibitors work in vitro, the bacteria is treated in combination with fosfomycin. Combination of the inhibitors with fosfomycin failed to change the observed MIC value of fosfomycin. This, in combination with the inability of the inhibitors to alter the growth of the bacteria on their own, likely indicates a cell impermeability of the inhibitors, preventing them from inhibiting FosB.
Table 3 MIC (μg mL−1) values of FosB inhibitors against S. aureus JE2
| Compound |
MIC |
MIC w/ Fos |
MICFos w/ 128 μg mL−1 inhibitor |
Synergy |
| Fos |
8 |
— |
— |
— |
|
1
|
>128 |
>128 |
8 |
None |
|
2
|
>128 |
>128 |
8 |
None |
|
3
|
>128 |
>128 |
8 |
None |
|
4
|
>128 |
>128 |
8 |
None |
|
5
|
>128 |
>128 |
8 |
None |
Analysis of cytotoxicity
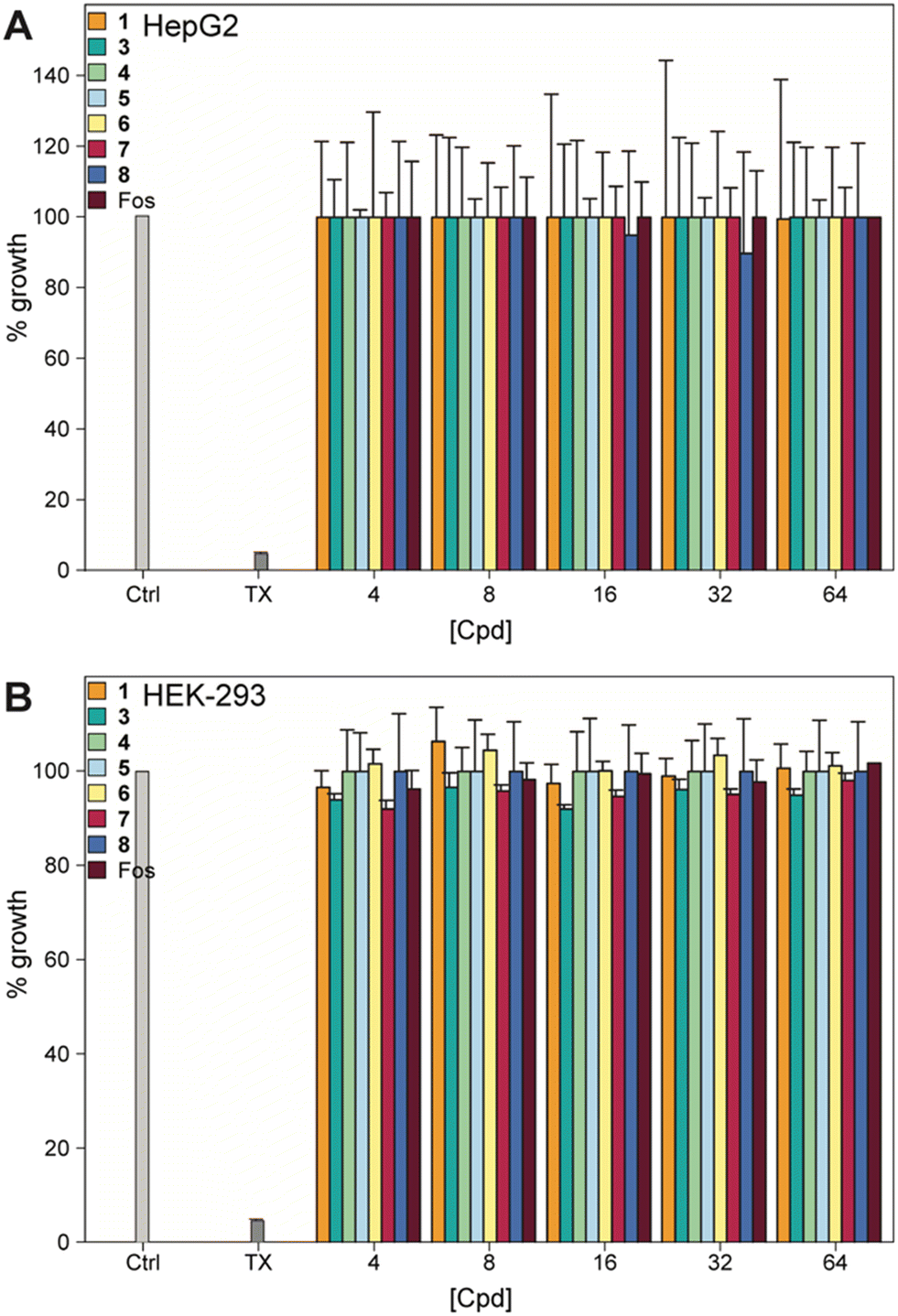
We examined the effect of the compounds on human embryonic kidney cells (HEK-293) and human hepatoma cells (HepG2), as many compounds are metabolized in the liver and/or excreted through the kidneys. None of the compounds, or fosfomycin, showed any toxicity to either cell type at 64 μg mL−1, indicating an excellent safety profile (Fig. 5).
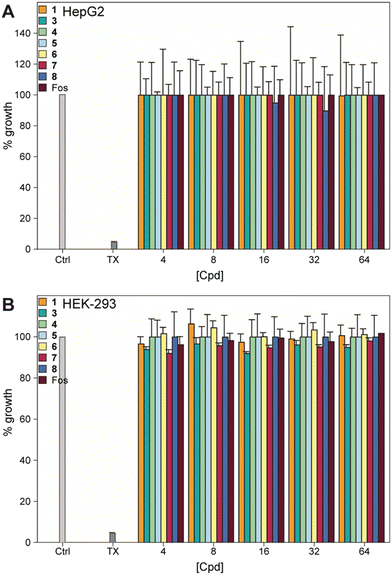 |
| | Fig. 5 Mammalian cell cytotoxicity of compounds 1, 3–8, and fosfomycin at varying concentrations against A) HepG2 and B) HEK-293 cell lines. Note: The corresponding non-normalized plots can be found in the ESI† (Fig. S14). | |
Conclusion
We have used HTVS to identify several new inhibitors of the FosB enzyme from S. aureus. We have characterized the inhibitors structurally and kinetically to determine which might serve as lead compounds in future combinatorial uses with fosfomycin. Our results indicate that molecules containing a phosphonate or sulfate-like moiety and the ability to coordinate the active site metal ion in a bidentate manner are a promising avenue for inhibition of the FosB enzymes. None of the identified compounds exhibited any appreciable cytotoxicity, but unfortunately, none of the compounds lowered the MIC of fosfomycin in cell-based assays with MRSA. Nevertheless, we will continue to investigate the prospects of these compounds in other Gram-positive strains, and we anticipate these preliminary HTVS results will inform on future structure-based drug design experiments in efforts to combat fosfomycin resistance.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was funded by The University of Alabama startup funds (to M. K. T.), the Cystic Fibrosis Foundation Grant #THOMPS2010 (to M. K. T.), and National Institutes of Health grant #1R15GM148956 (to M. K. T.). S. T. would like to acknowledge the Department of Education GAANN Grant #P200A150329. M. K. T would like to acknowledge the generosity of the Richard N. Armstrong family and Vanderbilt University Department of Biochemistry for initial laboratory equipment along with plasmids and reagents specific to this project. We thank the staff of the SER-CAT sector at the Advanced Photon Source at the Argonne National Laboratory for assistance with remote data collection and the University of Kentucky Center for Structural Biology for the synchrotron support. The Hek-293 are from ATCC (ATCC CRL-1573), the HepG2 from ATCC (ATCC HB-8065). Some NMR data reported in this publication were recorded on a Bruker AVANCE NEO 600 MHz high-performance digital NMR spectrometer supported by a NIH S10 grant S10OD028690 (to S. G.-T.). We thank the University of Kentucky PharmNMR Center (in the College of Pharmacy) for NMR support.
References
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. Kashef Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, N. Mturi, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, H. Vu, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- J. M. Munita and C. A. Arias, Microbiol. Spectrum, 2016, 4, 1–24 CAS.
- S. M. Drawz and R. A. Bonomo, Clin. Microbiol. Rev., 2010, 23, 160–201 CrossRef CAS PubMed.
- J. M. Hamilton-Miller, Microbios, 1992, 71, 95–103 CAS.
