
Oligoaniline-assisted self-assembly of polyaniline crystals†
Ian M.
Hill
a,
Di
Wu
a,
Bohao
Xu
a and
Yue
Wang
 *ab
*ab
aDepartment of Materials Science and Engineering, University of California, Merced, USA. E-mail: yuewang@ucmerced.edu
bDepartment of Chemistry and Biochemistry, University of California, Merced, USA
First published on 31st January 2023
Abstract
The conductivity and charge transport mobility of conjugated polymers (CPs) are largely correlated with their degree of crystallinity, rendering the crystallization of CPs an important endeavour. However, such tasks can be challenging, especially in the absence of sidechain functionalization. In this study, we demonstrate that the incorporation of a small amount of oligomers, specifically tetraaniline, can induce crystallization of the parent polymer, polyaniline, through a single-step self-assembly process. The resulting crystals are compositionally homogeneous because the oligomers and their parent polymer share the same repeating unit and are both electroactive. Mechanistic studies reveal that the tetraaniline forms a crystalline seed that subsequently guides the assembly of polyaniline due to their structural similarities. Applying this oligomer-assisted crystallization approach to polyaniline with defined molecular weights resulted in single crystalline nanowires for 5000 Da polyaniline, and nanowires with strong preferential chain orientation for those with molecular weights between 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 100000 Da. Absorption studies reveal that the polymer chains are in an expanded conformation, which likely contributed to the high degree of packing order. Two-probe, single nanowire measurements show that the crystals have conductivity as high as 19.5 S cm−1. This method is simple, general, and can potentially be applied to other CPs.
000 and 100000 Da. Absorption studies reveal that the polymer chains are in an expanded conformation, which likely contributed to the high degree of packing order. Two-probe, single nanowire measurements show that the crystals have conductivity as high as 19.5 S cm−1. This method is simple, general, and can potentially be applied to other CPs.
New conceptsCrystallization of unfunctionalized conjugated polymers (CPs) has traditionally been challenging due to the large free energy barrier for chain rearrangement. Here, we present a new concept: using monodispersed oligomers of CPs to “straighten out” the parent polymer chains, thus promoting crystallization. This oligomer-assisted self-assembly method offers two distinct advantages over existing methods, such as vapor phase polymerization or other in situ polymerization methods. (1) The resulting crystals are compositionally homogeneous because the oligomers and polymers share the same repeating unit. (2) This process decouples the control over the chemical properties and crystallinity of the CPs, allowing for greater tunability over individual material properties. For example, single crystals of polyaniline with a defined molecular weight have been produced. This concept is likely to be applicable to the crystallization of other CPs and even non-conjugated polymers because monodispersed oligomers can be synthesized for each polymer. Furthermore, while this work focuses on crystalline nanostructures, the general concept of oligomer-assisted crystallization may also lead to enhanced crystallinity in 2D thin films. |
1. Introduction
Advancement in the understanding of the fundamental structure–property relationships of conjugated polymers (CPs), including conducting and semiconducting polymers, has fuelled the development of high performance organic electronics.1,2 Crystalline CPs, especially single crystals, are fundamentally important for understanding the charge transport properties in relation to packing arrangement and orientation. From an application-driven perspective, crystalline CPs are important for achieving high efficiency in organic electronics because they provide enhanced and directional conductivity or charge carrier mobility compared to their disordered or poorly crystalline counterparts.3 Unfortunately, single crystals or crystals with strong preferential orientation of CPs are difficult to produce due to the rich free energy landscape compared to the soft chain polymer counterparts,4,5 but can be made possible through a few solution-based methods, such as side chain or backbone modification. Sidechain modification provides CPs with additional noncovalent interactions including van der Waals or solvophobic interactions through the interdigitation or aggregation of sidechains, thus leading to enhanced ordering.6,7 The steric effects from certain sidechains can also restrict the torsion angle between the repeating units along the polymer backbone, thus promoting ordered stacking. Directly increasing the backbone planarity such as by incorporating fused rings or alkyne groups can increase the persistence length of the polymer, and create enhanced intermolecular interactions.8,9 The same goal can be achieved by incorporating heteroatoms such as fluorine or sulfur along the polymer backbone to create additional intramolecular interactions that can “lock” the backbone conformation.10,11 In the absence of side chain or backbone engineering, ordered packing can be obtained by growing CPs from heterogeneous nucleation sites such as the surface of CNTs.12,13 Due to the preferential π–π packing orientation of CPs on the π-conjugated surface of CNTs, CPs can pack in a highly ordered fashion with the growth long axis perpendicular to the surface of the heterogeneous nucleation sites. Unfortunately, such methods only generate hetero-structures, essentially changing the chemical composition of the CPs.Compared to sidechain-functionalized or backbone-modified CPs, crystallization of unmodified CPs such as polyaniline (PANI), polypyrrole (PPy), polythiophene (PT), or poly(3,4-ethylenedioxythiophene) (PEDOT) is especially challenging due to the high torsion angle between the adjacent repeating units and the limited noncovalent interactions beyond those offered by the π-faces.14 Vapor phase polymerization (VPP) has been the most successful in generating crystalline entities for such materials, but the resulting structures are usually semicrystalline with small crystallites embedded in disordered regions.15,16 It has been shown that VPP in combination with nanoconfinement within lithographically-fabricated narrow channels can lead to single or polycrystalline PEDOT with very high conductivity.17 However, the degree of crystallinity is channel-width dependent. Also, channel fabrication requires multi-step lithography on a surface, limiting its scalability. Alternatively, through solution-based methods, crystals of unmodified CPs such as PANI can be synthesized through in situ polymerization at the air–water interface in the presence of surfactants.18 The resulting 2D crystals can be scaled up to wafer-size by increasing the air–water interfacial area, but the scalability pales in comparison to bulk solution- or vapor-based methods. Regardless of the specific method, a major limitation of all in situ polymerization approaches is that they do not offer the ability to separately control the degree of crystallinity and materials’ chemical properties such as molecular weight or polydispersity. On the contrary, the crystallization of pre-made CPs offers such explicit control because the chemical properties of the crystals are dictated through synthesis, whereas the packing order is controlled through crystallization.4,5 This distinct advantage makes post-synthetic crystallization approaches generally more favourable.
Here, we demonstrate a new concept for the crystallization of unmodified CPs. Using PANI as a model system, we achieve such a feat through a simple, one-step self-assembly process in the presence of a small amount of oligomers of PANI, such as tetraaniline (TANI). The easily crystallizable TANI lowers the free energy barrier for the organization of PANI chains into ordered states through noncovalent intermolecular interactions. Both single crystals and crystals with strong preferential chain orientation have been obtained. Furthermore, due to the identical repeating units in the oligomers and their parent polymer, the resulting crystals are compositionally homogeneous.
2. Results and discussion
2.1. Conceptual demonstration
The rationale behind this project is the following. (1) The difficulty in crystallizing CPs, especially for unmodified CPs such as PANI, lies in their rich free energy landscape and the entropic barriers for chain rearrangement from disordered to ordered states. (2) Unlike CPs such as PANI, their oligomers such as TANI can easily crystallize into single crystals.19,20 This is because the free energy barrier for oligomers to form long-range order is significantly lower than that for polymers owing to the short chain length of oligomers. (3) Oligomers and the parent polymers share the same chemical repeating units (e.g., TANI vs. PANI, shown in Fig. 1a and b), so the intermolecular interactions between them would be identical to those between adjacent oligomers or adjacent polymer chains. Combining these rationales, we hypothesized that adding a small amount of oligomers to the crystallization of the parent polymers may decrease the free energy barrier for “straightening out” the polymer chains and facilitating their packing in a similar manner to the oligomers. | ||
| Fig. 1 Chemical structures of (a) tetraaniline and (b) polyaniline. (c–e) SEM images of (c) PANI nanofibers, (d) TANI nanowires, and (e) self-assembled high aspect ratio PANI nanofibers containing 20% TANI (TAS-PANI). (f) TEM image of TAS-PANI. (g) TAS-PANI after acetone wash to remove TANI. (h) Photograph showing an aqueous dispersion of TAS-PANI. (i) Schematic diagram showing the mechanism behind TANI-assisted PANI assembly. | ||
To probe this hypothesis, we mixed 20% of TANI into PANI, both in the dedoped, emeraldine base (EB) oxidation state, and added the mixture to a self-assembly solvent environment composed of a 1:4 mixture of THF and 0.1 M HClO4 (aq.). The mixture is left undisturbed to self-assemble over the course of 5 days. Prior to self-assembly, the as-used PANI powder in the EB form has a low aspect-ratio, nanofibrous morphology (Fig. 1c), and the TANI EB powder has an irregularly shaped, agglomerated morphology (Fig. S7a, ESI†). When TANI by itself is subjected to the self-assembly solvent environment, rigid and crystalline nanowires are obtained (Fig. 1d), consistent with our previous findings.19,20 With the solid composition composed of 80% PANI and 20% TANI, high aspect ratio, nonwoven nanowires are obtained (Fig. 1e). The self-assembly process can be seen visually: the initial dark blue powder that settles at the bottom of the vial gradually turns green, indicative of TANI and/or PANI transitioning into the doped emeraldine salt (ES) form. The mixture slowly migrates into the surrounding medium through the self-assembly process, giving the precipitate a fluffy appearance. Upon purification via dialysis against deionized water, a stable dispersion can be formed by simple agitation (Fig. 1h). The resulting nanowires are typically tens of microns long with an average diameter of 20–30 nm (Fig. S3, ESI†). Selected area electron diffraction (SAED) of these nanowires shows weak scattering arcs along the wire long axis, suggesting that the polymer chains have a preferential orientation within the nanowires (Fig. S2, ESI†). The scattering arcs correspond to a d-spacing of 0.36 nm, characteristic of the intermolecular distance of π–π stacking along the [010] direction.4 This finding indicates that π–π stacking could be a main driving force for the self-assembly of TANI/PANI, leading the nanowires to preferentially grow along this direction. Hydrogen bonding between amine groups has also likely aided in stabilizing the packing arrangement. We thereafter refer to these crystalline nanostructures as tetraaniline-assisted self-assembled polyaniline, abbreviated as TAS-PANI.
2.2. Mechanistic understanding
To understand the role of TANI in assisting the self-assembly of PANI, we soaked the isolated TAS-PANI nanowires in a copious amount of acetone overnight. TANI is soluble in acetone, but PANI has no solubility in it due to the significantly higher molecular weight. Therefore, the acetone wash would selectively remove the TANI component and leave behind PANI. Transmission electron microscopy (TEM) shows that the pristine TAS-PANI exhibits a solid interior (Fig. 1f). However, a thin cavity can be clearly observed under TEM for the TAS-PANI nanowires after the solvent wash (Fig. 1g). This result indicates that the small amount of TANI molecules likely first assemble into a crystalline core due to their propensity to crystallize (Fig. 1i). PANI does not self-assemble in the absence of TANI (Fig. 2i). Subsequently, due to the structural similarity between TANI and PANI, the disordered PANI chains would interact with the ordered TANI core, leading the PANI chains to assemble along the same preferential stacking direction of TANI (Fig. 1i). | ||
| Fig. 2 (a–i) SEM images of TANI and TAS-PANI self-assembled at different TANI:PANI ratios in a solvent mixture of 20% THF and 80% 0.1 M HClO4. (j–l) Schematic diagrams illustrating the proposed mechanism behind the relationship between the TANI:PANI ratio and packing order. | ||
These experimental insights depict the following self-assembly mechanism. Neither TANI nor PANI is soluble in aqueous acidic solutions, but both can be doped in this environment. In the dedoped EB form, TANI is highly soluble whereas PANI is moderately soluble in polar organic solvents such as THF. Therefore, the solvent mixture used here offers partial solubility for PANI EB and TANI EB, which is generally considered a desirable condition for crystallization via self-assembly.21 At the same time, the HClO4 acid in the solvent mixture can dope PANI and TANI into their emeraldine salt (ES) forms, which have no and limited solubility in THF, respectively, and no solubility in water. Therefore, both PANI and TANI gradually lose solubility in the solvent mixture as increasing portions of them get protonated into the ES states, leading to interchain association due to solvophobic effects.
Due to the different solubilities of TANI and PANI in the EB states in the solvent mixture, there is likely a difference in the degree or rate of solvation and self-assembly, making the assembly of TANI and PANI sequential rather than concurrent. TANI has higher solubility in THF than PANI, so the 20% THF in the solvent mixture would first solvate mostly TANI. Once the TANI molecules are brought into the solvent mixture, they would get doped by the HClO4 acid and transition into the protonated ES form that has marginal solubility in THF/water mixtures. Therefore, supersaturation is quickly reached, causing the TANI molecules to aggregate through solvophobic, π–π, electrostatic, and hydrogen bonding interactions to minimize their interaction with solvent molecules.4,19,22 Ordered TANI cores are therefore formed and subsequently serve as nucleation centres for PANI assembly. Due to the lower solubility of PANI in THF, the same solvation–doping–precipitation–aggregation process occurs at a slower rate for PANI than that of TANI. As doped PANI chains are precipitated out from the solvent mixture, it is more energetically favourable for them to adhere to the existing heterogeneous nucleation centres of TANI “seed crystals” than spontaneously forming new homogenous nucleation centres. Therefore, PANI assembles around the TANI core. The π–π and other noncovalent interactions between the chemically identical oligomer and polymer lead PANI to adopt an extended chain conformation, facilitating PANI to stack along the same [010] long axis as TANI, leading to 1D nanowires with preferential ordering. We found that using other polar organic solvents such as dimethyl sulfoxide (DMSO) or other acids such as diluted nitric acid also leads to similar results (Fig. S5 and S6, ESI†). This proposed mechanism is analogous to that behind crystallization by self-seeding. Self-seeding is mostly used in the molten state for soft chain polymers such as polyethylene,23 but has been applied to creating single crystals of poly(3-hexylthiophene) (P3HT) from its solution.24 This process takes advantage of the slightly different solubility of amorphous versus crystalline domains of the same polymer in marginal solvents at elevated temperatures. When the solution is heated to such a temperature, the disordered domains of P3HT would dissolve in the solvent, but the highly ordered crystallites would remain intact because the ordered, tight packing makes solvent diffusion more difficult. Therefore, these undissolved crystallites serve as heterogeneous nucleation sites for crystal growth upon the cooling of the solution, leading to controlled growth of crystals. While mechanistically similar, our oligomer-assisted approach is conceptually and experimentally different. The addition of predetermined percentages of monodispersed oligomers allows our process to create such heterogeneous nucleation sites at room temperature using mostly water, an environmentally friendly solvent, and incorporate the dopant acid in the crystallization process. This is particularly useful for unmodified CPs like PANI or PEDOT because of their low solubility in common solvents.
To further understand the self-assembly mechanism, we systematically varied the ratio between TANI and PANI in the self-assembly process. We anticipated that a higher percentage of TANI would lead to better-defined structures due to TANI's high propensity to crystallize. To our surprise, the system with the highest TANI content (90% TANI and 10% PANI) only leads to irregularly shaped agglomerates (Fig. 2b). The morphology of the nanowires gradually becomes better-defined as the PANI content is increased, with the 4:6 and 2:8 TANI:PANI ratio leading to the most well-defined morphologies (Fig. 2f and g). Further increasing the PANI content to 90% generates agglomerates and poorly defined structures again (Fig. 2h). The same trend is observed in TAS-PANI systems using other organic/acidic aqueous solvent combinations (Fig. S5 and S6, ESI†).
Based on these observations, we conjecture that when there is a low concentration of PANI and a high concentration of TANI (e.g., 9:1 TANI:PANI ratio, Fig. 2b), PANI serves as “impurities” that drastically increase the polydispersity of TANI, thus disrupting the packing of TANI, leading to disordered structures with no defined supramolecular morphology (Fig. 2k). On the contrary, when the PANI content is beyond a certain concentration (i.e., beyond 50%), PANI becomes the major phase rather than impurities to TANI, which allows TANI to self-assemble into the “seed crystals” through the aforementioned mechanism, facilitating the ordered assembly of PANI (Fig. 2l). Furthermore, it is possible that in addition to TANI cores serving as heterogeneous nucleation sites for PANI assembly, TANI is playing a more direct role in “straightening out” the PANI chains. Unlike TANI, which can easily rotate and adopt lattice sites, the doped PANI chains have to overcome a cascade of free energy barriers such as disentanglements and intramolecular interactions in order to transition to an extended chain conformation to form crystals.4,5 With the addition of a small amount of TANI, some of the TANI molecules that are still in solution can interact with PANI through π–π and other noncovalent interactions. This interaction essentially “dilutes” the PANI chains, shielding them from intramolecular interactions, leading to a more expanded chain conformation through disentanglement. The TANI-decorated segments of PANI would also lead the resulting complexes to become more rigid than PANI due to the increased segmental rigidity and diameter of the complexes. In fact, in examining the TEM images of TAS-PANI nanowires made using a 2:8 TANI:PANI ratio that have been washed with acetone post-assembly (Fig. 1g), the diameter ratio between the cavity left by the removal of TANI and the remaining PANI shell is about 1:8, supporting our proposed mechanism that some of the TANI forms the nucleation centre, while some other TANI molecules “solubilize” the PANI chains and may have been co-crystallized with PANI in the nanowire shells, facilitating their ordered packing. This mechanism is analogous to work in parallel fields such as using graphene oxide to disperse carbon nanotubes (CNTs) or surfactants to induce the assembly of molecular semiconductors.25,26 However, unlike these studies, no impurities are introduced in our system due to the identical chemical repeating units of TANI and PANI.
The most well-defined nanowires with the highest aspect ratios are obtained with TANI:PANI ratios of 4:6 and 2:8 (Fig. 2f and g). Since the goal of this project is to use TANI to assist the crystallization of PANI, we use the 2:8 TANI:PANI ratio for the subsequent studies to maximize the PANI content in the system.
2.3. TANI-assisted crystallization of PANI with defined molecular weights
Compared to other methods, such as vapor phase polymerization, that can generate crystalline structures for unmodified CPs,15–17 the most significant advantage of using self-assembly to achieve crystalline structures is that we can separately control the chemical properties and the degree of crystallinity of the polymers. The former can be controlled through synthesis, whereas the latter can be tuned by the assembly or crystallization process. As a proof-of-concept, we choose molecular weight (MW) as an example of chemical property.PANIs with relatively well-defined weight average molecular weights (MW) of 5000, 10000, 20000, 50000, 65000, and 100000 Da are commercially available in the EB form. Subjecting them to the self-assembly process with the same solvent environment in the absence of TANI only yields large, irregularly-shaped agglomerates (Fig. S7, ESI,† insets to Fig. 3). However, when 20% TANI is incorporated into the system, PANI of all six MW values self-assembles into well-defined 1D nanostructures (Fig. 3). These TANI-assisted self-assembled nanostructures are abbreviated as TAS-PANI-5k, TAS-PANI-10k, TAS-PANI-20k, TAS-PANI-50k, TAS-PANI-65k, and TAS-PANI-100k, with the last component in the acronym indicating the average MW of PANI.
 | ||
| Fig. 3 SEM images of 20% TANI-assisted self-assembled nanostructures of PANI with defined molecular weight: (a) 5000 Da, (b) 10000 Da, (c) 20000 Da, (d) 50000 Da, (e) 65000 Da, (f) 100000 Da. The inset to each SEM shows PANI of the same molecular weight subjected to an identical self-assembly environment in the absence of TANI. The scale bars in the insets are 1 μm. | ||
To better understand the correlation between the MW of PANI and crystallinity of the nanostructures, SAED was performed on each sample (Fig. 4). CPs are known to be electron-beam sensitive and typically suffer from heating damage during SAED collection.27,28 Doped CPs such as PANI in this study are positively charged, making them additionally prone to ionization damage from the negatively charged electron beam. Such radiation damage would cause bond breakage, which in turn results in radical formation and loss of packing periodicity.27,28 Unsurprisingly, our initial attempts at collecting SAED on the TAS-PANI samples under ambient conditions did not generate any diffraction patterns. To overcome electron beam damage, cryogenic conditions were employed in combination with low dose exposure in the imaging and diffraction of all TAS-PANI nanostructures. The samples remain beam sensitive under such conditions, but the diffraction pattern would last for a few seconds before gradually fading away, which is sufficient for data collection.
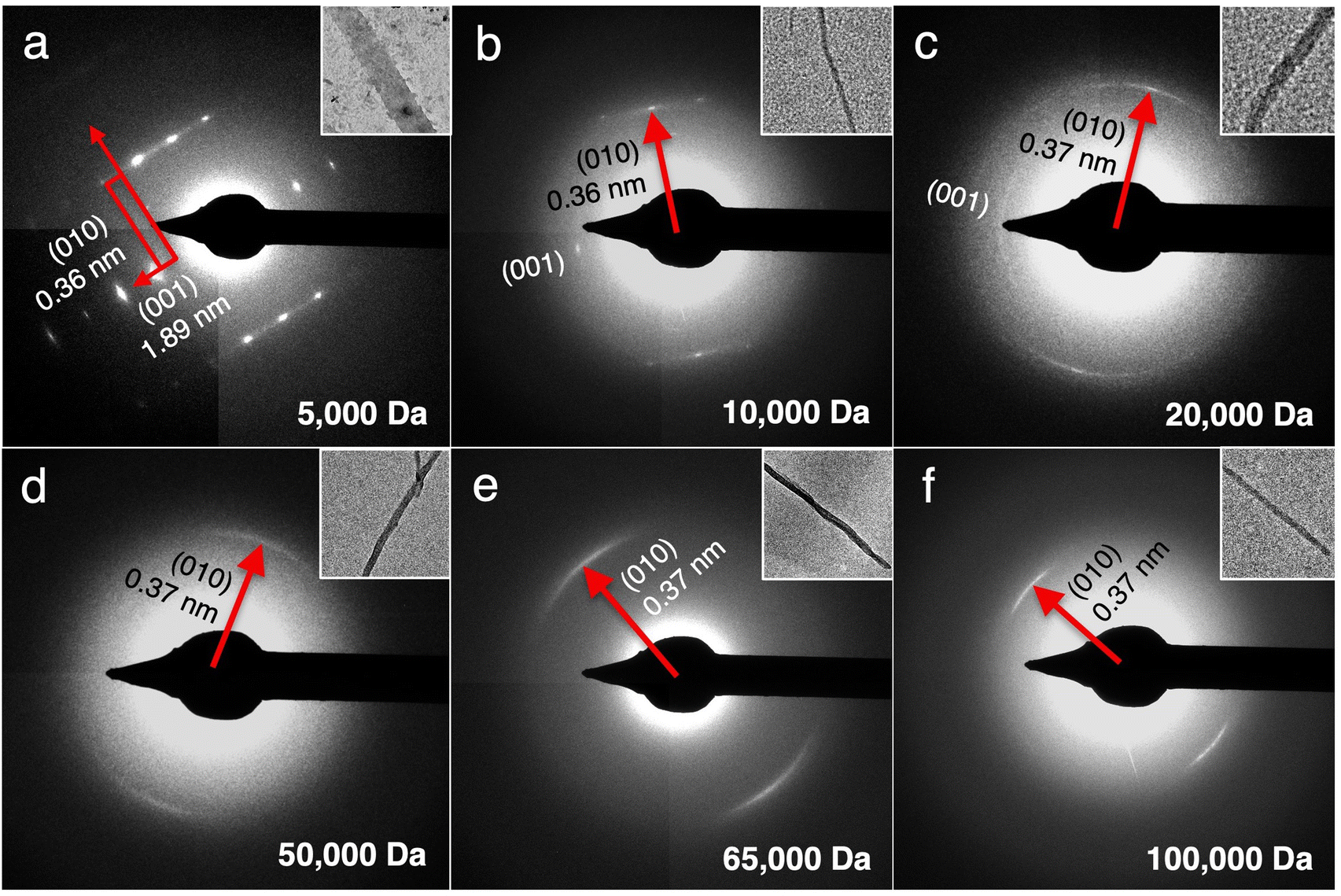 | ||
| Fig. 4 Select area electron diffraction (SAED) patterns of 20% TANI-assisted self-assembled nanostructures of PANI with defined molecular weight: (a) 5000 Da, (b) 10000 Da, (c) 20000 Da, (d) 50000 Da, (e) 65000 Da, (f) 100000 Da. These patterns are collected under cryogenic conditions. The inset to each SAED shows the TEM image of the nanoribbon/nanowire that corresponds to the SAED. | ||
Comparison of the SAED patterns to their corresponding bright-field TEM images in the insets reveals that the preferential crystal growth direction, regardless of PANI MW, is always along the [010] direction, indicating that the growth rate is the fastest along this facet. The spacing along [010] corresponds to π–π stacking,20 suggesting that the π–π interaction between the repeating aniline units is likely a main mechanism in driving the self-assembly process. The π–π stacking distance increases slightly from 0.36 to 0.37 nm with increasing PANI MW, likely due to the increasing entropic barriers to rearrange longer polymer chains, thus making the intermolecular packing less efficient. Most notably, clearly-defined Bragg diffraction spots along both the (010) and (001) planes are observed in the SAED pattern for the TAS-PANI-5k nanoribbons, indicating that they are single crystalline with consistent and regular packing conformation (Fig. 4a). SAED patterns collected over larger areas containing multiple TAS-PANI-5k nanoribbons show spotty ring patterns (Fig. S8, ESI†), which is a result of overlapping Bragg spot patterns in various orientations. This further confirms the single crystalline nature of the TAS-PANI-5k nanoribbons. The d-spacing between the (00l) planes is 1.89 nm, corresponding to the approximate length of 4 repetitions of aniline units along the PANI backbone.20 The systematic odd absences along the (00l) planes are consistent with glide symmetry, which indicates a possible alternating packing arrangement of the repeating units.
With increasing PANI MW, the SAED patterns gradually transition from Bragg diffraction spots to diffraction arcs, indicative of the preferential orientation of polymer chains.4,5 For TAS-PANI-10k, Bragg diffraction spots for (010) are still present, but the intensity is streaked between the adjacent spots, indicating some degree of disorder in an otherwise well-packed crystal. As the PANI MW is increased to 20000 Da, spotty arcs are observed, suggesting that packing within TAS-PANI-20k is less ordered than in the 10k counterpart. An increase in π–π stacking spacing from 0.36 to 0.37 nm along (010) is also observed between the MW transition here, further corroborating the decreasing degree of packing order. With an additional increase in PANI MW to 50k or beyond, only diffraction arcs are observed (Fig. 4d–f). These results indicate a decrease in the crystallinity of these nanostructures compared to the lower MW PANI counterparts. However, these sharp arcs reveal a high degree of long range, preferential orientation of the repeating aniline units along the π–π stacking direction that forms the crystal long axis. The decrease in crystallinity with increasing PANI MW is consistent with the general finding that the free energy barrier to reorganizing the conformations of the polymer chain into ordered states increases with increasing polymer MW.29
In addition to packing orientation and degree of crystallinity, previous studies have suggested that the diameters of well-ordered polymer crystals can be additional indications of chain conformation such as fully extended or folded. TAS-PANI-5k forms well-defined nanoribbons with an average diameter around 200 nm, whereas the other samples with a PANI MW of 10k, 20k, 50k, 65k, and 100k Da form nanowires with average diameters of around 25, 33, 90, 50, and 50 nm, respectively (Fig. S4, ESI†). Previous studies on the CP crystallization have noted a relationship between CP MW and crystal diameter.29 A fully extended PANI chain of 5k Da would be approximately 20–25 nm in length,30,31 which is significantly smaller than 200 nm, meaning the nanoribbons are composed of many PANI chains along the width direction. Interestingly, earlier work has shown that lower MW P3HT (i.e., 6k Da) has fully extended chains in its crystals, and nanofibers with diameter consistent with the length of the P3HT chains would transform into nanoribbons, likely to minimize surface energy.29 Previous studies on the crystal packing of oligoanilines have shown similar results possibly due to Ostwald ripening.20 It was found that the higher order structures are formed from the merging of smaller structures, i.e., nanoplates are the result of merged stacks of nanoribbons, and nanoribbons are the result of merged parallel arrays of nanowires. The nanowire diameters for TAS-PANI with PANI MW between 10k and 100k Da are significantly smaller than the length of fully extended polymer chains, indicating the presence of chain folding in the nanowires. It is interesting that TAS-PANI-65k and TAS-PANI-100k exhibit smaller diameter than the TAS-PANI-50k counterpart. This could suggest that PANI chains of higher MW fold more,29 likely due to the higher entropic barriers associated with “straightening out” longer chains, which typically have more entanglements and intramolecular interactions over extended distances.32
2.4. Spectroscopic and electrical characterization
To gain further insights into the effect of crystallographic ordering on the relationship between chain conformation and conductivity of the TAS-PANI nanowires, UV-vis-near infrared (NIR) spectroscopy was employed. Fig. 5a shows the solution UV-vis-NIR spectra of TANI and PANI of various MW subjected to the same self-assembly environment as TAS-PANI, whereas Fig. 5b shows those for the TAS-PANI nanowire dispersions. Three peaks centred at around 280 nm, 470 nm, and 780 nm can be observed in both collections of spectra, characteristic of doped polyaniline or oligoaniline.19,33 The 280 nm peak can be assigned to the π → π* transition, the 470 nm peak to the polaron → π* transition, and the 780 nm peak to the π → polaron transition.19,33 The 780 nm peaks for all the PANI-containing samples in both collections of spectra are highly asymmetrical and steadily increase into the NIR region, indicative of expanded PANI chain conformation and more ordered interchain packing.33,34 The intensity of the free carrier tail is associated with the delocalization of the polaron band, making it spread across the gap between the π and polaron bands.33,34 As a result, a decrease in intensity of the 280 nm peak typically accompanies the formation of the free carrier tail. Note that oligoanilines such as TANI do not exhibit free carrier tails despite the relative chain linearity and high crystallographic order because the chain length is not sufficiently long for extensive charge delocalization along the backbone to occur.19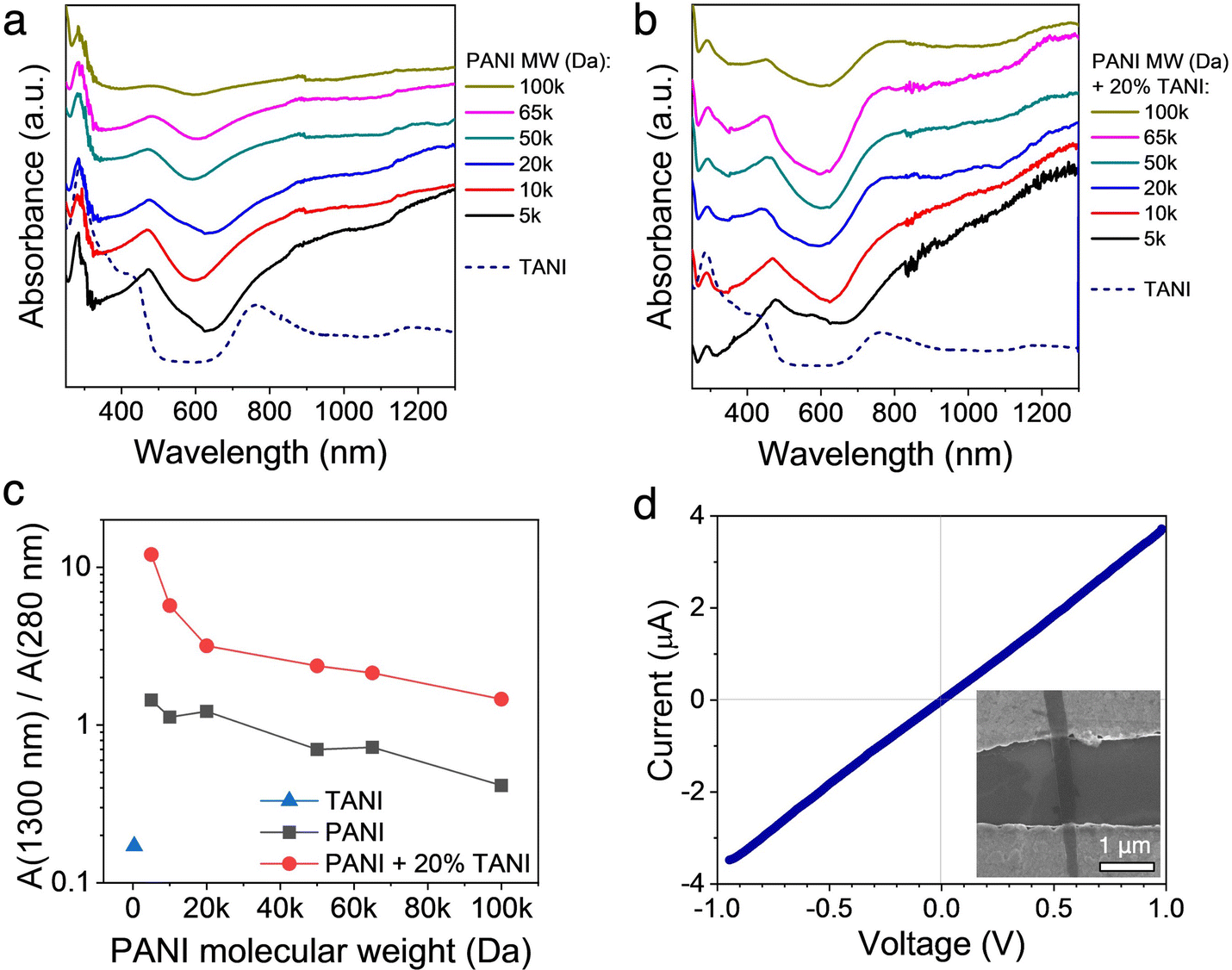 | ||
| Fig. 5 UV-vis-NIR spectra of (a) self-assembled TANI and PANI of different molecular weights, and (b) TAS-PANI of different PANI molecular weights. (c) The absorbance peak ratio between the ca. 1300 nm free-carrier tail and the ca. 280 nm π → π* transition for TAS-PANI with different PANI molecular weights, extracted from the spectra in (a and b). (d) Current–voltage curve of a two-probe, single nanowire device of TAS-PANI-5k. The inset shows the SEM image of a representative device. | ||
For the control experiments of PANI with various MW subjected to the same self-assembly environment as TAS-PANI (Fig. 5a), despite the presence of the free-carrier tail, the 280 nm peak that corresponds to the π → π* transition exhibits high intensity for all PANI MW. As a result, the absorbance ratio between the 1300 nm free-carrier tail and the 280 nm peaks is around or below 1 for all these samples (Fig. 5c), indicating a high degree of compact coil chain conformation and strong aggregation.19,34 This result is consistent with the SEM observations for these samples where only agglomerates are observed (Fig. S7, ESI†). The results here suggest that the self-assembly solvent environment alone is effective in doping PANI and partially expanding some coil conformation; however, the extent of such functions is limited and most of the chains remain in the highly coiled and aggregated state.
In contrast, the 280 nm peak associated with π → π* transition in the TAS-PANI samples shows significantly decreased intensity compared to the other peaks. This decrease is accompanied by an increase in the intensity of the 1300 nm free-carrier tail peaks (Fig. 5b). The collective effect of these changes in intensities is the significantly higher absorbance ratios between the 1300 nm and the 280 nm peaks (Fig. 5c), confirming the nature of expanded chain conformation in TAS-PANI, which leads to the delocalization of the polaron band and thus charges.34,35 This “straightening out” of the PANI chain conformation facilitates ordered interchain packing, and thus leads to crystallization of the PANI chains in the TAS-PANI nanowires (Fig. 3 and 4). In particular, TAS-PANI with the lowest MW, 5000 Da, exhibits a very intense free carrier tail, leading to a 1300-to-280 nm peak ratio of over 12, suggesting a high degree of chain linearity and hence interchain ordering. This is consistent with the SAED data of TAS-PANI-5k, which shows discrete diffraction spots, indicative of the single crystalline nature of these nanowires (Fig. 4a). As the PANI MW increases, the free-carrier tail weakens consistently. This is not only reflected by the gradually decreasing ratios between the 1300-to-280 nm peaks (Fig. 5c), but also the more obvious appearance of the localized 780 nm peak from TANI in the UV-vis-NIR spectra of TAS-PANI with PANI MW above 20000 Da (Fig. 5b). However, even with the highest MW PANI of 100000 Da, a 1300-to-280 nm peak ratio of 1.5 is obtained, significantly higher than the counterparts without TANI (Fig. 5c), illustrating the effectiveness of TANI in shaping PANI into a more extended chain conformation, promoting their preferential ordering (Fig. 4f).
The presence of a steadily increasing free-carrier tail in the NIR region is characteristic of highly conductive polymers with expanded chain conformation. It has been shown that a strong free-carrier tail is typically associated with metallic materials36 and correlates directly with high conductivity in conducting polymers. Hence, the UV-vis-NIR results (Fig. 5b) qualitatively suggest that the TAS-PANI nanowires have high conductivity.
Bulk conductivity measurements such as those based on casted films or pressed pellets do not offer meaningful assessment of the conductivity of crystalline nanostructures because the values are more reflective of the high contact resistance between the numerous nanostructure–nanostructure junctions rather than the intrinsic conductivity of the nanostructures.19,37 Therefore, we attempted single wire measurements for the TAS-PANI nanostructures by depositing them across two-contact microelectrodes with a gap size of 2–5 μm. Due to the tangled, web-like structure of the TAS-PANI nanowires with PANI MW between 10000 and 100000 Da (Fig. 3b–f), it was challenging to isolate enough of these MWs for reproducible measurements. However, the straight nanoribbon morphology of TAS-PANI-5k allowed us to isolate over 10 nanoribbons and bridge them across bottom-contact, Cr/Au microelectrodes for current–voltage (I–V) measurements.
Fig. 5d shows a representative I–V curve, with the SEM image of a representative device shown in the inset. The linearity of the I–V curve indicates ohmic contact between the nanoribbons and the microelectrodes. Conductivity values ranging from 4.3 to 19.5 S cm−1 are obtained from these devices. Given the very intense free carrier tail observed in the UV-vis-NIR spectra, the conductivity values of the TAS-PANI-5k nanoribbons are likely much higher than our results indicate here. The apparent conductivity from our measurements is limited by the following factors: (1) The short length of the nanoribbons only allowed us to carry out 2-point, rather than 4-point probe measurements, meaning the measured resistance values include contributions from contact resistance. (2) These are bottom-contact devices, meaning the contact quality between the nanostructures and the electrodes is not optimal. Solvents or other impurities are often trapped between the sample and electrodes, impeding charge transport. Potentially fabricating top-contact devices may solve this issue, but such feats are challenging given the small nanoribbon size and conducting polymers’ high sensitivity to the processing conditions required for lithography. These factors revolving around contact quality have likely also resulted in the spread of the measured conductivity values across the 4.3 to 19.5 S cm−1 range. Since the focus of this work is on demonstrating a new concept in crystallizing conjugated polymers, and not on device optimization, we reserve more elaborate measurements for future studies.
Despite the likely underestimation of the conductivity of TAS-PANI, it is worth emphasizing that these values are much higher than typical PANI processed from water, which generally has a conductivity around 1 S cm−1.38 Similarly intense free carrier tails from UV-vis-NIR and conductivity on the order of 101 S cm−1 or above typically require large dopant acids such as camphorsulfonic acid in combination with harsh and toxic “secondary dopant” processing solvents like m-cresol,34,35 which are undesirable and not compatible with flexible or stretchable substrates required for next-generation organic electronics.
3. Conclusions
In summary, we have developed a simple, one-step self-assembly method towards highly crystalline PANI nanostructures with the assistance of a small amount of the corresponding, monodispersed oligomers such as tetraaniline. Since TANI and PANI share the same repeating units and are both electroactive, no impurities are introduced into the material through this process. The synthetic component that controls the chemical structures and the self-assembly process that dictates morphology and crystal packing are decoupled into separate steps in this approach. Therefore, it can be applied to pre-made PANI with well-defined properties such as molecular weights, leading to their single crystals or crystals with preferential chain orientation. The presence of very strong free carrier tails in their UV-vis-NIR spectra indicates that these materials are likely highly conductive.The broader impact of this work lies in its simplicity and potential generality. The new concept demonstrated here is likely generally applicable to many other CPs or other polymers because oligomers of well-defined numbers of repeating units can be synthesized for each parent polymer. For the self-assembly of PANI, our preliminary results show that aniline dimer, which can also readily self-assemble into crystalline nanostructures, can play a similar role as TANI in assisting the self-assembly of PANI into ordered structures (Fig. S9, ESI†). This insight reveals that a variety of oligomers with different numbers of repeating units can potentially be used to induce the crystallization of their parent polymers, which will be the topic of a future study. Furthermore, being able to grow highly crystalline structures from a mostly aqueous solvent is environmentally friendly. Finally, even though this work focuses on crystalline nanostructures, we conjecture that the concept of oligomer-induced crystallization of the parent polymer can likely be extended to increasing the crystallinity of polymer thin films as well, potentially leading to enhanced performance in thin film electronics.
4. Experimental
Materials
Phenyl/amine-capped tetraaniline was synthesized via a previously reported procedure,19 which is described in detail in the ESI.† Polyaniline without defined MW was synthesized using a rapid mixing method39 and purified by dialysis. Polyanilines of defined molecular weights (5000, 10000, 20000, 50000, 65000, and 100000 Da) in the emeraldine base form were purchased from Sigma-Aldrich. All organic solvents, acids, and dialysis tubing used in this study were obtained from Fisher Scientific.
Self-assembly
Tetraaniline and polyaniline were ground into a fine powder using an agate mortar and pestle. In a typical process, 2 mg of powders composed of TANI and PANI at the desired ratio were individually weighed out using a microbalance and subsequently combined. The powder mixture is then added to a solvent mixture composed of 1 mL of organic solvent such as THF and 4 mL of an aqueous acidic solution such as 0.1 M HClO4. The mixture is swirled rapidly for a few seconds and left undisturbed for 5 days for the self-assembly process to occur. At the end of the process, dialysis was performed against deionized water for approximately 1 day to purify the products.Electron microscopy
Scanning electron microscopy (SEM) analysis was performed using a JEOL JSM-6700 SEM or Zeiss Gemini 500 SEM on samples drop casted on silicon wafers. Transmission electron microscopy (TEM) samples were prepared by depositing a drop of the nanomaterial dispersion onto a TEM grid placed on top of filter paper. These samples were loosely covered and left to air dry overnight. Due to the high sensitivity of PANI to the electron beam due to ionization and heating damage, cryogenic TEM and SAED were performed. TEM samples were kept under dynamic vacuum overnight to completely remove moisture. They were rapidly frozen using liquid nitrogen and kept at liquid nitrogen temperature using a cryogenic sample holder during the duration of imaging. TEM images and SAED patterns were collected on either an FEI/PHILIPS CM 120 TEM or a Talos F200C G2 TEM, operated under cryogenic conditions with an accelerating voltage of 200 kV. Au standards were used for SAED calibration.Spectroscopic characterization
UV-vis-NIR of the dispersions were collected on a Shimadzu UV-3600 Plus. A quartz cuvette with a 1 mm path length was used to minimize water absorbance in the NIR region. ATR-IR spectra were obtained on a Bruker Vertex 70 spectrometer (ATR mode, diamond window).Single wire measurement
Microelectrodes were fabricated via photolithography on a Si/SiO2 wafer with 300 nm of SiO2 layer followed by thermal evaporation of 5 nm of Cr and 90 nm of Au. A few drops of the dispersion were deposited on the microelectrode array. After a few seconds for the nanowires to settle, the wafer was tilted and the excess droplet was removed and blown dry by gently flowing nitrogen over the surface with a pressurized nitrogen gun. Measurements were carried out on freshly prepared samples using a Lakeshore semiconductor probe station under ambient conditions.Author contributions
Y. W. conceived and supervised the project. I. M. H. synthesized materials. D. W. led the mechanistic understanding. B. X. carried out characterization. D. W. and Y. W. wrote the manuscript draft. All authors contributed to data analysis and manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Adekunle Adewole for assistance with certain steps of the tetraaniline synthesis. Cleanroom access was provided by the UCM Stem Cell Instrumentation Foundry and the UCLA Nanofabrication Laboratory. Access to electron microscopes was provided by the UCM Imaging and Microscopy Facility and the UCLA Electron Imaging Center for Nanomachines. Financial support for this project was provided by the National Science Foundation CAREER Award DMR-1945664, the Arnold and Mabel Beckman Foundation Beckman Young Investigator Award, the University of California, Merced, Start-up Fund.References
- Z. F. Yao, J. Y. Wang and J. Pei, Chem. Sci., 2021, 12, 1193–1205 RSC.
- X. T. Zhang, H. L. Dong and W. P. Hu, Adv. Mater., 2018, 30, 1801048 CrossRef PubMed.
- J. Y. Kim, M. H. Kwon, J. T. Kim, S. Kwon, D. W. Ihm and Y. K. Min, J. Phys. Chem. C, 2007, 111, 11252–11258 CrossRef CAS.
- J. A. Lim, F. Liu, S. Ferdous, M. Muthukumar and A. L. Briseno, Mater. Today, 2010, 13, 14–24 CrossRef CAS.
- X. X. Cao, K. F. Zhao, L. Chen, J. G. Liu and Y. C. Han, Polym. Cryst., 2019, 2, e10064 Search PubMed.
- D. H. Kim, J. T. Han, Y. D. Park, Y. Jang, J. H. Cho, M. Hwang and K. Cho, Adv. Mater., 2016, 18, 719–723 CrossRef.
- X. L. Xiao, Z. J. Hu, Z. B. Wang and T. B. He, J. Phys. Chem. B, 2009, 113, 14604–14610 CrossRef CAS PubMed.
- A. L. Briseno, S. C. B. Mannsfeld, P. J. Shamberger, F. S. Ohuchi, Z. N. Bao, S. A. Jenekhe and Y. N. Xia, Chem. Mater., 2008, 20, 4712–4719 CrossRef CAS.
- H. L. Dong, S. D. Jiang, L. Jiang, Y. L. Liu, H. X. Li, W. P. Hu, E. J. Wang, S. K. Yan, Z. M. Wei, W. Xu and X. Gong, J. Am. Chem. Soc., 2009, 131, 17315–17320 CrossRef CAS PubMed.
- H. Hui, Y. Lei, F. Antonio and J. M. Tobin, Chem. Rev., 2017, 117, 10291–10318 CrossRef PubMed.
- Y. Q. Zheng, T. Lei, J. H. Dou, X. Xia, J. Y. Wang, C. J. Liu and J. Pei, Adv. Mater., 2016, 28, 7213–7219 CrossRef CAS PubMed.
- J. H. Liu, J. Moo-Young, M. McInnis, M. A. Pasquinelli and L. Zhai, Macromolecules, 2014, 47, 705–712 CrossRef CAS.
- Y. Q. Luo, F. A. Santos, T. W. Wagner, E. Tsoi and S. J. Zhang, J. Phys. Chem. B, 2014, 118, 6038–6046 CrossRef CAS PubMed.
- S. Agbolaghi, S. Abbaspoor and F. Abbasi, Prog. Polym. Sci., 2018, 81, 22–79 CrossRef CAS.
- J. M. D’Arcy, M. F. El-Kady, P. P. Khine, L. Zhang, S. H. Lee, N. R. Davis, D. S. Liu, M. T. Yeung, S. Y. Kim, C. L. Turner, A. T. Lech, P. T. Hammond and R. B. Kaner, ACS Nano, 2014, 8, 1500–1510 CrossRef PubMed.
- S. Majumdar, K. Sarmah and D. Mahanta, ACS Appl. Polym. Mater., 2020, 2, 1933–1942 CrossRef CAS.
- B. Cho, K. S. Park, J. M. Baek, H. S. Oh, Y. E. Koo Lee and M. M. Sung, Nano Lett., 2014, 14, 3321–3327 CrossRef CAS PubMed.
- T. Zhang, H. Qi, Z. Liao, Y. D. Horev, L. A. Panes-Ruiz, P. S. Petkov, Z. Zhang, R. Shivhare, P. Zhang, K. Liu, V. Bezugly, S. Liu, Z. Zheng, S. Mannsfeld, T. Heine, G. Cuniberti, H. Haick, E. Zschech, U. Kaiser, R. Dong and X. Feng, Nat. Commun., 2019, 10, 4225 CrossRef PubMed.
- Y. Wang, H. D. Tran, L. Liao, X. F. Duan and R. B. Kaner, J. Am. Chem. Soc., 2010, 132, 10365–10373 CrossRef CAS PubMed.
- Y. Wang, J. Liu, H. D. Tran, M. Mecklenburg, X. N. Guan, A. Z. Stieg, B. C. Regan, D. C. Martin and R. B. Kaner, J. Am. Chem. Soc., 2012, 134, 9251–9262 CrossRef CAS PubMed.
- Z. Y. Ma, Y. H. Geng and D. H. Yan, Polymer, 2007, 48, 31–34 CrossRef CAS.
- L. Zang, Y. K. Che and S. M. Jeffrey, Acc. Chem. Res., 2008, 41, 1596–1608 CrossRef CAS PubMed.
- J. J. Xu, Y. Ma, W. B. Hu, M. Rehahn and G. Reiter, Nat. Mater., 2009, 8, 348–353 CrossRef CAS PubMed.
- J. Qian, G. Guerin, Y. Lu, G. Cambridge, I. Manners and M. A. Winnik, Angew. Chem., Int. Ed., 2011, 50, 1622–1625 CrossRef CAS PubMed.
- Y. Yan, R. Wang, X. Qiu and Z. Wei, J. Am. Chem. Soc., 2010, 132, 12006–12012 CrossRef CAS PubMed.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- H. F. Greer and W. Zhou, Crystallogr. Rev., 2011, 17, 163–185 CrossRef CAS.
- D. C. Martin and E. L. Thomas, Polymer, 1995, 36, 1743–1759 CrossRef CAS.
- J. H. Liu, M. Arif, J. H. Zou, S. I. Khondaker and L. Zhai, Macromolecules, 2009, 42, 9390–9393 CrossRef CAS.
- J. P. Pouget, M. E. Jozefowicz, A. J. Epstein, X. Tang and A. G. MacDiarmid, Macromolecules, 1991, 24, 779–789 CrossRef CAS.
- R. H. Baughman, J. F. Wolf, H. Eckhardt and L. W. Shacklette, Synth. Met., 1988, 25, 121–137 CrossRef CAS.
- L. H. Sperling, Introduction to Physical Polymer Science, Wiley, Hobokon, New Jersey, 4th edn, 2006, pp. 29–70 Search PubMed.
- Y. N. Xia, J. M. Wiesinger, A. G. MacDiarmid and A. J. Epstein, Chem. Mater., 1995, 7, 443–445 CrossRef CAS.
- A. G. MacDiarmid and A. J. Epstein, Synth. Met., 1995, 69, 85–92 CrossRef CAS.
- A. G. MacDiarmid and A. J. Epstein, Synth. Met., 1994, 65, 103–116 CrossRef CAS.
- A. P. Monkman and P. Adams, Synth. Met., 1991, 41, 627–633 CrossRef CAS.
- Y. Wang, J. A. Torres, A. Z. Stieg, S. Jiang, M. T. Yeung, Y. Rubin, S. Chaudhuri, X. Duan and R. B. Kaner, ACS Nano, 2015, 9, 9486–9496 CrossRef CAS PubMed.
- D. Li, J. Huang and R. B. Kaner, Acc. Chem. Res., 2009, 42, 135–145 CrossRef CAS PubMed.
- J. Huang and R. B. Kaner, J. Am. Chem. Soc., 2004, 126, 851–855 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2mh01344d |
| This journal is © The Royal Society of Chemistry 2023 |