
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOriented growth of stacking α-cobalt hydroxide salt continuous films and their topotactic-like transformation to oriented mesoporous films of Co3O4 and CoO†
Tsutomu
Shinagawa
 *a,
Natsuko
Kotobuki
b and
Atsushi
Ohtaka
b
*a,
Natsuko
Kotobuki
b and
Atsushi
Ohtaka
b
aElectronic Materials Research Division, Morinomiya Center, Osaka Research Institute of Industrial Science and Technology (ORIST), 1-6-50 Morinomiya, Joto, Osaka, 536-8553, Japan. E-mail: tshina@orist.jp
bDepartment of Applied Chemistry, Faculty of Engineering, Osaka Institute of Technology, 5-16-1 Ohmiya, Asahi, Osaka, 535-8585, Japan
First published on 18th October 2022
Abstract
Mesoporous metal oxide films composed of nanocrystal assemblies with an aligned crystallographic orientation are key nanostructures for efficient interfacial reactions; however, the development of a simple and versatile method for their formation on substrates still constitutes a challenge. Here we report the template-free centimetre-scale formation of novel cobalt oxide films of Co3O4 and CoO with a [111]-oriented mesoporous structure starting from stacking cobalt hydroxide continuous films. The cobalt hydroxide precursor is formed electrochemically on conductive substrates from a Co(NO3)2 aqueous solution at room temperature. A thorough characterization by means of scanning electron microscopy, X-ray diffraction, X-ray photoelectron spectroscopy, UV-vis-NIR spectroscopy, IR spectroscopy and Raman spectroscopy analyses reveals that the precursor film is an α-type layered cobalt hydroxide salt (α-Co-LHS) containing interlayer nitrate and hydrated water, i.e., α-Co(OH)x(NO3)y·nH2O, with a [001]-oriented stacking film structure. Heat treatment of the [001]-α-Co-LHS films using different conditions, i.e., under air at 550 °C or under vacuum at 500 °C, results in the selective formation of Co3O4 or CoO mesoporous films, respectively. A plausible explanation for the observed centimetre-scale topotactic-like transformation from α-Co-LHS[001] to Co3O4[111] or CoO[111] is given according to the atomic framework similarity between the hydroxide precursor and the final oxides.
Introduction
Metal oxide-based mesoporous structures consisting of numerous nanopores have attracted increasing attention for application as (photo)electrocatalysts, chemical sensors and adsorbents because of the synergistic combination of their diverse physical and chemical properties and large surface area easily accessible from the outside.1–5 Mesoporous oxides are generally synthesized using templates such as surfactants and polymers.1,6 However, the formation and removal of templates complicates the synthesis process and generates residues. Consequently, the use of layered metal hydroxides (LMHs) as precursors has emerged as an alternative simple and template-free method for synthesizing mesoporous oxides.7–9LMHs have a layered structure comprising two-dimensional (2D) edge-shared octahedral hydroxide sheets and can be divided into layered double hydroxides with a composition of MII1−xM′IIIx(OH)2(Am−)x/m·nH2O containing divalent (Mg, Co, Ni, Cu, Zn, etc.) and trivalent (Al, Cr, Co, Fe, In, etc.) cations and layered hydroxide salts (LHSs) MIIx(OH)2x−myAm−y·nH2O consisting of only divalent cations.10,11 Anions (Am−) such as Cl−, NO3−, SO42− and CO32− are often incorporated between the hydroxide layers and participate in anion exchange reactions. Moreover, thermal decomposition of LMHs proceeds via anion elimination and dehydration reactions, usually without an external oxygen source, to form the corresponding metal oxides consisting of numerous oxide nanocrystals. Interestingly, some LMHs yield nanoporous oxides during the decomposition process while maintaining their apparent atomic arrangement and original crystal shape, that is, they exhibit topotactic-like pseudomorphic transformation (TPT) property.8,12–14 This TPT feature allows the template-free facile formation of crystallographically oriented nanoporous oxides.
Cobalt oxides (CoxOy), including cobalt(II, III) oxide (Co3O4) and cobalt(II) oxide (CoO), are among the most studied oxides prepared from LHS precursors because of their application in anode materials for lithium ion batteries,15,16 electrochemical capacitors17,18 and (photo)electrocatalysts for water splitting reactions (both hydrogen and oxygen evolution reactions)19–22 and for CO2 reduction.23,24 Several types of cobalt-based LHSs (Co-LHSs), e.g., Co(OH)2, Co2(NO3)(OH)3 and Co7(NO3)2(OH)12, have been synthesized by means of alkaline precipitation and hydrothermal methods.25–30 Furthermore, thermal decomposition of Co-LHSs to CoxOy has been reported to proceed via TPT with a crystallographic orientation relationship of Co-LHS[001]//Co3O4[111] and Co-LHS[001]//CoO[111].14,29,31,32
By taking advantage of this TPT property, mesoporous CoxOy films with a preferred growth orientation can be formed on substrates without using templates. In fact, Ma et al. reported the formation of [111]-oriented Co3O4 and CoO thin films on substrates via Langmuir–Blodgett coating of separately synthesized Co-LHS platelets to ensure a [001] orientation, followed by heat treatment.32 As an alternative to this stepwise procedure, the direct formation of oriented Co-LHS films on a substrate could dramatically simplify the formation process of oriented mesoporous CoxOy films.
In addition to the large surface area, oriented mesoporous CoxOy films are expected to (i) enhance catalytic activity due to the exposure of specific crystalline planes and (ii) improve charge transport efficiency due to lattice matching between CoxOy nanocrystals.33–36 In particular, the (111) plane has been reported to be more catalytically active for water splitting and CO oxidation than the (110) and (100) planes.37–39 Therefore, due to the topotactic relationship of Co-LHS[001]//CoxOy[111], [001]-oriented Co-LHS films are an attractive candidate to spontaneously provide a myriad of oriented CoxOy nanocrystals with the (111) exposed plane. Co-LHSs can be grown directly on substrates via electrochemical deposition in aqueous solution.40–44 However, unoriented Co-LHSs with a flake-like morphology are usually obtained, and epitaxial techniques using single-crystal substrates have to be adopted for their orientation.45 Thus, growing oriented films of Co-LHSs readily without using templates or epitaxial techniques is still a challenge.
Recently, we demonstrated the potential of electrochemical deposition for the growth of oriented LHS films. Specifically, under the appropriate conditions, even simple electrodeposition in aqueous solution was shown to produce directly [001]-oriented continuous Mg- and Ni-LHS films, which then undergo a TPT process to yield [111]-oriented nanoporous MgO and NiO films.46,47 Herein, we report the first successful formation of [001]-oriented Co-LHS continuous films via direct electrodeposition on glass substrates coated with F-doped SnO2 (FTO) and their heat treatment under different conditions to selectively yield mesoporous Co3O4 or CoO films with a [111] crystal orientation (Scheme 1). In addition, we discuss the observed centimetre-scale topotactic-like transformation on the basis of the similarity of the atomic frameworks between the hydroxide precursor and the final oxides.
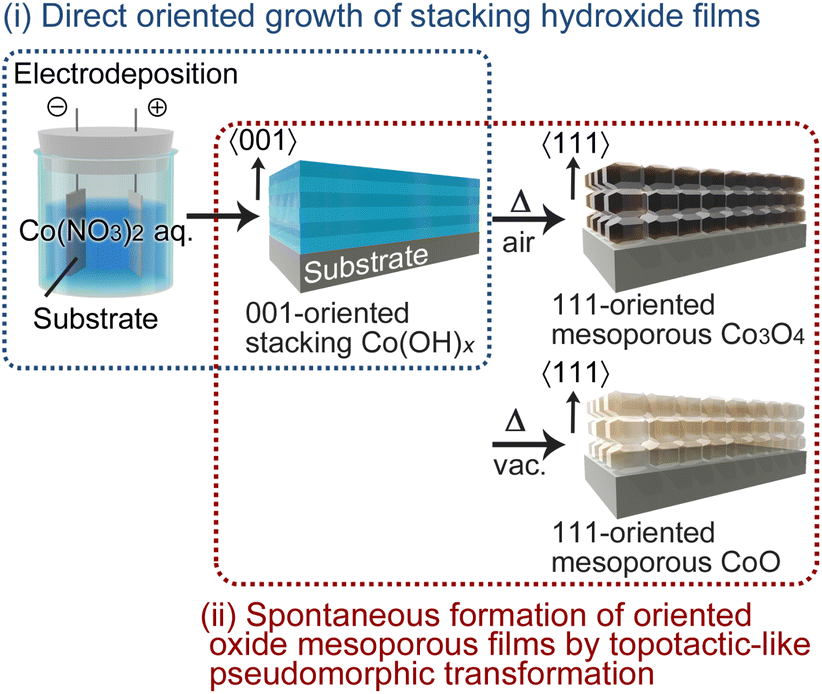 | ||
| Scheme 1 Schematic illustration of the template-free synthesis of oriented cobalt oxide mesoporous films. | ||
Results and discussion
Electrodeposition and characterization of Co-LHS films
Co-LHS was electrochemically deposited on an FTO substrate from a simple aqueous solution containing 10 mM Co(NO3)2 without pH adjustment (pH ∼ 5.9). A constant cathodic current of −1.0 mA cm−2 was applied on the FTO substrate using a platinum plate counter electrode at room temperature. The deposition process can be described by the following reactions:48| NO3− + H2O + 2e− → NO2− + 2OH− | (1a) |
| 2H2O + 2e− → H2 + 2OH− | (1b) |
| Co2+ + 2OH− → Co(OH)2 | (2a) |
| Co2+ + xOH− + yNO3− + nH2O → Co(OH)x(NO3)y·nH2O | (2b) |
The cathodic reactions of NO3− and H2O occurs on the electrolyte/FTO interface to give OH− ions (eqn 1a and 1b), causing an increase in local pH near the FTO surface that induces the precipitation of cobalt hydroxides (eqn 2a and 2b). The pH at which precipitation occurs can be estimated to be above 7.3 using a thermodynamic Pourbaix diagram, according to which further dehydration of hydroxides to form CoO (Co(OH)2 → CoO + H2O) is thermodynamically unfavorable at room temperature.49
Electrodeposition by passing a total electricity of −0.2 C cm−2 (deposition time = 3.3 min) resulted in pale light blue deposits on the substrate, which was rinsed with deionized water and then vacuum dried. Fig. 1a shows field-emission scanning electron microscopy (FESEM) images of the deposits, in which a lamellar continuous film structure with a thickness of ∼400 nm can be observed. In the 1.5 cm × 2.5 cm deposition area, the film thickness distribution within the central 1.0 cm × 1.5 cm area was evaluated to be approximately ±6.5% based on FESEM observation. The cross-sectional image shows approximately 10 layers with a thickness of 30–40 nm stacked parallel to the substrate, and the plan-view image displays continuous film with no distinct grain boundaries and a slightly wavy surface (FESEM images taken at low magnification are shown in Fig. S1 in the ESI†). This film structure is similar to that of the [001]-oriented Mg-LHS films reported in our previous work.46
 | ||
| Fig. 1 Characterization of [001]-oriented cobalt hydroxide films electrodeposited on F-doped SnO2 (FTO) substrates in Co(NO3)2 aqueous solution: (a) cross-sectional (top) and plan-view (bottom) FESEM images, (b) out-of-plane XRD patterns, (c) XPS wide spectra, (d) UV-vis-NIR transmittance spectrum and (e) ATR-FTIR spectrum. ICDD data for β-Co(OH)2 (no. 30-0443) and simonkolleite (no. 7-0155) and a photograph of the sample are presented in (b) and (d), respectively. | ||
Fig. 1b shows the θ–2θ X-ray diffraction (XRD) pattern of the as-deposited film. A distinct broad peak with a full width at half maximum (FWHM) of 1.28° was observed at 2θ = 10.71° with a lattice plane distance of 8.3 Å, and no other peaks were detected except for a secondary diffraction at 2θ = 21.9° and diffractions from the FTO substrate. This suggests that the obtained films consist of [001]-oriented Co-LHS with an interlayer distance (di) of 8.3 Å stacked parallel to the substrate surface. In addition, a similar distinct broad peak at 2θ = 9.9° (di = 8.9 Å) appeared in a film that was dried naturally instead of vacuum dried after electrodeposition, confirming that the interlayer distance became slightly larger (the XRD pattern of the naturally dried film is shown in Fig. S2†). These interlayer distances are nearly twice than that of β-Co(OH)2 (di = 4.6 Å, ICDD no. 30-0443) and close to that of Zn5(OH)8Cl2·H2O (di = 7.9 Å, simonkolleite, ICDD no. 7-0155) and Zn5(OH)8(NO3)2·2H2O (di = 9.7 Å, ICDD no. 72-0627). The large FWHM (1.28°) suggests that a crystallite smaller than 10 nm with low crystallinity was obtained.
Wide-scan X-ray photoelectron spectroscopy (XPS) confirmed that the resulting Co-LHS films contained nitrogen as well as cobalt and oxygen atoms, and no impurities other than adventitious carbon were detected (Fig. 1c). Fine XPS measurements in the N 1s region showed a peak at 406.5 eV that can be attributed to NO3− ions,50 suggesting the presence of NO3− ions in the interlayer of the obtained Co-LHS (Fig. S3a†). In the O 1s and Co 2p regions, typical peaks for metal hydroxides and Co(II) were observed at 531.2 and 780.5 eV, respectively,51 and no peaks originating from oxides, Co(0), or Co(III) species were detected (Fig. S3a†). On the basis of the XPS peak areas, the atomic ratio was estimated to be Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O:N = 32.8:65.1:2.1.
:O:N = 32.8:65.1:2.1.
The interlayer distance of LHSs generally depends on interlayer substances as well as the structure of the 2D M(OH)x sheets. Fig. 2 shows the structure model of five representative types of LHSs, namely, a β-type (Fig. 2a), two hydroxy double salts (Fig. 2b and c) and two α-type LHSs (Fig. 2d and e); these models were drawn using the VESTA software.52 Furthermore, the structure and composition of typical LHSs of each type, their interlayer distance, and characteristic IR bands (lattice OH− and interlayer NO3−) are summarized in Table 1. β-Type and hydroxy double salt LHSs can be categorized as ordered layered hydroxides because they often show well-faceted single-crystal grains, and α-type LHSs can be assigned as disordered layered hydroxides due to their low crystallinity and turbostratic stacking. The β-type brucite structure shown in Fig. 2a has 2D M(OH)x sheets consisting of edge-sharing octahedrally coordinated (Oh) metal hydroxides and the smallest interlayer distance of ∼4.6 Å due to no interlayer substances. In the hydroxy double salt shown in Fig. 2b, anions are coordinated to the Oh hydroxide sheets and the interlayer distance is typically ∼6.9 Å. The hydroxy double salt depicted in Fig. 2c comprises 2D M(OH)x sheets consisting of Oh hydroxide sheets partially sandwiched with tetrahedrally coordinated (Td) hydroxides, anions coordinated to or intercalated between the Oh–Td hydroxide sheets and a relatively large interlayer distance of 7–15 Å that varies depending on the anion species. The structure shown in Fig. 2d is typical of α-nickel hydroxides, which are composed of Oh hydroxide sheets similar to those of β-Ni(OH)2 but with a wider interlayer distance of ∼8 Å due to their turbostratic structure with unsettled interlayer H2O. This type of LHS often contains interlayer anions.53 Finally, in the α-type structure depicted in Fig. 2e, the Oh hydroxide sheets are replaced by Oh–Td hydroxide sheets, and α-cobalt hydroxide with a typical interlayer distance of 8–9 Å is a known example.54,55
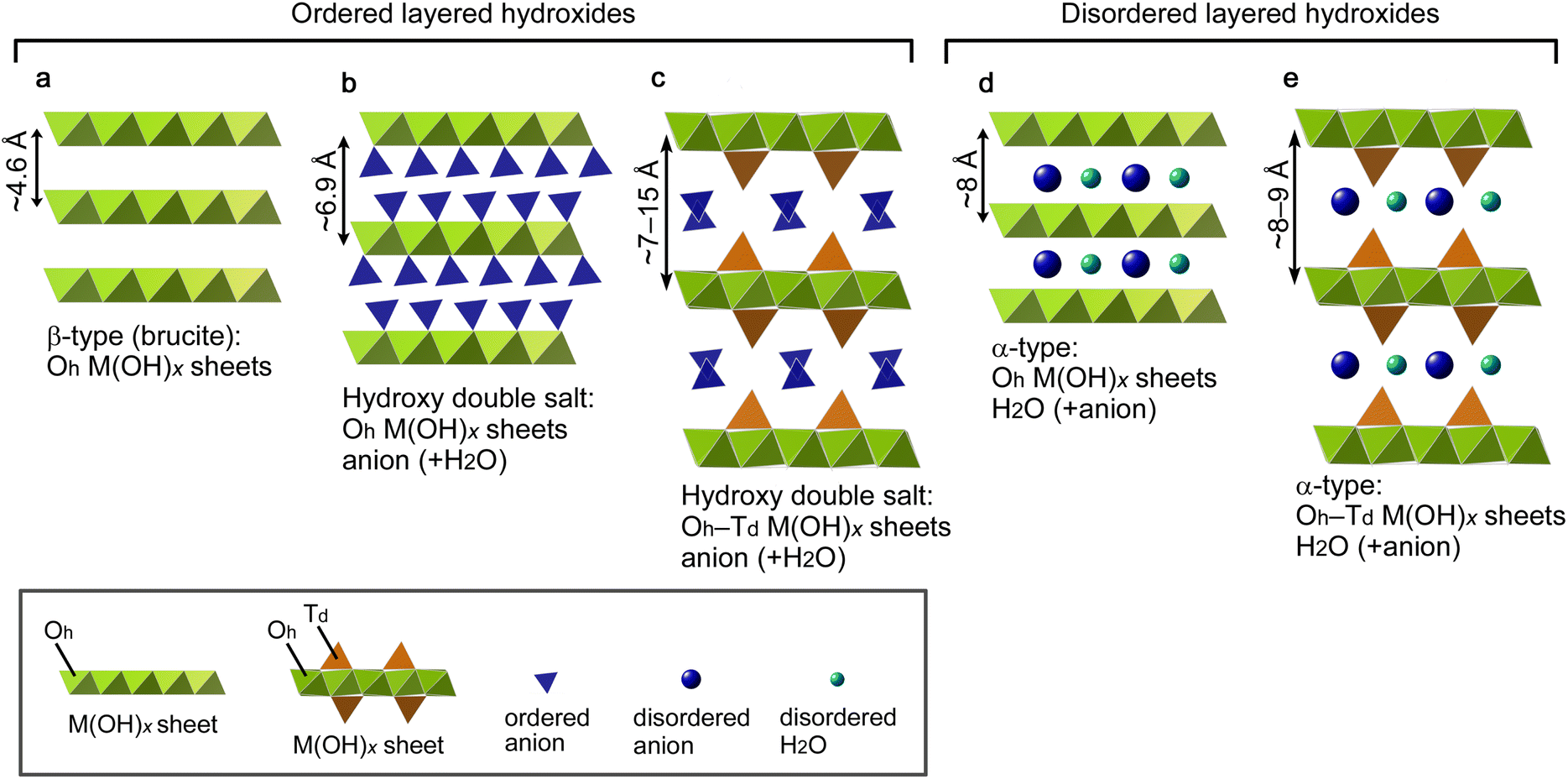 | ||
| Fig. 2 Structure models of five representative types of layered hydroxide salts: (a) β type, (b and c) hydroxy double salts with (b) octahedrally coordinated (Oh) and (c) Oh-tetrahedrally coordinated (Td) M(OH)x sheets, and (d and e) α types with (d) Oh and (e) Oh–Td M(OH)x sheets. | ||
| Structure type (sheet type) | Cation | Chemical composition | Interlayer [Å] | Characteristic IR banda [cm−1] | Ref. | |
|---|---|---|---|---|---|---|
| Lattice OH− | NO3− region | |||||
| a Notation: s, strong; m, medium; w, weak; sh, sharp; br, broad. | ||||||
| β (Oh sheet) | Co2+ | Co(OH)2 | 4.7 | 3632 (s, sh) | — | 27 |
| Ni2+ | Ni(OH)2 | 4.6 | 3650 (s, sh) | — | 56 | |
| Double salt (Oh sheet) | Co2+ | Co2(NO3)(OH)3 | 6.9 | 3611 (m, sh) | 1491 (s) | 57 |
| 3527 (m, br) | 1275 (s) | |||||
| 986 (s) | ||||||
| Ni2+ | Ni2(NO3)(OH)3 | 6.9 | ∼3600 (m, sh) | 1503 (s) | 58 | |
| ∼3400 (m, br) | 1316 (s) | |||||
| 997 (m) | ||||||
| Cu2+ | Cu2(NO3)(OH)3 | 6.9 | ∼3560 (m, sh) | 1428 (s) | 58 | |
| ∼3450 (m, br) | 1341 (s) | |||||
| 1047 (m, sh) | ||||||
| Double salt (Oh–Td sheet) | Zn2+ | Zn5(NO3)2(OH)8·2H2O | 9.8 | 3569 (w, sh) | 1370 (s) | 59 |
| 3465 (m, br) | ||||||
| α (Oh sheet) | Ni2+ | Ni(NO3)x(OH)2−x·nH2O | ∼8 | ∼3630 (s, br) | ∼1340 (s) | 60 |
| 1310 (s) | ||||||
| 1280 (s) | ||||||
| 1042 (m) | ||||||
| 992 (m) | ||||||
| α (Oh–Td sheet) | Co2+ | Co(NO3)0.2(OH)1.8·0.67H2O | 8.9 | ∼3450 (m, br) | 1491 (m) | 54 |
| 1393 (s, sh) | ||||||
| 1357 (m) | ||||||
| Co2+ | Co(NO3)x(OH)y·nH2O | 8.7–8.9 | ∼3500 (m, br) | 1464 (m) | This work | |
| ∼1348 (s, br) | ||||||
| 1046 (w) | ||||||
The structural features observed in the XRD and XPS measurements, i.e., low crystallinity, a large interlayer distance of 8.3–8.9 Å, and the presence of NO3− ions, suggest that the resulting [001]-stacking lamellar Co-LHS film is of the α-type. To confirm this, UV-vis-NIR, attenuated total reflection (ATR)-FTIR and Raman spectroscopy analyses were performed.
As shown in Fig. 1d, the as-deposited Co-LHS film showed visible transparency as high as ∼90%. Absorption peaks characteristic of Td–Co2+ were observed at wavelengths of 591 and 642 nm in addition to a less prominent peak at ∼520 nm due to Oh–Co2+,44 indicating that the 2D M(OH)x sheet has an Oh–Td structure (Fig. 2e). The ATR-FTIR spectrum (Fig. 1e) confirms the absence of a sharp ν(OH) band around 3600 cm−1 that would be attributed to β-type lattice –OH and the presence of H2O molecules (δ(H2O) at 1600 cm−1) and NO3− ions (∼1348 cm−1) in the interlayer, which are typical α-type features.
As shown in Table 1, the position and shape of the NO3− IR bands provide information on their state in the interlayer. Monodentate NO3− coordinated to metals (M–O–NO2) in regularly stacking 2D M(OH)x sheets (Fig. 2b) such as Co2(NO3)(OH)3, Ni2(NO3)(OH)3 and Cu2(NO3)(OH)3 usually show sharp doublet peaks at 1428–1503 cm−1 and 1275–1341 cm−1,57,58 which can be attributed to the asymmetric (ν4) and symmetric (ν1) NO2 stretching modes, respectively, with C2v symmetry. A sharp peak at 986–1047 cm−1 corresponding to the O–N stretching (ν2) mode is also characteristic for monodentate NO3−. Meanwhile, nearly free-state NO3− ions regularly packed in the interlayer with hydrogen bonds (Fig. 2c), such as those in Zn5(NO3)2(OH)8·2H2O,58,59 show a characteristic intense singlet peak at ∼1370 cm−1 similar to that of solid NaNO3, which can be attributed to antisymmetric N–O stretches (ν3) with D3h symmetry. In α-type LHSs with interlayer NO3−, due to the low stacking regularity of the 2D M(OH)x sheets, the interlayer NO3− is in an indeterminate state that depends on the synthesis conditions, which may result in a broad peak overlapped with multiple IR bands. Indeed, the FTIR spectra of α-type Ni-LHSs reported by Hall et al.60 suggest the presence of both free and monodentate NO3− in the interlayer.
The α-Co-LHS obtained in this study (Fig. 2e type) showed a broad strong IR peak at ∼1348 cm−1 accompanied by weak peaks at 1464 and 1046 cm−1. The latter two peaks can be attributed to the ν4 and ν2 modes characteristic of monodentate NO3−, while the former broad peak is likely a combination of the monodentate ν1 mode and the ν3 mode of free NO3−, suggesting the presence of both monodentate and free interlayer NO3−.
The Raman spectrum of the obtained α-Co-LHS films showed two broad peaks at 520 and 458 cm−1 (Fig. S3b†). According to the literature, β-Co(OH)2 exhibits several sharp Raman peaks, the most intense at 523 cm−1 and the second most intense at 457 cm−1, which can be attributed to Co–O symmetric stretching mode (Ag) and the O–Co–O bending mode, respectively.51 The Raman peaks observed in the present study exhibited similar positions but were broader, suggesting the low crystallinity of the obtained α-Co-LHS.
The results shown in Fig. 1 and S3† indicate that the electrodeposited Co-LHS is Co(NO3)x(OH)y·nH2O of an α-(Oh–Td hydroxide sheet) type with a [001]-oriented continuous film structure. To the best of our knowledge, this is the first example of directly formed oriented continuous films of Co-LHS.
Formation of [111]-oriented mesoporous Co3O4 films via TPT
The conversion of the [001]-oriented α-Co-LHS films to Co3O4 was conducted by heat treatment at 550 °C for 1 h in air. Co3O4 is a p-type semiconductor with a cubic spinel structure, where Co2+ cations occupy the Td sites and Co3+ cations occupy the Oh sites, [Co2+]Td[2Co3+]OhO4. Upon heating, the colour of the sample changed from pale light blue to dark brown (Fig. S4a†). As shown in Fig. 3a, the continuous lamellar film was converted to a continuous film (∼300 nm in thickness) with a mesoporous structure containing numerous particles with a size of ∼70 nm and nanopores having a diameter of ∼30 nm.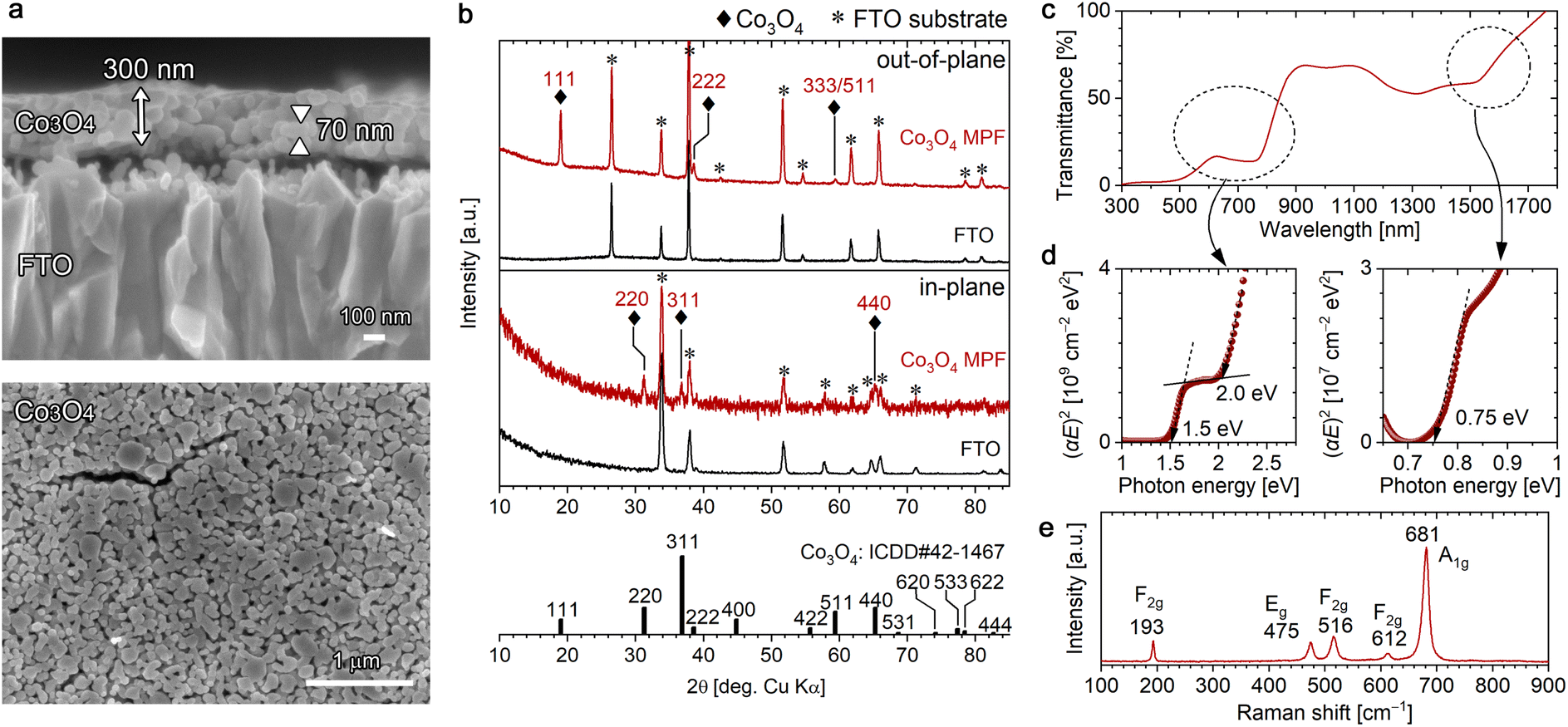 | ||
| Fig. 3 Characterization of the [111]-oriented Co3O4 mesoporous films (MPFs) obtained by heating [001]-oriented Co-LHS at 550 °C for 1 h in air: (a) cross-sectional (top) and plan-view (bottom) FESEM images, (b) out-of-plane (top) and in-plane (bottom) XRD patterns, (c) UV-vis-NIR transmittance spectrum, (d) Tauc plots and (e) Raman spectrum. ICDD data (no. 42-1467) for Co3O4 with a cubic spinel structure is presented in (b). | ||
All diffraction peaks observed in the out-of-plane (θ–2θ) XRD pattern of the mesoporous film (MPF) were ascribable to the FTO substrate or Co3O4 with a cubic spinel structure (ICDD no. 42-1467); no other phases were detected (Fig. 3b). Peaks attributed to Co3O4 were observed at 18.99°, 38.57° and 59.42° and assigned to 111, 222 and 333/511 diffractions, respectively, indicating that the resulting Co3O4 MPF has a [111] preferred orientation. The crystallite size of the resulting Co3O4 was estimated to be 31 nm using the Scherrer equation. To further confirm the crystal orientation, in-plane XRD measurements were performed to detect crystal planes perpendicular to the substrate surface. In-plane diffraction peaks stemming from Co3O4 were observed at 31.2°, 36.8° and 65.2°, which can be assigned to 220, 311 and 440 diffractions. For the 220 and 440 diffractions, since the angle between the (111) and (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) planes is 90° in the cubic spinel system, the results are consistent with the preferred [111] orientation. For the 311 diffraction, the angle between the (111) and (3) planes is 80°, suggesting the presence of some Co3O4 nanograins with a slightly tilted [111] direction. The fine XPS spectra of the O 1s and Co 2p regions (Fig. S5a†) showed an O 1s peak at 530.0 eV and Co 2p3/2 peaks at 779.6 and 780.6 eV derived from Co(III) and Co(II), respectively, which are in good agreement with the literature values.51
0) planes is 90° in the cubic spinel system, the results are consistent with the preferred [111] orientation. For the 311 diffraction, the angle between the (111) and (3) planes is 80°, suggesting the presence of some Co3O4 nanograins with a slightly tilted [111] direction. The fine XPS spectra of the O 1s and Co 2p regions (Fig. S5a†) showed an O 1s peak at 530.0 eV and Co 2p3/2 peaks at 779.6 and 780.6 eV derived from Co(III) and Co(II), respectively, which are in good agreement with the literature values.51
The UV-vis-NIR transmittance spectrum of the [111]-oriented Co3O4 MPFs (Fig. 3c) showed three absorption edges at wavelengths of ∼1600, ∼800 and ∼600 nm, and the optical bandgaps were estimated to be 0.75, 1.5 and 2.0 eV from the direct-transition Tauc plots shown in Fig. 3d. These values are in good agreement with the literature values,61 although the bandgap energy of 0.75 eV in the near-infrared region is not described in some of the reported studies. According to the literature,61 these peaks originate from crystal field 4A2(F) → 4T1(F) (0.75 eV) transitions and ligand–metal charge transfer involving O2−–Co3+ (1.5 eV) and O2−–Co2+ (2.0 eV); however, the electronic structure of spinel Co3O4 is not simple and is still under investigation from both theoretical and experimental aspects.62,63
ATR-FTIR spectrum of the [111]-Co3O4 MPFs confirmed the presence of Co–O vibrations at 678 cm−1 and the disappearance of NO3− and H2O (Fig. S5b†).64 The formation of Co3O4 with high crystallinity without residues was supported by the corresponding Raman spectra (Fig. 3e), where five sharp Raman bands characteristic of spinel Co3O4 were observed at 193(F2g), 475(Eg), 516(F2g), 612(F2g) and 681(A1g) cm−1.65
Formation of [111]-oriented mesoporous CoO films via TPT
Interestingly, by subjecting the [001]-oriented α-Co-LHS films to heat treatment under different conditions from those affording Co3O4, i.e., at 500 °C for 1 h under vacuum, CoO films were obtained. Cubic rocksalt-type CoO is known to exhibit Mott insulator property owing to its strongly correlated electron system.66 As shown in Fig. 4a, the [001]-oriented lamellar Co-LHS film was thermally transformed into a 200 nm-thick mesoporous film composed of a myriad of particles of 30 nm in diameter.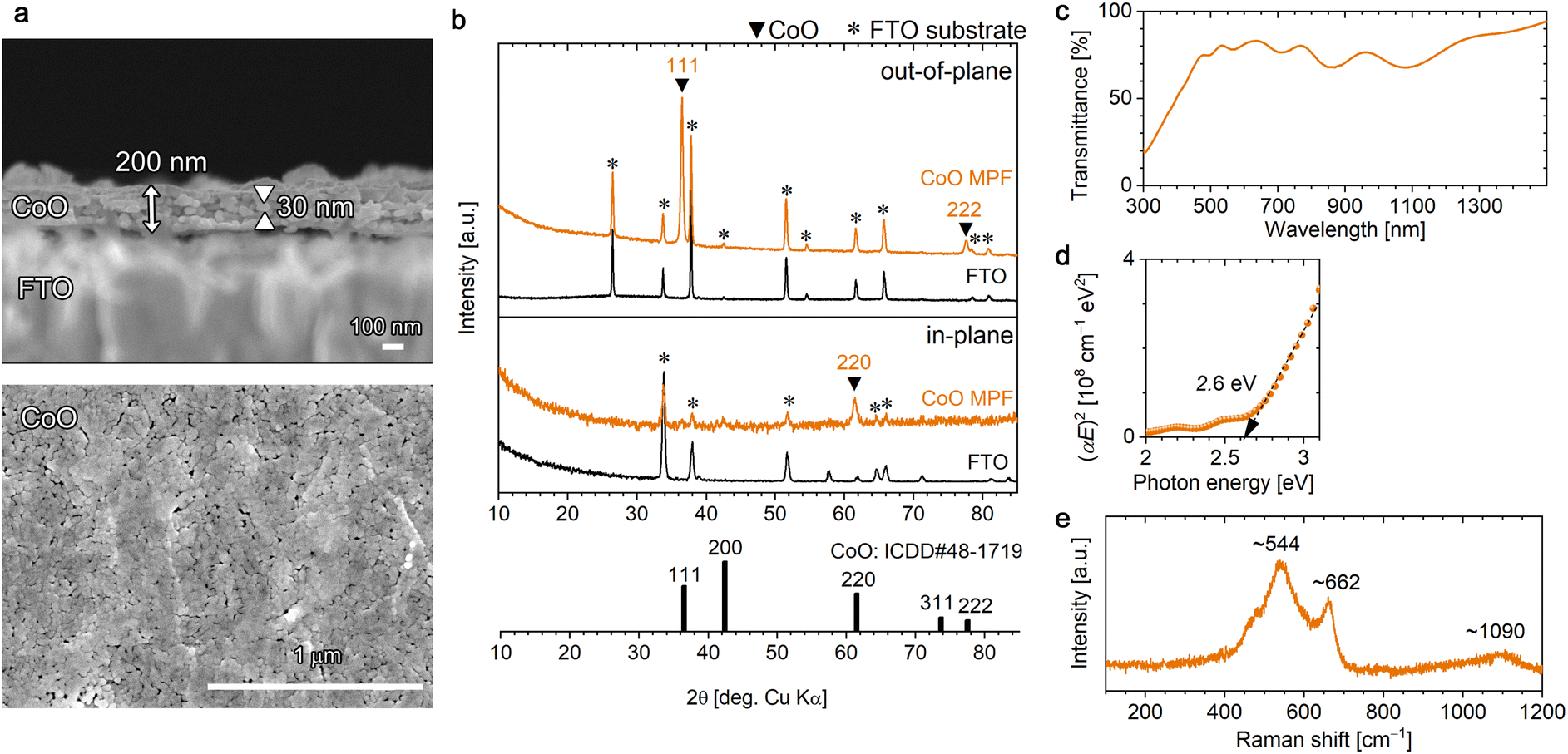 | ||
| Fig. 4 Characterization of the [111]-oriented CoO mesoporous films (MPFs) obtained by heating [001]-oriented Co-LHS films at 500 °C for 1 h under vacuum: (a) cross-sectional (top) and plan-view (bottom) FESEM images, (b) out-of-plane (top) and in-plane (bottom) XRD patterns, (c) UV-vis-NIR transmittance spectrum, (d) Tauc plots and (e) Raman spectrum. ICDD data (no. 48-1719) for CoO with a cubic rocksalt structure is presented in (b). | ||
Fig. 4b (top) shows the out-of-plane XRD pattern of the resulting mesoporous film, which revealed the formation of CoO with a cubic rocksalt structure (ICDD no. 48-1719). No other phases were detected. A sharp intense peak due to CoO was observed at 2θ = 36.48° accompanied by a weak peak at 77.53°, which can be assigned to 111 and 222 diffractions, respectively. Furthermore, a 220 diffraction peak was observed in the in-plane XRD pattern (Fig. 4b bottom), and the angle between the (111) and (10) planes is 90° in the cubic system, confirming the [111] crystal orientation of the resulting CoO MPF. The crystallite size of the CoO was estimated to be 23.6 nm in diameter using the Scherrer equation, which is consistent with the grain size observed in the FESEM images and suggests that each particle is single-crystal CoO. In the fine XPS measurements (Fig. S6a†), O 1s and Co 2p3/2 peaks were observed at 529.8 and 780.4 eV, respectively, which are in agreement with the literature.67
The obtained [111]-oriented CoO MPFs exhibited a pale olive colour (Fig. S4b†) and 70–80% optical transmittance in the visible region (Fig. 4c). The bandgap energy was estimated from the direct-transition Tauc plots to be 2.6 eV, which is consistent with the literature value (Fig. 4d).32,67 In the ATR-FTIR spectrum (Fig. S6b†), no significant bands except for those of the substrate were detected, confirming the negligible presence of –OH or H2O. A broad peak due to Co–O vibrations should appear around 510 cm−1,64 but this value is outside the measurement range used in this study.
The Raman spectrum of the obtained CoO MPF, which differed considerably from that of Co3O4, exhibited a broad peak at ∼544 cm−1 with a shoulder at ∼662 cm−1 (Fig. 4e). The rocksalt structure with a centrosymmetric lattice is generally a weak Raman scatter, where a first-order phonon scattering is forbidden due to the selection rules, whereas a two-phonon Raman scattering is allowed.68 Rivas-Murias et al. reported similar Raman spectra with a broad peak at 530 cm−1 and a weak shoulder at 680 cm−1 for octahedrally faceted CoO nanocrystals with a size of ∼60 nm.69 According to this, the observed broad peak at ∼544 cm−1 can be assigned to cobalt or structural defects inducing one-phonon longitudinal optical Raman scattering, and the shoulder peak is attributable to Co3O4 slightly generated upon laser irradiation. Thereby, the weak broad peak observed at ∼1090 cm−1 is assignable to second-order two-phonon Raman scattering.64 The appearance of inherently forbidden Raman scattering with more intensity than the allowed one has been previously reported for rocksalt-type NiO nanocrystals (∼36 nm diameter).47
Plausible mechanism for the transformation from [001]-α-Co-LHS to [111]-Co3O4 and [111]-CoO
The above results demonstrate that [111]-oriented MPFs of Co3O4 and CoO can be successfully obtained from [001]-oriented α-Co-LHS continuous films. Up to now, topotactic-like formation of oriented cobalt oxide on substrates has been limited to using microcrystal hydroxides as precursors.32,45 To the best of our knowledge, this is the first example of a centimetre-size continuous film with indistinct grain boundaries undergoing oriented transformation to yield cobalt oxide films. Nevertheless, the observed orientation relationship, that is, α-Co-LHS[001]//Co3O4[111] and α-Co-LHS[001]//CoO[111], must be explained from a microscopic view point. In fact, even if the starting hydroxide is single-crystal Mg(OH)2 with a simple brucite structure, aggregates of uniform MgO nanoparticles with a size of several nanometers are initially produced, suggesting that the TPT process occurs on a nanodomain unit.70 Thus, in the present case, the TPT process occurring in the numerous nanodomains of the [001]-stacking α-Co-LHS layers likely produced a myriad of [111]-oriented CoxOy nanocrystals, resulting in centimetre-scale [111]-oriented mesoporous oxide films as a whole. Here, the conversion processes are discussed in terms of the nanoregion structure, i.e., the atomic framework, of the starting hydroxides and product oxides.The structure of the α-Co-LHS films obtained in this study consists of 2D Oh–Td Co(OH)x sheets and the interlayer NO3− and H2O (Fig. 5a). The Oh/Td ratio depends on the metal and anion species,71 and the proposed structure was constructed on the basis of previously reported Zn5(OH)8Cl2·H2O with definite crystallographic information. The cross-sectional structure of the (001) plane is shown in Fig. 5d, where the 2D Oh–Td Co(OH)x sheets are stacked in the [001] direction. When the interlayer NO3− and H2O molecules disappear upon heating, the 2D Oh–Td Co(OH)x sheets approach each other to form an interlocked structure as shown in Fig. 5e, whose components are indicated by A–C. The corresponding top view images, i.e., the atomic framework of the (001) planes of A, B and C layers, are extracted in Fig. 5h. In A(001), there are regular vacancies in the 2D planar layer composed of edge-sharing Oh-Co2+ layers. B(001) is composed of isolated Td-Co2+ arranged in a three-fold symmetry, and C(001) has a structure in which the vacancies in A are filled with Td-Co2+.
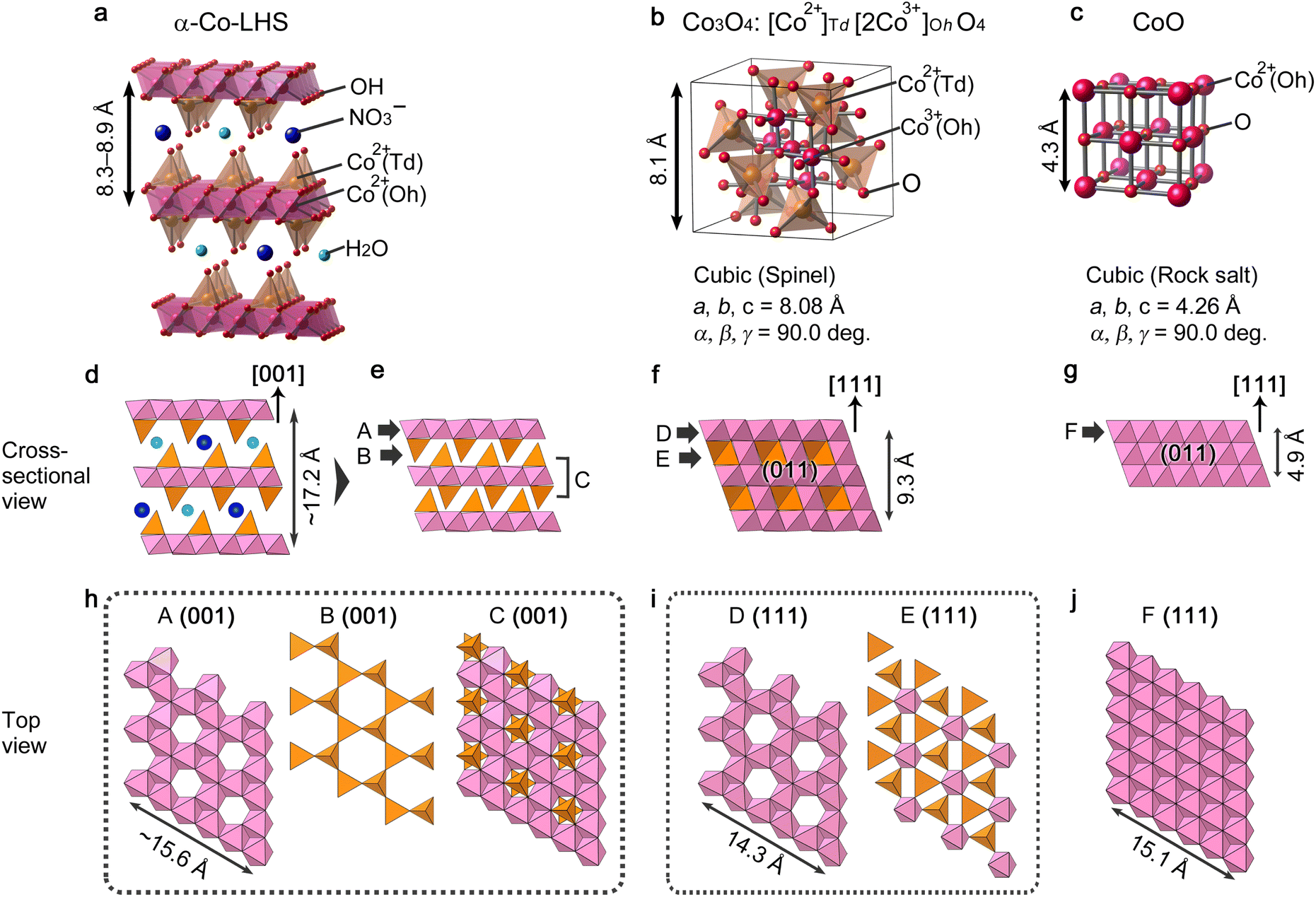 | ||
| Fig. 5 Crystal model of (a) α-Co-LHS (drawn on the basis of Zn5(OH)8Cl2·H2O with modifications), (b) Co3O4 and (c) CoO. (d–g) Cross-sectional view of crystal models parallel in the (d, e) α-Co-LHS[001], (f) Co3O4[111] and (g) CoO[111] directions. (h–j) Top view of crystal models for the (h) α-Co-LHS (001), (i) Co3O4(111) and (j) CoO(111) planes corresponding to the A–F components indicated in (e–g). The length of the atomic framework of A(001) is calculated on the basis of α-Co-LHS containing Cl and H2O in the interlayer (lattice constant a = 3.14 Å),55 which has the same space group as that of Zn5(OH)8Cl2·H2O. | ||
The crystal structures of Co3O4 and CoO are shown in Fig. 5b and c, respectively. Spinel-type Co3O4 is composed of Td-Co2+ and Oh-Co3+ and can be formally represented as [Co2+]Td[2Co3+]OhO4. Fig. 5f displays a crystal structure of the Co3O4(011) plane with the [111] direction pointing upward and two layers denoted by D and E alternately stacked. The corresponding (111) planes of these D and E layers are depicted in Fig. 5i. Interestingly, the atomic framework of D(111) is identical to that of A(001), and the slightly smaller dimension of D(111) is likely due to the difference in ionic radii of Co2+ (0.65 Å) and Co3+ (0.55 Å) for A and D, respectively.72 Thus, in the transformation, the α-Co-LHS A layer is most likely the main component forming the Co3O4D layer. Meanwhile, Co3O4E(111) consists mainly of Td-Co2+ with a three-fold symmetry and could be formed by a slight rearrangement of the α-Co-LHS B layer. Indeed, magnetic moment studies suggested that Td-Co2+ in α-Co-LHS is maintained during the conversion to Co3O4.26
The other cobalt oxide, rocksalt-type CoO, has a simple atomic framework consisting only of Oh-Co2+ (Fig. 5c). As shown in Fig. 5g, the (011) plane with the [111] direction pointing upward is a stack of Oh-Co2+ layers denoted as F. The F(111) structure shown in Fig. 5j has an atomic framework similar to that of A(001) but without vacancies. Thus, while the formation of the F(111) layer can be explained by considering a C(001) layer with vacancies filled with Td-Co2+, an excess of cobalt atoms might be present.
A comparison of the atomic frameworks reveals that the [001]-direction of α-Co-LHS and the [111]-direction of CoxOy have an apparent common structure with alternating layers of cobalt and oxygen atoms, as well as highly similar crystal plane structures, i.e., A–F. This 3D similarity in the atomic frameworks may provide a topotactic-like transformation from the [001]-oriented α-Co-LHS film to [111]-oriented Co3O4 and CoO films with minimal atomic rearrangements. In other words, the experimental results suggest that even low-crystalline α-type LHSs have a regular structure that satisfies topotactic transformation in their nanodomains.
Experimental
Materials
Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, ≥98.0%) and platinum plate (99.95%) were purchased from Nacalai Tesque Inc. and Nilaco Co. and used as received. The aqueous solution for electrodeposition was prepared using deionized water (>10 MΩ cm) purified using a Millipore Elix Advanced5 system. FTO-coated glass (∼10 Ω/sq, Asahi Glass) was used as a substrate.Synthesis of [001]-oriented α-Co-LHS films
Before electrodeposition, FTO substrates were treated with a UV-ozone cleaner for 10 min and then rinsed with deionized water. α-Co-LHS films were electrochemically deposited using a potentiostat/galvanostat (Hokuto Denko HABF5001) and a conventional two-electrode glass vessel consisting of the FTO substrate (1.5 × 4.0 cm in size with a deposition area of 3.75 cm2) as the working electrode and the platinum plate as the counter electrode. Galvanostatic electrolysis was performed in an aqueous solution containing 10 mM Co(NO3)2 using a cathodic constant current of −1.0 mA cm−2 for 3.3 min (total quantity of electricity −0.2 C cm−2) at room temperature. After deposition, the deposits were gently rinsed with deionized water and dried at room temperature under vacuum.Synthesis of [111]-oriented Co3O4 and CoO MPFs
[111]-Oriented Co3O4 MPFs were prepared by heating the electrodeposited [001]-oriented α-Co-LHS films in air at 550 °C for 1 h at a heating rate of 10 °C min−1 using an electric muffle furnace. [111]-Oriented CoO MPFs were prepared by heating the [001]-oriented α-Co-LHS films under vacuum (approximately 1 × 10−3 Pa) at 500 °C for 1 h at a heating rate of 5 °C min−1 using a quartz tube furnace with an oil diffusion pump.Characterization
Cross-sectional and surface FESEM images of films deposited on the FTO substrates were obtained using a JEOL JSM-6700F microscope with an acceleration voltage of 5.0 kV. Out-of-plane (θ–2θ) and in-plane (ω – 2θχ/φ) XRD patterns were obtained using a Rigaku SmartLab diffractometer with Cu Kα radiation operated at 6 kW. XPS spectra were recorded with a Kratos AXIS-Ultra DLD spectrometer using monochromated Al Kα radiation operated at 60 W. UV-vis-NIR transmittance spectra were recorded on a Shimadzu UV-3150 spectrophotometer using an integrating sphere with respect to the FTO substrate. ATR-FTIR spectra were recorded using a Thermo Nicolet 4700 spectrophotometer with a diamond reflection crystal unit (DuraSample IRII). Raman spectra were recorded using a microscopic laser Raman spectrometer (Horiba LabRAM HR Evolution) with a laser wavelength of 532 nm.Conclusions
In summary, we have demonstrated the successful formation of [111]-oriented mesoporous films of Co3O4 and CoO as a result of the TPT of [001]-oriented stacking films of α-Co-LHS. [001]-α-Co-LHS having a unique stacking continuous film structure was prepared on FTO substrates using a simple electrodeposition method from Co(NO3)2 aqueous solution at room temperature. A detailed characterization revealed that [001]-α-Co-LHS with an interlayer distance of 8.3–8.9 Å contained NO3− and H2O in the interlayer, and its 2D Co(OH)x sheets consisted of edge-shared Oh-Co2+ sheets partially sandwiched with Td-Co2+. [111]-oriented Co3O4 and CoO films were obtained upon calcination of the [001]-α-Co-LHS films at different conditions, i.e., in air at 550 °C and under vacuum at 500 °C, respectively. The resulting Co3O4 and CoO have a mesoporous structure consisting of an aggregation of numerous [111]-oriented nanocrystals with a diameter of ∼70 and ∼30 nm, respectively. The formation of the [111]-oriented Co3O4 and CoO films from the [001]-α-Co-LHS continuous films suggests that TPT proceeded on a centimetre-scale. A comparison between the atomic frameworks of α-Co-LHS and the cobalt oxides, Co3O4 and CoO, revealed similarities in the atomic arrangement of the α-Co-LHS(001) and cobalt oxide(111) planes. It can be concluded that the assembly of regular transformations in the nanoregion results in the centimetre-scale TPT.Author contributions
This work was designed and directed by T. S. with the support of A. O. T. S. and N. K. performed the experiments and analysed these data. The manuscript was prepared by T. S. supported by discussions with all the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A part of this work was supported by Grants-in-Aid for Scientific Research (C) (JSPS KAKENHI no. 22K05281).Notes and references
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1, 16023 CrossRef CAS.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS.
- T. Wagner, S. Haffer, C. Weinberger, D. Klaus and M. Tiemann, Chem. Soc. Rev., 2013, 42, 4036–4053 RSC.
- L.-S. Zhong, J.-S. Hu, L.-J. Wan and W.-G. Song, Chem. Commun., 2008, 1184–1186 RSC.
- S.-W. Bian, J. Baltrusaitis, P. Galhotra and V. H. Grassian, J. Mater. Chem., 2010, 20, 8705–8710 RSC.
- N. Dahal, I. A. Ibarra and S. M. Humphrey, J. Mater. Chem., 2012, 22, 12675–12681 RSC.
- R. Q. Song, A. W. Xu, B. Deng, Q. Li and G. Y. Chen, Adv. Funct. Mater., 2007, 17, 296–306 CrossRef CAS.
- W. Zhang and K. Yanagisawa, Chem. Mater., 2007, 19, 2329–2334 CrossRef CAS.
- X. Li, S. Xiong, J. Li, J. Bai and Y. Qian, J. Mater. Chem., 2012, 22, 14276–14283 RSC.
- G. G. C. Arizaga, K. G. Satyanarayana and F. Wypych, Solid State Ionics, 2007, 178, 1143–1162 CrossRef CAS.
- S. Nakagaki, G. S. Machado, J. F. Stival, E. Henrique dos Santos, G. M. Silva and F. Wypych, Prog. Solid State Chem., 2021, 64, 100335 CrossRef CAS.
- R. R. Balmbra, J. S. Clunie and J. F. Goodman, Nature, 1966, 209, 1083–1084 CrossRef CAS.
- M. Figlarz, B. Gérand, A. Delahaye-Vidal, B. Dumont, F. Harb, A. Coucou and F. Fievet, Solid State Ionics, 1990, 43, 143–170 CrossRef CAS.
- Y. Li, B. Tan and Y. Wu, J. Am. Chem. Soc., 2006, 128, 14258–14259 CrossRef CAS.
- J. Zhu, L. Bai, Y. Sun, X. Zhang, Q. Li, B. Cao, W. Yan and Y. Xie, Nanoscale, 2013, 5, 5241–5246 RSC.
- X. Wang, X. L. Wu, Y. G. Guo, Y. Zhong, X. Cao, Y. Ma and J. Yao, Adv. Funct. Mater., 2010, 20, 1680–1686 CrossRef CAS.
- K. K. Lee, W. S. Chin and C. H. Sow, J. Mater. Chem. A, 2014, 2, 17212–17248 RSC.
- N. Tang, W. Wang, H. You, Z. Zhai, J. Hilario, L. Zeng and L. Zhang, Catal. Today, 2019, 330, 240–245 CrossRef CAS.
- J. Ma, H. Wei, Y. Liu, X. Ren, Y. Li, F. Wang, X. Han, E. Xu, X. Cao and G. Wang, Int. J. Hydrogen Energy, 2020, 45, 21205–21220 CrossRef CAS.
- F. Tang, S. Guo, Y. Sun, X. Lin, J. Qiu and A. Cao, Small Struct., 2022, 3, 2100211 CrossRef CAS.
- S. L. Zhang, B. Y. Guan, X. F. Lu, S. Xi, Y. Du and X. W. Lou, Adv. Mater., 2020, 32, 2002235 CrossRef CAS PubMed.
- A. Gasparotto, D. Barreca, D. Bekermann, A. Devi, R. A. Fischer, P. Fornasiero, V. Gombac, O. I. Lebedev, C. Maccato and T. Montini, J. Am. Chem. Soc., 2011, 133, 19362–19365 CrossRef CAS PubMed.
- X. Huang, T. Cao, M. Liu and G. Zhao, J. Phys. Chem. C, 2013, 117, 26432–26440 CrossRef CAS.
- M. Ronda-Lloret, Y. Wang, P. Oulego, G. Rothenberg, X. Tu and N. R. Shiju, ACS Sustainable Chem. Eng., 2020, 8, 17397–17407 CrossRef CAS.
- P. Rabu, S. Angelov, P. Legoll, M. Belaiche and M. Drillon, Inorg. Chem., 1993, 32, 2463–2468 CrossRef CAS.
- L. Markov, K. Petrov and V. Petkov, Thermochim. Acta, 1986, 106, 283–292 CrossRef CAS.
- Z. Liu, R. Ma, M. Osada, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2005, 127, 13869–13874 CrossRef CAS.
- Z.-A. Hu, Y.-L. Xie, Y.-X. Wang, L.-J. Xie, G.-R. Fu, X.-Q. Jin, Z.-Y. Zhang, Y.-Y. Yang and H.-Y. Wu, J. Phys. Chem. C, 2009, 113, 12502–12508 CrossRef CAS.
- L. Tian, H. Zou, J. Fu, X. Yang, Y. Wang, H. Guo, X. Fu, C. Liang, M. Wu, P. K. Shen and Q. Gao, Adv. Funct. Mater., 2010, 20, 617–623 CrossRef CAS.
- M. Mansournia and N. Rakhshan, Ceram. Int., 2017, 43, 7282–7289 CrossRef CAS.
- X. W. Lou, D. Deng, J. Y. Lee, J. Feng and L. A. Archer, Adv. Mater., 2008, 20, 258–262 CrossRef CAS.
- R. Ma, M. Osada, L. Hu and T. Sasaki, Chem. Mater., 2010, 22, 6341–6346 CrossRef CAS.
- R. O. Da Silva, R. H. Gonçalves, D. G. Stroppa, A. J. Ramirez and E. R. Leite, Nanoscale, 2011, 3, 1910–1916 RSC.
- Q. Zhang, K. Zhang, D. Xu, G. Yang, H. Huang, F. Nie, C. Liu and S. Yang, Prog. Mater. Sci., 2014, 60, 208–337 CrossRef CAS.
- O. Elbanna, M. Fujitsuka and T. Majima, ACS Appl. Mater. Interfaces, 2017, 9, 34844–34854 CrossRef CAS.
- C. Nguyen Van, T. H. Do, J.-W. Chen, W.-Y. Tzeng, K.-A. Tsai, H. Song, H.-J. Liu, Y.-C. Lin, Y.-C. Chen and C.-L. Wu, NPG Asia Mater., 2017, 9, e357 CrossRef CAS.
- R. I. Balderas, C. V. Ciobanu and R. M. Richards, Cryst. Growth Des., 2022, 22, 6296–6322 CrossRef CAS.
- Y. Zeng, H. Li, Y. Xia, L. Wang, K. Yin, Y. Wei, X. Liu and S. Luo, ACS Appl. Mater. Interfaces, 2020, 12, 44608–44616 CrossRef CAS PubMed.
- L. Liu, Z. Jiang, L. Fang, H. Xu, H. Zhang, X. Gu and Y. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 27736–27744 CrossRef CAS PubMed.
- M. Dixit and P. V. Kamath, J. Power Sources, 1995, 56, 97–100 CrossRef CAS.
- V. Gupta, T. Kusahara, H. Toyama, S. Gupta and N. Miura, Electrochem. Commun., 2007, 9, 2315–2319 CrossRef CAS.
- J. B. Wu, Y. Lin, X. H. Xia, J. Y. Xu and Q. Y. Shi, Electrochim. Acta, 2011, 56, 7163–7170 CrossRef CAS.
- U. M. Patil, M. S. Nam, J. S. Sohn, S. B. Kulkarni, R. Shin, S. Kang, S. Lee, J. H. Kim and S. C. Jun, J. Mater. Chem. A, 2014, 2, 19075–19083 RSC.
- J. R. Brownson and C. Lévy-Clément, Electrochim. Acta, 2009, 54, 6637–6644 CrossRef CAS.
- C. M. Hull, J. A. Koza and J. A. Switzer, J. Mater. Res., 2016, 31, 3324–3331 CrossRef CAS.
- T. Shinagawa, M. Chigane and M. Izaki, ACS Omega, 2021, 6, 2312–2317 CrossRef CAS.
- T. Shinagawa, M. Chigane and M. Takahashi, Cryst. Growth Des., 2022, 22, 4122–4132 CrossRef CAS.
- G. H. A. Therese and P. V. Kamath, Chem. Mater., 2000, 12, 1195–1204 CrossRef CAS.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, NACE, Houston, TX, 1974 Search PubMed.
- T. Shinagawa, M. Watanabe, T. Mori, J.-i. Tani, M. Chigane and M. Izaki, Inorg. Chem., 2018, 57, 13137–13149 CrossRef CAS PubMed.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef PubMed.
- R. S. Jayashree and P. Vishnu Kamath, J. Mater. Chem., 1999, 9, 961–963 RSC.
- R. Ma, Z. Liu, K. Takada, K. Fukuda, Y. Ebina, Y. Bando and T. Sasaki, Inorg. Chem., 2006, 45, 3964–3969 CrossRef CAS PubMed.
- M. Rajamathi, P. V. Kamath and R. Seshadri, J. Mater. Chem., 2000, 10, 503–506 RSC.
- T. N. Ramesh, Inorg. Chem. Commun., 2011, 14, 419–422 CrossRef CAS.
- S. P. Newman and W. Jones, J. Solid State Chem., 1999, 148, 26–40 CrossRef CAS.
- T. Biswick, W. Jones, A. Pacuła, E. Serwicka and J. Podobinski, J. Solid State Chem., 2007, 180, 1171–1179 CrossRef CAS.
- D. S. Hall, D. J. Lockwood, S. Poirier, C. Bock and B. R. MacDougall, J. Phys. Chem. A, 2012, 116, 6771–6784 CrossRef CAS PubMed.
- D. Barreca, C. Massignan, S. Daolio, M. Fabrizio, C. Piccirillo, L. Armelao and E. Tondello, Chem. Mater., 2001, 13, 588–593 CrossRef CAS.
- V. Singh, M. Kosa, K. Majhi and D. T. Major, J. Chem. Theory Comput., 2015, 11, 64–72 CrossRef CAS PubMed.
- V. Singh and D. T. Major, Inorg. Chem., 2016, 55, 3307–3315 CrossRef CAS PubMed.
- Y. Li, W. Qiu, F. Qin, H. Fang, V. G. Hadjiev, D. Litvinov and J. Bao, J. Phys. Chem. C, 2016, 120, 4511–4516 CrossRef CAS.
- V. G. Hadjiev, M. N. Iliev and I. V. Vergilov, J. Phys. C: Solid State Phys., 1988, 21, L199 CrossRef.
- H. Jiang, R. I. Gomez-Abal, P. Rinke and M. Scheffler, Phys. Rev. B, 2010, 82, 045108 CrossRef.
- P. Prieto, J. F. Marco, A. Serrano, M. Manso and J. de la Figuera, J. Alloys Compd., 2019, 810, 151912 CrossRef CAS.
- R. K. Singh, Phys. Rep., 1982, 85, 259–401 CrossRef CAS.
- B. Rivas-Murias and V. Salgueiriño, J. Raman Spectrosc., 2017, 48, 837–841 CrossRef CAS.
- U. Dahmen, M. Kim and A. Searcy, Ultramicroscopy, 1987, 23, 365–370 CrossRef CAS.
- J. R. Neilson, B. Schwenzer, R. Seshadri and D. E. Morse, Inorg. Chem., 2009, 48, 11017–11023 CrossRef CAS PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC press, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FESEM images, XPS spectra, XRD patterns, Raman spectra, ATR-FTIR spectra and photographs of the obtained samples. See DOI: https://doi.org/10.1039/d2na00594h |
| This journal is © The Royal Society of Chemistry 2023 |