
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ study of Au nanoparticle formation in a mechanochemical-aging-based method†
Austin J.
Richard‡
 a,
Michael
Ferguson‡
ab,
Blaine G.
Fiss
a,
Hatem M.
Titi
a,
Jesus
Valdez
ac,
Nikolas
Provatas
de,
Tomislav
Friščić
*ab and
Audrey
Moores
*af
a,
Michael
Ferguson‡
ab,
Blaine G.
Fiss
a,
Hatem M.
Titi
a,
Jesus
Valdez
ac,
Nikolas
Provatas
de,
Tomislav
Friščić
*ab and
Audrey
Moores
*af
aCentre in Green Chemistry and Catalysis, Department of Chemistry, McGill University, 801 Sherbrooke Street West, Montréal, Québec H3A 0B8, Canada. E-mail: audrey.moores@mcgill.ca
bSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: t.friscic@bham.ac.uk
cFacility for Electron Microscopy Research, McGill University, 3640 University Street, Montréal, Québec H3A0C7, Canada
dDepartment of Physics, McGill University, 3600 University Street, Montréal, Québec H3A 2T8, Canada
eMcGill High Performance Computing Centre, École de Technologie Supérieure (ETS), 1100 Notre Dame Street West, Montréal, Québec H3C 1K3, Canada
fDepartment of Materials Engineering, McGill University, 3610 University Street, Montréal, Québec H3A 0C5, Canada
First published on 3rd April 2023
Abstract
As we strive to perform chemical transformations in a more sustainable fashion, enabling solid-state reactions through mechanochemistry has emerged as a highly successful approach. Due to the wide-ranging applications of gold nanoparticles (AuNPs), mechanochemical strategies have already been employed for their synthesis. However, the underlying processes surrounding gold salt reduction, nucleation and growth of AuNPs in the solid state are yet to be understood. Here, we present a mechanically activated aging synthesis of AuNPs, through a solid-state Turkevich reaction. Solid reactants are only briefly exposed to input of mechanical energy before being aged statically over a period of six weeks at different temperatures. This system offers an excellent opportunity for an in situ analysis of both reduction and nanoparticle formation processes. During the aging period, the reaction was monitored using a combination of X-ray photoelectron spectroscopy, diffuse reflectance spectroscopy, powder X-ray diffraction and transmission electron microscopy, to gain meaningful insights into the mechanisms of solid-state formation of gold nanoparticles. The acquired data allowed for the establishment of the first kinetic model for solid-state nanoparticle formation.
Introduction
Mechanochemistry, defined as chemical reactions activated or driven by mechanical energy, has emerged as a highly successful alternative to conventional solution-based techniques. With benefits including the avoidance of bulk solvent, reduced reaction times, and higher yields,1–3 mechanical activation enables more efficient, green, and sustainable chemical processes.4–6 As such, mechanochemistry has been applied with success to a broad range of materials, including coordination polymers,7 pharmaceutical co-crystals,8 perovskites,9 small molecule organics,10 solid-state electrolytes,11 and nanoparticles.12,13With applications ranging from catalysis and electronics to photodynamic therapies, there is increasing interest in developing sustainable synthesis routes to gold nanoparticles (AuNPs).14–16 In 2009, Debnath et al. reported a pioneering mechanosynthesis of AuNPs by reducing KAuCl4 with NaBH4 in the presence of polyvinylpyrrolidone (PVP), with particle size control in the range of 6–30 nm.17 Previous work from our group showed that ultrasmall AuNPs, with sizes ∼ 1.5–2 nm, are produced by galvanic reduction of Au(III) in the presence of long chain amines.18 The Camargo group then developed a strategy to grow nanotadpoles in solution from such mechanochemically accessed AuNPs.19
The mechanisms of nanoparticle nucleation and growth in solution are well studied, and several different theories have been proposed.20 Examples include the LaMer theory of burst nucleation,21 Ostwald's ripening,22 the Finke–Watzky two-step model of simultaneous nucleation and growth,23 the Brust–Shiffrin method,24 and a four-step mechanism for AuNP synthesis.25 However, the scope of these mechanisms is limited to solution synthesis and cannot be directly applied to processes in the solid state.26,27 Conversely, in the solid state, there is a good understanding of the kinetics of mechanochemical gold reduction, from in-depth in situ studies by powder X-ray diffraction (PXRD)28 and X-ray absorption spectroscopy (XAS).29
Our group has also studied the way in which HAuCl4 NPs, as precursors to Au NPs, evolve and react at the onset of such reactions.30 We have also presented the first theoretical model for AuNP nucleation under mechanochemical conditions, which introduced a ballistic term as a way to parametrise the role of mechanical energy in the formation of amine-stabilised AuNPs.31 Yet, despite such efforts, no study has permitted the tracking of NP formation and transformation in a mechanochemical set-up.
We now provide a direct, time-resolved, multi-scale study of the solid-state growth of AuNPs. This is achieved through a mechanically activated aging process based on the brief introduction of mechanical energy followed by static equilibration. Such mechanochemically activated aging reactions32 have recently been used for the synthesis of metal–organic frameworks,33 organometallic complexes,34,35 organic minerals,36 and polyaniline–clay nanocomposites,37 as well as nanoparticle systems, including Bi2S3 (ref. 38) and Ag39 NPs.
The use of a mechanically activated aging methodology enabled the observation of the solid-state growth process at the nanoparticle level, which is currently not possible using in situ methodologies while continuously milling. This granted us the opportunity to extract invaluable information on the mechanically activated synthesis of amine-stabilised AuNPs using a modified solid-state Turkevich approach40 (Fig. 1), using X-ray photoelectron spectroscopy (XPS), diffuse reflectance spectroscopy (DRS), PXRD, transmission electron microscopy (TEM), and energy dispersive X-ray spectroscopy (EDXS). The judicious choice of the reducing agent and modification of the environment allowed direct monitoring of AuNPs over a period of weeks. The data acquired through variable temperature (VT) PXRD and DRS allowed for the determination of the NP formation activation energy, as well as the establishment of a kinetic model. This work represents the first extended nanoscale monitoring of AuNP growth in the solid state. This work also provides key insights for the understanding of mechanochemical reactivity.
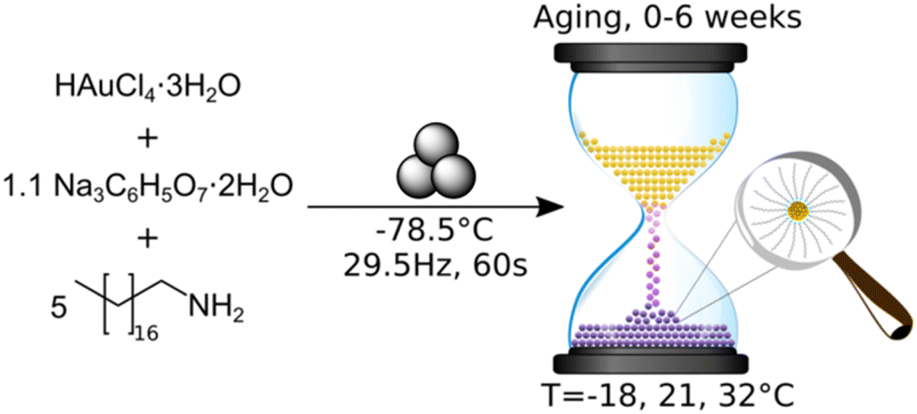 | ||
| Fig. 1 Synthesis of amine-stabilised AuNPs through mechanical activation and subsequent aging. | ||
Results and discussion
In developing the optimal approach to AuNP synthesis by mechanically activated aging, we explored ascorbic acid, hydroquinone, NaBH4, and sodium citrate (NaCt) as reducing agents,28 which were combined with the gold precursor HAuCl4·3H2O and octadecylamine (ODA) as the NP stabilizing ligand.18 The reagents were milled for 90 minutes, aged for 1 day at room temperature (21 °C), and analysed by TEM. The best performing reducing agent was NaCt, providing the ability to reliably produce AuNPs after milling for 90 min and aging for one day at room temperature, in a reaction reminiscent of the solution-based Turkevich method.40 To capture the initial stages of the reaction, the milling time was reduced to 60 s to induce the reaction without driving the rapid formation of AuNPs. During aging, the reaction is considered to be thermally driven. To understand the effect of temperature on such a process, freshly milled samples were stored in the dark at three different temperatures: −18 °C (low temperature, LT), 21 °C (room temperature, RT), and 32 °C (elevated temperature ET). The solid reaction mixtures were sampled on days, 0, 1, 4, 7, 15, 22, and 43 after milling, with each reaction performed in triplicate to ensure reproducibility.Color changes of the milled powder provided qualitative information on reaction progress. Specifically, freshly milled powders exhibited a yellow color, which persisted for the LT aging sample (ESI Fig. S1a†). The RT sample exhibited a gradual fading of the initial yellow color, and after seven days it exhibited a red/brown tinge (ESI Fig. S1b†). After two weeks, RT samples were purple in color, which deepened upon further aging. Such purple color is characteristic of the localised surface plasmon resonance (LSPR) exhibited by AuNPs.41 For the ET sample, the yellow color had completely disappeared after one day, and after four days the characteristic purple color of AuNPs was attained (ESI Fig. S1c†). The purple color intensified upon aging, and after two weeks the powder was almost black. This indicates that the reaction was significantly accelerated with increased temperature.
XPS was employed to monitor the reduction of Au(III) to Au(0) by tracking intensity changes in the Au 4f doublets. Raw XPS spectra were deconvoluted into the 4f7/2 and 4f5/2 doublets for Au(III) and Au(0), normalised to the Au(III) 4f7/2 peak at ∼87 eV of freshly milled powder. The Au(I) signals were also considered during deconvolution, but did not appear significant at any point during the reaction. While the standard major-peak binding energy value for bulk Au(III) and Au(0) is 86 and 84 eV respectively, the peak position may vary with the particle size and surroundings of the Au species.42 Knowing that our XPS Au signal is up-shifted between 0.2 and 1 eV from the bulk standards, and knowing from DRS and TEM analyses that AuNPs are present in the milled powder, it may be concluded that the slight shift observed in XPS may be due to the fact that gold is present as nanoparticles. The RT samples showed a consistent, gradual decrease in the Au(III) signal accompanied by a proportional increase in the Au(0) signal (Fig. 2a), suggesting a slow, continuous reaction of HAuCl4·3H2O during the aging process. Little change is observed for the LT sample, whereas the ET sample exhibited a much faster progression of the Au(III) reduction (ESI Fig. S6†).
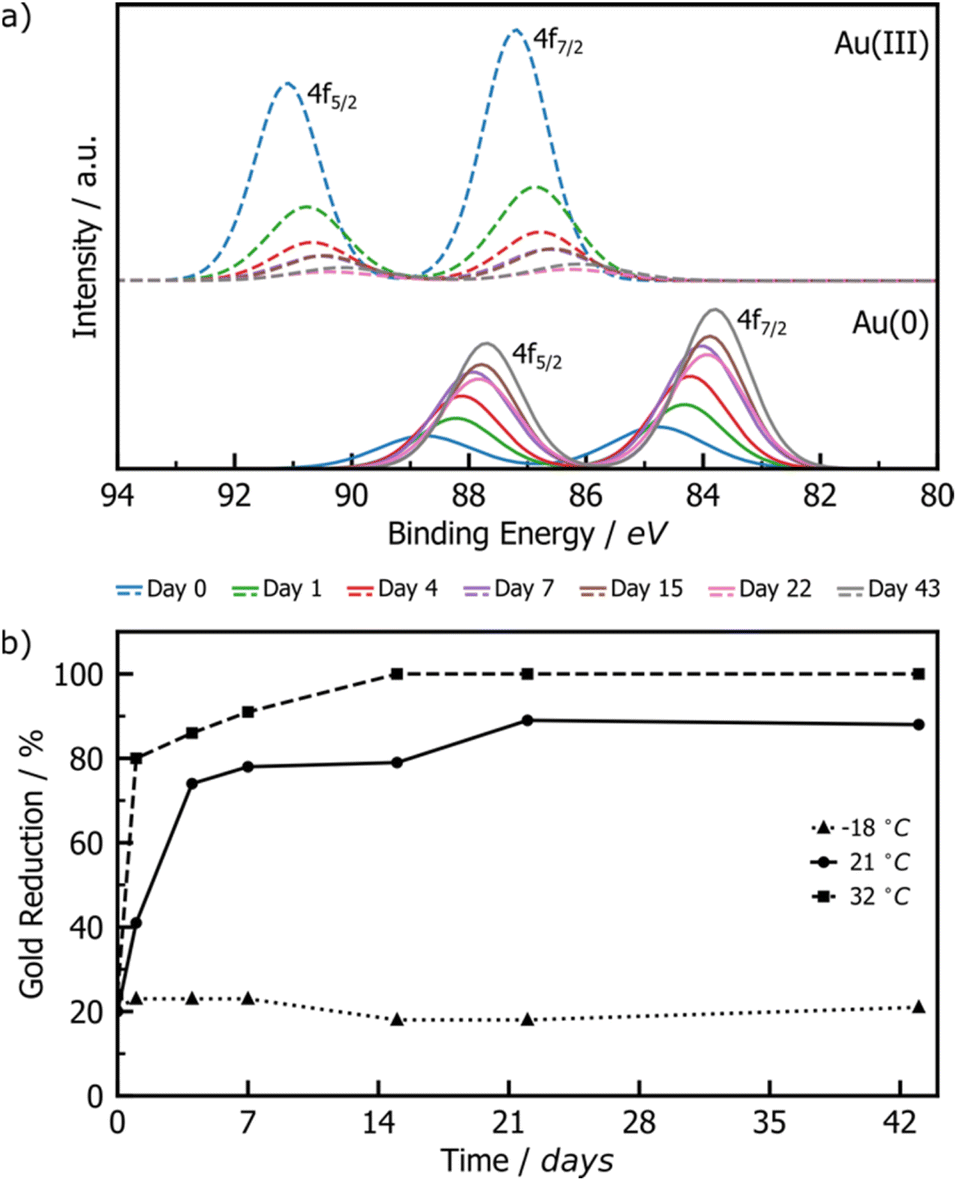 | ||
| Fig. 2 (a) Comparison of deconvoluted XPS data for Au(III) (dashed line) and Au(0) (solid line) 4f doublets of product AuNPs after aging at room temperature for 0 (blue), 1 (green), 4 (red), 7 (violet), 15 (brown), 22 (pink), and 43 (grey) days. Full deconvoluted envelop data are provided in Fig. S4–S6.† (b) Plot of the percentage of Au(III) reduced to Au(0) upon aging at −18 °C (triangles), 21 °C (circles), and 32 °C (squares). | ||
Integration of the normalised doublets gave access to the Au(III)/Au(0) ratio by which the percentage reduction could be quantified (Fig. 2b). For the ET sample, the reduction is initially rapid, reaching completion after 15 days, while the RT sample achieved ca. 90% reduction over the entire explored aging period of 43 days. Consistent with the visual inspection of the powders, little to no reduction occurred in the LT sample. All samples show ca. 20% reduction immediately after milling, which can be attributed to the previously reported photoreduction of Au(III) salts upon XPS analysis43,44 (ESI Section 2b†).
The growth of AuNPs in the bulk powder during aging was followed by DRS and PXRD. Specifically, DRS was used to follow changes in reflectance (converted from % R, ESI Section 1†) in the region 500–600 nm, where AuNPs, of a minimum diameter of 2 nm, exhibit characteristic absorption due to LSPR.45–47 The LT samples did not show significant reflectance in the visible region throughout the aging period (ESI Fig. S8†). Both RT and ET samples featured strong reflectance bands at ca. 550 nm, which appeared during aging (Fig. 3a and b). The normalised reflectance intensity increased gradually for RT samples during the full 43-day aging period, whereas for the ET sample the intensity rose sharply, reaching its maximum on day four. The final position of the reflectance maximum for ET samples (545 nm) was found to be red-shifted by 4 nm compared to RT samples (541 nm), a shift that may be caused by one or multiple different sources including larger average NP sizes, agglomeration, a higher polydispersity, and a changing refractive index.48–50 The ET samples also exhibited a broadening of the reflectance band for up to 15 days and were found by TEM to contain larger and more polydisperse AuNPs (ESI Fig. S22†). While nothing can currently be said on agglomeration or refractive index, they cannot be excluded as potential sources of red-shifting. These results demonstrate that NaCt and ODA are efficient in causing the reduction of Au(III) and NP formation. We ran two blanks at RT to understand the role of each, and found that ODA alone did cause the formation of AuNPs, but more slowly than with NaCt. NaCt alone did not afford any measurable LSPR signal upon 43 days of aging (Fig. S11†).
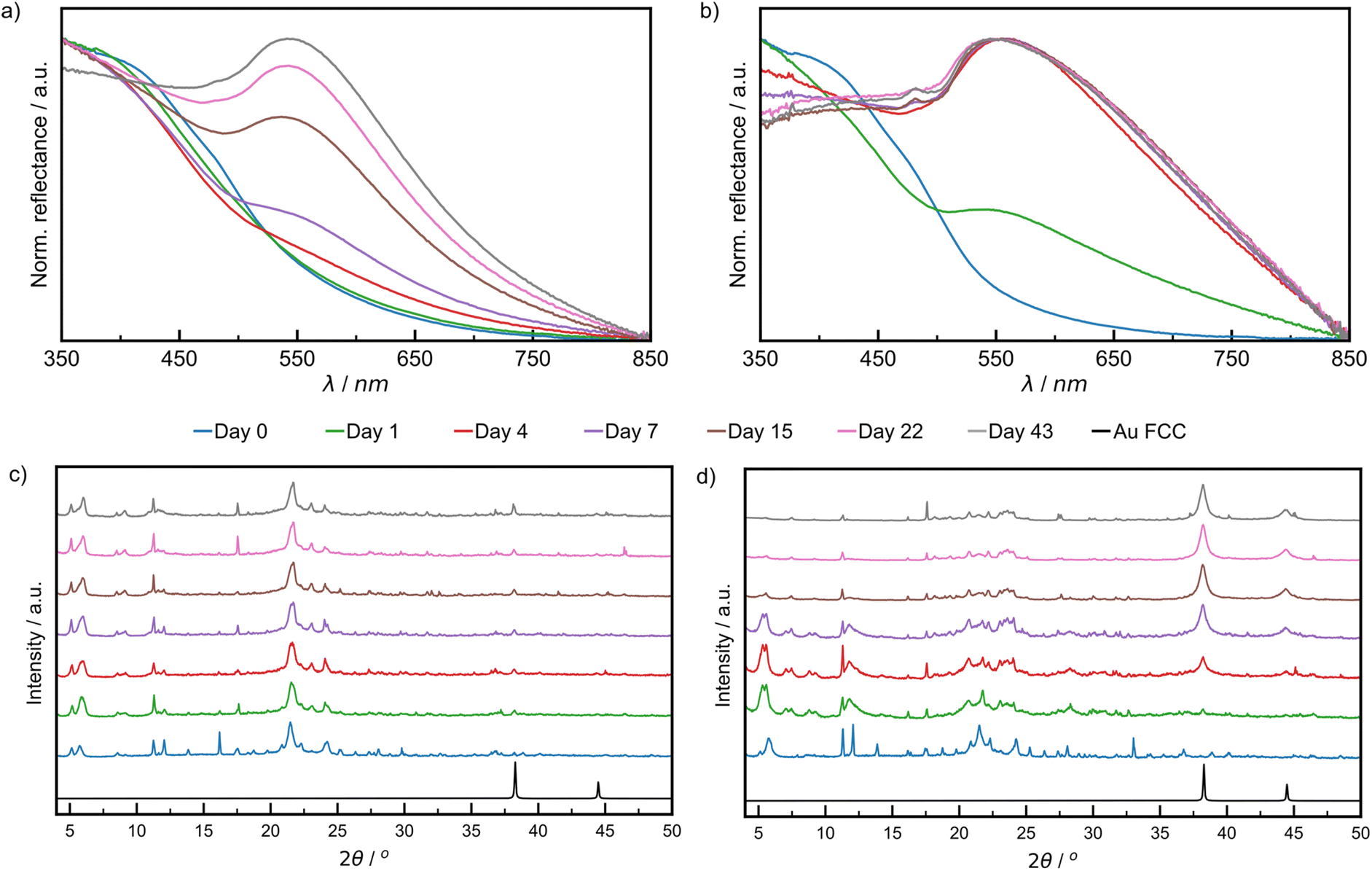 | ||
| Fig. 3 Comparison of DRS data of: (a) RT sample aged at 21 °C, (b) ET sample aged at 32 °C. Comparison of PXRD patterns for: (c) RT sample and (d) ET sample. All samples were analysed after 0 (blue), 1 (green), 4 (red), 7 (violet), 15 (brown), 22 (pink), and 43 (grey) days of aging. The simulated pattern for elemental gold in the FCC lattice in (c) and (d) is shown in black,50 where the reflection at 2θ of 38.2 corresponding to the (111) was used to monitor reaction progress. | ||
Similar trends for AuNP growth were observed by PXRD, following the intensity of the Au(111) X-ray reflection at 2θ = 38.2°.51 This characteristic X-ray reflection was not evident for the LT sample (ESI Fig. S13†) indicating a lack of NP growth. For the RT samples (Fig. 3c), the Au(111) X-ray reflection was observable after one day of aging and increased in intensity during the remaining six weeks of aging (zoomed diffractograms are available in ESI Fig. S12†). The Au(111) reflection was observable after one day for ET samples and continued to increase in intensity substantially for the remainder of the aging period (Fig. 3d). Consistent with visual inspection, XPS, DRS, and PXRD observations indicate a strong temperature dependence of the growth of NPs in a mechanically activated aging process.
Notably, the Au(200) X-ray reflection (2θ = 44.4°) was observable for ET samples, permitting the investigation of particle morphology via the ratio of the intensities of (200) and (111) reflections (I(200)/I(111)).52 It was previously shown that Au metal exhibits an I(200)/I(111) ratio of 0.53, while nanocubes, icosahedrons and tetrahedrons exhibit values of 1.93, 0.31, and 0.25, respectively. The herein calculated I(200)/I(111) ratio was 0.33, suggesting predominantly spherical AuNPs.52 This morphology was directly observed under a high resolution TEM in fully aged samples (ESI Fig. S17†). Therefore, the broadening of reflectance bands observed in DRS data is more likely due to polydisperse size distributions.
We then turned to TEM analyses to probe the effect of aging temperature on the size and morphology of AuNPs to be investigated. We first selected the solution drop-casting method to prepare the TEM samples, because it is a common, simple methodology that often leads to clean images through efficient particle spreading. Specifically, samples produced by solid phase aging were dispersed in toluene and the resulting suspension was rapidly drop-cast onto the TEM grid. Toluene was chosen because neither of our starting materials were soluble in this solvent, yet we needed to test if this sample preparation method led, by itself, to the formation of Au(0) NPs (for more details, see ESI Discussion S1†). To our surprise it did, as NPs of around 4 nm diameter were observed. The samples were identified as Au(0) by using scanning transmission electron microscopy coupled with a high-angle annular dark field detector (STEM-HAADF). As this blank test was featuring Au(0) NPs, the particle sizing and analyses were contextualised with the trends observed in the solid-state analyses, namely by XPS, DRS, and PXRD, and we used the latter methods as the sole reliable sources of information on the redox state of our samples (Fig. 2 and 3).
For freshly milled and one-day aged samples, TEM images (Fig. 4a and b) revealed two populations of NPs: a small number of large non-spherical particles whose diameter is approximately 20 nm (ESI Fig. S20†), as well as smaller spherical NPs (Ø = 7.0 ± 1.9 nm for freshly milled, and 8.2 ± 2.6 nm for particles aged over one day). STEM-HAADF imaging coupled with EDXS elemental mapping confirmed that particles of Ø ≥ 15 nm featured overlapping Au and Cl signals that correlate with the particles observed in the dark field image (ESI Fig. S21†). Consequently, these Ø ≥ 15 nm NPs are likely composed of HAuCl4 precursors, and their absence in blank tests is consistent with particle comminution taking place during mechanochemical activation. This effect and the way precursor NPs evolved over time were addressed in depth previously.30 The smaller NPs, made of Au(0), were very similar to the blank test NPs and because solid state techniques (especially PXRD and DRS) did not indicate the presence of Au(0) NPs, we attributed their presence to this sampling artefact.
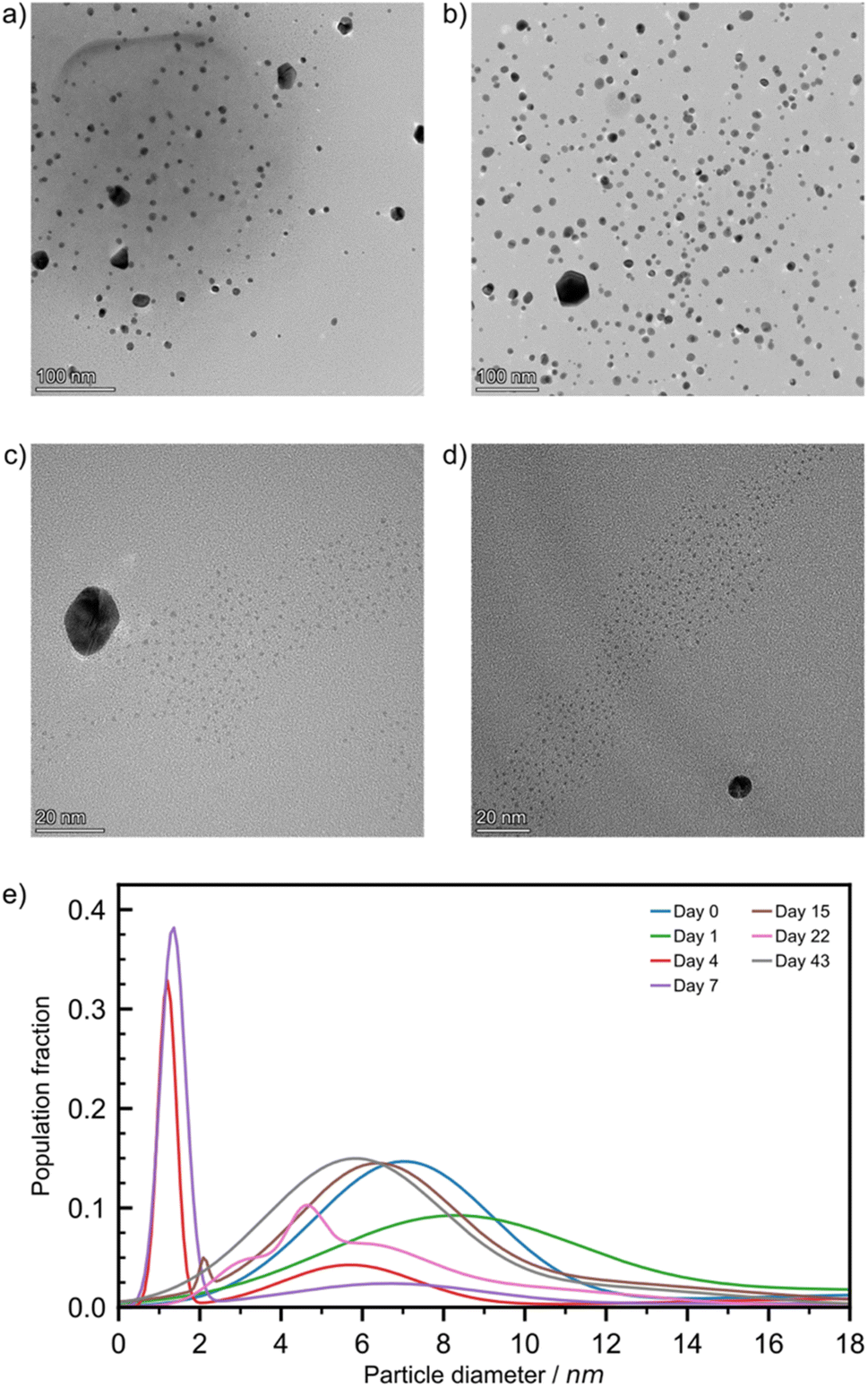 | ||
| Fig. 4 Bright field TEM images from solution drop-cast preparation of the RT sample when aged for: (a) 0, (b) 1, (c) 4, and (d) 7 days at magnifications of ×120k, ×94k, ×500k, and ×390k, respectively. (e) The AuNP size population distributions for the RT sample after aging for 0 (blue), 1 (green), 4 (red), 7 (violet), 15 (brown), 22 (pink), and 43 (grey) days. | ||
Looking at four-day aged samples, ultrasmall AuNPs, Ø = 1.2 ± 0.2 nm, were observed by TEM for the first time (Fig. 4c). Such NPs are absent from any blank tests. PXRD analysis revealed a small reflection for metallic gold after just one day of aging. Therefore, we believe that ultrasmall AuNPs are present in the sample after one day of aging but in such low concentration that we could not spot them reliably by TEM. Between one and four days of aging the amount of Au(0) in the sample increases by approximately 35% (Fig. 2b). This causes the formation of ultrasmall AuNPs in sufficient quantity to be encountered during TEM studies. After one week of aging, the ultrasmall NPs are still visible, exhibiting a slightly larger diameter, and account for a larger fraction of the overall population (Fig. 4d and e). The representative TEM images for RT samples (Fig. 4) taken during the first week of aging confirm the AuNP icosahedral geometry suggested by the I(200)/I(111) ratio calculated from the PXRD data. After two weeks of aging, the ultrasmall NP population was greatly reduced while accompanied by an increase in particle diameter to Ø = 2.1 ± 0.1 nm. At the same time, the population with Ø ≈ 20 nm disappeared, which is tentatively ascribed to the consumption of HAuCl4. After 22 days of aging RT samples showed a multimodal distribution of AuNPs with Ø ≤ 6.5 nm. Previously observed ultrasmall particles had grown since their first appearance in a non-uniform fashion. Finally, after six weeks the AuNPs were observed in a bimodal distribution with Ø = 5.8 ± 1.9 nm and 11.9 ± 2.6 nm.
For ET samples, TEM analysis confirms the acceleration of the reaction suggested by XPS, DRS and PXRD data. Particles after one day of aging were slightly smaller, Ø of 4.3 ± 0.9 nm, than those visible immediately after milling, Ø of 6.8 ± 1.8 nm (ESI Table S3†). We believe that these particles are the result of ultrasmall AuNPs that had formed and already grown between sampling times. Additionally, the secondary distribution at Ø ≈ 20 nm, attributed to the HAuCl4 precursor disappeared, consistent with the reduction indicated by XPS (Fig. 2b). Throughout the remainder of the aging period the particles continued to grow with multi-modal distributions. After six weeks a quad modal distribution was observed where the smallest particles had an average Ø of 6.6 ± 0.2 nm while the largest particles had a Ø of 20.3 ± 7.5 nm. Therefore, the broadening of the reflectance band in DRS spectra is caused by the polydispersity of the resulting AuNPs. While aging at elevated temperatures is advantageous for the rate of NP growth, it appears to be offset by a loss of size control. Particle size population profiles exhibited by LT aged samples did not show any significant growth compared to the day 0 starting point (ESI Fig. S21†). It appears that the reaction is effectively paused at these temperatures.
While the drop-casting method was useful for statistical NP size analysis, we showed it could lead to artefact formation. We thus turned to solid-state TEM sampling to confirm that AuNPs were formed in the absence of bulk solvent. Samples were prepared by impregnating a SiOx substrate bearing (30 nm thick) copper grid with freshly milled powder. Impregnation was achieved by dragging the grid through the loose powder, after which the grid was cleaned with pressured air to remove excess material.
The resulting samples were studied in the solid-phase using STEM-HAADF. A freshly milled powder sample featured large particles in the 100 nm range. Fig. 5a shows a representative example, as a large, non-spherical particle of a maximum diameter ≈ 125 nm. Elemental maps of Au and Cl were constructed for this image using EDXS. The overlapping signals for Au and Cl in the EDXS analysis (Fig. 5b and c) revealed it as the HAuCl4 precursor, consistent with previous analyses.
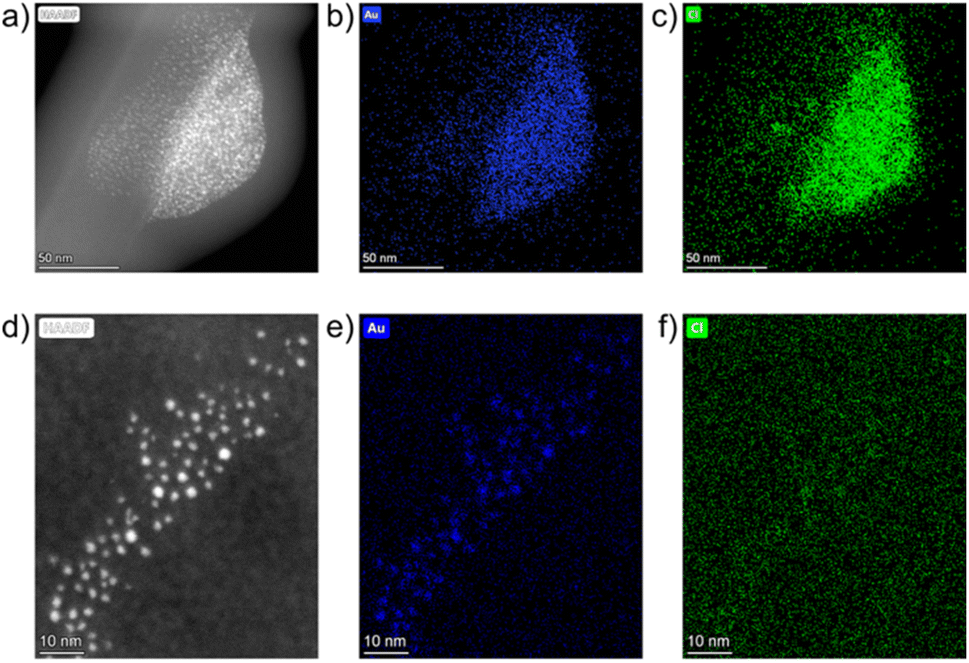 | ||
| Fig. 5 Solid-state STEM-HAADF images and EDXS elemental maps for Au and Cl (left to right, respectively) for: (a–c) freshly milled powder at ×186k magnification and (d–f) for the powder after seven days of aging at room temperature at ×740k magnification. | ||
After one week of aging another grid was prepared and ultrasmall AuNPs were observed in a monodisperse distribution, with a Ø of 1.5 ± 0.4 nm. Elemental mapping showed localised concentrations of Au, consistent with the bright spots in the STEM-HAADF image (Fig. 5d and e). Cl signals (Fig. 5f) were dispersed throughout the region with minimal localised concentrations. This confirms that ultrasmall AuNPs have formed in the solid phase, and not as a result of the drop-casting process. The XPS, DRS, PXRD and TEM studies demonstrate that AuNPs can be synthesised in the solid phase through an aging process and that this reaction is temperature dependent.
The aging process was readily followed in situ, providing access to kinetics of AuNP formation in the solid state. We have plotted the intensity of the 541 nm LSPR peak observed by DRS for samples aged at 21 °C over 43 days (Fig. 6a). At the onset of the reaction, between days 0 and 22, the LSPR signal grew in a linear fashion at a rate of 3.43 × 10−7 s−1. This is comparable to the initial rate of 4.48 × 10−7 s−1 at 24 °C observed during the initial 2.5 hours of the variable temperature (VT) PXRD experiment in Fig. 6c. After day 22, the curve saturated, as the reaction came to completion. This overall sigmoidal curve is reminiscent of solution-based nanoparticle growth mechanisms, whereby autocatalytic effects are at play.53 The curve is also a close match to the sigmoidal curve observed by the James group for mechanochemical reactions, in which feedback between rheological/mechanical effects and chemistry influence reaction kinetics.54 Details on the fitting of the sigmoidal curve can be found in ESI Section 2c.† To deepen our understanding, we focused on the initial stage of the reaction and performed two variable temperature (VT) PXRD studies, focusing on the intensity of the Au(111) X-ray reflection of metallic gold (2θ = 38.2°). First, we acquired 121 PXRD patterns over 60 hours for three separate experiments performed with the thermo-coupled stage held at a constant temperature (30, 35, and 40 °C).
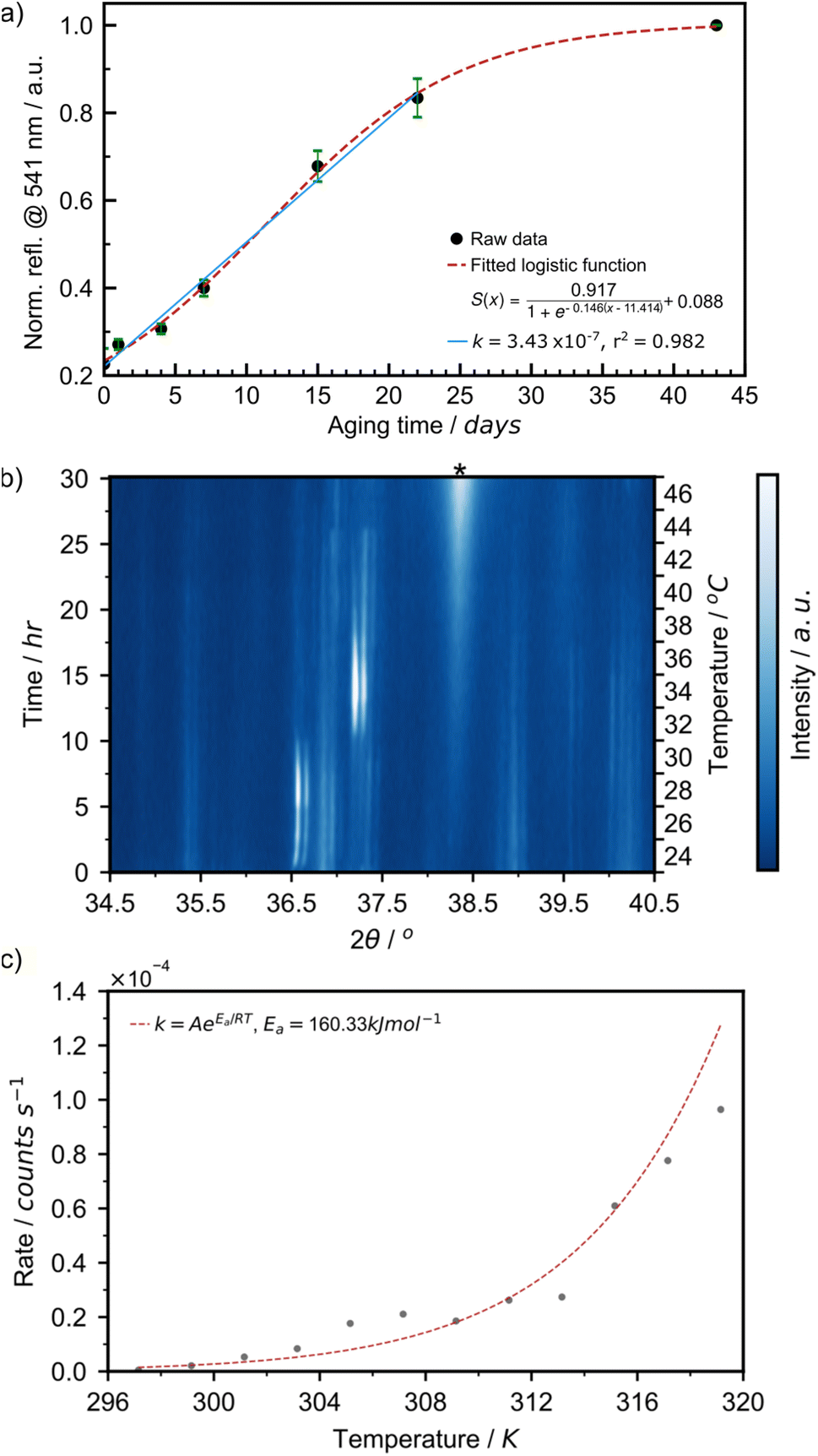 | ||
| Fig. 6 (a) Plot of normalised reflectance resulting from localised surface plasmon resonance at 541 nm for samples aged at 21 °C (black circles with green standard error bars) with a fitted logistic function (red dashed lines). Inset shows the linear fitting (solid blue line) of data points between 0 and 22 days of aging functions to obtain an initial growth rate. (b) In situ variable temperature PXRD analysis of AuNP formation between 24 and 46 °C where * denotes the reflection corresponding to the (111) lattice plane of elemental gold. (c) Plot of the rate of increase in intensity of the reflection at 2θ = 38.2° for each temperature (grey circles) with the model Arrhenius plot with parameters taken from the resolved data. | ||
For each temperature, the intensity of the 38.2° reflection grew linearly, as suggested in the DRS experiment, and the rate of AuNP growth increased with temperature (ESI Fig. S24a†). The activation energy for AuNP growth in the presented mechanically activated aging process was found to be 158.4 kJ mol−1.
A second VT-PXRD study was performed to confirm the exponential relationship between AuNP growth rate and aging temperature. Therein, the thermo-coupled stage was initially set to 24 °C and increased by 2 °C every 2.5 hours during a period of 30 hours with five measurements performed at each temperature. The intensity of the Au(111) X-ray reflection, indicated by ‘*’, initially increased gradually and proceeded to intensify more rapidly with an increase in temperature (Fig. 6a). The intensity of the X-ray reflection was measured using five scans at each temperature and, thus, 12 rates were obtained. In this case, the activation energy for amine-stabilised AuNP growth in the solid state was found to be equal to 160.3 kJ mol−1 (ESI Fig. S25†). The solved Arrhenius equation was in good agreement with the 12 individually obtained growth rates (Fig. 6b). This confirmed the exponential increase in AuNP growth rate with aging temperature. Further details on the VT-PXRD studies performed can be found in the ESI (Section 2f).† Given the low discrepancy between the two studies (1.2%) we calculated the average activation energy of AuNP growth in a solid-state aging process to be 159.4 kJ mol−1. Such activation energies are fairly high and explain that the reaction under these conditions happens over a few weeks. This study confirms that AuNPs can be grown under aging conditions and that the kinetics of the reaction follow a sigmoidal pattern. Initial reaction rates can be measured and feature an exponential relationship to temperature.
Conclusion
Herein, we have presented the mechanically activated aging synthesis of AuNPs using a Turkevich method that was modified for the solid state. Fig. 7 provides a schematic representation of the various reaction steps of this reaction. Using octadecylamine as the stabilizing agent, HAuCl4·3H2O and trisodium citrate dihydrate were milled together for 60 s at −78.5 °C using a vibrational ball mill to initiate the reaction. After brief milling, the resulting powders were aged in the dark at −18, 21, and 32 °C for a period of six weeks, during which the powders were frequently analysed using XPS, DRS, PXRD, and TEM. Aging at low temperature (−18 °C) effectively froze the reaction, with no observed changes for the duration of the aging period. Strong reflectance of visible light and the appearance of metallic gold lattice plane reflections confirmed that AuNPs were produced in samples aged at room temperature (21 °C) and elevated temperature (32 °C). At room temperature, the process was gradual and occurred throughout the whole aging period whereas at elevated temperature the process was significantly accelerated. This was confirmed by TEM images of the samples with grids made using the drop-casting technique. Ultrasmall AuNPs were found after four days of aging at room temperature while at elevated temperatures AuNPs were observed after just one day of aging. The production of AuNPs in the absence of suspension in bulk solution was confirmed using a solid-state impregnation preparation of TEM grids that were analyzed using STEM-HAADF coupled with EDXS. Therein, the formation of ultrasmall AuNPs was confirmed after a seven-day aging period. Following the reaction progress by DRS showed a sigmoidal behaviour, reminiscent of the solution-based NP formation models. Diffusion in the solid phase should play a role in this reaction, and future work will focus on this aspect. Multiple in situ temperature-controlled PXRD experiments were performed to investigate the temperature dependence of the reaction. The Arrhenius equation was solved from each of the in situ PXRD aging studies leading to the determination of the activation energy for AuNP formation in a solid-state aging process. With a 1.2% discrepancy between the results, the average activation energy for the process was 159.4 kJ mol−1. Thus, by extending the timeframe of the reaction, the mechanically activated aging approach has demonstrated an ability to provide previously not accessible, macroscopic and nanoscopic details on the course of mechanically activated AuNP synthesis.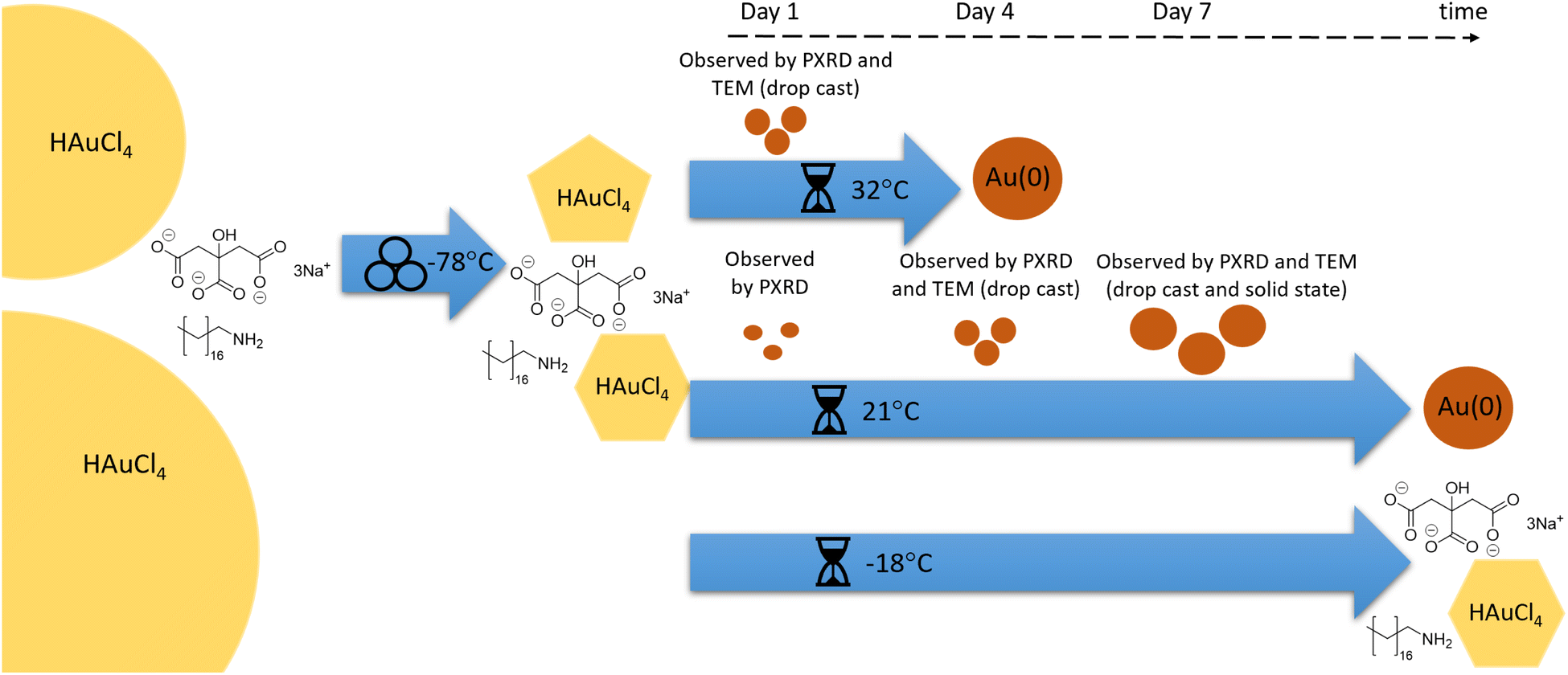 | ||
| Fig. 7 Schematic representation of the synthetic sequence: precursors HAuCl4, NaCt and ODA were milled at −78 °C, leading to HAuCl4 comminution. Aging at −18 °C (LT) led to no reaction; aging at 21 °C (RT) led to gradual reduction and nanoparticle growth, as observed by different techniques; aging at 32 °C (ET) led to a similar outcome in an accelerated sequence. | ||
Experimental
Tetrachloroauric(III) acid trihydrate (ACS reagent, ≥49.0% Au basis), 1-octadecylamine (technical grade, 90%), and trisodium citrate dihydrate (ACS reagent, ≥99.0%) were obtained from Sigma-Aldrich and used without further purification. A 15 ml zirconia milling jar (Form-Tech Scientific), a 10 mm (3.0 g) diameter zirconia milling ball, tetrachloroauric(III) acid trihydrate (0.143 g, 0.363 mmol), trisodium citrate dihydrate (0.118 g, 0.400 mmol), and 1-octadecylamine (0.490 g, 1.816 mmol) were chilled separately over dry ice for 60 minutes to ensure a consistent starting point for each reaction. The milling jar was charged with the milling ball and solid reactants and subsequently set to shake on a Retsch MM400 mixer mill for 60 s at 29.5 Hz. All millings were performed under air. The resulting free-flowing yellow powder was transferred to a sample vial, covered and stored at the desired temperature. Note: given the potential for galvanic reduction, tetrachloroauric(III) acid trihydrate and the post-milling powder were manipulated with a PTFE coated spatula. For room temperature aging, 21 °C, samples were stored inside an opaque closed box in the research lab. Low temperature aging samples were stored in a freezer with an internal temperature of −18 °C. For aging at elevated temperature, the samples were covered and stored in an oven with an internal temperature of 32 °C (oven thermostat set to 45 °C). All reactions were performed in triplicate to ensure reproducibility. See the ESI† for details of XPS, DRS, PXRD, and TEM analysis.Author contributions
Austin Richard: conceptualization, investigation, data curation, visualization – review and editing; Michael Ferguson: conceptualization, investigation, data curation, visualization, writing – original draft, review and editing; Blaine Fiss: conceptualization, investigation; Hatem Titi: investigation, data curation; Jesus Valdez: investigation, data curation; Nikolas Provatas: conceptualization; Tomislav Friščić: conceptualization, supervision, writing – review & editing; Audrey Moores: conceptualization, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Science and Engineering Research Council of Canada (NSERC) Discovery Grant and Discovery Accelerator Supplement, the Canada Foundation for Innovation (CFI), the McGill Sustainability Systems Initiative (MSSI), the Fonds de Recherche du Québec – Nature et Technologies (FRQNT) – Centre for Green Chemistry and Catalysis (CGCC), Walter C. Sumner Memorial Fellowship (B. G. F.), and McGill University for their financial support. We thank the Facility for Electron Microscopy Research of McGill University for help in acquiring electron microscopy data, the MC2 facility at McGill University for help in acquiring PXRD data, the McGill Institute for Advanced Materials and the Mining and Materials Engineering Department at McGill University for the use of their X-ray photoelectron spectroscopy (XPS) instrument and associated Avantage processing software, and Galyna Shul from NanoUQAM at the Université du Québec à Montréal for help in acquiring the diffuse reflectance spectroscopy (DRS) data. T. F. and M. F. gratefully acknowledge the Leverhulme Trust and the University of Birmingham.Notes and references
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC
.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed
- E. Colacino, F. Delogu and T. Hanusa, ACS Sustainable Chem. Eng., 2021, 9, 10662–10663 CrossRef CAS
-
IUPAC Compendium of Chemical Terminology, ed. A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn (The “Gold Book”), 1997 Search PubMed
- L. Takacs, Acta Phys. Pol., A, 2014, 126, 1040–1043 CrossRef CAS
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC
- T. Friščić, I. Halasz, V. Štrukil, M. Eckert-Maksić and R. E. Dinnebier, Croat. Chem. Acta, 2012, 85, 367–378 CrossRef
- M. Solares-Briones, G. Coyote-Dotor, J. C. Páez-Franco, M. R. Zermeño-Ortega, C. M. de la O Contreras, D. Canseco-González, A. Avila-Sorrosa, D. Morales-Morales and J. M. Germán-Acacio, Pharmaceutics, 2021, 13(6), 790 CrossRef CAS PubMed
- F. Palazon, Y. El Ajjouri and H. J. Bolink, Adv. Energy Mater., 2019, 10(13), 1902499 CrossRef
- J. M. Andersen and J. Mack, Chem. Sci., 2017, 8, 5447–5453 RSC
- R. Schlem, C. F. Burmeister, P. Michalowski, S. Ohno, G. F. Dewald, A. Kwade and W. G. Zeier, Adv. Energy Mater., 2021, 11(30), 2101022 CrossRef CAS
- P. Baláž, M. Achimovičová, M. Baláž, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutková, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and K. Wieczorek-Ciurowa, Chem. Soc. Rev., 2013, 42, 7571–7637 RSC
- P. F. M. de Oliveira, R. M. Torresi, F. Emmerling and P. H. C. Camargo, J. Mater. Chem. A, 2020, 8, 16114–16141 RSC
- N. Elahi, M. Kamali and M. H. Baghersad, Talanta, 2018, 184, 537–556 CrossRef CAS PubMed
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS PubMed
- S. Guo and E. Wang, Anal. Chim. Acta, 2007, 598, 181–192 CrossRef CAS PubMed
- D. Debnath, S. H. Kim and K. E. Geckeler, J. Mater. Chem., 2009, 19, 8810–8816 RSC
- M. J. Rak, N. K. Saadé, T. Friščić and A. Moores, Green Chem., 2014, 16, 86–89 RSC
- P. F. M. de Oliveira, J. Quiroz, D. C. de Oliveira and P. H. C. Camargo, Chem. Commun., 2019, 55, 14267–14270 RSC
- N. T. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 2002, 72, 4847–4854 CrossRef
-
W. Ostwald, Lehrbruck der Allgemeinen Chemie, Engelmann, Leipzig, Germany, 1896 Search PubMed
- M. A. Watzky and R. G. Finke, J. Am. Chem. Soc., 1997, 119, 10382–10400 CrossRef CAS
- P. J. Goulet and R. B. Lennox, J. Am. Chem. Soc., 2010, 132, 9582–9584 CrossRef CAS PubMed
- J. Polte, T. T. Ahner, F. Delissen, S. Sokolov, F. Emmerling, A. F. Thunemann and R. Kraehnert, J. Am. Chem. Soc., 2010, 132, 1296–1301 CrossRef CAS PubMed
- C. D. De Souza, B. R. Nogueira and M. E. C. M. Rostelato, J. Alloys Compd., 2019, 798, 714–740 CrossRef
- M. Sengani, A. M. Grumezescu and V. D. Rajeswari, OpenNano, 2017, 2, 37–46 CrossRef
- P. F. M. de Oliveira, A. A. L. Michalchuk, J. Marquardt, T. Feiler, C. Prinz, R. M. Torresi, P. H. C. Camargo and F. Emmerling, CrystEngComm, 2020, 22, 6261–6267 RSC
- P. F. M. de Oliveira, A. A. L. Michalchuk, A. G. Buzanich, R. Bienert, R. M. Torresi, P. H. C. Camargo and F. Emmerling, Chem. Commun., 2020, 56, 10329–10332 RSC
- M. Ferguson, A. J. Richard, J. Valdez, B. G. Fiss, H. M. Titi, N. Provatas, T. Friščić and A. Moores, Faraday Discuss., 2023, 241, 278–288 RSC
- L. Yang, A. Moores, T. Friščić and N. Provatas, ACS Appl. Nano Mater., 2021, 4, 1886–1897 CrossRef CAS
- C. Mottillo and T. Friščić, Molecules, 2017, 22(1), 144 CrossRef PubMed
- C. A. O'Keefe, C. Mottillo, J. Vainauskas, L. Fábián, T. Friščić and R. W. Schurko, Chem. Mater., 2020, 32, 4273–4281 CrossRef
- K. Budny-Godlewski, I. Justyniak, M. K. Leszczyński and J. Lewiński, Chem. Sci., 2019, 10, 7149–7155 RSC
- K. Budny-Godlewski, M. K. Leszczyński, A. Tulewicz, I. Justyniak, D. Pinkowicz, B. Sieklucka, K. Kruczała, Z. Sojka and J. Lewiński, ChemSusChem, 2021, 14, 3887–3894 CrossRef CAS PubMed
- S. Li, I. Huskić, N. Novendra, H. M. Titi, A. Navrotsky and T. Friščić, ACS Omega, 2019, 4, 5486–5495 CrossRef CAS PubMed
- I. Bekri-Abbes and E. Srasra, Mater. Sci. Semicond. Process., 2015, 40, 543–549 CrossRef CAS
- M. Y. Malca, H. Bao, T. Bastaille, N. K. Saadé, J. M. Kinsella, T. Friščić and A. Moores, Chem. Mater., 2017, 29, 7766–7773 CrossRef CAS
- S. S. M. Ali, W. H. Eisa and A. Abouelsayed, Composites, Part B, 2019, 176, 107289 CrossRef CAS
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC
- P. M. Adam, S. Benrezzak, J. L. Bijeon and P. Royer, J. Appl. Phys., 2000, 88, 6919–6921 CrossRef CAS
- S. Peters, S. Peredkov, M. Neeb, W. Eberhardt and M. Al-Hada, Surf. Sci., 2013, 608, 129–134 CrossRef CAS
- M. Sankar, Q. He, M. Morad, J. Pritchard, S. J. Freakley, J. K. Edwards, S. H. Taylor, D. J. Morgan, A. F. Carley, D. W. Knight, C. J. Kiely and G. J. Hutchings, ACS Nano, 2012, 6, 6600–6613 CrossRef CAS PubMed
- J. Radnik, C. Mohr and P. Claus, Phys. Chem. Chem. Phys., 2003, 5, 172–177 RSC
- W. Haiss, N. T. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed
- R. Ankri and D. Fixler, Nanophotonics, 2017, 6, 1031–1042 CrossRef CAS
- S. Liu and Y. J. Xu, Sci. Rep., 2016, 6, 22742 CrossRef CAS PubMed
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef CAS
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 8410–8426 CrossRef CAS
- T. Shimizu, T. Teranishi, S. Hasegawa and M. Miyake, J. Phys. Chem. B, 2003, 107, 2719–2724 CrossRef CAS
- W. P. Davey, Phys. Rev., 1925, 25, 753–761 CrossRef CAS
- F. Kim, S. Connor, H. Song, T. Kuykendall and P. Yang, Angew. Chem., Int. Ed., 2004, 43, 3673–3677 CrossRef CAS PubMed
- C. B. Whitehead, S. Özkar and R. G. Finke, Chem. Mater., 2019, 31, 7116–7132 CrossRef CAS
- B. P. Hutchings, D. E. Crawford, L. Gao, P. Hu and S. L. James, Angew. Chem., Int. Ed., 2017, 56, 15252–15256 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Photographs of milled and aged powders, XPS spectra, DRS spectra, PXRD patterns, TEM micrographs and population distributions over the six-week aging period of AuNPs, and details of the thermally coupled PXRD measurement for the determination of the activation energy of the AuNP formation. See DOI: https://doi.org/10.1039/d2na00759b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |