
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mixing ligands to enhance gas uptake in polyMOFs†
Matthew A.
Pearson
,
Sachin
Bhagchandani
,
Mircea
Dincă
 and
Jeremiah A.
Johnson
*
and
Jeremiah A.
Johnson
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. E-mail: jaj2109@mit.edu
First published on 25th January 2023
Abstract
Polymer-metal organic frameworks (polyMOFs) offer a pathway toward processable polymer–MOF hybrid materials; however, because polymeric ligands are incorporated throughout the MOF lattice, polyMOFs have an inherent shortcoming of reduced pore accessability and surface area compared to traditional MOFs. Herein, a strategy for altering the degree of polymer incorporation in polyMOFs by mixing a multivalent polymer ligand containing MOF-forming linkers with “free” linkers is investigated as a means to tune the properties of polyMOFs, resulting in polyMOFs with superior N2 and CO2 uptake. The mixed ligand approach is further extended to distinct MOF-forming polymer ligands to create multivariate (MTV)-polyMOFs, which provides a method for incorporating low-dispersity polymer ligands with complex architectures into polyMOF lattices without the addition of small molecule components.
Design, System, ApplicationMOFs synthesized from multivalent polymer ligands (polyMOFs) generally suffer from limited scope and decreased permanent porosity as a result of pore filling by the polymer backbones. Here, we employ a new design strategy that involves modifying the relative incorporation of polymer ligands in multi-component polyMOFs by mixing the polymer ligand with small molecule ligands. Increasing the amount of free 1,4-benzenedicarboxylic acid (H2bdc) relative to polymer-bound H2bdc lowers the amount of polymer and provides a concomitant increase in surface area of the polyMOF. This strategy allows for record BET surface areas and CO2 uptake even compared to native MOF-5. The strategy can be extended to mixed polymer ligand polyMOFs, which expands the scope of possible polyMOFs and opens an avenue for the development of polyMOFs that incorporate more complex macromolecular ligands. The strategy also illustrates that the addition of even small amounts of polymer ligand can have drastic impacts on the properties and morphologies of MOFs. |
Introduction
Metal–organic frameworks (MOFs), a class of crystalline, porous coordination polymers composed of inorganic metal-containing clusters connected by multitopic organic ligands, have received notable attention in recent years.1 Their many attractive features, including high surface areas, tunable pore sizes, and modular and addressable chemical functionalities have made them promising platforms for catalysis,2 gas storage and separation,3,4 sensing5,6 and drug delivery.7 Macroscopically, MOFs are microcrystalline powders that generally cannot be melted without destroying the crystal structure. As a result, for industrial applications in separations and catalysis, MOFs are processed into bulk samples by mechanical pressing or mixing with a suitable binder that can then be extruded.8–10 Improving the processability of these materials is required to further their industrial applications and expand potential device fabrication. In recent years, there has been growing interest in the development of hybrid materials of flexible organic polymers and MOFs11,12 with the ultimate goal of combining the porosity, tunability, and functionality of MOFs with the processability and mechanical flexibility of polymers.13–17One approach to polymer/MOF hybridization is the assembly of MOFs from multivalent polymeric ligands, providing “polyMOFs.” The polymer ligands of polyMOFs are generally composed of MOF-forming organic linkers that are connected through a flexible macromolecular backbone.18 Unlike with physical blends of polymers and MOFs, which can generate macrovoids due to chemical incompatibilities at the MOF–polymer interface, in polyMOFs the polymer ligands weave through the MOF lattice and become fully incorporated into the framework during crystal growth. Despite the potential to improve the processability and mechanical stability of MOFs, however, polyMOFs have an inherent drawback of reduced accessible surface areas due to pore filling by the polymer ligands.19 For the isoreticular polyMOF analogues of MOF-5, the highest BET surface area achieved has reached only 1100 m2 g−1, which is approximately one third of the surface area of native MOF-5.18 We recently reported the synthesis of polyMOFs from low dispersity polymers synthesized from reversible addition fragmentation chain transfer (RAFT) polymerization, which allows for greater control over the composition, molar mass, and dispersity of the polymer ligand and enables further expansion of the scope of potential polyMOF materials.20 The utility of these polyMOFs is limited, however, by their low surface areas (only up to 140 m2 g−1). In order to realize the advantages of polyMOFs, the development of new strategies to circumvent this limitation is imperative.
