
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow temperature in situ immobilization of nanoscale fcc and hcp polymorphic nickel particles in polymer-derived Si–C–O–N(H) to promote electrocatalytic water oxidation in alkaline media†
Roberta Karoline
Morais Ferreira
ab,
Marwan
Ben Miled
a,
Rafael Kenji
Nishihora
ab,
Nicolas
Christophe
ac,
Pierre
Carles
a,
Günter
Motz
 d,
Assil
Bouzid
a,
Ricardo
Machado
b,
Olivier
Masson
a,
Yuji
Iwamoto
e,
Stéphane
Célérier
c,
Aurélien
Habrioux
*c and
Samuel
Bernard
*a
d,
Assil
Bouzid
a,
Ricardo
Machado
b,
Olivier
Masson
a,
Yuji
Iwamoto
e,
Stéphane
Célérier
c,
Aurélien
Habrioux
*c and
Samuel
Bernard
*a
aUniv. Limoges, CNRS, IRCER, UMR 7315, F-87000, Limoges, France. E-mail: samuel.bernard@unilim.fr
bChemical Engineering, Federal University of Santa Catarina, 88010-970 Florianópolis, Brazil
cInstitut de Chimie des Milieux et Matériaux de Poitiers (IC2MP), Université de Poitiers, CNRS, F-86073 Poitiers, France
dUniversity of Bayreuth, Ceramic Materials Engineering (CME), Bayreuth, Germany
eGraduated School of Engineering, Department of Life Science and Applied Chemistry, Nagoya Institute of Technology, Gokiso-cho, Showa-ku, Nagoya, Aichi 466-8555, Japan. E-mail: aurelien.habrioux@univ-poitiers.fr
First published on 6th December 2022
Abstract
We synthesized nickel (Ni) nanoparticles (NPs) in a high specific surface area (SSA) p-block element-containing inorganic compound prepared via the polymer-derived ceramics (PDC) route to dispatch the obtained nanocomposite towards oxygen evolution reaction (OER). The in situ formation of Ni NPs in an amorphous silicon carboxynitride (Si–C–O–N(H)) matrix is allowed by the reactive blending of a polysilazane, NiCl2 and DMF followed by the subsequent thermolysis of the Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :organosilicon polymer coordination complex at a temperature as low as 500 °C in flowing argon. The final nanocomposite displays a BET SSA as high as 311 m2 g−1 while the structure of the NPs corresponds to face-centred cubic (fcc) Ni along with interstitial-atom free (IAF) hexagonal close-packed (hcp) Ni as revealed by XRD. A closer look into the compound through FEG-SEM microscopy confirms the formation of pure metallic Ni, while HR-TEM imaging reveals the occurrence of Ni particles featuring a fcc phase and surrounded by carbon layers; thus, forming core–shell structures, along with Ni NPs in an IAF hcp phase. By considering that this newly synthesized material contains only Ni without doping (e.g., Fe) with a low mass loading (0.15 mg cm−2), it shows promising OER performances with an overpotential as low as 360 mV at 10 mA cm−2 according to the high SSA matrix, the presence of the IAF hcp Ni NPs and the development of core–shell structures. Given the simplicity, the flexibility, and the low cost of the proposed synthesis approach, this work opens the doors towards a new family of very active and stable high SSA nanocomposites made by the PDC route containing well dispersed and accessible non-noble transition metals for electrocatalysis applications.
:organosilicon polymer coordination complex at a temperature as low as 500 °C in flowing argon. The final nanocomposite displays a BET SSA as high as 311 m2 g−1 while the structure of the NPs corresponds to face-centred cubic (fcc) Ni along with interstitial-atom free (IAF) hexagonal close-packed (hcp) Ni as revealed by XRD. A closer look into the compound through FEG-SEM microscopy confirms the formation of pure metallic Ni, while HR-TEM imaging reveals the occurrence of Ni particles featuring a fcc phase and surrounded by carbon layers; thus, forming core–shell structures, along with Ni NPs in an IAF hcp phase. By considering that this newly synthesized material contains only Ni without doping (e.g., Fe) with a low mass loading (0.15 mg cm−2), it shows promising OER performances with an overpotential as low as 360 mV at 10 mA cm−2 according to the high SSA matrix, the presence of the IAF hcp Ni NPs and the development of core–shell structures. Given the simplicity, the flexibility, and the low cost of the proposed synthesis approach, this work opens the doors towards a new family of very active and stable high SSA nanocomposites made by the PDC route containing well dispersed and accessible non-noble transition metals for electrocatalysis applications.
Introduction
Owing to the considerable attention brought to microstructural design and the wide range of element combinations available, as well as their remarkable stability in air and harsh environments, inorganic materials including metal oxides and non-oxide materials are at the core of both established and emerging energy technologies.1The PDC route – via in situ nano-structuring2–20 – could represent an effective precursor approach for synthesizing conductive (because of the presence of sp2 carbon) and high surface area (due to the evolution of gaseous species in the low temperature thermolysis regime) inorganic materials – acting as substrates – containing in situ generated crystalline, accessible and dispersible earth-abundant (non-noble) 3d block transition metal (TM) nanoparticles. Non-noble TM are extensively studied as pre-catalysts for providing attractive HER and/or OER performances occurring in anion exchange membrane electrolyzers (AEMEs)21–25 and Ni is regarded as a promising pre-catalyst for OER;26–34 one of the half-reaction of water electrolysis involving a four-electron transfer hampered by kinetic factors. Hence, the PDC design strategy can allow the design of supported pre-catalysts for water oxidation in alkaline medium. As-obtained materials are expected to: (i) display long-term stability due to the strong nanocrystal–matrix interaction and to its expected corrosion resistance;35 (ii) reduce interface resistance; (iii) expose more active sites; (iv) avoid active sites aggregation during electrochemical process leading thereby to a constant catalytic activity; and (v) have practical applicability. However, such compounds remain still underexplored toward direct production of hydrogen fuel by the more technologically mature low-temperature electrochemical water splitting in alkaline medium, involving H2 evolution reaction (HER) and O2 evolution reaction (OER). This is mainly related to the lack of a material design strategy that could boost their catalytic activity, increase their conductivity, and develop their specific surface area for these reactions while being stable in aggressive environments.
Herein, we report the first polymer-derived Si–C–O–N(H) compound containing in situ generated and dispersed fcc- and IAF hcp-Ni particles (0.15 mg cm−2), produced at low temperature (500 °C), with high specific surface area (SSA, 311 m2 g−1) and able to promote OER activity (1.59 V vs. RHE at 10 mA cm−2) in alkaline medium. Although OER activity remains rather moderate in comparison with those reported in literature, it corresponds to one of the best performances ever reported for materials containing only nickel as an active phase, thus validating our approach and the prospects that will result from it. The key step in our approach lies in the use of a polysilazane working as a N-donor preformed polymer and leading to the pre-catalyst precursor labelled PSZNi2.5 by reactive blending (Fig. 1).
 | ||
| Fig. 1 Schematic diagram of the two-step general process of designing Si–C–O–N(H) immobilized nanoscaled Ni pre-catalysts. | ||
Results and discussion
Precursor preparation occurred effectively in N,N-dimethylformamide (DMF). DMF was used for mixing the polysilazane Durazane™ 1800 labeled PSZ – containing highly reactive Si–H and N–H bonds (see Fig. 1) – with nickel chloride (NiCl2) – acting as a Ni source – while promoting PSZ crosslinking to form a solid compound. The synthesis was carried out with a predetermined atomic Si:Ni ratio of 2.5 in order to achieve a Ni content sufficiently high in the final compound for the targeted reaction. Fig. S1 (ESI†) illustrates the different reaction steps based on the solution color evolution, changing from orange to dark green a few minutes after adding PSZ to NiCl2 and ended owing a general blackish tone with some blue highlights after DMF extraction. The elemental analysis data of PSZNi2.5 provided in Table 1 allowed the determination of an empirical formula of Si1.0C3.6N1.5O1.2H9.3Ni0.4Cl0.8 (referenced to Si1.0) to be compared to PSZ (Si1.0C1.5N1.1O0.0H5.6). Because the atomic Cl:Ni ratio calculated in PSZNi2.5 is identical to that of NiCl2 and the measured Si:Ni ratio is 2.5, we can conclude that no significant release of Cl and Si-containing species occurs during the reactive blending. However, a relatively high incorporation of oxygen atoms into the backbone of PSZNi2.5 occurs, especially compared to that measured in PSZ, which can be correlated to the use of DMF as a solvent during the precursor synthesis. We suggested that NiCl2 catalyzes the reduction of the amido group of DMF by the Si–H groups bearing by PSZ affording amines and a PSZ containing Si–O units, i.e., PSZNi2.5; the oxygen atoms coming from the carbonyl group of DMF. Such a transition metal-catalyzed reduction of amides by hydrosilanes is well reported to synthesize amines along with the appropriate disiloxane.36
| Sample | C (w%) | H (w%) | N (w%) | O (w%) | Ni (w%) | Si (w%) | Cl (w%) |
|---|---|---|---|---|---|---|---|
| PSZ | 27.3 | 8.3 | 22.7 | 0.4 | — | 41.3 | — |
| PSZNi2.5 | 24.1 | 5.3 | 11.8 | 10.8 | 14.8 | 15.8 | 17.4 |
| PSZNi2.5_5 | 19.3 | 2.9 | 3.6 | 14.7 | 41.3 | 18.2 | — |
A comparison of the empirical formula between PSZNi2.5 and PSZ also indicates that the presence of residual DMF (C3H7O2N) trapped in the PSZNi2.5 sample is the major reason to explain the increased nitrogen, carbon, oxygen and hydrogen content in PSZNi2.5 compared to PSZ. Elemental analysis results are highly reflected in the FTIR spectroscopy data (Fig. 2).
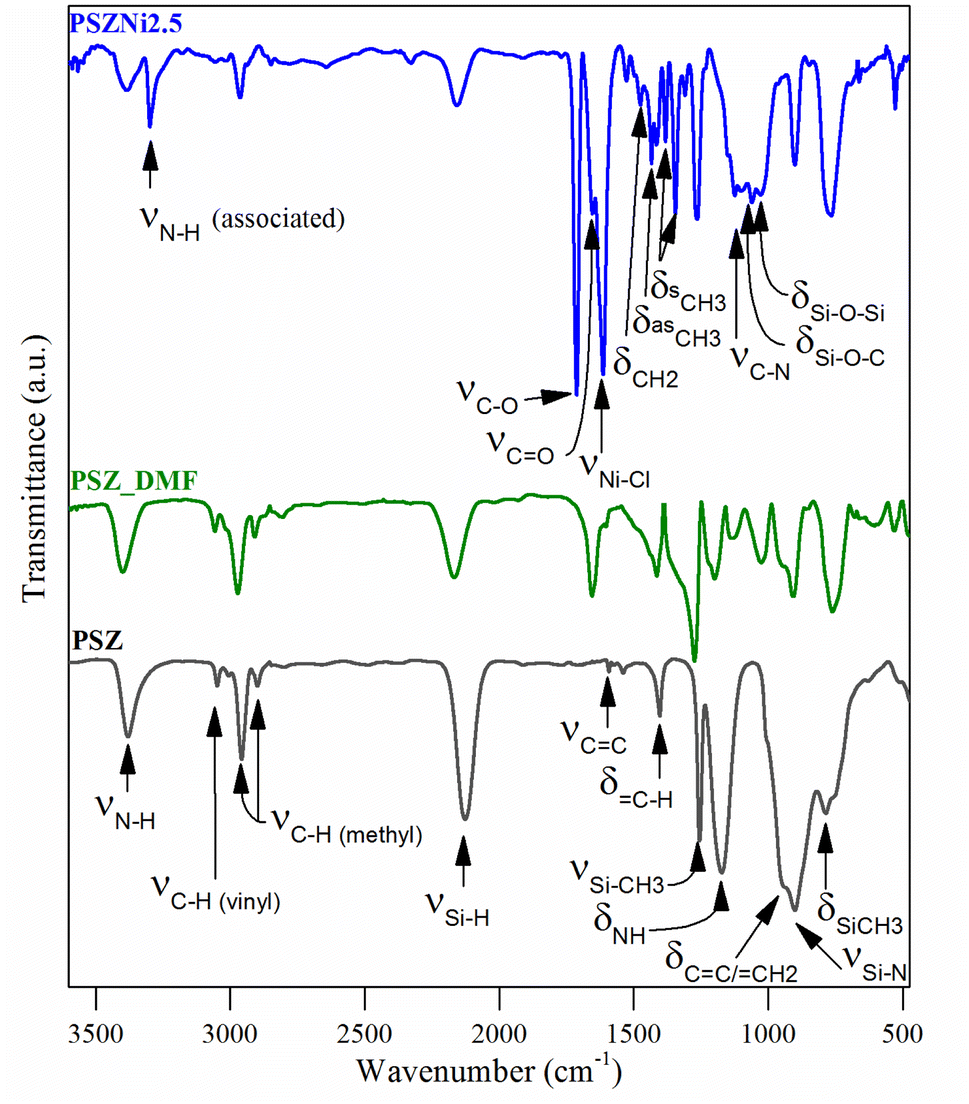 | ||
| Fig. 2 FTIR spectra of the PSZNi2.5, PSZ and PSZ_DMF samples. | ||
(1) The spectrum of PSZNi2.5 exhibits one intense band attributed to NiCl2 at 1612 cm−1 along with the presence of three characteristic bands assigned to DMF signature at 1658 (appearing as a shoulder on the left of the NiCl2 band), 1385, and 1122 cm−1, assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, –CH3 bending, and C–N stretching, respectively. Bands related to DMF identified in the spectrum of PSZNi2.5 also appear in the spectrum of the PSZ solubilized in DMF – labelled PSZ_DMF – that underwent the same synthesis procedure as PSZNi2.5 but without NiCl2 (Fig. 2).
O stretching, –CH3 bending, and C–N stretching, respectively. Bands related to DMF identified in the spectrum of PSZNi2.5 also appear in the spectrum of the PSZ solubilized in DMF – labelled PSZ_DMF – that underwent the same synthesis procedure as PSZNi2.5 but without NiCl2 (Fig. 2).
(2) The spectrum contains a series of bands – not present in both the PSZ and PSZ_DMF spectra – at 3301 cm−1 and 1715 cm−1. Furthermore, the single N–H band at 1169 cm−1 present in the spectrum of PSZ becomes a quadruplet (including the C–N stretching band at 1122 cm−1 as discussed above) in wavenumbers ranging from 1168 cm−1 to 980 cm−1. These bands are attributed to the formation of hydrogen-bonded N–H (3301 cm−1), –C–O– (1715 cm−1) units as well as Si–O–Si and/or Si–O–C units (from 1168 cm−1 to 980 cm−1).37,38 These findings indicate that N–H groups from PSZ are associated with CO groups via N–H⋯OC hydrogen bonding in PSZNi2.5 and they confirm the occurrence of crosslinking via the formation of Si–O linkages, hence, turning PSZ into a N- and O-donor organosilicon polymer.
(3) The large bands39 attributed to Si–N stretching in Si–N–Si units centered at 904 cm−1 and Si–C bond stretching at 779 cm−1 (PSZ and PSZ_DMF spectra) sharpen in the spectrum of PSZNi2.5 while the typical absorption bands (PSZ and PSZ_DMF spectra) in the wavenumber positions of 3388–3382 (νN–H), 3045 cm−1 (νC–H in vinyl groups), 2978–2894 cm−1 (νC–H in methyl groups), 2170–2127 (νSi–H), 1595 cm−1 (vCC) and 1174–1169 (δN–H in Si–N–Si units) decrease in intensity or almost vanish (3045 and 1595 cm−1) while those of CH2 and CH3 groups in the wavenumber range 1530–1340 cm−1 appear. These evolutions pinpoint the occurrence of dehydrocoupling reactions (N–H + Si–H) and hydrosilylation reactions (addition of Si–H across –CC–) to deliver PSZNi2.5.
It has been already reported that TM compounds can act as catalysts to remarkably increase the hydrosilylation rate and lower its temperature of occurrence.21 Furthermore, they are active toward bond formation between groups 14 and 15 elements – such as Si–N bonds in PSZ – resulting from dehydrocoupling reaction between Si–H and N–H in the temperature range 20–90 °C.40 Considering our synthesis conditions (DMF reflux) combined with the color evolution of the solution during precursor preparation along with above-discussed results, the incorporation of Ni as a salt in the PSZ network most probably occurs via the silylamino and/or siloxane ligands in PSZ (see Fig. 1); that is Ni2+ mainly coordinate the favourable silylamino and/or siloxane ligands in the sidegroup of PSZNi2.5 as indicated in Fig. 1. Thus, PSZNi2.5 represents a Ni:organosilicon polymer coordination complex in which Ni-catalyzed crosslinking linkages occur via Si–O–Si (DMF's reduction by Si–H), Si–C–C–Si (hydrosilylation) and Si–N–Si (dehydrocoupling) units formation. As a consequence, chain mobility is reduced and a solid compound is generated. It has been heat-treated in flowing argon to generate a catalyst active towards OER.
Considering that preceramic polymers display an intrinsic ability to form micropores in the low-temperature regime of the thermolysis,41–43PSZNi2.5 has been exposed at a temperature as low as 500 °C in flowing argon to maintain a relatively high level of SSA while transforming into an inorganic compound with a low portion of residual ‘organic’ groups. As a result, the thermolysis led to a compound, hereafter labelled PSZNi2.5_5, with an empirical formula of Si1.0C2.5N0.4O1.4H4.4Ni1.1 (referenced to Si1.0, see Table 1). The strong decrease of the Si:Ni ratio (0.9) compared to the one fixed in the precursor (2.5) is inherently due to the release of chlorosilane during the thermolysis.10 It allowed to (fully) eliminate chlorine (Cl) elements in the samples (a Cl content as low as 0.5 wt% has been measured). The high content of carbon with respect to Si and N indicates the presence of sp2 C as commonly found in polysilazane-derived ceramics.44 N2 sorption isotherm of the catalyst (Fig. 3) showed a well-defined type II isotherm45 having a distinct H3 hysteresis loop covering the 0.2–1.0 range of relative pressure. The latter indicates the formation of aggregates of plate-like particles giving rise to slit-shaped pores.46 A BET surface area of 311 m2 g−1 has been measured while t-plot analysis indicates the presence of a high amount of micropores; a typical t-plot micropore area of 201 m2 g−1 has been measured. This high intrinsic microporosity results from the evolution of residual DMF and gaseous species in the low-temperature regime of the thermolysis.41–43 Accordingly, these characteristics should allow the active sites of the PSZNi2.5_5 sample, Ni, to be easily accessible for OER.
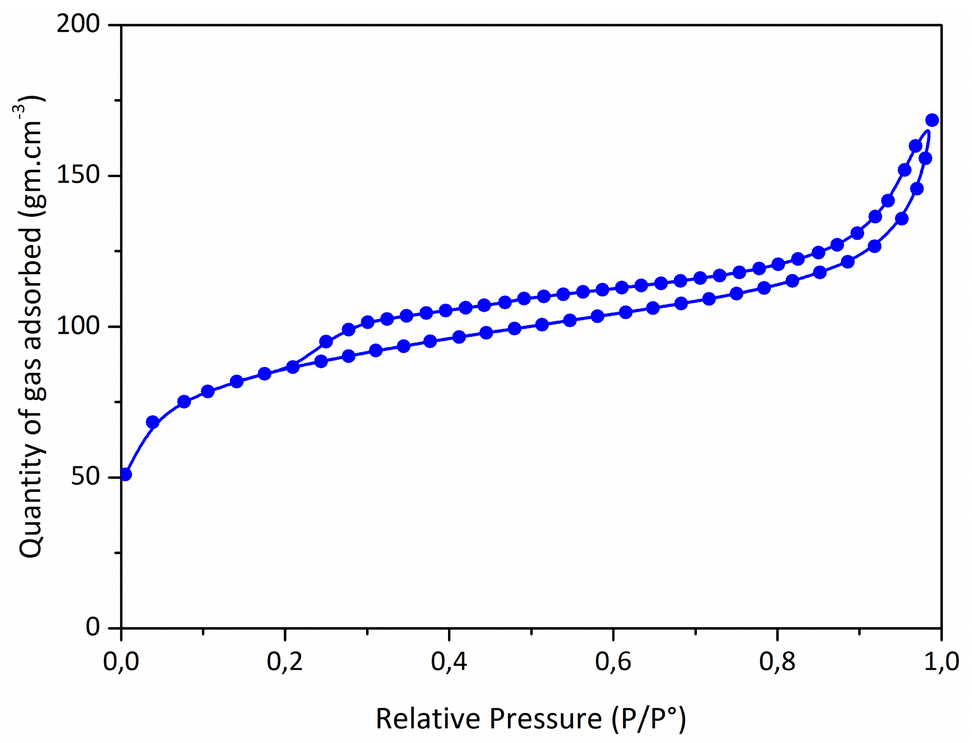 | ||
| Fig. 3 N2 sorption isotherms of the PSZNi2.5_5 sample. | ||
The XRD pattern of the PSZNi2.5_5 sample allows the identification of the crystalline phases causing such active sites. It exhibits dominant diffraction peaks at 2θ = 44.5°, 51.8°, and 76.4° (Fig. 4, see the original XRD pattern in Fig. S2 in ESI†), which can be unambiguously assigned to the (111), (200) and (220) lattice planes of the stable face-centered cubic (fcc) structure of Ni, with cell parameter a = 0.352 nm (JCPDS 04-0850). There are also four peaks of lower intensities at 41.9°, 47.6°, 62.5° and 84.1°. They can correspond to the 100, 101, 102, and 103 lines of the interstitial-atom-free (IAF) hexagonal close-packed (hcp)-Ni phase.47 This metastable phase has indeed been identified as a possible intermediate phase during the conversion of Ni3C into fcc-Ni.48 This is confirmed by a Rietveld refinement using the two fcc-Ni and hcp-Ni phases. The refined cell parameters are a = 0.3523 nm for the fcc-Ni phase and a = 0.2491 nm and c = 0.4083 nm for the hcp-Ni phase. These cell parameters are clearly distinguishable from those of hcp Ni3N (a = 0.2667 nm and c = 0.4312 nm)49 and Ni3C (a = 0.2628 nm and c = 0.4306 nm)50 as possible phases identifiable in Ni-modified PSZ. They are closer to the theoretically predicted cell parameters for IAF hcp-Ni (a = 0.2500 ± 0.001 nm and c = 0.3980 ± 0.006 nm)51 and similar to those identified for a hexagonal close-packed Ni phase from Ni(II) glycinate.52 Furthermore, they are also consistent with the cell constants of the hcp Co analog (JCPDS 89-7373; a = 0.251 nm and c = 0.407 nm), which is generally considered as a structural reference to justify the formation of the IAF hcp-Ni phase.53 The Rietveld refined relative weight proportion are about 85–15% for the fcc-Ni and hcp-Ni phases respectively. In addition, the refined microstructural parameters lead to clearly different apparent crystallite sizes for the two phases: about 42 nm for fcc-Ni (as well as an amount of apparent microstrains of 0.17%) and about 16 nm for hcp-Ni. These results also show that nickel was produced without oxidation in nanometric size and that no silicide phases are identified, as is commonly reported when mixing Ni complexes with organosilicon polymers.13,14 To further investigate the formation of fcc–hcp polymorphic Ni, the PSZNi2.5_5 sample has been subjected to a thorough FEG-SEM (Fig. 5) and HRTEM (Fig. 6) analysis. Elemental mapping (Fig. 5) of the PSZNi2.5_5 sample confirms a reasonable distribution of the five major elements in the micrometer and nanometer ranges.
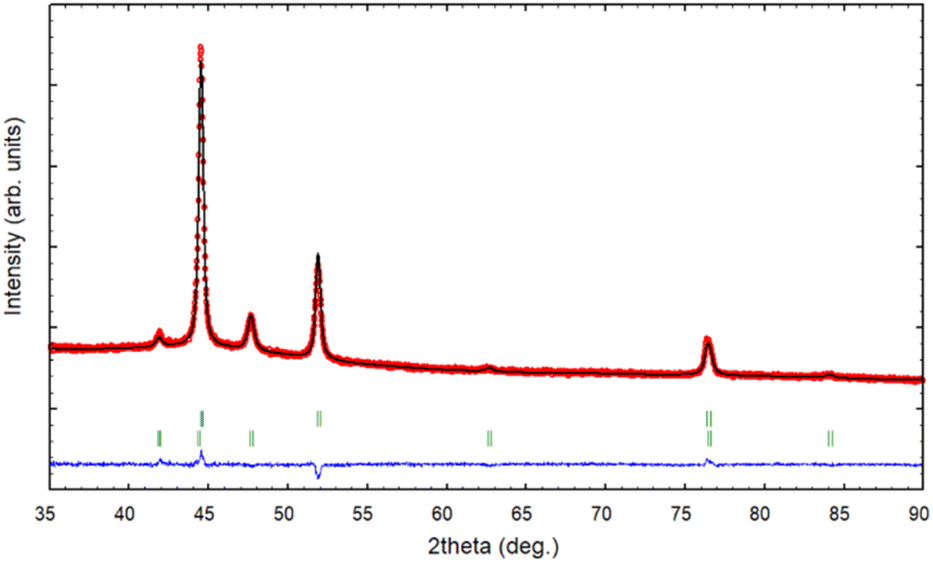 | ||
| Fig. 4 XRD pattern (red circles) and Rietveld refinement (black line) of the PSZNi2.5_5 sample, with the green vertical tick marks for fcc-Ni (upper) and hcp-Ni (lower). | ||
 | ||
| Fig. 5 FEG-SEM micrograph and corresponding elemental mapping images for the PSZNi2.5_5 sample. | ||
 | ||
| Fig. 6 Low magnification TEM images of the PSZNi2.5_5 sample (a–c), (b) SAED pattern corresponding to image (a), and high magnification images of nanoparticles isolated in the PSZNi2.5_5 (d and e). | ||
It is clearly observable that the Ni-rich domains (see in Fig. 5 arrows pointing out a highly Ni-rich domain) do not appear in the mapping of Si, C, N and O; thus, demonstrating that Ni can be considered as a single-phase anchored upon the Si–C–O–N(H) matrix. In addition, the Si- and O-rich domains (blue circles in Fig. 5) do not appear in the mapping of Ni; this is a further confirmation that Ni is not linked to Si or O and that the latter are linked to each other. As shown in the low magnification TEM images (Fig. 6a and associated insert), the sample consists of homogeneously dispersed small nuclei appearing as dark dots embedded in an amorphous matrix. The associated SAED pattern (Fig. 6b) – composed of spots indicating the high crystallinity of the analyzed phase – shows that the sample's structure is consistent with XRD results: the calculation of the d-spacing from the SAED gives values corresponding to the (111), (200), (220), and (311) lattice planes of fcc Ni, as well those corresponding to the lattice planes of the proposed IAF hcp-Ni phase.
We can observe in other areas of the sample (Fig. 6c) that the Ni size is relatively heterogeneous locally; in particular, two populations of Ni nanoparticles can co-exist in our system. One (green square) of the relatively big particles (from ∼40 to ∼70 nm) shown in Fig. 6c has been isolated and the corresponding high-resolution TEM (HRTEM) image is presented in Fig. 6d. It has been investigated to measure the lattice distance and a value of 0.202 nm has been calculated; it therefore relates to the (111) planes of fcc-Ni. Furthermore, Fig. 6d shows that such particles are coated with carbon layers based on the interlayer distance (0.34 nm); thereby forming core–shell nanostructures. One (blue square) of the smallest particles identified in Fig. 6c (they display an average particle size as low as 5.9 ± 1.2 nm (Fig. S3 in ESI†)) has been also isolated. Based on the investigation of the corresponding high magnification image in Fig. 6e, the fringes have a reduced spacing of 0.19 nm, corresponding to the (101) planes of IAF hcp Ni. Interestingly, the carbon layers surrounding the biggest nanoparticles seem to be not present as a shell around the smallest nanoparticles. These observations support the results from XRD, namely the population of the smallest Ni particles corresponds to the metastable hcp-Ni phase. To confirm this, we measured the SAED pattern from an area containing relatively large (100–200 nm) particles (Fig. S4 in ESI†): they are only composed of the fcc phase. Besides this, we recorded a TEM image from an area made of particles with a reduced size (Fig. S5 in ESI†): the hcp Ni phase could be indexed in addition to the fcc phase in the associated SAED pattern. Finally, higher magnification imaging (Fig. S6 in ESI†) allowed measuring the d-spacing of the Ni core in core–shell nanoparticles (Fig. S6a in ESI†). It agrees well with the fcc-Ni structure. They also reveal that smaller grains (Fig. S6b in ESI†) exhibit an hcp structure.54 We suggested that the passivating effect of the PSZ matrix leading to the nanoconfinement of Ni particles in a covalently-bonded Si–C–O–N(H) matrix is most likely to restrain the slippage of the close-packed layers, prompting the formation of the IAF hcp Ni phase, as previously suggested in ref. 48 for carbon layers surrounding Ni particles.
The high specific surface area of the PSZNi2.5_5 sample combined with the immobilization of well-dispersed nanoscale Ni particles, the presence of a hcp-Ni phase which has been reported to be more active than fcc-Ni towards OER,30 the development of core–shell nanostructures55 and the presence of sp2 carbon has motivated us to explore its OER electrocatalytic properties.
We have assessed the electrocatalytic properties for OER in a basic electrolyte (1 M KOH, Fig. 7). The electroactivity of the PSZNi2.5_5 sample was investigated using a rotating disk electrode set at 1600 rpm. Prior to recording polarization curves presented in Fig. 7a, voltammetric cycles were performed between 0.8 and 1.55 V vs. RHE.
 | ||
| Fig. 7 (a) Polarization curves recorded at 5 mV s−1 after different numbers of voltammetric cycles in nitrogen saturated 1 M KOH electrolyte, (b) schematic showing removal of the carbon shell surrounding fcc-Ni particles to be redistributed within the Si–C–O–N(H) matrix under working conditions in a 1 M KOH electrolyte. | ||
Upon electrochemical cycling, activation of the catalytic surface is observed and the coulometry associated with the Ni2+/Ni3+ oxidation wave increases (inset of Fig. 7a).56 This indicates an increase in high valence electrochemically active Ni atoms. Hence, it can be postulated that a surface reconstruction57 of the Ni particles occurs upon electrochemical cycling via the formation of a lamellar oxy(hydr-)oxide. Moreover, it is usually admitted that the slight shift of the position of the Ni2+/Ni3+ redox transition towards higher electrode potentials may be associated to the incorporation of iron from the 1 M KOH electrolyte.58 Although the exact role of iron on OER activity enhancement remains a matter of debate,59 the iron concentration determined from ICP measurements is as low as 8 ppb which clearly limits a possible boost of the OER performance by Fe.
During the activation phase, OER activity increases and, after 200 voltammetric cycles, the catalytic surface is stabilized requiring an electrode potential as low as 1.59 V vs. RHE to drive a current density of 10 mA cm−2. The Tafel slope calculated from polarization curve recorded after 200 voltammetric cycles in the 1.5–1.56 V vs. RHE potential range is of 66 mV dec−1. This value agrees with a rate determining step corresponding to the second step of the OER process, assuming no surface blocking and fast electron transfer.60 The PSZNi2.5_5 sample exhibits an activity surpassing than those obtained with Ni/SiOC and Co/SiOC nanocomposites (1.62 V and 1.67 V vs. RHE, respectively),61 and with a commercial IrO2 catalyst (1.64 V vs. RHE). It was previously mentioned that particles crystallizing into the hcp-Ni phase are largely smaller than fcc Ni particles; therefore, most of the electroactive surface area can be ascribed to the presence of hcp-Ni nanoparticles; a phase which has been reported to be more active than fcc-Ni towards OER.30 Hence, we can suggest that the surface reconstruction of the Ni particles discussed above efficiently occurs at the surface of the hcp-Ni NPs. In contrast, the formation of the lamellar oxy(hydr-)oxide phase proceeds with redistribution of carbon surrounding fcc-Ni particles according to Fig. 7b: we suggested that the carbon shell is gradually removed from fcc-Ni particles – allowing the oxidation of the fcc-Ni particle surface – to be redistributed within the Si–C–O–N(H) matrix; thereby increasing its conductivity (resistance decrease). Taken together, these results demonstrate that PSZNi2.5_5 material is a promising ‘pre-catalyst’ whose surface reconstruction and carbon redistribution under working conditions in a 1 M KOH electrolyte leads to a promising OER catalyst, especially considering that it is solely composed of Ni with a Ni mass loading as low as 0.15 mg cm−2.
To shed more light on interfacial processes such as charge transfer and adsorption of reaction intermediates, occurring during OER, electrochemical impedance spectra were recorded after different numbers of voltammetric cycles (Fig. 8a). All spectra were fitted using the equivalent circuit model proposed in Fig. 8b (Armstrong–Henderson equivalent circuit)62 in which, Rs corresponds to the cell resistance, which considers electrical connections, electrolyte, and the catalytic deposit. CPEdl models the double-layer capacitance (Cdl). To account for the catalyst surface roughness and the nonuniform distribution of active sites, the classical capacitance was replaced by a constant phase element.63R1 and R2 allow simulating the kinetics of the charge transfer reaction. To describe the kinetics of the OER, the total faradaic resistance (Rfar) must be considered, which is defined as the sum of R1 and R2.64 The values of CPEads in parallel with R2 allow mimicking the charge associated with the reversible adsorption of OER intermediates.59
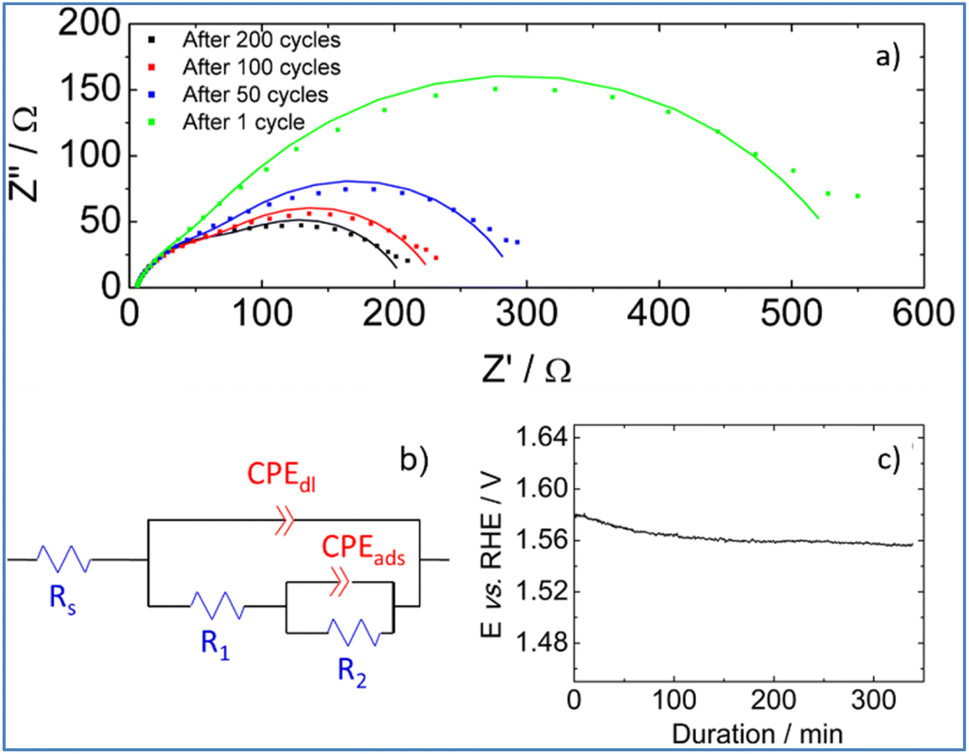 | ||
| Fig. 8 (a) Impedance spectroscopy measurements carried out after different numbers of voltammetric cycles in a 1 M KOH electrolyte at 1.7 V vs. RHE. Simulated curves are presented with solid lines, (b) equivalent circuit model used to fit experimental electrochemical impedance spectra and (c) chronopotentiometric curve recorded for the PSZNi2.5_5 catalyst immobilized onto a Ni foam in nitrogen saturated 1 M KOH electrolyte at a current density of 10 mA cm−2. | ||
Values obtained for Rs, Rfar, and Cdl, as a result of the best fit of spectra recorded at 1.7 V vs. RHE after 1, 50, 100 and 200 cycles, are reported in Table 2. We note that equivalent double-layer capacitance (Cdl) was obtained using Brug's formula.65,66
| Number of cycles | R s/Ω | R far/Ω | C dl/μF cm−2 |
|---|---|---|---|
| 1 | 5.9 | 567 | 104 |
| 50 | 5.7 | 335 | 265 |
| 100 | 5.8 | 277 | 318 |
| 200 | 5.9 | 270 | 351 |
Table 2 shows that the cell resistance (Rs) remains nearly constant and is hence unaffected by the potential cycling. The value of Cdl rises as the number of voltammetric cycles increases, confirming that electrochemical active surface area increases upon cycling; thereby confirming surface reconstruction and consistent with the formation of a high surface area lamellar structure. The drop of R2 value upon cycling explains the decrease of Rfar since R1 value was nearly kept constant. This is clearly related to a strong increase in the rate of intermediates formation/desorption at the reconstructed catalytic surface as schematically explained in Fig. 7b.
Finally, the stability of PSZNi2.5_5 catalyst is assessed by chronopotentiometric measurement carried out at 10 mA cm−2 in nitrogen saturated 1 M KOH electrolyte. To perform this test, the catalyst was immobilized onto a Ni foam, which is known for enhancing the electrocatalytic activity in comparison to glassy carbon substrates.67 Two distinct stages can be detected by examining the chronopotentiometric curve (Fig. 8c). Because of the surface reconstruction phenomena, the electrode potential drops within the first 100 minutes, resulting in an improved catalytic performance. Then, the electrode potential remains at a constant value of ca. 1.56 V vs. RHE for 250 minutes, demonstrating the promising stability of the PSZNi2.5_5 sample during OER process.
Experimental
Materials
The synthesis of the precursor is carried out in a purified argon atmosphere successively passing through a column of phosphorus pentoxide and then a vacuum/argon line by means of standard Schlenk techniques. Before the introduction of reactants, the cleaned glassware is stored in an oven at 95 °C overnight before being connected to the vacuum/argon line, assembled and pumped under vacuum for 30 min and then filled with argon. All chemical products are handled in an argon-filled glove box (Jacomex, Campus-type; O2 and H2O concentrations kept at ≤ 0.1 ppm and ≤ 0.8 ppm, respectively). The Durazane™ 1800 labeled here PSZ was provided in a 5 L container by durXtreme GmbH, Ulm (Germany), stored in a fridge and used as-received. Anal. Found (wt%): Si, 41.3; C, 27.3; N, 22.7; H, 8.3; O, 0.4. [Si1.0C1.5N1.1H5.5]n (referenced to Si1.0 and oxygen content was omitted in the empirical formulae). FTIR (ATR/cm−1): ν(N–H) = 3400 (s), ν(C–H in vinyl) = 3058 (m), ν(C–H in methyl) = 2954 (s) and 2898 (m), ν(Si–H) = 2123 (s), δ(CC) = 1594 (w), δ(C–H) = 1405 (w), δ(Si–CH3) = 1251 (m), δ(N–H): 1169 (s), δ(CC/ = CH2) = 947 (w), ν(N–Si–N) = 897 (vs.), δ(Si–CH3) = 787 (m); 1H NMR (300 MHz, CDCl3, δ/ppm): 0.4–0.1 (br, SiCH3), 1.1–0.5 (br, NH), 4.9–4.4 (br, SiH), 6.3–5.7 (br, vinyl). N,N-Dimethylformamide (DMF), 99.8%, extra sec over Molecular Sieve, AcroSeal™ was purchased from Fisher scientific and systematically used under argon. The nickel(II) chloride anhydrous for synthesis (98% purity), was obtained from Merck and stored in the glove-box.
Reactive blending
The reactive blending consists to add under argon the PSZ to a suspension of previously weighed (in the glove-box) NiCl2 in DMF in a three-neck round-bottom flask then stirred the mixture for 3 h at room temperature. In a typical experiment, 2.0 g of PSZ (30.52 mmol referred to the theoretical monomeric unit of the polymer) are quickly added with a syringe under flowing argon to a suspension made of 1.61 g of NiCl2 (12.42 mmol) in 50 mL of DMF at RT under vigorous stirring (350 rpm) (Si:Ni ratio of 2.5). Then, the temperature is increased up to DMF reflux under static argon and under vigorous stirring overnight. After cooling down, the solvent is extracted via an ether bridge (100 °C/1.5 10−1 mbar) to release a solid precursor labelled PSZNi2.5 (2.5 being the Si:Ni ratio). Anal. Found (wt%): Si 15.8, C 24.1, N 11.8, O 10.8, H 5.3, Ni 14.8, Cl 17.4. [Si1.0C3.6N1.5O1.2H9.3Ni0.4Cl0.8]n (referenced to Si1.0). FTIR (KBr/cm−1): ν(N–H) = 3388 (m), ν(N–H⋯OC) = 3301 (w), ν(C–H in methyl): 2965 (s), 2894 (s), ν(Si–H) = 2156 (m), ν(C–O) = 1715 (vs.), ν(CO) = 1658 (s), ν(Ni–Cl) = 1612 (vs.), δ(CH2) = 1457 (s), δas(CH3) = 1417 (m), δs(CH3) = 1385 (m), δ(Si–CH3) = 1254 (s), δ(N–H): 1169 (m), δ(C–N) = 1122 (m), δ(Si–O–C) = 1059 (m), δ(Si–O–Si) = 1024 (m), ν(Si–N in Si–N–Si units) = 904 (m), ν(Si–C) = 779 (s).
Thermolysis
The as-synthesize compound PSZNi2.5 is directly thermolyzed at 500 °C in flowing argon to be transformed into an inorganic compound labelled PSZNi2.5_5. For that, the sample is handled in the glove-box and the procedure consists to introduce a controlled quantity in an alumina boat to be put in a sealed tube to prevent any oxygen contamination of the sample during the transfer from the glove-box to the furnace. The sealed tube is introduced and then open into a silica tube from a horizontal furnace (Carbolite BGHA12/450B) under argon and the alumina boat containing the precursor powders is pushed to the heat zone of the furnace. The tube is then evacuated (0.1 mbar) for 30 min and refilled with argon (99.995%) to atmospheric pressure. Subsequently, the sample is subjected to a cycle of ramping of 5 °C min−1 to 500 °C in flowing argon (dwelling time of 2 h). A constant flow (120 mL min−1) of argon is passed through the tube during the thermolysis cycle. After cooling under argon atmosphere, the compound labelled PSZNi2.5_5 is recovered under argon to be transferred in the sealed tube to the glove-box to be stored for further characterization.Characterization
The chemical bonds present in the different compounds are investigated using FTIR in transmission mode over a spectral range of 490 to 3600 cm−1 (Thermo Scientific Nicolet 6700). The solid compounds are mixed with potassium bromide (KBr) in the glovebox to be then pressed under 5 T in the form of pellets. Liquid compounds are analyzed using two KBr commercial windows. OMNIC software is used for data acquisition and processing and the band intensities reported in this manuscript are ‘relative’ to Si–CH3 and are normalized by the integral of the Si–CH3 band. The crystalline phases of the thermolyzed sample – ground as fine powders – were analysed from XRD data obtained with a Bruker D8 Advance diffractometer. The scan was performed using the CuKα1/2 radiations, from 20 to 90° 2θ with a step size of 0.02° and acquisition time of 0.9 s per step. The diffraction patterns were analyzed using the Diffrac + EVA software with the JCPDS-ICDD database and the program Fullprof68 for the Rietveld refinements. Scale factor, cell parameters, Ni isotropic displacement parameter and line width parameters were refined for each phase. The profile shapes were modelled with the pseudo-Voigt function within the Thompson–Cox–Hastings formulation. C, as well as N, O and H contents of the thermolyzed powders, are measured by hot gas extraction techniques using a Horiba Emia-321V for carbon content and using a Horiba EMGA-830 for oxygen, nitrogen and hydrogen contents. The Si and Ni contents of the materials are determined by Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) using a PerkinElmer Optima 2000DV instrument. The Brunauer–Emmett–Teller (BET) method has been used to calculate the specific surface area of the thermolyzed sample. The pore-size distribution is derived from the desorption branches of the isotherms using the Barrett–Joyner–Halenda (BJH) method whereas the total pore volume is estimated from the amount of N2 adsorbed at a relative pressure (P/P0) of 0.97. The data are collected using a Micrometrics ASAP 2020 apparatus by N2 adsorption–desorption. Before adsorption measurements, samples were outgassed overnight at 150 °C in the degas port of the adsorption analyzer. Further observations of the nanoparticle microstructure and morphology as well as element distribution have been done using a field emission gun scanning electron microscope (FEG-SEM) 7900F from JEOL and by transmission electron microscopy (TEM) with a JEOL JEM 2100F. SEM is equipped with an Energy Dispersive X-ray Spectrometer (EDS) from Brücker (with Esprit software) allowing the element mapping. Electrochemical measurements have been carried out at room temperature using a homemade three-electrode Teflon cell connected to a potentiostat (Biologic SP-300). A reference hydrogen electrode (RHE) (Hydroflex purchased by Gaskatel) and a counter electrode made of glassy carbon (GC) slab is used. The working electrode is a 5 mm GC disk. A catalytic ink is prepared by dispersing 10 mg of catalytic powder into a solution composed of 500 μL ultra-pure water (Millipore), 500 μL of isopropanol (Sigma Aldrich, 99.5%) and 100 μL Nafion (Sigma Aldrich, 5 wt% in lower aliphatic alcohols). The ink is homogenized by ultrasonication for 10 min. 8 μL of catalytic ink is then deposited onto the glassy carbon disc and then allowed to dry under N2 (Air liquid, U) atmosphere at room temperature. The catalyst loading was 0.15 mg cm−2 for all experiments. All measurements are performed in a nitrogen saturated 1 mol L−1 KOH electrolyte (flakes, 90%, Merck to prevent iron contamination). Cyclic voltammograms and polarization curves are respectively recorded between 0.8 and 1.55 V vs. RHE and between 0.8 and 1.65 V vs. RHE. Polarization curves are recorded by applying a rotating rate of 1600 rpm to the rotating disc electrode. IR drop correction of polarization curves has been performed. For each measurement, cell resistance is determined using electrochemical impedance spectroscopy in the capacitive region and then further used for IR drop correction. For all measurements, a 10 mV amplitude was applied. Impedance spectra are acquired between 100 kHz and 10 Hz region using a potentiostat (Biologic SP-300). Acquired spectra were fitted using Biologic software (Zfit). Stability tests are performed using the previously described mounting except for the working electrode. This latter was a nickel foam (Goodfellow) connected to the potentiostat using a gold wire. Stability tests are performed in a 1 M nitrogen saturated KOH electrolyte. Chronopotentiometric measurements are performed by applying a constant current density of 10 mA cm−2.Conclusions
As a conclusion, a newly inorganic compound made out of p-block elements, namely silicon (Si), carbon (C), oxygen (O) and nitrogen (N) elements, surrounding nickel (Ni) nanoparticles has demonstrated promising OER activity. We exploited the high reactivity of polysilazanes toward DMF in the presence of NiCl2 to prepare a Ni:organosilicon polymer coordination complex that could deliver via a single-step process at a temperature as low as 500 °C under flowing argon a nanocomposite made of fcc and hcp polymorphic nanoscale Ni particles immobilized in a high SSA PDC matrix. The findings of our work are summarized as follows:
1. FTIR and elemental analyses demonstrated that the precursor represents an organosilicon polymer in which Ni-catalyzed crosslinking linkage formation occurs via Si–O–Si (DMF's reduction by Si–H), Si–C–C–Si (hydrosilylation) and Si–N–Si (dehydrocoupling) unit formation; Ni2+ coordinating silylamino and/or siloxane ligands.
2. N2 adsorption–desorption isotherms highlighted the large specific surface area of the thermolyzed compound and its high micropore content.
3. X-ray diffraction coupled with FEG-SEM and HRTEM observations revealed the formation of pure nickel with a structure corresponding to both fcc Ni (core (Ni)–shell (sp2 C)) and interstitial-atom free (IAF) hcp Ni; the latter being preferentially formed in low size particles.
4. Electrochemical measurements revealed an outstanding OER activity with an overpotential as low as 360 mV at 10 mA cm−2 and a Tafel slope of 66 mV dec−1 for the titled compound, especially considering that this newly synthesized material contains only Ni without doping (e.g., Fe) with a low Ni mass loading (0.15 mg cm−2).
5. We strongly suggested that OER performance is ascribed to the large specific surface area of the sample, allowing easier access to the active sites and the potential induced surface reconstruction of the Ni particles. It occurs via (i) the direct formation of a lamellar oxyhydroxide phase at the surface of hcp-Ni nanoparticles and (ii) the removal of the carbon shell surrounding fcc-Ni particles to be redistributed within the Si–C–O–N(H) matrix increasing its conductivity (resistance decrease) and rendering fcc-Ni nanoparticles accessible.
Although the OER activity of the studied catalyst is not currently the highest reported in literature in an alkaline electrolyte, it is among the best performances reported with Ni active phases. This study therefore did not aim to find the most efficient material for OER but leaves the door open to the development of future very active catalysts synthesized using a simple method a as well as cheap precursors.
There is still room for further studies on optimizing the OER properties and extend them toward HER by considering specific process parameters that can affect the structure of Ni, the chemical composition of the inorganic matrix, and the specific surface area of the final compound. We can also easily consider the use of other low-cost transition metals such as Fe or Mn (or mixture of them) using the PDC design strategy, known to be active catalysts, allowing to modify drastically and improve the catalytic properties towards the targeted reactions. Considering the process's simple and flexible chemistry as well as the readily available and low-cost precursors, these polymer-derived high SSA Si–C–O–N(H) compounds immobilized nanosized transition metal catalysts are expected to have a significant impact on electrocatalytic processes for clean energy applications.
Author contributions
RKMF, MBM and RKN contributed equally to this work by synthesizing the materials, carrying out ATR-FTIR, XRD and BET characterizations, analyzed and discussed the experimental data, prepared and drew the images and graphics for the publication and wrote the first draft of the manuscript. NC conducted material syntheses and performed selected characterization and PC was involved in the TEM observations and interpretations. PC, GM, YI, AB, OM (co-supervisor of MBM), and RM (co-supervisor of RKMF and RKN) reviewed the paper before submission while SC (co-supervisor of NC), AH (co-supervisor of MBM and NC) and SB (co-supervisor of RKMF, MBM, RKN and NC) supervised this research project, assisted in the electrocatalytic investigations (AH) and material syntheses (SB), contributed to the experimental design and data interpretation (SC, AH and SB with AB, YI and OM) and reviewed the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Campus France/CAPES (COFECUB Project N°PhC/956-19) coordinated by Dr S. Bernard (SB) and Prof. R. Machado (RM), which are also acknowledged for the financial support for the PhD of R. K. M. Ferreira (RKMF, CAPES, 88887.364640/2019-00) and the post-doctoral fellowship of Dr R. K. Nishihora (RKN, CAPES, 88887.364636/2019-00). In addition, SB, Dr A. Habrioux (AH) and Dr S. Célérier (SC) gratefully acknowledge the financial contribution from universities of Limoges and Poitiers (ARIC 2019 project; Master training of N. Christophe), from CNRS (SYNCOPE PEPS Energie 2020 project), Nouvelle-Aquitaine region and IRCER (SYNERGY 2021 project; PhD of M. Ben Miled (MBM)). This work was partially supported by CNRS via the International Research Project (IRP, SB and Prof Y. Iwamoto (YI)) ‘Ceramics materials for societal challenges’ as well as by the ANR (Grant project Number ANR-21-CE08-0036-01; Dr A. Bouzid (AB) and Number ANR-19-MRS3-0023; SB) and DFG (DFG project number 490841897 (MO 851/22; Dr G. Motz (GM))). MBM benefits from an institutional grant from the National Research Agency under the programme “Investments for the future”, EUR TACTIC with the reference ANR-18-EURE-0017. This work also pertains to the French government program “Investissements d'Avenir” (EUR INTREE, reference ANR-18-EURE-0010). The authors acknowledge financial support from the European Union (ERDF) and Région Nouvelle Aquitaine. M. Soustres (IRCER), R. Mayet (IRCER), L. Mazière (IC2MP) and J. Rousseau (IC2MP) are also acknowledged for BET, XRD, ICP-OES and FEG-SEM analyses respectively.Notes and references
- P. Feng and Z. Zou, Dalton Trans., 2017, 46, 10528 RSC.
- M. Zaheer, J. Hermannsdörfer, W. P. Kretschmer, G. Motz and R. Kempe, ChemCatChem, 2014, 6, 91–95 CrossRef CAS.
- D. Schumacher, M. Wilhelm and K. Rezwan, J. Am. Ceram. Soc., 2020, 103, 2991–3001 CrossRef CAS.
- M. S. Bazarjani, H. Kleebe, M. M. Mathis, C. Fasel, M. B. Yazdi, A. Gurlo and R. Riedel, Chem. Mater., 2011, 23, 4112–4123 CrossRef CAS.
- D. Forberg, T. Schwob and R. Kempe, Nat. Commun., 2018, 9, 1751–1757 CrossRef PubMed.
- D. Forberg, T. Schwob, M. Zaheer, M. Friedrich, N. Miyajima and R. Kempe, Nat. Commun., 2016, 7, 1–6 Search PubMed.
- G. Glatz, T. Schmalz, T. Kraus, F. Haarmann, G. Motz and R. Kempe, Chem.–Eur. J., 2010, 16, 4231–4238 CrossRef CAS PubMed.
- M. Zaheer, T. Schmalz, G. Motz and R. Kempe, Chem. Soc. Rev., 2012, 41, 5102–5116 RSC.
- D. Forberg, J. Obenauf, M. Friedrich, S. M. Hühne, W. Mader, G. Motz and R. Kempe, Catal. Sci. Technol., 2014, 4, 4188–4192 RSC.
- G. Hahn, J. K. Ewert, C. Denner, D. Tilgner and R. Kempe, ChemCatChem, 2016, 8, 2461–2465 CrossRef CAS.
- M. Kamperman, A. Burns, R. Weissgraeber, N. Van Vegten, S. C. Warren, S. M. Gruner, A. Baiker and U. Wiesner, Nano Lett., 2009, 9, 2756–2762 CrossRef CAS PubMed.
- S. M. Sachau, M. Zaheer, A. Lale, M. Friedrich, C. E. Denner, U. B. Demirci, S. Bernard, G. Motz and R. Kempe, Chem.–Eur. J., 2016, 22, 15508–15512 CrossRef CAS PubMed.
- T. Schwob and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 15175–15179 CrossRef CAS PubMed.
- C. Bäumler and R. Kempe, Chem.–Eur. J., 2018, 24, 8989–8993 CrossRef PubMed.
- A. Lale, V. Proust, M. C. Bechelany, A. Viard, S. Malo and S. Bernard, J. Eur. Ceram. Soc., 2017, 37, 5167–5175 CrossRef CAS.
- M. Balestrat, A. Lale, A. V. A. Bezerra, V. Proust, E. W. Awin, R. A. F. Machado, P. Carles, R. Kumar, C. Gervais and S. Bernard, Molecules, 2020, 25, 5236–5258 CrossRef CAS PubMed.
- C. Zhou, A. Ott, R. Ishikawa, Y. Ikuhara, R. Riedel and E. Ionescu, J. Eur. Ceram. Soc., 2020, 40, 6280–6287 CrossRef CAS.
- S. Tada, M. D. Mallmann, H. Takagi, J. Iihama, N. Asakuma, T. Asaka, Y. Daiko, S. Honda, R. K. Nishihora, R. A. F. Machado, S. Bernard and Y. Iwamoto, Chem. Commun., 2021, 57, 2057–2060 RSC.
- A. Lale, M. D. Mallmann, S. Tada, A. Bruma, S. Özkar, R. Kumar, M. Haneda, R. A. Francisco Machado, Y. Iwamoto, U. B. Demirci and S. Bernard, Appl. Catal., B, 2020, 272, 118975–118984 CrossRef CAS.
- M. C. Bechelany, V. Proust, A. Lale, P. Miele, S. Malo, C. Gervais and S. Bernard, Chem.–Eur. J., 2017, 23, 832–845 CrossRef CAS PubMed.
- H. Wan, X. Liu, H. Wang, R. Ma and T. Sasaki, Nanoscale Horiz., 2019, 4, 789–808 RSC.
- S. Li, E. Li, X. An, X. Hao, Z. Jiang and G. Guan, Nanoscale, 2021, 13, 12788–12817 RSC.
- K. Wang, X. Wang, Z. Li, B. Yang, M. Ling, X. Gao, J. Lu, Q. Shi, L. Lei, G. Wu and Y. Hou, Nano Energy, 2020, 77, 105162–105180 CrossRef CAS.
- K. Karthick, S. S. Sankar, S. Kumaravel, A. Karmakar, R. Madhu, K. Bera and S. Kundu, Dalton Trans., 2021, 50, 13176–13200 RSC.
- H. J. Kim, H. Y. Kim, J. Joo, S. H. Joo, J. S. Lim, J. Lee, H. Huang, M. Shao, J. Hu, J. Y. Kim, B. J. Min, S. W. Lee, M. Kang, K. Lee, S. Choi, Y. Park, Y. Wang, J. Li, Z. Zhang, J. Ma and S.-I. Choi, J. Mater. Chem. A, 2022, 10, 50–88 RSC.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 1–26 Search PubMed.
- S. De, J. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347 RSC.
- Y. Xu, W. Tu, B. Zhang, S. Yin, Y. Huang, M. Kraft and R. Xu, Adv. Mater., 2017, 29, 1605957 CrossRef PubMed.
- G. Zhong, S. Li, S. Xu, W. Liao, X. Fu and F. Peng, ACS Sustainable Chem. Eng., 2018, 6, 15108–15118 CrossRef CAS.
- C. Wang, Y. Wang, H. Yang, Y. Zhang, H. Zhao and Q. Wang, Small, 2018, 14, 1802895 CrossRef PubMed.
- P. Wang, R. Qin, P. Ji, Z. Pu, J. Zhu, C. Lin, Y. Zhao, H. Tang, W. Li and S. Mu, Small, 2020, 16, 2001642 CrossRef CAS PubMed.
- Y. Liu, H. Jiang, Y. Zhu, X. Yang and C. Li, J. Mater. Chem. A, 2016, 4, 1694–1701 RSC.
- Ł. Harynski, K. Grochowska, J. Karczewski, J. Ryl, J. Rysz and K. Siuzdak, RSC Adv., 2021, 11, 219–228 RSC.
- S. Öztürk, Y. Xiao, D. Dietrich, B. Giesen, J. Barthel, J. Ying, X. Yang and C. Janiak, Beilstein J. Nanotechnol., 2020, 11, 770–781 CrossRef PubMed.
- R. Riedel, G. Mera, R. Hauser and A. Klonczynski, J. Ceram. Soc. Jpn., 2006, 114, 425–444 CrossRef CAS.
- R. Arias-ugarte, H. K. Sharma, A. L. C. Morris and K. H. Pannell, J. Am. Chem. Soc., 2012, 134, 848–851 CrossRef CAS PubMed.
- H. Lu, X. Wang, Y. Yao, J. Gou, D. Hui, B. Xu and Y. Q. Fu, Synergistic effect of siloxane modified aluminum nanopowders and carbon fiber on electrothermal efficiency of polymeric shape memory nanocomposite, Composites, Part B, 2015, 80, 1–6 CrossRef CAS.
- D. Su, Y.-L. Li, H.-J. An, X. Liu, F. Hou, J.-Y. Li and X. Fu, J. Eur. Ceram. Soc., 2010, 30, 1503–1511 CrossRef CAS.
- M. Fedel, F. J. R. Gómez, S. Rossi and F. Deflorian, Coatings, 2019, 9, 680–693 CrossRef CAS.
- T. J. Clark, K. Lee and I. Manners, Chem.–Eur. J., 2006, 12, 8634–8648 CrossRef CAS PubMed.
- J. Lipowitz, J. A. Rabe, L. K. Frevel and R. L. Miller, J. Mater. Sci., 1990, 25, 2118–2124 CrossRef CAS.
- J. P. Dismukes, J. W. Johnson, J. S. Bradley and J. M. Millar, Am. Chem. Soc., 1997, 9, 699–706 CAS.
- M. Wilhelm, C. Soltmann, D. Koch and G. Grathwohl, J. Eur. Ceram. Soc., 2005, 25, 271–276 CrossRef CAS.
- G. Mera, A. Navrotsky, S. Sen, H.-J. Kleebe and R. Riedel, J. Mater. Chem. A, 2013, 1, 3826–3836 RSC.
- M. Thommes, K. Kaneko, A. V Neimark, J. P. Olivier, F. Rodriguez-reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- G. Mason, J. Colloid Interface Sci., 1982, 88, 36–46 CrossRef CAS.
- A. S. Bolokang and M. J. Phasha, Mater. Lett., 2011, 65, 59–60 CrossRef CAS.
- R.-T. Chiang, R.-K. Chiang and F.-S. Shieu, RSC Adv., 2014, 4, 19488 RSC.
- K. H. Jack, Acta Crystallogr., 1950, 3, 392–394 CrossRef CAS.
- S. Nagakura, J. Phys. Soc. Jpn., 1957, 12, 482–494 CrossRef CAS.
- J. G. Wright and J. Goddard, Phil. Mag., 1965, 11, 485–493 CrossRef CAS.
- V. Rodríguez-González, E. Marceau, P. Beaunier, M. Che and C. Train, J. Solid State Chem., 2007, 180, 22–30 CrossRef.
- S. Illy, O. Tillement, F. Machizaud, J. M. Dubois, F. Massicot, Y. Fort and J. Ghanbaja, Philos. Mag. A, 1999, 79, 1021–1031 CAS.
- M. K. Zakaryan, S. M. Estalaki, S. Kharatyan, A. M. Matzner, A. S. Mukasyan, T. Luo and K. V. Manukyan, J. Phys. Chem. C, 2022, 126, 12301–12312 CrossRef CAS.
- Y. Song, S. Casale, A. Miche, D. Montero, C. Laberty-Robert and D. Portehault, J. Mater. Chem. A, 2022, 10, 1350–1358 RSC.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- Y. Zheng, G. Zhang, P. Zhang, S. Chu, D. Wu, C. Sun, B. Qian, S. Chen, S. Tao and L. Song, Chem. Eng. J., 2022, 429, 132122 CrossRef CAS.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514–105519 CrossRef CAS.
- I. Spanos, J. Masa, A. Zeradjanin and R. Schlögl, Catal. Lett., 2021, 151, 1843–1856 CrossRef CAS.
- L. Köhler, M. E. Abrishami, V. Roddatis, J. Geppert and M. Risch, ChemSusChem, 2017, 10, 4479–4490 CrossRef PubMed.
- S. Abinaya, P. Moni, V. Parthiban, A. K. Sahu and M. Wihelm, ChemElectroChem, 2019, 6, 3268–3278 CrossRef CAS.
- R. D. Armstrong and M. Henderson, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 81–90 CrossRef CAS.
- J. Ruhl, L. M. Riegger, M. Ghidiu and W. G. Zeier, Adv. Energy Sustain. Res., 2021, 2, 200077–200086 Search PubMed.
- R. L. Doyle and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 5224–5237 RSC.
- V. A. Alves, L. A. Silva and J. F. C. Boodts, Electrochim. Acta, 1998, 44, 1525–1534 CrossRef CAS.
- L. Negahdar, F. Zeng, S. Palkovits, C. Broicher and R. Palkovits, ChemElectroChem, 2019, 6, 5588–5595 CrossRef CAS.
- J. N. Hausmann, R. Beltrán-suito, S. Mebs, V. Hlukhyy, T. F. Fässler, H. Dau, M. Driess and P. W. Menezes, Adv. Mater., 2021, 33, 2008823–2008831 CrossRef CAS PubMed.
- J. Rodriguez-Carvajal, Phys. B, 1993, 192, 55 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00821a |
| This journal is © The Royal Society of Chemistry 2023 |