
Medicinal chemistry approaches to target the MNK–eIF4E axis in cancer
Ann
Fernandez
a,
Paige J.
Monsen
a,
Leonidas C.
Platanias
bcd and
Gary E.
Schiltz
 *abe
*abe
aDepartment of Chemistry, Northwestern University, Evanston, IL 60208, USA. E-mail: gary-schiltz@northwestern.edu
bRobert H. Lurie Comprehensive Cancer Center, Chicago, IL 60611, USA
cDivision of Hematology-Oncology, Department of Medicine, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA
dDepartment of Medicine, Jesse Brown Veterans Affairs Medical Center, Chicago, IL 60612, USA
eDepartment of Pharmacology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA
First published on 9th May 2023
Abstract
Aberrant translation of proteins that promote cell proliferation is an essential factor that defines oncogenic processes and cancer. The process for ribosomal translation of proteins from mRNA requires an essential initiation step which is controlled by the protein eIF4E, which binds the RNA 5′-cap and forms the eIF4F complex that subsequently translates protein. Typically, eIF4E is activated by phosphorylation on Ser209 by MNK1 and MNK2 kinases. Substantial work has shown that eIF4E and MNK1/2 are dysregulated in many cancers and this axis has therefore become an active area of interest for developing new cancer therapeutics. This review summarizes and discusses recent work to develop small molecules that target different steps in the MNK–eIF4E axis as potential cancer therapeutics. The aim of this review is to cover the breadth of different molecular approaches being taken and the medicinal chemistry basis for their optimization and testing as new cancer therapeutics.
1. Introduction
An aggressive and constantly evolving tumor microenvironment (TME) plays a key role in tumor progression and cancer cell survival.1,2 It is established that integrated intracellular and extracellular signaling cues allows for rapid shifts in cancer cell phenotypes.3 Within the TME, processes such as proliferation rates, cellular growth, and gene expression are tightly regulated via mRNA translation into various oncogenic proteins.4,5 Control of mRNA translation has also evolved into a convergent mechanism crucial at various stages of cancer development.6–9 In multiple cancer types, overexpression of the eukaryotic translation initiation factor 4E (eIF4E) enables the translation of oncogenes, with increasing levels of eIF4E correlating with increasing severity of the disease.10–13 Phosphorylation of eIF4E by mitogen-activated protein kinase (MAPK)-interacting kinases (MNKs) is an essential modification required for activating eIF4E-dependant cancer cell transformation.14–17 This aberrant phosphorylation to synthesize or activate oncogenic proteins is a common cause of many cancers, ultimately leading to tumorigenesis and metastasis.18–20 Additionally, the translation of mRNAs regulated by the eIF4E complex also contributes to disease resistance.11,21 While eIF4E phosphorylation has been shown to be critical for downstream signaling and mRNA translation in many circumstances, there have been some reports that this phosphorylation is dispensable for protein synthesis in some situations.22 Over the past decade, a number of small molecule approaches targeting the MNK–eIF4E signaling complex have been developed to address translational dysregulation in an effort to restore sensitivity to cancer therapies. Herein, we seek to review the structure and function of MNK and eIF4E and provide an update on the recent progress of small molecule medicinal chemistry approaches targeting the MNK–eIF4E axis in cancer.MNK1 and MNK2 are ubiquitously expressed enzymes involved in transcription regulation due to their ability to phosphorylate eIF4E at Serine 209.15,17,23 MNK1/2 are characterized as serine/threonine kinases from the group of Ca2+/calmodulin-dependent kinases (CaMKs), whereby the MNK kinase domain shares about 80% of sequence identity with other kinases.24 There are two MNK genes in humans, both of which encode two transcript splice variants, resulting in the expression of MNK1a, MNK1b, MNK2a, and MNK2b.25–28 The a-isoforms are primarily cytoplasmic, while the b-isoforms are localized within the nucleus.26,29 MNKs can be activated via extracellular signal-regulated kinase (ERK) or p38 MAPKs.23,30 The p38 pathway is activated by upstream stress inducers, thereby converting cell stress to transcriptional regulation.3,31–41 The ERK pathway is primarily mediated by pro-growth stimuli.42,43 The presence of a MAPK binding domain in the C terminus is present in the longer alternate MNK splice forms, MNK1a and MNK2a.26 MNK isoforms also display varying basal activities and regulatory capabilities. For example, MNK1a possesses low basal activity that is stimulated by upstream ERK,23,26 while MNK1b has higher basal activity and remains unresponsive to external stimuli. MNK2a demonstrates high basal activity and is activated solely by ERK, while MNK2b displays basal activity independent of ERK and p38 MAPK activity. Furthermore MNK2a has been reported to remain bound to ERK after ERK activation, which partly explains its high basal activity. While most studies propose that MNKs are modulated by the upstream ERK and p38 proteins, several have suggested that MNK is activated independently of MAPKs by other known and unknown kinases.25,43 In the context of eIF4E activation, it was recently shown that eIF4E phosphorylation (p-eIF4E) was mediated by MNK2 and not MNK1.43 Specifically, it was observed that phosphorylation occurs at residues distinct from those phosphorylated by MAPKs, whereby Ser437 is phosphorylated via an unidentified mechanism.
Both MNK1 and MNK2 contain several structural features that make them unique among other kianses.23,30 They are known to possess a unique DFD (Asp–Phe–Asp) motif, as opposed to the structurally conserved DFG (Asp–Phe–Gly) motif found in other kinases (Fig. 1).44 The MNK catalytic domain is also altered by the presence of two short inserts providing further structural distinction from other protein kinases. As a result of this altered DFD motif, adenosine triphosphate (ATP) access to the binding domain is restricted. There are several resolved crystal structures of MNK1 and MNK2.44–48 The magnesium-binding loop (residues 191–225 in MNK1 and 226–260 in MNK2) and activation segments are involved in substrate recognition and phosphatase activity.44
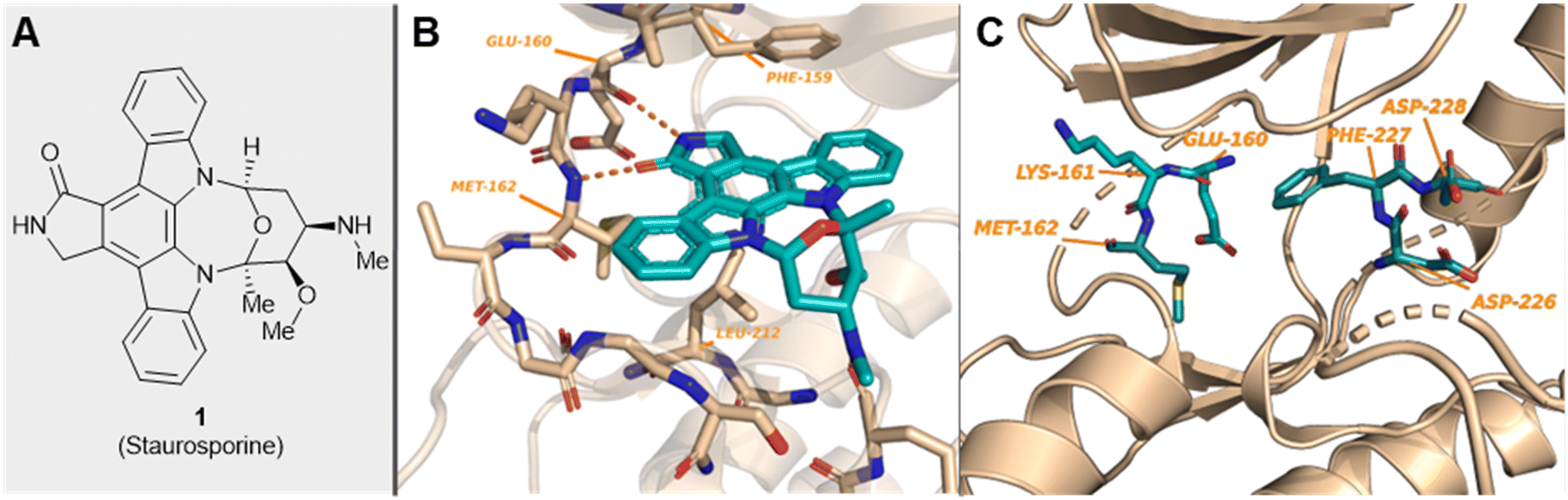 | ||
| Fig. 1 (A) Structure of staurosporine, 1; (B) MNK2 protein showing the hinge-binding residues interacting with ligand 1 (PDB 2HW7); and (C) location of the unique MNK DFD sequence and hinge-binding residues in MNK2 (PDB 1AC3). | ||
eIF4E is a potent oncoprotein that is well characterized as an in vivo MNK substrate that undergoes MNK-mediated phosphorylation at Ser209.15,24 eIF4E is a cap-binding protein that facilitates mRNA translation in eukaryotic cells and binds to both the 5′-cap structure eIF4A, an ATP-dependent RNA helicase, and eIF4G, a modular scaffolding protein.49,50 The three substrates, eIF4E, eIF4A, and eIF4G form the eIF4F complex to allow for ribosome recruitment and initiation of cap-dependent translation, which is a predominant form of eukaryotic translation (Fig. 2). Activation of eIF4E and its complexation with eIF4G is regarded as the rate-limiting factor in translation initiation. Overall, the rates of translation are higher in tumors, and it is now understood that certain mRNA subsets are regulated at the translational level.8,12,51 As the central step in translation initiation, eIF4F is tightly regulated under basal conditions and is regulated via a variety of extracellular stimuli.15,52 Currently, eIF4E activation by mechanistic target of rapamycin complex 1 (mTORC1) and MNK is of significant interest, due to its role in cancer development and progression.53,54 Activity of eIF4E is further regulated by mTORC1 phosphorylation of the eukaryotic initiation factor-4E binding proteins (4E-BPs). Binding of phosphorylated 4E-BPs to eIF4E inhibits interaction with eIF4G (Fig. 3).50,55 However, 4E-BP hyperphosphorylation by mTORC1 leads to its dissociation from eIF4E, and enables its interaction with eIF4G to form the eIF4F complex. Alternatively, MNK can directly phosphorylate eIF4E at a single site, Ser209, which enables eIF4F complex formation.56,57 The formation of this eIF4F complex is known to contribute to aberrant signaling, apoptotic resistance, angiogenesis, metastasis, and programmed cell death resistance.58–67
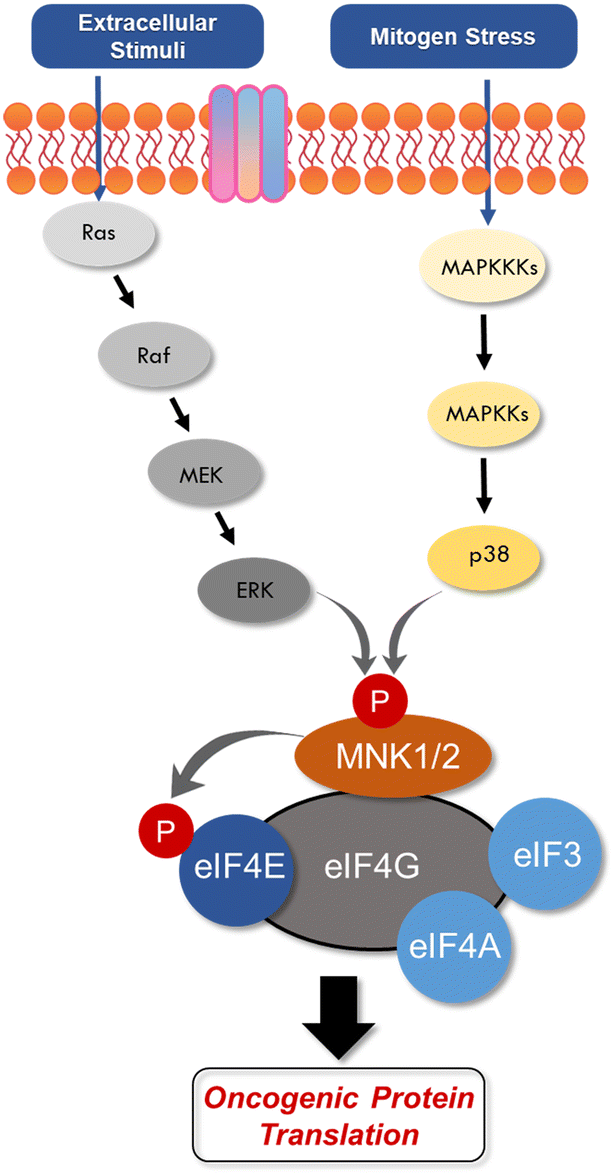 | ||
| Fig. 2 Activation of translation initiation MNK by the ERK p38 pathways via eIF4G. | ||
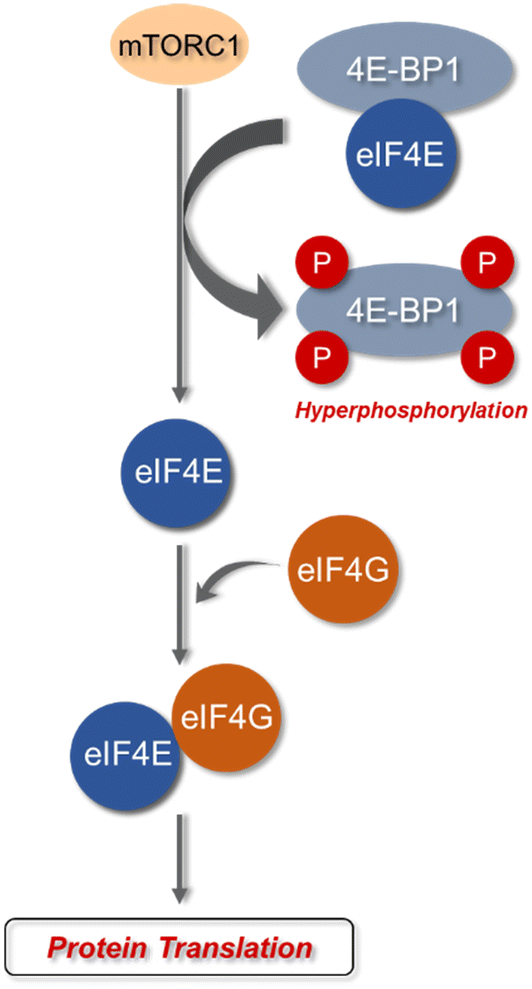 | ||
| Fig. 3 mTORC-mediated phosphorylation pathway that controls modulation of 4E-BPs and protein translation. Phosphorylation of 4E-BP1 by mTORC results in the release of eIF4E from the inactive 4E-BP1–eIF4E complex to be able to from the eIF4E–eIF4G active complex. This active complex can then form additional interactions to lead to translation. | ||
eIF4E overexpression and aberrant mRNA translation is seen in many tumor types such as those occurring in the prostate, brain, lung, bladder, colon, thyroid, cervix, ovary, head, neck, and hematological malignancies such as leukemia.68–74 High expression levels of eIF4E are associated with an increase in tumorigenesis, abnormal proliferation, and metastasis through the increased translation of certain mRNAs that sustains cell growth and angiogenesis. Although high expression of eIF4E in multiple cancers has been reported since the 1990s it was only until 2004, when Topisirovic and coworkers investigated the role phosphorylation of eIF4E plays in its ability to transform cells and assist in mRNA translation.75 While eIF4E is present in both the nucleus and the cytoplasm,75 it appears to plays different roles in each compartment. In the cytoplasm, eIF4E functions as the rate-limiting step in cap-dependent protein translation. However, in the nucleus, eIF4E facilitates increased nuclear export of tumorigenic mRNAs, including cyclin D1,76 at least in part through its interactions with promyelocytic leukemia protein77 and proline-rich homeodomain protein.78 Through the use of mutagenesis and eIF4E phosphorylation inhibition studies, Borden and colleagues were able to demonstrate that phosphorylation of eIF4E was required to enhance mRNA transport function and is critical to its oncogenic transformation capability.75 The complete mechanisms by which eIF4E mediates cancer formation remains incompletely understood, while there are complex coordination pathways through which growth factors and binding partners promote oncogenic activity through eIF4E activation.62,75,79
eIF4E overexpression in mice has shown to increase cancer susceptibility and promote the development of lymphomas, hepatomas, and lung carcinomas.80 In glioblastoma (GBM), for example, pharmacological inhibition of MNK1 and MNK2, and MNK1 knockdown, lead to the sensitization of GBM cells to existing treatment methods.81 MNK1 is expressed in both primary GBM and glioma cell lines and the phosphorylation of MNK1 and eIF4E are associated with increased tumor size and recurrence of the disease. It is widely believed that cancer formation occurs due to enhanced translation of a subset of mRNAs, leading to an overexpression in cyclin-dependent kinases (CDKs), c-MYC, myeloid cell leukemia 1 (Mcl-1), B-cell lymphoma 2 (Bcl-2), survivin, vascular endothelial growth factor (VEGF), fibroblast growth factor 2 (FGF2), matrix metalloproteinase 9 (MMP9) and heparanase, resulting in cell proliferation, apoptosis, evasion, angiogenesis and metastasis.82,83 It is worth noting that eIF4E is only dysregulated in certain subsets of each cancer type and is usually associated with high levels of MNK1 and MNK2 and the phosphorylated form of eIF4E.84
In addition to eIF4E, there are several downstream targets of MNK involved in cancer cell survival, proliferation, and differentiation including the polypyrimidine tract-binding protein-associated splicing factor (PSF), heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1), cytosolic phospholipase A2 (cPLA2), and Sprouty2 (Spry2) (Fig. 4).29,85–87 PSF is characterized as an RNA-binding protein that plays a role in multiple aspects of RNA metabolism such as pre-mRNA splicing, mRNA stability, and translation.85 Abnormal splicing and dysregulated translation are common contributors to tumor development and growth in cancer. In 2008, Buxadé and coworkers demonstrated PSF is phosphorylated at two sites (Ser8 and Ser283) by MNK kinases. Interestingly, PSF phosphorylation at Ser8 was primarily achieved by MNK2, providing evidence of substrate specificity of MNK1 and MNK2. MNK-mediated phosphorylation lead to an increase in binding of PSF to the proinflammatory cytokine tumor necrosis factor α (TNF-α) mRNA in T-cells.85 In addition to MNK-promoted phosphorylation of PSF, Buxadé and coworkers reported that MNK phosphorylated Ser192 and Ser310 on hnRNPA1, a nuclear protein involved in mRNA translation and inflammatory response.29 Phosphorylation of hnRNPA1 leads to a decrease in binding of hnRNPA1 to TNF-α AU-rich elements (AREs), causing T-cell activation.15 The activation of the MAPK cascade results in MNK-mediated phosphorylation of target substrate hnRNPA1 and subsequent dissociation from the TNF-α ARE to ultimately activate translation of the TNF-α mRNA.15,29 Increased levels of TNF-α is implicated in the promotion of tumor growth, invasion and metastasis. Therefore, the mechanisms regulating TNF-α expression, such as the MNK pathway, have important therapeutic relevance. Activated by the increase of cytosolic calcium, cPLA2 is an enzyme that promotes the release of arachidonate acid from glycerophospholipids to obtain the precursor of eicosanoids.87,88 Eicosanoids are a group of signaling molecules that play important roles in several physiological and pathological processes including inflammation, immunity, and CNS regulation.87,88 cPLA2 has been linked to the MNK signaling pathway, specifically the phosphorylation at Ser727 by MNK1. This process results in the enhancement of cPLA2 enzymatic activity, leading to the increased release of arachidonic acid and subsequent production of eicosanoids.87,88 Therefore, inhibiting MNK1 and the phosphorylation of cPLA2 is an attractive therapeutic target. Another target of MNK kinase is Spry2, a protein that acts as a negative regulator of several receptor tyrosine kinase pathways, specifically the ERK MAPK pathway. Yusoff and coworkers demonstrated Spry2 phosphorylation by MNK on Ser112 and Ser121 leads to increased stability of Spry2 and enhances its ability to inhibit the ERK pathway.89 Overall, the MNK kinases play an important role in regulating a variety of biological processes through phosphorylating downstream substrates which can lead to cell growth, proliferation and metastasis, suggesting that inhibition of the MNK pathway is a promising strategy for cancer therapy. At the same time, the transcription of oncogenic mRNAs and their translation into tumorigenic proteins is a highly complex process with eIF4E being only one important pathway. It is conceivable that inhibition of MNK and eIF4E phosphorylation will be insufficient to completely block the translation of oncoproteins. Further work to define the specific roles of MNK/eIF4E and other translation pathways involved in cancer will be needed to fully elucidate the best mechanism by which to target translation in cancer.
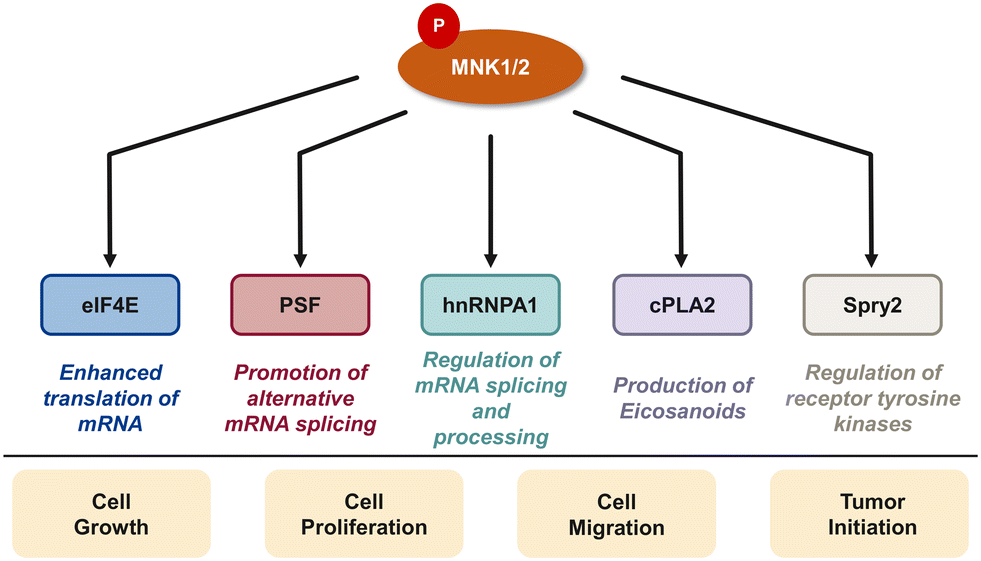 | ||
| Fig. 4 MNK-phosphorylated substrates critical for the development of cancers. | ||
The previous sections highlight the catalytic function of MNK. In addition to this, certain non-catalytic functions have also been reported for MNK1/2. In one study on metabolic disease by Merrett and coworkers, MNKs were implicated in mediating diet-induced obesity by regulating adipose tissue expansion.90,91 To study the effect of MNK, a small molecule MNK inhibitor was introduced to prevent adipogenesis; however, it failed to impair lipid accumulation in animal models. On the other hand, knockdown of MNK2 led to reduced lipid accumulation and down-regulation of several lipid regulatory genes. These consequences suggest that MNK can engage with other proteins non-catalytically and the observed results can be attributed to the loss of MNK entirely and not just its catalytic activity. While the mechanisms of MNK function beyond its catalytic activity remain unclear, it is likely that MNK may possess non-catalytic activity in the context of eIF4E-mediated translation initiation.
Although MNK activity determines diverse cellular functions, dysregulation of the MNK–eIF4E axis has direct implications on cancer biology. Specifically, the development of drug resistance to cancer therapeutics can occur through initiation of cap independent translation and mediation of pro-inflammatory cytokine production and cytokine signaling. Furthermore, mouse models confirm a correlation between eIF4E phosphorylation by MNK and tumorigenesis.56,92–96 However, Ser209 phosphorylation of eIF4E is non-essential in normal cells but is required in malignant cells.62 In 2009 Assouline et al. demonstrated for the first time the therapeutic benefit of targeting eIF4E, specifically for patients diagnosed with M4/M5 acute myeloid leukemias (AML) with elevated levels of eIF4E and poor prognosis.97 In this study, Ribavirin, a broad-spectrum antiviral that blocks eIF4E activity through mimicking the mRNA 7-methyl guanosine (m7G) cap structure, was administered to 11 patients in a phase II clinical trial (NCT00559091). Overall, inhibition of eIF4E with ribavirin resulted in a clinical benefit for patients diagnosed with AML and correlated with clinical remissions, providing the first proof-of-concept that eIF4E is a promising target for cancer therapeutics. Furthermore, in vivo studies have reported the lack of apparent phenotype changes observed in mice after MNK knockdown. Therefore, MNK is non-essential to growth and development and as a result, targeting of MNK could be an ideal therapeutic approach to selectively target cancer cells. Additional studies targeting eIF4E and the MNK–eIF4E axis is of great relevance for the development of potential anti-cancer drug candidates.
2. Inhibitors of MNK kinase
Since the report of Staurosporine (1) and its analogs by Tschopp and coworkers in 2000, the identification of selective and potent MNK inhibitors as potential cancer therapeutics has seen growing interest.98,99 In 2018, eFFECTOR Therapeutics published a report describing their development of eFT508 (2, Fig. 5), a potent and selective inhibitor of both MNK1 and MNK2 (MNK1/2 inhibitor) that is currently in phase II clinical trials for the treatment of several types of cancer.48 Through a highly structure-guided design approach, Reich and coworkers exploited the unique active site of MNK1/2 to identify favorable stereoelectronic interactions with residues distinctive to MNK1/2.48 The MNK1/2-specific active site led to a resulting sensitivity to distance and orientation of the ligand–protein interactions and were exploited to improve the inhibitor kinase selectivity.100 Therefore, utilizing cocrystal structures of MNK2 with various ligands, the identification of a hit chemotype with high ligand efficiency to be utilized as the starting scaffold for further derivatizations was the initial focus.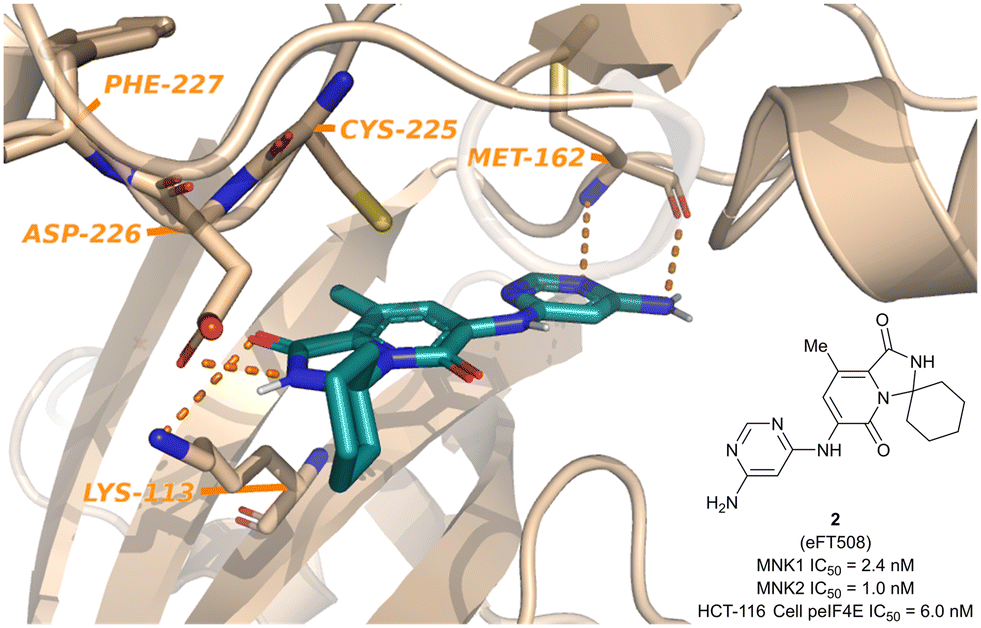 | ||
| Fig. 5 Structure of MNK1/2 inhibitor eFT508 (2) and the cocrystal structure bound to MNK active site. | ||
The ATP binding site of MNK1/2 possesses several distinct residues with two, Phe159 and Cys225, capable of providing unique ligand interactions.46 The presence of this atypical residue pattern is noted in only cyclin-dependent kinase-like (CDKL)1–5, fms-like tyrosine kinase 3 (FLT3), FLT4, pre-mRNA processing factor 4B (PRP4), MAPK7, and MNK across the kinome. Therefore, targeting an interaction with these residues was hypothesized to lead to MNK-selective inhibitors. Using a structure-guided approach to focus on creating distinctive interactions with the MNK1/2 active site-specific residues Phe159 and Cys225, Reich and coworkers discovered several chemotypes (3–8) displaying good potency for MNK as measured by their MNK1 IC50 (Fig. 6).48 Cocrystal structures obtained of compounds 3 and 4 bound to the MNK2 active site demonstrated potential interactions with both key residues, Phe159 and Cys225, suggesting further optimization of both 3 and 4 would be of interest. A lack of interaction with the key residues and the kinase-hinge region was noted in the cocrystal structures of 5 and 6 with MNK2. Although cocrystal structures could not be obtained for compounds 7 and 8, they were found to inhibit MNK1 with IC50 values of 4.7 μM and 5.5 μM, respectively, and were considered for further optimization. Utilizing compounds 3–8 as fragment-like scaffolds, a small, diverse library of analogs were designed and analyzed for their potency for MNK1/2. Results from this initial screen concluded derivatives of the carboxamide scaffold 3 possessed the highest ligand efficiencies and low nanomolar binding to MNK. Therefore, hit analog 3 underwent further optimization and structure–activity relationship (SAR) analysis to improve both potency and physicochemical properties (Fig. 7).
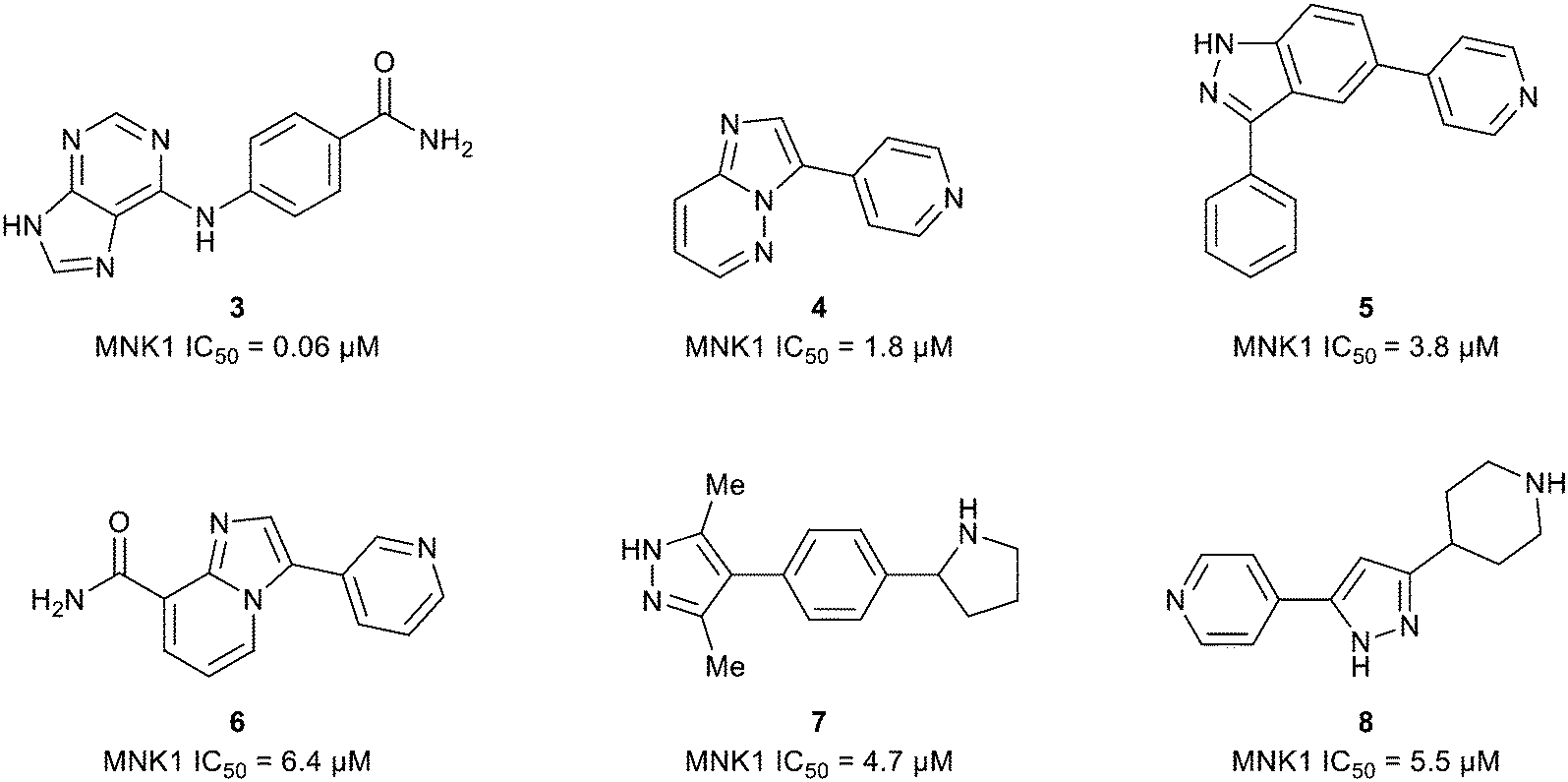 | ||
| Fig. 6 Representative fragments identified in initial structure-guided design of MNK inhibitors. | ||
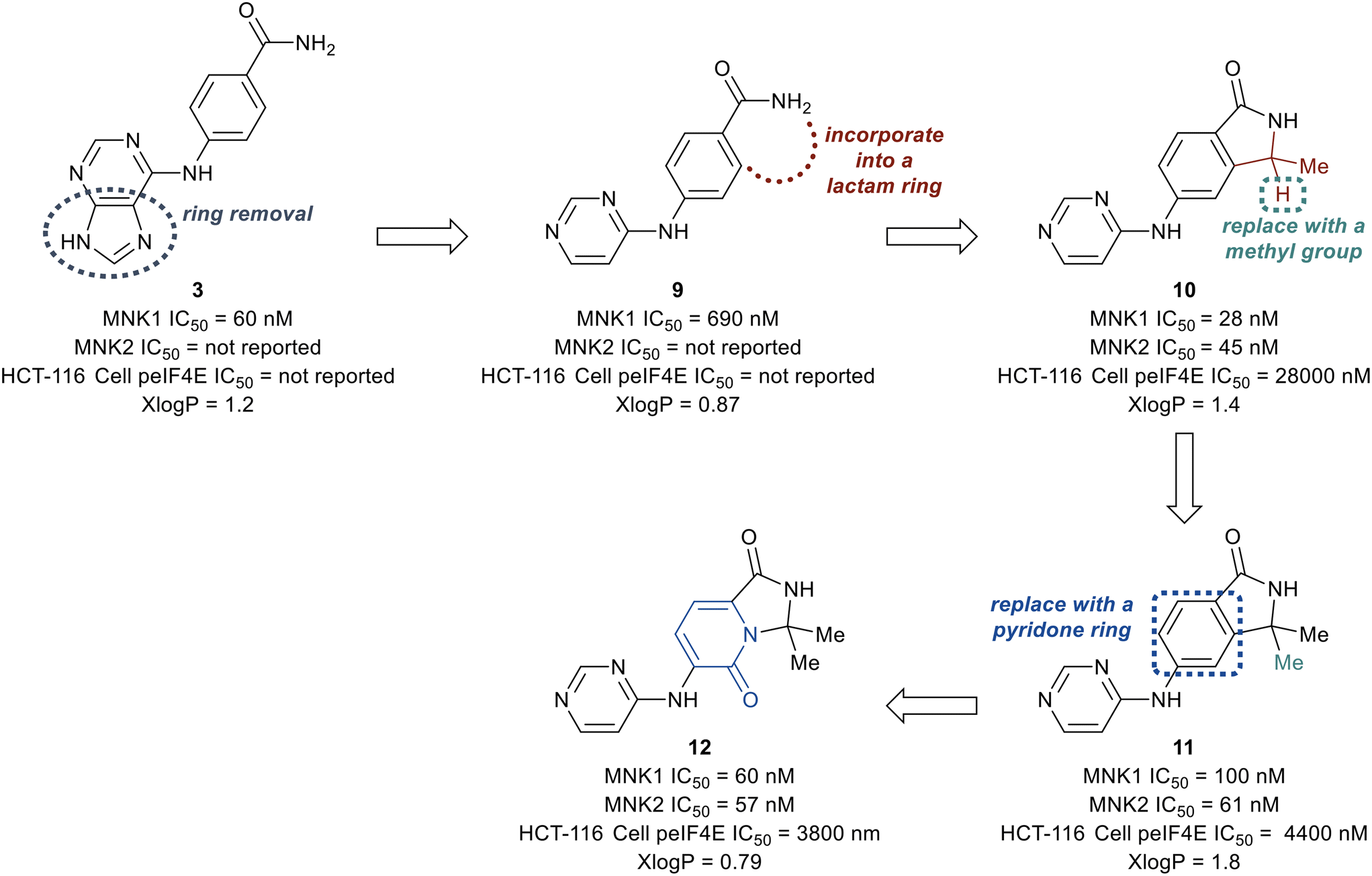 | ||
| Fig. 7 Initial optimization of hit fragment 3. | ||
The deconstruction of the purine ring in 3 provided compound 9 possessing a 10-fold loss in potency (MNK1 IC50 = 690 nM), though a reduction in molecular weight and x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP (calculated logarithm of octanol–water patrician coefficient) compared to 3 was achieved. Conversion of the amide in 9 into a substituted lactam to remove a hydrogen bond donor was then investigated. The newly formed lactam 10 was found to be 25-times more potent (MNK1 IC50 = 25 nM) compared to the uncyclized amide 9 (MNK1 IC50 = 690 nM). The addition of a second methyl group to provide the gem-dimethyl analog 11, resulted in a loss of biochemical potency for MNK1 (MNK1 IC50 = 100 nM) compared to 10, but possessed an increase in cellular potency when analyzed for inhibition of eIF4E phosphorylation in solid tumor cell line HCT-116 (HCT-116 cell p-eIF4E IC50 = 4.4 μM for 11vs. HCT-116 cell p-eIF4E IC50 = 28 μM for 10). Replacement of the benzene ring with a pyridone ring system to provide an interaction with Cys225 to yield analog 12 was found to be equipotent (MNK1 IC50 = 60 nM, HCT-116 cell p-eIF4E IC50 = 3800 nM) and less lipophilic than 11 (xlogP = 0.79 for 12vs. xlogP = 1.8 for 11). Furthermore, the pyridone ring system was found to provide selectivity for MNK1/2 over the benzene ring when comparing the kinome profiles (>400 kinases) of both 11 and 12. Analysis of the kinase active site showed the presence of an empty pocket near the key residue Phe159 which was hypothesized to be ideal for the accommodation of a heavy atom substituted at the 5-position of the pyridone ring of 12. Therefore, three substituted analogs of 12 were synthesized to possess a fluorine (13), chlorine (14), or methyl (15) substituent at the 5-position (Fig. 8). The newly substituted derivatives all exhibited increased potency, though the 5-chlorine derivative (14) was superior, displaying a greater than 50-fold increase in potency (MNK1 IC50 = 3.3 nM, HCT-116 cell p-eIF4E IC50 = 73 nM) compared to the unsubstituted pyridone 12.
logP (calculated logarithm of octanol–water patrician coefficient) compared to 3 was achieved. Conversion of the amide in 9 into a substituted lactam to remove a hydrogen bond donor was then investigated. The newly formed lactam 10 was found to be 25-times more potent (MNK1 IC50 = 25 nM) compared to the uncyclized amide 9 (MNK1 IC50 = 690 nM). The addition of a second methyl group to provide the gem-dimethyl analog 11, resulted in a loss of biochemical potency for MNK1 (MNK1 IC50 = 100 nM) compared to 10, but possessed an increase in cellular potency when analyzed for inhibition of eIF4E phosphorylation in solid tumor cell line HCT-116 (HCT-116 cell p-eIF4E IC50 = 4.4 μM for 11vs. HCT-116 cell p-eIF4E IC50 = 28 μM for 10). Replacement of the benzene ring with a pyridone ring system to provide an interaction with Cys225 to yield analog 12 was found to be equipotent (MNK1 IC50 = 60 nM, HCT-116 cell p-eIF4E IC50 = 3800 nM) and less lipophilic than 11 (xlogP = 0.79 for 12vs. xlogP = 1.8 for 11). Furthermore, the pyridone ring system was found to provide selectivity for MNK1/2 over the benzene ring when comparing the kinome profiles (>400 kinases) of both 11 and 12. Analysis of the kinase active site showed the presence of an empty pocket near the key residue Phe159 which was hypothesized to be ideal for the accommodation of a heavy atom substituted at the 5-position of the pyridone ring of 12. Therefore, three substituted analogs of 12 were synthesized to possess a fluorine (13), chlorine (14), or methyl (15) substituent at the 5-position (Fig. 8). The newly substituted derivatives all exhibited increased potency, though the 5-chlorine derivative (14) was superior, displaying a greater than 50-fold increase in potency (MNK1 IC50 = 3.3 nM, HCT-116 cell p-eIF4E IC50 = 73 nM) compared to the unsubstituted pyridone 12.
 | ||
| Fig. 8 Final optimization of MNK1/2 inhibitors to obtain 2. | ||
Additional optimization of the MNK1/2 inhibitors focused on improving cellular potency, maintaining good physicochemical properties, and obtaining a desired pharmacokinetic/pharmacodynamic (PK/PD) profile. The incorporation of the gem-dimethyl group into a spirocyclic scaffold, specifically with a six-membered ring, provided greater kinome selectivity. Unfortunately, the presence of spirocarbocycles resulted in undesired metabolism, however, this effect could be mitigated via the addition of a free amino group at the 4-position of the pyrimidine. Taking into account the above findings, the final optimizations were performed in parallel with multispecies PK (mouse, rat, dog, and monkey) analysis and ultimately allowed for the identification of compound 2, in which the 5-chlorine atom was ultimately replaced with a methyl group. Compound 2 was reported to have the most ideal overall profile and was therefore utilized as the lead candidate for additional studies.
MNK inhibitor 2 (eFT508) was tested in TMD8 cells to analyze the impact of MNK1/2 inhibition. A dose-dependent reduction on pro-inflammatory cytokines [interleukin-6 (IL-6), interleukin-8 (IL-8), and TNF-α] was observed and correlated with a decrease in mRNA stability, a key phenomenon consistent with literature.29,85 Because 2 was characterized as a potent inhibitor of eIF4e phosphorylation, select mRNA stability and cytokine production, the efficacy of 2 in a TMD8 xenograft tumor model was explored. Treatment with 2 resulted in a significant tumor growth inhibition (TGI) when dosed orally at 1 and 10 mg kg−1 once daily (QD), leading to an average TGI of 71% and 75%, respectively. Furthermore, 2 was well tolerated at doses of 1 and 10 mg kg−1 QD, as measured by lack of body weight loss. Assessment of MNK inhibition in TMD8 xenografts at three dose levels (0.3, 1, and 10 mg kg−1) through PK/PD measurements of p-eIF4E (Ser209) showed an oral dose of 2 at 1 mg kg−1 QD produced maximal efficacy and exhibited a reduction of p-eIF4E of 80% for 8 hours. Lastly, because eIF4E phosphorylation has been reported in breast cancer tumors and is correlated to a decrease in survival outcome, 2 was tested in the MDA-MB-231 breast cancer xenograft model.101 Excitingly, dosing animals with 1 or 10 mg kg−1 of 2 QD resulted in significant in vivo TGI (TGI >100%). Overall, the work reported by Reich and coworkers demonstrates the application of structure-based design as a strategy to yield novel, selective, and potent MNK1/2 inhibitors.48 The identification and characterization of 2 (eFT508) with in vivo activity in multiple tumor models provides proof of concept that inhibition of MNK1 and MNK2 has the potential in the treatment of cancer.
In 2018, Yang et al. developed MNK inhibitors for blast crisis chronic myeloid leukemia (BC-CML) patients who did not respond to FDA-approved break point cluster Ableson 1 (BCR-ABL1) kinase inhibitors.102 The MNK–eIF4E axis is believed to promote treatment resistance, self-renewal, and proliferation of leukemic stem cells.103,104 Since the inhibition of eIF4E phosphorylation by either MNK kinase inhibitors or MNK knockdown is known to suppress leukemic stem cells (LSC) activity, studies were carried out to develop selective MNK inhibitors from MNK-binding fragments as published by Oyarzabal and coworkers.105 From these studies, an imidazopyrazine scaffold (16) was established as the key chemotype and docking studies were performed to further understand its binding to MNK. Molecular modeling revealed several interaction sites that could be modified to improve the MNK inhibitor and SAR studies were undertaken.
Initial studies were performed on analogs in which the substituents at the C6 position of 16 were varied (Fig. 9). The acetamide 17 and the phenyl-substituted compound 18 had similar potencies for both MNK1 and MNK2 (0.79 vs. 0.53 μM). The addition of an amide substituent at the para position of the added phenyl ring in 18 to yield compound 19 resulted in improved inhibitory activity (0.3 μM). This was reasoned to be due to a favorable interaction between the electron-rich π-cloud of the phenyl ring and the partial positive charge of the amide N–H of Ser166. Interestingly, when the methyl group of amide 17 was replaced by a phenyl ring to give compound 20, a significant decrease in potency from 0.79 μM to 3.4 μM was observed. This reduction in potency was rationalized to be due to an unfavorable interaction between Leu212 and the phenyl ring in 20. The primary amide in 19 was converted into tertiary heterocyclic amides to produce the N-methyl and N-acetyl piperazines (21 and 22), and the morpholine (23). These structural changes only slightly improved potency relative to the primary amide 17 (∼3–6-fold) but displayed improved permeability and solubility. Overall, the methyl piperazine analog (22) and the morpholine benzamide (23) were identified as the best overall C6 substituents.
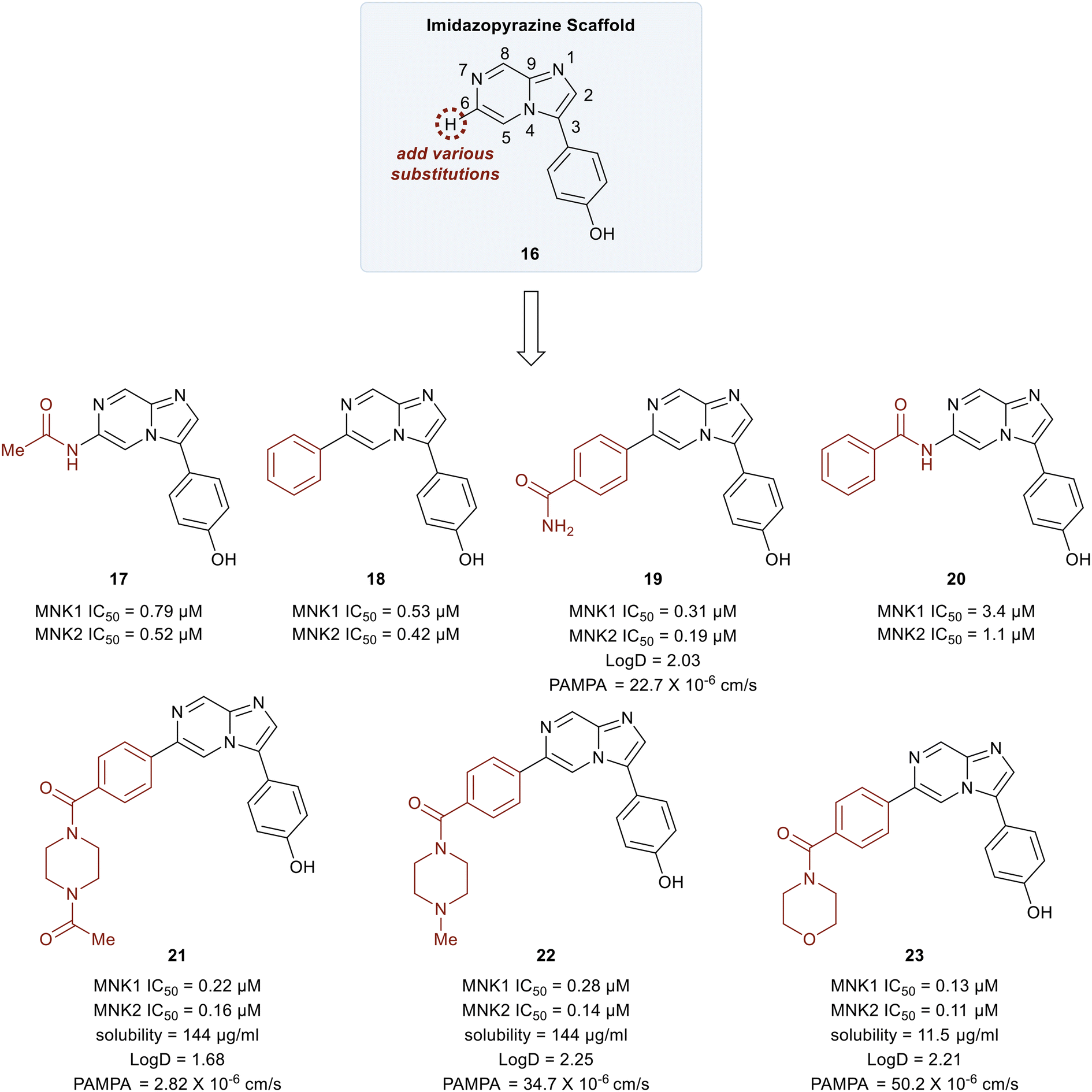 | ||
| Fig. 9 Initial SAR studies investigating substitutions at the imidazopyrazine 6-position. | ||
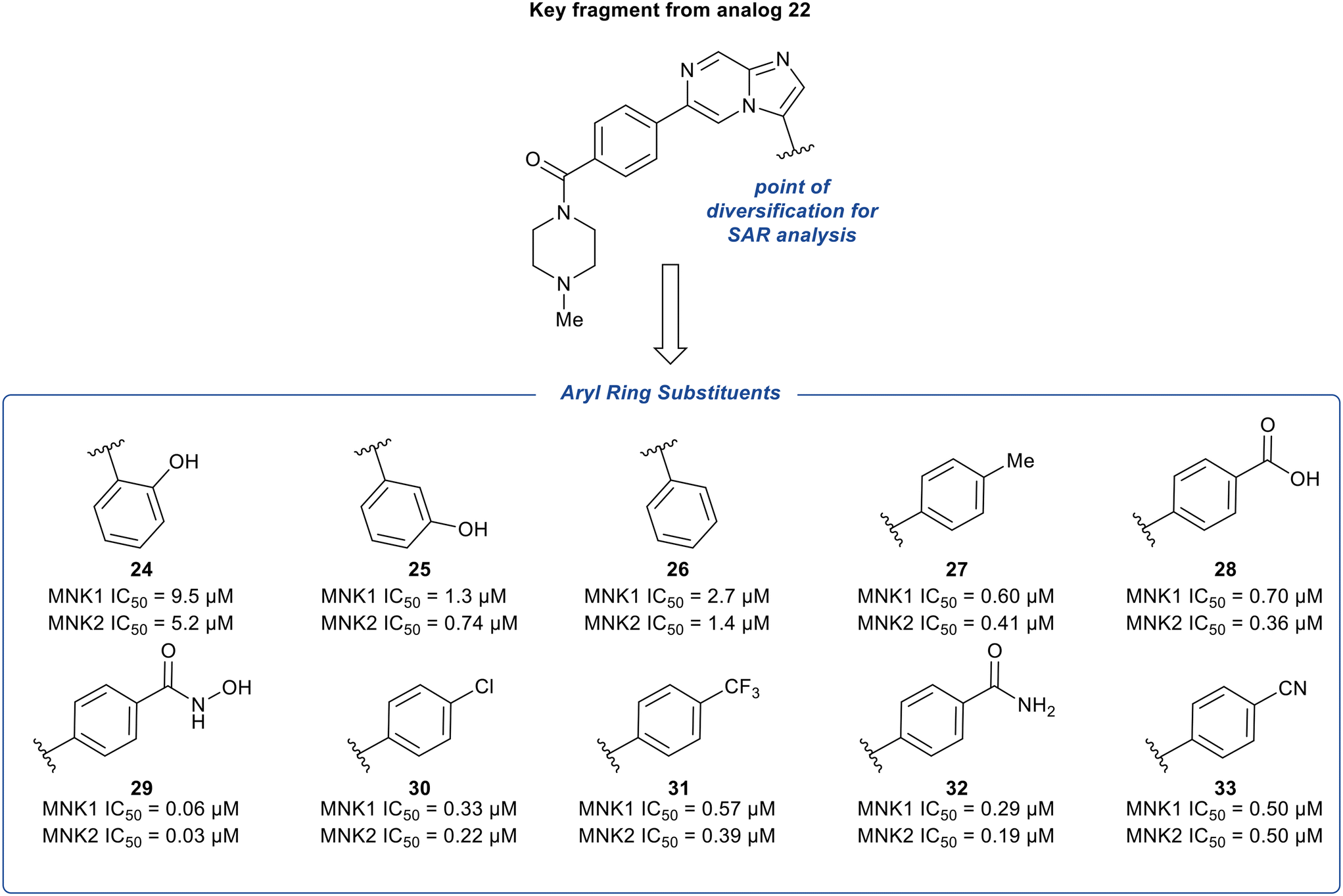
The phenyl substituent at the 3-position of the imidazopyridazine was hypothesized to be favored due to the hydrophobic nature of the binding site it occupies. To further explore the SAR around substituents on this phenyl ring, additional analogs of 22 were synthesized and tested for inhibition of MNK1 and MNK2 (Fig. 10). The hydroxyl group was moved to the 2- and 3-position of the phenyl ring to provide compounds 24 and 25, respectively. Altering the substitution position of the hydroxyl group resulted in significantly less potent analogs. Specifically, a 34-fold decrease in potency for 24 and a 4-fold decrease in potency for 25 was observed, compared to 22. Both the removal of the hydroxyl group to provide derivative 26 and the replacement of the hydroxyl group with a hydrophobic methyl substituent to provide derivative 27, were found to be less potent than 22, displaying MNK1 IC50 values of 2.7 μM and 0.60 μM, respectively. The addition of acidic groups was also tested and the carboxylic acid analog (28) showed an approximately 2-fold decrease in potency compared to phenol 22. Interestingly, replacing the hydroxyl group in 22 with a hydroxamic acid group yielded a nearly 5-times more potent MNK1 inhibitor 29 (0.06 μM vs. 0.28 μM). To rationalize this, docking studies suggested that the –NH and –OH groups of the hydroxamic acid formed hydrogen-bonds with Asp226 and Lys113, respectively. While other electron-withdrawing substituents were tested (30–32), their MNK inhibitory potencies were found to be similar or worse than compound 22. Replacement of the hydroxyl group in 22 with a cyano group (33) resulted in a slight change in potency (0.50 μM) for MNK1. However, compared to analog 22, MNK inhibitor 33 possessed improved absorption, distribution, metabolism, and excretion (ADME) properties, and therefore due to a favorable balance of potency and PK properties, 33 was selected for further optimization.
 | ||
| Fig. 10 SAR of imidazopyridazine 3-aryl group modifications. | ||
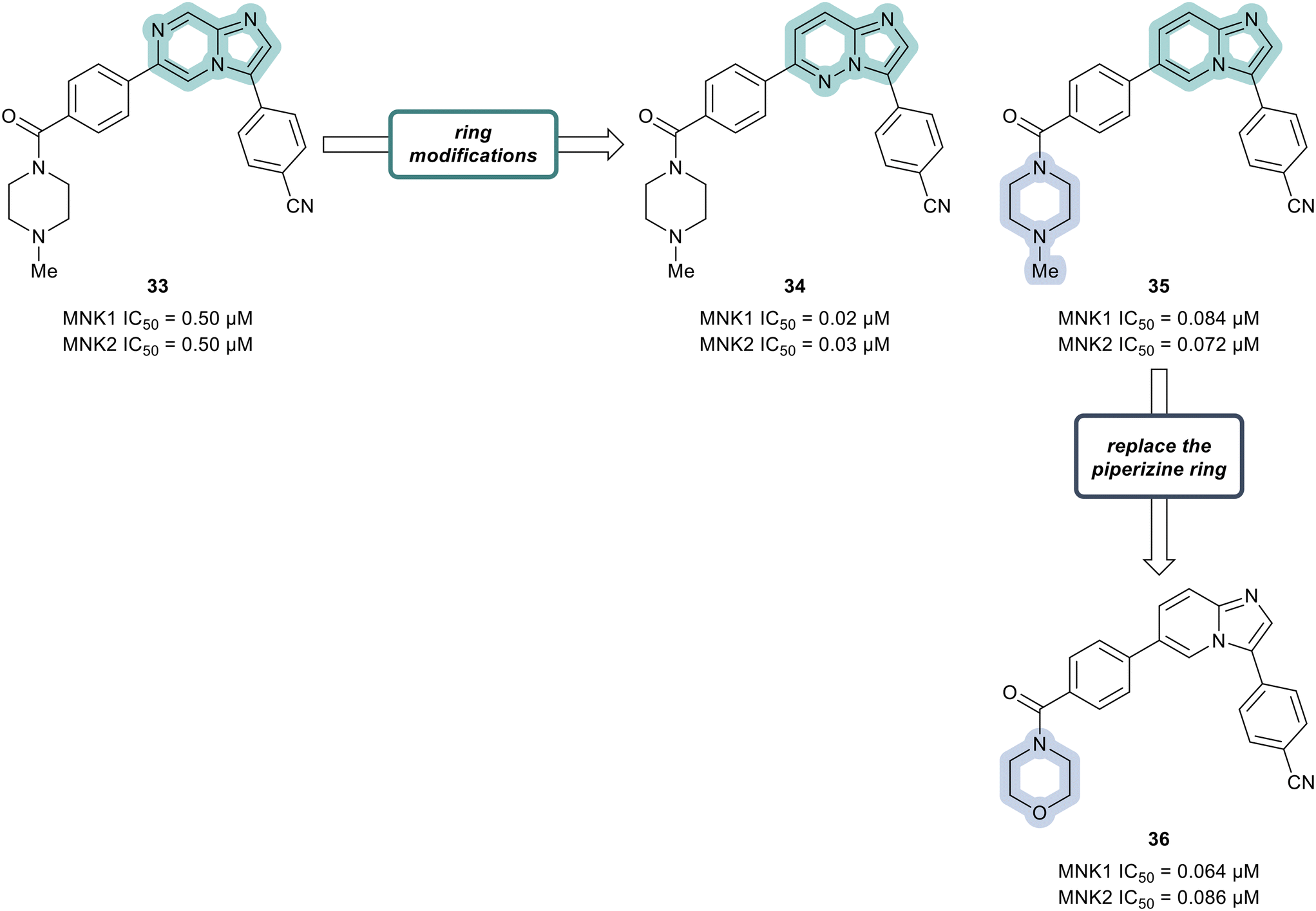
Additional SAR analysis was focused on investigating several fused heterocyclic cores as possible replacements of the imidazopyrazine present in lead compound 33. As the N-1 of the imidazole moiety was expected to form a critical interaction with the hinge group based on docking studies, the imidazole was left intact. Therefore, both the imidazopyridazine 34 and imidazopyridine 35 were prepared (Fig. 11). Relative to compound 33 (0.50 μM), analogs 34 and 35 displayed markedly improved potencies against both MNK1 (0.02 μM in 34 and 0.084 μM in 35) and MNK2 (0.03 μM in 34 and 0.072 μM in 35). Replacing the piperazine ring in derivative 35 with a morpholine ring system to provide analog 36, resulted in a MNK inhibitor with comparable potency (MNK1 IC50 = 0.064 μM and MNK2 IC50 = 0.086 μM) and favorable physiochemical and ADME properties. Oral administration of 36 to mice at 5 mg kg−1 demonstrated that 36 possessed favorable to good exposure and PK properties: F (%) = 64, Cmax = 2481 ng mL−1, AUC0−t (last) = 5484 ng h mL−1. Furthermore, 36 was well tolerated (up to 200 mg kg−1 dosage) with no weight loss or clinical adverse effects in healthy mice being observed.
 | ||
| Fig. 11 SAR of fused heterocyclic core and piperazine replacement to obtain 36. | ||
Compound 36 displayed excellent kinase selectivity against a panel of 104 kinases, in which only one other kinase was inhibited more than 65% other than the desired MNK1 and MNK2 targets, the mitogen-activated protein kinase kinase (MEK). Only MNK was inhibited greater than 90% further demonstrating the high kinase selectivity of inhibitor 36. Compound 36 was tested for in vivo anti-tumor efficacy after oral administration alone or in combination with dasatinib, an ATP-competitive protein tyrosine kinase inhibitor, in a K562 overexpressing (o/e) eIF4E mouse xenograft model. Alone, 36 at the highest administered dose (100 mg kg−1) yielded only a TGI of 23%, though the combination of 36 with 2.5 mg kg−1 of dasatinib increased TGI in a dose-dependent manner (TGI = 107% for 25 mg kg−1 dose of 36 and TGI = 110% for 50 mg kg−1 dose of 36). Excitingly the combination approach with 2.5 mg kg−1 of dasatinib and 100 mg kg−1 of 36 resulted in 8 out of 8 mice administered to be tumor-free at the end of the study. Pharmacodynamic studies showed that the combination of dasatinib and 36 inhibited the phosphorylation of eIF4E (MNK substrate) and CRKL (BCR-ABL1 substrate) by their respective kinases, MNK1/2 and BCR-ABL1. Combination treatment is expected to reduce dosage in patients and reduce dasatinib toxicity. Generally, this can also be potentially extended to current and future therapies where resistance develops due to increased expression of p-eIF4E. Inhibitor 36 entered Phase I clinical trials to evaluate its effect in BC-CML patients treated with dasatinib.
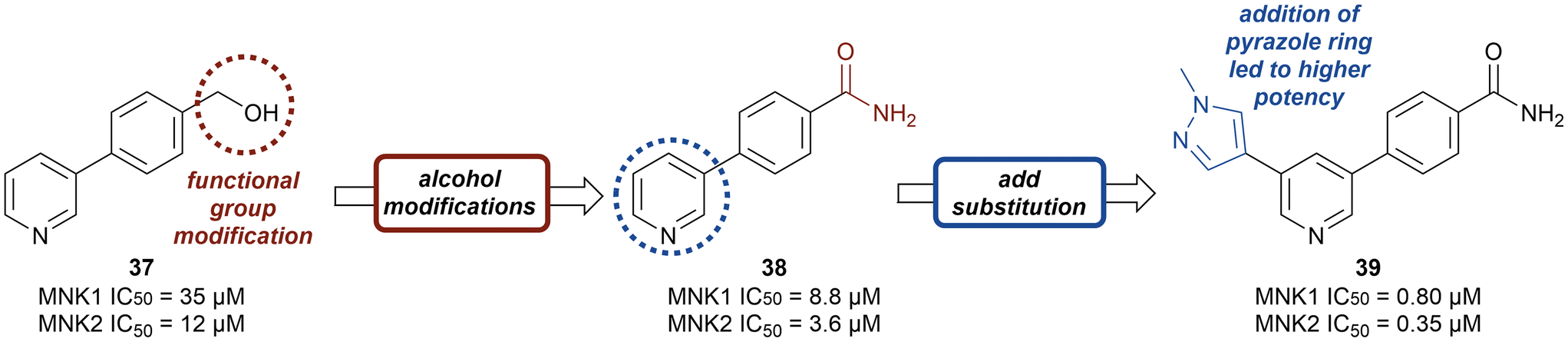
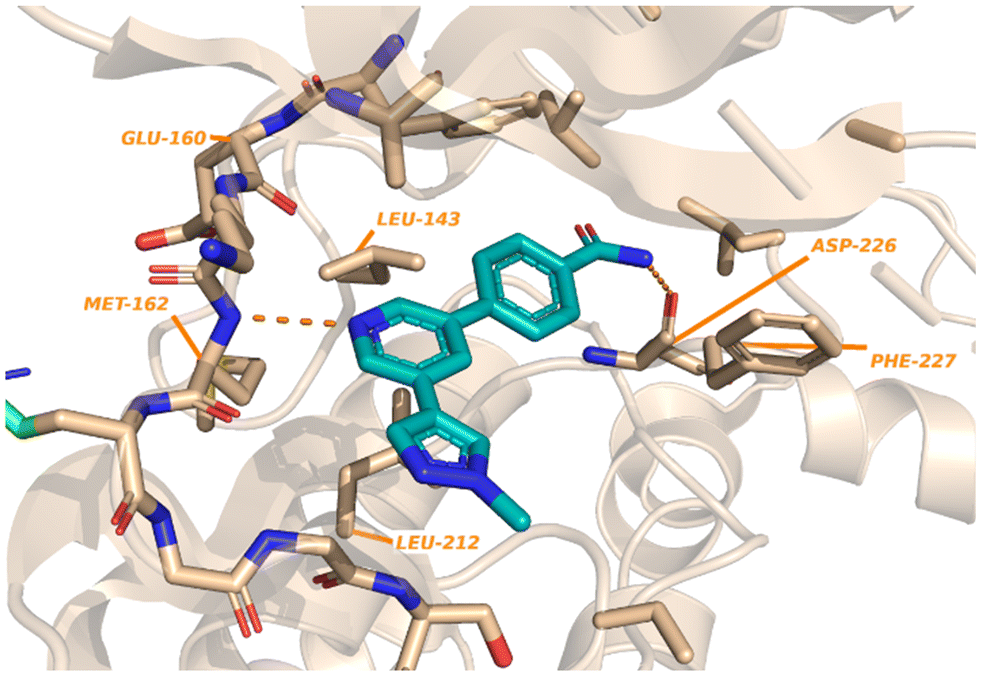
Kwiatkowski et al. described a fragment-based discovery of novel MNK1/2 inhibitors which began by screening a library of 1700 fragments.45 Among the initial hits was compound 37 containing a meta-substituted pyridine with an aryl linker (Fig. 12). Conversion of the primary alcohol of 37 into the primary amide (38) led to a slight improvement in potency compared to initial hit 37 for MNK1 (8.8 μM vs. 35 μM) and MNK2 (3.6 μM vs. 12 μM). Further optimization of 38 was investigated in which the functionalization of the amide resulted in a decrease in potency, but the addition of a pyrazole ring on the pyridine core to provide analog 39, resulted in a 10-fold increase in potency (0.8 μM in MNK1 and 0.35 μM in MNK2) compared to 38. Analog 39 was used to obtain the X-ray cocrystal structure bound to MNK2 (Fig. 13). Based on this structure, it is apparent that the central pyridine ring binds to the hinge Met162 residue. The terminal primary amide group forms a hydrogen-bond to Asp226 and Phe159 forms a pi–pi interaction with the phenyl ring to fix the structure in place. Additional SAR modifications were performed on the amide derivative 38 due to its ability to bind with the hinge region of MNK. Specifically, the introduction of various substituents on the aromatic ring was of interest (Fig. 14). First, the effect of adding a substituent at the ortho position of the phenyl ring was examined. The introduction of a hydroxyl group (40) at the ortho position was well-tolerated (MNK1 IC50 = 4.3 μM), while a chlorine (41, MNK1 IC50 = 11 μM) or a methyl (42, MNK1 IC50 = 22 μM) group resulted in decreased potency. This was hypothesized to be due to a favorable intramolecular hydrogen bond between the hydroxyl and amide groups. Interestingly, adding a methoxy group at C2 ortho position of 38 to yield analog 43 resulted in a 2–3-fold improvement in potency for both MNK1 (IC50 = 2.8 μM) and MNK2 (IC50 = 1.0 μM). Further modification through movement of the methoxy group from the C2 to C3 position (44) resulted in a loss of activity, likely due to the change in the dihedral angle between the pyridine and benzamide. Replacement of the methyl of the methoxy group with larger and more rigid groups such as the ethyl (45), isopropyl (46), cyclobutyl (47), cyclohexyl (48), pyrrolidine (49) and piperidine (50) all resulted in analogs with improved potency. Notably, the isopropyl substituted analog 46 displayed the greatest enhancement in potency possessing IC50 values for MNK1 and MNK2 of 0.88 μM and 0.34 μM, respectively. The introduction of an ethylamino group (51) resulted in a significant decrease in activity against both isoforms compared to the ether-isopropyl derivative 46. The addition of an aminoethyl group to the ether (52) yielded a MNK inhibitor with comparable activity to 46. Since compound 46 exhibited the highest potency, the ADME and PK properties were assessed. Utilizing the Caco-2 assay, 46 exhibited good solubility [Sol. (7.4) = 45 μg mL−1] with good permeability. Furthermore, MNK inhibitor 46 was studied for its in vivo PK profile. Notably, at a 5 mg kg−1 oral dosage, 46 reached a maximum concentration of 615 ng mL−1 in 10 minutes and displayed decent oral bioavailability (F = 61%).
 | ||
| Fig. 12 Initial optimization of meta-substituted pyridine 37. | ||
 | ||
| Fig. 13 Crystal structure of 42 with MNK2 (PDB:6LJR). | ||
 | ||
| Fig. 14 SAR of the phenyl ring substitutions to the core amide scaffold. | ||
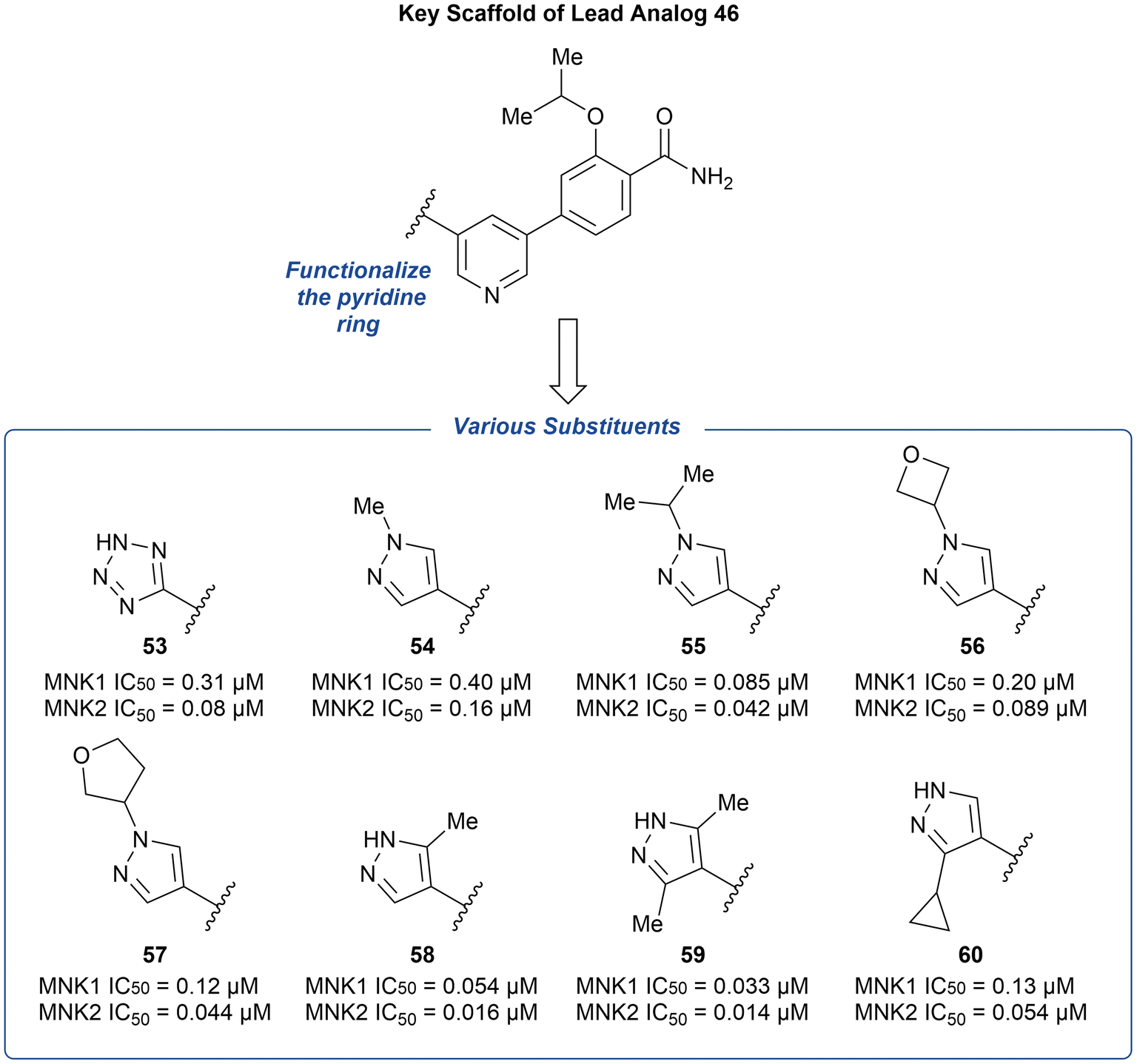
The preliminary optimization of 38 involving the addition of the pyrazole ring (39) provided evidence that attachment of a nitrogen heterocycle to the pyridine core can increase potency. Thus, the lead analog 46 underwent additional derivatization via the substitution of the pyridine ring with various heterocyclic motifs to provide a novel group of MNK inhibitors (Fig. 15). Potencies were improved by the addition of either a tetrazole (53) and pyrazole (54) ring. However, due to the lack of ability to further modify the tetrazole ring, the pyrazole ring system was used to further explore substituents. Upon increasing N-substituent size from a methyl (54) to isopropyl (55), there was a 4-fold improvement in potency. However, upon further increase in substituent size, such as an oxetane or furan ring to provide 56 and 57, respectively, the effect plateaued. Upon examining additional pyrazole substitutions, mono- (58) and bis-methylation (59) were more favored in comparison to 60 containing a cyclopropyl substitution. In vitro ADME screening of the more potent pyridine-substituted analogs revealed that 56 had a balanced ADME profile, which translated into good mouse pharmacokinetics, with low clearance (2.1 L h−1 kg−1), high exposure (2079 ng h mL−1), and good oral bioavailability (F = 73%) after a 5 mg kg−1 oral dose. Furthermore, the selectivity of 56 across the kinome was measured and was found to inhibit only three other kinases over 90% [ABL1, Janus Kinase 3 (JAK3), and death-associated protein kinase-related apoptosis-inducing kinase 1 (DRAK1)].
 | ||
| Fig. 15 SAR via substitution of the pyridine ring in 46. | ||
In another recent study, novel inhibitors of MNK1/2 and BCR-ABL1 were developed that display good oral bioavailability.106 To carry out these studies, a known compound, AST-487 (61) was used as the starting scaffold and optimized to improve the bioactivity of the known compound. To improve the kinase selectivity of AST-487 (61), a brief SAR was carried out by first modifying the urea linker present in 61 (Fig. 16). Among the various linkers, it was found that the replacement of the urea linker with an amide linker was tolerated (62). Conversion of the N-methylaminopyrimidine in 62 to the N-methylpyridine-2-amine to yield 63 resulted in an improvement in potency. Further modifications to the linker moiety and pyridine scaffold to obtain 64 and 65 led to compounds with comparable MNK inhibition.
 | ||
| Fig. 16 SAR of modified AST-487 (61) analogs. | ||
Although derivatives 64 and 65 were potent MNK1/2 inhibitors, they were found to display relatively little activity for ABL1 (IC50 > 10 μM), a key goal of this work. Therefore, the researchers utilized molecular modeling of 64 with MNK2 and ABL1 enzyme models to provide elucidation on further manipulations that could be investigated to increase the ABL1 potency. Docking studies revealed hydrogen bonding between the protonated piperazine and the carbonyl of Ala202 and His203 in the active site of MNK2 (Ile360 and His361 in ABL1). In addition, the trifluoromethyl functionality binds to a hydrophobic pocket of MNK2 and ABL1. The nitrogen in the pyridine partakes in a hydrogen-bonding interaction with the hinge residue of MNK2 with Met162 (Met318 in ABL1) and the amide group in the linker interacts with the backbone nitrogen of Glu129 in MNK2 (Glu286 in ABL1). Overall, the molecular docking studies provided evidence that optimization of 64via introducing groups on the pyridine ring to interact with the hinge binding region could be beneficial for both MNK1/2 and ABL1 potency. Therefore, the introduction of an acetamide substituent to the pyridine ring system and subsequent replacement of the oxygen in the linker scaffold with a nitrogen to provide 66 was achieved (Fig. 17). The combination of modifications resulted in a derivative (66) exhibiting 10-fold weaker activity against MNK1 compared to 64, though dramatically increased the activity for ABL1 (IC50 = 0.38 μM). Replacing the acetamide with a cyclopropanamide group (67) led to an increase in activity for both MNK1/2 and ABL1. Further derivatization was performed by the combination of the addition of a fluorine to the di-substituted phenyl ring and truncating the N-ethyl piperazine to an N-methyl piperazine to provide 68. Derivative 68 exhibited improved MNK1 and MNK2 potencies with IC50 values of 0.20 μM and 0.010 μM, respectively, and an improved ABL1 potency with an IC50 value of 0.01 μM. Against HeLa cells overexpressing eIF4E, compounds 67 and 68 showed strong antiproliferative activity and furthermore showed a reduction in phosphorylation of eIF4E at Ser209. Both lead analogs inhibited the phosphorylation of eIF4E and CRKL in K562 cells overexpressing eIF4E as well. Against a panel of CML cell lines, compound 68 showed greater antiproliferative activity (1–13 nM) compared to 67 (15–59 nM). The selectivity of 67 and 68 were assessed in a kinome panel (104 kinases) and revealed that at 1 μM 67 and 68 inhibited 12 and 15 kinases (90% activity inhibited), respectively. The greatest inhibition was found against the rearranged during transfection (RET) kinase, FLT3 and VEGF receptor 2 (VEGFR2) with IC50 values of less than 24 nM. There was also comparatively weaker inhibition of tyrosine–protein kinase KIT, platelet derived growth factor receptor (PDGFR)α, PDGFRβ, and FGF receptor 2 (FGFR2). Furthermore, when dosed at 50 mg kg−1 orally, 67 and 68 were reported to present a favorable PK profile and oral bioavailability of 80 and 31%, respectively (Table 1). Inhibitors 67 and 68 were tested for TGI in an in vivo K562 over expressing eIF4E xenograft model. Inhibitor 67 displayed a dose dependent TGI at 100 and 200 mg kg−1 when dosed orally once daily. Additionally, inhibitor 68 showed complete tumor regression at 100 mg kg−1.
 | ||
| Fig. 17 Optimization of 64 through strategic functional group modifications. | ||
| IV (5 mg kg−1 for 67 and 10 mg kg−1 for 68) | PO (50 mg kg−1) | ||||
|---|---|---|---|---|---|
| Compound | Cl (L h−1 kg−1) | T 1/2 (h) | C max (ng mL−1) | AUC (ng h mL−1) | F (%) |
| 67 | 2.7 | 2.1 | 2490 | 14337 |
80 |
| 68 | 3 | 2.2 | 1212 | 5128 | 31 |
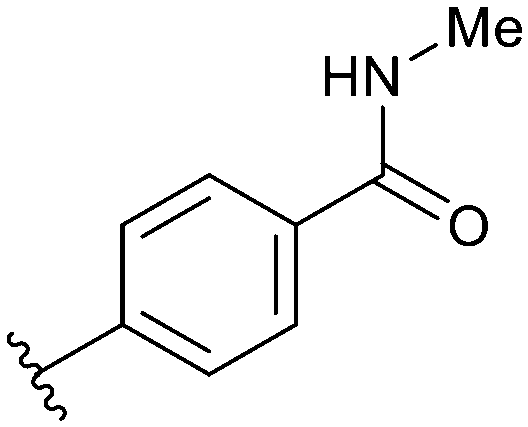
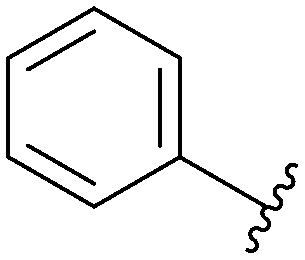
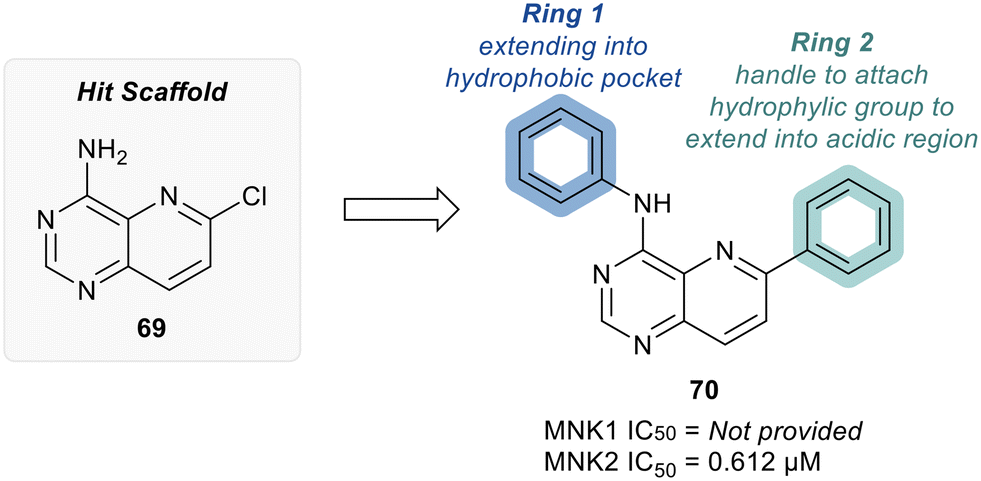
MNKs and Maloney murine leukemia virus kinases (PIMs) have overlapping and complementary effects in regard to regulating cap-dependent protein translation and both are over expressed in a variety of cancers.107 Therefore, in 2021 Han et al. reported the identification of dual MNK and PIM inhibitors using the cocrystal structure of staurosporine (1) bound to MNK2 (pdb file 2HW7 as shown in Fig. 1) in a structure-guided approach.108 Based on their evaluation of the crystal structure, they observed that MNK's gatekeeper residues, Phe159 and Cys225, and the hydrophobic pocket, consisting of Glu92, Glu209, Asn210 and Asp226, being acidic in nature, suggest that basic amine groups would be complementary for binding (Fig. 1). The crystal structure 2HW7 was used to screen an in-house fragment library to obtain the lead compound, 69 (Table 2). Docking studies revealed that the nitrogen atoms at the 3- and 4-positions interact with Met162 and Glu160 in the MNK2 hinge region. Introduction of a benzene ring (ring 1) at the 4-position amino group resulted in the formation of a pi–pi interaction with Phe159. To further fill the MNK active site, a 6-position aromatic ring (ring 2) was introduced (70) which could also provide a scaffold handle to attach hydrophilic substituents to reach into the acidic region. To optimize analog 70, functionalization of ring 2 was first investigated, in which the introduction of an N-methylamide group (71) and a piperazine amide group (72) was performed. The addition of the hydrophilic substituents assisted in reaching into the acidic region and provided compounds with increased potency for MNK2 (IC50 = 0.058 μM for 71 and IC50 = 0.036 μM for 72). The incorporation of the larger piperazine group led to enhanced potency compared to the N-methylamide group and therefore underwent further optimization. Functionalization of ring 1 of 72 provided the substituted derivatives, 73 (MNK2 IC50 = 0.01 μM) and 74 (MNK1 IC50 = 0.001 μM and MNK2 IC50 = 0.007 μM) which both displayed enhanced potency compared to 72 (MNK2 IC50 = 0.036 μM). The functionalization of both ring 1 and ring 2 on the preliminary inhibitor 70 led to analogs with improved potency for both MNK1 and MNK2, ultimately providing 74, a low nanomolar MNK1/2 inhibitor.
|
|
|||
|---|---|---|---|
| Functionalization of ring 1 and ring 2 | |||
| Compound | Ring 1 | Ring 2 | MNK1/MNK2 inhibition (IC50) |
| 71 |
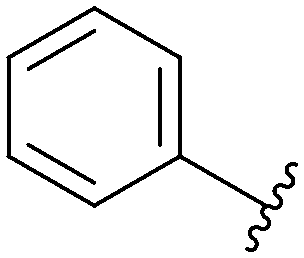
|
|
Not provided/0.058 μM |
| 72 |
|
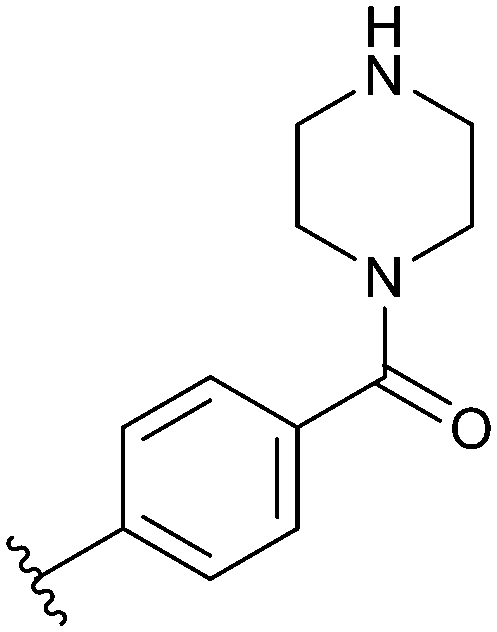
|
Not provided/0.036 μM |
| 73 |
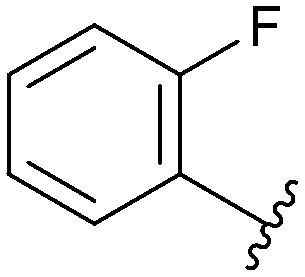
|

|
Not provided/0.01 μM |
| 74 |
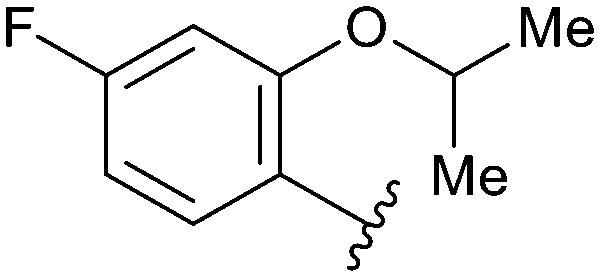
|
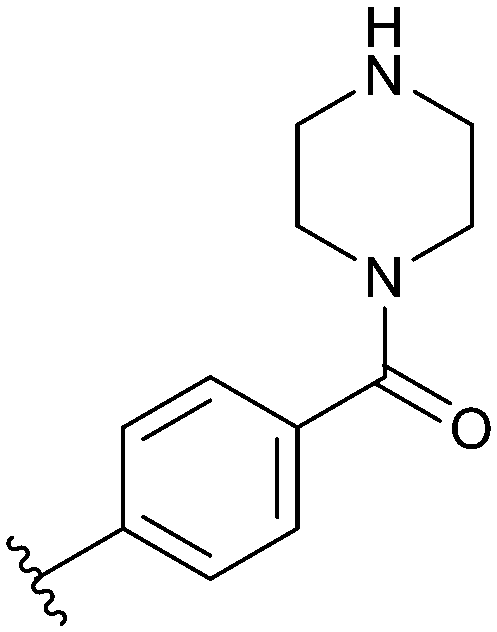
|
0.001 μM/0.007 μM |
Because myeloid leukemia cells have increased MNK activities, acute myeloid leukemia MOLM-13 cells with FLT3-ITD mutation were used to analyze the cell growth inhibition (GI50) effects. Compound 74 was found to inhibit MOLM-13 cell growth inhibition with a GI50 of 1.23 ± 0.18 μM. The inhibitory activity against PIM kinases were also measured and 74 was found to be a potent inhibitor of PIM1, PIM2 and PIM3 displaying IC50 values of 43, 232, and 774 nM, respectively. Thus, demonstrating the ability of 74 to act as a dual MNK/PIM inhibitor. The antileukemia effect of 74 was also tested in several leukemia cell lines with different mutation profiles: K562 cells (BCR-ABL mutation), MOLM-13 cells (FLT3-ITD mutation), and HL-60 cells (no mutations). Using western blot analysis, the effect 74 had on downstream substrates such as eIF4E (MNK1/2 substrate), Bcl2-asssociated death promoter (BAD), and eIF4E binding protein (4EBP1) (PIM substrates) in MOLM13-cells was analyzed. 74 was treated at 1 μM and 2 μM concentrations and showed a reduction in the levels of the substrates of both MNK (p-eIF4E) and PIM (p-BAD and p-4EBP1) in MOLM-13 cells, suggesting inhibition of MNK and PIM results in inhibition of downstream targets. The kinase selectivity of 74 was investigated using DiscoveryX KINOMEscan technology from Eurofins, in which 30 MNK homologous kinases and other common kinases were tested for inhibition rates of 74 (at 1 μM). Other than MNK1/2 and PIM1/3, only kinases BRAF, EGFR, and ERBB2 exhibited >30% inhibition. Interestingly, the MNK/PIM dual inhibitor, 74, was found to induce apoptosis in MOLM-13 cells as measured with Annexin V, though when treated with MNK inhibitor alone, apoptosis was not observed, suggesting inhibiting both MNK/PIM is required for the induction of apoptosis. 74 was also found to have favorable antileukemia effects in MOLM-13 xenografts when treated for two weeks daily at doses of 5 mg kg−1 and 10 mg kg−1 intraperitoneal (IP), displaying TGI of 41.2% and 57.7%, respectively at the two doses. At the lower dose, there was an apparent decrease of p-4EBP1, while at the higher dose, there was a decrease in cyclin D1, p-eIF4E, p-BAD, and p-4EBP1. Pharmacokinetics evaluation of 74 showed a favorable profile and supported further in vivo testing (Table 3). Compound 74 was orally bioavailable (F = 35%) with good exposure.
| Dose (mg kg−1) | 2 | 10 | 10 |
|---|---|---|---|
| Route | IV | IP | PO |
| C 0 (ng mL−1) | 765 | — | — |
| C max (ng mL−1) | 652 | 405 | 224 |
| T max (h) | 0.083 | 0.67 | 8.00 |
| T 1/2 (h) | 6.71 | 4.40 | NA |
| AUC0−t (last) (ng h mL−1) | 1609 | 1661 | 2852 |
| AUC0-inf (ng h mL−1) | 1715 | 1682 | NA |
| F (%) | — | 21 | 35 |
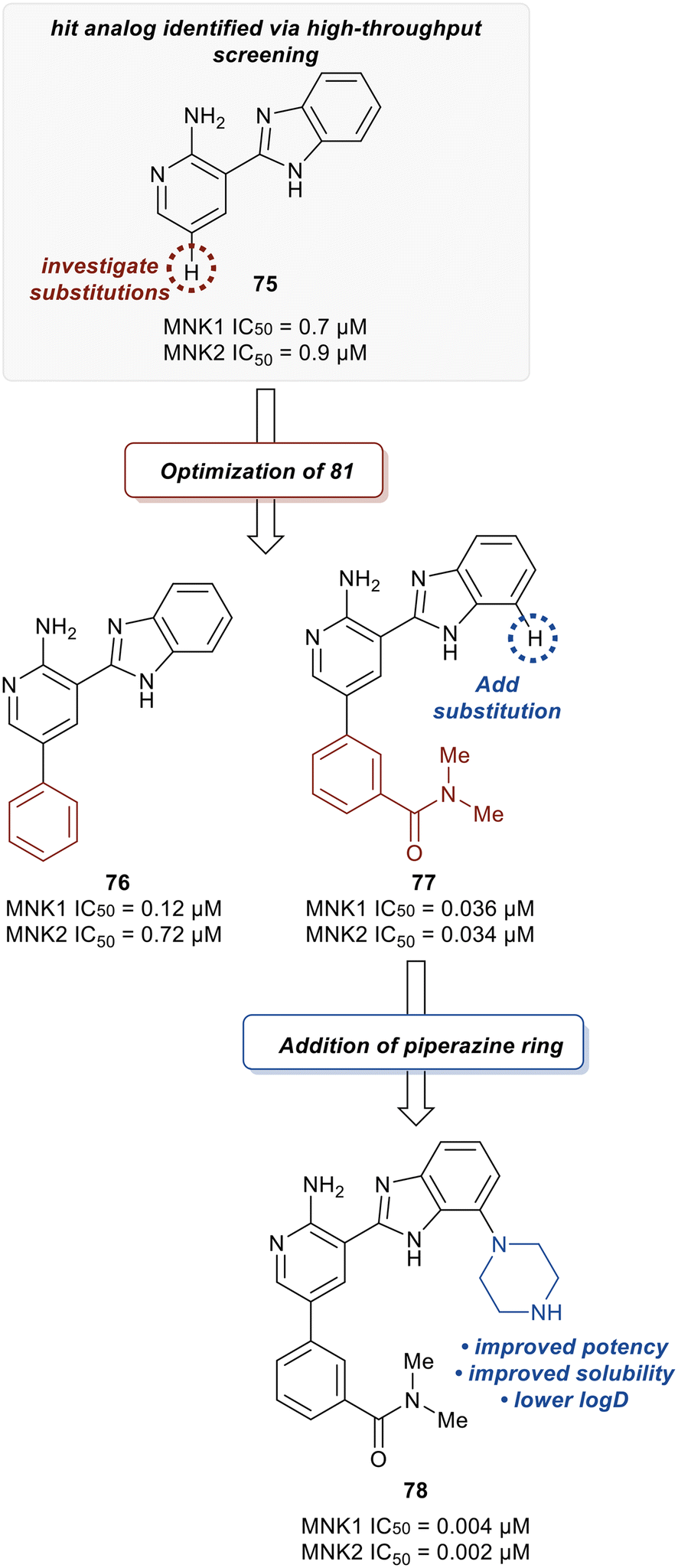
Han et al. in 2016 described the optimization of a series of non-covalent MNK1/2 inhibitors.109 MNK1 and MNK2 share very high similarity, with amino acid sequence similarity of 72–78% in the active site. Among the initial hits identified via high-throughput screening using a cocrystal structure of staurosporine (1) bound to MNK2 (Fig. 1) was an aminopyridine benzimidazole (75), which possessed an IC50 < 1 μM against both MNK1 and MNK2, with significant selectivity against identified off-targets CDK1 (IC50 = 10 μM) and CDK2 (IC50 = 25 μM) (Fig. 18). In designing improved analogs, analysis of docking studies with 75 suggested that additional favorable interactions with the hinge region could be generated by the addition of an aryl or heteroaryl ring at C5 of 75. Following up on this result, the authors incorporated a phenyl ring at C5 (76), which showed improvement in biochemical.
 | ||
| Fig. 18 SAR of non-covalent MNK1/2 inhibitor 75. | ||
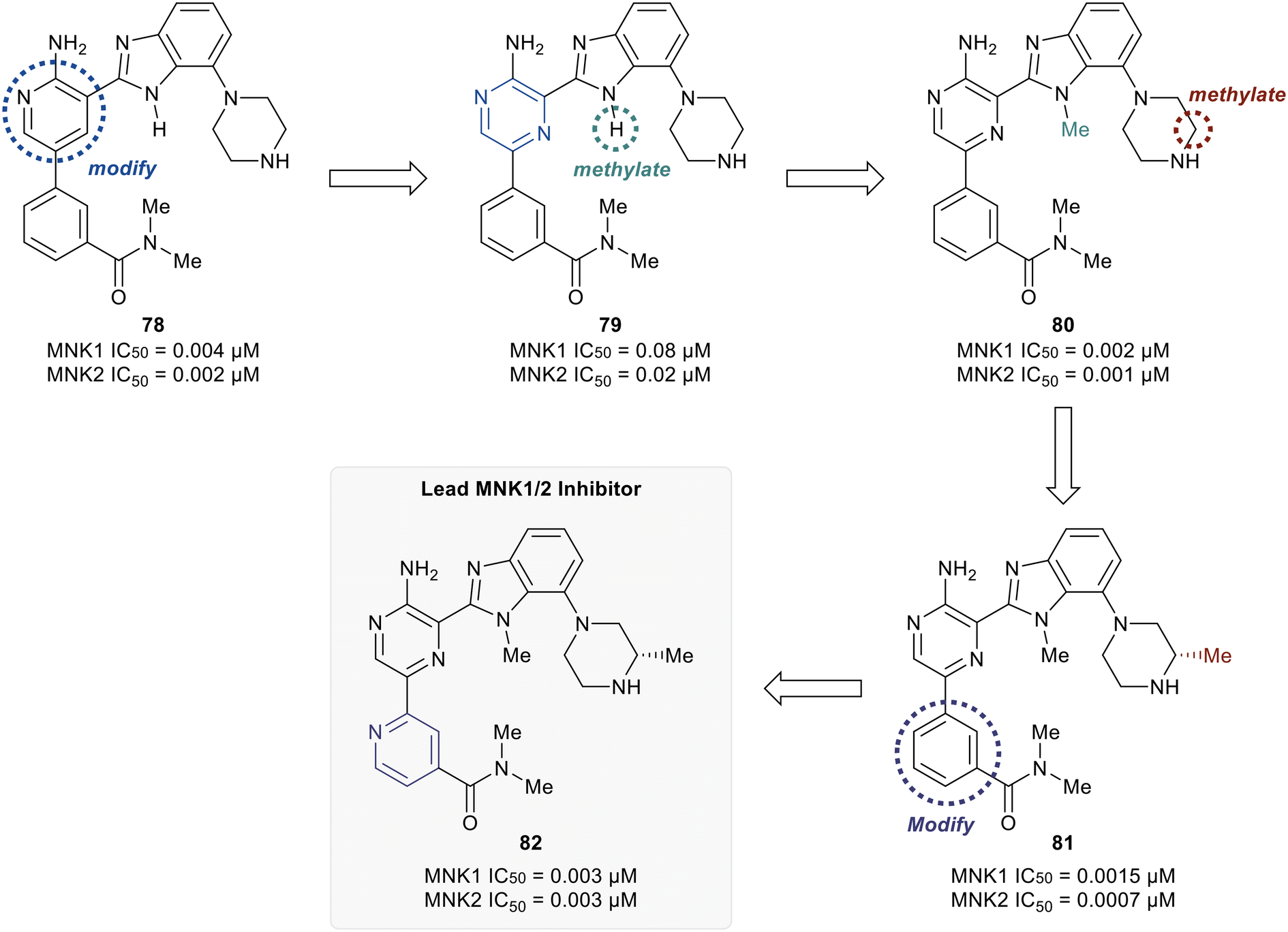
MNK1 and MNK2 potencies with IC50 values of 0.12 μM and 0.72 μM, respectively. However, there was increased binding with CDK1 (0.16 μM) and CDK2 (13 μM) compared to 75. To address this undesired activity, a dimethyl amide at the meta-position of the phenyl ring was introduced (77). This reduced CDK1/2 binding (1.0/5.7 μM), while also improving MNK binding (0.035/0.034 μM). Molecular modeling of 77 showed the interaction of the carbonyl oxygen in the amide with the backbone N–H of Ser166 along with hydrophobic interactions with the P-loop residues. Due to the unfavorably high logD value of 4.2 for 77, the effect of adding basic amine groups at the 7′-position of the benzimidazole was tested. The addition of a piperazine ring to obtain 78 led to >10-fold increase in potencies against MNK1 and MNK2 compared to 77 along with an improvement in solubility without inhibiting the undesired CDK1/2 (1.0/4.0 μM). Based on the promising results of the benzimidazole aminopyridines, SAR investigation into this group of compounds was performed, however, additional modifications to the aminopyridine series led to no improvements in cellular activity. Consequentially, the researchers began investigating aminopyrazine-type analogs. The conversion of aminopyridine 78 to aminopyrazine 79 resulted in a significant loss of potency against MNK1 and MNK2 (Fig. 19). This decrease in activity was hypothesized to be a result of a difference in torsional energy between the aminopyridine and aminopyrazine benzimidazole (∼1.4 kcal mol−1). To lower the torsional energy barrier, present in the aminopyrazine-type scaffold, a methyl group was added on to the nitrogen of the benzimidazole ring in 79 to provide analog 80. The planarity disruption resulted in a perpendicular orientation of the piperazine relative to the benzimidazole ring, thereby allowing a hydrophobic interaction of the piperazine with the P-loop of MNK (bioactive conformation), leading to a potency improvement for MNK1/2 (0.002/0.001 μM). However, off-target effects with CDK1/2 (0.50/1.4 μM) were observed due to the interaction of the piperazine with the P-loop in CDK. To eliminate this interaction, a methyl group was added to the piperazine ring to obtain compound 81, which had similar biochemical MNK1/2 potency (0.0015/0.007 μM) as 80, but improved selectivity against CDK1/2 (1.0/3.4 μM). Upon exchanging the phenyl ring in 81 with a pyridyl ring to obtain 82, a highly MNK-selective inhibitor was obtained (0.003 μM for MNK and >25 μM for CDK). This high selectivity was attributed to the interaction of the pyridyl nitrogen with Leu83 in CDK2. Due to the nitrogen's proximity to the residue, the lone pair of electrons on nitrogen were also seen to cause a repulsive effect with the oxygen of the carbonyl on Leu83. Phosphorylation of eIF4E at Ser209 by 82 was measured with an EC50 value of 0.6 nM in KMS11-luc human multiple myeloma tumor cell line. Compound 82 was also observed to have excellent selectivity against 53 other kinases with only four being inhibited with IC50 values of less than 1 μM (CaMK2D, FLT3, PIM2, and ROCK2). Furthermore, in Caco-2 assay, 82 displayed favorable cell permeability and modest efflux. Other physicochemical properties such as logD and solubility were also measured to be desirable values for 82. This molecule is being further evaluated for in vivo anti-cancer effects through MNK1/2 inhibition.
 | ||
| Fig. 19 SAR of benzimidazole aminopyrazine series to obtain lead MNK1/2 inhibitor 82. | ||
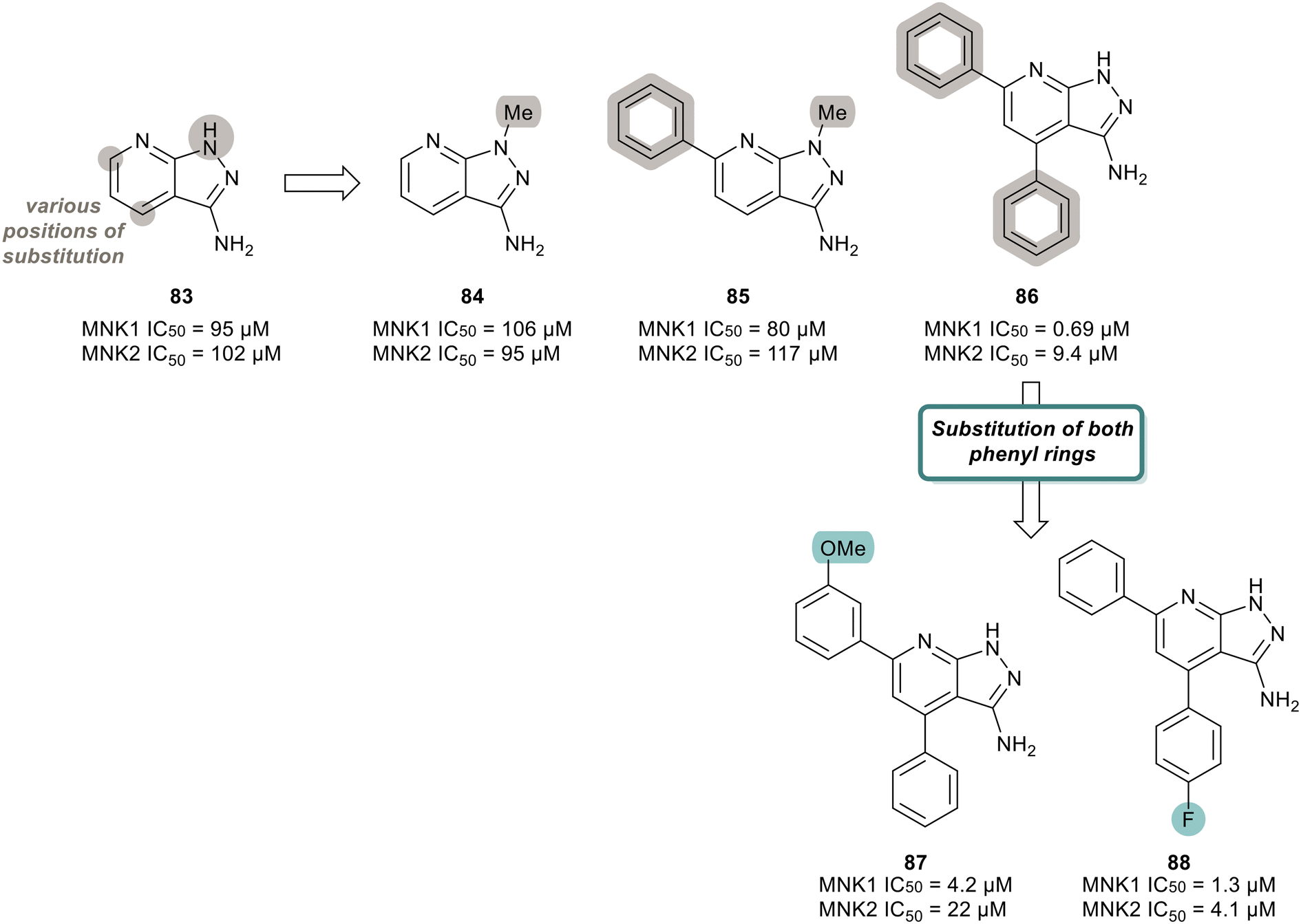
MNK inhibitors such as those currently in clinical trials are classified as type-I kinase inhibitors and can activate the non-catalytic functions of the kinase.90,91,110–112 To identify non-ATP-competitive MNK inhibitors, Bou-Petit and coworkers looked into alternate scaffolds without the ability to directly interact at the ATP binding site.113 They reported a new pyrazolo[3,4-b]pyridine-3-amine scaffold with potent MNK inhibitory activity, which was identified from a radiometric protein kinase assay.113 Their initial efforts were focused on the development of a series of pyrazolo[3,4-b]pyridine-6-one compounds. However, since these were inactive, a series of pyrazolo[3,4-b]pyridine-3-amine compounds were synthesized (Fig. 20). The effect of a combination of substituents added to the pyridine and pyrazole rings, such as various methyl and phenyl groups were tested. Beginning first with simple substitutions to the ring systems of 83, substitution of a methyl group on the pyrazole ring was unfavored as displayed in analogs 84 and 85 with potencies of 106 μM and 80 μM, respectively, indicating that a H-bond donor is essential for activity. The addition of two phenyl rings at C5 and C7 on the pyridine ring to provide 86 resulted in significantly enhanced activity with a potency of 0.69 μM for MNK1. The effect the incorporation of the phenyl rings has on potency is apparent when comparing the MNK1/2 IC50 values obtained for the bi-aryl analog 86 and the unsubstituted analog 83 (MNK1 IC50 = 95 μM for 83vs. MNK1 IC50 = 0.69 μM for 86). Functionalization of both phenyl groups in 86 was investigated in which the addition of a methoxy group on the C5 phenyl ring in 86 to produce 87 resulted in reduced potency (0.69 μM for 86 and 4.2 μM for 87). Similarly, a fluorine substituent on the C7 ring (88) reduced MNK1 and MNK2 potencies (1.3 μM and 4.1 μM, respectively). Since 86 possessed the greatest activity, further biological characterization was performed. Inhibitor 86 was tested in MDA-MB-231 (breast cancer), MCF7 (breast cancer), 22RV1 (prostate cancer), A375M (melanoma) and MV4-11 (leukemia) cells for its ability to inhibit phosphorylation of eIF4E and exclude any cell-line effects on inhibition. It was observed that 86 dose-dependently inhibited eIF4E phosphorylation in all cell lines. Molecular dynamics studies revealed that 86 is a type II inhibitor and binds to the inactive form of MNK. Upon comparing its activity to other known inhibitors, 86 showed greater activity than known MNK inhibitor, CGP57380, but lower potency than eFT508 (2). However, unlike CGP57380 and eFT508 (2), 86 did not induce MNK hyperphosphorylation and its paradoxical activation. It was found that hyper-phosphorylation did not occur upon 86 binding, thereby preventing MNK to adopt an active conformation to further bind to eIF4G. Furthermore, when MDA-MB-231 cells were treated with 86, cell growth was inhibited in a dose dependent manner with a maximal reduction to 60% at 20 μM. Normal IMR90 cells were unaffected by the treatment of 86, suggesting cell growth inhibition is limited to tumor cells.
 | ||
| Fig. 20 Represented analogs of pyrazolo[3,4-b]pyridine-3-amine MNK1/2 inhibitors. | ||
The development of potent and selective MNK1/2 inhibitors has been a growing field of interest over the last decade. The utilization of high-throughput screening and structure-guided design to lay the groundwork for the discovery of diverse inhibitors and subsequent SAR analysis has greatly enhanced the medicinal chemistry field. Specifically, the characterization of novel lead MNK1/2 inhibitors as described above has provided new insight into structural chemotypes to be further explored. Summarized in Table 4 are the lead MNK1/2 inhibitors and their corresponding potencies, kinase selectivity profiles, and downstream/biological effects. Overall, the MNK1/2 inhibitors reported herein are potent with the majority possessing IC50 values less than or equal to 200 nM. The inhibition of the phosphorylation of eIF4E and other substrates discussed by the MNK1/2 inhibitors, demonstrates that targeting MNK1/2 results in the inhibition of downstream targets that play roles in the regulation of the translation of mRNA, cell proliferation, and tumor growth.
| Inhibitor | MNK1/2 IC50 | Selectivity of MNK1/2 inhibitors (% inhibition, IC50) | Downstream effects measured |
|---|---|---|---|
| a HCT-116 cell line. b HeLa cell line. c Measured with western blot analysis of tumor samples excised from mice with 2.5 mg kg−1 of dasatinib and 100 mg kg−1 of 36. d Western blot of K562 cells treated with 100–1000 nM dose of 68. e Western blot of MOLM-13 cells treated with 2 μM of 74. f KMS11-luc human multiple myeloma tumor cell line. g Western blot of MDA-MB-231 cells with 20 μM treatment of 86. | |||
| 2 (EFT508) | 2.4 nM/1.0 nM | MNK1 (>85%, NA), MNK2 (>85%, NA), DRAK1 (82%, 131 nM), CLK4 (60%, 787 nM) | p-eIF4E IC50 = 6 nMa |
| Reduction of pro-inflammatory cytokine production (TNF-α, IL-6, and IL-8) | |||
| Reduction of mRNA stability | |||
| 36 | 64 nM/86 nM | MNK2 (>90%, NA), MEK (>65%, NA) | p-eIF4E IC50 = 0.321 μMb |
| p-CRKLc | |||
| 56 | 200 nM/89 nM | MNK1, MNK2, ABL1 (>90%, NA), JAK3 (>90%, NA), DRAK1 (>90%, NA) | p-eIF4E IC50 = 0.2 μMb |
| 68 | 200 nM/10 nM | MNK1, MNK2, RET (>90%, 0.001 μM), FLT3 (>90%, 0.020 μM), VEGFR2 (>90%, 0.021 μM), PDGFRα (>90%, 1.280 μM), PDGFRβ (>90%, 1.055 μM), FGFR2 (>90%, 0.272 μM), cKIT (>90%, 1.509 μM), CSF1R (>90%, NA), TIE2 (>90%, NA), AXL (>90%, NA), ABL1 (>90%, NA), RSK2 (>90%, NA), TRKA (>90%, NA), CDK7 (>90%, NA) | ABL1 IC50 = 0.01 μMb |
| p-eIF4E IC50 =0.28 μMb | |||
| p-CRKLd | |||
| 74 | 1 nM/7 nM | MNK1 (100%, 1 nM), MNK2 (100%, 7 nM), PIM1 (96%, 43 nM), PIM3 (90%, 774 nM), BRAF (87%, 2217 nM), EGFR (73%, 1970 nM), ERBB2 (60%, 7491 nM) | Reduced levels of p-eIF4E, p-BAD, p-4EBP1, c-myc, cyclin D1, and Mcl-1 levelse |
| 82 | 3 nM/3 nM | MNK1, MNK2, CaMK2D (NA, 0.69 μM), FLT3 (NA, 0.28 μM), PIM2 (NA, 0.73 μM), ROCK2 (NA, 0.37 μM) | p-eIF4E (EC50 = 0.6 nM)f |
| 86 | 690 nM/9400 nM | Not provided | p-eIF4Eg |
3. Small molecule approaches targeting the eIF4F complex
While the previous section discussed various approaches to target the MNK–eIF4E axis through the development of MNK inhibitors, another strategy that has been recently described is to target the eIF4F complex to block the interaction of eIF4E with its partners and repress the translation of mRNA. One approach that was taken was the development of antisense oligonucleotides (ASOs) targeting eIF4E. In one particularly noteworthy report, eIF4E ASOs were developed that reduced eIF4E protein and its downstream targets, and had strong anti-tumor efficacy in breast cancer xenografts.114 However, in a clinical trial, these eIF4E ASOs failed to produce an anti-tumor response even though a reduction in eIF4E was observed.115 There are a handful of eIF4A small molecule inhibitors with preliminary results showing the ability to treat cancers. Examples include the widely studied Silvesterol, Hippuristanol, Pateamine A, and Flavagline analogs which were discovered several years ago, although the precise mechanisms of each of these compounds may be complex.116–121 The discovery and activity of these compounds have been reviewed elsewhere.122–124 This review focuses on the more recent updates within the last 8–10 years.3.1. Inhibitors of eIF4A
A study in 2021 by Zerio et al. described a series of direct eIF4A inhibitors with a novel mechanism of action.125 The study was initiated with a biochemical screen of more than 100000 compounds, which revealed eighteen hits, and the most potent being 2-(5-(4-butylphenyl)furan-2-yl)quinoline-4-carboxylic acid (89) (Fig. 21). Compound 89 inhibited the ATPase activity of eIF4A and was therefore expected to be an ATP-competitive inhibitor. However, competition studies revealed that it was non-competitive with ATP. Inhibitor 90, the ethyl ester derivative of 89, was five-fold less potent (27 μM vs. 129 μM) suggesting the importance of the acid functionality due to the formation of a salt bridge with Arg311 of eIF4A. The bromine substitution at the 6-position was also essential for the potency of 90. The presence of iodine or chlorine produced molecules with lower potency, hypothesized to be due to the unfavorable size of the halogens. The substitution of hydrocarbons such as methyl, ethyl, isopropyl, phenyl and methoxy revealed that size did not affect the trend in potency. The effect of adding a trifluoromethyl group was also tested and found that substitution at the 7-position (91) lead to an analog with less potency than 89 and substitution at the 8-position (92) resulted in an inhibitor with comparable potency to 89. Electron-donating amino and acetamido at the 6-position failed to increase potency. Replacement of the bromine with a nitro group at the 6-position produced the most potent molecule, 93, exhibiting an IC50 = 8.60 μM.
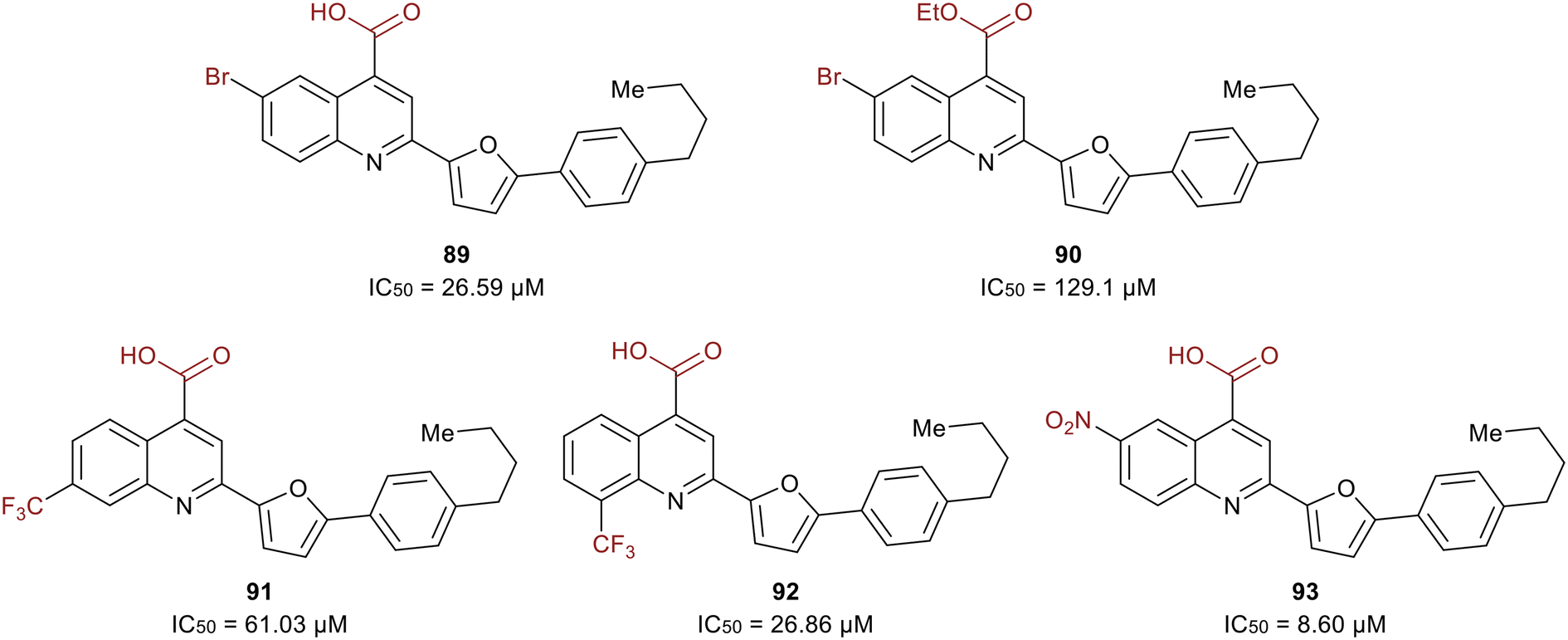 | ||
| Fig. 21 SAR of eIF4A inhibitors. | ||
Compound 93 was found to be an RNA-competitive eIF4A inhibitor, but did not compete with ATP, suggesting it possessed a novel inhibitory mechanism of action. As an ATP-uncompetitive inhibitor, 93 was found to bind to eIF4A. With both 93 and ATP bound, RNA binding to eIF4A occurs in a conformation that inactivates eIF4A. For unknown reasons, 93 also completely competes off RNA at high concentrations. However, at lower concentrations, RNA is still bound but forced to bind eIF4A in a pseudo-closed ATP-bound state with 93, preventing ATP hydrolysis and RNA unwinding. Hence, this work presents a unique eIF4A inhibitor due to its ability to preferentially inhibit cap-dependent translation by engaging with eIF4A in cells and operating through a novel mechanism of action.
Work published in 2020 by Ernst et al. describes the work that led to the development of eFT226 (Zotatifin, 94) (Fig. 22).126 An analog of a flavagline natural product, rocaglamide A (95), was utilized as the chemotype scaffold to optimize and identify eFT226 (94). During the design of 94, the crystal structure of rocaglamide A (95) was not available. A molecular model based on PDB 2HYI was used to understand the binding between the RNA, rocaglamide A (95), and eIF4A1. The model suggested the A and D phenyl rings of 95 formed a pi-stacking arrangement with the RNA bases at the RNA/eIF4A1 interface. It was also postulated that the D and E rings fit into two hydrophobic pockets of eIF4A1. A hydrogen bond was also predicted between the N–H side group of Gln195 and the carbonyl carbon of the dimethyl amide group in 95. This hydrogen bond provided additional ligand–protein stabilization. Since this was still considered low resolution, additional computational methodologies were used to assess compound designs. Torsional modeling revealed that the preferred angle between rings D and E was 40° and this criterion was used to design molecules for further testing. Additionally, a hydrogen bond was suspected between the tertiary hydroxyl of 95 and N-7 of the adjacent RNA guanine base, which was hypothesized to impart stabilization of the purine sequences to eIF4A1.
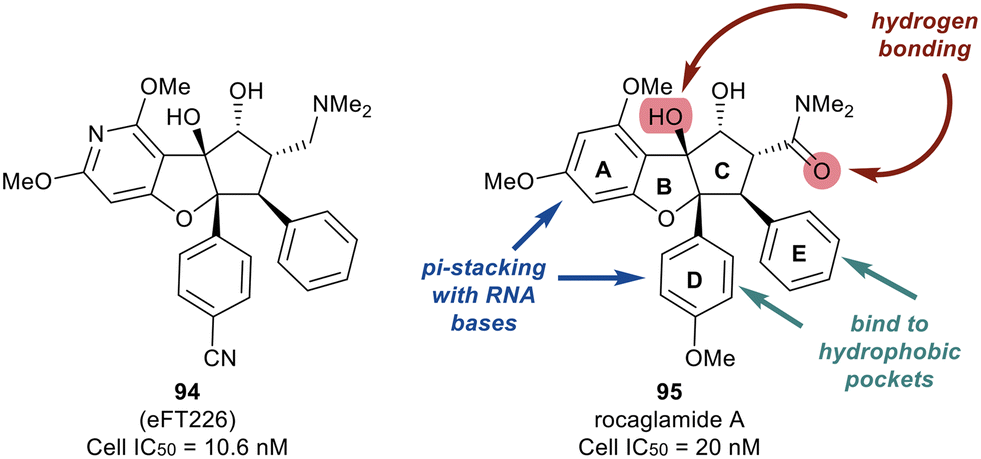 | ||
| Fig. 22 Representative structures of eFT226 (94) and rocaglamide A (95). | ||
Replacing the phenyl ring A with a heterocycle in rocaglamide A (95) to enhance the pi-stacking interaction with the RNA base was expected to stabilize binding, while also lowering the lipophilicity of the core. Therefore, a pyridine ring was first incorporated to provide a nitrogen at the 8-position (96) of the core (Fig. 23). This provided the ability to replace the hydrogen bond acceptor predicted from the methoxy group present in the parental compound 95. However, this modification had an unfavorable potency of 990 nM. Replacement of the methoxy on the D ring with a nitrile group led to derivative 97 which displayed improved potency compared to 96 (159 nM vs. 990 nM). Modeling of 97 revealed the nitrile functionality fit into a narrow groove formed by the RNA and eIF4A and interacts with Asn167.
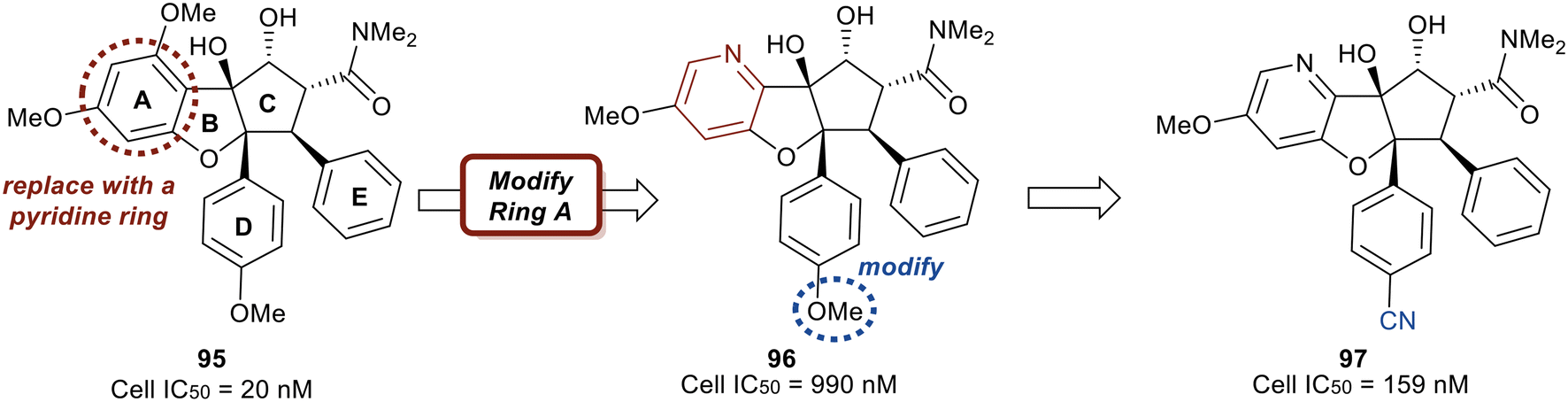 | ||
| Fig. 23 Initial structural modifications made to 95. | ||
An exploration of various pyridyl regioisomers of derivative 97 showed a difference in potencies (Fig. 24). The 7-position and 6-position pyridyl regioisomers (98 and 99) were more potent compared to the 5-position isomer (100), showing the effect of subtle changes in the chemical makeup of ring A on potency. Further modification of 98via addition of a secondary methoxy group onto ring A of the 2-position led to 94 (eFT226), demonstrating favorable potency and high selectivity (10.6 nM). The selectivity of 94 to target eIF4A was analyzed by mutating the assumed binding site of eIF4A1, specifically converting Phe163 to Leu163 (F163L), in the HAP1 cell line. The mutated cell line was treated with 94 at various doses and western blot analysis was performed to visualize the effect of 94 in the unaltered HAP1 cells and the mutated eIF4A1 F163L cells had on protein levels of downstream substrates (MYC, MCl-1 and Cyclin D1). In the unaltered cell line, 94 demonstrated a dose dependent decrease in the protein levels of the downstream targets whereas in the mutated cell line no changes in protein levels were observed. The results from this study indicate that binding to eIF4A is essential for 94 to induce a decrease in protein levels of downstream targets and that eIF4A is the specific cellular target of 94. Compound 94 also possessed favorable pharmacokinetics, with a half-life in mice of 10.9 hours and modest clearance (15.8 mL min−1 kg−1). Inhibitor eFT226 (94) was tested in a triple negative breast cancer xenograft model (MDA-MB-231) with once-a-week 1 mg kg−1 dosing IV (intravenous) and showed 122% TGI or regression throughout the study.
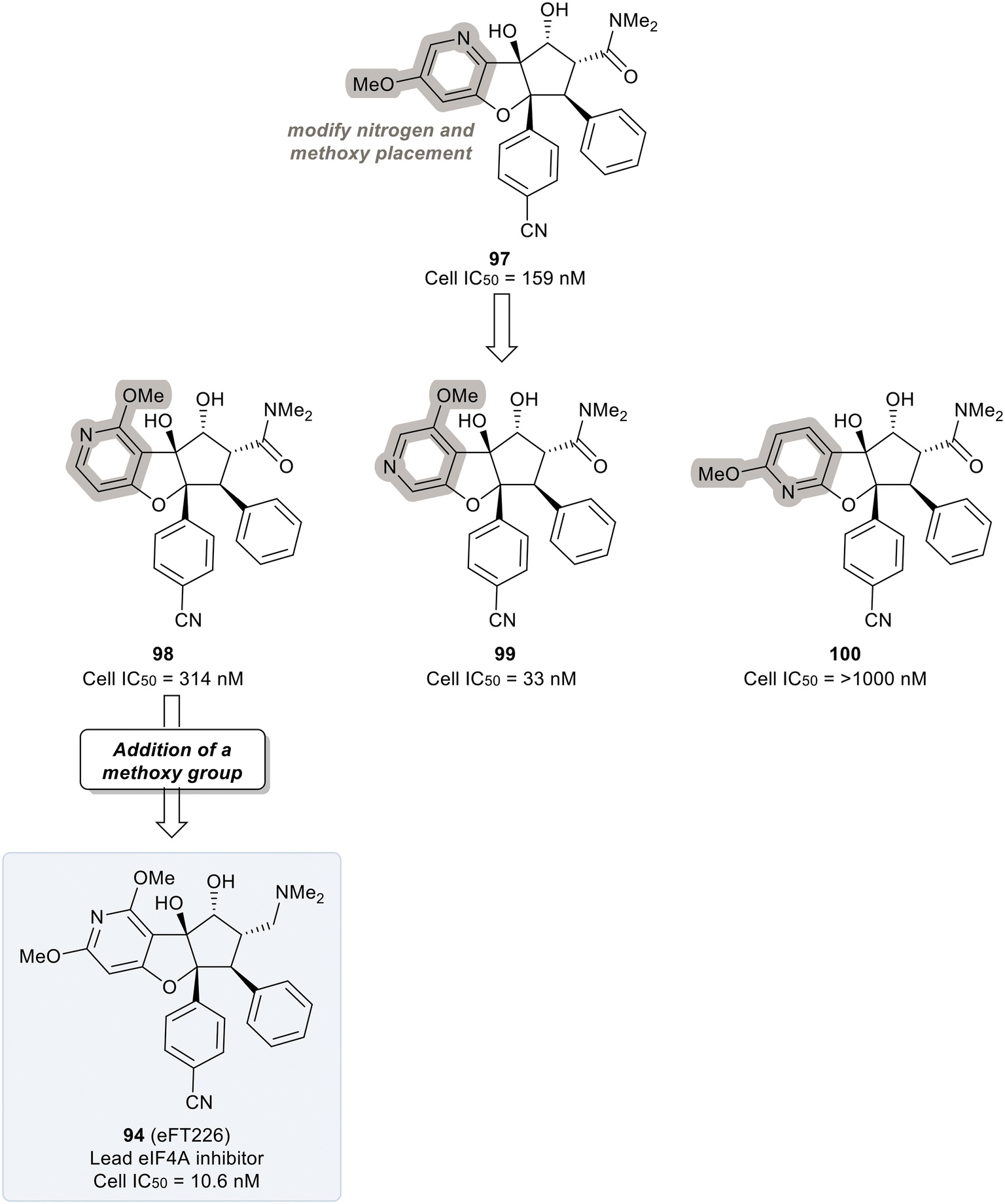 | ||
| Fig. 24 Final optimization of 97 to identify eIF4a inhibitor 94. | ||
3.2. Allosteric regulation of the eIF4F complex
One of the earliest inspirations for identifying and developing small molecule inhibitors of the eIF4F complex was the observation that the antiviral drug ribavirin directly binds eIF4E and disrupts eIF4E–RNA binding.127 Ribavirin was found to bind eIF4E with a KD = 8.4 μM and inhibited the oncogenic transformation of NIH 3T3 cells. Importantly, ribavirin was found to significantly inhibit FaDu tumor growth in a mouse model, supporting the potential for eIF4E inhibitors as an anti-cancer strategy.High throughput screening led to the discovery of small molecule eIF4E allosteric modulators such as 4EGI-1 (101), 4E1Rcat and 4E2Rcat.128,129 As mentioned in the prior sections, 4E-BPs are proteins with the ability to sequester eIF4E. Upon 4E-BP phosphorylation, there is displacement of eIF4E from 4E-BP, initiating the eIF4F complex formation and subsequent mRNA translation. Allosteric modulation of eIF4E is hypothesized to reduce 4E-BP binding with eIF4E, preventing eIF4F formation. It is likely that an allosteric inhibitor would also interfere with cap binding, and thereby further disrupt protein synthesis. For example, 4EGI-1 is an allosteric modulator that enhances 4E-BP–eIF4E binding while also preventing eIF4E binding to eIF4G through interaction with a specific hydrophobic pocket of eIF4E. Furthermore, when 4E-BP is phosphorylated, 4EGI-1(101) replaces 4E-BP and hinders eIF4E–eIF4D interaction. There is also an allosteric dislocation of eIF4G from eIF4E and without this binding, eIF4G is no longer able to compete with 4E-BP, leading to an overall increase in 4E-BP binding to eIF4E. As a follow-up to the work on 4EGI-1 (101) the effect of structural rigidification was explored to increase specificity, potency, and pharmacokinetics via conformational constraints.130 To better understand the effect of the essential pharmacophore in 4E-BP, Mahalingam et al. carried out SAR studies by using (E/Z)-4EGI-1 (101) as the starting chemotype (Fig. 25).128,130 In many systems, it is known that structural rigidification results in higher activity, bioavailability and stability.131–134 Based on these studies, the authors sought to rigidify 101 by joining position 1 of the 3,4-dichlorophenyl ring and the 5-position of thiazolidine. The formation of a rigid scaffold with ethylene, methylene oxide, methylenesulfide, methylenesulfoxide, and methylene sulfone connectors were examined. Overall, rigidification improved potencies of the compounds. The presence of the ethylene bridge displayed an increase in binding by 1.5–2-fold compared to the unrigid parent. The presence of the oxymethylene linkage further increased binding affinity 3-fold. However, the bridges containing thiomethylene, methylenesulfoxide or methylenesulfone did not contribute to binding affinity enhancement. Overall, rigidification with a methylenesulfide bridge (102) improved potencies by 3-fold compared to the parent inhibitor (15.5 ± 0.70 μM for 102vs. 43.5 ± 1.52 μM for 101). It was confirmed that this compound disrupts the eIF4E/eIF4G complex effectively and increases binding of 4E-BP1 to eIF4E. Cell proliferation inhibition of human cancer cell lines CRL-2813 (melanoma) and CRL-2351 (breast) were examined. It was found that compound 102 inhibited proliferation of CRL-2813 cells and exhibited a cellular IC50 of 5.1 ± 0.1 μM. The authors have suggested that 102 is likely to be interacting with eIF4E through an allosteric mechanism and further studies are under investigation to optimize the rigidification modes to introduce more conformational and configurational constraints to confirm allosteric mechanism of action.
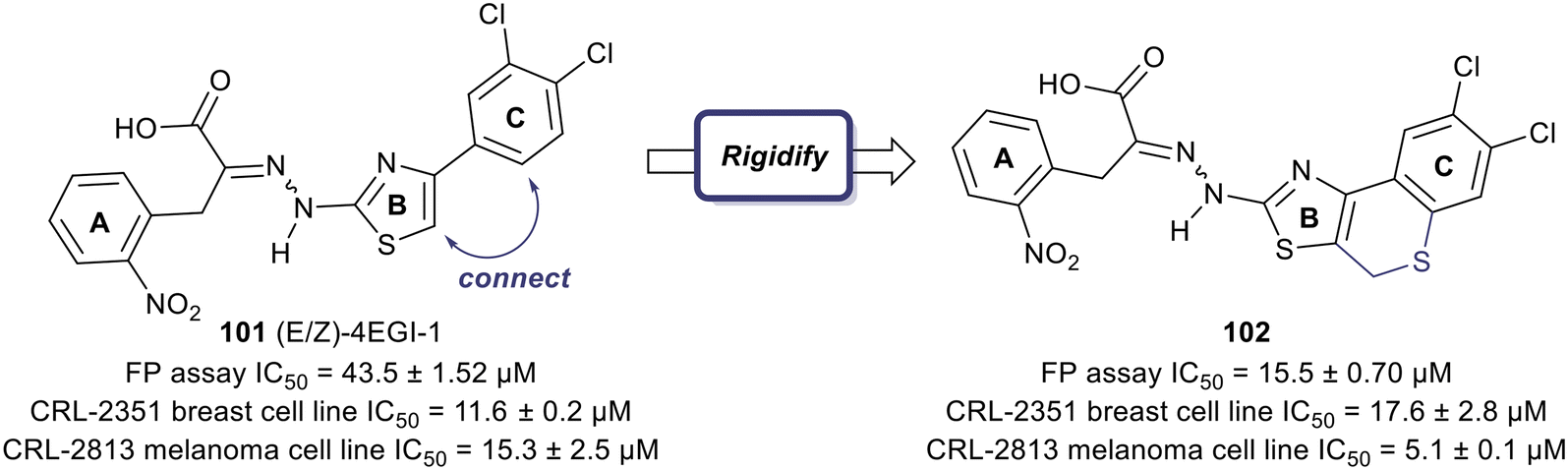 | ||
| Fig. 25 Structural rigidification of 101. | ||
3.3. eIF4E degradation approaches
4EGI-1 (101) is the first small molecule inhibitor of eIF4E that was reported in 2007 with a potency of 57 μM by Moerke et al.128 The X-ray crystal structure of eIF4E with inhibitor 4EGI-1 (101) was later reported in 2014.135 In 2021, Fischer et al., in an effort to improve the properties of 4EGI-1 (101), discovered i4EG-BiP (103) and solved the X-ray crystal structure of 103 bound to eIF4E (Fig. 26).136 Interestingly, even though 4E-BP1 and eIF4G contain the same binding motif, 4EGI-1 (101) and i4EG-BiP (103) can only displace eIF4G and not 4E-BP1 in cells. It was proposed that this effect was caused by the inhibitors displacing helices in eIF4E and eIF4G which in turn created a steric between the sidechain of Ser82 in eIF4E and Leu641 in eIF4G. In contrast, with 4E-BP1, this clash was not produced due to the stabilization of a hydrogen bond between the backbone of Ser82 in eIF4E and the backbone of Ser83 in 4E-BP1. By leveraging these structural insights obtained from the cocrystal structures of inhibitors 4EGI-1 (101) and i4EG-BiP (103) with eIF4E, eIF4E degraders were able to be designed (Fig. 27). The initial set of degraders were designed using 4EGI-1 (101), by first surveying for an ideal location to incorporate flexible linkers attached to an E3-ligase recruiting ligand, thalidomide. Initially, the carboxylic acid unit was found to be a suitable position for the connection of an exit vector; however, modeling revealed that the acid formed a critical salt bridge with eIF4E Lys49. As a result, the solvent exposed thiazole ring was chosen as the point of attachment in which various alkyl-linker motifs were used to connect the eIF4E inhibitor to the E3-ligase ligand to provide the first proteolysis targeting chimeras (PROTACs) of eIF4E (Fig. 27). PROTAC 104 was confirmed to engage with eIF4G using a fluorescence polarization assay, however, it was unable to engage with cereblon (CRBN), the E3-ligase required for protein degradation, in cellular studies. Based on previous reports on poor cellular permeability of negatively charged cap analogues,137 failure of 105 to engage with CRBN was attributed to its net negative charge. To negate this effect, the carboxylic acid was converted to a pivaloyloxymethyl ester (105). Cellular permeability was increased by removing the 2-nitrophenyl group from parental inhibitor 4EGI-1 (101) to provide 106, as well as the free acid derivative of 106 (107) which was used as a control. When the activity of the compounds was tested, the protected acids were able to engage with CRBN, supporting the hypothesis that a net negative charge adversely affects activity. To expand on the degrader library, i4EG-BiP (103) was used as a scaffold for synthesizing new eIF4E degraders. For i4EG-BiP (103), the carboxylic acid did not form the same interaction with the protein as the acid group in 4EGI-1 (101) did. Furthermore, modeling suggested that the nitrophenyl in i4EG-BiP (103) plays a minor role in binding and could be eliminated from the PROTAC structure without hindering the ligand–protein interactions. Therefore, in some of the i4EG-BiP-type PROTAC designs, this nitrophenyl motif was eliminated. Improvement in CRBN engagement was also targeted by substituting the ether connectivity to thalidomide present in the 4EGI-1-type PROTACs with an amine connection. Thus, four PROTACs were made with these changes to provide 108–111. Unfortunately, with these changes, degradation was not observed even though engagement with the CRBN was present. This was postulated to be attributed to the relatively low binding affinity of the parent compounds 4EGI-1 (101) and i4EG-BiP (103). PROTACs 108 and 109 were tested for engagement of the E3-ligase (CRBN) in cells with 108 displaying an IC50 value of 1.6 μM, and 109 possessing significantly less potent activity with an IC50 value of 13 μM. This correlation was likely due to the detrimental effect of the 2-nitrophenyl group on cell permeability. Interestingly, PROTACs 108–111 exhibited cytotoxicity at 10 μM, killing over 50% of HeLa cells. Although a successful degrader was not obtained, there is room for improvement by using medicinal chemistry approaches to design new analogs of the inhibitor and increase the affinity of the compounds to engage with eIF4E.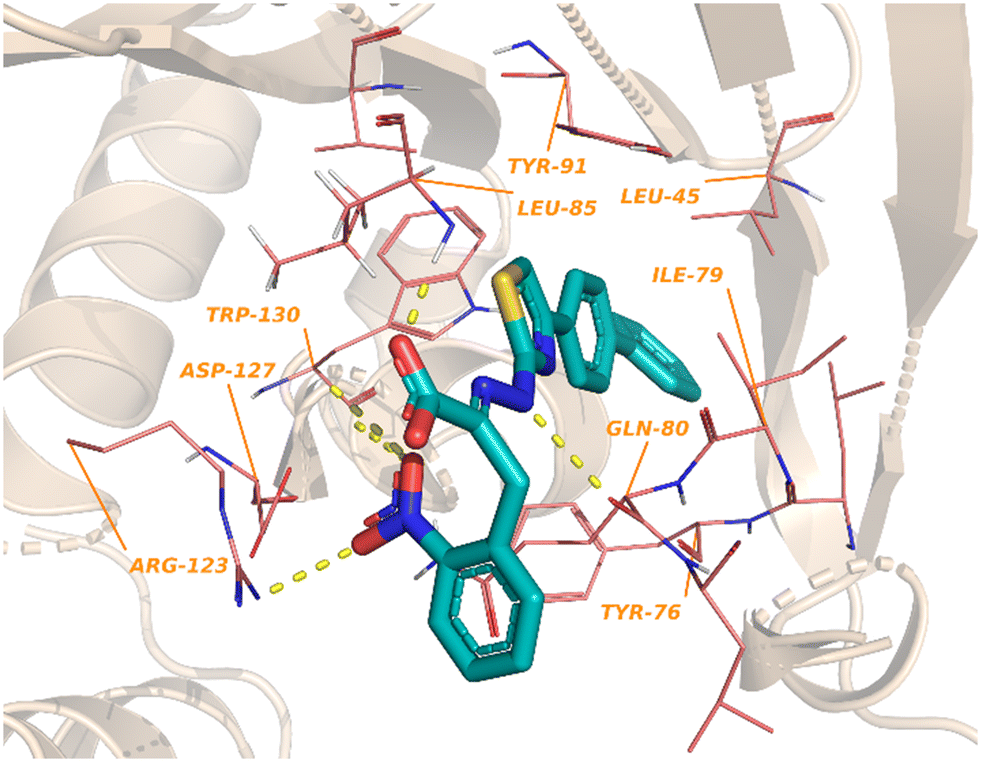 | ||
| Fig. 26 X-ray crystal structure of eIF4E with inhibitor i4EG-BiP (103) showing key interactions and residues (PDB:4TPW). | ||
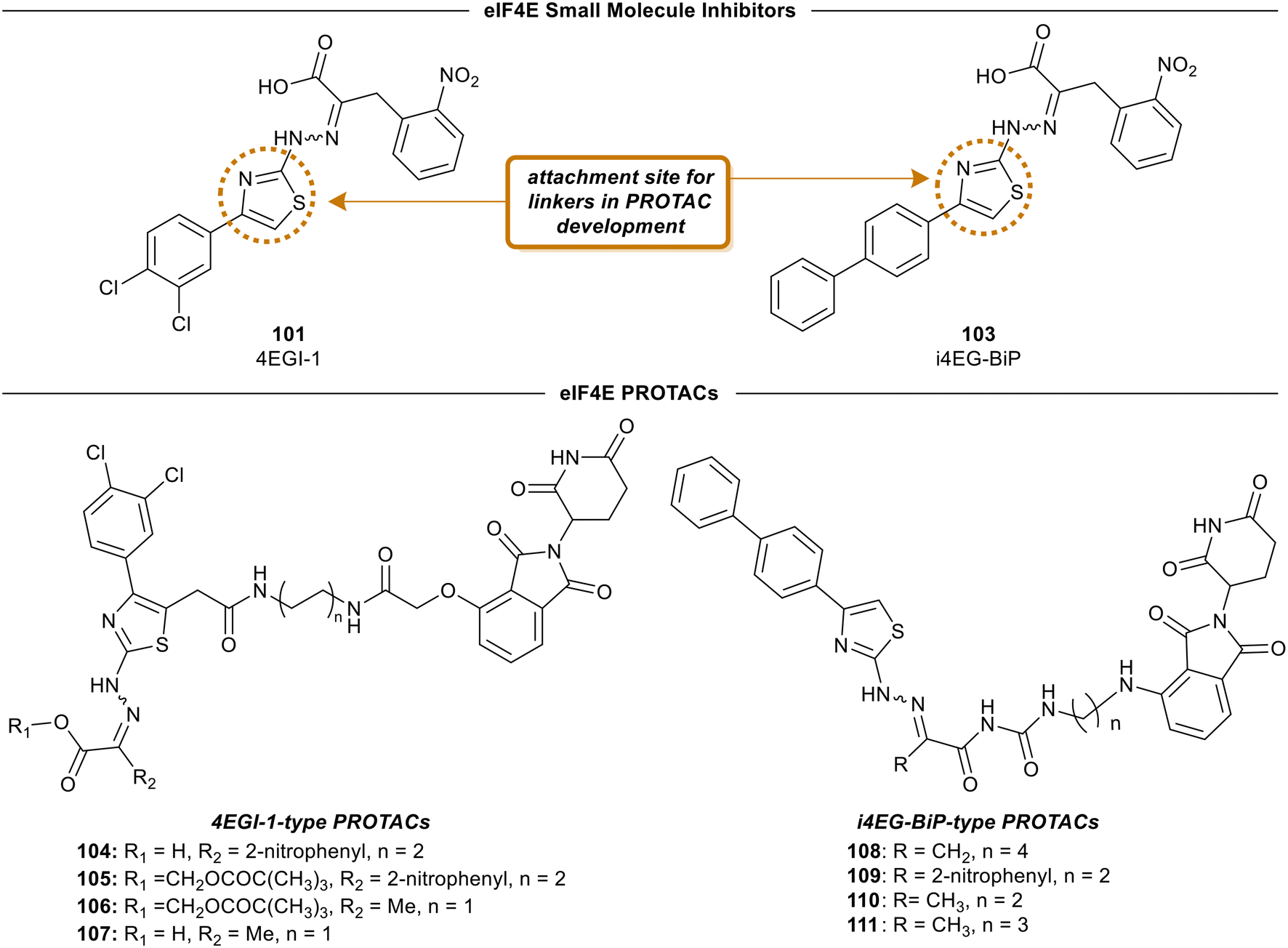 | ||
| Fig. 27 Representative structures of eIF4E inhibitors and eIF4E PROTACs. | ||
4. Conclusion
The MNK–eIF4E axis has been the subject of increased attention in the recent years due its role in the development and progression of various diseases. New advances in various fields such as single-cell sequencing will provide new insights into the activity of MNK–eIF4E and opportunities for the rational design of new drug candidates to modulate activity. Recent medicinal chemistry approaches have led to MNK- and eIF4E-directed small molecules that have entered clinical trials. These molecules bind to MNK by competing with ATP to decrease phosphorylation on effector proteins. Although several candidates have entered clinical trials, none have been approved so far due to their lack of efficacy.138–141 One potential explanation for the weak efficacy observed is that even though many of these compounds potently inhibit phosphorylation of eIF4E, levels of eIF4E protein itself remain unchanged. It is therefore possible that MNK may have other tumorigenic substrates, and/or the non-enzymatic activity of MNK contributes to tumor growth. Similarly, other non-enzymatic protein–protein interactions may be activated by MNK that could modulate different cell-signaling pathways to initiate tumorigenesis.As an alternative mechanism to inhibit MNK–eIF4E function, allosteric targeting has been proposed; however, molecules that target the allosteric domain of MNK have not been developed to-date. Targeting a non-catalytic domain in MNK would be advantageous to facilitate high potency and kinase selectivity for reduced side effects. A drawback, however, with MNK inhibitors is that it only affects the levels of phosphorylated eIF4E. Endogenous levels of eIF4E are not affected, and an overexpression of eIF4E is also known to induce oncogenesis.
The use of new modalities such as molecular glues and degraders capable of recruiting E3 ligases to regulate the function of the MNK–eIF4E axis is also attractive since non-catalytic functions and paradoxical activation of the target can be prevented. In addition to this, prolonged pharmacodynamic effects can be expected with the use of these strategies. The discovery of CNS-specific E3 ligases can also be useful for the development of targeted degradation molecules suitable for the treatment of CNS diseases such as Alzheimer's and glioblastoma.
Author contributions
Gary Schiltz conceived and designed the review article, generated the molecular modeling figures, revised the general pathway figures, and reviewed and edited the entire manuscript. Ann Fernandez conducted literature review, drafted chemistry and schematic figures, and wrote the main drafts of most of the manuscript. Paige Monsen reviewed literature, drafted several sections and figures, and reviewed the manuscript. Leonidas Platanias provided input on the design of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Research reported in this publication was supported by the National Institute Of Neurological Disorders And Stroke of the National Institutes of Health under Award Number R01NS113425. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- R. Baghban, L. Roshangar, R. Jahanban-Esfahlan, K. Seidi, A. Ebrahimi-Kalan, M. Jaymand, S. Kolahian, T. Javaheri and P. Zare, Cell Commun. Signaling, 2020, 18, 59 CrossRef PubMed.
- R. Jinka, R. Kapoor, P. G. Sistla, T. A. Raj and G. Pande, Int. J. Cell Biol., 2012, 2012, 219196 CrossRef PubMed.
- J. C. Lee, J. T. Laydon, P. C. McDonnell, T. F. Gallagher, S. Kumar, D. Green, D. McNulty, M. J. Blumenthal, J. R. Heys, S. W. Landvatter, J. E. Strickler, M. M. McLaughlin, I. R. Siemens, S. M. Fisher, G. P. Livi, J. R. White, J. L. Adams and P. R. Young, Nature, 1994, 372, 739–746 CrossRef CAS PubMed.
- R. A. Cardone, V. Casavola and S. J. Reshkin, Nat. Rev. Cancer, 2005, 5, 786–795 CrossRef CAS PubMed.
- G. Charras and E. Sahai, Nat. Rev. Mol. Cell Biol., 2014, 15, 813–824 CrossRef CAS PubMed.
- B. Gao and P. P. Roux, Biochim. Biophys. Acta, 2015, 1849, 753–765 CrossRef CAS PubMed.
- P. P. Roux and I. Topisirovic, Cold Spring Harbor Perspect. Biol., 2012, 4, 1–23 Search PubMed.
- C. A. Rubio, B. Weisburd, M. Holderfield, C. Arias, E. Fang, J. L. DeRisi and A. Fanidi, Genome Biol., 2014, 15, 476 CrossRef PubMed.
- P. Song, F. Yang, H. Jin and X. Wang, Signal Transduction Targeted Ther., 2021, 6, 68 CrossRef CAS PubMed.
- M. Carroll and K. L. Borden, J. Interferon Cytokine Res., 2013, 33, 227–238 CrossRef CAS PubMed.
- C. Gong, H. Tsoi, K. C. Mok, J. Cheung, E. P. S. Man, K. Fujino, A. Wong, E. W. F. Lam and U. S. Khoo, Oncogene, 2020, 39, 3206–3217 CrossRef CAS PubMed.
- A. C. Hsieh and D. Ruggero, Clin. Cancer Res., 2010, 16, 4914–4920 CrossRef CAS PubMed.
- S. Manier, D. Huynh, Y. J. Shen, J. Zhou, T. Yusufzai, K. Z. Salem, R. Y. Ebright, J. Shi, J. Park, S. V. Glavey, W. G. Devine, C. J. Liu, X. Leleu, B. Quesnel, C. Roche-Lestienne, J. K. Snyder, L. E. Brown, N. Gray, J. Bradner, L. Whitesell, J. A. Porco Jr. and I. M. Ghobrial, Sci. Transl. Med., 2017, 9, eaal2668 CrossRef PubMed.
- S. Diab, M. Kumarasiri, M. Yu, T. Teo, C. Proud, R. Milne and S. Wang, Chem. Biol., 2014, 21, 441–452 CrossRef CAS PubMed.
- S. Joshi and L. C. Platanias, World J. Biol. Chem., 2014, 5, 321–333 CrossRef PubMed.
- M. Shveygert, C. Kaiser, S. S. Bradrick and M. Gromeier, Mol. Cell. Biol., 2010, 30, 5160–5167 CrossRef CAS PubMed.
- T. Ueda, M. Sasaki, A. J. Elia, I. I. Chio, K. Hamada, R. Fukunaga and T. W. Mak, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13984–13990 CrossRef CAS PubMed.
- H. Abramczyk, A. Imiela, B. Brozek-Pluska, M. Kopec, J. Surmacki and A. Sliwinska, Cancers, 2019, 11 DOI:10.3390/cancers11122017.
- P. He, A. Bian, Y. Miao, W. Jin, H. Chen, J. He, L. Li, Y. Sun, J. Ye, M. Liu, Z. Yi, W. Zhou and Y. Chen, J. Med. Chem., 2022, 65, 15487–15511 CrossRef CAS PubMed.
- Q. Zhang, Z. Fan, L. Zhang, Q. You and L. Wang, J. Med. Chem., 2021, 64, 8916–8938 CrossRef CAS PubMed.
- S. Karaki, C. Andrieu, H. Ziouziou and P. Rocchi, Adv. Protein Chem. Struct. Biol., 2015, 101, 1–26 CAS.
- S. J. Morley and S. Naegele, J. Biol. Chem., 2002, 277, 32855–32859 CrossRef CAS PubMed.
- A. J. Waskiewicz, A. Flynn, C. G. Proud and J. A. Cooper, EMBO J., 1997, 16, 1909–1920 CrossRef CAS PubMed.
- C. G. Proud, Biochim. Biophys. Acta, 2015, 1849, 766–773 CrossRef CAS.
- S. Goto, Z. Yao and C. G. Proud, Biochem. J., 2009, 423, 279–290 CrossRef CAS PubMed.
- G. C. Scheper, J. L. Parra, M. Wilson, B. Van Kollenburg, A. C. Vertegaal, Z. G. Han and C. G. Proud, Mol. Cell. Biol., 2003, 23, 5692–5705 CrossRef CAS PubMed.
- A. O'Loghlen, V. M. Gonzalez, D. Pineiro, M. I. Perez-Morgado, M. Salinas and M. E. Martin, Exp. Cell Res., 2004, 299, 343–355 CrossRef PubMed.
- K. Slentz-Kesler, J. T. Moore, M. Lombard, J. Zhang, R. Hollingsworth and M. P. Weiner, Genomics, 2000, 69, 63–71 CrossRef CAS PubMed.
- M. Buxadé, J. L. Parra, S. Rousseau, N. Shpiro, R. Marquez, N. Morrice, J. Bain, E. Espel and C. G. Proud, Immunity, 2005, 23, 177–189 CrossRef.
- R. Fukunaga and T. Hunter, EMBO J., 1997, 16, 1921–1933 CrossRef CAS PubMed.
- I. N. Foltz, J. C. Lee, P. R. Young and J. W. Schrader, J. Biol. Chem., 1997, 272, 3296–3301 CrossRef CAS PubMed.
- N. W. Freshney, L. Rawlinson, F. Guesdon, E. Jones, S. Cowley, J. Hsuan and J. Saklatvala, Cell, 1994, 78, 1039–1049 CrossRef CAS PubMed.
- J. Han, J. D. Lee, L. Bibbs and R. J. Ulevitch, Science, 1994, 265, 808–811 CrossRef CAS PubMed.
- M. R. Junttila, S. P. Li and J. Westermarck, FASEB J., 2008, 22, 954–965 CrossRef CAS PubMed.
- A. Pietersma, B. C. Tilly, M. Gaestel, N. de Jong, J. C. Lee, J. F. Koster and W. Sluiter, Biochem. Biophys. Res. Commun., 1997, 230, 44–48 CrossRef CAS PubMed.
- J. Raingeaud, S. Gupta, J. S. Rogers, M. Dickens, J. Han, R. J. Ulevitch and R. J. Davis, J. Biol. Chem., 1995, 270, 7420–7426 CrossRef CAS PubMed.
- J. Rouse, P. Cohen, S. Trigon, M. Morange, A. Alonso-Llamazares, D. Zamanillo, T. Hunt and A. R. Nebreda, Cell, 1994, 78, 1027–1037 CrossRef CAS PubMed.
- T. Shalom-Barak, J. Quach and M. Lotz, J. Biol. Chem., 1998, 273, 27467–27473 CrossRef CAS PubMed.
- L. Shapiro, A. J. Puren, H. A. Barton, D. Novick, R. L. Peskind, R. Shenkar, Y. Gu, M. S. Su and C. A. Dinarello, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12550–12555 CrossRef CAS PubMed.
- X. S. Wang, K. Diener, C. L. Manthey, S. Wang, B. Rosenzweig, J. Bray, J. Delaney, C. N. Cole, P. Y. Chan-Hui, N. Mantlo, H. S. Lichenstein, M. Zukowski and Z. Yao, J. Biol. Chem., 1997, 272, 23668–23674 CrossRef CAS PubMed.
- M. M. McKay and D. K. Morrison, Oncogene, 2007, 26, 3113–3121 CrossRef CAS PubMed.
- E. Rozengurt, J. Cell. Physiol., 2007, 213, 589–602 CrossRef CAS PubMed.
- R. L. Stead and C. G. Proud, FEBS Lett., 2013, 587, 2623–2628 CrossRef CAS PubMed.
- R. Jauch, M. K. Cho, S. Jakel, C. Netter, K. Schreiter, B. Aicher, M. Zweckstetter, H. Jackle and M. C. Wahl, EMBO J., 2006, 25, 4020–4032 CrossRef CAS PubMed.
- J. Kwiatkowski, B. Liu, S. Pang, N. H. B. Ahmad, G. Wang, A. Poulsen, H. Yang, Y. R. Poh, D. H. Y. Tee, E. Ong, P. Retna, N. Dinie, P. Kwek, J. L. K. Wee, V. Manoharan, C. B. Low, P. G. Seah, V. Pendharkar, K. Sangthongpitag, J. Joy, N. Baburajendran, A. E. Jansson, K. Nacro, J. Hill, T. H. Keller and A. W. Hung, J. Med. Chem., 2020, 63, 621–637 CrossRef CAS PubMed.
- R. Jauch, S. Jakel, C. Netter, K. Schreiter, B. Aicher, H. Jackle and M. C. Wahl, Structure, 2005, 13, 1559–1568 CrossRef CAS PubMed.
- Y. Matsui, I. Yasumatsu, K. I. Yoshida, S. Iimura, Y. Ikeno, T. Nawano, H. Fukano, O. Ubukata, H. Hanzawa, F. Tanzawa and T. Isoyama, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2018, 74, 156–160 CrossRef CAS PubMed.
- S. H. Reich, P. A. Sprengeler, G. G. Chiang, J. R. Appleman, J. Chen, J. Clarine, B. Eam, J. T. Ernst, Q. Han, V. K. Goel, E. Z. R. Han, V. Huang, I. N. J. Hung, A. Jemison, K. A. Jessen, J. Molter, D. Murphy, M. Neal, G. S. Parker, M. Shaghafi, S. Sperry, J. Staunton, C. R. Stumpf, P. A. Thompson, C. Tran, S. E. Webber, C. J. Wegerski, H. Zheng and K. R. Webster, J. Med. Chem., 2018, 61, 3516–3540 CrossRef CAS PubMed.
- R. Duncan, S. C. Milburn and J. W. Hershey, J. Biol. Chem., 1987, 262, 380–388 CrossRef CAS.
- A. C. Gingras, B. Raught and N. Sonenberg, Annu. Rev. Biochem., 1999, 68, 913–963 CrossRef CAS PubMed.
- M. Barna, A. Pusic, O. Zollo, M. Costa, N. Kondrashov, E. Rego, P. H. Rao and D. Ruggero, Nature, 2008, 456, 971–975 CrossRef CAS PubMed.
- R. A. Saxton and D. M. Sabatini, Cell, 2017, 169, 361–371 CrossRef CAS PubMed.
- A. Batool, S. T. Majeed, S. Aashaq, R. Majeed, N. N. Bhat and K. I. Andrabi, Mol. Cell. Biochem., 2020, 465, 13–26 CrossRef CAS PubMed.
- A. Batool, S. T. Majeed, S. Aashaq, R. Majeed, G. Shah, N. Nazir and K. I. Andrabi, Int. J. Biol. Macromol., 2019, 125, 651–659 CrossRef CAS PubMed.
- A. Pause, G. J. Belsham, A. C. Gingras, O. Donze, T. A. Lin, J. C. Lawrence Jr. and N. Sonenberg, Nature, 1994, 371, 762–767 CrossRef CAS PubMed.
- C. N. Chen, F. J. Hsieh, Y. M. Cheng, P. H. Lee and K. J. Chang, J. Surg. Oncol., 2004, 86, 22–27 CrossRef CAS PubMed.
- L. Furic, L. Rong, O. Larsson, I. H. Koumakpayi, K. Yoshida, A. Brueschke, E. Petroulakis, N. Robichaud, M. Pollak, L. A. Gaboury, P. P. Pandolfi, F. Saad and N. Sonenberg, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14134–14139 CrossRef CAS PubMed.
- A. De Benedetti and A. L. Harris, Int. J. Biochem. Cell Biol., 1999, 31, 59–72 CrossRef CAS PubMed.
- V. L. Greenberg and S. G. Zimmer, Oncogene, 2005, 24, 4851–4860 CrossRef CAS PubMed.
- J. W. Hershey, Annu. Rev. Biochem., 1991, 60, 717–755 CrossRef CAS PubMed.
- A. Lazaris-Karatzas, K. S. Montine and N. Sonenberg, Nature, 1990, 345, 544–547 CrossRef CAS PubMed.
- H. G. Wendel, E. De Stanchina, J. S. Fridman, A. Malina, S. Ray, S. Kogan, C. Cordon-Cardo, J. Pelletier and S. W. Lowe, Nature, 2004, 428, 332–337 CrossRef CAS PubMed.
- D. Rousseau, A. C. Gingras, A. Pause and N. Sonenberg, Oncogene, 1996, 13, 2415–2420 CAS.
- V. A. Polunovsky, A. C. Gingras, N. Sonenberg, M. Peterson, A. Tan, J. B. Rubins, J. C. Manivel and P. B. Bitterman, J. Biol. Chem., 2000, 275, 24776–24780 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- A. Tan, P. Bitterman, N. Sonenberg, M. Peterson and V. Polunovsky, Oncogene, 2000, 19, 1437–1447 CrossRef CAS PubMed.
- S. Li, T. Takasu, D. M. Perlman, M. S. Peterson, D. Burrichter, S. Avdulov, P. B. Bitterman and V. A. Polunovsky, J. Biol. Chem., 2003, 278, 3015–3022 CrossRef CAS PubMed.
- S. Fan, S. S. Ramalingam, J. Kauh, Z. Xu, F. R. Khuri and S. Y. Sun, Cancer Biol. Ther., 2009, 8, 1463–1469 CrossRef PubMed.
- T. Shukla, J. B. de la Pena, J. M. Perish, J. E. Ploski, C. R. Stumpf, K. R. Webster, C. A. Thorn and Z. T. Campbell, Neurotherapeutics, 2021, 18, 624–639 CrossRef CAS PubMed.
- A. Ghosh, K. Mizuno, S. S. Tiwari, P. Proitsi, B. Gomez Perez-Nievas, E. Glennon, R. T. Martinez-Nunez and K. P. Giese, Transl. Psychiatry, 2020, 10, 192 CrossRef CAS PubMed.
- P. Martin, V. Wagh, S. A. Reis, S. Erdin, R. L. Beauchamp, G. Shaikh, M. Talkowski, E. Thiele, S. D. Sheridan, S. J. Haggarty and V. Ramesh, Mol. Autism, 2020, 11, 2 CrossRef CAS PubMed.
- C. G. Gkogkas, A. Khoutorsky, R. Cao, S. M. Jafarnejad, M. Prager-Khoutorsky, N. Giannakas, A. Kaminari, A. Fragkouli, K. Nader, T. J. Price, B. W. Konicek, J. R. Graff, A. K. Tzinia, J. C. Lacaille and N. Sonenberg, Cell Rep., 2014, 9, 1742–1755 CrossRef CAS PubMed.
- I. S. Amorim, S. Kedia, S. Kouloulia, K. Simbriger, I. Gantois, S. M. Jafarnejad, Y. Li, A. Kampaite, T. Pooters, N. Romano and C. G. Gkogkas, J. Neurosci., 2018, 38, 2118–2133 CrossRef CAS PubMed.
- I. Topisirovic, M. L. Guzman, M. J. McConnell, J. D. Licht, B. Culjkovic, S. J. Neering, C. T. Jordan and K. L. Borden, Mol. Cell. Biol., 2003, 23, 8992–9002 CrossRef CAS PubMed.
- I. Topisirovic, M. Ruiz-Gutierrez and K. L. Borden, Cancer Res., 2004, 64, 8639–8642 CrossRef CAS PubMed.
- D. Rousseau, R. Kaspar, I. Rosenwald, L. Gehrke and N. Sonenberg, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1065–1070 CrossRef CAS PubMed.
- N. Cohen, M. Sharma, A. Kentsis, J. M. Perez, S. Strudwick and K. L. Borden, EMBO J., 2001, 20, 4547–4559 CrossRef CAS.
- I. Topisirovic, B. Culjkovic, N. Cohen, J. M. Perez, L. Skrabanek and K. L. Borden, EMBO J., 2003, 22, 689–703 CrossRef CAS PubMed.
- A. Romagnoli, M. D'Agostino, C. Ardiccioni, C. Maracci, S. Motta, A. La Teana and D. Di Marino, Cell. Mol. Life Sci., 2021, 78, 6869–6885 CrossRef CAS PubMed.
- D. Ruggero, L. Montanaro, L. Ma, W. Xu, P. Londei, C. Cordon-Cardo and P. P. Pandolfi, Nat. Med., 2004, 10, 484–486 CrossRef CAS PubMed.
- J. B. Bell, F. Eckerdt, H. D. Dhruv, D. Finlay, S. Peng, S. Kim, B. Kroczynska, E. M. Beauchamp, K. Alley, J. Clymer, S. Goldman, S. Y. Cheng, C. D. James, I. Nakano, C. Horbinski, A. P. Mazar, K. Vuori, P. Kumthekar, J. Raizer, M. E. Berens and L. C. Platanias, Mol. Cancer Res., 2018, 16, 32–46 CrossRef CAS PubMed.
- B. W. Konicek, C. A. Dumstorf and J. R. Graff, Cell Cycle, 2008, 7, 2466–2471 CrossRef CAS PubMed.
- D. Silvera, S. C. Formenti and R. J. Schneider, Nat. Rev. Cancer, 2010, 10, 254–266 CrossRef CAS PubMed.
- V. Venturi, T. Masek and M. Pospisek, Physiol. Res., 2018, 67, 363–382 CAS.
- M. Buxadé, N. Morrice, D. L. Krebs and C. G. Proud, J. Biol. Chem., 2008, 283, 57–65 CrossRef PubMed.
- J. DaSilva, L. Xu, H. J. Kim, W. T. Miller and D. Bar-Sagi, Mol. Cell. Biol., 2006, 26, 1898–1907 CrossRef CAS PubMed.
- Y. Hefner, A. G. Borsch-Haubold, M. Murakami, J. I. Wilde, S. Pasquet, D. Schieltz, F. Ghomashchi, J. R. Yates, C. G. Armstrong, A. Paterson, P. Cohen, R. Fukunaga, T. Hunter, I. Kudo, S. P. Watson and M. H. Gelb, J. Biol. Chem., 2000, 275, 37542–37551 CrossRef CAS.
- H. Harizi, J. B. Corcuff and N. Gualde, Trends Mol. Med., 2008, 14, 461–469 CrossRef CAS PubMed.
- P. Yusoff, D. H. Lao, S. H. Ong, E. S. Wong, J. Lim, T. L. Lo, H. F. Leong, C. W. Fong and G. R. Guy, J. Biol. Chem., 2002, 277, 3195–3201 CrossRef CAS PubMed.
- J. E. Merrett, J. Xie, P. J. Psaltis and C. G. Proud, Biochem. J., 2020, 477, 2735–2754 CrossRef CAS PubMed.
- C. E. Moore, J. Pickford, F. R. Cagampang, R. L. Stead, S. Tian, X. Zhao, X. Tang, C. D. Byrne and C. G. Proud, Sci. Rep., 2016, 6, 23476 CrossRef CAS PubMed.
- H. Malka-Mahieu, M. Newman, L. Desaubry, C. Robert and S. Vagner, Clin. Cancer Res., 2017, 23, 21–25 CrossRef CAS PubMed.
- I. B. Rosenwald, J. J. Chen, S. Wang, L. Savas, I. M. London and J. Pullman, Oncogene, 1999, 18, 2507–2517 CrossRef CAS PubMed.
- Z. Salehi, F. Mashayekhi and F. Shahosseini, Cell Biol. Int., 2007, 31, 1400–1404 CrossRef CAS PubMed.
- R. Wang, J. Geng, J. H. Wang, X. Y. Chu, H. C. Geng and L. B. Chen, Lung Cancer, 2009, 66, 237–244 CrossRef PubMed.
- S. Wang, I. B. Rosenwald, M. J. Hutzler, G. A. Pihan, L. Savas, J. J. Chen and B. A. Woda, Am. J. Pathol., 1999, 155, 247–255 CrossRef CAS PubMed.
- S. Assouline, B. Culjkovic, E. Cocolakis, C. Rousseau, N. Beslu, A. Amri, S. Caplan, B. Leber, D. C. Roy, W. H. Miller Jr. and K. L. Borden, Blood, 2009, 114, 257–260 CrossRef CAS PubMed.
- C. Tschopp, U. Knauf, M. Brauchle, M. Zurini, P. Ramage, D. Glueck, L. New, J. Han and H. Gram, Mol. Cell Biol. Res. Commun., 2000, 3, 205–211 CrossRef CAS PubMed.
- R. K. Mishra, M. R. Clutter, G. T. Blyth, E. M. Kosciuczuk, A. Z. Blackburn, E. M. Beauchamp, G. E. Schiltz and L. C. Platanias, Chem. Biol. Drug Des., 2019, 94, 1813–1823 CrossRef CAS PubMed.
- C. Fufezan, Proteins, 2010, 78, 2831–2838 CrossRef CAS PubMed.
- M. J. Wheater, P. W. Johnson and J. P. Blaydes, Cancer Biol. Ther., 2010, 10, 728–735 CrossRef CAS PubMed.
- H. Yang, L. R. Chennamaneni, M. W. T. Ho, S. H. Ang, E. S. W. Tan, D. A. Jeyaraj, Y. S. Yeap, B. Liu, E. H. Ong, J. K. Joy, J. L. K. Wee, P. Kwek, P. Retna, N. Dinie, T. T. H. Nguyen, S. J. Tai, V. Manoharan, V. Pendharkar, C. B. Low, Y. S. Chew, S. Vuddagiri, K. Sangthongpitag, M. L. Choong, M. A. Lee, S. Kannan, C. S. Verma, A. Poulsen, S. Lim, C. Chuah, T. S. Ong, J. Hill, A. Matter and K. Nacro, J. Med. Chem., 2018, 61, 4348–4369 CrossRef CAS PubMed.
- S. Lim, T. Y. Saw, M. Zhang, M. R. Janes, K. Nacro, J. Hill, A. Q. Lim, C. T. Chang, D. A. Fruman, D. A. Rizzieri, S. Y. Tan, H. Fan, C. T. Chuah and S. T. Ong, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2298–E2307 CAS.
- E. Y. Cai, M. N. Kufeld, S. Schuster, S. Arora, M. Larkin, A. A. Germanos, A. C. Hsieh and S. Beronja, Cell Stem Cell, 2020, 27, 270–283 e277 CrossRef CAS PubMed.
- J. Oyarzabal, N. Zarich, M. I. Albarran, I. Palacios, M. Urbano-Cuadrado, G. Mateos, I. Reymundo, O. Rabal, A. Salgado, A. Corrionero, J. Fominaya, J. Pastor and J. R. Bischoff, J. Med. Chem., 2010, 53, 6618–6628 CrossRef CAS PubMed.
- J. Cherian, K. Nacro, Z. Y. Poh, S. Guo, D. A. Jeyaraj, Y. X. Wong, M. Ho, H. Y. Yang, J. K. Joy, Z. P. Kwek, B. Liu, J. L. Wee, E. H. Ong, M. L. Choong, A. Poulsen, M. A. Lee, V. Pendharkar, L. J. Ding, V. Manoharan, Y. S. Chew, K. Sangthongpitag, S. Lim, S. T. Ong, J. Hill and T. H. Keller, J. Med. Chem., 2016, 59, 3063–3078 CrossRef CAS PubMed.
- J. H. Schatz and H. G. Wendel, Cell Cycle, 2011, 10, 3830–3833 CrossRef CAS PubMed.
- Y. Han, H. Zhang, S. Wang, B. Li, K. Xing, Y. Shi, H. Cao, J. Zhang, T. Tong, J. Zang, L. Guan, X. Gao, Y. Wang, D. Liu, M. Huang, Y. Jing and L. Zhao, J. Med. Chem., 2021, 64, 13719–13735 CrossRef CAS PubMed.
- W. Han, Y. Ding, Y. Xu, K. Pfister, S. Zhu, B. Warne, M. Doyle, M. Aikawa, P. Amiri, B. Appleton, D. D. Stuart, A. Fanidi and C. M. Shafer, J. Med. Chem., 2016, 59, 3034–3045 CrossRef CAS PubMed.
- O. Hantschel, ACS Chem. Biol., 2015, 10, 234–245 CrossRef CAS PubMed.
- S. V. Frye and G. L. Johnson, Nat. Chem. Biol., 2009, 5, 448–449 CrossRef CAS PubMed.
- S. J. Cook, J. A. Tucker and P. A. Lochhead, Biochem. Soc. Trans., 2020, 48, 1859–1875 CrossRef CAS PubMed.
- E. Bou-Petit, S. Hummer, H. Alarcon, K. Slobodnyuk, M. Cano-Galietero, P. Fuentes, P. J. Guijarro, M. J. Munoz, L. Suarez-Cabrera, A. Santamaria, R. Estrada-Tejedor, J. I. Borrell and Y. C. S. Ramon, J. Med. Chem., 2022, 65, 6070–6087 CrossRef CAS PubMed.
- J. R. Graff, B. W. Konicek, T. M. Vincent, R. L. Lynch, D. Monteith, S. N. Weir, P. Schwier, A. Capen, R. L. Goode, M. S. Dowless, Y. Chen, H. Zhang, S. Sissons, K. Cox, A. M. McNulty, S. H. Parsons, T. Wang, L. Sams, S. Geeganage, L. E. Douglass, B. L. Neubauer, N. M. Dean, K. Blanchard, J. Shou, L. F. Stancato, J. H. Carter and E. G. Marcusson, J. Clin. Invest., 2007, 117, 2638–2648 CrossRef CAS PubMed.
- D. S. Hong, R. Kurzrock, Y. Oh, J. Wheler, A. Naing, L. Brail, S. Callies, V. Andre, S. K. Kadam, A. Nasir, T. R. Holzer, F. Meric-Bernstam, M. Fishman and G. Simon, Clin. Cancer Res., 2011, 17, 6582–6591 CrossRef CAS PubMed.
- D. M. Lucas, R. B. Edwards, G. Lozanski, D. A. West, J. D. Shin, M. A. Vargo, M. E. Davis, D. M. Rozewski, A. J. Johnson, B. N. Su, V. M. Goettl, N. A. Heerema, T. S. Lin, A. Lehman, X. Zhang, D. Jarjoura, D. J. Newman, J. C. Byrd, A. D. Kinghorn and M. R. Grever, Blood, 2009, 113, 4656–4666 CrossRef CAS PubMed.
- R. Cencic, F. Robert, G. Galicia-Vazquez, A. Malina, K. Ravindar, R. Somaiah, P. Pierre, J. Tanaka, P. Deslongchamps and J. Pelletier, Blood Cancer J., 2013, 3, e128 CrossRef CAS PubMed.
- M. E. Bordeleau, R. Cencic, L. Lindqvist, M. Oberer, P. Northcote, G. Wagner and J. Pelletier, Chem. Biol., 2006, 13, 1287–1295 CrossRef CAS PubMed.
- R. Cencic, M. Carrier, G. Galicia-Vazquez, M. E. Bordeleau, R. Sukarieh, A. Bourdeau, B. Brem, J. G. Teodoro, H. Greger, M. L. Tremblay, J. A. Porco Jr. and J. Pelletier, PLoS One, 2009, 4, e5223 CrossRef PubMed.
- M. E. Bordeleau, F. Robert, B. Gerard, L. Lindqvist, S. M. Chen, H. G. Wendel, B. Brem, H. Greger, S. W. Lowe, J. A. Porco Jr. and J. Pelletier, J. Clin. Invest., 2008, 118, 2651–2660 CAS.
- W. K. Low, Y. Dang, T. Schneider-Poetsch, Z. Shi, N. S. Choi, W. C. Merrick, D. Romo and J. O. Liu, Mol. Cell, 2005, 20, 709–722 CrossRef CAS PubMed.
- L. Pan, U. M. Acuna, J. Li, N. Jena, T. N. Ninh, C. M. Pannell, H. Chai, J. R. Fuchs, E. J. Carcache de Blanco, D. D. Soejarto and A. D. Kinghorn, J. Nat. Prod., 2013, 76, 394–404 CrossRef CAS PubMed.
- L. Pan, J. L. Woodard, D. M. Lucas, J. R. Fuchs and A. D. Kinghorn, Nat. Prod. Rep., 2014, 31, 924–939 RSC.
- S. Uttam, C. Wong, T. J. Price and A. Khoutorsky, Front. Genet., 2018, 9, 470 CrossRef CAS PubMed.
- C. J. Zerio, T. A. Cunningham, A. S. Tulino, E. A. Alimusa, T. M. Buckley, K. T. Moore, M. Dodson, N. C. Wilson, A. J. Ambrose, T. Shi, J. Sivinski, D. J. Essegian, D. D. Zhang, S. C. Schurer, J. H. Schatz and E. Chapman, J. Med. Chem., 2021, 64, 15727–15746 CrossRef CAS PubMed.
- J. T. Ernst, P. A. Thompson, C. Nilewski, P. A. Sprengeler, S. Sperry, G. Packard, T. Michels, A. Xiang, C. Tran, C. J. Wegerski, B. Eam, N. P. Young, S. Fish, J. Chen, H. Howard, J. Staunton, J. Molter, J. Clarine, A. Nevarez, G. G. Chiang, J. R. Appleman, K. R. Webster and S. H. Reich, J. Med. Chem., 2020, 63, 5879–5955 CrossRef CAS PubMed.
- A. Kentsis, I. Topisirovic, B. Culjkovic, L. Shao and K. L. Borden, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 18105–18110 CrossRef CAS PubMed.
- N. J. Moerke, H. Aktas, H. Chen, S. Cantel, M. Y. Reibarkh, A. Fahmy, J. D. Gross, A. Degterev, J. Yuan, M. Chorev, J. A. Halperin and G. Wagner, Cell, 2007, 128, 257–267 CrossRef CAS PubMed.
- R. Cencic, D. R. Hall, F. Robert, Y. Du, J. Min, L. Li, M. Qui, I. Lewis, S. Kurtkaya, R. Dingledine, H. Fu, D. Kozakov, S. Vajda and J. Pelletier, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1046–1051 CrossRef CAS PubMed.
- P. Mahalingam, K. Takrouri, T. Chen, R. Sahoo, E. Papadopoulos, L. Chen, G. Wagner, B. H. Aktas, J. A. Halperin and M. Chorev, J. Med. Chem., 2014, 57, 5094–5111 CrossRef CAS PubMed.
- S. Lucas, M. Negri, R. Heim, C. Zimmer and R. W. Hartmann, J. Med. Chem., 2011, 54, 2307–2319 CrossRef CAS PubMed.
- Q. Hu, M. Negri, K. Jahn-Hoffmann, Y. Zhuang, S. Olgen, M. Bartels, U. Muller-Vieira, T. Lauterbach and R. W. Hartmann, Bioorg. Med. Chem., 2008, 16, 7715–7727 CrossRef CAS PubMed.
- S. Chowdhury, M. Chafeev, S. Liu, J. Sun, V. Raina, R. Chui, W. Young, R. Kwan, J. Fu and J. A. Cadieux, Bioorg. Med. Chem. Lett., 2011, 21, 3676–3681 CrossRef CAS PubMed.
- B. W. Caprathe, J. C. Jaen, L. D. Wise, T. G. Heffner, T. A. Pugsley, L. T. Meltzer and M. Parvez, J. Med. Chem., 1991, 34, 2736–2746 CrossRef CAS PubMed.
- E. Papadopoulos, S. Jenni, E. Kabha, K. J. Takrouri, T. Yi, N. Salvi, R. E. Luna, E. Gavathiotis, P. Mahalingam, H. Arthanari, R. Rodriguez-Mias, R. Yefidoff-Freedman, B. H. Aktas, M. Chorev, J. A. Halperin and G. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3187–E3195 CrossRef CAS PubMed.
- P. D. Fischer, E. Papadopoulos, J. M. Dempersmier, Z. F. Wang, R. P. Nowak, K. A. Donovan, J. Kalabathula, C. Gorgulla, P. P. M. Junghanns, E. Kabha, N. Dimitrakakis, O. I. Petrov, C. Mitsiades, C. Ducho, V. Gelev, E. S. Fischer, G. Wagner and H. Arthanari, Eur. J. Med. Chem., 2021, 219, 113435 CrossRef CAS PubMed.
- T. Kaur, A. Menon and A. L. Garner, Eur. J. Med. Chem., 2019, 166, 339–350 CrossRef CAS PubMed.
- S. Santag, F. Siegel, A. M. Wengner, C. Lange, U. Bomer, K. Eis, F. Puhler, P. Lienau, L. Bergemann, M. Michels, F. von Nussbaum, D. Mumberg and K. Petersen, Cancer Lett., 2017, 390, 21–29 CrossRef CAS PubMed.
- A. M. Abdelaziz, M. Yu and S. Wang, Pharm. Pat. Anal., 2021, 10, 25–35 CrossRef CAS PubMed.
- V. Teneggi, V. Novotny-Diermayr, L. H. Lee, M. Yasin, P. Yeo, K. Ethirajulu, S. B. H. Gan, S. E. Blanchard, R. Nellore, D. N. Umrani, R. Gomeni, D. L. W. Teck, G. Li, Q. S. Lu, Y. Cao and A. Matter, Clin. Transl. Sci., 2020, 13, 57–66 CrossRef PubMed.
- W. Xu, S. Kannan, C. S. Verma and K. Nacro, J. Med. Chem., 2022, 65, 983–1007 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |