
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the potential of tamoxifen-based copper(II) dichloride in breast cancer therapy†‡
Aleksandr
Kazimir
 a,
Benedikt
Schwarze
b,
Peter
Lönnecke
a,
Sanja
Jelača
c,
Sanja
Mijatović
c,
Danijela
Maksimović-Ivanić
*c and
Evamarie
Hey-Hawkins
*a
a,
Benedikt
Schwarze
b,
Peter
Lönnecke
a,
Sanja
Jelača
c,
Sanja
Mijatović
c,
Danijela
Maksimović-Ivanić
*c and
Evamarie
Hey-Hawkins
*a
aInstitute of Inorganic Chemistry, Faculty of Chemistry and Mineralogy, Leipzig University, Leipzig, Germany. E-mail: hey@uni-leipzig.de
bInstitute of Medical Physics and Biophysics, Faculty of Medicine, Leipzig University, Germany
cDepartment of Immunology, Institute for Biological Research “Siniša Stanković”, National Institute of the Republic of Serbia, University of Belgrade, Bulevar despota Stefana 142, 11060 Belgrade, Serbia. E-mail: nelamax@ibiss.bg.ac.rs
First published on 12th September 2023
Abstract
For decades, tamoxifen-based hormone therapy has effectively addressed oestrogen receptor positive (ER+) luminal A breast cancer. Nonetheless, the emergence of tamoxifen resistance required innovative approaches, leading to hybrid metallodrugs with several therapeutic effects besides the inhibition of oestrogen receptor α (ERα). Drawing inspiration from tamoxifen metabolite structures (4-hydroxytamoxifen and 4,4′-dihyroxytamoxifen), a phenyl ring was replaced by a bidentate 2,2′-bipyridine donor moiety to give 4-[1,1-bis(4-methoxyphenyl)but-1-en-2-yl]-2,2′-bipyridine (L), enabling coordination of bioactive transition metal compounds such as copper(II) dichloride, yielding [CuCl(μ-Cl)(L-κ2N,N′)]2 (1). Notably, copper(II) complex 1 exhibited remarkable activity within the low micromolar concentration range against ER+ human glioblastoma U251, as well as breast carcinomas MDA-MB-361 and MCF-7, surpassing the efficacy of previously reported palladium(II) and platinum(II) dichloride analogs against these cell lines. The pronounced efficacy of complex 1 against triple-negative MDA-MB-231 cells highlights its potential multitherapeutic approach, evident through induction of apoptosis and antioxidant activity. This study evaluates the potential of copper–tamoxifen hybrid complex 1 as a potent therapeutic candidate, highlighting its diverse mechanism of action against challenging breast cancer subtypes.
1. Introduction
Breast cancer is the most prevalent form of cancer in women with an estimated ∼2.3 million diagnosed cases in 2020.1 Hormone receptor-positive (HR+) breast cancers feature the oestrogen receptors (ER) and/or the progesterone receptor (PR), while hormone receptor-negative (HR−) cancer cells have neither of these receptors.2 Additionally, in breast cancer diagnosis, the determination of the human epidermal growth factor receptor 2 (HER2) status is important as this growth factor is responsible for faster proliferation of cancer cells.3 HER2-positive cancers are, however, susceptible to anti-HER2 treatment.4 The luminal A breast cancer subtype (ER+/PR+) is associated with the overexpression of ERα,5 and tamoxifen (I, Scheme 1), a selective oestrogen receptor modulator (SERM), is widely applied for treatment.6 As a prodrug, tamoxifen is metabolised in the liver by cytochrome P450 enzymes to produce several metabolites, including 4-hydroxytamoxifen (II, Scheme 1) and 4,4′-dihydroxytamoxifen (III, Scheme 1), which exhibit up to 25–50 times higher affinity than their parent molecule.7 The metabolites bind non-covalently to ERα and consequently inhibit its activity.6,8 However, long-term treatment with tamoxifen often leads to resistance due to the development of oestrogen-independent mechanisms by the breast cancer.5,9 Furthermore, treatment of triple-negative breast cancer (TNBC), expressing neither HR nor HER2, is a very problematic task.10 Therefore, new approaches such as combinations of inhibitors and metallodrugs (hybrid drugs) may be one way to cope with these challenges due to their multifaceted modes of action, rather than acting on one single target only.11,12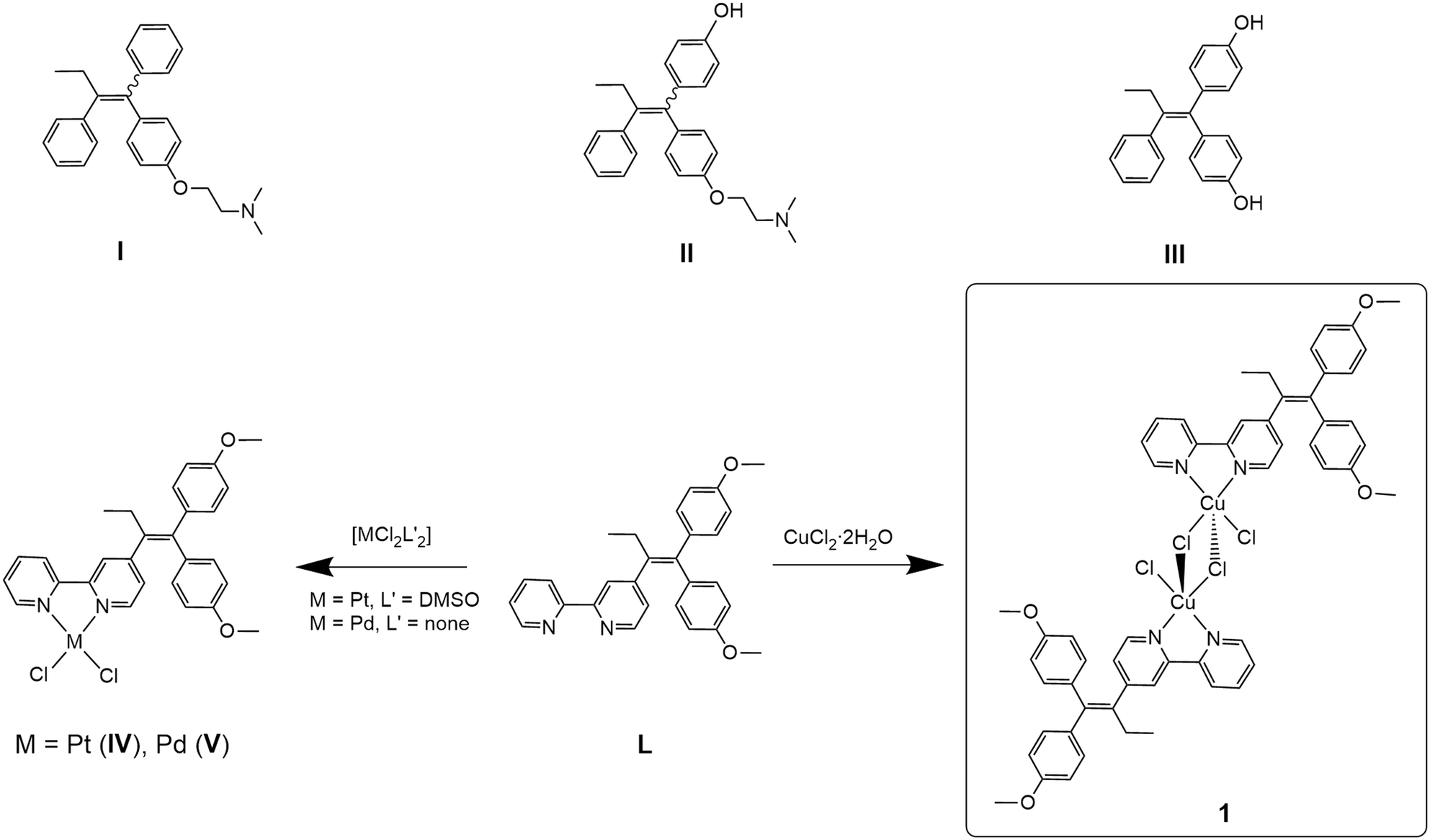 | ||
| Scheme 1 Tamoxifen (I) and the tamoxifen metabolites6 4-hydroxytamoxifen (II), 4,4′-dihydroxytamoxifen (III); tamoxifen-based ligand 4-[1,1-bis(4-methoxyphenyl)but-1-en-2-yl]-2,2′-bipyridine (L), previously reported complexes [PtCl2(L-κ2N,N′)] (IV), [PdCl2(L-κ2N,N′)] (V)13,14 and the dimeric copper(II) dichloride complex [CuCl(μ-Cl)(L-κ2N,N′)]2 (1) reported here. | ||
Recently, we reported our approach for a new class of hybrid drugs by combining 2,2′-bipyridine-modified tamoxifen (L) and transition metals such as molybdenum,13 platinum (IV) or palladium (V).14 Most of the metal complexes and the ligand alone exhibit cytotoxic activities in the low micromolar range against three ER+ (U251, MCF-7, MDA-MB-361) and HR− (MDA-MB-231) cancer cell lines in vitro. Further mechanistic investigation on MCF-7 cells revealed that cellular death processes, such as caspase-dependent apoptosis, oxidative stress or autophagy, are characteristically altered depending on the nature of the transition metal. This allows for the modular assembly of ligand L and transition metals to exploit their properties for specific needs in the treatment of (different) cancers regarding the cytotoxicity, selectivity and mechanism of action.14
The transition metal copper is widely used as active centre in metallodrugs for breast cancer therapy. Like the majority of platinum-based drugs, CuI and CuII are able to interact with DNA, causing damage.15 Therefore, potential copper therapeutics have been considered as an economical substitute for platinum drugs. Furthermore, since copper is involved in many cellular processes, its high redox activity requires tight regulation inside the cell, necessitating an elaborated copper homeostasis system.16 Interestingly, copper homeostasis16,17 is significantly altered in tumour tissues, and the concentration of copper was found to be higher in tissues of several cancer types (e.g., breast, lung, prostate and brain).18,19 Numerous studies have evidenced that copper plays an essential role in the growth and progression of tumours by initiating angiogenesis (at earlier stages of cancer) and promoting metastatic processes.18,20–22 Therefore, current clinical therapy predominantly targets Cu using copper-chelating ligands (e.g., penicillamine, trientine, tetrathiomolybdate) that bind to the metal inside the cell, facilitating its removal.23,24 However, the increased uptake of copper by tumour cells provides the opportunity to accumulate Cu-based anticancer agents with bioactive components (hybrid drugs) inside the cancer tissue, making the design and investigation of copper-containing therapeutics relevant.18,19 In parallel with our studies, Scalcon et al. recently reported on the copper(II) dichloride and gold(III) chloride complexes of ligand L.25 The copper(II) complex was shown to induce oxidative damage of mitochondria in two breast adenocarcinoma cell lines (MCF-7 and MDA-MB-231) by increasing the level of the reactive oxygen species (ROS).25–27
Herein, we report the synthesis of the dinuclear copper(II) complex with ligand L, compound 1, and discuss the potential of 1 against ER+ and ER− breast cancer cell lines, expanding the collection of hybrid metallodrugs with distinct modes of action.
2. Results and discussion
2.1. Synthesis and characterisation
The synthesis of ligand L was previously reported by us13 (characterisation is given in the ESI‡). L was already employed as a ligand in various transition metal complexes.14 The copper(II) complex 1 was synthesised by reacting ligand L with CuCl2·2H2O under a nitrogen atmosphere. In contrast to the procedure reported by Scalcon et al., who prepared the copper(II) complex in dry DCM,25 here the reaction mixture was stirred for 12 hours in degassed ethanol. The green product precipitated and was isolated by filtration. The dimeric complex 1 was obtained in good yield (81%) and characterised by UV-vis and FT-IR spectroscopy, HR-ESI mass spectrometry, single crystal X-ray crystallography and elemental analysis.The FT-IR spectrum of complex 1 shows the vibrations of L (Fig. S6 and S7, ESI‡). HR-ESI-MS (Fig. S5, ESI‡) confirmed the presence of the dimeric complex 1 (Scheme 1), as well as monomeric species. Accordingly, single-crystal X-ray crystallography confirmed the formation of a dimeric structure in the solid state. Crystals of compound 1 were obtained from a concentrated DCM/n-pentane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution over a period of 14 days. Complex 1 crystallises in the monoclinic space group P21/c. 1 forms a centrosymmetric chlorido-bridged complex (Fig. 1). The copper atoms are five-coordinated and exhibit a square-pyramidal geometry (Fig. S9, ESI‡) with small deviations from the theoretical value of 90° for the bond angles. The coordination sites are occupied by two nitrogen atoms of the 2,2′-bpy moiety of ligand L, two bridging chlorides (Cl1 and Cl1′) and one terminal chloride (Cl2 or Cl2′). The Cu–Cl1 bond (229.87(6) pm) is slightly longer than Cu1–Cl2 (225.10(6) pm), while the long Cu1–Cl1′ bond (267.75(6) pm) indicates a weak coordinative interaction.
:1, v/v) solution over a period of 14 days. Complex 1 crystallises in the monoclinic space group P21/c. 1 forms a centrosymmetric chlorido-bridged complex (Fig. 1). The copper atoms are five-coordinated and exhibit a square-pyramidal geometry (Fig. S9, ESI‡) with small deviations from the theoretical value of 90° for the bond angles. The coordination sites are occupied by two nitrogen atoms of the 2,2′-bpy moiety of ligand L, two bridging chlorides (Cl1 and Cl1′) and one terminal chloride (Cl2 or Cl2′). The Cu–Cl1 bond (229.87(6) pm) is slightly longer than Cu1–Cl2 (225.10(6) pm), while the long Cu1–Cl1′ bond (267.75(6) pm) indicates a weak coordinative interaction.
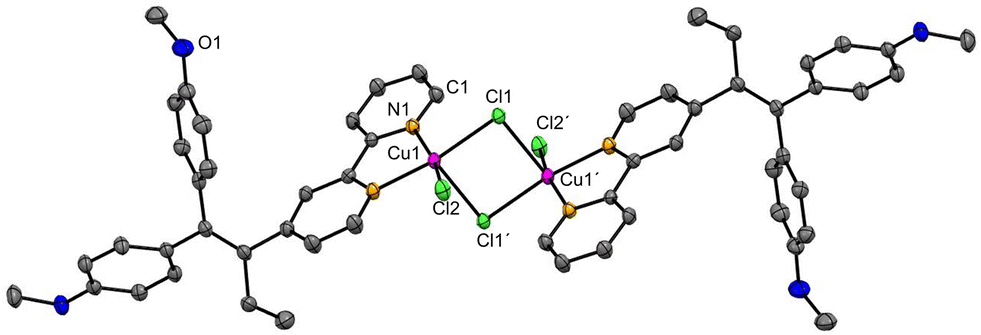 | ||
| Fig. 1 Molecular structure of [CuCl(μ-Cl)(L-κ2N,N′)]2. Hydrogen atoms are omitted for clarity. Displacement ellipsoids are presented at 50% probability level. Selected distances (pm) and bond angles (°): Cu⋯Cu′ 353.8, Cu1–Cl1 229.87(6), Cu1–Cl1′ 267.75(6), Cu1–N1 203.5(2), Cu1–Cl2 225.10(6), Cu1–N2 201.6(2), N2–Cu1–Cl2 93.61(6), N1–Cu1–Cl1 93.86(6), Cl2–Cu1–Cl1 91.33(2), N1–Cu1–N2 79.81(8), Cl2–Cu1–Cl1′ 100.0, Cl1–Cu1–Cl1′ 89.69(2). | ||
2.2. DFT calculations
Density functional theory (DFT) calculations were used to estimate the stability of compound 1 in water and proposed monomeric species 2–4 as a result of dissociation of 1 (Fig. 2). For this purpose, we optimised the geometries of the parent structure 1 and monomeric complexes 2–4.28 We implemented the conductor-like polarisable continuum model (CPCM)29 in our calculation in order to include the solvation effect, and considered water as the solvent because of its biological relevance.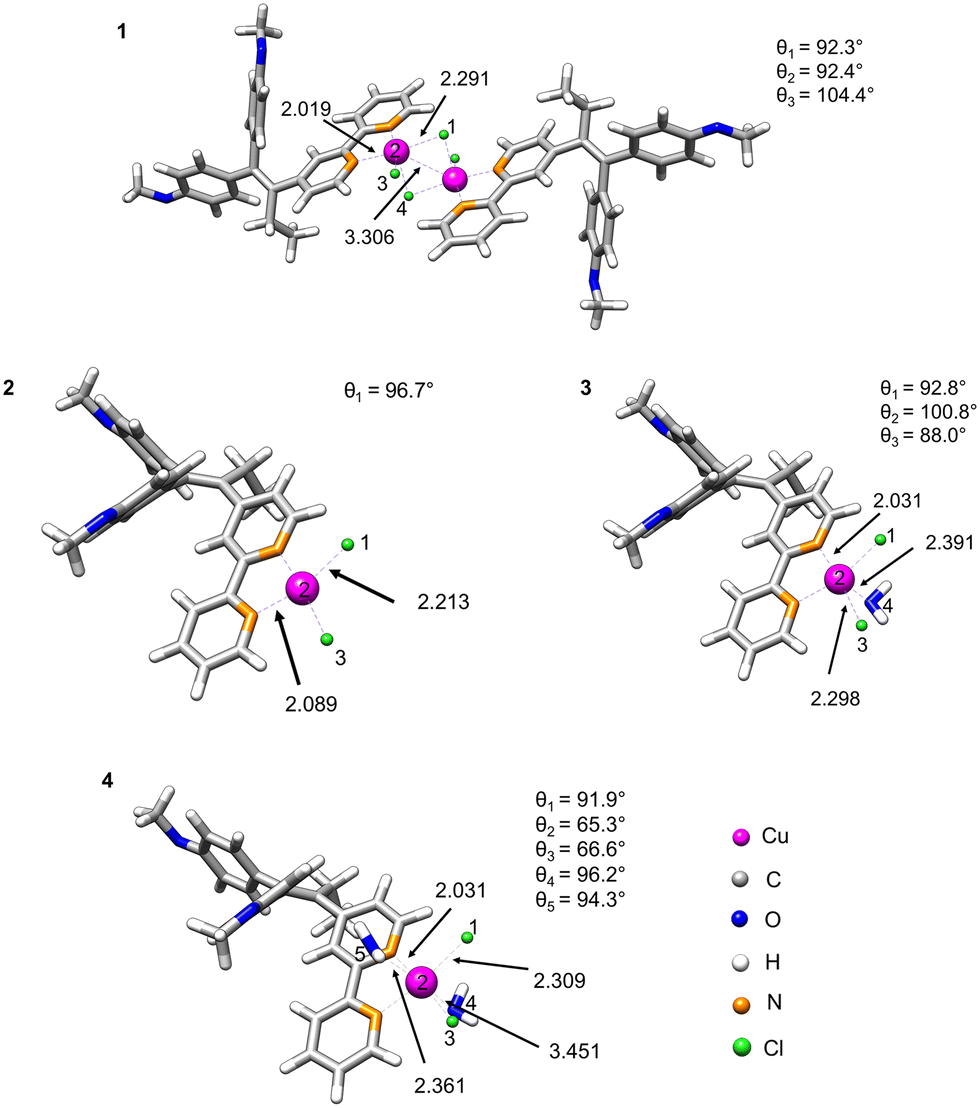 | ||
| Fig. 2 Potential arrangement of the ligands of optimised geometries of bimetallic and monometallic species 1–4: [CuCl(μ-Cl)(L-κ2N,N′)]2 (1), [CuCl2(L-κ2N,N′)] (2), [CuCl2(L-κ2N,N′)(H2O)] (3), [CuCl2(L-κ2N,N′)(H2O)2] (4). Geometry optimisation was performed using DFT at the PBE0 D3BJ/def2-TZVP level of theory, incorporating solvent (water) as a conductor-like polarisable model (CPCM). The distances are reported in angstrom. The angles θ1 = ∢1–2–3, θ2 = ∢1–2–4, θ3 = ∢3–2–4, θ4 = ∢1–2–5, θ5 = ∢3–2–5 are angles between the numbered Cu (pink), Cl (green) and O (blue) atoms. | ||
The fully DFT-optimised structures of the monomeric species [CuCl2(L-κ2N,N′)] in gas or water phase demonstrated a distorted square-planar configuration. However, the probability of dissociation of the dimer with the formation of two monomers [CuCl2(L-κ2N,N′)] (2) is quite low, as indicated by their high dissociation energies in both gas and water phases (3505 and 3440 kJ mol−1, respectively, Table 1). The situation, however, changes when one or two coordinating solvent molecules (H2O) are explicitly included in the coordination sphere of copper. A decrease of the dissociation energy is observed (12.6 kJ mol−1) for the formation of [CuCl2(L-κ2N,N′)(H2O)] (3).
| Possible monomeric species | Phase | ΔG of dissociation, kJ mol−1 |
|---|---|---|
| [CuCl2(L-κ2N,N′)] (2) | Gas | 3505 |
| Water | 3440 | |
| [CuCl2(L-κ2N,N′)(H2O)] (3) | Water | 12.6 |
| [CuCl2(L-κ2N,N′)(H2O)2] (4) | Water | −8.4 |
However, the positive dissociation energy still indicates a low probability of the formation of the monohydrate complex 3. A negative ΔG value of −8.4 kJ mol−1 was observed for the monomeric octahedral copper complex 4 involving two water molecules in the coordination sphere. Nevertheless, the observed small energy change could suggest the coexistence of dimeric and monomeric forms in an equilibrium depending on ambient conditions.
The presence of coordinated water molecules in monomeric complexes 2–4 was experimentally assessed by UV-vis spectroscopy by comparing the spectrum obtained for 1 in a non-coordinating solvent (dichloromethane (DCM)) with spectra recorded in DMSO/water (1% v/v DMSO). It was assumed that a change in the coordination sphere should be detectable if LMCT or MLCT would be involved. Furthermore, these transitions are sensitive to solvent changes (Fig. S11, ESI‡).
Both UV-vis spectra showed high similarity with three peaks at 250, 287 and 387 nm in DCM and at 247, 291 and 353 nm in DMSO/water. This might indicate the presence of only the dimeric complex, or both dimeric and monomeric species. Additionally, we have computed the UV-vis vertical excitation using TDDFT30 for monomeric [CuCl2(L-κ2N,N′)(H2O)2] (4) and dimeric [CuCl(μ-Cl)(L-κ2N,N′)]2 (1), and compared them to experimentally recorded spectra. The observed transitions for 1 and 4 exhibited a similar trend (Table S4, ESI‡). A computed LMCT at 270 nm is expected to involve water molecules and chlorides (Fig. S11, ESI‡). Additionally, ILCT (phenyl moiety (Ph) → 2,2′-bpy) at 340 nm was observed for this complex. Interestingly, complex 4 exhibited both ILCT and Cl → Cu charge transfer at 416 nm. The theoretically calculated dimeric species is expected to demonstrate ILCT and LMCT involving chloride at 304 nm. A Ph → 2,2′-bpy charge transfer occurred at 344 nm simultaneously at both tamoxifen moieties (see Fig. S15, ESI‡). An additional LMCT (Ph → [CuCl2]) was detected at 425 nm (Fig. S15, ESI‡). All calculated excitations for both compounds 1 and 4 were in the range of experimentally observed peaks. It is noteworthy that the d–d transitions were exclusively detected in DCM solution, but not in 1% DMSO/water. This finding supports the assumption that a dimeric complex is present in non-coordinating solvents, however the existence of both dimeric and monomeric species cannot be excluded. Nonetheless, the d–d transitions were also computationally assessed for the mononuclear configuration within the same spectral range, and their detection is dependent on the solvent (Fig. S16, ESI‡). The frontier orbitals supported the observed results from charge transfers (Fig. S17 and S18, ESI‡).
2.3. In vitro studies
For the biological assays, the stability of compound 1 in a DMSO/water solution (with up to 1% v/v of DMSO) was assessed by UV-vis spectroscopy, demonstrating its stability for more than 5 days (Fig. S10, ESI‡).In order to evaluate the cytotoxic behaviour of complex 1 in comparison to the reported compounds III and L, several breast cancer cell lines with different receptor status (Table 2) were exposed to 1. For the study, two breast adenocarcinoma lines (MCF-7, MDA-MB-361) were used; these cell lines expressed one or several receptors, such as oestrogen receptor (ER), progesterone receptor (PR) or human epidermal growth receptor 2 (HER2), triple negative breast cancer line MDA-MB-231 and triple positive U251 (Table 2).
| Cell line/receptor | ER | PR | HER2 |
|---|---|---|---|
| MCF-7 | + | + | − |
| MDA-MB-231 | − | − | − |
| MDA-MB-361 | + | − | o.e. |
| U251 | + | + | + |
Cells were cultivated with 1 for 72 hours, and the number of viable cells was quantified by measuring the cell respiration or the number of adherent cells per well using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) or CV (crystal violet) assays. The obtained data are presented as the half-maximal inhibitory concentration (IC50) in Table 3.
| Compound | Assay | U251 (μM) | MCF-7 (μM) | MDA-MB-361 (μM) | MDA-MB-231 (μM) |
|---|---|---|---|---|---|
| III 14 | MTT | 28.1 ± 1.4 | 14.3 ± 2.9 | 28.6 ± 0.2 | 24.5 ± 0.7 |
| CV | 20.1 ± 1.9 | 19.1 ± 1.3 | 36.2 ± 2.1 | 26.2 ± 3.7 | |
| L 14 | MTT | 4.1 ± 0.3 | 2.5 ± 0.4 | 4.8 ± 0.0 | 2.1 ± 0.0 |
| CV | 4.4 ± 0.3 | 2.8 ± 0.1 | 5.8 ± 0.2 | 2.3 ± 0.0 | |
| IV 14 | MTT | 2.4 ± 0.4 | 5.4 ± 0.4 | 5.5 ± 0.1 | 2.2 ± 0.1 |
| CV | 3.0 ± 0.3 | 7.3 ± 0.7 | 6.2 ± 0.0 | 2.6 ± 0.1 | |
| V 14 | MTT | 4.4 ± 0.3 | 2.7 ± 0.6 | 9.3 ± 0.2 | 2.0 ± 0.1 |
| CV | 5.9 ± 0.2 | 4.4 ± 0.4 | 11.4 ± 1.0 | 2.3 ± 0.1 | |
| 1 | MTT | 1.1 ± 0.1 | 0.8 ± 0.1 | 1.3 ± 0.1 | 1.2 ± 0.0 |
| CV | 1.4 ± 0.1 | 1.1 ± 0.1 | 1.6 ± 0.1 | 2.0 ± 0.1 |
Previous cytotoxicity studies showed that incorporating the 2,2′-bpy unit into the tamoxifen metabolite III increased the cytotoxicity by a factor of 7.6 across the panel of tested cell lines (Table 3, MTT assays).14 Upon coordination of L to platinum(II) (IV) or palladium(II) (V), the IC50 values are similar (up to a maximal factor of around two in both directions).14 However, using copper(II) as the metal resulted in significantly higher toxicity against all studied cell lines in a low micromolar concentration range (0.8 to 1.3 μM, MTT assay), exceeding the cytotoxic effect of the previously reported compounds by two or four times (Table 3).
The activity of compound 1 against the triple-negative cell line MDA-MB-231 suggests a hormone-independent mechanism of action in the IC50 range, similar to what has been previously reported for compounds L, IV and V.14
The sensitivity of all tested cell lines with different receptor status was almost equal. Thus, the mechanism of the drug's action is probably independent from ER expression. Numerous tamoxifen off-targets that can be relevant for the observed effect were already identified in the literature, like those involved in calcium homeostasis and lipid metabolism, protein kinase C, glucocorticoid receptor, as well as the transcription factors peroxisome proliferator-activated receptor gamma, signal transducer and activator of transcription 1 and nuclear factor erythroid 2-related factor 2.33
Further investigations on isolated peritoneal exudate cells, as a model for healthy primary cells, were conducted to assess the selectivity of 1 towards the studied cancer cell lines. The experiments gave an IC50 value of 6.2 μM, which is about three times higher than that for the cancer cells lines (Fig. 3).
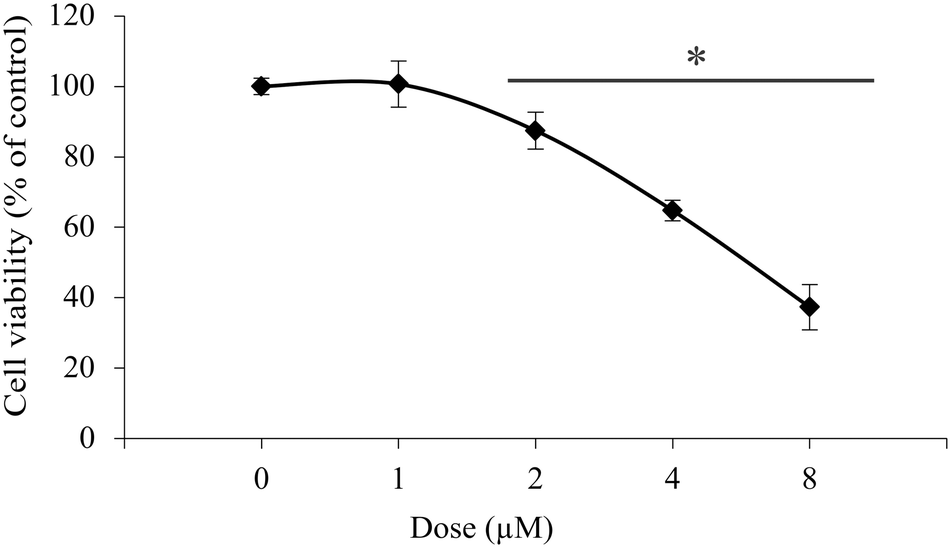 | ||
| Fig. 3 The effect of compound 1 on the viability of primary peritoneal exudate cells. Cells were treated with 1 within the indicated dose range for 72 h. Cell viability was determined by CV assay. The data are presented as the mean ± SD from one representative out of three independent experiments (*p < 0.05 compared to untreated cultures). | ||
2.4. Flow cytometry
In search of possible mechanisms of action of 1 for reducing the cell viability in MCF-7 breast cancer cells, the capacity of 1 to induce apoptotic cell death was evaluated. After treatment with 1, the accumulation of cells in the early and late stages of programmed cell death (Ann+/PI− and double-positive cells) (Fig. 4) was noted. Also, the analysis of DAPI (4′,6-diamidino-2-phenylindole)-stained cells under fluorescence microscopy revealed an abnormal shape of nuclei with condensed chromatin, which is characteristic of apoptotic cell morphology. This process coincided with caspase activation, as observed using the pan-caspase inhibitor Apostat. Interestingly, similar observations have been reported previously for metal-containing compounds IV (PtII) and V (PdII), while the ligand L led to caspase-independent apoptosis.14 Thus, the presence of transition metals seems to activate the caspase cell death pathway. Furthermore, measurement of cell proliferation by carboxyfluorescein diacetate succinimidyl ester (CFSE) indicated a strong arrest in cell division within the surviving fraction of cells. | ||
| Fig. 4 Complex 1 inhibits the proliferation of MCF-7 cells. MCF-7 cells were exposed to an IC50 dose of compound 1 for 60 h and analysed by flow cytometry: (A) apoptosis detection (Annexin V/PI staining), (B) morphology of nuclei: arrows show abnormal cell nuclei with condensed chromatin as a sign of apoptosis (4′,6-diamidino-2-phenylindole (DAPI) staining, 400× magnification), (C) Apostat for caspase detection, and (D) carboxyfluorescein diacetate succinimidyl ester (CFSE) for division rate quantification. (E) Compound 1 affected the level of ROS/RNS production in the MCF-7 cell line. MCF-7 cells were prestained with dihydrorhodamine 123 (DHR123) and treated with an IC50 dose of compound 1 for 60 h. Intracellular accumulation of ROS/RNS were measured by flow cytometry. Representative histograms from three independent experiments are shown. | ||
It is noteworthy that the literature-reported copper complexes have a tendency to elevate the presence of ROS within cells.19,34,35 Scalcon et al. also observed an increased level of ROS after treatment of MCF-7 and MDA-MB-231 cells with their tamoxifen-bipy-copper(II) complex, which might be related to the conditions of the experiment and time-dependency of ROS production.25 However, in contrast to these reports and the reported platinum-based drug IV,14 strong inhibition of ROS by copper complex 1 was detected during the same period of incubation using dihydrorhodamine 123 (DHR123) (Fig. 4E). Cancer cells generally exhibit higher levels of reactive oxygen species (ROS) compared to normal cells.36 In this regard, the antioxidant effect of copper therapeutics can also exert a beneficial influence,37 as their inhibition leads to restricted growth of cancer cells and cell death. Furthermore, inhibition of pro-survival amounts of superoxide anions at the membrane level induces caspase activation and aids the execution of the apoptotic process.38
3. Conclusion
Treatment of advanced cancer types, which for instance lack the usual drug targets (e.g., TNBC) or have developed resistance mechanisms against standard therapeutic schemes, requires new approaches. From the small molecule point of view, a combination of lead structures of known inhibitors and metallodrugs (hybrid drugs) may be a suitable approach to face these challenges due to their multifaceted modes of action, rather than acting on one single target.Tamoxifen modified with a 2,2′-bpy moiety (L) facilitates the coordination of bioactive transition metals, such as MoII,13 PtII or PdII.14 Most of these tamoxifen-inspired compounds showed cytotoxic activity in the low micromolar range with a diversity of cell death mechanisms on MCF-7 cell lines in vitro. Herein, we have extended the spectrum of transition metals to CuII.
Copper(II) complexes are extremely labile as a result of the d9 configuration and coordination numbers from four to six,39 facilitating interactions of copper complexes with biomolecules. Furthermore, in comparison to 2,2′-bpy platinum and palladium dichloride complexes, 2,2′-bpy copper dichloride has a tendency to form a dimeric chlorido-bridged structure.40 Accordingly, copper complex 1 forms a dimeric structure in the solid state. In aqueous solution, however, a dissociation into two monomers is conceivable as supported by HR-MS, UV-vis spectra and DFT calculations.
Compound 1 displayed substantially higher cytotoxic activity compared to previously reported complexes with a similar tamoxifen vector. The presence of two metal centers might have a positive impact on the biological properties, as has been shown for some bimetallic copper complexes (e.g., thiosemicarbazone complexes of copper(II)41,42). Importantly, the cytotoxic behaviour of 1 was independent of the HR status, as compound 1 exhibited activity against all studied cancer cell lines. Investigations of the mode of action revealed that 1 can induce caspase-dependent apoptosis, as reported for compounds IV and V. In contrast to the platinum analogue IV, the copper-based metallodrug induced apoptotic cell death by reducing the reactive oxygen species (ROS), as was previously also observed for palladium compound V.14 We assume that this important mechanism of action may be associated with the presence of the reduced form of CuI within cells, which is important for cuproptosis.43
Overall, this study demonstrates the potential of the copper–tamoxifen hybrid drug 1 as an intriguing alternative to platinum complexes commonly used in anticancer treatments. Incorporating copper into the tamoxifen-based structure L provides the opportunity to vary the mechanism of action in an HR-independent manner. Our next goal is to assess the safety and efficiency of the platinum-, palladium- and copper-based tamoxifen complexes in vivo.
4. Experimental section
4.1. Instrumentation
ESI mass spectra were recorded with a Bruker ESQUIRE 3000 (Benchtop LC Ion trap) mass spectrometer. The FT-IR spectra were obtained with a Nicolette IS5 (ATR) from ThermoFisher (Waltham, MA, USA) with a 4000–400 cm−1 scan range. A Heraeus VARIO EL oven was used to perform elemental analyses. UV-vis absorption spectra of DMSO/water solutions of 1 were measured with a PerkinElmer UV/vis/NIR Lambda 900 spectrometer, using quartz cuvettes (volume = ca. 1.2 cm3).4.2. Materials
The ligand L was prepared according to our previously published procedure.13 Copper(II) chloride dihydrate (97%) was obtained from Sigma-Aldrich and used as purchased. Solvents (acetonitrile (ACN), dimethyl sulfoxide (DMSO), dichloromethane (DCM) and n-pentane) were used as purchased. Ethanol (EtOH) was flushed with N2 for 15 min and kept over molecular sieves (3 Å).4.3. Synthesis of [CuCl(μ-Cl)(L-κ2N,N′)]2 (1)
CuCl2·2H2O (50 mg, 0.29 mmol, 1 eq.) was dissolved in 10 ml ethanol under nitrogen atmosphere. Ligand L (123 mg, 0.29 mmol, 1 eq.) was dissolved in 10 ml ethanol under N2, and added dropwise to the green solution of CuCl2·2H2O. The reaction mixture was stirred for 12 h during which the product precipitated as a green solid. The precipitate was isolated by filtration in air and washed with cold ethanol (3 × 10 ml). The green powder was dried under vacuum for several hours. Yield: 261 mg (0.25 mmol, 81%). Complex 1 (50 mg) was dissolved in DCM and the solution was layered with n-pentane (1:1 v/v), yielding green crystals over two weeks suitable for X-ray structure determination.
HR-ESI-MS (positive mode, acetonitrile), M = 1113.13 g mol−1, m/z [M–Cl]+ = 1077.1616 (calc.), 1077.1551 (found); m/z [(M/2)–Cl]+ = 520.0979 (calc.), 520.0975 (found); m/z [(M/2)–2Cl]+ = 485.1290 (calc.), 485.1278 (found); m/z [(M/2)–CuCl2]+ = 423.2073 (calc.), 423.2061 (found). IR, ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) in cm−1: 2968 (w, Calk–H), 2929 (w, Calk–H), 1604 (m, v(C
in cm−1: 2968 (w, Calk–H), 2929 (w, Calk–H), 1604 (m, v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C)), 1582 (w), 1538 (w), 1506 (m, Carom–H in-plane bending), 1457 (w, Carom–H in-plane bending), 1439 (m, Carom–H in-plane bending), 1412 (w), 1386 (w), 1290 (w), 1242 (m, Carom–O), 1171 (m, Carom–O), 1106 (w), 1067 (w), 1029 (m, Calk–O), 991 (w), 896 (w), 827 (m, Carom–H out-of-plane bending), 792 (m, Carom–H out-of-plane bending), 745 (w), 697 (w), 661 (w), 618 (w), 589 (w), 570 (w), 519 (w). UV-vis: λmax, nm (molar absorption coefficient ε, cm−1 M−1 in 1% DMSO/H2O) = 227 (24300), 300 (16700), 361 (5700); λmax, nm (molar absorption coefficient ε, cm−1 M−1 in DCM) = 250 (33300), 297 (21300), 383 (6700). Elemental analysis: C56H52Cl4Cu2N4O4, calc. (%) C 60.38, H 4.71, N 5.03; found (%) C 60.01, H 4.66, N 5.00.
C)), 1582 (w), 1538 (w), 1506 (m, Carom–H in-plane bending), 1457 (w, Carom–H in-plane bending), 1439 (m, Carom–H in-plane bending), 1412 (w), 1386 (w), 1290 (w), 1242 (m, Carom–O), 1171 (m, Carom–O), 1106 (w), 1067 (w), 1029 (m, Calk–O), 991 (w), 896 (w), 827 (m, Carom–H out-of-plane bending), 792 (m, Carom–H out-of-plane bending), 745 (w), 697 (w), 661 (w), 618 (w), 589 (w), 570 (w), 519 (w). UV-vis: λmax, nm (molar absorption coefficient ε, cm−1 M−1 in 1% DMSO/H2O) = 227 (24300), 300 (16700), 361 (5700); λmax, nm (molar absorption coefficient ε, cm−1 M−1 in DCM) = 250 (33300), 297 (21300), 383 (6700). Elemental analysis: C56H52Cl4Cu2N4O4, calc. (%) C 60.38, H 4.71, N 5.03; found (%) C 60.01, H 4.66, N 5.00.
4.4. Computational chemistry
The geometry optimisation of complex 1 was done based on the molecular structure from single crystal X-ray crystallography using density functional theory (DFT),44 and performed with the ORCA 5.0 package.45 The monomeric species 2–4 were generated from the molecular structure of the dimeric form. The functional PBE0 has been chosen based on the results of benchmark studies for a set of the transition metal complexes.28,46 Becke–Johnson dispersion correction of third order (D3BJ)47 was included in the functional. Additionally, the density fitting technique resolution-of-identity approximation (RI-J)48 and chain-of-sphere approximation (COSX)48 were applied in the geometry optimisation to speed up the calculations. The polarised basis set def2-TZVPP49 was applied for good convergence of the energy. The local minima of the optimised geometries were verified with numerical frequency analysis, where no imaginary frequencies were observed. The calculations of the excited states of both monomeric and dimeric complexes were performed using time-dependent DFT (TDDFT)30 at the similar level of theory, including water as the conductor-like polarisable model (CPCM).29 The calculated structures were visualised using UCSF ChimeraX software.504.5. Reagents and cells
Culture medium RPMI-1640 and fetal bovine serum (FBS) were obtained from Capricorn Scientific GmbH (Hessen, Germany). Propidium iodide (PI), dimethyl sulfoxide (DMSO), crystal violet (CV), phosphate-buffered saline (PBS), and carboxyfluorescein diacetate succinimidyl ester (CFSE) were purchased from Sigma Aldrich (St. Louis, MO, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from AppliChem (MO, USA) and paraformaldehyde (PFA) from Serva Electrophoresis GmbH (Heidelberg, Germany). Annexin V-FITC (AnnV) was obtained from BD (Pharmingen, San Diego, CA, USA) and ApoStat was from R&D Systems (Minneapolis, MN, USA). Penicillin–streptomycin solution was obtained from Biological Industries (Cromwell, CT, USA). Dihydrorhodamine 123 (DHR) was from ThermoFisher Scientific (Waltham, MA, USA). 4′,6-Diamidino-2-phenylindole (DAPI) Fluoromount-G® was purchased from Southern Biotech (Birmingham, AL, USA). Human breast adenocarcinoma (MCF-7, MDA-MB-361, and MDA-MB-231) and human malignant glioma (U251) were acquired from American Type Culture Collection (ATCC, Manassas, Virginia, USA).All human cell lines were cultivated in HEPES-buffered RPMI-1640 medium, which was previously supplemented with 10% heat-inactivated FBS, penicillin (100 units per mL) and streptomycin (100 μg mL−1). Cells were grown at 37 °C in a humidified atmosphere with 5% CO2. Cells were seeded at the following densities in 96-well plates for viability determination: MDA-MB-361 (7 × 103 cells per well), MCF-7 (10 × 103 cells per well), U251 (3 × 103 cells per well) and MDA-MB-231 (8 × 103 cells per well). The density of MCF-7 cells in 6-well plates for flow cytometric analyses was 2.5 × 105 cells per well.
Primary peritoneal exudate cells were isolated from C57BL/6 mice. Animals originated from the animal facility at the Institute for Biological Research “Siniša Stanković” – National Institute of the Republic of Serbia, University of Belgrade (Belgrade, Serbia). After isolation, the cells were routinely cultivated in HEPES-buffered RPMI-1640 medium previously supplemented with 5% heat-inactivated FBS, penicillin (100 units per mL) and streptomycin (100 μg mL−1) under standard growth conditions (37 °C, 5% CO2). Cells were seeded at a density of 1.5 × 105 cells per well in 96-well plates. After two hours, non-adherent cells were removed. After 72 h treatment, cell viability was determined using a CV assay. The study protocol and handling of animals were in agreement with the local guidelines and the European Community guidelines (EEC Directive of 1986; 86/609/EEC), and were approved by the local Institutional Animal Care and Use Committee (IACUC). The national licensing committee at the Department of Animal Welfare, Veterinary Directorate, Ministry of Agriculture, Forestry and Water Management of the Republic of Serbia granted approval for the experimental protocols (323-07-02147/2023-05).
4.6. Determination of cell viability (MTT and CV assays)
Cell lines were seeded overnight and subsequently treated with 1. After 72 h incubation time, the supernatant was discarded, and the cells were washed with PBS two times. Afterwards, MTT solution (0.5 mg mL−1) was added and incubated at 37 °C until purple formazan crystals were formed (approximately 30 min). The solubilised dye was then discarded, and DMSO was added in order to dissolve the formed formazan crystals. The absorbance was measured at λmax = 540 nm, with the reference/background wavelength of 670 nm. Results were expressed as a percentage of the control value (100%).For the CV assay, after 72 h of treatment, the cells were washed with PBS and fixed with 4% (w/v) of paraformaldehyde (PFA) for 10 min. After fixation, cells were stained with 1% (w/v) aqueous CV solution for 15 min at room temperature (rt). Finally, cells were washed with tap water and dried. Prior to absorbance measuring the dye was dissolved in 33% (v/v) acetic acid. The absorbance was measured at λmax = 540 nm, with the reference/background wavelength of 670 nm. Results were expressed as a percentage of the control value (100%).
4.7. Annexin V/propidium iodide (PI) and Apostat staining
For detection of apoptotic cell death, MCF-7 cells were treated with 1 μM of 1 (IC50 value) for 60 h. Afterwards, the cells were washed with PBS and stained with AnnV (15 μg mL−1) and PI (15 μg mL−1) for 15 min at rt, protected from light according to the manufacturer's protocol. Finally, the cells were resuspended in AnnV-binding buffer and analysed by flow cytometry (CyFlow® Space Partec using the PartecFloMax® software (Münster, Germany)). To determine whether apoptosis was mediated by caspase activation, the cells were incubated with pan-caspase inhibitor Apostat for 30 min at 37 °C. After the incubation period, the cells were washed with PBS and analysed by flow cytometry (CyFlow® Space Partec using the PartecFloMax® software (Münster, Germany)).4.8. CFSE staining
For detection of cell proliferation, CFSE staining was used. Prior to seeding, MCF-7 cells were stained with CFSE to a final concentration of 1 μM for 10 min at 37 °C. After incubation, the cells were washed, seeded, and treated with an IC50 dose (1 μM) of 1 for 60 h. Finally, the cells were trypsinised, washed, resuspended in PBS, and analysed by flow cytometry (CyFlow® Space Partec using the PartecFloMax® software (Münster, Germany)).4.9. Measurement of generation of reactive oxygen and nitrogen species (ROS/RNS)
For detecting the production of ROS/RNS species, the intensity of the green fluorescence emitted by the redox-sensitive dye dihydrorhodamine 123 (DHR123) was measured. MCF-7 cells were prestained with 1 μM DHR123 for 20 min at 37 °C, and treated with an IC50 dose (1 μM) of 1 for 60 h. Finally, the cells were washed, trypsinised and analysed using flow cytometry (CyFlow® Space Partec using the PartecFloMax® software (Münster, Germany)).4.10. DAPI staining on chamber slides
In order to evaluate the morphological signs of apoptotic cell death, MCF-7 cells were seeded in 8-chamber slides at a density of 1.5 × 104 cells per well, and treated with an IC50 dose (1 μM) of 1 for 60 h. After incubation, the cells were fixed with 4% (w/v) paraformaldehyde for 15 min at rt, washed, and covered with DAPI Fluoromount-G® mounting medium. Slides were analysed at 400× magnification using a Zeiss AxioObserver Z1 inverted fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany).4.11. Statistical analysis
All data are represented as mean ± SD of at least three independent replicates. Student t-test was used to evaluate the significance between groups. Two-sided p values of less than 0.05 were considered statistically significant.Author contributions
Conceptualisation, E. H.-H., D. M.-I., A. K. and B. S.; methodology, A. K., S. J., P. L., D. M.-I. and S. M.; validation, A. K., P. L., S. J., D. M.-I. and S. M.; formal analysis, A. K., S. J., D. M.-I., S. M. and P. L.; investigation, A. K., P. L., S. J., S. M. and D. M.-I.; resources, E. H.-H., D. M.-I. and S. M.; data curation, A. K., P. L., E. H.-H., D. M.-I. and S. M.; writing – original draft preparation, A. K.; writing – review and editing, A. K., P. L., B. S., E. H.-H., D. M.-I., S. J. and S. M.; visualisation, A. K., S. J., P. L., D. M.-I. and S. M.; supervision, E. H.-H., D. M.-I. and S. M.; project administration, E. H.-H., D. M.-I. and S. M.; funding acquisition, E. H.-H., D. M.-I. and S. M. All authors have read and agreed with the published version of the manuscript.Conflicts of interest
There is no conflict to declare.Acknowledgements
Ministry of Science, Technological Development and Innovation of the Republic of Serbia, grant number 451-03-47/2023-01/200007. A. K. thanks the DAAD for funding (funding program number: 57440919; funding program: Research Grants – Bi-national 2019/2020).Notes and references
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- C. K. Osborne and R. Schiff, Annu. Rev. Med., 2011, 62, 233–247 CrossRef CAS PubMed.
- N. Iqbal and N. Iqbal, Mol. Biol. Int., 2014, 2014, 852748 Search PubMed.
- A. A. Larionov, Front. Oncol., 2018, 8, 1–17 CrossRef PubMed.
- D. H. Anderson, in Biological Mechanisms and the Advancing Approaches to Overcoming Cancer Drug Resistance, ed. A. Freywald and F. J. Vizeacoumar, Academic Press, 2021, vol. 12, pp. 1–22 Search PubMed.
- C. K. Osborne, N. Engl. J. Med., 1998, 339, 1609–1618 CrossRef CAS PubMed.
- C. Fabian, L. Tilzer and L. Sternson, Biopharm. Drug Dispos., 1981, 2, 381–390 CrossRef CAS PubMed.
- D. J. Bentrem, R. C. Dardes, H. Liu, J. MacGregor-Schafer, J. W. Zapf and V. C. Jordan, Endocrinology, 2001, 142, 838–846 CrossRef CAS PubMed.
- Molecular mechanisms and mode of tamoxifen resistance in breast cancer, http://www.bioinformation.net/012/97320630012135.htm, (accessed 19 June 2023).
- S. A. Dass, K. L. Tan, R. S. Rajan, N. F. Mokhtar, E. R. M. Adzmi, W. F. W. A. Rahman, T. A. D. A.-A. T. Din and V. Balakrishnan, Medicina, 2021, 57, 62 CrossRef PubMed.
- E. Ortega, G. Vigueras, F. J. Ballester and J. Ruiz, Coord. Chem. Rev., 2021, 446, 214129 CrossRef CAS.
- I. Yousuf, M. Bashir, F. Arjmand and S. Tabassum, Coord. Chem. Rev., 2021, 445, 214104 CrossRef CAS.
- B. Schwarze, S. Jelača, L. Welcke, D. Maksimović-Ivanić, S. Mijatović and E. Hey-Hawkins, ChemMedChem, 2019, 14, 2075–2083 CrossRef CAS PubMed.
- A. Kazimir, B. Schwarze, P. Lönnecke, S. Jelača, S. Mijatović, D. Maksimović-Ivanić and E. Hey-Hawkins, Pharmaceutics, 2023, 15, 682 CrossRef CAS PubMed.
- M. Govindaraju, H. S. Shekar, S. B. Sateesha, P. Vasudeva Raju, K. R. Sambasiva Rao, K. S. J. Rao and A. J. Rajamma, J. Pharm. Anal., 2013, 3, 354–359 CrossRef CAS PubMed.
- L. Chen, J. Min and F. Wang, Signal Transduction Targeted Ther., 2022, 7, 1–16 CrossRef CAS PubMed.
- A. Bhattacharjee, K. Chakraborty and A. Shukla, Metallomics, 2017, 9, 1376–1388 CrossRef CAS PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- J. Rani J and S. Roy, ChemMedChem, 2023, 18, e202200652 CrossRef CAS PubMed.
- V. Oliveri, Front. Mol. Biosci., 2022, 9, 1–14 Search PubMed.
- E. Urso and M. Maffia, J. Vasc. Res., 2015, 52, 172–196 CrossRef CAS PubMed.
- O. Karginova, C. M. Weekley, A. Raoul, A. Alsayed, T. Wu, S. S.-Y. Lee, C. He and O. I. Olopade, Mol. Cancer Ther., 2019, 18, 873–885 CrossRef CAS PubMed.
- V. C. Shanbhag, N. Gudekar, K. Jasmer, C. Papageorgiou, K. Singh and M. J. Petris, Biochim. Biophys. Acta, Mol. Cell Res., 2021, 1868, 118893 CrossRef CAS PubMed.
- D. A. Da Silva, A. De Luca, R. Squitti, M. Rongioletti, L. Rossi, C. M. L. Machado and G. Cerchiaro, J. Inorg. Biochem., 2022, 226, 111634 CrossRef CAS PubMed.
- V. Scalcon, R. Bonsignore, J. Aupič, S. R. Thomas, A. Folda, A. A. Heidecker, A. Pöthig, A. Magistrato, A. Casini and M. P. Rigobello, J. Med. Chem., 2023, 66, 9823–9841 CrossRef CAS PubMed.
- X. Ma, C. Zhang, J. Hua, P. Ma, J. Wang and J. Niu, CrystEngComm, 2019, 21, 394–398 RSC.
- H. Arif, A. Sohail, M. Farhan, A. A. Rehman, A. Ahmad and S. M. Hadi, Int. J. Biol. Macromol., 2018, 106, 569–578 CrossRef CAS PubMed.
- C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757–10816 RSC.
- Y. Takano and K. N. Houk, J. Chem. Theory Comput., 2005, 1, 70–77 CrossRef PubMed.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- W. W. Hwang-Verslues, W.-H. Kuo, P.-H. Chang, C.-C. Pan, H.-H. Wang, S.-T. Tsai, Y.-M. Jeng, J.-Y. Shew, J. T. Kung, C.-H. Chen, E. Y.-H. P. Lee, K.-J. Chang and W.-H. Lee, PLoS One, 2009, 4, e8377 CrossRef PubMed.
- A. M. Hernández-Vega, A. Del Moral-Morales, C. J. Zamora-Sánchez, A. G. Piña-Medina, A. González-Arenas and I. Camacho-Arroyo, Cells, 2020, 9, 1930 CrossRef PubMed.
- C. Sfogliarini, G. Pepe, A. Dolce, S. Della Torre, M. C. Cesta, M. Allegretti, M. Locati and E. Vegeto, Front. Pharmacol., 2022, 13, 879020 CrossRef CAS PubMed.
- Q. Peña, G. Sciortino, J.-D. Maréchal, S. Bertaina, A. J. Simaan, J. Lorenzo, M. Capdevila, P. Bayón, O. Iranzo and Ò. Palacios, Inorg. Chem., 2021, 60, 2939–2952 CrossRef PubMed.
- P. Ji, P. Wang, H. Chen, Y. Xu, J. Ge, Z. Tian and Z. Yan, Pharmaceuticals, 2023, 16, 234 CrossRef CAS PubMed.
- H. Nakamura and K. Takada, Cancer Sci., 2021, 112, 3945–3952 CrossRef CAS PubMed.
- R. Olar, C. Maxim, M. Badea, M. Bacalum, M. Raileanu, S. Avram, N. Č. Korošin, T. Burlanescu and A. M. Rostas, Pharmaceutics, 2022, 14, 1692 CrossRef CAS PubMed.
- S. Pervaiz and M.-V. Clement, Curr. Pharm. Des., 2004, 10, 1969–1977 CrossRef CAS PubMed.
- V. Chaurin, E. C. Constable and C. E. Housecroft, New J. Chem., 2006, 30, 1740–1744 RSC.
- M. Hernández-Molina, J. González-Platas, C. Ruiz-Pérez, F. Lloret and M. Julve, Inorg. Chim. Acta, 1999, 284, 258–265 CrossRef.
- M. Muralisankar, S. Sujith, N. S. P. Bhuvanesh and A. Sreekanth, Polyhedron, 2016, 118, 103–117 CrossRef CAS.
- N. Balakrishnan, J. Haribabu, A. Krishnan Dhanabalan, S. Swaminathan, S. Sun, D. Fita Dibwe, N. Bhuvanesh, S. Awale and R. Karvembu, Dalton Trans., 2020, 49, 9411–9424 RSC.
- D. Tang, X. Chen and G. Kroemer, Cell Res., 2022, 32, 417–418 CrossRef PubMed.
- W. Kohn, in Density Functional Methods In Physics, ed. R. M. Dreizler and J. da Providência, Springer US, Boston, MA, 1985, pp. 1–9 Search PubMed.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- T. Weymuth, E. P. A. Couzijn, P. Chen and M. Reiher, J. Chem. Theory Comput., 2014, 10, 3092–3103 CrossRef CAS PubMed.
- H. Schröder, A. Creon and T. Schwabe, J. Chem. Theory Comput., 2015, 11, 3163–3170 CrossRef PubMed.
- S. Kossmann and F. Neese, J. Chem. Theory Comput., 2010, 6, 2325–2338 CrossRef CAS PubMed.
- A. Hellweg and D. Rappoport, Phys. Chem. Chem. Phys., 2014, 17, 1010–1017 RSC.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, E. C. Meng, G. S. Couch, T. I. Croll, J. H. Morris and T. E. Ferrin, Protein Sci., 2021, 30, 70–82 CrossRef CAS PubMed.
Footnotes |
| † These results were presented in part at the 16th European Biological Inorganic Chemistry Conference: EuroBIC-16; 17–21 July 2022, Grenoble, France; abstract no. BI1. https://radar.ibiss.bg.ac.rs/handle/123456789/5318 |
| ‡ Electronic supplementary information (ESI) available: Characterisation of 1: MS, IR, UV-vis, X-ray crystallography (CCDC 2281073). Computational chemistry: free energy of dissociation, transitions. Bioanalytical measurements (MTT, CV). CCDC 2281073. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3md00344b |
| This journal is © The Royal Society of Chemistry 2023 |