A variety of strategies have been employed to improve the surface area and gas uptake of MOFs, including the modification of organic linkers with specific functional groups,21 metal substitution,22 judicious design of MOF topology and structure,23 and the utilization of alternative methods of solvent evacuation such as activation by supercritical CO2.24 Additionally, the use of multiple organic linkers containing different functionalities to synthesize so-called multivariate (MTV) MOFs has been found to improve the porosity, catalytic activity, and optical properties of MOFs relative to their parent materials (Fig. 1A).25,26 Notably, the resulting properties have not necessarily been a linear combination of the constituent parts. In an early example illustrating this concept, Deng et al. synthesized MTV-MOFs with eight distinct dicarboxylate linkers containing different functionalities, with one “MTV-MOF-5” material exhibiting an increase in CO2 selectivity over CO four times that of the pure component MOFs.27 This work, among others, exemplifies that introducing heterogeneity into these otherwise ordered structures can have synergistic effects that improve the properties of the resulting MOF.
 | ||
| Fig. 1 Mixed ligand strategies used in conventional MOF syntheses and investigated here for polyMOFs. (A) Unique functionalized ligands have been used to synthesize MTV-MOFs. Comparably, we explored (B) a mixed ligand strategy with step-growth polymer ligands and (C) a mixed polymer ligand approach, employing both step-growth and RAFT-synthesized polymers to synthesize MTV-polyMOFs. | ||
In a related approach, we demonstrated that for polyMOFs employing RAFT-derived polymer ligands, the degree of polymer incorporation and properties of the resultant polyMOF could be tuned by including free linkers.20 While this method proved successful for tuning the crystallinity and hydrolytic stability of the polyMOFs, the surface areas, though still tunable, remained low even upon the addition of three equivalents of free linker. We hypothesized that the polyacrylamide structure, which placed the linkers on the polymer sidechains, likely results in greater pore filling relative to polymer ligands with linkers in the polymer backbone. Therefore, we envisioned expanding this strategy to polyMOFs synthesized from step-growth polymerizations that have already demonstrated appreciable surface areas (Fig. 1B). By tuning the relative amount of polymer incorporation, we could access polyMOFs with higher surfaces areas, while maintaining the favorable properties induced by incorporation of the polymer ligand. Additionally, by utilizing multiple unique polymer ligands, we can synthesize multivariate polyMOFs (MTV-polyMOFs) that enable incorporation of low dispersity polymer ligands without the need for small-molecule linkers (Fig. 1C).
Results and discussion
We began our investigation by synthesizing a polymer ligand that had previously been reported to form polyMOFs with high surface areas. We selected a polyether ligand (pbdc-7a) comprising 1,4-benzenedicarboxylic acid (H2bdc) linkers connected by a seven-carbon spacer, previously reported by Cohen et al. to form polyMOF analogues of both MOF-5 and UiO-66.18,28 This ligand has been shown to generate polyMOFs with the highest reported BET surface areas and highest CO2 uptake at 298 K to date.18 The polymer was synthesized in a two-step procedure (Scheme S1†): first, a condensation polymerization of diethyl 2,5-dihydroxyterephthalate and 1,7-dibromoheptane in DMF at 100 °C for 24 hours gave the ester form of the polymer (pbdc-7e), which was then hydrolyzed to obtain the polyacid pbdc-7a in 55% yield, as indicated by 1H NMR and 13C NMR (Fig. S1 and S2†). Analysis by gel permeation chromatography (GPC) showed that the polymer had a number- and weight-average molar mass of 7000 and 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 g mol−1, respectively, and a unimodal molar mass distribution with a high dispersity (Đ = 2.10), comparable to reported values18,29 (Fig. S3†).
700 g mol−1, respectively, and a unimodal molar mass distribution with a high dispersity (Đ = 2.10), comparable to reported values18,29 (Fig. S3†).
To determine the ability of pbdc-7a to form mixed ligand polyMOF-5, as opposed to independently forming polyMOF-5 and native MOF-5, we synthesized a set of polyMOFs with varying amounts of added free H2bdc linker. Free H2bdc, pbdc-7a, and Zn(NO3)2·6H2O were dissolved in DMF and placed in a preheated 100 °C oven for 24 h. For each of the reactions, the ratio of Zn(NO3)2·6H2O to total H2bdc (taken as the sum of polymer-bound ligands and free ligands) was kept constant at 3:1, while the ratio of free MOF-forming H2bdc linker to polymer-bound H2bdc ranged from 0.1 to 3. The concentration was also kept constant at 0.24 M with respect to Zn(NO3)2·6H2O. Each of the reactions produced an off-white precipitate. Interestingly, for the reactions with 0, 0.1, 0.5, and 1 equivalents of free linker added (notated as Zn-pbdc-7a-xeq, where x indicates the equivalents of free ligand added to the initial reaction solution), the precipitate took the form of a brittle film, whereas those with 2 and 3 equivalents of added free linker formed powders, as might be expected as the material becomes more resemblant of the native MOF-5 due to decreased polymer incorporation.
The materials were each characterized by powder X-ray diffraction (PXRD). All samples exhibited the diffraction patterns characteristic of MOF-5, as well as a broad peak centered around 2θ = 22°, indicative of the presence of an amorphous phase (Fig. 2). This amorphous phase was most prominent among Zn-pbdc-7a-0eq, Zn-pbdc-7a-2eq, and Zn-pbdc-7a-3eq, contrary to what might be expected for polyMOFs with a lower fraction of polymer incorporation. The latter two samples also more prominently displayed higher angle peaks not clearly visible in other polyMOF samples.
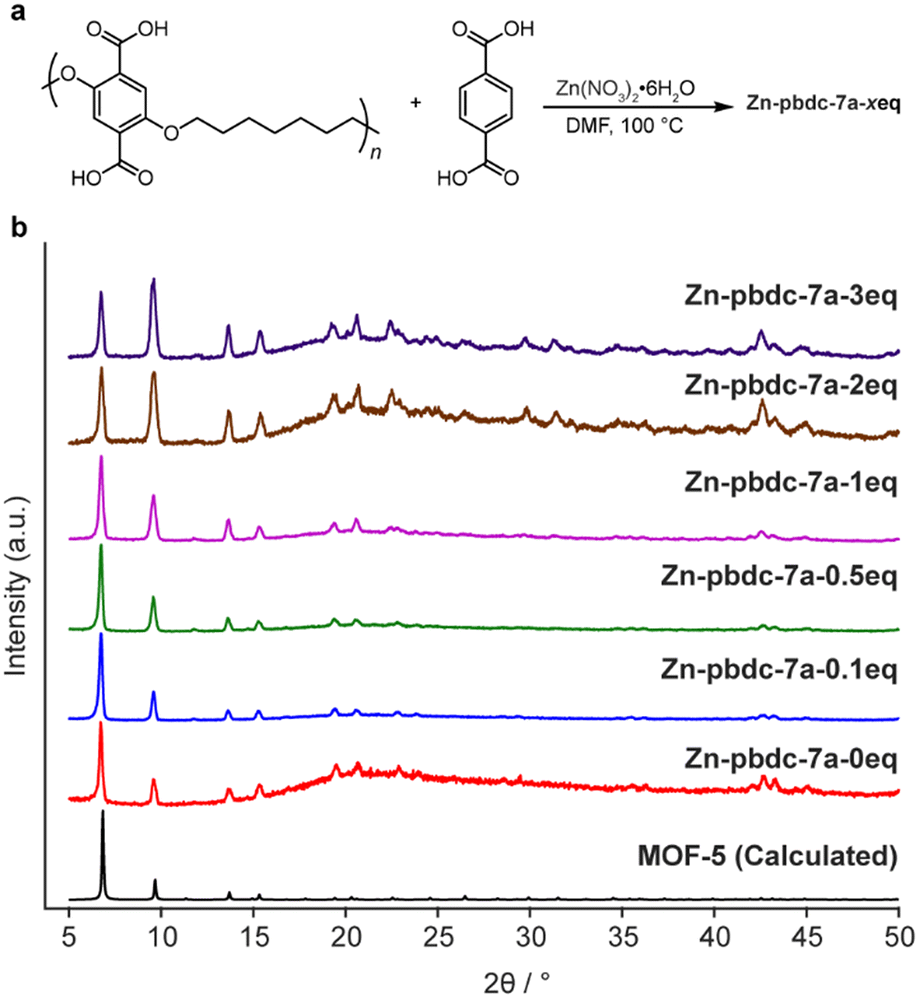 | ||
| Fig. 2 Synthesis of mixed ligand polyMOF Zn-pbdc-7a-xeq, polyMOFs formed from pbdc-7a and x equivalents of free H2bdc (a) and powder X-ray diffractogram (b) of Zn-pbdc-7a-xeq with varying equivalents of free H2bdc used in the synthesis. | ||
To determine the relative amount of polymer-bound linker and free linker in each of the crystalline polyMOFs, the samples were digested in DCl/DMSO-d6 and the ratios were quantified by 1H NMR analysis (Fig. S4–S9†). The results (Table S1†) show these ratios remain comparable to the initial reaction stoichiometries. To further probe the polymer ligand incorporation, we analysed a mixed ligand polyMOF synthesized with pbdc-7a and 2-aminobenzene-1,4-dicarboxylic acid (H2bdc-NH2) by transmission electron microscopy and electron-dispersive X-ray spectroscopy (TEM-EDX). The presence of the nitrogen atoms in the elemental mappings (Fig. 3 and S10†) corroborates the incorporation of free bdc-NH2 linkers. Altogether, these data suggest that both pbdc-7a and free H2bdc are incorporated comparably into the MOF lattices, allowing for the relative incorporation of these polymers to be rationally tuned by altering the initial reaction stoichiometry.
 | ||
| Fig. 3 (a) TEM image of Zn-pbdc-7a-2eq-NH2 and (b), Zn (c) O, (d) N EDX elemental mapping of Zn-pbdc-7a-2eq-NH2, indicating incorporation of free bdc-NH2 ligand into polyMOF lattice. | ||
Morphological differences between the particles are clearly evident in scanning electron microscopy (SEM) images (Fig. S11–S15†). Zn-pbdc-7a-0eq shows the formation of a film of interconnected cubic MOF crystals, which is not present in the polyMOFs with greater amounts of free linker added. Instead, particles of interconnected cubic MOF crystals form, with the size of these particles decreasing as the fraction of added free linker increases. In particular, Zn-pbdc-7a-1eq and Zn-pbdc-7a-3eq form particles with a wide range of sizes from 20–120 μm, while Zn-pbdc-7a-15eq and Zn-pbdc-7a-30eq (synthesized to probe the impact of further addition of free H2bdc ligand) ranged from 10–20 μm. As both the formation of the film and intergrowth of the MOF-5 crystals is most likely the result of multiple nucleation sites along individual polymer chains, it is reasonable that different morphologies are formed at lower polymer ligand concentrations.
We hypothesized that decreasing the polymer ligand concentration should have a measurable impact on the BET surface areas of the polyMOFs, as the occluded space in the pores should be reduced. We tested this hypothesis using N2 gas sorption measurements for a series of polyMOFs at 77 K. The polyMOFs all exhibited type-I isotherms and calculations based on the N2 adsorption isotherms for Zn-pbdc-7a-0eq, Zn-pbdc-7a-1eq, Zn-pbdc-7a-3eq, Zn-pbdc-7a-15eq, and Zn-pbdc-7a-30eq indicated BET surface areas of 1011, 1287, 1359, 1356, and 562 m2 g−1, respectively (Fig. 4a).30 These measurements provide evidence that the mixed ligand strategy can be used to tune the accessible surface of polyMOFs. Furthermore, using this method we were able to obtain a record high surface for polyMOF-5, which represents a 34% increase from the “pure” polyMOF measurement Zn-pbdc-a-0eq.18 Modifying these gravimetric BET surface areas to only account for the mass that potentially contributes to the MOF lattice (i.e., to only include the mass of the bdc linkers of the polymer ligands and not the spacers between each bdc group) (Fig. 4b, Table S2†) suggested that the source of the increase in BET surface area largely arose from a decrease in total mass of the polymer ligands. Previous work has found that isoreticular expansions of the Zr polyMOF polyUiO-66 with variants composed of biphenyl- and terphenyl-based ligands led to concomitant increases in BET surface area, attributable to a decrease in pore filling.31 As such, future work should examine the impact of decreased polymer incorporation into larger pore polyMOFs.
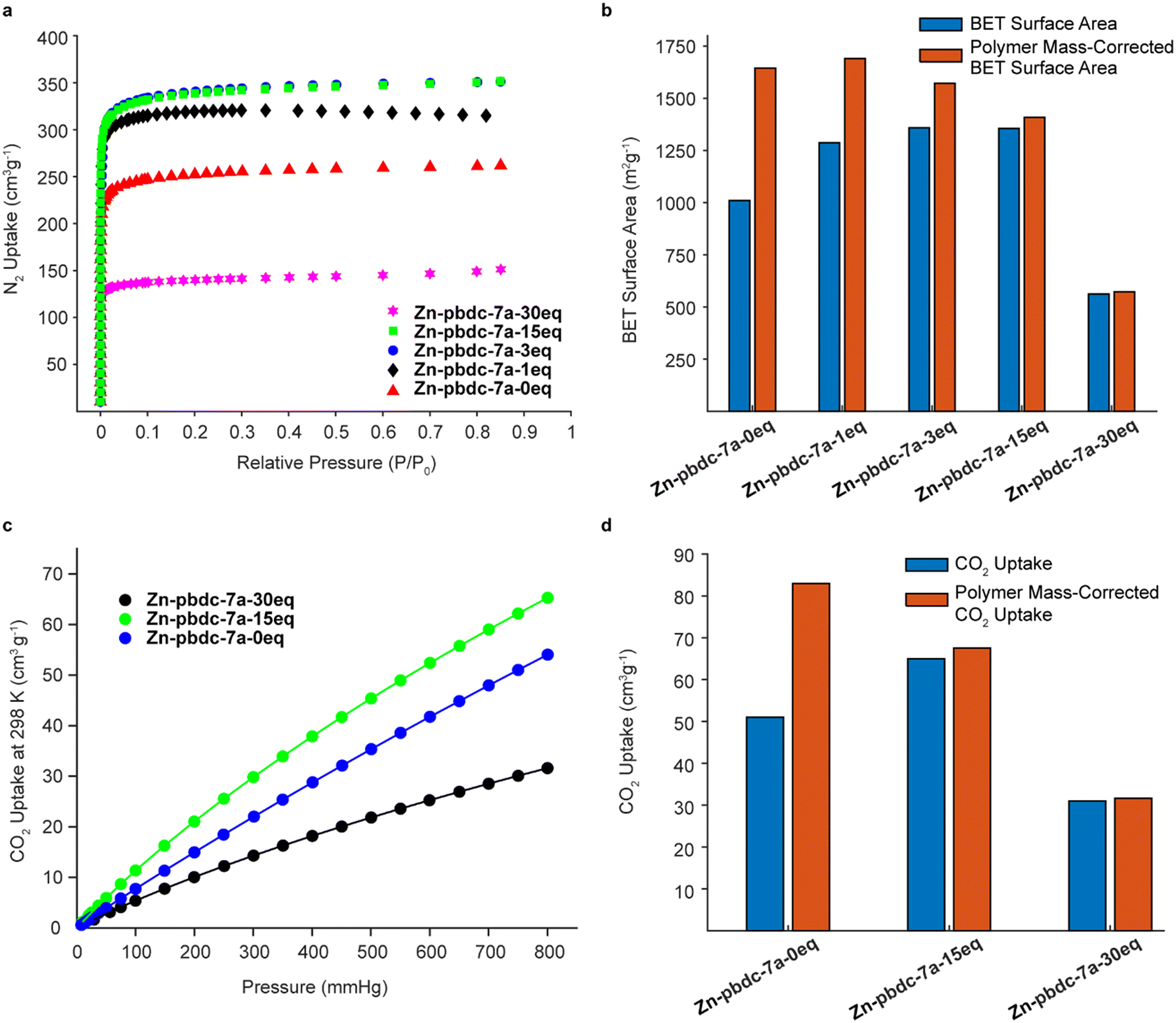 | ||
| Fig. 4 N2 (a) and CO2 (c) gas sorption isotherms for a series of polyMOF-5 materials synthesized from pbdc-7a and x equivalents of free H2bdc linkers and a comparison of the polyMOFs' BET surface areas (b) and total CO2 uptake at 298 K and 1 atm (d). BET surface areas and CO2 uptake capacities corrected to only account for the mass contributing to the framework are shown in red. | ||
We also investigated the CO2 uptake of several of these polyMOFs, as it has been shown that polyMOFs can have increased CO2 uptake relative to their native MOF counterparts. It was suggested by Cohen and co-workers that the observed higher heats of CO2 adsorption (Qst) originate from a decrease in pore size that results from the incorporation of polymers throughout the framework.18,29,32,33 We reasoned that a lower polymer fraction could maintain the higher heats of CO2 adsorption observed from polymer incorporation, while decreasing the mass of polymer ligand in the pores, leading to higher CO2 uptake. Measurements of CO2 uptake (Fig. 4c) for Zn-pbdc-7a-0eq, Zn-pbdc-7a-15eq, and Zn-pbdc-7a-30eq gave values of 51, 65, and 31 cm3 g−1 at 1 atm and 298 K, compared to 20 cm3 g−1 that has been previously measured for the native MOF-5.18 Interestingly, addition of free linker initially leads to an increase in CO2 uptake—27% over Zn-pbdc-7a-0eq and 325% over native MOF-5—but further increases result in reductions of CO2 uptake from the pure polymer Zn-pbdc-7a-0eq. One possible explanation for this trend could be that, past a certain point, the polyMOF becomes more structurally comparable to the native MOF-5 and thus leads to a relative decrease in CO2 uptake. Regardless, these results show that even small amounts of polymer incorporation can have positive impacts on the properties of the resulting polyMOF.
Encouraged by these results, we sought to determine whether a similar strategy could be utilized to form MTV-polyMOFs composed of two unique polymer ligands bearing the same organic linker. As a proof of concept, we synthesized a set of polyMOFs from pbdc-7a and a previously reported poly(acrylamide), pabdc-0a, that was synthesized via RAFT and that bears the H2bdc linkers on pendant sidechains (Fig. 5a).20 The dispersity of pabdc-0a was shown to play a significant role in the ability of the polymer to form polyMOFs. In particular, pabdc-0a was only able to form crystalline material when synthesized by an uncontrolled free radical polymerization, leading to high dispersity, or when free H2bdc linker was added to the reaction solution during MOF synthesis. The high dispersity polymer pbdc-7a could function similarly in enabling the integration of low dispersity pabdc-0a into the MOF framework, thereby allowing access to polyMOFs composed purely of polymers that incorporate well-defined polymers with complex architectures.
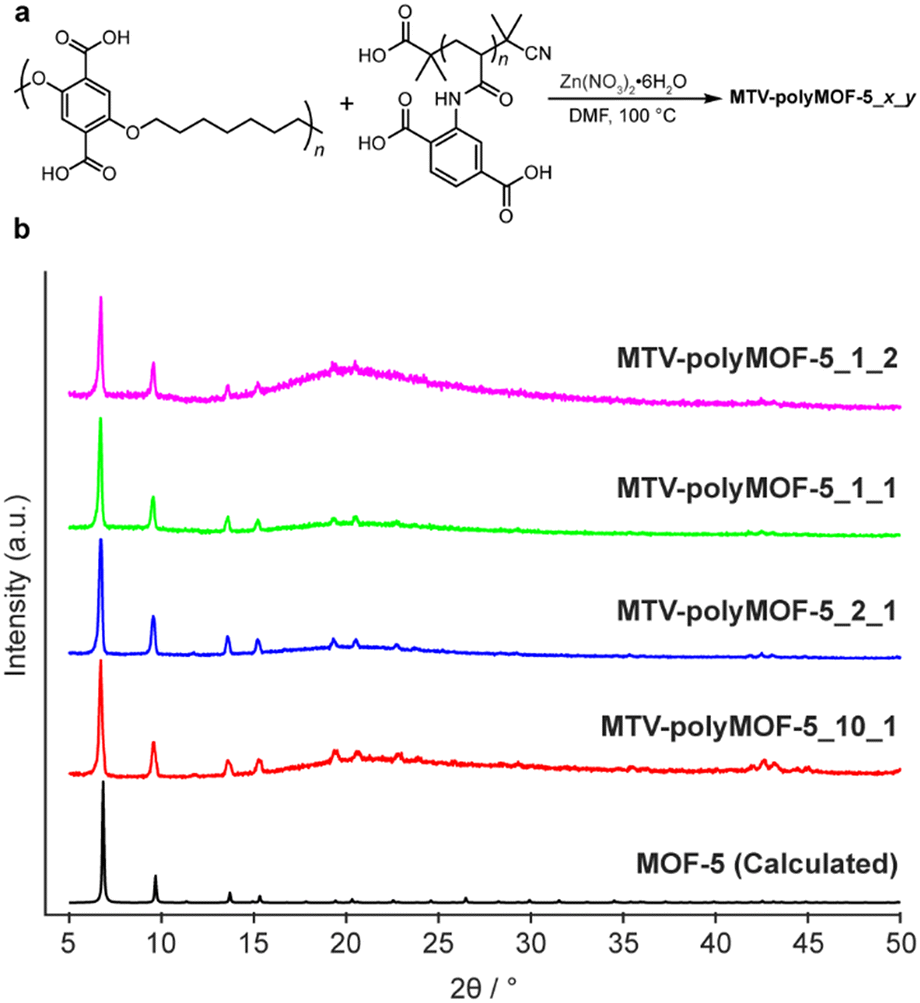 | ||
| Fig. 5 (a) Synthesis of mixed polymer ligand polyMOFs MTV-polyMOF-5_x_y from x equivalents of pbdc-7a and y equivalents of pabdc-0a and (b) powder X-ray diffractogram of series of MTV-polyMOF-5_x_y. | ||
We explored four different ligand ratios of pbdc-7a to pabdc-0a (10:1, 2:1, 1:1, and 1:2, based on the H2bdc bound to each polymer) and maintained the ratio of Zn2+ to total H2bdc constant at 3:1 for each synthesis. The polymers and Zn(NO3)2·6H2O were dissolved in DMF and placed in a 100 °C preheated oven for 24 hours, after which a precipitated solid was isolated. The solids each exhibited diffraction patterns matching that of MOF-5 (Fig. 5b). Additionally, 1H NMR of the digested materials (Fig. S16–S19†) showed polymer ratios comparable to the initial stoichiometric ratio, and elemental mapping by TEM-EDX (Fig. S20 and S21†) provided evidence of homogenous incorporation of pabdc-0a based on the nitrogen atoms of the acrylamide, suggesting that this strategy can facilitate the formation of MTV-polyMOFs over a range of polymer ratios.
We further explored this strategy as a means to incorporate more complex polymer architectures into the MOF framework by synthesizing MTV-polyMOFs from pbdc-7a and diblock copolymer PS-b-pabdc-0a (Scheme S2†) over the same range of polymer ratios. Brittle films were formed for each of the syntheses and again they each showed diffraction patterns corresponding to MOF-5 (Fig. S22†). Interestingly, however, samples with larger fractions of PS-b-pabdc-0a, as determined by 1H NMR of the digested polyMOFs (Fig. S23–S26†), showed the appearance of several new diffraction peaks (most prominently at 14.7°, 20.2°, and 21.7°) not typical of MOF-5, indicating the formation of a new crystalline phase in addition to that of MOF-5. These results highlight that the addition of even small amounts of a polymer ligand can have impacts on the structure of the material and opens up MTV-polyMOFs as a viable approach for incorporating low dispersity polymers with complex architectures and functionalities into polyMOFs.19,34,35
Conclusions
We have demonstrated that a mixed ligand strategy can be utilized to tune the relative incorporation of polymer ligands in polyMOFs. This approach was found to generate polyMOFs with improved BET surface areas and record high CO2 uptake, significantly outpacing the native MOF-5. This strategy was further shown to enable the synthesis of mixed polymer ligand MTV-polyMOFs. MTV-polyMOFs can enable the incorporation of low dispersity RAFT polymers into polyMOFs without the addition of small molecule free linker. Altogether, these results expand the scope of polyMOF materials by facilitating the precise tuning of their properties and the integration of complex polymer architectures.Author contributions
M. A. P., M. D., and J. A. J. conceived the research and designed experiments. M. A. P. and S. B. performed the research and collected data. M. A. P. analyzed the data. The manuscript was written through contributions of M. A. P., M. D., and J. A. J.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF Center for the Chemistry of Molecularly Optimized Networks (MONET), CHE-2116298. M. D. was supported through the Department of Energy (DE-SC0023288).Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341 DOI:10.1126/science.1230444.
- D. Yang and B. C. Gates, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- H. Li, K. Wang, Y. Sun, C. T. Lollar, J. Li and H. C. Zhou, Mater. Today, 2018, 21, 108–121 CrossRef CAS.
- P. Kumar, A. Deep and K. H. Kim, TrAC, Trends Anal. Chem., 2015, 73, 39–53 CrossRef CAS.
- I. Stassen, J.-H. Dou, C. Hendon and M. Dincă, ACS Cent. Sci., 2019, 5, 1425–1431 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- B. Yilmaz, N. Trukhan and U. Müller, Chin. J. Catal., 2012, 33, 3–10 CrossRef CAS.
- A. I. Spjelkavik, Aarti, S. Divekar, T. Didriksen and R. Blom, Chem. – Eur. J., 2014, 20, 8973–8978 CAS.
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- F. Jin, J. Liu, Y. Chen and Z. Zhang, Angew. Chem., 2021, 133, 14342–14355 CrossRef.
- M. Galizia, W. S. Chi, Z. P. Smith, T. C. Merkel, R. W. Baker and B. D. Freeman, Macromolecules, 2017, 50, 7809–7843 CrossRef CAS.
- Y. Cheng, Y. Ying, S. Japip, S.-D. Jiang, T.-S. Chung, S. Zhang, D. Zhao, Y. Cheng, Y. Ying, S. Japip, S. Jiang, T. Chung, S. Zhang and D. Zhao, Adv. Mater., 2018, 30, 1802401 CrossRef PubMed.
- H. B. Tanh Jeazet, C. Staudt and C. Janiak, Dalton Trans., 2012, 41, 14003–14027 RSC.
- V. J. Pastore and T. R. Cook, Chem. Mater., 2020, 32, 3680–3700 CrossRef CAS.
- M. Kalaj, K. C. Bentz, S. Ayala, J. M. Palomba, K. S. Barcus, Y. Katayama and S. M. Cohen, Chem. Rev., 2020, 120, 8267–8302 CrossRef CAS PubMed.
- Z. Zhang, H. T. H. Nguyen, S. A. Miller and S. M. Cohen, Angew. Chem., Int. Ed., 2015, 54, 6152–6157 CrossRef CAS PubMed.
- K. C. Bentz, K. Gnanasekaran, J. B. Bailey, S. Ayala, F. A. Tezcan, N. C. Gianneschi and S. M. Cohen, Chem. Sci., 2020, 11, 10523–10528 RSC.
- M. A. Pearson, M. Dincă and J. A. Johnson, Chem. Mater., 2021, 33, 9508–9514 CrossRef CAS.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS PubMed.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef CAS PubMed.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- A. P. Nelson, O. K. Farha, K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 458–460 CrossRef CAS PubMed.
- A. Helal, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Natl. Sci. Rev., 2017, 4, 296–298 CrossRef CAS.
- J. Jiao, W. Gong, X. Wu, S. Yang and Y. Cui, Coord. Chem. Rev., 2019, 385, 174–190 CrossRef CAS.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- P. G. M. Mileo, S. Yuan, S. Ayala, P. Duan, R. Semino, S. M. Cohen, K. Schmidt-Rohr and G. Maurin, J. Am. Chem. Soc., 2020, 142, 10863–10868 CrossRef CAS PubMed.
- Z. Zhang, H. T. H. Nguyen, S. A. Miller, A. M. Ploskonka, J. B. Decoste and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 920–925 CrossRef CAS PubMed.
- J. W. M. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider, R. Ameloot, J. Marreiros, C. Ania, D. Azevedo, E. Vilarrasa-Garcia, B. F. Santos, X. H. Bu, Z. Chang, H. Bunzen, N. R. Champness, S. L. Griffin, B. Chen, R. B. Lin, B. Coasne, S. Cohen, J. C. Moreton, Y. J. Colón, L. Chen, R. Clowes, F. X. Coudert, Y. Cui, B. Hou, D. M. D'Alessandro, P. W. Doheny, M. Dincă, C. Sun, C. Doonan, M. T. Huxley, J. D. Evans, P. Falcaro, R. Ricco, O. Farha, K. B. Idrees, T. Islamoglu, P. Feng, H. Yang, R. S. Forgan, D. Bara, S. Furukawa, E. Sanchez, J. Gascon, S. Telalović, S. K. Ghosh, S. Mukherjee, M. R. Hill, M. M. Sadiq, P. Horcajada, P. Salcedo-Abraira, K. Kaneko, R. Kukobat, J. Kenvin, S. Keskin, S. Kitagawa, K. ichi Otake, R. P. Lively, S. J. A. DeWitt, P. Llewellyn, B. v. Lotsch, S. T. Emmerling, A. M. Pütz, C. Martí-Gastaldo, N. M. Padial, J. García-Martínez, N. Linares, D. Maspoch, J. A. Suárez del Pino, P. Moghadam, R. Oktavian, R. E. Morris, P. S. Wheatley, J. Navarro, C. Petit, D. Danaci, M. J. Rosseinsky, A. P. Katsoulidis, M. Schröder, X. Han, S. Yang, C. Serre, G. Mouchaham, D. S. Sholl, R. Thyagarajan, D. Siderius, R. Q. Snurr, R. B. Goncalves, S. Telfer, S. J. Lee, V. P. Ting, J. L. Rowlandson, T. Uemura, T. Iiyuka, M. A. van der Veen, D. Rega, V. van Speybroeck, S. M. J. Rogge, A. Lamaire, K. S. Walton, L. W. Bingel, S. Wuttke, J. Andreo, O. Yaghi, B. Zhang, C. T. Yavuz, T. S. Nguyen, F. Zamora, C. Montoro, H. Zhou, A. Kirchon and D. Fairen-Jimenez, Adv. Mater., 2022, 34, 2201502 CrossRef CAS PubMed.
- G. E. M. Schukraft, S. Ayala, B. L. Dick and S. M. Cohen, Chem. Commun., 2017, 53, 10684–10687 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724–781 CrossRef PubMed.
- C. Trickett, A. Helal and B. Al-Maythalony, et al. , Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- M. J. Macleod and J. A. Johnson, Polym. Chem., 2017, 8, 4488–4493 RSC.
- S. Ayala, K. C. Bentz and S. M. Cohen, Chem. Sci., 2019, 10, 1746–1753 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2me00227b |
| This journal is © The Royal Society of Chemistry 2023 |