- J. Ruiz Ramos and M. Salavert Lletí, Rev. Esp. Quimioter., 2019, 32(Suppl 1), 45–54 Search PubMed.
- S. Eschenburg, W. Kabsch, M. L. Healy and E. Schonbrunn, J. Biol. Chem., 2003, 278, 49215–49222 CrossRef CAS PubMed.
- S. Eschenburg, M. Priestman and E. Schönbrunn, J. Biol. Chem., 2005, 280, 3757–3763 CrossRef CAS PubMed.
- D. Hendlin, E. O. Stapley, M. Jackson, H. Wallick, A. K. Miller, F. J. Wolf, T. W. Miller, L. Chaiet, F. M. Kahan, E. L. Foltz, H. B. Woodruff, J. M. Mata, S. Hernandez and S. Mochales, Science, 1969, 166, 122–123 CrossRef CAS PubMed.
- A. C. Dijkmans, N. V. O. Zacarías, J. Burggraaf, J. W. Mouton, E. B. Wilms, C. van Nieuwkoop, D. J. Touw, J. Stevens and I. M. C. Kamerling, Antibiotics, 2017, 6, 1–17 CrossRef PubMed.
- S. S. Patel, J. A. Balfour and H. M. Bryson, Drugs, 1997, 53, 637–656 CrossRef CAS PubMed.
- M. K. Thompson, M. E. Keithly, M. C. Goodman, N. D. Hammer, P. D. Cook, K. L. Jagessar, J. Harp, E. P. Skaar and R. N. Armstrong, Biochemistry, 2014, 53, 755–765 CrossRef CAS PubMed.
- M. K. Thompson, M. E. Keithly, J. Harp, P. D. Cook, K. L. Jagessar, G. A. Sulikowski and R. N. Armstrong, Biochemistry, 2013, 52, 7350–7362 CrossRef CAS PubMed.
- V. Wiltsie, S. Travis, M. R. Shay, Z. Simmons, P. Frantom and M. K. Thompson, Protein Sci., 2022, 31, 580–590 CrossRef CAS PubMed.
- Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics, WHO, 2017.
- S. Travis, K. D. Green, N. C. Gilbert, O. V. Tsodikov, S. Garneau-Tsodikova and M. K. Thompson, Biochemistry, 2023, 62, 109–117 CrossRef CAS PubMed.
- J. J. Irwin and B. K. Shoichet, J. Chem. Inf. Model., 2005, 45, 177–182 CrossRef CAS PubMed.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, J. Chem. Inf. Model., 2012, 52, 1757–1768 CrossRef CAS PubMed.
- T. Sterling and J. J. Irwin, J. Chem. Inf. Model., 2015, 55, 2324–2337 CrossRef CAS PubMed.
- D. K. Ekanayake, B. Andi, K. D. Bobyk, A. H. West and P. F. Cook, J. Biol. Chem., 2010, 285, 20756–20768 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- K. Shrestha Sanjib, Y. Fosso Marina, D. Green Keith and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2015, 59, 4861–4869 CrossRef CAS PubMed.
- M. S. Murgueitio, M. Bermudez, J. Mortier and G. Wolber, Drug Discovery Today: Technol., 2012, 9, e219–e225 CrossRef CAS PubMed.
- R.-L. Du, N. Sun, Y.-H. Fung, Y.-Y. Zheng, Y.-W. Chen, P.-H. Chan, W.-L. Wong and K.-Y. Wong, RSC Med. Chem., 2022, 13, 79–89 RSC.
- T. F. Vieira, R. P. Magalhães, M. Simões and S. F. Sousa, Antibiotics, 2022, 11, 1–22 CrossRef PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
-
Schrödinger, What is considered a good GlideScore?, (accessed January 16, 2023).
- W. R. Harris, C. E. Brook, C. D. Spilling, S. Elleppan, W. Peng, M. Xin and J. V. Wyk, J. Inorg. Biochem., 2004, 98, 1824–1836 CrossRef CAS PubMed.
- R. N. Armstrong, Biochemistry, 2000, 39, 13625–13632 CrossRef CAS PubMed.
- K. L. Fillgrove, S. Pakhomova, M. R. Schaab, M. E. Newcomer and R. N. Armstrong, Biochemistry, 2007, 46, 8110–8120 CrossRef CAS PubMed.
- C. L. Rife, R. E. Pharris, M. E. Newcomer and R. N. Armstrong, J. Am. Chem. Soc., 2002, 124, 11001–11003 CrossRef CAS PubMed.
- A. H. Lima, J. R. A. Silva, C. N. Alves and J. Lameira, ACS Omega, 2021, 6, 12507–12512 CrossRef CAS PubMed.
- A. A. Roberts, S. V. Sharma, A. W. Strankman, S. R. Duran, M. Rawat and C. J. Hamilton, Biochem. J., 2013, 451, 69–79 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Keith D.
Green
b,
Nishad
Thamban Chandrika
b,
Allan H.
Pang
a,
Keith D.
Green
b,
Nishad
Thamban Chandrika
b,
Allan H.
Pang
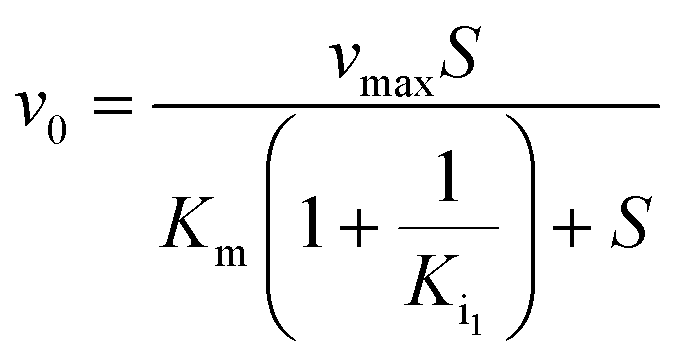
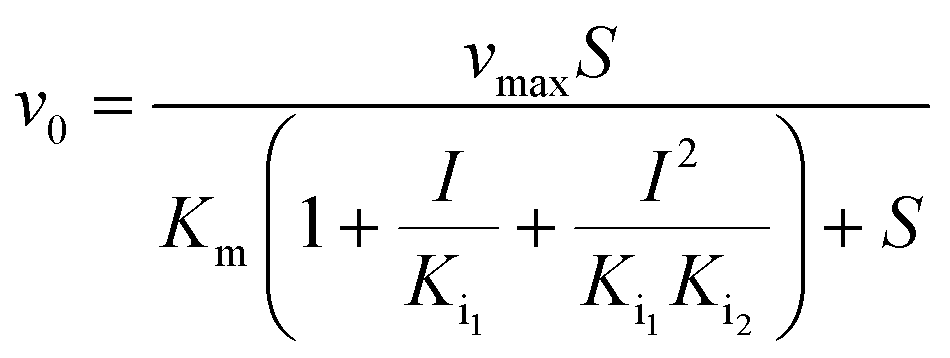
 ZINC1530427
ZINC1530427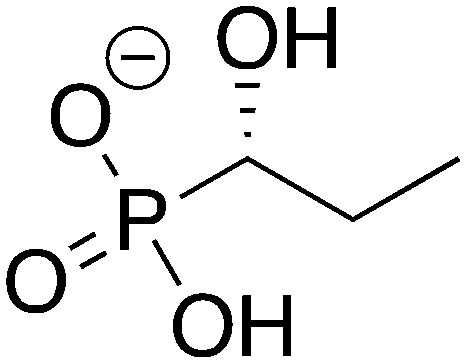 ZINC39255972
ZINC39255972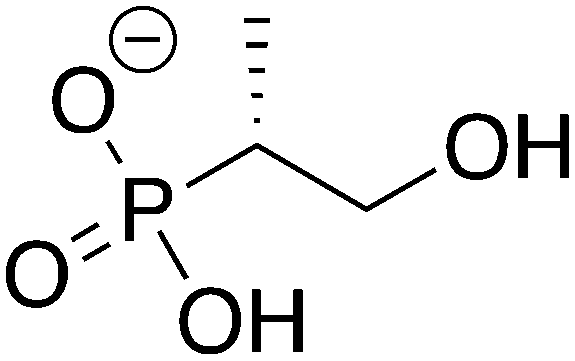 ZINC34127926
ZINC34127926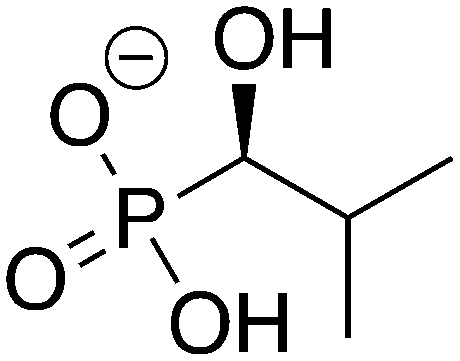 ZINC39275104
ZINC39275104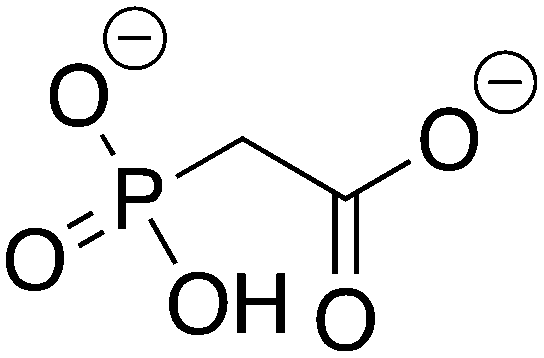 ZINC3869741
ZINC3869741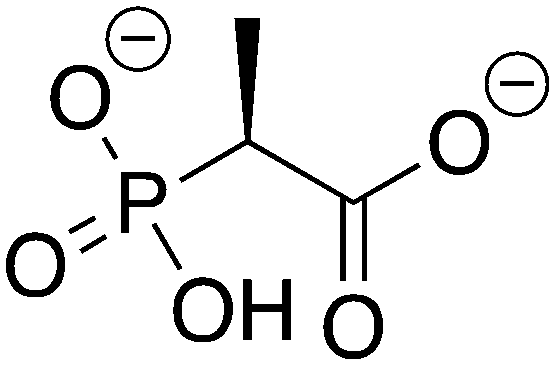 ZINC2008861
ZINC2008861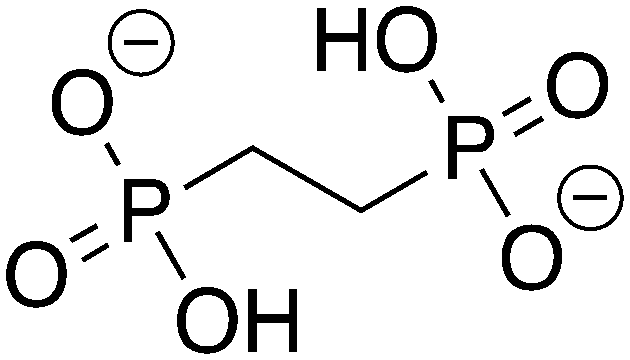 ZINC3953973
ZINC3953973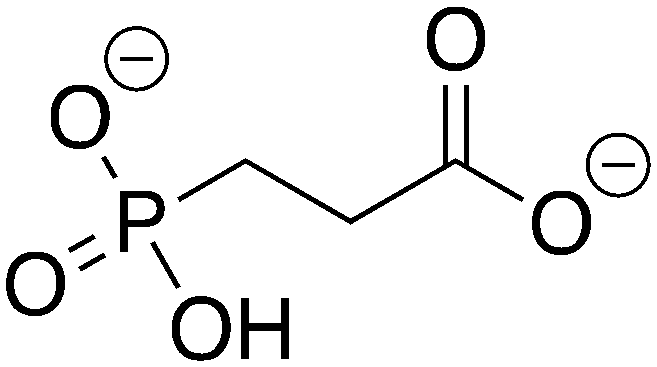 ZINC2008858
ZINC2008858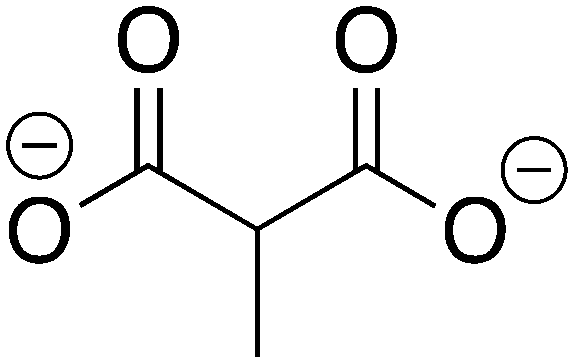 ZINC901289
ZINC901289