
Dual functional therapeutics: mitigating bacterial infection and associated inflammation
Yash
Acharya
 a,
Kashish Kumar
Taneja
a and
Jayanta
Haldar
*ab
a,
Kashish Kumar
Taneja
a and
Jayanta
Haldar
*ab
aAntimicrobial Research Laboratory, New Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur, Bengaluru 560064, Karnataka, India. E-mail: jayanta@jncasr.ac.in
bSchool of Advanced Materials, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur, Bengaluru 560064, Karnataka, India
First published on 24th May 2023
Abstract
The emergence of antimicrobial resistance, coupled with the occurrence of persistent systemic infections, has already complicated clinical therapy efforts. Moreover, infections are also accompanied by strong inflammatory responses, generated by the host's innate and adaptive immune systems. The closely intertwined relationship between bacterial infection and inflammation has multiple implications on the ability of antibacterial therapeutics to tackle infection and inflammation. Particularly, uncontrolled inflammatory responses to infection can lead to sepsis, a life-threatening physiological condition. In this review, we discuss dual-functional antibacterial therapeutics that have potential to be developed for treating inflammation associated with bacterial infections. Immense research is underway that aims to develop new therapeutic agents that, when administered, regulate the excess inflammatory response, i.e. they have immunomodulatory properties along with the desired antibacterial activity. The classes of antibiotics that have immunomodulatory function in addition to antibacterial activity have been reviewed. Host defense peptides and their synthetic mimics are amongst the most sought-after solutions to develop such dual-functional therapeutics. This review also highlights the important classes of peptidomimetics that exhibit both antibacterial and immunomodulatory properties.
 Yash Acharya | Yash Acharya completed his Bachelor's degree in Chemistry at Ramnarain Ruia College, Mumbai. He received his Master's degree in Chemistry from the New Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), and joined Prof. Jayanta Haldar's lab in 2020. At the Haldar lab, Yash is working on developing peptidomimetic small antimicrobial agents and semisynthetic glycopeptide derivatives to tackle antimicrobial resistance and complicated bacterial infections. He is also working on developing host-modulating antibacterial therapies to tackle infection-associated inflammation and intracellular infections. |
 Kashish Kumar Taneja | Kashish Taneja received his Bachelor's degree in Chemistry from Sri Venkateswara College, University of Delhi. He joined the New Chemistry Unit at JNCASR as a Master's student in September 2021. He is working on his master's thesis research under the supervision of Prof. Jayanta Haldar. His research project is focused on the development of novel amphiphilic molecules and membrane-perturbing adjuvants to rejuvenate and repurpose obsolete antibiotics against multi-drug resistant superbugs. Through his research, he aims to overcome acquired, intrinsic and phenotypic resistance in multi-drug resistant bacteria. |
 Jayanta Haldar | Jayanta Haldar received his Doctoral degree from the Indian Institute of Science in Bangalore. After completing his postdoctoral research at the Massachusetts Institute of Technology, he joined the New Chemistry Unit, JNCASR. He is currently a Professor at the institute. His lab works in the fields of Medicinal Chemistry, Chemical Biology and Biomaterials, for tackling and preventing antimicrobial resistance and infections. His group has developed novel therapeutics and new synergistic strategies for tackling infections caused by pathogenic bacteria, fungi and viruses, and infection-associated inflammation. His group has also developed smart biomaterials which aid in preventing the spread of infectious diseases, as well as cure infections and enhance wound healing. |
1. Introduction
The discovery of antibiotics was a game-changer in mitigating infectious diseases and public health. However, the increasing emergence of antimicrobial resistance is threatening to undo the gains of the last century.1 Along with increasing antimicrobial resistance, infection associated conditions, such as hyperimmune response, cytokine storm, and sepsis, lead to many complications in therapy. The co-occurrence of bacterial infection and associated inflammation severely jeopardises public health. The initial stages of bacterial infection are often associated with subdued inflammatory response, particularly from the innate component of host immunity.2 The various virulence factors possessed by bacterial pathogens are thought to be responsible for initial masking of the innate response.3 However, once an infection is established, and pathogens have reached dangerously high numbers, the immune system is activated due to the high burden.4 At such a stage, the significant pathogen number overwhelms the host immune system and leads to a disproportionately severe reaction from the host, known as sepsis. This host-response to pathogens comprises a basket of innate and humoral immunity components.5 It is commonly characterised by clinical features such as elevated body temperature, shivering, altered mental status, rapid breathing, increased heart rate, weak pulse/low blood pressure, low urine output, cyanotic or mottled skin, and extreme body pain or discomfort.6 This situation is highly unstable for the patient and can deteriorate very sharply.The presence of a strong inflammatory response in a patient with high pathogen load can lead to multi-organ failure, and eventual death, if uncontrolled (Fig. 1).7 Sepsis and hyperimmune response in systemic infections are a major cause of mortality, with approximately 49 million affected annual cases and over 11 million estimated deaths being caused by the syndrome, accounting for up to 19.7% of all deaths worldwide.8 An even more surprising statistic is that even today, up to 25% patients with sepsis and 40% patients with septic shock succumb to the syndrome.9 The ongoing COVID-19 pandemic highlighted the need to mitigate hyperimmune response, as many patients displayed elevated cytokine levels during the viral infection.10 A recent study indicated that almost 17% of total SARS-CoV-2 infections were accompanied by co-infections with bacterial pathogens, such as S. aureus, P aeruginosa, etc.11 Such polymicrobial infections can trigger further inflammation as well.12
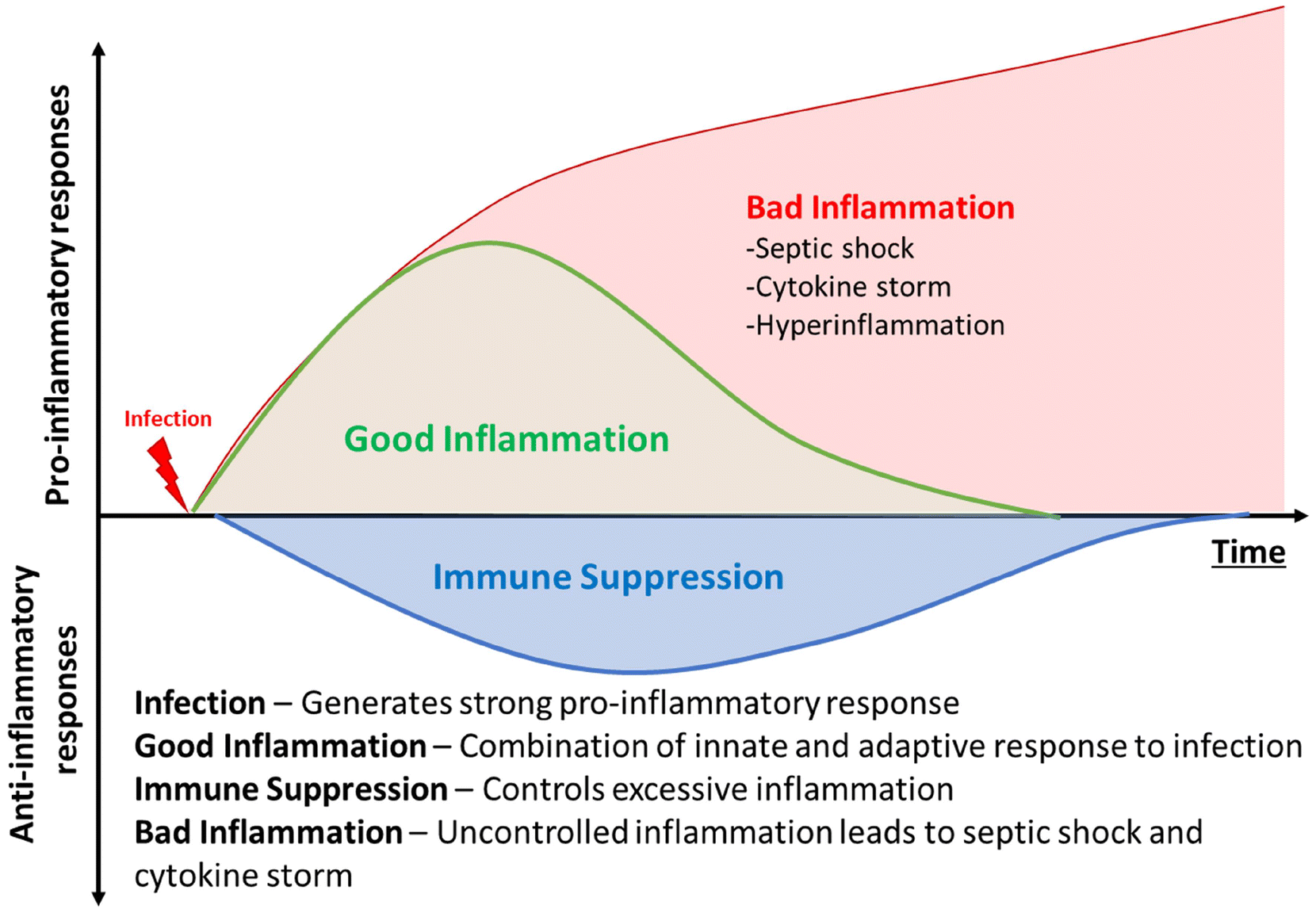 | ||
| Fig. 1 Infection, inflammation and sepsis. | ||
While the diagnostics of sepsis have evolved significantly, the therapeutic strategies are limited and nebulous. One of the mitigation strategies prescribed for sepsis patients in India is the SALVAGE therapy (Table 1).6 The acronym SALVAGE stands for Steroids, Antibiotics, Low-dose heparin, Ventilation with lung protective strategy, Activated protein C, Glucose homeostasis, and Early goal directed therapy.13 Some combinations or variations of these interventions are emulated for sepsis therapy globally. However, given the diverse physiology of sepsis, its treatment often fails to yield required results in time to save the patient.14 The broader problem also lies in the inability of this therapy to clear infection and significantly reduce immune activation. This calls for a holistic and multi-functional approach towards mitigating sepsis and associated inflammation in severe bacterial infections. In almost 87% of patients, sepsis starts before the patient is hospitalised.8 Upon hospitalisation, the immediate treatment strategies include reducing the infection burden through antibacterial therapy and reducing the excessive immune response. However, such therapy is seldom effective and can have various side effects associated with anti-inflammatory steroids. In such a situation, where there is a possibility of excessive immune response disrupting all therapeutic interventions, if an antibacterial therapeutic that is being used for reducing the bacterial burden can also have the additional ability to lower or suppress antigen-responsive inflammatory response, such a dual-functional therapeutic may greatly enhance our ability to tackle sepsis. It is already reported that many clinically prevalent antibiotics also possess immunomodulatory properties, and other reviews have covered them in significant detail.15 However, antibiotics can be limited by the presence of multi-drug resistant bacteria. Additionally, some broad-spectrum antibiotics such as β-lactams (penicillin, oxacillin, ceftazidime, etc.) and folic acid inhibitors (trimethoprim) are reported to further aggravate inflammation.6 Similarly, the emerging class of versatile membrane-targeting antimicrobial peptides (also known as host defense peptides) have been reported to show immunomodulatory properties.16 There are peptides, natural product metabolites, as well as antibiotics, which show only immunomodulatory properties, which have been covered in other reviews.15,17–21 Through this review, we attempt to summarize three important classes of antimicrobial therapeutics, namely, antibiotics, host defense peptides and peptidomimetics, which have the potential to show dual-functionality owing to their immunosuppressive properties. While some of the members of these classes have been reported to show pro-inflammatory effects, we have focused here on those inherently antibacterial compounds which reduce excessive inflammatory response and can have the potential to mitigate sepsis.
| Component | Purpose | |
|---|---|---|
| S | Steroids | Reduce inflammation |
| A | Antibiotics | Mitigate bacterial infection |
| L | Low-dose heparin | Prevents thromboembolisms |
| V | Ventilation with lung protective strategy | Ensures adequate tissue oxygen delivery |
| A | Activated protein C | Reduces clotting |
| G | Glucose homeostasis | Prevents hypoglycemia |
| E | Early goal directed therapy | Early resolution prevents mortality |
2. Infection and sepsis
The multi-faceted nature of infections and the ability of some antimicrobial agents to aggravate infection associated problems led to the development of structurally and functionally diverse novel antibacterial therapeutics.2 The primary component of the Gram-negative bacterial outer membrane, lipopolysaccharide (LPS), is the most potent and notorious bacterial factor, known for triggering strong inflammatory responses. LPS endotoxin is the strongest known stimulator of the immune system, causing the activation of inflammatory signalling pathways leading to expression of pro-inflammatory cytokines for the development and evolution of an inflammatory response. If this inflammation is uncontrolled – it can lead to sepsis, multi-organ failure and eventual death.Inflammation is a physiological response to disturbed tissue homeostasis due to stimuli such as infections or tissue injury. It is a complex and tightly regulated process in which blood derived products such as plasma proteins and leukocytes are recruited into the perturbed tissue. This process is governed by a number of mediators released by a variety of leukocytes (neutrophils, monocytes/macrophages, eosinophils, basophils, and lymphocytes). LPS from Gram-negative bacteria is a recognized pathogen associated molecular pattern (PAMP) responsible for induction of an inflammatory response in cases of bacterial infections.5 Various other bacterial antigens are known PAMPs, such as peptidoglycan, lipoteichoic acid (LTA) (a major Gram-positive bacterial antigen), triacyl lipoproteins, CpG rich DNA, etc. Out of these, LPS is the most well studied and reported PAMP. LPS binds to lipopolysaccharide binding protein (LBP) to give an LPS–LBP complex which binds to a CD14 (cluster of differentiation 14) receptor.22 CD14 is a protein made by macrophages and acts as a co-receptor for LPS detection. While macrophages and neutrophils are CD14 positive (they have membrane bound CD14, mCD14), some such as dendritic cells, fibroblasts, smooth muscle cells and vascular endothelium are CD14 negative, but they still respond to LPS via soluble CD14, sCD14. CD14 recruits single molecules of LPS and then transfers them to the TLR4–MD2 complex.5 The LPS-bound TLR4–MD2 heterodimer then dimerizes with another LPS-bound heterodimer, and this activates the downstream signalling pathway. The Gram-positive antigen LTA activates TLR2 signalling.22 Various kinases have been reported to play an important role in cellular signalling; however, their function is not properly understood.23 TLR4 activation leads to the activation of NF-κB and formation of subsequent pro-inflammatory cytokines. Classic cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumour necrosis factor-α (TNF-α) derived from host cells are pro-inflammatory in nature and play a key role in development of the innate immune response. Along with these, other mediators such as interleukin-8 (IL-8), interleukin-12 (IL-12), and interferon-γ (IFN-γ) are important for the manifestation of an inflammatory response. LPS also induces the expression of inducible nitric oxide synthase (iNOS) which increases the nitric oxide (NO) levels associated with hypotension.24 LPS activates the nuclear translocation of nuclear factor kappa B (NF-κB) by degradation of its inhibitory subunit IκB.5 This NF-κB is responsible for gene transcription of numerous pro-inflammatory cytokines and iNOS. Gram-positive bacteria do not have an endotoxin as such, but a similar cascade operates in activation of toll-like receptor 2 (TLR2) by LTA – a cell wall component.
3. Antibiotics as immunomodulators
Conventionally used antibiotics have been identified and employed over the years for their potent antibacterial activities. However, a closer look at the physiology of antibiotic action has revealed some moonlighting roles for some key antibiotics. Studies observed that apart from direct antibacterial activity, these antibiotics also display host-modulatory effects, involving particularly the suppression of the bacteria-associated immune response (Table 2). In this section, we have attempted to provide an overview of the various immunomodulatory effects of key antibiotics (Fig. 2). The existing studies on the immunomodulatory potential of antibiotics span across both the domains of pre-clinical research as well as clinical studies.| Antibiotic class | Antibiotics | Antibacterial activity spectrum | Immunomodulatory activity | Ref. |
|---|---|---|---|---|
| Macrolides | Azithromycin (1) | Gram-positive and Gram-negative | Lowers IL-1β, IL-8, NF-κB, IL-10, MMP-9 | 26–28 |
| Clarithromycin (2) | Gram-positive and Gram-negative | Lowers TNF-α. Restores the TNF-α/IL-10 ratio | 31 | |
| Tetracyclines | Tetracycline (3) | Gram-positive and Gram-negative | Prevents IL-1β and IL-18 release. Inhibits MMP-2 and MMP-9 | 34 and 35 |
| Doxycycline (4) | Gram-positive and Gram-negative bacteria | Lowers TNF-α, IL-1β, IL-6, IL-8, and IL-10 | 36 and 37 | |
| Tetracycline 3 (5) | Gram-positive | Lowers TNF-α, IL-1β, IL-6, IL-8, and IL-10 | 36–38 | |
| Oxazolidinones | Linezolid (6) | Gram-positive | Lowers TNF-α, IFN-γ, IL-1β, IL-6, IL-12, IL-17, MIP-2, MMP-9, and MMP-5 | 40–46 |
| Fluoroquinolones | Moxifloxacin (7) | Gram-positive and Gram-negative | Lowers KC, IL-1β, and IL-17A | 48–51 and 53 |
| Ciprofloxacin (8) | Gram-positive and Gram-negative | Lowers TNF-α, IL-1β, and CXCL2/MIP-2 | 49 and 50 | |
| Levofloxacin (9) | Gram-positive and Gram-negative | Reduces ROS, NOX | 49 and 52 | |
| Glycopeptides | Vancomycin (10) | Gram-positive | Lowers IL-1β | 42 |
| Polymyxins | Colistin (11) | Gram-negative | Lowers TNF-α, IL-1β | 57–60 |
 | ||
| Fig. 2 Antibacterial and immunomodulatory antibiotics. | ||
3.1 Macrolides
A 14 to 16-membered ring of macrolactone, bearing a broad-spectrum activity against bacteria, distinguishes the most often used macrolides. They attach reversibly to the bacterial ribosome's 50S subunit to prevent the start of protein synthesis.25 The macrolide class of antibiotics is amongst the most well-studied for its immunomodulatory potential. Macrolides inhibit NLRP3 inflammasome activation. Azithromycin (1) suppressed the induction of caspase-4 while not reducing NF-κB and blocked the LPS-triggered production of IL-1β cytokine in vitro and in vivo (Table 2).26 By the reduction of IL-1β and IL-18 that are dependent on inflammasome release, azithromycin reduced lung damage in a Pseudomonas aeruginosa-infected mouse model.27 In a mouse LPS-induced pulmonary damage model system, azithromycin dramatically reduced NF-κB activation in a concentration dependent manner.28 According to a number of preclinical investigations, macrolides suppress the innate immune response by preventing the generation of pro-inflammatory cytokines both in vitro and in vivo.29,30 Macrolides mitigate lung injury by lowering levels of pro-inflammatory cytokines (such as IL-1β, IL-5, IL-6, IL-17, IL-18, and TNF-α) and chemokines (such as C–X–C motif chemokine ligands CXCL8 and CXCL9).15 Macrolide therapy was linked to a 9–13% mortality decrease and a quicker time to successfully stop using mechanical breathing. In a multi-centre research study involving 200 patients with ventilator-associated pneumonia (VAP)-induced septic shock, those who took clarithromycin exhibited significantly reduced levels of TNF-α and enhanced levels of IL-6 compared to those in the control cohort. The TNF-α/IL-10 ratio, which represents quantitatively the equilibrium between pro- and anti-inflammatory cytokines, was also restored by clarithromycin treatment.31 A research study which involved community-acquired pneumonia patients who weren't responding to treatment found that macrolide treatment decreased levels of IL-6 and TNF-α in lung fluid and IL-8, IL-6, and IL-10 in plasma. Macrolide treated patients experienced shorter hospital stays and quicker clinical stabilisation.323.2 Tetracyclines
Tetracyclines are a type of broad-spectrum antibiotic used to treat a variety of conditions, including cholera, Lyme disease, sex-transmitted infections, and community-acquired pneumonia.33 They reversibly bind to the 30S ribosomal subunit to suppress protein synthesis and stop the charged tRNA from entering and binding to the ribosome's A site.33 They exhibit immunomodulatory activities in addition to their antibacterial ones (Table 2). Tetracyclines (3) prevent caspase-1 dependent IL-1β and IL-18 release, which lowers inflammasome-mediated lung damage.34 Tetracyclines can also prevent matrix metalloproteinases (MMPs) from working by chelating Zn2+ ions out of their active site.35 In animal models of secondary acute respiratory distress syndrome (ARDS), the lowered release of immune markers IL-1β, TNF-α, IL-8, IL-6 and IL-10 upon exposure to doxycycline (4) and chemically modified tetracycline 3 (5) increased survival.36,37 A recent study found that tetracycline greatly reduced lung damage in mice brought on by LPS and influenza, by decreasing the generation of IL-1β and IL-18 that is dependent on the inflammasome and caspase-1.34Tetracycline 3 (5) that has been chemically altered to lack antibacterial action against Gram-negative bacteria has been found to lessen neutrophil sequestration in a porcine model.38 Chemically modified tetracycline 3 (5) had a negligible impact on sequestration of mononuclear cells, but it prevented respiratory distress, providing evidence that neutrophil inflow is an important factor in the aetiology of ARDS.38 In animal infection models, doxycycline also reduced neutrophil influx. In multiple animal models of indirect ARDS, prophylactic chemically modified tetracycline 3 therapy decreased lung damage and improved survival by inhibiting neutrophil infiltration and lowering NE and MMP levels in bronchial alveolar lavage fluid (BALF). There is a report of a modified tetracycline displaying immunosuppressive properties upon sequential exposure, in a zebra fish model also. Tetracycline (3) reduced the generation of IL-1β and IL-18 by alveolar immune cells in a newly published study.34
3.3 Oxazolidinones
Linezolid (6), a prominent member of the oxazolidinone class, has antibacterial properties against Gram-positive pathogens by preventing the formation of bacterial proteins. It is mostly used as an antibiotic of last resort for mitigating infections caused by multi-drug resistant Gram-positive pathogens, including vancomycin-resistant enterococci (VRE) and methicillin-resistant Staphylococcus aureus (MRSA).39 Linezolid reduces the chemotaxis of immune cells into the airways by inhibiting the release of proinflammatory mediators. In particular, ARDS is characterised by restricted neutrophil movement. Linezolid also reduces macrophage phagocytosis while restoring neutrophil effector mechanisms including phagocytosis and pathogen killing.40 Linezolid increases natural apoptosis in neutrophils, which also contributes towards lowering inflammation.41In animal models of MRSA-induced lung injury, linezolid's effects on the production of inflammatory mediators have been examined. In a mouse model of MRSA-induced lung injury, it was demonstrated that linezolid (6) and vancomycin (10) exhibit equivalent effects in altering the production of MMP-9, MMP-5 and IL-6 in lung fluid and in controlling neutrophil entry (Table 2).42 Nevertheless, efferocytosis was unaffected by either antibiotic, which promoted neutrophil death.43 In contrast to glycopeptides, linezolid increased survival and reduced lung injury in animal models of MRSA pneumonia by lowering neutrophil infiltration and TNF-α levels. Inflammatory cytokines such as IL-1β, IFN-γ, IL-12, IL-17, IL-6, and chemokine MIP-2 were also reduced by linezolid (Table 2).41
There aren't many ex vivo studies that discuss linezolid's immunomodulatory effects on people. Human peripheral blood mononuclear cells (hPBMCs) stimulated with LPS endotoxin or MRSA secreted proinflammatory cytokines such as IL-1β, IL-1β receptor antagonist (IL-1RA), TNF-α, IL-6, IL-8, and CCL2.44,45 Linezolid dramatically reduced this secretion. In addition, linezolid reduced the ability of polymorphonuclear neutrophils (PMNs) to phagocytose S. aureus and P. aeruginosa, as opposed to two earlier trials in which linezolid had no effect on PMNs' ability to phagocytose Gram-positive cocci.15,46
3.4 Fluoroquinolones
As an important group of synthetic antimicrobials with a broad scope, fluoroquinolones target the enzymes DNA gyrase and topoisomerase IV to prevent the production of DNA.47 Gram-positive and Gram-negative bacteria are all included in the antimicrobial spectrum. In vitro and in vivo evidence of fluoroquinolones' anti-oxidative capabilities has been found.47 Moreover, they hinder the release of cytokines and chemokines that promote inflammation, which impairs neutrophil chemotaxis. Fluoroquinolones have been shown to have immunomodulatory effects for bacterial, viral, and fungal pathogen-induced lung damage in diverse animal models (Table 2).15 Not all fluoroquinolones seem to have immunomodulatory effects in bacterial-induced lung damage. Moxifloxacin (7) displays contradictory effects. It reduced neutrophil infiltration and proinflammatory markers (such as KC, IL-1β, and IL-17A) in a mouse model of lung infection of S. pneumoniae and P. aeruginosa.48 Ciprofloxacin, as opposed to moxifloxacin and levofloxacin, significantly reduced levels of TNF-α, IL-1β, and CXCL2/MIP-2 and improved lung injury severity and survival in LPS-induced lung injury.49 There are some conflicting reports on immunomodulatory properties of quinolones which require further investigation. Results in the aforementioned research may have been affected by dosing timing and frequency.Fluoroquinolones like ciprofloxacin (8) and moxifloxacin (7) that have a cyclopropyl-moiety at the N1 position in the quinolone ring have particularly strong immunomodulatory effects.50 Moreover, moxifloxacin did not reduce cytokine release in LPS-stimulated human monocytes but did in heat-killed S. aureus-stimulated human monocytes, indicating differing effects on Gram-positive and Gram-negative cytokines.51 Levofloxacin (9) was demonstrated to reduce oxidative and nitrative stress in a mouse model of ARDS brought on by the influenza virus (H1N1).52 Levofloxacin (9) demonstrated a scavenging ability against ROS produced by neutrophils, dramatically lowering lung damage and boosting survival.52 Moxifloxacin's effects on fungus-caused lung damage were studied, in addition to the effects of fluoroquinolones towards immunomodulation in LPS and influenza-induced pulmonary distress. TNF-α and KC/CXCL1 levels in immunocompromised mice pre-treated with moxifloxacin remained low, as compared to the control.53
3.5 Polymyxins
Polymyxins are an important class of lipopeptide antibiotics against Gram-negative bacteria. Colistin (11), a member of polymyxins, is a last resort antibiotic, often prescribed in cases of multi-drug resistant Gram-negative infections. These antibiotics interact strongly with bacterial LPS in inner and outer membranes, effectively killing bacteria.54 The ability of colistin to interact with endotoxin has spurred investigations into its immunomodulatory properties. Some of the early studies on the mode of action of colistin explored its binding to endotoxin. One study employed a fluorophore-conjugated probe, dansylcadaverin.55 This probe weakly interacts with the lipid A component of LPS, and its fluorescence is quenched in this state. When an LPS-interactor, like polymyxin B, displaces the probe, an increase in fluorescence is observed, which is used to quantify LPS interaction.55 These and other NMR and simulation-based studies suggested a strong binding between polymyxins and LPS, which prompted the investigation of colistin's immunomodulatory properties.55,56 Studies at the in cellulo level show that colistin can have concentration-dependent effects of different cytokine levels. In LPS-challenged rat macrophage primary cells, colistin treatment enhances TNF-α levels, while reducing IL-6 and IL-1β.57 It was hypothesized that this may be an effect of p38/MAPK pathway perturbation by colistin. This study was not very conclusive, as divergent effects were seen for different cytokines.57 However, another report demonstrated that colistin treatment in LPS-challenged CuFi-1 cell lines leads to a significant reduction in the production of the IL-8 pro-inflammatory cytokine.58 The study hypothesized the ability of colistin to interact with LPS to be the reason for this immunomodulatory activity. These conflicting reports suggest that the immunomodulatory effects may be cell- and concentration-specific.An in vivo study was performed for colistin in a rat model of infection, and it was observed that pre-administration of colistin led to lower levels of cytokines such as TNF-α, IL-6 and IL-1β in animal plasma, compared with the infected group without treatment.59 Colistin pre-treatment also led to reduced bacterial burden, strong reduction in cytokine levels and improved histopathological injury in infected lung tissues compared with the untreated animals. This study also demonstrated that a p38/MAPK inhibitor does not reverse the effects of colistin, thereby contradicting earlier reports about the mechanism of immunomodulation.59 Followed by these studies, a randomized, double-blind, placebo-controlled crossover trial was performed to evaluate the effect of colistin on inflammatory responses in LPS-challenged healthy volunteers in a human endotoxemia model.60 A single dose of colistin methanesulphonate through the intravenous route led to a significant reduction of the inflammatory cytokine response (IL-6, IL-8, and TNF-α) in healthy volunteers after 24 hours (Table 2). Studies have also investigated the LPS-binding and immunomodulatory properties of a non-active polymyxin nonapeptide and its various derivatives.61
Along with the examples discussed earlier, where colistin pre-administration shows immunosuppressive effects, there are slightly differing reports of colistin displaying reverse effects. Research groups have shown that colistin treatment can enhance the production of pro-inflammatory cytokines such as IL-6, TNF-α, etc. Hence, it is imperative to understand the mechanisms of immunomodulation in both cases to explain such divergent effects. Importantly, larger clinical trials, in non-septic patients and healthy patients, are necessary to determine the safety of colistin use in context of inflammation.
4. Immunomodulatory properties of antibacterial host defense peptides
Host defense peptides (HDPs) form an essential component of the innate and adaptive immunity of all life forms from insects to mammals.19 The term antimicrobial peptides (AMPs) is used interchangeably with HDPs because of their potent activity against bacteria, fungi, eukaryotic parasites, membrane bound viruses, etc. Apart from this, HDPs possess a host of other functions such as immune system modulation, inflammatory suppression, and immune enhancement.20 Increasing research has identified that HDPs can modulate pro- and anti-inflammatory responses, chemoattraction, extracellular and intracellular bacterial killing, cellular differentiation and activation of the innate and adaptive compartments, wound-healing, and autophagy as well as apoptosis and pyroptosis.17 These are just a few of the diverse immunomodulatory properties of HDPs that have been extensively studied and reviewed.17,18 Recently, the focus has shifted towards the multi-functional nature of HDPs. This section focuses on various classes of HDPs that exhibit direct antibacterial activity along with immunomodulatory properties (Table 3).| Peptide | Sequence | MIC (μM) | Immunomodulatory activity | Ref. |
|---|---|---|---|---|
| a f symbolizes phenylalanine peptoid. b Smaller case letters represent D-amino acids. c k symbolizes lysine peptoid. | ||||
| Papiliocin | RWKIFKKIEKVGRNVRDGIIKAGPAVAVVGQAATVVK–NH2 | 0.58 | Lowers NO, TNF-α, MIP-1, MIP-2, IL-1β, IL-6, TLR4, and NF-κB | 69 |
| Cathelicidin-PY | KCNFLCKLKEKLRTVITSHIDKVLRPQG | 1.37–10.95 | Lowers NO, TNF-α, MIP-1, IL-6, TLR4, and NF-κB | 70 |
| KR-12-a3 | KRIVKRIKKWLR–NH2 | 2.8 | Lowers TNF-α | 74 |
| Fowl-1 (8–26)-WRK | LVIRWVRAGYKLYRAIKKK–NH2 | 3.7 | Lowers TNF-α and NO | 76 |
| CR-163 | RRWVQRWIRRWRKWV | 5 | Lowers NO, TNF-α, IL-6, and IL-8 | 82 and 83 |
| A7 | ILKWKWKWWKWRR–NH2 | 7.5 | Lowers NO | 84 |
| MC1-1 | SAVGRHGRRFGLRKHRKH | 3.59–4.42 | Lowers TNF-α and IL-6 | 87 |
| TL-3 | FVRWWSRWLRRIL–NH2 | 5.7 | Lowers TNF-α and NO | 89 |
| K6L2W3 | KLWKKWKKWLK–NH2 | 1–2 | Lowers NO | 95 |
| KLW-f | KWKKLLKKfLKLfKKLLK–NH2a | 0.5–2 | Lowers NO | 97 |
| D-RR4 | wlrrikawlrrika–NH2b | 2–8 | Lowers TNF-α and IL-6 | 100 |
| Lf-KR | RRWQWRPKRIVKLIKKWLR–NH2 | 7 | Lowers TNF-α and NO | 103 |
| PapMa-k | RWKIFKKIkKFLHSAKKF–NH2c | 2–8 | Lowers TNF-α, IL-1β, MIP-1, and MIP-2 | 104 |
4.1 α-Helical and derived antibacterial host defense peptides
LL-37 (a cathelicidin) is widely distributed in nature.62 It can influence the ability of innate immune cells to produce chemokines and cytokines, guide immune cell chemoattraction, promote angiogenesis, and repair wounds. LPS-neutralizing activities of human CAP18 and guinea pig CAP11 peptides have been studied in detail. CAP18 and CAP11 have lipid A or LPS binding abilities, and they effectively prevent LPS from interacting with LBP, which is what delivers LPS to CD14 to initiate an inflammatory response.63 LL-37 has been reported to modulate the response of macrophages during Mycobacterium tuberculosis infection by control of the pro-inflammatory and anti-inflammatory cytokine expression.64 LL-37 has also been reported to selectively suppress the TLR-4 induced pro-inflammatory cytokine secretion from monocytes (Table 3).65 Cathelicidins are reported to have strong antibacterial activity along with plausible cell selectivity. Many HDPs in clinical trials are cathelicidins.66 Because of increased interest in development of dual-functional antibacterial and immunomodulatory therapeutic agents, even more HDPs continue to be identified, characterized, and isolated. Along with the identification of novel HDPs, many groups are designing short peptides inspired from their natural counterparts to achieve better activity and stability.67,68Papiliocin (37-meric peptide) was highly active against Gram-negative bacteria with a geometric mean of MIC equal to 0.58 μM (Table 3).69 RAW264.7 cells when challenged with LPS and treated with 10 μg ml−1 peptide showed almost complete inhibition in nitric oxide (NO) production (Fig. 3C). The production of pro-inflammatory cytokines TNF-α and macrophage inflammatory protein-2 (MIP-2) was also significantly inhibited (Fig. 3A and B). Upon total mRNA analysis, it was observed that the markers of inflammation TNF-α, iNOS, IL-1β, MIP-1, MIP-2, and IL-6 were also suppressed in LPS stimulated RAW264.7 cells treated with papiliocin. The expressions of TLR4 and nuclear translocation of factor NF-κB were also down-regulated (Fig. 3D).69 The addition of papiliocin also caused a dose-dependent increase in the fluorescence intensity of FITC–LPS suggesting that the interaction of papiliocin with LPS resulted in dissociation of LPS aggregates (Fig. 3E). This was further validated by the stronger blue-shift in the emission of the tryptophan (2Trp) residue within the peptide, upon interaction with negatively charged LPS, as compared to neutral lipids. The interaction of the peptide with LPS was probed further through saturation transfer difference NMR (STD-NMR), which suggested that the aromatic rings of 2Trp and 5Phe played a key role in the interaction with LPS, as their protons displayed the largest STD effect.
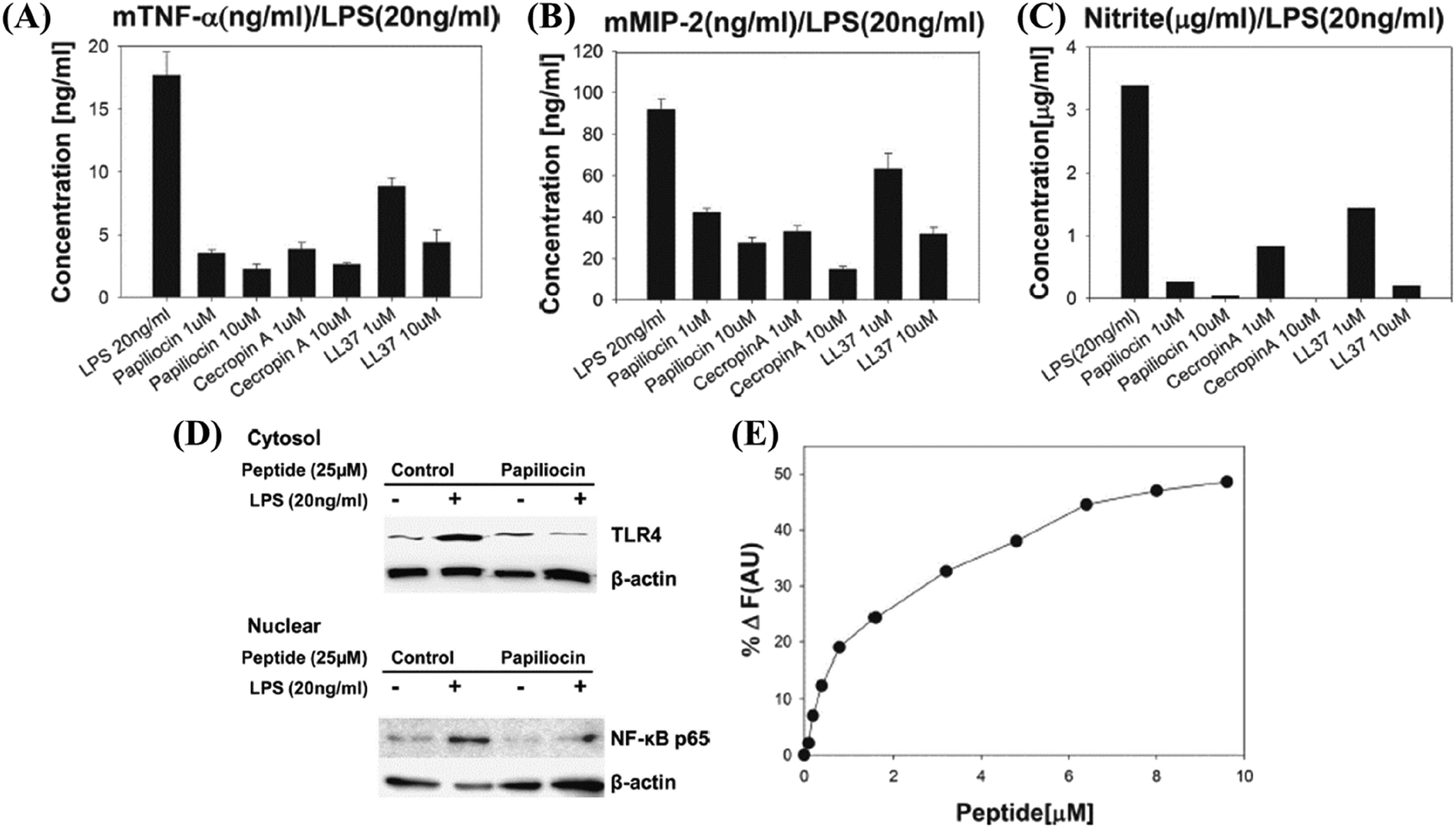 | ||
| Fig. 3 Papiliocin – antibacterial and immunomodulatory host defense peptide inhibition of (A) mTNF-α, (B) mMIP-2 and (C) nitrite production upon treatment with papiliocin in LPS-stimulated RAW264.7 cells. (D) Effect of TLR4 and nuclear NF-κB in LPS stimulated RAW264.7 cells. (E) Enhancement of fluorescence intensity of FITC–LPS upon addition of papiliocin. Reproduced from ref. 69 with permission from Elsevier, Copyright 2011. | ||
Cathelicidin-PY (from the skin secretions of the frog Paa yunnanensis) showed good antibacterial activity against Gram-positive and Gram-negative bacteria (sensitive and drug-resistant strains, MIC = 4.69–37.5 μg ml−1).70 A 70% inhibition in NO production was observed in RAW264.7 cells challenged with LPS on treatment with 20 μg ml−1 cathelicidin-PY (Table 3). The production of pro-inflammatory cytokines TNF-α, IL-6, and MCP-1 was also inhibited by 60%, 86%, and 68%, respectively.70 The interaction between cathelicidin-PY and LPS was investigated by NMR titration; LPS bound to cathelicidin-PY with medium affinity.
Despite the immense potential that HDPs offer, their development as therapeutics is met with difficulty because of their bulky nature, poor cell selectivity, toxicity and high cost of development.71 Along with the identification of novel natural peptides, major research is performed to develop short, cost-effective, and efficient derivatives of HDPs.72 The 12-meric peptide sequence (from 18 to 29) of LL-37 labelled KR-12 is reported to be responsible for its antimicrobial activity.73 A series of short KR-12 analogues to optimize the hydrophobic–hydrophilic amino acid ratio essential for cell selectivity have been reported.74 The geometric mean of MIC determined against a host of Gram-positive and Gram-negative bacteria was 2.8 μM for KR-12-a3. It was 22-fold more selective than the parent LL-37. RAW264.7 cells challenged with LPS showed 74.2% inhibition in TNF-α production in the presence of 10 μM peptide.74
Fowlicidin-1 (Fowl-1), a 26-meric antimicrobial peptide active against both Gram-positive and Gram-negative bacteria with the ability to reduce LPS-induced expression of pro-inflammatory cytokines, has been previously reported. However, its further development as a therapeutic agent was hindered because of extreme cytotoxicity.75 Based on this work, a series of short N- and C-terminal truncated 19-meric Fowl-1 peptides have been reported. Fowl-1 (8–26) was the most potent (geometric mean of MIC = 6.7 μM) with the highest therapeutic index (TI) of 35.6 (3.5 fold higher than Fowl-1).76 Despite Fowl-1 (8–26) being the most potent and cell selective antibacterial peptide, it only reduced the NO and TNF-α production by 16.2% and 13.8%, respectively, on LPS challenge.76 In order to improve the immunomodulatory properties of Fowl-1 (8–26) while maintaining its cell selectivity, the sequence was altered such that the fifth threonine was replaced by tryptophan, the seventh isoleucine was replaced by arginine, and the eleventh asparagine was replaced by lysine. Fowl-1 (8–26)-WRK had reduced cell selectivity (TI = 28.4) compared to the parent Fowl-1 (8–26) but improved immunomodulatory activity. It suppressed the NO and TNF-α production by 94.2 and 75.2%, respectively (Table 3).76 Fowl-1 (8–26)-WRK (16 μM) showed a concentration dependent increase in fluorescence intensity of BODIPY-TR-cadaverine with almost complete LPS neutralization.
The octadecapeptide Amyl-1-18 from α-amylase of rice showed poor to moderate antibacterial activity (IC50 > 50 μM for most bacterial pathogens).77 A continuing study from the same group has reported the LPS neutralizing activity of this peptide.78 A dose-dependent decrease in NO production was observed in LPS/lipid A challenged RAW264.7 cells treated with the peptide. Amyl-1-18 was also found to reduce LPS–LBP interaction in a dose-dependent manner.78 CM4 isolated from Chinese silkworm Bombys mori has been shown to have broad-spectrum antibacterial activity.79 TNF-α and IL-6 mRNA levels were reduced by CM4, and TNF-α and NO release from RAW264.7 was also suppressed in LPS stimulated cells.
A synthetic peptide GW-A2 showed good to moderate antibacterial activity (average MIC against Gram-positive bacteria = 42.29 μg ml−1 and average MIC against Gram-negative bacteria = 64 μg ml−1).80 In a following study, GW-A2 inhibited the NO production in a dose-dependent manner in LPS challenged RAW264.7 cells.81 The phosphorylation levels of ERK1/2, JNK1/2, and p38 (important in the inflammatory signaling pathway) were also found to be reduced by GW-A2. GW-A2 also inhibited NF-κB transcriptional activity in LPS-stimulated macrophages in a dose-dependent manner. It also inhibited ATP-mediated activation of NLPR3 inflammasome resulting in reduced IL-1β secretion. The in vivo immunomodulatory activity of the peptide was also assessed in mice where the levels of IL-1β, IL-6, and TNF-α were significantly reduced in sera collected after 4 h of LPS injection. The expression levels of iNOS, COX-2, and NLRP3 were elevated in the lungs and liver of LPS-injected animals, and these effects were mitigated by GW-A2.81 A study developed cathelicidin-related helical peptides, based on a core 12-amino acid motif “RRWVQRWIRRWR”, known as PepBiotics, with antibacterial activity against Gram-positive and Gram-negative bacteria.82 It screened a series of PepBiotics with various amino acid modifications, additions and truncations, for antibacterial activity and immunomodulatory properties. Many of the peptides displayed significant antimicrobial activity, as well as immunomodulatory properties, as they reduced nitrite levels in LPS-challenged RAW264.7 cells.82 The ability of the lead peptides to exothermically bind to LPS and displace polymyxin, along with its immunomodulatory properties against gentamicin treated and heat-killed bacteria, was also demonstrated in a following study.83 These synthetic cathelicidin-related peptides bear potential for dual-functional antibacterial and immunomodulatory activity.83
4.2 Extended-structure bearing antibacterial host defense peptides
Indolicidin (IN) is a naturally occurring tryptophan rich 13-meric peptide with potent antimicrobial activity but its high toxicity towards mammalian cells hindered its development as a therapeutic agent. Indolicidin analogues such that one, two, or three prolines were replaced by lysine residue(s) have been reported.84 The analogue A7 was the most potent and cell selective (geometric mean of MIC = 7.5 μg ml−1). A7 significantly inhibited the nitric oxide (NO) production at 20 μg ml−1 peptide concentration in LPS challenged RAW264.7 cells (Table 3).Chensinin-1, an 18 amino acid residue peptide, showed conclusive results to be developed further as a potent antimicrobial and anti-inflammatory agent.85 Novel mutant chensinin-1 peptides have been reported.86 MC1-1 had glycine residues replaced by tryptophan. Tryptophan has been reported to facilitate anchoring of the peptide to the lipid bilayer.87 RAW264.7 cells treated with MC1-1 showed significant reduction (>50%) in the levels of TNF-α and IL-6. To assess the immunomodulatory nature of the mutated peptides, dynamic light scattering studies were performed. A bimodal size distribution existed for LPS aggregates in the absence of the peptide but a unimodal size distribution that corresponded to smaller LPS aggregates was observed on addition of MC1-1. The peptide–LPS interactions were exothermic (ΔH = −39.8 kcal mol−1, Kd = 10 μM). MC1-1 was selected for the in vivo anti-inflammatory study and mice were challenged with a lethal amount of LPS and the observed survival rates were 10%, 70%, and 75% for 40, 80, and 160 μg kg−1 MC1-1 administered intraperitoneally, respectively. The levels of pro-inflammatory cytokines TNF-α and IL-6 were also reduced by 75% and 46%, respectively (Table 3).86
Temporin-1Tl is a known amphibian antimicrobial peptide with reported anti-inflammatory activity against LPS-stimulated macrophage cells.88 Novel TL analogues with systematic substitution of tryptophan in the TL hydrophobic region are reported.89 The MIC of the TL analogues determined against a range of Gram-positive and Gram-negative bacteria ranged from 4–32 μM; all lower than the parent TL. The immunomodulatory nature of TL analogues was cemented by RT-PCR as mRNA expression of iNOS and TNF-α in LPS challenged RAW264.7 cells was suppressed in cells treated with TL analogues (Table 3). The immunomodulatory nature of TL analogues could be due to their ability to bind the endotoxin LPS.89
4.3 Specific amino-acid enriched antibacterial host defense peptides
Current HDP research has focused on ways to search through a constellation of known or expected peptide sequences – either experimentally or computationally – for peptides with specified qualities. Utilizing simple design and optimization procedures, several improved variations have been generated and are evaluated for their therapeutic potential.90–94A series of short 11-meric tryptophan rich antimicrobial peptides with varying ratios of leucine and lysine/arginine residues have been reported.95 The antibacterial activities of all peptides and indolicidin were determined against a range of Gram-positive and Gram-negative bacteria. K6L2W3 was the most potent candidate (MIC = 1–2 μM, therapeutic index = 400) with 2–4 fold higher activity than indolicidin (IN). K6L2W3 inhibited 50% NO production in RAW264.7 cells challenged with LPS at 10 μM peptide concentration and completely suppressed iNOS gene expression (Table 3). K6L2W3 also showed nearly 100% LPS neutralizing ability. Conversely, the D-amino acid containing analogue of this peptide displayed lower selectivity, but more potent immunomodulatory properties.95
There are reports of introduction of a hinge region (such as proline) in a peptide sequence which leads to enhanced cell selectivity.96 Similar to proline, peptoid residues also lack an amide proton and can lead to cell selectivity. In another study, a novel leucine/lysine rich antimicrobial peptide with two phenyl peptoid (f) residues in its middle position, KLW-f, has been developed.97 The peptide showed plausible antibacterial activity with MIC in the range of 0.5–2.0 μM. A dose dependent inhibition of nitric oxide production was observed in LPS challenged RAW264.7 cells when treated with 10 μg ml−1 or higher peptide concentration.97
A small synthetic peptide, RR, with potent in vitro and in vivo antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA) was reported.98 In another study, the bacterial burden in MRSA skin lesions was demonstrated to be significantly reduced by topical administration of RR. Additionally, these peptides lowered the release of TNF-α and IL-6 (Table 3).99 However, RR did not show any activity against Gram-negative pathogens. To expand on the antibacterial spectrum of RR, novel RR derivatives are reported in a following report.100 The derivatives were tested against a range of multidrug-resistant strains of P. aeruginosa and A. baumannii which impose a serious threat to global health. D-RR4 exhibited promising antibacterial activity against the tested strains with MIC values that ranged from 2–4 μM. These values were far more superior to those of the parent peptide RR and naturally occurring LL-37 and indolicidin. D-RR4 inhibited the pro-inflammatory cytokine production in macrophages on exposure to LPS.100 D-RR4 was better than RR at 10 μM because it reduced the production of TNF-α and IL-6 by 64.7% and 96.4%, respectively, whereas RR, at 10 μM, decreased TNF-α and IL-6 levels by 24.4% and 75.4%, respectively (Table 3). The capacity of the proposed peptides to lower endotoxin-mediated generation of pro-inflammatory cytokines offers a viable route for their development as standalone antibacterial medicines or as an adjuvant therapy to antibiotics to treat sepsis.100 KLK is a synthetic antimicrobial peptide produced from sapecin B, an antibacterial protein found in the flesh fly Sarcophaga peregrina.101 The KLK peptide was discovered to exhibit moderate microbicidal activity against drug-sensitive Staphylococcus aureus, E. coli, and methicillin-resistant S. aureus (MRSA).101 KLK has been reported to inhibit the production of nitric oxide (NO) by approximately 91% in LPS stimulated RAW264.7 cells.102 The levels of inflammatory mediators IL-1β, TNF-α, and PGE2 were also reduced upon peptide treatment in LPS challenged RAW264.7 cells. The mRNA expression levels of iNOS, COX-2, IL-1β, and TNF-α were also reduced.
4.4 Hybrid antibacterial host defense peptides
An attractive approach to develop new and enhanced antibacterial agents is to link the active parts of maternal peptides with an appropriate hinge to create a hybridized peptide. Lf-KR is a novel hybrid peptide composed of LfcinB6 and KR-12-a4 linked with a proline hinge.103 In the design of Lf-KR, LfcinB6 (the N-terminus) was linked by proline to KR-12-a4 (the C-terminus). The geometric mean of MIC values for Lf-KR was 7 μM. RAW264.7 cells challenged with 200 ng ml−1 LPS and treated with 4 μM peptide concentration of Lf-KR showed significant inhibition in production of inflammatory mediators such as NO and TNF-α. The mRNA expression for iNOS and TNF-α was also suppressed (Table 3).103Another 18-residue hybrid peptide of the natural peptides papiliocin and magainin 2, PapMA-k, showed broad spectrum antibacterial activity, superior to parent peptides papiliocin and magainin 2 (geometric mean of MIC values against Gram-positive and Gram-negative bacteria was 5.3 μM and 5.7 μM, respectively).104 PapMA-k demonstrated good immunomodulatory activity since in RAW264.7 cells challenged with LPS, a significant inhibition in nitric oxide (NO) production was observed at peptide concentrations of 5–10 μM (Table 3). The gene expression of pro-inflammatory mediators such as TNF-α, IL-1β, MIP-1, and MIP-2 was considerably suppressed in LPS challenged RAW264.7 cells treated with 10 μM PapMA-k.104
One study reports the development of an AMP conjugated to a chitosan nanoparticle (CNM).105 This nanoconjugate, having the AMP microcin J25, was found to have potent activity against various bacterial strains, including MRSA, E. coli, tetracycline (Tet)-resistant enterotoxigenic E. coli (ETEC), etc.106 This nanoconjugate also reduced LPS-induced damage in RAW264.7 macrophages. Enhanced inhibition of production of proinflammatory cytokines TNF-α, IL-6, IL-1β, IL-8, TLR4, p-NF-κB, and p-p38 and the transcript and protein level was also observed upon treatment with CNM.106 This study indicates the potential of such conjugates to act as dual-functional agents. In another study, a peptide was developed from the LPS-binding structural motifs of limulus anti-LPS factor (LALF) protein, constituting amino acids 28 to 54 from the entire protein.107 This peptide fragment displayed moderate antibacterial activity in vitro, and also displayed LPS-neutralising ability, comparable to the parent protein.107 However, the study did not look at the mechanism of immunomodulation.
5. Antibacterial peptidomimetics as immunomodulators
One major roadblock in the development of HDPs is their poor protease stability apart from high production cost.67 The basic residues imparting the cationic charge to HDPs are indispensable for their activity. Due to the presence of chymotrypsin-like enzymes that cleave the peptide at basic residues, most clinical trials have been focused on topical infections.67 To expand on the development of HDPs as therapeutic agents, several solutions have been suggested. This has encouraged research into the creation of synthetic HDPs or non-natural peptide mimics of HDPs, as well as synthetic macromolecules with the goal of duplicating important HDP biophysical properties including cationic charge and the amphiphilic structure.108–110 A number of compounds with direct antibacterial action and decreased cytotoxicity have been developed as a result of systematic structure–activity relationship (SAR) research; some of these compounds are already in the clinical development stage.90,111Our group has recently developed a new class of antibacterial peptidomimetics: amino-acid conjugated small antibacterial molecules (ASAMs). ASAM-10 (12) exhibited good antibacterial activity and cell selectivity (MIC = 0.9–7.8 μg ml−1 and HC50 = 758 μg ml−1) and was selected as the lead molecule (Fig. 5A).112 It was observed that ASAM-10 was able to down-regulate the expression of genes corresponding to IL-6, IL-8, 1L-1β, and TNF-α in ARPE-19 cells infected with S. aureus and P. aeruginosa (Table 4) (Fig. 5D).112 Representative data for Gram-negative bacteria are provided (Fig. 5). A bimodal to unimodal size distribution of LPS and LTA aggregates upon treatment with 25 μg ml−1 ASAM-10 was seen (Fig. 5C). This inherent change in the nature of native LPS and LTA aggregates, coupled with enhanced stability of compound-treated aggregates (evidenced by the higher mean count rate), was hypothesized to be responsible for stalled overexpression of pro-inflammatory cytokines (Fig. 5C). Another molecule to tackle the problems associated with staphylococcal infections (MRSA) has been reported by our group.113 This compound 2 (13), bearing a biphenyl-lipidated lysine structure, was identified through structure–activity relationship studies (Fig. 4). The MIC value of the molecule against isolates of Gram-positive S. aureus is 3.1–6.2 μg ml−1. The compound significantly suppressed TNF-α production (13-fold relative to untreated case) at a concentration of 10 μg ml−1 in LTA challenged hPBMCs (Table 4).113
| Compounds | MIC | Immunomodulatory activity | Ref. |
|---|---|---|---|
| a μg mL−1. b μM. | |||
| ASAM-10 (12) | 0.9–7.8a | Lowers TNF-α, IL-6, IL-8, and IL-1β | 112 |
| Compound 2 (13) | 3.1–6.2a | Lowers TNF-α | 113 |
| NCK-10 (14) | 4.1–5.7b | Lowers TNF-α and IL-6 | 120 |
| SMAMP 3 (15) | 3.13–12.5a | Lowers TNF-α | 121 |
| HDAMP-1 (16) | 4.3b | Lowers TNF-α and NO | 122 |
| YL-36 (17) | 1–5b | Lowers NO | 123 |
| Compound 3 (18) | 0.6–2.4b | Lowers TNF-α and NF-κB | 124 |
20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 Bu:DM (19) :80 Bu:DM (19) |
3.13–25a | Modulates MCP-1, TNF-α, IL-1β, IL-10, and IL-1RA levels in a concentration dependent manner | 126 |
| QCybuAP and Qn-prAP (20) | 8–16a | Lowers TNF-α and IL-6 | 131 |
| LPEI (21) | 17.8a | Lowers TNF-α and IL-6 | 132 |
 | ||
| Fig. 4 Antibacterial and immunomodulatory peptidomimetics. | ||
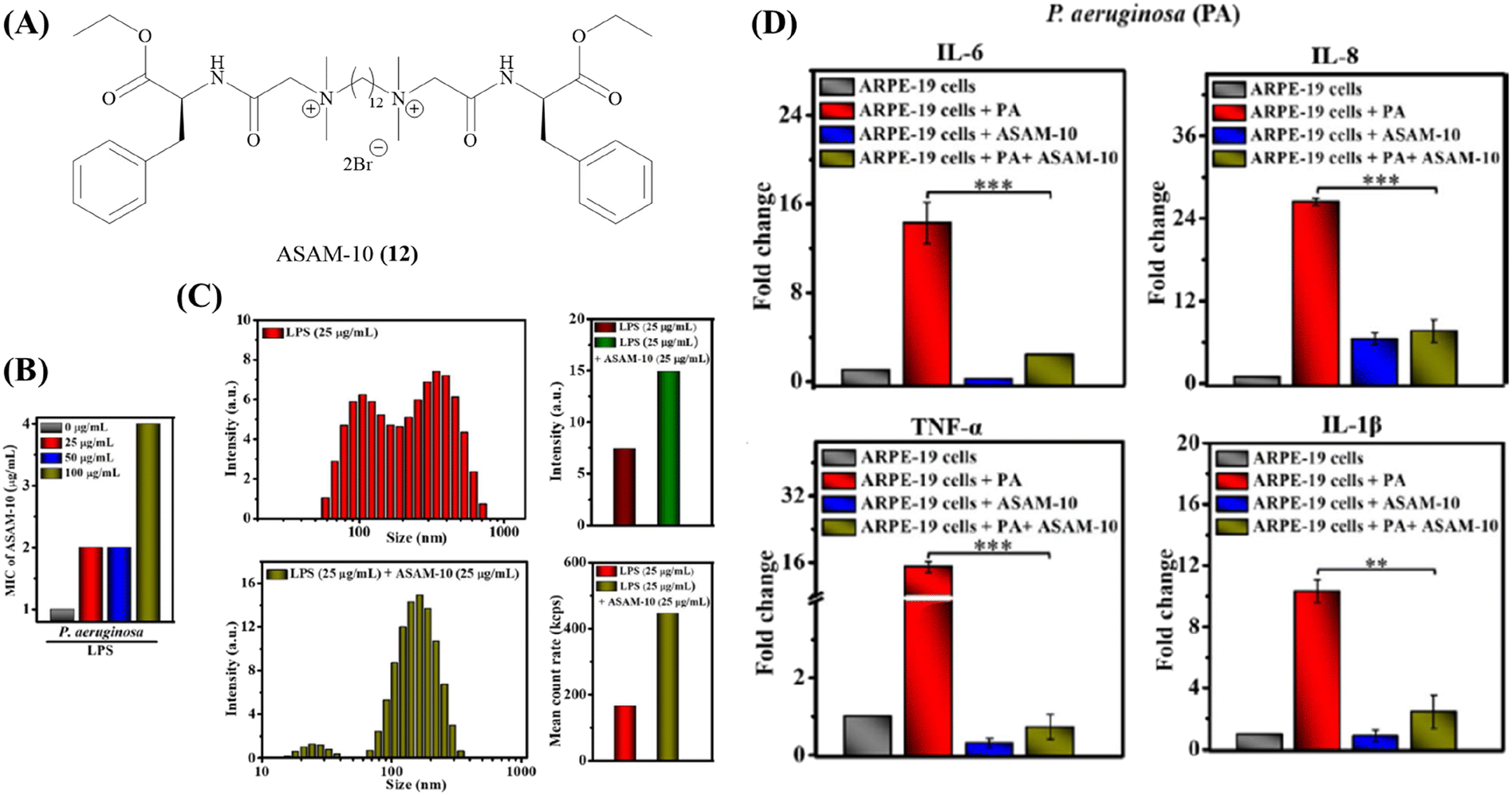 | ||
| Fig. 5 ASAM-10 – antibacterial and immunomodulatory peptidomimetic small molecule. (A) Structure of ASAM-10. (B) MIC of ASAM-10 in the presence of exogenous LPS. (C) LPS binding with ASAM-10 through DLS, size distribution and mean count rate. (D) Pro-inflammatory cytokine (IL-6, IL-8, TNF-α, IL-1β) levels upon ARPE-19 cell line infection by P. aeruginosa, in ASAM-10 treated and untreated cells. Reproduced from ref. 112 with permission from John Wiley and Sons, Copyright 2022. | ||
Our group has developed membrane-active naphthalene-based aryl–alkyl lysines with varying hydrophobicity, which inhibit the growth of disease-causing pathogens.114–119 NCK-10 (14) was the most potent against Gram-positive bacterial pathogens with MIC values that ranged from 4.1–5.7 μM (Fig. 4).120 It also showed a concentration dependent breakdown of the endotoxin LPS. NCK-10 (10 μg ml−1) also significantly inhibited the TNF-α and IL-6 production in hPBMCs challenged with LPS (Table 4).120
Yet another class of aryl-based synthetic mimics of antimicrobial peptides (SMAMPs) has been reported.121 The lead molecule, SMAMP 4, was the most selective and potent against both S. aureus and E. coli (HC50 = 656 μg ml−1 and MIC = 3.13 μg ml−1) (Fig. 4). However, RAW 264.7 macrophage cells pre-incubated with SMAMP 4 showed an increased production of pro-inflammatory markers TNF-α and IL-6 along with suppression of anti-inflammatory marker IL-10 upon LPS challenge (Table 4).121 However, SMAMP 3 (15), with a structural modification involving a pendant phenyl group attached to the benzyl core and a reduced number of charged amine groups, showed good antibacterial activity (HC50 = 537 μg ml−1 and MIC = 3.13–12.5 μg ml−1) and marginally suppressed LPS induced TNF-α production.
Another class of molecules is the ultra-short histidine derived antimicrobial peptides (HDAMPs).122 HDAMP-1 (16) was the most potent molecule (geometric mean of MIC = 4.3 μM). It showed a dose-dependent inhibition in nitric oxide (NO) production in RAW264.7 cells challenged with LPS. It also showed significant inhibition in TNF-α cytokine production (Table 4).122
The antimicrobial and immunomodulatory activities of a previously reported class of peptidomimetics, lipidated cyclic γ-AApeptides, have been evaluated in this study. YL-36 (17) exhibited broad spectrum antimicrobial activity (MIC = 1–5 μM) (Fig. 4).123 RAW 264.7 cells challenged with LPS were treated with different concentrations of the peptide to monitor the nitric oxide production (Table 4). YL-36 proved to be the most potent anti-TLR4 signalling agent (100% inhibition). The pre-treatment of cells with YL-36 (10 μM) prior to LPS challenge did not abolish the anti-TLR4 activity which suggested that the mechanism of inhibition of the TLR induced inflammatory response is different from binding of YL-36 to LPS. The production of TNF-α in LPS challenged HEK293 cells was also inhibited with an increase in YL-36 concentration.123 The same group has reported novel cyclic lipo-α-AApeptides.124 The trimeric peptide with a hexadecyl chain, compound 3 (18), was the most potent with MIC values between 0.6 and 2.4 μM (Fig. 4). A significant decrease in NF-κB activation (in HEK293 cells) and TNF-α production (in RAW264.7 cells) was observed in LPS challenged cells treated with the peptide (Table 4).124
HDP mimicking poly-β-peptides composed of β-amino acids have been previously reported.125 These peptides (average length of 20-mer) harboured hydrophobicity (butyl side chain, Bu) and cationic nature (primary amine side chain, DM) in variable ratios. The β-peptide 20:80 Bu:DM (19) was the most potent polymer (MIC = 3.13–25 μg ml−1), and it also showed good selectivity (HC50 = 200 μg ml−1).126 The immunomodulatory activity of 20:80 Bu:DM was assessed in terms of induction or release of chemokines or cytokines from peripheral blood mononuclear cells (PBMCs) with or without the challenge of P. aeruginosa lipopolysaccharide (LPS). An increased production of monocyte chemoattractant protein 1 (MCP-1) was observed in PBMCs treated with an increasing concentration of 20:80 Bu:DM (12.5–100 μg ml−1) both in the presence and absence of LPS. This peptide displayed concentration-dependent effects, with opposing features at high and low concentrations. While resulting in an enhanced pro-inflammatory cytokine response at high peptide concentrations, low concentrations of 12.5 and 25 μg ml−1 led to modest, but non-significant reduction in LPS-induced TNF-α. A similar effect was observed for IL-1β, with lower concentration of 25 μg mL−1 showing significant reduction in the levels of this key pro-inflammatory cytokine (Table 4). More importantly, the polymer significantly increased the release of anti-inflammatory mediator IL-1RA. These in vitro immunological studies suggested the possibility of using 20:80 Bu:DM to selectively activate, augment, or decrease innate immune responses.126
A new class of amphiphilic macromolecules based on poly-(isobutylene-alt-N-alkyl maleimide) with quaternary nitrogen bearing various groups (alkyl amides, cyclic-aliphatic chain amides, unsaturated aliphatic amides, PEG-like) hanging as pendants from the polymer backbone is reported by our group.127–130 The antibacterial activity of the polymers was tested against E. coli, S. aureus, and highly pathogenic MRSA and VRE.131 The side-chain structural variations significantly influenced the antibacterial properties of these polymers with the amide moiety being important for optimization of this activity. Immunomodulatory studies performed for all the polymers revealed an important role of the side chain (Fig. 6E). While some polymers possessed minimal antibacterial activity and immunomodulatory properties, two polymers, namely, QCybuAP (20a) and Qn-prAP (20b) with sufficient hydrophobicity and hydrogen bonding were potent antibacterial agents (MIC = 8–16 μg ml−1) and immunomodulators (Fig. 4).131 The hPBMCs stimulated with LPS in the presence and absence of 20 μg ml−1 QCybuAP showed an 80% inhibition of pro-inflammatory cytokines TNF-α and IL-6 compared with LPS alone (Table 4) (Fig. 6E). This study also investigated the LPS-binding ability of these polymers though fluorescent LPS probes. The overall amphiphilicity influenced LPS-neutralizing ability. Ester-bearing and unsaturation-containing polymers, with lower overall hydrophobicity, demonstrated low binding affinity for LPS, and thereby, lesser inhibition of cytokines (Fig. 6E). It was also found through DLS studies that these polymers disrupted the inherent LPS aggregates and induced formation of pseudoaggregates of LPS-polymers (Fig. 6B and C). The light scattering intensity and mean count rate data suggested that the pseudoaggregates formed by the immunomodulatory LPS-binding polymers were more stable (Fig. 6D). It was hypothesized that the formation of such non-natural aggregates may interfere with the recognition of LPS and prevent immune responses.131
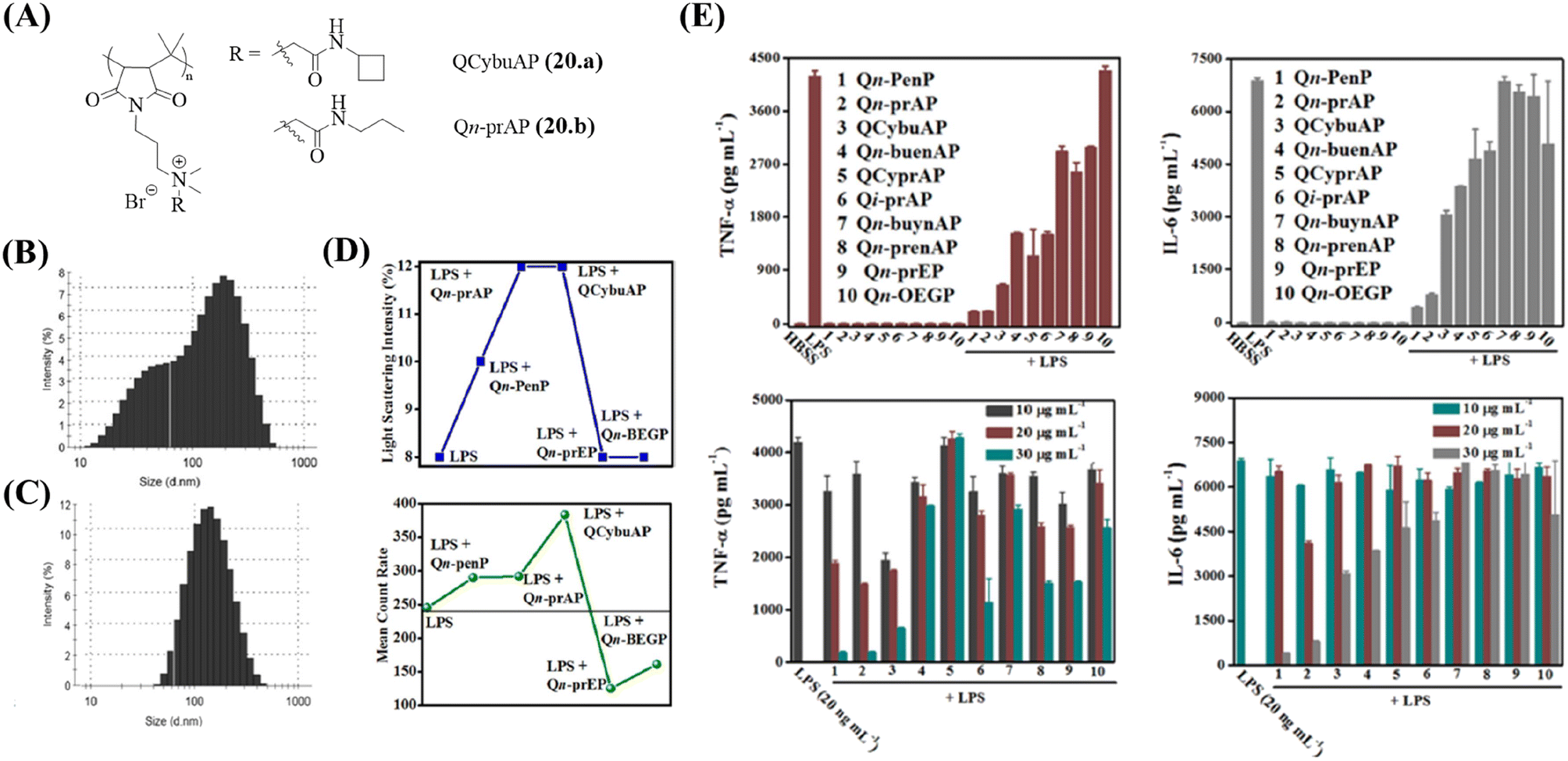 | ||
| Fig. 6 Cationic macromolecules – antibacterial and immunomodulatory macromolecules. (A) Structures of QCybuAP and Qn-prAP. Size distribution of (B) LPS aggregates and (C) LPS + QCybuAP. (D) Light scattering intensities and mean count rate of LPS in the presence and absence of macromolecules. (E) Secretion of pro-inflammatory cytokines (TNF-α, IL-6) after stimulation of hPMBCs with LPS. QCybuAP (20a) is represented as 3 and Qn-prAP (20b) is represented as 2 in the graphs. Reproduced from ref. 131 with permission from American Chemical Society, Copyright 2016. | ||
The therapeutic potential of linear polyethyleneimine (LPEI) (21) has been explored (Fig. 4).132 It showed moderate antibacterial activity against a panel of multi-drug resistant MRSA strains (mean MIC = 17.8 μg ml−1). LPEI at high concentration (125 μg ml−1) completely inhibited the production of pro-inflammatory cytokines TNF-α and IL-6 on challenge with bacterial secretomes (Table 4).132
6. Conclusions and future perspectives
The problem of infection is complicated by the occurrence of a hyperimmune response and eventual sepsis in its later stages. The nebulous therapeutic strategy and the high mortality rate of sepsis warrant investigations towards a holistic redressal strategy, which takes into consideration the closely linked relationship between infection and inflammation. Towards this goal, we have attempted here to collate examples of key antibacterial therapeutics, namely, antibiotics, antimicrobial peptides and peptidomimetics, which possess additional anti-inflammatory properties, along with inherent antibacterial activity. The various members of these classes bear diverging immunomodulatory effects. However in this review, we focused on those candidates which can show immunosuppressive effects. The goal of such dual-functional antibacterial–immunosuppressive therapeutics would be, in addition to bacterial clearance, to mitigate the effects of excessive inflammatory response arising from severe infections and to prevent the advent of sepsis.One of the major classes of antibacterial agents which have known immunomodulatory properties is the conventionally used antibiotics. There is a lot of diversity in the immunomodulatory effects of different antibiotic classes. A lot of the available information of immunomodulatory properties of antibiotics first originated from clinical reports as well as clinical trials performed for the antibacterial activity of the antibiotic. However, this information was not investigated in detail for many of the classes. More importantly, there is a lacuna in the mechanistic understanding of the immunomodulatory properties of antibiotics. The understanding is further muddled by the opposing effects observed for some antibiotics, in different experiments or in different model systems such as mice, clinical trials, etc. To address this confusion, the actual host targets of these antibiotics and the interplay between their antibacterial properties and immunomodulatory activity need to be investigated in pre-clinical and clinical studies. As mentioned earlier, some antibiotics can also aggravate inflammatory cascades and may prove detrimental in tackling systemic infections. Already, a significant amount of clinical data on immunomodulatory effects of commonly prescribed antibiotics is available, through some clinical trials. These data should be analysed, and protocols and SOPs should be developed for specifying antibiotic prescription for sepsis patients for doctors.
The emerging class of antibacterial agents, host defense peptides (HDPs), has also been thoroughly investigated for immunomodulatory properties. Robert Hancock and co-workers have been contributing extensively to these studies and have identified HDPs with both pro-inflammatory and anti-inflammatory properties. The extent of immunomodulatory studies for HDPs, however, has been limited to the laboratories. Wide-ranging studies in higher animal models need to be performed for better understanding these effects. Also, synthetic antibacterial peptidomimetics can be the best choice, particularly for multi-drug resistant infections. Given their potent activity and low propensity of resistance development, if such peptidomimetics also possess immunomodulatory properties, their therapeutic potential can be greatly enhanced. There are already reports of LPS-interacting peptidomimetics, which show anti-inflammatory properties in endotoxin-challenged macrophages. Many studies have investigated the interaction of peptides/peptidomimetics with LPS through a myriad of techniques, such as fluorescence spectroscopy, circular dichroism, NMR, etc. However, the mechanistic effects of this binding on recognition of LPS and the eventual resultant immunomodulatory activity need to be investigated in greater detail. The benefit of LPS-interactors is that they can show an antigen-responsive anti-inflammatory response, rather than leading to a block in host immune function. Additionally, small molecules can also have effects on the down-stream processes of immune responses. These effects can also help mitigate excessive inflammation. The host-modulating ability of such dual-functional therapeutics is of particular significance in cases of polymicrobial infections, where a single therapeutic has direct antibacterial efficacy only against a limited pathogen group. A host-modulating therapeutic can also mitigate excessive inflammatory response originating from other microbial species. However, the effect of this on pathogen clearance should be considered while proceeding with therapy. Hence, it is important to make efforts towards developing such dual-functional therapeutics. More importantly, the challenges which can be encountered in taking forward such dual-functional antibacterial and immunomodulatory therapeutics need to be factored in during the development process. Starting from commercial value, interest from the pharmaceutical industry, and availability of integrated animal model studies, which assess both the parameters of infection and inflammation simultaneously, as well as feasibility of clinical trials for such lead compounds, are just a few of the points which will require extensive support from all segments of the research and development community, if such dual-functional therapeutics are to be translated to clinical use.
Through this review, it was our endeavour to collate reports of dual-functional antibacterial and immunomodulatory molecules. Currently, the understanding of infection and inflammation, particularly in the context of complicated bacterial infections, is limited, contradictory in some cases, and inconsistent. Hence, the effect of the therapeutic strategy on inflammatory responses of the host is similarly much more ambiguous. Persistent efforts to understand effects of antibacterial therapy on inflammation and anti-inflammatory therapy on infection, particularly in clinically relevant model systems and settings, are of urgent need. However, the current in vitro and animal study results provide some hope for enhanced efficacy of existing dual-functional therapeutics, particularly in mitigating excessive inflammation and cytokine storm in complicated infections. Similarly, clinical reports of effects of antibiotics on inflammation also provide directions for upcoming laboratory and clinical research. These results encourage us to consider the possibility of dual-functional therapeutics replacing conventional antibiotic and steroid therapy in sepsis management. Hence, we believe that this review compilation is timely and will prompt detailed mechanistic investigations into broader effects of such dual-functional therapeutics.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
J. H. acknowledges DBT (BT/PR31801/MED) and SERB (CRG/2020/003118), Govt. of India. Y. A. acknowledges CSIR for a research fellowship.Notes and references
- G. Dhanda, Y. Acharya and J. Haldar, ACS Omega, 2023, 8, 10757–10783 CrossRef CAS PubMed.
- K. Kubelkova and A. Macela, Front. Cell. Infect. Microbiol., 2019, 9, 241 CrossRef CAS PubMed.
- K. Van Avondt, N. M. van Sorge and L. Meyaard, PLoS Pathog., 2015, 11, e1004644 CrossRef PubMed.
- B. Albiger, S. Dahlberg, B. Henriques-Normark and S. Normark, J. Intern. Med., 2007, 261, 511–528 CrossRef CAS PubMed.
- J. Cohen, Nature, 2002, 420, 885–891 CrossRef CAS PubMed.
- D. S. Uppu, C. Ghosh and J. Haldar, Microb. Pathog., 2015, 80, 7–13 CrossRef PubMed.
- D. Jarczak, S. Kluge and A. Nierhaus, Front. Med., 2021, 8, 628302 CrossRef PubMed.
- Centre for Disease Control and Prevention, Sepsis – https://www.cdc.gov/sepsis/what-is-sepsis.html (Accessed on April 5, 2023) Search PubMed.
- R. S. Hotchkiss, L. L. Moldawer, S. M. Opal, K. Reinhart, I. R. Turnbull and J. L. Vincent, Nat. Rev. Dis. Primers, 2016, 2, 16045 CrossRef PubMed.
- L. Y. Tan, T. V. Komarasamy and V. R. Balasubramaniam, Front. Immunol., 2021, 12, 742941 CrossRef CAS PubMed.
- B. J. Langford, M. So, S. Raybardhan, V. Leung, D. Westwood, D. R. MacFadden, J. R. Soucy and N. Daneman, Clin. Microbiol. Infect., 2020, 26, 1622–1629 CrossRef CAS PubMed.
- W. H. Tay, K. K. Chong and K. A. Kline, J. Mol. Biol., 2016, 428, 3355–3371 CrossRef CAS PubMed.
- “It's time we “SALVAGE” patients with sepsis!!!” – https://journals.lww.com/lungindia/Fulltext/2005/22020/IT_S_TIME_WE__SALVAGE__PATIENTS_WITH_SEPSIS____.1.aspx (Acessed on April 5, 2023) Search PubMed.
- J. M. Cavaillon, M. Singer and T. Skirecki, EMBO Mol. Med., 2020, 12, e10128 CrossRef CAS PubMed.
- A. Sauer, K. Peukert, C. Putensen and C. Bode, Eur. Respir. Rev., 2021, 30, 210093 CrossRef PubMed.
- D. M. Easton, A. Nijnik, M. L. Mayer and R. E. Hancock, Trends Biotechnol., 2009, 27, 582–590 CrossRef CAS PubMed.
- S. V. Guryanova and T. V. Ovchinnikova, Int. J. Mol. Sci., 2022, 23, 2499 CrossRef CAS PubMed.
- R. E. Hancock, E. F. Haney and E. E. Gill, Nat. Rev. Immunol., 2016, 16, 321–334 CrossRef CAS PubMed.
- R. E. Hancock and G. Diamond, Trends Microbiol., 2000, 8, 402–410 CrossRef CAS PubMed.
- K. L. Brown and R. E. Hancock, Curr. Opin. Immunol., 2006, 18, 24–30 CrossRef CAS PubMed.
- S. C. Mansour, O. M. Pena and R. E. Hancock, Trends Immunol., 2014, 35, 443–450 CrossRef CAS PubMed.
- S. J. Kim and H. M. Kim, BMB Rep., 2017, 50, 55–57 CrossRef CAS PubMed.
- E. M. Palsson-McDermott and L. A. O'Neill, Immunology, 2004, 113, 153–162 CrossRef CAS PubMed.
- R. Karima, S. Matsumoto, H. Higashi and K. Matsushima, Mol. Med. Today, 1999, 5, 123–132 CrossRef CAS PubMed.
- G. Kapoor, S. Saigal and A. Elongavan, J. Anaesthesiol., Clin. Pharmacol., 2017, 33, 300–305 CrossRef CAS PubMed.
- G. A. Gualdoni, T. Lingscheid, K. G. Schmetterer, A. Hennig, P. Steinberger and G. J. Zlabinger, Sci. Rep., 2015, 5, 12016 CrossRef PubMed.
- L. C. Fan, J. L. Lin, J. W. Yang, B. Mao, H. W. Lu, B. X. Ge, A. M. K. Choi and J. F. Xu, Am. J. Physiol., 2017, 313, L677–L686 Search PubMed.
- F. F. Stellari, A. Sala, G. Donofrio, F. Ruscitti, P. Caruso, T. M. Topini, K. P. Francis, X. Li, C. Carnini, M. Civelli and G. Villetti, Pharmacol. Res. Perspect., 2014, 2, e00058 Search PubMed.
- J. Altenburg, C. S. de Graaff, T. S. van der Werf and W. G. Boersma, Respiration, 2011, 81, 67–74 CrossRef CAS PubMed.
- M. Shinkai, M. O. Henke and B. K. Rubin, Pharmacol. Ther., 2008, 117, 393–405 CrossRef CAS PubMed.
- A. Spyridaki, M. Raftogiannis, A. Antonopoulou, T. Tsaganos, C. Routsi, F. Baziaka, V. Karagianni, M. Mouktaroudi, P. Koutoukas, A. Pelekanou, A. Kotanidou, S. E. Orfanos, J. W. van der Meer, M. G. Netea and E. J. Giamarellos-Bourboulis, Antimicrob. Agents Chemother., 2012, 56, 3819–3825 CrossRef CAS PubMed.
- M. J. Lorenzo, I. Moret, B. Sarria, E. Cases, J. Cortijo, R. Mendez, J. Molina, A. Gimeno and R. Menendez, Respir. Res., 2015, 16, 15 CrossRef PubMed.
- I. Chopra and M. Roberts, Microbiol. Mol. Biol. Rev., 2001, 65, 232–260 CrossRef CAS PubMed ; second page, table of contents.
- K. Peukert, M. Fox, S. Schulz, C. Feuerborn, S. Frede, C. Putensen, H. Wrigge, B. M. Kummerer, S. David, B. Seeliger, T. Welte, E. Latz, D. Klinman, C. Wilhelm, F. Steinhagen and C. Bode, Am. J. Respir. Crit. Care Med., 2021, 204, 53–63 CrossRef CAS PubMed.
- L. M. Golub, H. M. Lee, M. E. Ryan, W. V. Giannobile, J. Payne and T. Sorsa, Adv. Dent. Res., 1998, 12, 12–26 CrossRef CAS PubMed.
- A. Patel, H. Khande, H. Periasamy and S. Mokale, Inflammation, 2020, 43, 1035–1043 CrossRef PubMed.
- J. Steinberg, J. Halter, H. Schiller, L. Gatto, D. Carney, H. M. Lee, L. Golub and G. Nieman, Shock, 2005, 24, 348–356 CrossRef CAS PubMed.
- U. G. McCann, L. A. Gatto, B. Searles, D. E. Carney, C. J. Lutz, A. L. Picone, H. J. Schiller and G. F. Nieman, J. Extra-Corpor. Technol., 1999, 31, 67–75 Search PubMed.
- S. M. R. Hashemian, T. Farhadi and M. Ganjparvar, Drug Des., Dev. Ther., 2018, 12, 1759–1767 CrossRef CAS PubMed.
- S. Yoshizawa, K. Tateda, T. Saga, Y. Ishii and K. Yamaguchi, Antimicrob. Agents Chemother., 2012, 56, 1744–1748 CrossRef CAS PubMed.
- J. Chen, G. Feng, Y. Song, J. B. Wardenburg, S. Lin, I. Inoshima, M. Otto and R. G. Wunderink, PLoS One, 2013, 8, e67994 CrossRef CAS PubMed.
- M. E. Akinnusi, A. Hattemer, W. Gao and A. A. El-Solh, Crit. Care Med., 2011, 39, 1944–1952 CrossRef CAS PubMed.
- C. Jacqueline, A. Broquet, A. Roquilly, M. Davieau, J. Caillon, F. Altare, G. Potel and K. Asehnoune, J. Infect. Dis., 2014, 210, 814–823 CrossRef CAS PubMed.
- P. Garcia-Roca, J. Mancilla-Ramirez, A. Santos-Segura, M. Fernandez-Aviles and E. Calderon-Jaimes, Arch. Med. Res., 2006, 37, 31–35 CrossRef CAS PubMed.
- Z. Franks, R. A. Campbell, A. V. de Abreu, J. T. Holloway, J. E. Marvin, B. F. Kraemer, G. A. Zimmerman, A. S. Weyrich and M. T. Rondina, Thromb. Haemostasis, 2013, 109, 684–695 CrossRef CAS PubMed.
- T. Gruger, T. Schmidt, N. Schnitzler, S. Nidermajer, K. Brandenburg and J. Zundorf, Chemotherapy, 2012, 58, 206–211 CrossRef PubMed.
- K. J. Aldred, R. J. Kerns and N. Osheroff, Biochemistry, 2014, 53, 1565–1574 CrossRef CAS PubMed.
- C. Beisswenger, A. Honecker, A. Kamyschnikow, M. Bischoff, T. Tschernig and R. Bals, Respir. Res., 2014, 15, 82 CrossRef PubMed.
- H. C. Huang, C. C. Shieh, W. L. Yu, K. C. Cheng, C. C. Chen, S. T. Chang and Y. C. Chuang, Respirology, 2008, 13, 47–52 CrossRef PubMed.
- A. Dalhoff, Infection, 2005, 33(Suppl 2), 55–70 CrossRef CAS PubMed.
- F. G. Araujo, T. L. Slifer and J. S. Remington, Clin. Microbiol. Infect., 2002, 8, 26–30 CrossRef CAS PubMed.
- Y. Enoki, Y. Ishima, R. Tanaka, K. Sato, K. Kimachi, T. Shirai, H. Watanabe, V. T. Chuang, Y. Fujiwara, M. Takeya, M. Otagiri and T. Maruyama, PLoS One, 2015, 10, e0130248 CrossRef PubMed.
- I. Shalit, L. Horev-Azaria, I. Fabian, H. Blau, N. Kariv, I. Shechtman, H. Alteraz and Y. Kletter, Antimicrob. Agents Chemother., 2002, 46, 2442–2449 CrossRef CAS PubMed.
- A. Sabnis, K. L. Hagart, A. Klockner, M. Becce, L. E. Evans, R. C. D. Furniss, D. A. Mavridou, R. Murphy, M. M. Stevens, J. C. Davies, G. J. Larrouy-Maumus, T. B. Clarke and A. M. Edwards, eLife, 2021, 10, e65836 CrossRef CAS PubMed.
- S. A. David, K. A. Balasubramanian, V. I. Mathan and P. Balaram, Biochim. Biophys. Acta, 1992, 1165, 147–152 CrossRef CAS PubMed.
- P. Pristovsek and J. Kidric, J. Med. Chem., 1999, 42, 4604–4613 CrossRef CAS PubMed.
- J. Wang, W. Shao, H. Niu, T. Yang, Y. Wang and Y. Cai, Front. Pharmacol., 2019, 10, 729 CrossRef CAS PubMed.
- Z. Sheikh, P. Bradbury, T. A. Reekie, M. Pozzoli, P. D. Robinson, M. Kassiou, P. M. Young, H. X. Ong and D. Traini, Eur. J. Pharmacol., 2021, 902, 174098 CrossRef CAS PubMed.
- H. Niu, T. Yang, J. Wang, R. Wang and Y. Cai, Front. Pharmacol., 2020, 11, 602054 CrossRef CAS PubMed.
- P. Matzneller, S. Strommer, C. Drucker, K. Petroczi, C. Schorgenhofer, E. Lackner, B. Jilma and M. Zeitlinger, Clin. Pharmacol. Ther., 2017, 101, 773–781 CrossRef CAS PubMed.
- H. Tsubery, I. Ofek, S. Cohen, M. Eisenstein and M. Fridkin, Mol. Pharmacol., 2002, 62, 1036–1042 CrossRef CAS PubMed.
- E. M. Kosciuczuk, P. Lisowski, J. Jarczak, N. Strzalkowska, A. Jozwik, J. Horbanczuk, J. Krzyzewski, L. Zwierzchowski and E. Bagnicka, Mol. Biol. Rep., 2012, 39, 10957–10970 CrossRef CAS PubMed.
- I. Nagaoka, S. Hirota, F. Niyonsaba, M. Hirata, Y. Adachi, H. Tamura and D. Heumann, J. Immunol., 2001, 167, 3329–3338 CrossRef CAS PubMed.
- F. Torres-Juarez, A. Cardenas-Vargas, A. Montoya-Rosales, I. Gonzalez-Curiel, M. H. Garcia-Hernandez, J. A. Enciso-Moreno, R. E. Hancock and B. Rivas-Santiago, Infect. Immun., 2015, 83, 4495–4503 CrossRef CAS PubMed.
- N. Mookherjee, K. L. Brown, D. M. Bowdish, S. Doria, R. Falsafi, K. Hokamp, F. M. Roche, R. Mu, G. H. Doho, J. Pistolic, J. P. Powers, J. Bryan, F. S. Brinkman and R. E. Hancock, J. Immunol., 2006, 176, 2455–2464 CrossRef CAS PubMed.
- Y. J. Gordon, E. G. Romanowski and A. M. McDermott, Curr. Eye Res., 2005, 30, 505–515 CrossRef CAS PubMed.
- R. E. Hancock and H. G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed.
- Y. Huan, Q. Kong, H. Mou and H. Yi, Front. Microbiol., 2020, 11, 582779 CrossRef PubMed.
- J. K. Kim, E. Lee, S. Shin, K. W. Jeong, J. Y. Lee, S. Y. Bae, S. H. Kim, J. Lee, S. R. Kim, D. G. Lee, J. S. Hwang and Y. Kim, J. Biol. Chem., 2011, 286, 41296–41311 CrossRef CAS PubMed.
- L. Wei, J. Yang, X. He, G. Mo, J. Hong, X. Yan, D. Lin and R. Lai, J. Med. Chem., 2013, 56, 3546–3556 CrossRef CAS PubMed.
- R. E. Hancock and R. Lehrer, Trends Biotechnol., 1998, 16, 82–88 CrossRef CAS PubMed.
- A. K. Marr, W. J. Gooderham and R. E. Hancock, Curr. Opin. Pharmacol., 2006, 6, 468–472 CrossRef CAS PubMed.
- G. Wang, J. Biol. Chem., 2008, 283, 32637–32643 CrossRef CAS PubMed.
- B. Jacob, I. S. Park, J. K. Bang and S. Y. Shin, J. Pept. Sci., 2013, 19, 700–707 CrossRef CAS PubMed.
- Y. Xiao, Y. Cai, Y. R. Bommineni, S. C. Fernando, O. Prakash, S. E. Gilliland and G. Zhang, J. Biol. Chem., 2006, 281, 2858–2867 CrossRef CAS PubMed.
- G. Rajasekaran, S. D. Kumar, S. Yang and S. Y. Shin, Eur. J. Med. Chem., 2019, 182, 111623 CrossRef CAS PubMed.
- M. Taniguchi, A. Ochiai, K. Takahashi, S. Nakamichi, T. Nomoto, E. Saitoh, T. Kato and T. Tanaka, Biopolymers, 2015, 104, 73–83 CrossRef CAS PubMed.
- M. Taniguchi, A. Ochiai, K. Matsushima, K. Tajima, T. Kato, E. Saitoh and T. Tanaka, Peptides, 2016, 75, 101–108 CrossRef CAS PubMed.
- Q. P. Lin, L. F. Zhou, N. N. Li, Y. Q. Chen, B. C. Li, Y. F. Cai and S. Q. Zhang, Eur. J. Pharmacol., 2008, 596, 160–165 CrossRef CAS PubMed.
- H. T. Chou, T. Y. Kuo, J. C. Chiang, M. J. Pei, W. T. Yang, H. C. Yu, S. B. Lin and W. J. Chen, Int. J. Antimicrob. Agents, 2008, 32, 130–138 CrossRef CAS PubMed.
- L. H. Li, T. C. Ju, C. Y. Hsieh, W. C. Dong, W. T. Chen, K. F. Hua and W. J. Chen, PLoS One, 2017, 12, e0182057 CrossRef PubMed.
- M. van Eijk, A. van Dijk, C. K. van der Ent, H. G. M. Arets, E. Breukink, N. van Os, R. Adrichem, S. van der Water, R. L. Gomez, M. Kristensen, M. Hessing, S. Jekhmane, M. Weingarth, R. A. W. Veldhuizen, E. J. A. Veldhuizen and H. P. Haagsman, Biochim. Biophys. Acta, Gen. Subj., 2021, 1865, 129951 CrossRef CAS PubMed.
- N. van Os, A. Javed, F. Broere, A. van Dijk, M. D. Balhuizen, M. van Eijk, S. H. M. Rooijakkers, B. W. Bardoel, D. A. C. Heesterbeek, H. P. Haagsman and E. Veldhuizen, J. Global Antimicrob. Resist., 2022, 30, 406–413 CrossRef CAS PubMed.
- Y. H. Nan, J. K. Bang and S. Y. Shin, Peptides, 2009, 30, 832–838 CrossRef CAS PubMed.
- D. Shang, Y. Sun, C. Wang, S. Wei, L. Ma and L. Sun, Appl. Microbiol. Biotechnol., 2012, 96, 1551–1560 CrossRef CAS PubMed.
- W. Dong, X. Mao, Y. Guan, Y. Kang and D. Shang, Sci. Rep., 2017, 7, 40228 CrossRef CAS PubMed.
- H. Sun, D. V. Greathouse, O. S. Andersen and R. E. Koeppe, J. Biol. Chem., 2008, 283, 22233–22243 CrossRef CAS PubMed.
- S. Srivastava and J. K. Ghosh, Antimicrob. Agents Chemother., 2013, 57, 2457–2466 CrossRef CAS PubMed.
- G. Rajasekaran, R. Kamalakannan and S. Y. Shin, J. Pept. Sci., 2015, 21, 779–785 CrossRef CAS PubMed.
- C. D. Fjell, J. A. Hiss, R. E. Hancock and G. Schneider, Nat. Rev. Drug Discovery, 2011, 11, 37–51 CrossRef PubMed.
- S. E. Blondelle and R. A. Houghten, Biochemistry, 1992, 31, 12688–12694 CrossRef CAS PubMed.
- D. I. Chan, E. J. Prenner and H. J. Vogel, Biochim. Biophys. Acta, 2006, 1758, 1184–1202 CrossRef CAS PubMed.
- B. Deslouches, K. Islam, J. K. Craigo, S. M. Paranjape, R. C. Montelaro and T. A. Mietzner, Antimicrob. Agents Chemother., 2005, 49, 3208–3216 CrossRef CAS PubMed.
- B. Deslouches, J. D. Steckbeck, J. K. Craigo, Y. Doi, T. A. Mietzner and R. C. Montelaro, Antimicrob. Agents Chemother., 2013, 57, 2511–2521 CrossRef CAS PubMed.
- K. H. Park, Y. H. Nan, Y. Park, J. I. Kim, I. S. Park, K. S. Hahm and S. Y. Shin, Biochim. Biophys. Acta, 2009, 1788, 1193–1203 CrossRef CAS PubMed.
- C. B. Park, K. S. Yi, K. Matsuzaki, M. S. Kim and S. C. Kim, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8245–8250 CrossRef CAS PubMed.
- Y. H. Nan, K. H. Park, Y. J. Jeon, Y. Park, I. S. Park, K. S. Hahm and S. Y. Shin, Protein Pept. Lett., 2007, 14, 1003–1007 CrossRef CAS PubMed.
- M. F. Mohamed, M. I. Hamed, A. Panitch and M. N. Seleem, Antimicrob. Agents Chemother., 2014, 58, 4113–4122 CrossRef PubMed.
- M. F. Mohamed and M. N. Seleem, Drug Des., Dev. Ther., 2014, 8, 1979–1983 CrossRef PubMed.
- M. F. Mohamed, A. Brezden, H. Mohammad, J. Chmielewski and M. N. Seleem, Sci. Rep., 2017, 7, 6953 CrossRef PubMed.
- J. Alvarez-Bravo, S. Kurata and S. Natori, Biochem. J., 1994, 302(Pt 2), 535–538 CrossRef CAS PubMed.
- P. Jantaruk, S. Roytrakul, S. Sitthisak and D. Kunthalert, PLoS One, 2017, 12, e0183852 CrossRef PubMed.
- C. Ajish, S. Yang, S. D. Kumar, E. Y. Kim, H. J. Min, C. W. Lee, S. H. Shin and S. Y. Shin, Sci. Rep., 2022, 12, 4365 CrossRef CAS PubMed.
- A. Shin, E. Lee, D. Jeon, Y. G. Park, J. K. Bang, Y. S. Park, S. Y. Shin and Y. Kim, Biochemistry, 2015, 54, 3921–3931 CrossRef CAS PubMed.
- H. Yu, Z. Ma, S. Meng, S. Qiao, X. Zeng, Z. Tong and K. C. Jeong, Carbohydr. Polym., 2021, 253, 117309 CrossRef CAS PubMed.
- Y. Haitao, C. Yifan, S. Mingchao and H. Shuaijuan, Front. Immunol., 2021, 12, 811381 CrossRef PubMed.
- C. A. Weiss, K. R. Wasiluk, T. A. Kellogg and D. L. Dunn, Surgery, 2000, 128, 339–344 CrossRef PubMed.
- C. Ghosh and J. Haldar, ChemMedChem, 2015, 10, 1606–1624 CrossRef CAS PubMed.
- C. Ghosh, P. Sarkar, R. Issa and J. Haldar, Trends Microbiol., 2019, 27, 323–338 CrossRef CAS PubMed.
- M. M. Konai, B. Bhattacharjee, S. Ghosh and J. Haldar, Biomacromolecules, 2018, 19, 1888–1917 CrossRef CAS PubMed.
- M. Lesiuk, M. Paduszynska and K. E. Greber, Antibiotics, 2022, 11, 1062 CrossRef CAS PubMed.
- S. Barman, G. Dhanda, P. Naik, R. Mukherjee, L. Jolly, J. Joseph and J. Haldar, Adv. Ther., 2022, 5, 2100234 CrossRef CAS.
- C. Ghosh, P. Sarkar, S. Samaddar, D. Uppu and J. Haldar, Chem. Commun., 2017, 53, 8427–8430 RSC.
- C. Ghosh, G. B. Manjunath, P. Akkapeddi, V. Yarlagadda, J. Hoque, D. S. Uppu, M. M. Konai and J. Haldar, J. Med. Chem., 2014, 57, 1428–1436 CrossRef CAS PubMed.
- C. Ghosh, G. B. Manjunath, M. M. Konai, D. S. Uppu, J. Hoque, K. Paramanandham, B. R. Shome and J. Haldar, PLoS One, 2015, 10, e0144094 CrossRef PubMed.
- C. Ghosh, G. B. Manjunath, M. M. Konai, D. S. Uppu, K. Paramanandham, B. R. Shome, R. Ravikumar and J. Haldar, ACS Infect. Dis., 2016, 2, 111–122 CrossRef CAS PubMed.
- S. D. Dowall, K. Bewley, R. J. Watson, S. S. Vasan, C. Ghosh, M. M. Konai, G. Gausdal, J. B. Lorens, J. Long, W. Barclay, I. Garcia-Dorival, J. Hiscox, A. Bosworth, I. Taylor, L. Easterbrook, J. Pitman, S. Summers, J. Chan-Pensley, S. Funnell, J. Vipond, S. Charlton, J. Haldar, R. Hewson and M. W. Carroll, Viruses, 2016, 8, 277 CrossRef PubMed.
- C. Ghosh, S. Chaubey, U. Tatu and J. Haldar, MedChemComm, 2017, 8, 434–439 RSC.
- C. Ghosh, V. Yadav, W. Younis, H. Mohammad, Y. A. Hegazy, M. N. Seleem, K. Sanyal and J. Haldar, ACS Infect. Dis., 2017, 3, 293–301 CrossRef CAS PubMed.
- C. Ghosh, N. Harmouche, B. Bechinger and J. Haldar, ACS Omega, 2018, 3, 9182–9190 CrossRef CAS PubMed.
- H. D. Thaker, A. Som, F. Ayaz, D. Lui, W. Pan, R. W. Scott, J. Anguita and G. N. Tew, J. Am. Chem. Soc., 2012, 134, 11088–11091 CrossRef CAS PubMed.
- R. N. Murugan, B. Jacob, M. Ahn, E. Hwang, H. Sohn, H. N. Park, E. Lee, J. H. Seo, C. Cheong, K. Y. Nam, J. K. Hyun, K. W. Jeong, Y. Kim, S. Y. Shin and J. K. Bang, PLoS One, 2013, 8, e80025 CrossRef PubMed.
- Y. Li, C. Smith, H. Wu, S. Padhee, N. Manoj, J. Cardiello, Q. Qiao, C. Cao, H. Yin and J. Cai, ACS Chem. Biol., 2014, 9, 211–217 CrossRef CAS PubMed.
- S. Padhee, C. Smith, H. Wu, Y. Li, N. Manoj, Q. Qiao, Z. Khan, C. Cao, H. Yin and J. Cai, ChemBioChem, 2014, 15, 688–694 CrossRef CAS PubMed.
- Q. Zhang, P. Ma, J. Xie, S. Zhang, X. Xiao, Z. Qiao, N. Shao, M. Zhou, W. Zhang, C. Dai, Y. Qian, F. Qi and R. Liu, Biomater. Sci., 2019, 7, 2144–2151 RSC.
- H. Etayash, Y. Qian, D. Pletzer, Q. Zhang, J. Xie, R. Cui, C. Dai, P. Ma, F. Qi, R. Liu and R. E. W. Hancock, J. Med. Chem., 2020, 63, 12921–12928 CrossRef CAS PubMed.
- D. S. Uppu, P. Akkapeddi, G. B. Manjunath, V. Yarlagadda, J. Hoque and J. Haldar, Chem. Commun., 2013, 49, 9389–9391 RSC.
- D. S. Uppu, S. Samaddar, J. Hoque, M. M. Konai, P. Krishnamoorthy, B. R. Shome and J. Haldar, Biomacromolecules, 2016, 17, 3094–3102 CrossRef CAS PubMed.
- D. Uppu, M. M. Konai, U. Baul, P. Singh, T. K. Siersma, S. Samaddar, S. Vemparala, L. W. Hamoen, C. Narayana and J. Haldar, Chem. Sci., 2016, 7, 4613–4623 RSC.
- D. S. Uppu, S. Samaddar, C. Ghosh, K. Paramanandham, B. R. Shome and J. Haldar, Biomaterials, 2016, 74, 131–143 CrossRef CAS PubMed.
- D. S. Uppu and J. Haldar, Biomacromolecules, 2016, 17, 862–873 CrossRef CAS PubMed.
- V. Mayandi, S. Sridhar, M. Fazil, E. T. L. Goh, H. M. Htoon, G. Orive, Y. K. Choong, R. Saravanan, R. W. Beuerman, T. M. S. Barkham, L. Yang, M. Baskaran, V. Jhanji, X. J. Loh, N. K. Verma and R. Lakshminarayanan, ACS Infect. Dis., 2019, 5, 1411–1422 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |