
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAluminium substitution in Sb2S3 nanorods enhances the stability of the microstructure and high-rate capability in the alloying regime†
Akshay Kumar
Budumuru‡
ab,
Lokeswararao
Yelamnchi‡
a and
Chandran
Sudakar
 *ab
*ab
aMultifunctional Materials Laboratory, Department of Physics, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: csudakar@iitm.ac.in; Tel: +91-44-2257-4895
bCenter for Advanced Materials and Microscopy, Indian Institute of Technology Madras, Chennai 600036, India
First published on 6th March 2023
Abstract
Alloy anodes, with twice the capacity of graphite, are promising for next-generation lithium-ion batteries (LIBs). However, poor rate-capability and cycling stability, mainly due to pulverization, have limited their application. By constraining the cutoff voltage to the alloying regime (1 V to 10 mV vs. Li/Li+), we show that Sb1.9Al0.1S3 nanorods provide excellent electrochemical performance, with an initial capacity of ∼450 mA h g−1 and excellent cycling stability with 63% retention (capacity ∼240 mA h g−1 after 1000 cycles at 5C-rate), unlike 71.4 mA h g−1 after 500 cycles observed in full-regime cycling. When conversion cycling is also involved the capacity degrades faster (<20% retention after 200 cycles) irrespective of Al doping. The contribution of alloy storage to total capacity is always larger than the conversion storage indicating the superiority of the former. The formation of crystalline Sb(Al) is noted in Sb1.9Al0.1S3, unlike amorphous Sb in Sb2S3. Retention of the nanorod microstructure in Sb1.9Al0.1S3 despite the volume expansion enhances the performance. On the contrary, the Sb2S3 nanorod electrode gets pulverized and the surface shows microcracks. Percolating Sb nanoparticles buffered by the Li2S matrix and other polysulfides enhance the performance of the electrode. These studies pave the way for high-energy and high-power density LIBs with alloy anodes.
Introduction
Lithium-ion batteries (LIBs) are omnipresent and are expected to play an important role in the fourth industrial revolution by powering portable augmented reality systems, smart mobile robots and advanced human–machine interfaces.1,2 However, LIBs with larger energy and power densities are needed for the full utilization of the microprocessors involved in these portable smart machines.3 This is possible with the right choice of electrode materials that exhibit larger capacity and support quick lithium insertion and removal.4 For most of these applications, an anode that can replace commercially used graphite is the need of the hour.5 Many materials are being explored as an alternative anode for LIBs.6 Examples include graphene, silicon, antimony, Sb2S3, MoS2, etc.7,8 Of these, Sb2S3 is considered a promising next-generation anode for lithium-ion batteries.9,10Sb2S3 has a theoretical capacity of 946 mA h g−1 with 12 moles of lithium stored for a unit mole of Sb2S3, which is more than twice the capacity of commercially used graphite (372 mA h g−1).5,11 Sb2S3 has a layered structure which enables easy lithium diffusion. Furthermore, it has reasonably good operating voltage and relatively less volume expansion upon lithiation compared to other conversion and alloying anodes.12,13 The relatively low phase formation temperature (∼300 °C) also makes it an attractive alternative.14 The main issues with Sb2S3 are the low rate-capability and serious capacity loss during extended high-current cycling.15,16 These bottlenecks need to be addressed to make it an attractive alternative anode.
Sb2S3 crystallizes in an orthorhombic structure (space group Pnmb) with lattice parameters a = 11.3107 Å, b = 3.8363 Å, and c = 11.2285 Å.17 In a unit cell of Sb2S3, there are two structurally distinct Sb atoms and three distinct S atoms. The first Sb atom is in square pyramidal coordination with S atoms sharing edges with other square pyramids.17 The second Sb atom is in trigonal coordination, sharing one corner with a square polyhedron and two corners with adjacent trigonal sulfur atoms.17 Two such trigonal SbS3 and two square pyramidal SbS5 extend along the crystallographic c-axis forming (Sb4S6)n chains.18 Within a chain, the bonds between Sb and S are covalent and, across the chains, the attraction is due to weak van der Waal forces.18
Lithium storage in Sb2S3 happens via a conversion reaction followed by an alloying mechanism.19 During the conversion reaction (see eqn (1)), which occurs in the voltage range 2.5 to 1 V (vs. Li/Li+), Sb2S3 reacts with lithium ions and electrons, forming metallic Sb and Li2S with an estimated theoretical capacity of 473 mA h g−1.20
Conversion reaction
| Sb2S3 + 6Li+ + 6e− ⇔ 2Sb + 3Li2S | (1) |
Alloying reaction
| 2Sb + 6Li+ + 6e− ⇔ 2Li3Sb | (2) |
Upon further lithiation (see eqn (2)), metallic Sb left over from the conversion reaction alloys with lithium forming Li3Sb, with an estimated capacity of 473 mA h g−1.21 The Li2S left over from the conversion reaction is inactive during alloying and is expected to have no major influence on the alloying electrochemical reaction.22 During delithiation, Li3Sb dealloys to form metallic Sb.5 However, due to the difference in the reaction mechanism during alloying and dealloying, the latter reaction happens between 10 mV and 1.2 V vs. Li/Li+, unlike the former reaction, which takes place between 1 V and 10 mV vs. Li/Li+.23 When delithiated above 1.2 V, the Li2S reconverts and reacts with Sb to form Sb2S3. Unlike many other sulfides, including MoS2 and WS2, where the decomposition during the first lithiation is irreversible, Sb2S3 reforms at the end of every delithiation.24,25
The volume of the Sb2S3 electrode is estimated to increase by >230% during lithiation.26 In contrast, the volume expansion in graphite upon lithiation is ∼10%.27 Such huge volume expansion in Sb2S3 pulverizes the active material destroying the microstructure of the active material and increasing the impedance.28 Such changes are predominantly responsible for the low rate capability and the abysmal capacity retention of Sb2S3.5 The commonly used methodologies in the literature to alleviate these issues are either to decrease the particle size of the active material, tailor the morphology by incorporating voids to accommodate the volume expansion, or form composites, predominantly with carbonaceous materials, to accommodate the volume expansion and improve the conductivity.29–32 Such approaches have improved the performance of Sb2S3 to various degrees. Amorphous Sb2S3 synthesized via reactive radio frequency magnetron sputtering has a capacity of 678.6 mA h g−1 at 0.1C and a capacity of 495.6 mA h g−1 at 5C.11 When cycled at 0.2C for 250 cycles, a capacity of 585.4 mA h g−1 is observed. The enhanced charge storage properties are attributed to the amorphous phase and 3D structure of the anode.11 Sb2S3 hollow microspheres were reported to have an initial capacity of 1379 mA h g−1 when cycled at 50 mA g−1.29 At higher current rates, 5 A g−1, a capacity of 541 mA h g−1 is observed. Cycling stability studies carried out at 200 mA h g−1 show a capacity of 674 mA h g−1 after 50 cycles.29 Ultrathin Sb2S3 nanosheets were shown to exhibit a reversible capacity of 607 mA h g−1 at 2 A g−1 and a reversible capacity of 800 mA h g−1 after 200 cycles at 0.2 A g−1.15 Similar enhancements were also observed in composite Sb2S3 anodes.9,33
Though the performance of these anodes has been improved, its full utilization in LIBs is beset due to the required performance being far below the commercialization needs.9,34–36 For a next-generation anode material, high rate capability (current rate > 1C) for over 1000 cycles with a capacity retention of >70% is a necessary (though not sufficient) requirement.37 To realize this goal, other strategies need to be explored. One well-known strategy to improve the performance of the electrode materials is cation substitution.38,39
Aluminum substitution in nickel-rich layered oxide is a successful example.40 Aluminium substitution in layered oxides is a promising way to make LIBs with high power densities and a long cycle life, required for electric vehicle application.41 Aluminium substitution in Ni-rich layered oxides tunes the phase transformation process minimizing the mechanical degradation and improves the performance of the layered oxide cathode.42 While there are few reports which study the influence of cation substitution43 and its influence on energy storage properties of Sb2S3,44 a comprehensive study on aluminium substitution and its influence on lithium storage properties are missing.
Aluminium sulfide (Al2S3) occurs in nature and has been tested as an anode for lithium-ion batteries.45,46 The lithium storage mechanism in Al2S3 is similar to that of Sb2S3, i.e., the conversion reaction followed by alloying.47 Despite a similar mechanism, the voltages at which these reactions happen are different due to the difference in the electron energy levels of Al and Sb. Also, Al2S3 predominantly crystallizes in the hexagonal or tetragonal crystal system.48,49 Since Al2S3 does not exist in the orthorhombic phase with lattice parameters similar to that of Sb2S3, it is not possible for these sulfides to form solid solutions over the entire compositional range.50–52 But, consider the Al substitution in layered oxides. LiAlO2 predominantly crystallizes in a tetragonal structure, whereas LiNiO2 crystallizes in a trigonal structure, but it is still possible to get a solid solution when the concentration of Al substituted is less (∼5 at%).53,54 Hence, it might be possible to synthesize a solid solution of Al and Sb sulfides if the quantity of Al substituted is little. Since both Sb2S3 and Al2S3 exist in nature, it is quite possible for the resulting composition to be stable, when Al is substituted in small at% (<10%). The strength of the Al–S bond is less than the bond strength of Sb–S, so the resultant antimony aluminum sulfide should accommodate the lithium ions more easily compared to the pristine Sb2S3.55 Also, the presence of Al nanoparticles in addition to Sb nanoparticles might improve the electronic conductivity of the electrode after lithiation.
In this manuscript, we present the structural, compositional, and electrochemical properties of pristine and Al (5 at%)-substituted Sb2S3 [Sb1.9Al0.1S3] nanorods. We investigate the lithium storage properties in the conversion (2.5 V to 1 V vs. Li/Li+) and alloying (1 V to 10 mV vs. Li/Li+) reaction regimes. Cycling studies are performed with the reactions involved from both the regimes and as well as examining the cycling properties independently in the respective voltage zones. Pristine and Al-substituted Sb2S3 anodes show similar charge–discharge profiles and capacity at low current rates. However, Sb1.9Al0.1S3 nanorods exhibit significantly improved rate-capability at higher current rates having a capacity of 468 mA h g−1 at 10C (9.4 A g−1) and a capacity of 211 mA h g−1 at 25C (23.5 A g−1). Pristine Sb2S3 nanorods, on the other hand, have a capacity of 80 mA h g−1 at 10C (9.4 A g−1). Studies on the conversion and alloying regimes separately indicate that the latter has better electrochemical properties and Al substitution in Sb2S3 further enhances them. Alloying storage of Al-substituted Sb2S3 has a capacity of 383 mA h g−1 at 5C with an outstanding capacity retention of 63% after 1000 cycles. Post-electrochemical microstructural studies indicate that the Al substitution stabilizes the morphology of the nanorods in the alloying regime, facilitating better rate-capability and cycling stability. Pristine Sb2S3 nanorods, on the other hand, undergo pulverization resulting in the loss of the microstructure and rapid capacity fading. Based on the postmortem studies, it is contemplated that Sb nanoparticles form a percolating network in Li2S polysulfides formed at the end of conversion cycling, facilitating better conductivity during cycling in the alloying regime. The lower operating voltage (∼0.7 V vs. Li/Li+) of alloying storage, together with its excellent rate-capability and cycling stability, makes it a promising alternative anode for next-generation LIBs.
Experimental
Materials
Antimony chloride (SbCl3, 99%), aluminium nitrate (Al(NO3)3.9H2O, 98%), and 3-mercaptopropionic acid (3-MPA, C3H6O2S, 99%) were obtained from Alfa Aesar. Ethanol (99.9%) was obtained from Changshu Hongsheng Fine Chemicals. All the chemicals were used as procured without further purification.Synthesis
Sb2S3 nanorods are synthesized by following a hydrothermal method.56 Initially, 0.6379 g of SbCl3 is dissolved in 30 ml of ethanol. 0.9372 g of 3-MPA is added to this ethanol solution. After homogenizing the solution, it is transferred into a 50 ml Teflon-lined stainless steel autoclave and kept in a hot air oven at 180 °C for 12 h. After heating, the autoclave is cooled down to room temperature naturally. The precipitate obtained from the autoclave is filtered, washed with ethanol several times, and dried in air. The resulting powder is annealed at 330 °C for 1 h in high pure argon ambient. The obtained sample is referred to as Sb2S3. To synthesize 5 at% aluminium substituted Sb2S3 (Sb1.9Al0.1S3), the same procedure is followed except that both SbCl3 and Al(NO3)3·9H2O are taken in the 0.95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.05 metal cation ratio. The aluminum substituted sample is referred to as Sb1.9Al0.1S3.
:0.05 metal cation ratio. The aluminum substituted sample is referred to as Sb1.9Al0.1S3.
Compositional and structural characterization
Powder X-ray diffraction patterns of Sb2S3 and Sb1.9Al0.1S3 samples are collected using a Rigaku SmartLab X-ray diffractometer employing a Cu anode operating at 4 kW (Cu Kα λ = 1.5406 Å) between 2θ angles 10° and 60°, with a step size of 0.02°. Raman spectral studies are performed using a Horiba Jobin-Yvon spectrometer working with an excitation laser source of wavelength 632 nm and dispersing the scattering light with 1800 lines per mm grating. X-ray photoelectron spectroscopy is performed on a PHI 5000 Versa Probe III, Physical Electronics, using an Al Kα source. The spectra are corrected by aligning the C 1s peak to 284.6 eV. Field emission scanning electron microscopy (FESEM) and energy dispersive X-ray spectral (EDS) studies are performed using an FEI Inspect F50 operating at 30 kV. Transmission electron microscopy (TEM) and selected area electron diffraction (SAED) studies are carried out using an FEI Tecnai G20 operating at 200 kV, and high-resolution TEM (HRTEM) and EDS mapping studies in scanning transmission electron microscopy (STEM) mode are carried out using an image corrector equipped FEI Titan operating at 300 kV.Electrochemical characterization
The electrodes for electrochemical characterization are fabricated by mixing the active material, acetylene black (MTI Corp) and carboxymethyl cellulose (Sigma-Aldrich), in the ratio of 7:2:1 in an agate motor. A slurry is prepared by adding a few ml of DI water followed by grinding. Later the viscous slurry is cast onto a copper foil laid flat on a glass plate. The slurry is spread using a doctor blade with 100 μm clearance. The slurry is dried and heated at 70 °C for 12 h in a vacuum oven maintained at 10−2 bar. A 15 mm disc is punched from the foil, and coin cells are fabricated using lithium metal foil (Alfa Aeser) as a counter electrode, polypropylene membrane as a separator and a 1 M LiPF6 in a mixture of dimethyl carbonate and fluoroethylene carbonate (FEC) (4:1 vol.%) as electrolyte. Galvanostatic charge–discharge measurements are carried out at different current rates from 0.1C to 40C rate, assuming a theoretical capacity of 946 mA h g−1 (1C = 946 mA g−1) irrespective of the cutoff voltages. Cyclic voltammetry studies were carried out at scan rates ranging between 0.05 and 1 mV s−1. Electrochemical impedance spectroscopy studies are performed with the cells in the discharged state by applying a sinusoidal excitation voltage of 10 mV between frequencies 10 mHz and 1 MHz. All the electrochemical measurements are carried out on a Biologic VSP 300 workstation.
Results and discussion
Structural, microstructural and compositional characterization
Powder X-ray diffraction patterns of Sb2S3 and Sb1.9Al0.1S3 are shown in Fig. 1a and b along with a standard diffraction pattern of Sb2S3 (42-1393) from the inorganic crystal structure database (ICSD). All the peaks present in the diffraction pattern are indexed to the orthorhombic stibnite structure (space group: Pbnm). Both Sb2S3 and Sb1.9Al0.1S3 samples are devoid of impure phases, as discerned from the absence of diffraction peaks attributable to possible secondary phases like antimony oxides. Rietveld refinement further confirms that the structure remains Stibnite and phase pure with the estimated lattice parameters as given in Fig. 1a and b. The lattice parameters are similar for Sb2S3 and Sb1.9Al0.1S3, suggesting that Al substitution does not change the crystal structure drastically. This suggests that most of the Al3+ dopants have been substituted at Sb3+ sites in the lattice. A slight reduction in the lattice parameters of the orthorhombic structure can be attributed to the smaller ionic radius of Al3+ (∼54 pm) compared to Sb3+ (∼76 pm), reducing the lattice dimension slightly without modifying the structure.57 The crystallite size estimated using the Scherrer equation from the most intense peak (hkl) is ∼90 nm for Sb2S3 and ∼60 nm for Sb1.9Al0.1S3.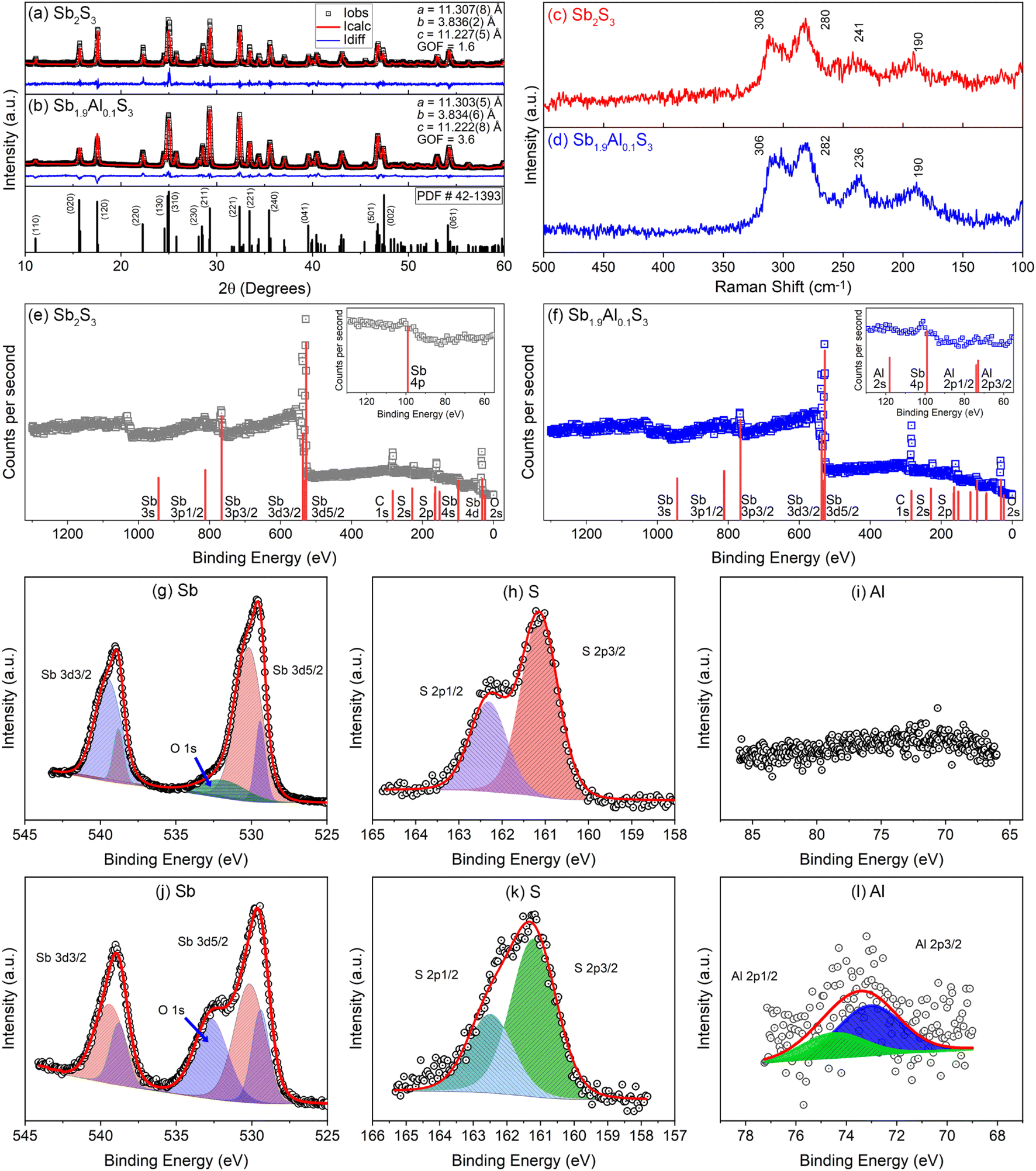 | ||
| Fig. 1 Rietveld refined powder X-ray diffraction patterns of (a) Sb2S3 and (b) Sb1.9Al0.1S3 along with the standard Sb2S3 ICSD powder diffraction pattern (PDF # 42-1393). Raman spectra of (c) Sb2S3 and (d) Sb1.9Al0.1S3. X-ray photoelectron survey spectra of (e) Sb2S3 and (f) Sb1.9Al0.1S3 anodes. The panels in the inset show the enlarged region between the binding energies 55 and 130 eV. Panels (g) – (i) and panels (j) – (l) are the high-resolution spectra collected on Sb2S3 and Sb1.9Al0.1S3 anode in the energy range corresponding to Sb, S and Al elements, respectively. | ||
Raman spectra of Sb2S3 and Sb1.9Al0.1S3 samples are shown in Fig. 1c and d. Both the spectra show four broad peaks centered approximately at 190 cm−1, 241 cm−1, 280 cm−1, and 308 cm−1.58 The first two peaks are attributed to the B1g asymmetric and symmetric bending modes of S–Sb–S, respectively. Two strong modes centered at 280 cm−1 and 308 cm−1 are due to asymmetric Ag and B2g S–Sb–S stretching vibrational modes.59 Al substitution has resulted in a small peak shift (∼4 cm−1) in the Sb1.9Al0.1S3 anode. Such peak shifts were reported in the literature when cations were substituted at the Sb lattice site in Sb2S3.60,61 Raman spectroscopic studies further confirm the phase purity of the Sb2S3 and indicate that Al substitution does not significantly alter the structural features of Sb2S3.
X-ray photoelectron spectroscopic studies on Sb2S3 and Sb1.9Al0.1S3 anodes are shown in Fig. 1e–l. The survey scan of the anodes predominantly shows S and Sb spectral features [Fig. 1e and f]. In Sb1.9Al0.1S3, the survey scan shows peaks attributable to Al 2s/2p orbitals [see inset Fig. 1f]. In addition to these, photoelectron peaks attributable to surface-adsorbed oxygen and adventitious carbon are also observed. Spectral calibration is done using the carbon 2p peak (284.8 eV). Two distinct peaks are present in the Sb 3d core–shell spectra of Sb2S3 with the separation of ∼9 eV between Sb 3d3/2 (∼537 eV) and Sb 3d5/2 (∼528 eV) core levels (Fig. 1g and j). The peaks' position, separation due to the spin–orbit coupling and the intensity ratio between peaks are consistent with the reports on Sb2S3.62,63 Each of the Sb 3d3/2 and Sb 3d5/2 peaks, in fact, can be resolved into two sets of peaks separated by a binding energy difference of ∼9 eV.64 This is attributable to the two structurally different Sb ions present in the lattice.63 The difference in the electronic levels between trigonal and square pyramidal coordinated Sb is the reason for the doublets seen in the 3d core levels. In addition to core-level spectra from Sb atoms, another broad hump is observed at ∼530 eV. The peak could be attributed to the surface-adsorbed oxygen.65 This hump is more prominent in Al-doped Sb2S3 samples, possibly due to oxidation of Al exposed to the surface. Two sets of peaks which can be deconvoluted to S 2p1/2 and S 2p3/2 are observed in the high-resolution sulfur spectra [Fig. 1h and k].63 High-resolution spectra acquired in the binding energy range between 66 and 86 eV show a broad peak in Sb1.9Al0.1S3 attributable to Al 2p [Fig. 1i and l].66 The relatively low signal-to-noise ratio is attributed to the low concentration of aluminium substitution.
Scanning electron micrographs of the Sb2S3 and Sb1.9Al0.1S3 anodes are shown in Fig. 2a and b. Both the materials show rod-like features with a length of ∼10 μm and a width of ∼200 nm. EDS indicates that composition is close to the expected stoichiometric composition in both Sb2S3 and Sb1.9Al0.1S3 (Fig. S1 and Table S1, ESI†). Bright-field TEM images of the rods are shown in Fig. 2c and d. The rods are weakly electron transparent, suggesting a thickness greater than 100 nm. The estimated width of the rods is ∼160 nm. Selected area electron diffraction patterns show symmetrical spots confirming the single crystalline nature of these nanorods [insets of Fig. 2c and d]. These patterns are indexed to the stibnite phase. HRTEM images of representative samples show lattice fringes from highly crystalline nanorods [Fig. 2e and f]. The lattice spacing is attributable to the d-spacing of Sb2S3 (hkl) planes observed in the powder X-ray diffraction patterns. High-angle annular dark field images obtained from the STEM are shown in Fig. 2g and j for Sb2S3 and Sb1.9Al0.1S3 nanorods, respectively. Corresponding elemental maps for Sb2S3 and Sb1.9Al0.1S3 are shown in Fig. 2h, i and k–m, respectively. The images show a uniform distribution of elements without any discernible phase segregation. These studies confirm that the rods are phase pure, and aluminium has been substituted in the Sb2S3 lattice homogeneously.
 | ||
| Fig. 2 Scanning electron micrographs of (a) Sb2S3 and (b) Sb1.9Al0.1S3 revealing the nanorod morphology of the anodes. Bright-field transmission electron micrographs of (c) Sb2S3 and (d) Sb1.9Al0.1S3. Insets in (c) and (d) show the diffraction patterns acquired from the respective nanorods. High-resolution transmission electron micrographs of representative Sb2S3 and Sb1.9Al0.1S3 nanorods are shown in panels (e) and (f), respectively. High angle annular dark field image of Sb2S3 is shown in (g), and element maps of Sb and S are shown in panels (h) and (k), respectively. High angle annular dark field image of Sb1.9Al0.1S3 is shown in (j), and element maps of Sb, S and Al are shown in panels (k), (l) and (m), respectively. | ||
Electrochemical characterization
The electrochemical properties of Sb2S3 and Sb1.9Al0.1S3 anodes are tested in half-cell configuration, initially between 2.5 V and 10 mV (vs. Li/Li+) and later in the conversion (2.5 V to 1 V vs. Li/Li+) and alloying (1.2 V to 10 mV vs. Li/Li+) regimes separately. The first three cycles of galvanostatic charge–discharge profiles of Sb2S3 and Sb1.9Al0.1S3 are shown in Fig. 3a and b, respectively. In the case of Sb2S3, the first lithiation and delithiation capacities are 1221.3 mA h g−1 and 898.8 mA h g−1, respectively, yielding a coulombic efficiency of 73.6%. Sb1.9Al0.1S3 nanorod anodes exhibited first lithiation (1272.0 mA h g−1) and delithiation (940.3 mA h g−1) capacity with a resulting coulombic efficiency of 73.9%. Both the nanorods exhibited similar coulombic efficiency, suggesting an initial irreversible loss of lithium. | ||
| Fig. 3 Galvanostatic charge–discharge profiles of (a) Sb2S3 and (b) Sb1.9Al0.1S3 at 0.05C after the fabrication of the coin cell. Charge–discharge profiles of (c) Sb2S3 and (d) Sb1.9Al0.1S3 at different current rates. (e) Capacity as a function of the number of repeat cycles performed at different C-rates for both lithiation and delithiation cycles of Sb2S3 and Sb1.9Al0.1S3 anodes. Capacity vs. cycle number plots of cycling studies carried out at 5C for 500 cycles are shown in (f). | ||
Al2S3 also stores lithium by conversion and alloying reactions, described using eqn (3) and (4).47 However, the voltages at which these reactions occur are different. The absence of any features attributable to Al2S3 indicates that Al has not segregated in Sb2S3, and Al substitution does not significantly alter the lithium storage mechanism in Sb2S3.
ca. 1.0 V vs. Li/Li+
| Al2S3 + 6Li+ + 6e− ↔ 2Al + 3Li2S | (3) |
ca. 0.5 V vs. Li/Li+
| Al + xLi+ + xe− ↔ LixAl | (4) |
In the lithiation profiles of both the anodes, two-step flat potential features are seen. The initial step (ca.1.7 V vs. Li/Li+) is attributed to the conversion reaction upon lithium insertion into the Sb2S3 lattice. This results in the formation of metallic Sb nanoparticles and Li2S.10 This is represented using eqn (1). The experimental lithiation capacity for the conversion reaction, i.e., the capacity between 2.5 and 1 V is 646.0 mA h g−1 in the case of Sb2S3 and 685.8 mA h g−1 for Sb1.9Al0.1S3 anodes. These values are higher than the estimated theoretical capacity (473.4 mA h g−1), suggesting that in addition to the contribution from the conversion reaction, lithium adsorbed at the defects or on the surface due to the nanostructured nature of Sb2S3 can contribute to the lithium storage.15 Sb2S3-based supercapacitors making use of such non-faradaic and faradaic contributions have been investigated.67,68 Presumably, such a mechanism is responsible for the increased lithiation capacity. Upon further lithiation, another step-like profile is observed at ∼0.9 V vs. Li/Li+. This is attributed to the alloying of lithium with the metallic antimony formed during the conversion reaction, resulting in the formation of Li3Sb alloy.19 This reaction is represented in eqn (2). The estimated theoretical capacity of the alloying reaction is 473.4 mA h g−1. The obtained lithiation capacity for the alloying reactions, i.e., capacity between 1 V and 10 mV is 575.1 mA h g−1 for Sb2S3 and 586.2 mA h g−1 for Sb1.9Al0.1S3. Some of this capacity can be attributed to the decomposition of the fluoroethylene carbonate in the electrolyte, which results in forming a solid electrolyte interphase (SEI).69
During the first delithiation, two-step profiles are observed. The first step between 10 mV and 1.2 V can be attributed to the dealloying of Li3Sb, resulting in the formation of metallic Sb (eqn (1)). The second step can be attributed to the reformation of Sb2S3 during the delithiation of Li2S (eqn (2)). It should be noted that the voltage at which delithiation completes during dealloying (1.2 V vs. Li/Li+) is higher than the voltage at which it begins during lithiation (1 V vs. Li/Li+). Such features are usually attributed to different lithiation/delithiation mechanisms, polarization or slower kinetics during delithiation.39 In metallic Sb, the lithiation and delithiation do not follow a similar reaction pathway. During lithiation of Sb, a crystalline intermediate Li2Sb is initially formed, which upon further lithiation results in the formation of Li3Sb.5 During delithiation, no intermediate phases are formed, and Li3Sb reverts to crystalline Sb in a single step.5 As a consequence of this asymmetry, a voltage hysteresis of 0.2-0.3 V is usually observed in the delithiation profiles. This results in higher upper cutoff voltage even at very low current rates.23 While these features are observed in metallic Sb, we expect these to hold true for the profiles noted for the Sb2S3 anode as well because the end product from the conversion reaction, i.e., Li2S, is inactive in this voltage range.
The delithiation capacity observed during the alloying reaction is 451.1 mA h g−1 for Sb2S3 and 457.4 mA h g−1 for Sb1.9Al0.1S3. This is less than the theoretical capacity (473.4 mA h g−1) of the alloying reaction suggesting irreversible losses, with some of these losses attributable to the formation of the SEI layer during the first lithiation.69 The coulombic efficiency of alloying storage during the first cycle is 78.4% for Sb2S3 and 78% for Sb1.9Al0.1S3. Upon further delithiation, another sloping profile is observed attributable to the delithiation of Li2S and the reformation of Sb2S3. This occurs between 1.2 and 2.5 V (vs. Li/Li+) and is not as flat as the lithiation profiles, suggesting serious polarization losses even at low current rates.39 The observed delithiation capacities are 447.5 mA h g−1 for Sb2S3 and 482.9 mA h g−1 for Sb1.9Al0.1S3. Coulombic efficiency of conversion storage during the first cycle is 69.3 and 70.4% for Sb2S3 and Sb1.9Al0.1S3, respectively. It should be noted that the coulombic efficiency is larger for alloying storage than for conversion storage. Also, polarization losses are lesser (curves are flatter) for alloying storage than for conversion storage.
During the second lithiation, two flat profiles like the ones observed in the first lithiation are present in both the anodes. However, one interesting feature is that the contribution from conversion storage during the second lithiation is much lesser than in the first (Fig. 3a and b). Also, the decrease is primarily observed in the flat potential region, suggesting that material unavailability following the changes during the first cycle is the likely cause. Since the lithiation profiles are similar, the second lithiation can be assumed to follow the exact reaction mechanism of the first, albeit with lesser active mass available for interaction. The conversion lithiation capacity during the second cycle (i.e., capacity between 2.5 and 1 V) is 399.9 mA h g−1 for Sb2S3 and 438.8 mA h g−1 for Sb1.9Al0.1S3. The ratio of second cycle lithiation capacity to the first lithiation capacity in the conversion regime is similar for both anodes, ∼63%, suggesting that it might be a property intrinsic to the conversion reaction in Sb2S3. The second lithiation capacity of the alloying is 524.2 mA h g−1 for Sb2S3 and 530.9 mA h g−1 for Sb1.9Al0.1S3, with the second lithiation capacity ∼91% of the first lithiation capacity (in the alloying regime). This suggests that alloying storage has less capacity loss and has better lithiation capacity in the second cycle compared to conversion storage.
During the second delithiation, between 10 mV and 1.2 V, the capacity of Sb2S3 is 461.7 mA h g−1, and the capacity of Sb1.9Al0.1S3 is 470.9 mA h g−1. Between 1.2 and 2.5 V, 431.4 mA h g−1 was observed for Sb2S3, and 468.4 mA h g−1 was observed for Sb1.9Al0.1S3. It should be noted that if conversion and alloying regimes are defined by the cutoff voltages, the delithiation capacity of the conversion regime is more than the lithiation capacity. This indicates that a clear distinction between conversion and alloying storage in terms of cutoff voltage is not always possible, very likely because of the shift in the potential caused by polarization losses. In a further analysis where individual contributions due to the conversion and alloying are estimated, the change in profile shape is used to identify the contributions. The third cycle profile closely overlaps with the second, indicating that side reactions are minimal after the first lithiation cycle.
The cells were cycled at different current rates to test the charge storage properties as a function of the current rate, and the representative profiles of each charge–discharge cycle are shown in Fig. 3c and d. The capacity vs. cycle number plots of these profiles are shown in Fig. 3e. Sb2S3 has a delithiation capacity of 37.4 mA h g−1 at 20C, whereas Sb1.9Al0.1S3 has a delithiation capacity of 292.1 mA h g−1 at 20C. Also, at a high current rate of 40C, Sb1.9Al0.1S3 has a capacity of 57 mA h g−1, indicating superior electrochemical performance.
When cycled at a low current rate after the high current studies, the Sb2S3 anode exhibited only a fraction of its initial capacity (Fig. 3e). Such capacity loss is attributed to either irreversible reactions, material unavailability, or both. Since initial cycling at 0.05C has a larger capacity, the irreversible reaction during a high current rate is the likely cause of the capacity decrease. Sb1.9Al0.1S3 anode, on the contrary, regains most of the initial capacity, albeit it exhibits capacity degradation over a few cycles, very likely attributable to increased polarization and side reactions that occurred during high current cycling. The same cells were cycled at a 5C rate (4.73 A g−1) for 500 cycles to test the cycling stability. The capacity vs. cycle number plots of these studies are shown in Fig. 3f. Sb2S3 has an initial capacity of 48.1 mA h g−1 and a final capacity of 31.7 mA h g−1, with a capacity retention of 65.9%. Sb1.9Al0.1S3 has an initial capacity of 320.0 mA h g−1 and a final capacity of 71.4 mA h g−1. It should be noted that even though the capacity retention of Sb1.9Al0.1S3 is less than the capacity retention observed in the Sb2S3 anode, the absolute value of capacity at the end of 500 cycles is twice the capacity observed in Sb2S3. Also, in both Sb2S3 and Sb1.9Al0.1S3, a decrease in capacity is observed in the initial few cycles. This is followed by a momentary increase in capacity for a few tens of cycles and later a continuous gradual decrease in capacity. Such features have been observed in electrodes with large irreversible lithium loss during the first cycle.70 The increase in capacity is sometimes referred to as activation and is attributed to the shuttling of lithium trapped in the electrode from the first lithiation. The decrease in capacity is largely attributed to the structural degradation of the system due to accumulated stresses from volume expansion and contraction.70
To elucidate the influence of aluminium substitution on the conversion and alloying of lithium storage mechanisms separately, a fresh set of Sb2S3 and Sb1.9Al0.1S3 cells were fabricated and were cycled within the conversion (2.5 to 1 V vs. Li/Li+) and alloying (1.2 V to 10 mV vs. Li/Li+) storage regimes. The first two galvanostatic charge–discharge profiles measured with a 0.05C rate of the cells are shown in Fig. S2, ESI.† The cells were cycled at different current rates to understand the charge storage properties as a function of the current rate, and the observed profiles are shown in Fig. 4a–d. Capacity vs. cycle number plots of the measurements are shown in Fig. 4e.
 | ||
| Fig. 4 Charge–discharge profiles of Sb2S3 when cycled in the (a) conversion regime and (b) alloying regime. Charge discharge profiles of Sb1.9Al0.1S3 when cycled in (c) conversion and (d) alloying regimes. (e) Capacity vs. cycle number plot of charge–discharge studies carried out on Sb2S3 and Sb1.9Al0.1S3 at different current rates. (f) Capacity vs. cycle number plot of cycling stability studies carried out at 5C for over 500 cycles. | ||
Capacity values at 0.1C are similar for Sb2S3 and Sb1.9Al0.1S3 anodes in the conversion and alloying regimes (Fig. 4e). Sb2S3 shows a capacity of 366.6 mA h g−1 for the conversion reaction and a capacity of 408.9 mA h g−1 for the alloying reaction. The conversion and alloying reaction capacities for Sb1.9Al0.1S3 are 337.2 mA h g−1 and 425.7 mA h g−1, respectively. At higher current rates, a clear pattern in rate capability is observed. The rate-capability performance of the Sb1.9Al0.1S3 anode is better than the Sb2S3 anode in the order, Sb1.9Al0.1S3-alloy > Sb1.9Al0.1S3-conversion > Sb2S3-alloy > Sb2S3-conversion. It is interesting to note that Sb2S3-conversion and Sb1.9Al0.1S3-conversion showed an increase in capacity when cycled between 1 and 15C, with the increase clearly appearing in the case of Sb1.9Al0.1S3. This is similar to the activation-like feature observed in the cycling stability studies when a cell is cycled in both conversion and alloying regimes (Fig. 3e and f). This indicates that the lithium storage involving the conversion mechanism of the Sb2S3 has activation-like features and not the alloy storage, possibly due to the larger lithium loss observed during conversion storage (Fig. 3a and S2a and c, ESI†). During the conversion reaction, Sb2S3 decomposes resulting in the formation of a host of lithium-sulfur compounds (polysulfides) with Li2S formation at the end of conversion. It is quite probable that some of these reactions are incomplete and facilitate lithium shuttling across the anode resulting in the activation.71
At a current rate of 10C (9.4 A g−1), the capacity values of Sb2S3 for the conversion and alloying reactions are 28.1 mA h g−1 and 62.8 mA h g−1, respectively. In contrast, the capacity values are four to five times higher for the Sb1.9Al0.1S3 anode, i.e. conversion and alloying capacities are 150.3 mA h g−1 and 231.6 mA h g−1, respectively, for Sb1.9Al0.1S3 anode. At further higher current rates, say 40C (37.6 A g−1), Sb1.9Al0.1S3 only shows measurable capacity. Conversion and alloying reactions retain close to one-tenth of the initial capacity, i.e. 32.6 mA h g−1 and 49.3 mA h g−1. Such high capacity retention as the current increased from 0.1C to 40C indicates the excellent rate capability of the Sb1.9Al0.1S3 anode.
It should be noted that the alloying capacity of Sb2S3 is less compared to that of Sb1.9Al0.1S3. In electrode materials, the microstructure plays an important role in deciding the electrochemical performance. Post-electrochemical scanning electron microscopy studies (discussed in the following section) show that Sb1.9Al0.1S3 retains the nanorod morphology whereas Sb2S3 nanorods disintegrate upon lithiation. The retention of nanorod morphology enhances the easy lithium diffusion in the system facilitating large capacity at high current rates.
A fresh set of coin cells was fabricated to understand the cycling stability aspects, and after a few cycles at 0.1C-rate, the cells were cycled at 5C rate for 1000 cycles. The capacity vs. cycle number plots of the same are shown in Fig. 4f. It should be noted that the conversion reactions in both Sb2S3 and Sb1.9Al0.1S3 anodes showed an initial increase for a few tens of cycles followed by a decrease in capacity. On the contrary, the alloying storage reactions showed an almost continuous decrease in capacity. In the case of Sb1.9Al0.1S3 alloying reactions, the initial capacity is 383.5 mA h g−1, and the final capacity after 1000 cycles at 5C-rate is 239.8 mA h g−1. This gives capacity retention of 63%. However cycling stability corresponding to the conversion reaction of Sb2S3 and Sb1.9Al0.1S3 has a capacity retention of only 12–13%. Similarly, the alloying reaction of Sb2S3 has capacity retention of only 18%. These studies suggest the importance of Al doping and limiting the alloying regime to harness the best high-rate capacity and cyclic stability.
To better understand the electrochemical performance in electrodes cycled in both alloying and conversion regimes, individual contributions from conversion and alloying storage are estimated and are plotted as a function of the current rate in Fig. 5a. Even though the theoretical contributions of each storage mechanism to the total capacity are the same, it can be noted that alloying, in general, contributes more to the capacity as the current rate increases. For example, the contribution from alloying storage is 64.4 mA h g−1 in Sb2S3 at 10C-rate, whereas the contribution from the conversion reaction is only 29.9 mA h g−1. This contrast is well observed in the Sb1.9Al0.1S3 anode. In the Sb1.9Al0.1S3 anode, alloying and conversion contributed 458.4 mA h g−1 and 459.9 mA h g−1, respectively, to total capacity (at 0.1C-rate). But at all higher current rates, alloying reactions contributed a higher fraction to the total capacity. For example, at 20C-rate, alloying contributed 230.1 mA h g−1 (∼85% of total capacity), whereas conversion contributed only 41.3 mA h g−1 (∼15% of total capacity). Capacity vs. current rate plots of the cells cycled in conversion or alloying separately are shown in Fig. 5b. The capacity values of Sb2S3 (conversion) and Sb2S3 (alloying) show a steep decrease with the current rate. In the case of Sb1.9Al0.1S3, a steep drop is noted initially between 0.1C and 2C rate, after which there is an almost gradual linear decrease in capacity values. The decrease is estimated to be ∼8 (∼5) mA h g−1 per C-rate for the alloying (conversion) reaction. These studies unequivocally establish that a small fraction of Al in Sb2S3 causes beneficial changes to the electrochemical performance.
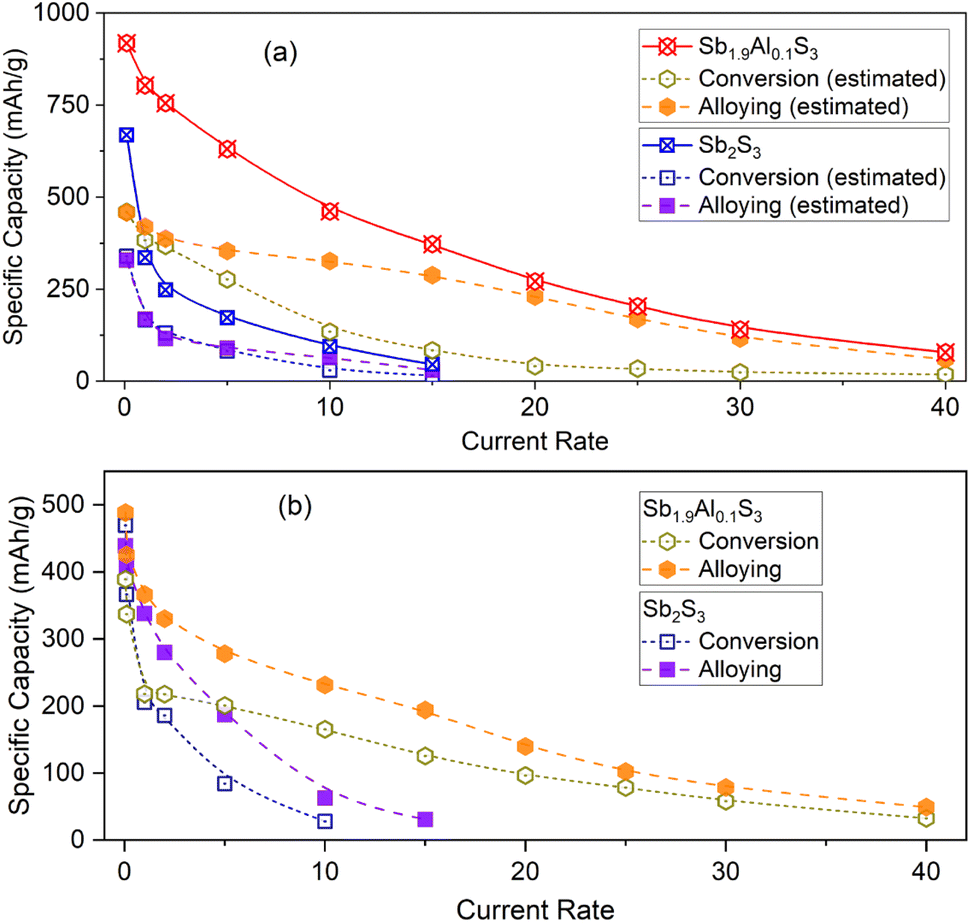 | ||
| Fig. 5 (a) Estimated individual contributions of conversion (open symbols) and alloying (closed symbols) storage mechanisms to total delithiation capacity (open symbols with a cross) of Sb2S3 and Sb1.9Al0.1S3 anodes. (b) The capacity of conversion and alloying storage systems in Sb2S3 and Sb1.9Al0.1S3 anodes plotted as a function of the current rate. | ||
It is noteworthy that the electrolyte's composition influences the anode's cycling stability significantly. Cells fabricated with the best performing Sb1.9Al0.1S3-alloy anode using commercially used 1 M LiPF6 in ethylene carbonate, dimethyl carbonate, and ethyl methyl carbonate (1:1:1 vol.%) electrolyte show a final capacity of 33.3 mA h g−1 with a capacity retention of 19% (Fig. S3, ESI†). These results indicate that the electrolyte composition also plays an important role in deciding the electrochemical performance of the system.
In alloy anodes, where a large volume expansion (>100%) occurs, the SEI layer formed during the first lithiation is not stable. During the delithiation, due to large volume change, the surface of the SEI layer cracks up exposing a new anode surface to the electrolyte. Fluoroethylene carbonate (FEC) decomposes at ∼1 V vs. Li/Li+, forming a lithium-ion conducting cross-linked polymer as a part of the solid electrolyte interphase.69 During delithiation, lithium from this polymer is extracted, and the thickness of the SEI layer decreases. Because of the changing thickness of the SEI layer during lithiation and delithiation, these are also called breathing SEI layers.72 FEC acts as a sacrificial component in the electrolyte reacting with the fresh layers and forming a new SEI layer. However, as the cycling continues, the amount of FEC in the electrolyte reduces and when FEC is exhausted, side reactions occur adversely affecting the battery performance. As long as FEC is present in the system, the SEI layer can reform and prevent the side reactions with freshly exposed anode surface, making it suitable for alloy systems that otherwise undergo a huge volume expansion and pulverization upon lithiation. Commercial electrolytes without such an advantage showed larger capacity loss and capacity fading.
Cyclic voltammetry and electrochemical impedance spectroscopy (Fig. S4–S6, ESI†) studies indicate that alloying in Sb1.9Al0.1S3 has much better rate capability and cycling stability than the charge storage properties of Sb2S3.
Post-electrochemical studies
Post-electrochemical structural and microstructural studies were carried out on Sb2S3 and Sb1.9Al0.1S3 anode foils in the lithiated state to gain further insight into the influence of Al on the structural and microstructural properties.Powder X-ray diffraction of the anodes, recovered from the cells at the end of conversion, alloying and combined storage mechanisms in the lithiated state are shown in Fig. S7, ESI.† These anodes correspond to cells which have undergone several cycles of charging-discharging at different C-rates. For comparison, the X-ray diffraction pattern of pristine foils is also included. In cycled Sb2S3 anodes, the XRD patterns show only an amorphous phase after the cycling. In the Sb1.9Al0.1S3 anode, amorphous phases are observed after cycling in conversion and both regimes. However, in the case of the alloy anode, a sharp peak is noticed. The peak position matches the strongest peak observed in metallic Sb.73 This indicates the presence of crystalline Sb after cycling. It should be noted that after lithiation in the alloying regime, the expected composition is Li3Sb. The presence of metallic Sb indicates that the signal is from the Sb nanoparticles, which have not participated in the electrochemical reaction, as can be understood from the reduced capacity at the end of cycling. In the literature, the formation of amorphous antimony after the delithiation of Sb is reported.74 This corroborates well with XRD studies from the Sb2S3-alloying anode. However, the presence of Al in Sb2S3 results in the formation of crystalline Sb or AlSb nanoparticles after lithiation.
Raman spectroscopic studies also indicate the presence of metallic Sb (145 cm−1 and 105 cm−1) in all the anodes (Fig. S8, ESI†).75 This suggests that the Sb is in the amorphous phase except in the case of the Sb1.9Al0.1S3-alloying anode. This corroborates with in situ high-resolution transmission electron microscopy observations made when alkali metal ions were intercalated into Sb2S3.76 In the report, electron diffraction patterns attributable to the formation of amorphous Sb were observed.76
Scanning electron microscopy studies of the pristine and anodes cycled in both regimes and independently in the conversion and alloying are shown in Fig. 6. The electrode films of pristine Sb2S3 and Sb1.9Al0.1S3 are flat and continuous without any cracks on the surface (Fig. 6a and e). The insets in Fig. 6a and e show high-resolution images of the Sb2S3 and Sb1.9Al0.1S3 showing the nanorod morphology after the electrode film fabrication. Fig. 6b shows the Sb2S3 anode after cycling in both conversion and alloying regimes. Cracks on the surface of the film are evident, and the microstructure is mostly pulverized. It should be noted that a few swollen or expanded rod-like features are present. These are very likely the Sb2S3 nanorods which have not participated in the electrochemical storage after a couple of cycles. Fig. 6f shows Sb1.9Al0.1S3 after cycling in both conversion and alloying regimes. The micro-cracks are also present on the surface, and the image in the inset confirms the loss of nanorod morphology and the formation of nanoparticles due to pulverization, even in this case.
 | ||
| Fig. 6 Scanning electron micrographs of pristine (a) Sb2S3 and (e) Sb1.9Al0.1S3 anodes coated on Cu foil. Insets in respective panels show the nanorod morphology of the anodes. Scanning electron micrographs of (b) Sb2S3 and (f) Sb1.9Al0.1S3 anodes after cycling in conversion and alloying regimes for a large number of cycles. The surface of electrode films shows cracks from volume expansion. Insets in the respective figures show the absence of nanorod morphology. The nanorods present in the inset of (b) can be attributed to the incomplete reaction of Sb2S3 with lithium. Similar observations are also made from cyclic voltammetry. SEM images of (c) Sb2S3 and (g) Sb1.9Al0.1S3 after cycling in the conversion regime. Inset in (c) shows an absence of nanorod morphology. Inset in (g) shows nanorods which have expanded due to lithiation. SEM images of (d) Sb2S3 and (h) Sb1.9Al0.1S3 show the morphology of the anode after cycling in the alloying regime. (d) Shows two-dimensional lithium dendrites growing on the surface of the electrode in which nanorod morphology is completely lost due to pulverization. Inset in (h) shows Sb1.9Al0.1S3, which retained nanorod morphology, albeit with expanded volume. | ||
When the pristine Sb2S3 anode was cycled only in the conversion regime, the electrode had cracks on the film, and the microstructure (Fig. 6c and inset) was lost. However with the Sb1.9Al0.1S3 lesser number of cracks on the electrode film is observed after the conversion reaction. Interestingly, the nanorod morphology is retained in this electrode, highlighting the role of Al doping in the retention of microstructure. Cycling in the alloying regime of pristine Sb2S3 resulted in the formation of flake-like structures obscuring the microstructure of the electrode coating (Fig. 6d and inset). These are very likely the lithium dendrite growths formed on the surface of the film due to discharge to low voltages. It should be noted that due to the body centred cubic structure of metallic lithium, most of the dendritic structures in lithium have 1D topology with aggressive piercing capability.77 2D plate-like dendritic features have not been reported in lithium-ion batteries, to the best of our knowledge. The formation of 2D hexagonal-like plates has been reported in Zn-based batteries, and the morphology is attributed to the hexagonal closed packing structure of zinc.78 Alloys of Li and Sb are known to exist in the hexagonal crystal structure.79–81 So, it is possible that the dendritic growth has resulted from such an alloy phase, which leads to the 2D morphology of dendrites. Also, lithium dendrites are well known to have a non-uniform distribution on a surface, both in terms of their presence over an area and length. The presence of mostly uniform 2D dendrites over all the anodes suggests that growth from the Li–Sb alloy phase is very likely. The uniform height of the dendrites suggests that the phase is possibly a metastable state which cannot support itself beyond a certain height. Such features are of interest for making better SEI layers.82 Interestingly, Sb1.9Al0.1S3 electrodes do not show such dendritic growth. Despite the cracks on the surface film, the electrode retains its nanorod morphology, albeit with thicker nanorods of size ∼1.2 μm, very likely due to expansion upon lithiation (Fig. 6h). The post-electrochemical structural and microstructural studies, along with the electrochemical performance, strongly suggest that Al substitution in Sb2S3 improves the performance by mainly retaining the nanorod morphology.
It is noteworthy that the nanorod morphology is retained in the Sb1.9Al0.1S3 anode cycled in alloying and conversion regimes separately (Fig. 6g and h), whereas the Sb1.9Al0.1S3 electrode cycled in both regimes underwent pulverization and lost the microstructure (Fig. 6f). During conversion, the electrode volume increases by 1.64 times. During alloying, the volume expands by 1.4 times, resulting in a total volume increase of ∼230%.26 When cycled only in conversion or alloying, the volume expansion is lower, and changes from Al substitution could accommodate the excess volume. On the other hand, the volume expansion from cycling in conversion and alloying is too large to be mitigated by Al substitution.
Post-electrochemical transmission electron microscopy studies carried out on the anodes after cycling in conversion and alloying regimes separately for a large number of cycles are shown in Fig. 7. Bright-field TEM images of the pristine anodes are also shown for comparison (Fig. 7a and f). When pristine Sb2S3 is cycled in the conversion (Fig. 7b and c) or alloying regime (Fig. 7d and e), the microstructure of the nanorods is completely lost and in the HRTEM studies, thickness contrast indicates the formation of amorphous-to-crystalline like nanoparticle structures, very likely Sb nanoparticles. In Sb1.9Al0.1S3, upon conversion (Fig. 7g and h) and alloying cycling (Fig. 7i and j), the nanorod microstructure is predominantly retained. Also, in both cases, thickness contrast indicated the formation of Sb nanoparticles. The high-resolution TEM studies indicate that the particles are mostly crystalline. The lattice fringes are much clearer in the alloy cycled Sb1.9Al0.1S3 anode. The measured d-spacing can be attributed to the Sb metal, indicating that these are crystalline Sb nanoparticles.83 Also these nanoparticles are observed to be present close to one another forming a percolation network in Sb1.9Al0.1S3. The formation of such nanoparticles very likely mitigates the volume expansion while enhancing the electronic conductivity improving the electrochemical performance of Sb1.9Al0.1S3.
 | ||
| Fig. 7 (a) Bright-field TEM image of pristine Sb2S3 nanorods. HRTEM images at different magnifications of (b) and (c) Sb2S3 cycled in the conversion regime; (d) and (e) Sb2S3 cycled in the alloying regime. (f) Bright-field TEM image of pristine Sb1.9Al0.1S3 nanorod. HRTEM images at different magnifications of (g) and (h) Sb1.9Al0.1S3 cycled in the conversion regime; (i) and (j) Sb1.9Al0.1S3 cycled in the alloying regime. All the cycled electrodes are studied in the lithiated state. | ||
A schematic summarizing the influence of Al substitution on the morphology of the Sb2S3 nanorods is shown in Fig. 8. When Sb2S3 nanorods are cycled in the conversion regime, the nanorod morphology is lost, and dendritic structures of lithium form on the electrode surface during cycling in the alloying regime. In the case of Sb1.9Al0.1S3 electrodes, cycling in the conversion or alloying regime preserves the nanorod morphology to a significant extent, albeit with an expansion in the size of the nanorods.
 | ||
| Fig. 8 Schematic depiction of the effect of Al substitution on the microstructure of the Sb2S3 anode during conversion and alloying storage mechanisms. In pristine Sb2S3, the volume expansion due to lithiation completely pulverizes the electrode resulting in a loss of microstructure. Al substitution (Sb1.9Al0.1S3) mitigates the growth of Sb nanoparticles and helps maintain the nanorod microstructure when cycled in alloying or conversion regimes. | ||
Aluminium is known to form alloys with both Li and Sb.84,85 During charge–discharge, it is quite possible for the Sb1.9Al0.1S3 anode to have Li–Al–Sb and Al–Sb phases in the alloys along with Sb–Li and metallic Sb phases.86 It has been observed that alkali metal ion insertion into Sb2S3 results in the formation of amorphous Sb particles.76 The presence of Al in Sb2S3 during such growth results in the formation of crystalline Sb nanoparticles in the alloying regime, as observed from the post-electrochemical XRD studies. The particle size of the Sb nanoparticles as estimated from the post-electrochemical transmission electron microscopy is ∼10 nm. It is surmised that the Sb nanoparticles percolate the Li2S formed at the end of the conversion regime. Since the Li2S is insulating, such a percolating network plays a vital role in improving the electrical conductivity of the electrode.70 Since the conversion electrodes undergo structural degradation, there will not be a host structure through which lithium can diffuse. The lithium mostly diffuses from the electrolyte into the electrode. As such, the electron transfer becomes the bottleneck which can be alleviated by the percolated Sb nanoparticle network.
Performance enhancement is well observed in nanostructured Sb2S3 (shown in Table S5, ESI†). The improvement is mainly attributed to the reduced diffusion lengths and enhanced access to lithium diffusion pathways facilitated by the nanostructured nature. In Sb1.9Al0.1S3 nanorods, the Al substitution preserves the nanostructured nature, viz nanorod morphology, upon lithiation facilitating easy access to diffusing lithium ions. Also, the crystalline Sb nanoparticles formed upon lithiation improve the electronic conductivity enhancing the performance of the electrode at high current rates. In the alloying reaction, the enhancement is much more pronounced, indicating that electronic conductivity is the likely limiting factor.
Many strategies have been adopted in the literature to improve the electrochemical performance of Sb2S3. Microstructural tailoring is one strategy which was well explored. Amorphous Sb2S3, bulk Sb2S3, colloidal Sb2S3etc. were explored as anodes with performance improvement to various degrees. Cation substitution, though frequently adopted for cathodes, is less used in anodes. Here, we show that cation substitution is another promising strategy to achieve significant enhancement in performance. Table S6, ESI,† shows the performance of a few anodes reported in the literature, and the performance of the Sb1.9Al0.1S3 nanorod anode in the alloying regime is better or comparable to the anodes reported in the literature. These results show a promising strategy to mitigate structural degradation and improve the performance of conversion anodes.
Conclusion
This work details the aluminum substitution strategy together with cycling only in the alloying regime to realize high rate capability and cycling stability over 1000 cycles at 5C (4.7 A g−1) in an antimony trisulfide anode. Aluminium substitution (5 at%) does not alter the crystal structure or the morphology of the Sb2S3 nanorods. Aluminium substituted Sb2S3 has a rate capability of 471.9 mA h g−1 at 10C (9.4 A g−1) compared to the 89.2 mA h g−1 at 10C of the pristine Sb2S3. Sb1.9Al0.1S3 also had better capacity after 500 cycles at 5C. Even though the enhancement in the performance is good, significantly better performance can be obtained by cycling the anode only within the alloying regime. Apart from the excellent rate capability (139.8 mA h g−1 at 20C) and cycling stability (63% retention after 1000 cycles at 5C), alloy storage also has a smaller operating voltage making it an attractive alternative anode for next-generation lithium-ion batteries. Aluminium is expected to tune the antimony nanoparticle formation in the system, resulting in the formation of larger crystallites which help improve the mechanical stability of the microstructure. Though further studies are needed to better elucidate the influence of aluminum, these results show that Al substitution is a reliable and facile way to tune the anode properties and improve the electrochemical performance significantly.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
LY and CS conceptualized the study; LY and AKB developed the methodology and performed experimental work under the supervision of CS; AKB wrote the original draft and LY and CS reviewed and edited the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. S. acknowledges the support of the Ministry of Education, India, through the Institutes of Eminence (vide SP20210777DRMHRDDIRIIT) for the research initiatives on establishing the Research Centre for Advanced Materials and Microscopy (vide SB20210844MMMHRD008277). CS acknowledges the Department of Science and Technology-Materials for Energy Storage 2016 supported facilities (vide DST/TMD/MES/2K16/68(G)) for carrying out this work. The authors acknowledge the Central Electron Microscopy Facility, IIT Madras and the FESEM facility at the Department of Physics, IIT Madras.References
- K. Schwab, The Fourth Industrial Revolution: what it means and how to respond, https://www.weforum.org/agenda/2016/01/the-fourth-industrial-revolution-what-it-means-and-how-to-respond/, accessed 2 May 2022 Search PubMed.
- T. Philbeck and N. Davis, J. Int. Aff., 2018, 72, 17–22 Search PubMed.
- X. Li, X. Zhang, K. Chen and S. Feng, in ICIST 2014 - Proceedings of 2014 4th IEEE International Conference on Information Science and Technology, 2014, pp. 242–245 Search PubMed.
- G. Zubi, R. Dufo-López, M. Carvalho and G. Pasaoglu, Renewable Sustainable Energy Rev., 2018, 89, 292–308 CrossRef.
- J. He, Y. Wei, T. Zhai and H. Li, Mater. Chem. Front., 2018, 2, 437–455 RSC.
- R. Borah, F. R. Hughson, J. Johnston and T. Nann, Mater. Today Adv., 2020, 6, 100046 CrossRef.
- J. Lu, Z. Chen, F. Pan, Y. Cui and K. Amine, Electrochem. Energy Rev., 2018, 1, 35–53 CrossRef CAS.
- P. U. Nzereogu, A. D. Omah, F. I. Ezema, E. I. Iwuoha and A. C. Nwanya, Appl. Surf. Sci. Adv., 2022, 9, 100233 CrossRef.
- Y. Dong, S. Yang, Z. Zhang, J.-M. Lee and J. A. Zapien, Nanoscale, 2018, 10, 3159–3165 RSC.
- D. Y. W. Yu, H. E. Hoster and S. K. Batabyal, Sci. Rep., 2014, 4, 4562 CrossRef PubMed.
- Q. Wang, Y. Lai, F. Liu, L. Jiang and M. Jia, Energy Technol., 2019, 7, 1900928 CrossRef CAS.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- W. J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- R. Parize, T. Cossuet, O. Chaix-Pluchery, H. Roussel, E. Appert and V. Consonni, Mater. Des., 2017, 121, 1–10 CrossRef CAS.
- S. Yao, J. Cui, Y. Deng, W. G. Chong, J. Wu, M. Ihsan-Ul-Haq, Y. W. Mai and J. K. Kim, Energy Storage Mater., 2019, 20, 36–45 CrossRef.
- S. Wang, Y. Cheng, H. Xue, W. Liu, Z. Yi, L. Chang and L. Wang, J. Mater. Chem. A, 2021, 9, 7838–7847 RSC.
- P. BAYLISS and W. NOWACKI, Z. fur Krist. - Cryst. Mater., 1972, 135, 308–315 CrossRef CAS.
- R. Kondrotas, C. Chen and J. Tang, Joule, 2018, 2, 857–878 CrossRef CAS.
- K. V. Kravchyk, M. V. Kovalenko and M. I. Bodnarchuk, Sci. Rep., 2020, 10, 2554 CrossRef CAS PubMed.
- C.-M. Park, Y. Hwa, N.-E. Sung and H.-J. Sohn, J. Mater. Chem., 2010, 20, 1097–1102 RSC.
- H. Yang, X. Su and A. Tang, Mater. Res. Bull., 2007, 42, 1357–1363 CrossRef CAS.
- Y. Yang, G. Zheng, S. Misra, J. Nelson, M. F. Toney and Y. Cui, J. Am. Chem. Soc., 2012, 134, 15387–15394 CrossRef CAS PubMed.
- D. Chang, H. Huo, K. E. Johnston, M. Ménétrier, L. Monconduit, C. P. Grey and A. Van Der Ven, J. Mater. Chem. A, 2015, 3, 18928–18943 RSC.
- X. Fang, C. Hua, C. Wu, X. Wang, L. Shen, Q. Kong, J. Wang, Y. Hu, Z. Wang and L. Chen, Chem. - Eur. J., 2013, 19, 5694–5700 CrossRef CAS PubMed.
- L. Wang, Z. Xu, W. Wang and X. Bai, J. Am. Chem. Soc., 2014, 136, 6693–6697 CrossRef CAS PubMed.
- J. Xie, J. Xia, Y. Yuan, L. Liu, Y. Zhang, S. Nie, H. Yan and X. Wang, J. Power Sources, 2019, 435, 226762 CrossRef CAS.
- Y. Qi and S. J. Harris, J. Electrochem. Soc., 2010, 157, A741 CrossRef CAS.
- L. Dashairya, D. Das and P. Saha, J. Alloys Compd., 2021, 883, 160906 CrossRef CAS.
- J. Xie, L. Liu, J. Xia, Y. Zhang, M. Li, Y. Ouyang, S. Nie and X. Wang, Nano-Micro Lett., 2018, 10, 1–12 CrossRef PubMed.
- W. Luo, X. Ao, Z. Li, L. Lv, J. Li, G. Hong, Q. H. Wu and C. Wang, Electrochim. Acta, 2018, 290, 185–192 CrossRef CAS.
- H. Yin, K. S. Hui, X. Zhao, S. Mei, X. Lv, K. N. Hui and J. Chen, ACS Appl. Energy Mater., 2020, 3, 6897–6906 CrossRef CAS.
- J. Ma, X. Duan, J. Lian, T. Kim, P. Peng, X. Liu, Z. Liu, H. Li and W. Zheng, Chem. - Eur. J., 2010, 16, 13210–13217 CrossRef CAS PubMed.
- J. Xia, X. Zhang, Y. Yang, X. Wang and J. Yao, Chem. Eng. J., 2021, 413, 127400 CrossRef CAS.
- H. Ye, Z. Wang, J. Yan, Z. Wang, J. Chen, Q. Dai, Y. Su, B. Guo, H. Li, L. Geng, C. Du, J. Wang, Y. Tang, L. Zhang, L. Zhu and J. Huang, Adv. Funct. Mater., 2022, 32, 2204231 CrossRef CAS.
- W. Ni, X. Li, L.-Y. Shi and J. Ma, Nanoscale, 2022, 14, 9609–9635 RSC.
- Z. Wei, L. Wang, M. Zhuo, W. Ni, H. Wang and J. Ma, J. Mater. Chem. A, 2018, 6, 12185–12214 RSC.
- A. Schmidt, A. Smith and H. Ehrenberg, J. Power Sources, 2019, 425, 27–38 CrossRef CAS.
- M. Salah, C. Hall, P. Murphy, C. Francis, R. Kerr, B. Stoehr, S. Rudd and M. Fabretto, J. Power Sources, 2021, 506, 230194 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- W. Yan, S. Yang, Y. Huang, Y. Yang and G. Yuan, J. Alloys Compd., 2020, 819, 153048 CrossRef CAS.
- Y.-S. Duh, Y. Sun, X. Lin, J. Zheng, M. Wang, Y. Wang, X. Lin, X. Jiang, Z. Zheng, S. Zheng and G. Yu, J. Energy Storage, 2021, 41, 102888 CrossRef.
- J. Li, W. Li, S. Wang, K. Jarvis, J. Yang and A. Manthiram, Chem. Mater., 2018, 30, 3101–3109 CrossRef CAS.
- P. Myagmarsereejid, M. Ingram, M. Batmunkh and Y. L. Zhong, Small, 2021, 17, 2100241 CrossRef CAS PubMed.
- P. Sharvanti, K. Abinaya and N. R. Yogamalar, Opt. Mater., 2020, 109, 110319 CrossRef CAS.
- Y. Guo, Z. Hu, Y. Cui, J. Wang, S. Jin, Z. Peng and H. Ji, Batteries Supercaps, 2022, 5, e202100355 CAS.
- S. Shao, W. Zhu, J. Lv, Y. Wang, Y. Chen and Y. Ma, npj Comput. Mater., 2020, 6, 11 CrossRef CAS.
- H. Senoh, T. Takeuchi, H. Kageyama, H. Sakaebe, M. Yao, K. Nakanishi, T. Ohta, T. Sakai and K. Yasuda, J. Power Sources, 2010, 195, 8327–8330 CrossRef CAS.
- B. Krebs, A. Schiemann and M. Läge, Z. Anorg. Allg. Chem., 1993, 619, 983–988 CrossRef CAS.
- P. C. Donohue, J. Solid State Chem., 1970, 2, 6–8 CrossRef CAS.
- R. Ruffo, C. M. Mari, F. Morazzoni, F. Rosciano and R. Scotti, Ionics, 2007, 13, 287–291 CrossRef CAS.
- R. Qing, M. C. Yang, Y. S. Meng and W. Sigmund, Electrochim. Acta, 2013, 108, 827–832 CrossRef CAS.
- A. K. Budumuru, M. Viji, A. Jena, B. R. K. Nanda and C. Sudakar, J. Power Sources, 2018, 406, 50–62 CrossRef CAS.
- D. Wiedemann, S. Nakhal, J. Rahn, E. Witt, M. M. Islam, S. Zander, P. Heitjans, H. Schmidt, T. Bredow, M. Wilkening and M. Lerch, Chem. Mater., 2016, 28, 915–924 CrossRef CAS.
- H. H. Ryu, G. T. Park, C. S. Yoon and Y. K. Sun, J. Mater. Chem. A, 2019, 7, 18580–18588 RSC.
- Y.-R. Luo, J. Am. Chem. Soc., 2004, 126, 982 CrossRef PubMed.
- V. Sharma, T. K. Das, P. Ilaiyaraja, A. C. Dakshinamurthy and C. Sudakar, Mater. Res. Bull., 2020, 131, 110980 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- V. Sharma, P. Ilaiyaraja, A. C. Dakshinamurthy and C. Sudakar, Mater. Sci. Semicond. Process., 2021, 121, 105330 CrossRef CAS.
- M. Medles, N. Benramdane, A. Bouzidi, K. Sahraoui, R. Miloua, R. Desfeux and C. Mathieu, J. Optoelectron. Adv. Mater., 2014, 16, 726–731 CAS.
- U. Chalapathi, B. Poornaprakash, C.-H. Ahn and S.-H. Park, Mater. Sci. Semicond. Process., 2018, 84, 138–143 CrossRef CAS.
- H. Ning, H. Guo, J. Zhang, X. Wang, X. Jia, J. Qiu, N. Yuan and J. Ding, Sol. Energy Mater. Sol. Cells, 2021, 221, 110816 CrossRef CAS.
- J. Grigas, E. Talik and V. Lazauskas, Phase Transitions, 2002, 75, 323–337 CrossRef CAS.
- V. P. Zakaznova-Herzog, S. L. Harmer, H. W. Nesbitt, G. M. Bancroft, R. Flemming and A. R. Pratt, Surf. Sci., 2006, 600, 348–356 CrossRef CAS.
- D. J. Morgan, Surf. Sci. Spectra, 2017, 24, 024004 CrossRef.
- T. Li, T. Cai, H. Hu, X. Li, D. Wang, Y. Zhang, Y. Cui, L. Zhao, X. Wei and Z.-F. Yan, J. Mater. Chem. A, 2022, 10(20), 10829–10836 RSC.
- J. A. Rotole and P. M. A. Sherwood, Surf. Sci. Spectra, 1998, 5, 4–10 CrossRef CAS.
- B. N. Reddy, M. Deepa and A. G. Joshi, Phys. Chem. Chem. Phys., 2014, 16, 2062–2071 RSC.
- R. K. Sahoo, S. Singh, J. M. Yun, S. H. Kwon and K. H. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 33966–33977 CrossRef CAS PubMed.
- R. Jung, M. Metzger, D. Haering, S. Solchenbach, C. Marino, N. Tsiouvaras, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, A1705–A1716 CrossRef CAS.
- A. K. Budumuru, B. Rakesh and C. Sudakar, Nanoscale, 2019, 11, 8882–8897 RSC.
- S. Li, W. Zhang, J. Zheng, M. Lv, H. Song and L. Du, Adv. Energy Mater., 2021, 11, 2000779 CrossRef CAS.
- G. M. Veith, M. Doucet, R. L. Sacci, B. Vacaliuc, J. K. Baldwin and J. F. Browning, Sci. Rep., 2017, 7, 6326 CrossRef PubMed.
- Y. Mozharivskyj, A. O. Pecharsky, S. Bud’ko and G. J. Miller, Chem. Mater., 2004, 16, 1580–1589 CrossRef CAS.
- N. E. Drewett, I. M. Aldous, J. Zou and L. J. Hardwick, Electrochim. Acta, 2017, 247, 296–305 CrossRef CAS.
- P. Makreski, G. Petruševski, S. Ugarković and G. Jovanovski, Vib. Spectrosc., 2013, 68, 177–182 CrossRef CAS.
- Y. Cheng, Z. Yao, Q. Zhang, J. Chen, W. Ye, S. Zhou, H. Liu and M. S. Wang, Adv. Funct. Mater., 2020, 30, 2005417 CrossRef CAS.
- G. Zheng, C. Wang, A. Pei, J. Lopez, F. Shi, Z. Chen, A. D. Sendek, H.-W. Lee, Z. Lu, H. Schneider, M. M. Safont-Sempere, S. Chu, Z. Bao and Y. Cui, ACS Energy Lett., 2016, 1, 1247–1255 CrossRef CAS.
- Q. Yang, Q. Li, Z. Liu, D. Wang, Y. Guo, X. Li, Y. Tang, H. Li, B. Dong and C. Zhi, Adv. Mater., 2020, 32, 2001854 CrossRef CAS PubMed.
- A. Beutl, D. Cupid and H. Flandorfer, J. Alloys Compd., 2017, 695, 1052–1060 CrossRef CAS.
- W. Müller, Z. Naturforsch., B: J. Chem. Sci., 1977, 32, 357–359 CrossRef.
- G. Brauer and E. Zintl, Z. Phys. Chem., 1937, 37B, 323–352 CrossRef.
- X. Wu, K. Song, X. Zhang, N. Hu, L. Li, W. Li, L. Zhang and H. Zhang, Front. Energy Res., 2019, 7, 65 CrossRef.
- C. S. Barrett, P. Cucka and K. Haefner, Acta Crystallogr., 1963, 16, 451–453 CrossRef CAS.
- N. Kumagai, Y. Kikuchi, K. Tanno, F. Lantelme and M. Chemla, J. Appl. Electrochem., 1992, 22, 728–732 CrossRef CAS.
- W. Weppner and R. A. Huggins, J. Electrochem. Soc., 1977, 124, 1569–1578 CrossRef CAS.
- L. Baggetto, M. Marszewski, J. Górka, M. Jaroniec and G. M. Veith, J. Power Sources, 2013, 243, 699–705 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Energy dispersive X-ray spectroscopy of anodes, electrochemical studies and post-electrochemical studies of anodes. See DOI: https://doi.org/10.1039/d2na00695b |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |