
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBimetallic oxide Cu2O@MnO2 with exposed phase interfaces for dual-effect purification of indoor formaldehyde and pathogenic bacteria†
Jia Yu
Zheng‡
a,
Hao
Zhang‡
a,
Jun Da
He‡
b,
Bo Hai
Tian
a,
Chang Bao
Han
 *a,
Zhixiang
Cui
*c and
Hui
Yan
a
*a,
Zhixiang
Cui
*c and
Hui
Yan
a
aThe Key Laboratory of Advanced Functional Materials, Ministry of Education of China, Faculty of Materials and Manufacturing, Beijing University of Technology, Beijing 100124, People's Republic of China. E-mail: cbhan@bjut.edu.cn
bKey Laboratory of Beijing on Regional Air Pollution Control, Beijing University of Technology, Beijing 100124, People's Republic of China
cCollege of Materials Science and Engineering, Fujian University of Technology, Fuzhou, Fujian 350118, People's Republic of China. E-mail: cuizhixiang2006@126.com
First published on 8th March 2023
Abstract
The combination of materials with different functions is an optimal strategy for synchronously removing various indoor pollutants. For multiphase composites, exposing all components and their phase interfaces fully to the reaction atmosphere is a critical problem that needs to be solved urgently. Here, a bimetallic oxide Cu2O@MnO2 with exposed phase interfaces was prepared by a surfactant-assisted two-step electrochemical method, which shows a composite structure of non-continuously dispersed Cu2O particles anchored on flower-like MnO2. Compared with the pure catalyst MnO2 and bacteriostatic agent Cu2O, Cu2O@MnO2 respectively shows superior dynamic formaldehyde (HCHO) removal efficiency (97.2% with a weight hourly space velocity of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1) and pathogen inactivation ability (the minimum inhibitory concentration for 104 CFU mL−1Staphylococcus aureus is 10 μg mL−1). According to material characterization and theoretical calculation, its excellent catalytic-oxidative activity is attributable to the electron-rich region at the phase interface which is fully exposed to the reaction atmosphere, inducing the capture and activation of O2 on the material surface, and then promoting the generation of reactive oxygen species that can be used for the oxidative-removal of HCHO and bacteria. Additionally, as a photocatalytic semiconductor, Cu2O further enhances the catalytic ability of Cu2O@MnO2 under the assistance of visible light. This work will provide efficient theoretical guidance and a practical basis for the ingenious construction of multiphase coexisting composites in the field of multi-functional indoor pollutant purification strategies.
000 mL g−1 h−1) and pathogen inactivation ability (the minimum inhibitory concentration for 104 CFU mL−1Staphylococcus aureus is 10 μg mL−1). According to material characterization and theoretical calculation, its excellent catalytic-oxidative activity is attributable to the electron-rich region at the phase interface which is fully exposed to the reaction atmosphere, inducing the capture and activation of O2 on the material surface, and then promoting the generation of reactive oxygen species that can be used for the oxidative-removal of HCHO and bacteria. Additionally, as a photocatalytic semiconductor, Cu2O further enhances the catalytic ability of Cu2O@MnO2 under the assistance of visible light. This work will provide efficient theoretical guidance and a practical basis for the ingenious construction of multiphase coexisting composites in the field of multi-functional indoor pollutant purification strategies.
1. Introduction
Since the end of the 20th century, great changes have taken place in people's lifestyles, and numerous health problems caused by indoor environmental pollution have become one of the most critical issues concerning people all over the world.1 Indoor environmental pollutants mainly exist in the air, which can be divided into two categories: volatile organic pollutants that are released from building materials, furniture and other industrial products;2,3 pathogenic bacteria that are generated by the natural metabolism of organisms in humid environments.4 As a highly toxic volatile organic gaseous pollutant, formaldehyde (HCHO) is commonly present in indoor environments, and it was listed as a class I carcinogen by the International Agency for Research on Cancer as early as 2004.5,6 In addition, indoor air conditioning systems, carpets, heating systems and pet hair are all hotbeds of microbial reproduction, and when the concentrations of pathogenic microorganisms such as E. coli and S. aureus in the air increase to a sufficient extent, the incidence of pneumonia, cold, tuberculosis and other diseases will rise abruptly.7,8 Therefore, in order to protect people from health damage caused by polluted indoor air in their long-term indoor lives, it is of unprecedented social value and economic benefit to develop a material that can synchronously remove various indoor pollutants.Manganese-based materials have the advantages of high catalytic-oxidative activity, non-toxicity, and low cost, and they can catalyze HCHO molecules to mineralize into non-toxic final products H2O and CO2 at room temperature, showing great potential in the field of removing indoor organic pollutants.9,10 For instance, Wang et al.11 reported a palygorskite-supported MnO2 catalyst (MnO2–palygorskite), which could achieve 100% dynamic removal efficiency for HCHO with a gaseous hourly space velocity of 150000 h−1 at room temperature. However, the dynamic removal efficiency decreased to 72.1% after this catalyst had only be reused five times, which was mainly attributed to catalyst inactivation caused by the accumulation of intermediates that were not oxidized and decomposed in time. In order to solve the problem of catalyst inactivation at room temperature, many reported studies have attempted to modify the original MnO2 catalyst to improve its ability to activate O2 and H2O molecules into reactive oxygen species (O* and ·OH), promoting the decomposition of intermediates during catalysis.12–14 In particular, by combining with transition metal oxide photocatalysts, the modified MnO2 catalyst could generate more reactive oxygen species under the action of excited carrier transitions and the interface effect of bimetallic oxides, which is regarded as a feasible strategy to improve the activity and stability of an MnO2-based catalyst for catalytic HCHO oxidation at room temperature.15,16 Cuprous oxide (Cu2O), as a P-type Cu-based semiconductor photocatalyst with a forbidden bandwidth of 2.17 eV, has recently begun to be applied in the field of environmental purification.17,18 Under visible light, the photoexcited electrons generated by a Cu2O catalyst could capture O2 and H2O in the air and activate them into reactive oxygen species to promote the catalytic oxidation process.19 Thus, it is reasonable to infer that an ingenious combination of MnO2 and Cu2O could enhance the catalytic activity and stability of the Mn-based catalyst for catalytic-oxidative HCHO degradation at room temperature with the assistance of visible light. More importantly, Cu2O is also an excellent Cu-based bactericidal material, which always shows highly-efficient toxic metamorphic ability for pathogenic bacteria such as E. coli and S. aureus.20,21 Therefore, a bimetallic oxide composite of Cu2O–MnO2 is expected to achieve efficient purification of indoor HCHO and pathogenic bacteria synchronously.
For bimetallic oxide composites, a large number of defects is often present on the interface, such as oxygen vacancies and metal atom vacancies.22–24 According to previous reports, these vacancies are widely considered to be the critically active sites for the generation of active oxygen species (O* and ·OH) in the process of catalytic oxidation.25 For instance, the generation of active oxygen species on a metal oxide composite MgO/Al2O3 was significantly increased under the action of the interface effect, and showed higher capacity than pure metal oxide catalysts in catalytic-oxidative HCHO degradation.26 In addition, the existence of phase interfaces will enhance the capacity of electron gain-and-loss or reduce the barrier to O2 adsorption and activation to some extent on the catalyst surface, promoting the catalytic oxidation capacity of the catalyst by improving the amount of reactive oxygen species produced.9 Therefore, it could be inferred that creating an interface of a bimetallic oxide to further enhance the catalytic-oxidative activity for organic pollutant degradation is a feasible scheme. Moreover, in order to realize the unique functions of the two metal oxides and synchronously take full advantage of the interface effect between them, it is necessary to find a reasonable design for the compounded form of the two components on the basis of maintaining their double-phase coexistence. In previous studies, bimetallic oxides were usually synthesized by conventional or single-step reflux hydrothermal methods,27,28 but it is difficult to prepare a composite with exposed phase interfaces on the premise of maintaining double-phase coexistence via this method. Based on previously reported work by our group,29 two-step heating-assisted electrodeposition technology could regulate the proportion of different components in the composite on the basis of maintaining their independent phases by changing the deposition voltage, current and time. Considering that nonionic surfactants could efficiently control the size, morphology and dispersion in the process of material synthesis, it is speculated that a bimetallic oxide catalyst with exposed double phases and a phase interface could be obtained by the method of two-step electrodeposition assisted by surfactants.30
In this work, a bimetallic oxide Cu2O@MnO2 was prepared by the two-step heated electrochemical process, and non-continuously dispersed Cu2O nanoparticles were anchored on self-supporting MnO2 nanoflowers under the action of surfactant modification (polyethylene glycol, PEG-6000). In order to regulate the loading amount, particle size and dispersion of Cu2O particles, different electrodeposition times were set in the preparation process to explore the optimal synthesis conditions for the bimetallic oxide. To evaluate the dual-effect purification function of indoor HCHO and pathogenic bacteria, HCHO removal efficiency tests and bactericidal performance tests were performed in this work, and the efficiency of pure catalyst MnO2 and bacteriostatic agent Cu2O were compared. Further, by characterization of the materials and theoretical calculations based on first principles, the phase interface effect and adsorptive activity of O2 on the surface of the bimetallic oxide Cu2O@MnO2 were further studied to explore the mechanism of improving its dual-effect purification capacity. This work not only provides a feasible strategy for the construction of a bimetallic oxide with exposed phase interfaces, but also innovatively provides an idea for the design of a dual-effect purification material for indoor formaldehyde and pathogenic bacteria.
2. Results and discussion
2.1 Phase structure and morphology of bimetallic oxide Cu2O–MnO2
Two-step electrochemical deposition was used to prepare the composite to ensure the coexistence of two phases in the bimetallic oxide, and the process of formation of Cu2O anchored on MnO2 in the second-step electrodeposition is shown in Fig. 1a. Specifically, positively charged Cu2+[C3H6O3]n complexes rapidly moved to the side of the MnO2–CC sheet under the action of an electric field to form Cu+[C3H6O3]n by electron transfer.31 Then, in the alkaline electrolyte, the phase of Cu2O generated by the redox reactions between Cu+[C3H6O3]n and free hydroxyl ions (–OH) was in situ anchored on the surface of MnO2–CC to obtain the bimetallic oxide Cu2O@MnO2–CC (abbreviated to Cu2O@MnO2). Meanwhile, during the deposition of Cu2O on MnO2, as shown in Fig. 1b, the surface morphology and dispersion of Cu2O particles were controlled by introducing the surfactant PEG-6000.32,33 Under the action of surfactant modification, the original Cu2O cubes (Fig. 1c) were transformed into polycrystalline stacked nanospheres (Fig. 1d). Since the introduction of surfactants could promote the crystallization of Cu2O grains by reducing the surface tension during the growth process to improve their dispersion,34,35 the addition of surfactants into the deposition process is expected to introduce the second phase on the basis of preventing the substrate from being completely covered. The loading amount and dispersion of Cu2O anchored on MnO2 were controlled by controlling the deposition time, and the samples prepared with electrodeposition times of 15, 30, 60, 120 and 240 min were named “15 min Cu2O@MnO2”, “30 min Cu2O@MnO2”, “60 min Cu2O@MnO2”, “120 min Cu2O@MnO2” and “240 min Cu2O@MnO2” respectively. In the scanning electron microscopy (SEM) images of the samples with various Cu2O deposition times (Fig. 1e–i), the loading amount and particle size of Cu2O anchored on MnO2 gradually increase with an increase in the second-step electrodeposition time. In particular, the Cu2O particles of “60 min Cu2O@MnO2” with a particle size of about 10 nm realized non-continuous dispersion on the flower-like MnO2 substrate material without completely covering it, showing a structure with exposed phase interfaces in the reaction atmosphere (Fig. S1†). However, with a further increase in the deposition time, the original morphology and phase component of MnO2 could not be observed because it had been completely covered by the deposited Cu2O particles. Here, it could be preliminarily inferred that bimetallic oxide Cu2O@MnO2 with exposed phase interfaces based on the flower-like MnO2 structure could be prepared by surfactant-assisted two-step electrodeposition technology with an appropriate deposition time.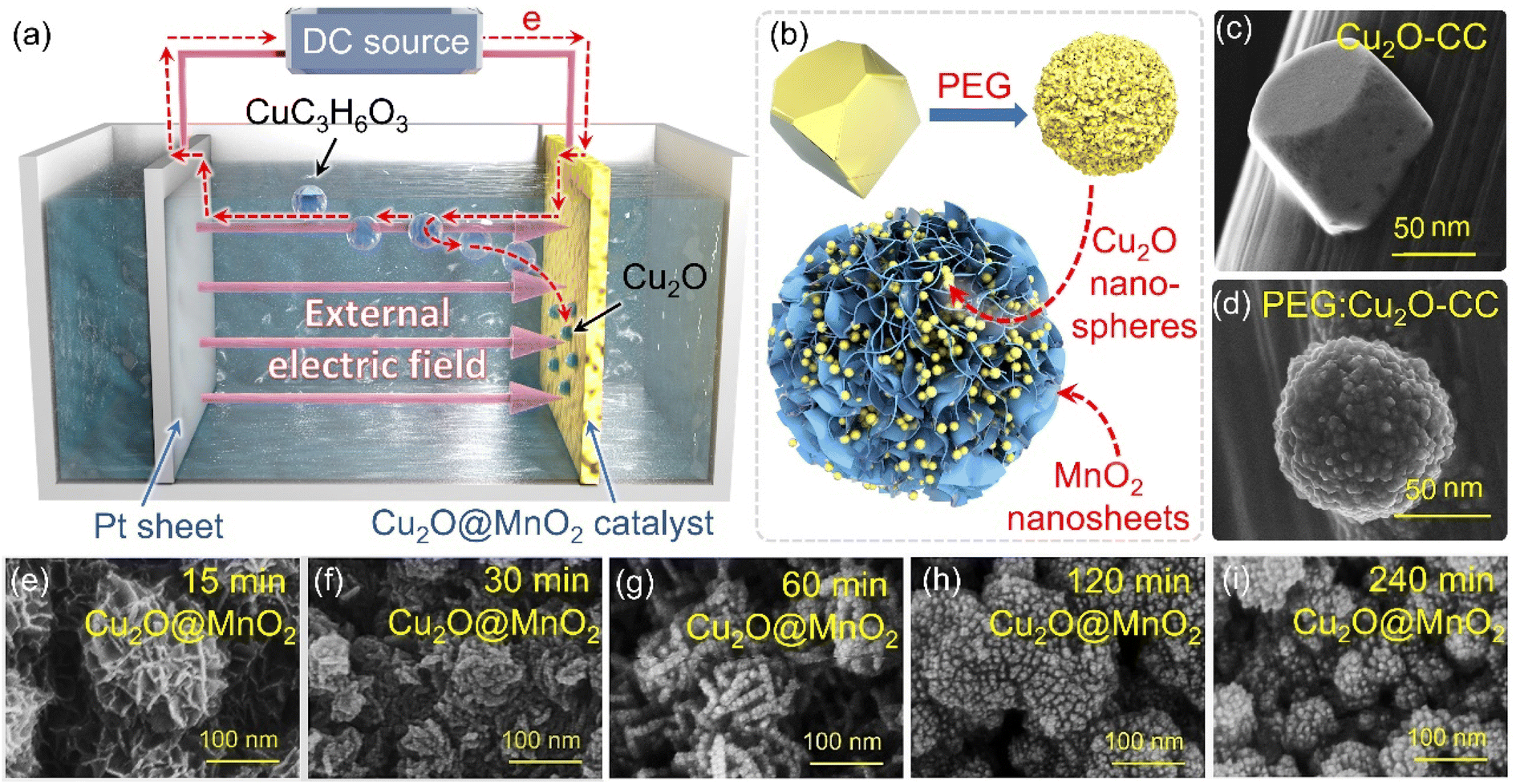 | ||
| Fig. 1 (a) Schematic diagram of the electrodeposition process of Cu2O anchored to MnO2–CC. (b) Diagrammatic drawing of Cu2O surface modification under the action of a surfactant. The SEM images of Cu2O particles (c) without PEG modification and (d) with PEG modification. SEM images of Cu2O@MnO2 prepared during different deposition times: (e) 15 min, (f) 30 min, (g) 60 min, (h) 120 min, (i) 240 min. | ||
In the X-ray diffractometer (XRD) characterization shown in Fig. 2a, the diffraction peaks located at 12.3°, 24.6° and 36.5° corresponding to δ-MnO2 (JCPDS 80-1098) were found in all the samples,36,37 meaning that the phase structure of MnO2 in the bimetallic oxides had been not changed significantly by the deposition of Cu2O. The diffraction peaks at 36.4°, 42.3° and 61.3° become gradually obvious with the deposition time of Cu2O, which correspond to the crystal planes of (111), (200) and (220), respectively, in cubic phase Cu2O.38 Considering that “60 min Cu2O@MnO2” not only has the morphology of the two-phase interface exposed to the reaction environment, but also shows the structure of the coexistence of two phases, it is necessary to study it further. In Fig. 2b, the Raman spectrum of “60 min Cu2O@MnO2” shows the characteristic peaks corresponding to the bonds of Cu–O and Mn–O at the same time, further confirming that it is composed of two metal oxides. Moreover, in the enlarged transmission electron microscopy (TEM) image (Fig. 2c), lattice spacings of 2.43 nm and 2.45 nm were found in two related regions, corresponding to (−111) of MnO2 and (111) of Cu2O. This result is consistent with the conclusions based on XRD and Raman data, indicating that the prepared sample is indeed a bimetallic oxide co-constructed from the two phases of Cu2O and MnO2.
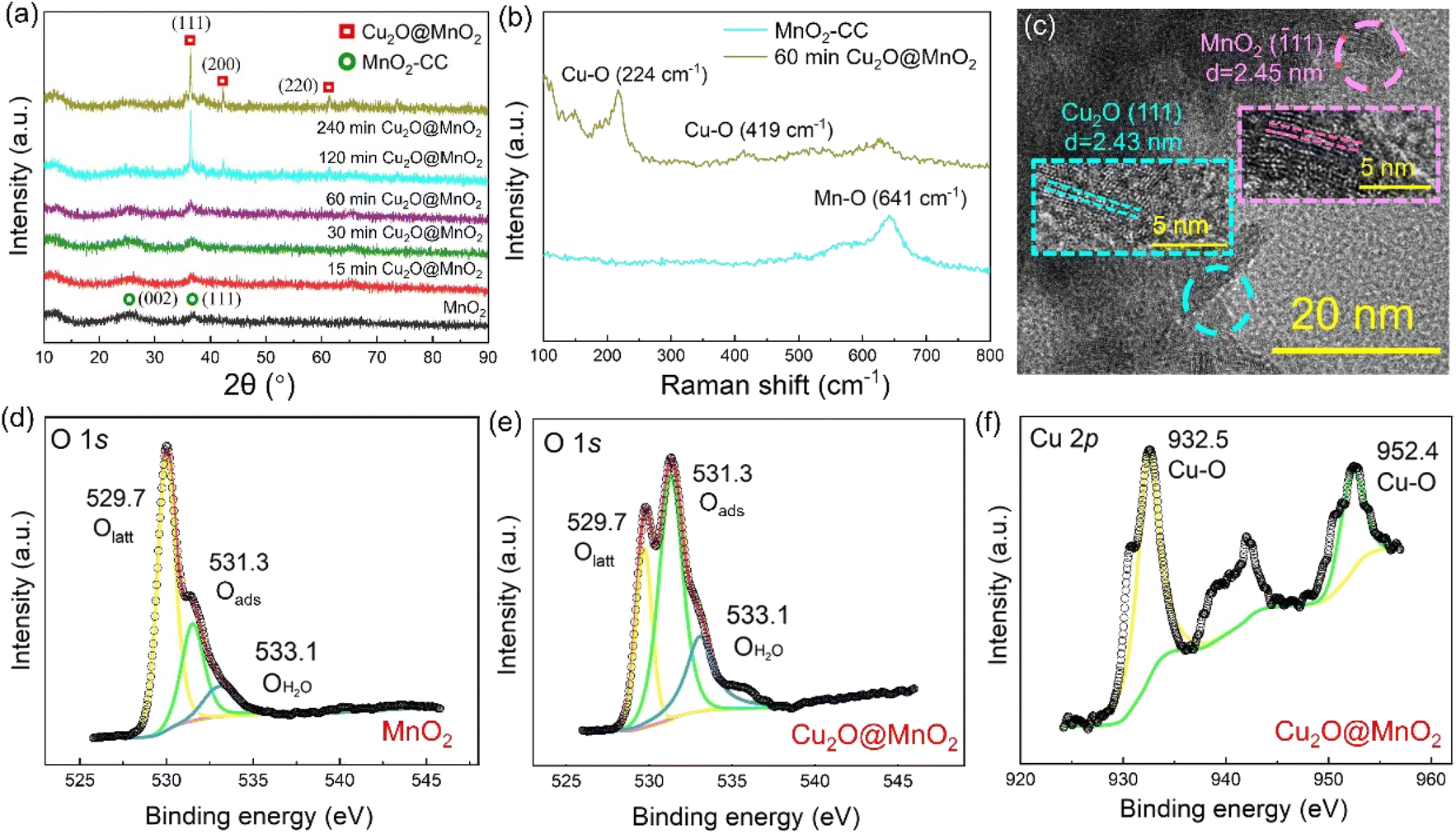 | ||
| Fig. 2 (a) XRD patterns of MnO2–CC and Cu2O@MnO2 prepared during different Cu2O deposition times. (b) Raman spectra of MnO2–CC and the “60 min Cu2O@MnO2” sample. (c) TEM image of the “60 min Cu2O@MnO2” sample. XPS spectra of (d) “MnO2” and (e) “60 min Cu2O@MnO2” in O 1s. (f) XPS spectra of “60 min Cu2O@MnO2” in Cu 2p. | ||
The changes in element types and valence states in the bimetallic oxide caused by the introduction of the Cu2O phase were further characterized by performing XPS tests on “60 min Cu2O@MnO2” and the initial MnO2 catalyst. Compared with pure MnO2, the XPS survey spectrum of Cu2O@MnO2 adds the characteristic peak of the Cu element (Fig. S2†), indicating that Cu2O is successfully anchored on MnO2. As shown in the Mn 3s patterns (Fig. S3†), the average oxidation states (AOSs) of Mn in MnO2 and Cu2O@MnO2 are 3.19 and 3.69, respectively, according to the calculation based on the empirical formula (AOS = 8.956 − 1.126 × ΔE), meaning the manganese oxides in both samples show the characteristics of mixed-valence compounds. Moreover, according to the Mn 2p spectra (Fig. S4†) and the ratio of different elements in the samples (Table S1†), the proportion of Mn(III) increases as that of Mn(IV) decreases after modification with Cu2O. The increase in the valence state of Mn element and the proportion of Mn(III) in the bimetallic oxide Cu2O@MnO2 could even enhance the oxidation of HCHO and intermediates by the efficient transfer and transformation of electrons. In Fig. 2d and e, the content of adsorbed oxygen in oxygen vacancies (Oads) in Cu2O@MnO2 is obviously higher than that for pure catalyst MnO2. In the electron paramagnetic resonance (EPR) tests shown in Fig. S5,† the signal strength of Cu2O@MnO2 is higher than that of MnO2, from which it could be inferred that MnO2 modified by Cu2O particles may have more abundant oxygen vacancies and adsorbed oxygen species.39 Considering that Oads are the critically active sites in metal oxide catalysts to convert O2 into active oxygen species,40,41 the introduction of Cu2O into MnO2 promotes the amount of reactive oxygen species produced on the surface of the catalyst by converting the lattice oxygen (Olatt) in MnO2 into Oads, thus improving its catalytic activity at room temperature. Besides, in Fig. 2f, the existence of Cu–O bonds between Cu atoms and lattice oxygen further verifies the successful preparation of bimetallic oxide Cu2O@MnO2.
2.2 Light-assisted catalytic oxidation for HCHO removal
Static HCHO removal efficiency tests were conducted in dark and light environments. The change in HCHO concentration in a closed test chamber with time under the action of different samples is shown in Fig. 3a and b. It was found that all of the Cu2O@MnO2 catalysts can decrease the concentration of HCHO to a much lower level in the light-assisted environment compared with that in the dark environment. In particular, the “60 min Cu2O@MnO2” sample reduces the concentration of HCHO to less than 1 ppm in the shortest possible time compared to the pure MnO2 catalyst and all the other bimetallic oxide samples, showing the best HCHO removal capacity with the assistance of light. Furthermore, in order to explore the differences in catalytic-oxidative activity of bimetallic oxides in an environment with or without light, the enhancement value for HCHO static removal efficiency under the action of light has been determined in Fig. 3c and S6.† The improvement in static efficiency first gradually increases as the Cu2O deposition time for Cu2O@MnO2 is prolonged, but it shows a downward trend when the Cu2O deposition time is longer than 120 min. It can be inferred that this phenomenon may be due to the fact that overloading with Cu2O particles is caused by too long a deposition time, resulting in MnO2 being completely covered and unable to participate in the catalytic-oxidative reaction. This inference is consistent with the analysis of the SEM data shown in Fig. 1e–i. In conclusion, although the anchored Cu2O particles in the bimetallic oxide could improve its catalytic ability for HCHO oxidation with the assistance of visible light, an excessive supporting amount of Cu2O will lead to failure of the main catalytic phase of MnO2 to come into contact with HCHO gas molecules, resulting in a reduction in catalyst activity. Therefore, adequate exposure of Cu2O and MnO2 and their interface in the reaction atmosphere while maintaining the coexistence of both in Cu2O@MnO2 is critical to exerting the unique advantages of bimetallic oxides in light-assisted catalytic HCHO oxidation.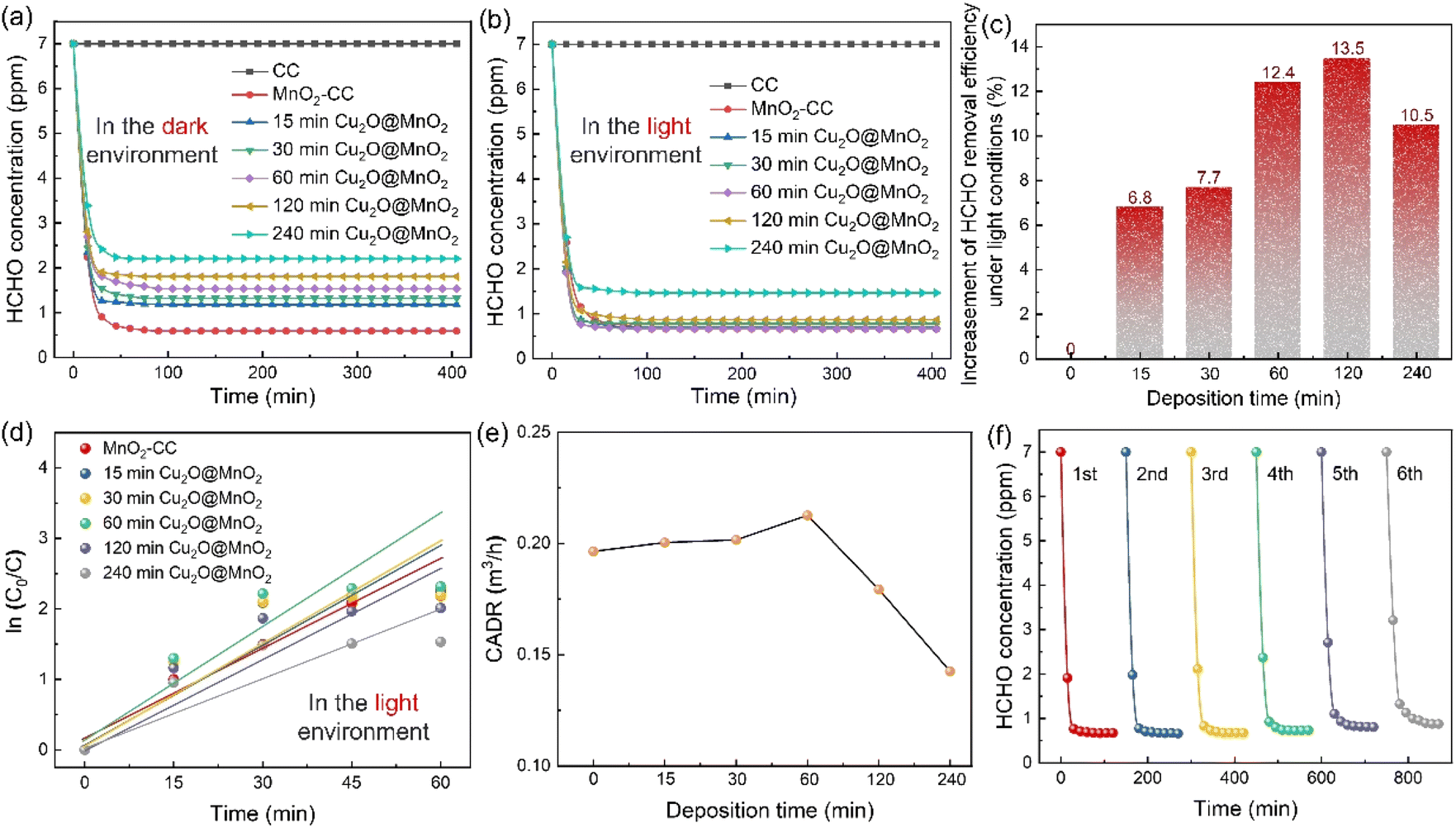 | ||
| Fig. 3 HCHO concentration changes with time under the action of CC, MnO2–CC and Cu2O@MnO2 (a) in the dark state and (b) in the light environment. (c) Efficiency increase, (d) first-order reaction fitting and (e) the available clean air volume (CADR) calculation values for HCHO static removal efficiency by different catalysts. (f) Thermal regeneration tests of “60 min Cu2O@MnO2” over 6 cycles. | ||
Additionally, in order to quantify the HCHO removal capacity of the samples prepared with different Cu2O deposition times and make a preliminary evaluation of the potential of the catalyst in practical applications, the static removal efficiency curves were fitted with a first-order equation to calculate the corresponding CADR values (Fig. 3d and e).42 In particular, “60 min Cu2O@MnO2” shows the highest CADR value of 0.229 m3 h−1 compared to all Cu2O@MnO2 samples and pure MnO2 catalyst in the light environment. Furthermore, in light-assisted dynamic tests (Fig. S7†), the variation trend of dynamic efficiency over Cu2O deposition time is consistent with that of static efficiency, in which Cu2O@MnO2 with a deposition time of 60 min shows the highest efficiency of 97.2% under a weight hourly space velocity (WHSV) of 120000 mL g−1 h−1. Thus, it can be further confirmed that the modification of Cu2O on MnO2 improves the catalytic-oxidative degradation ability of the initial pure MnO2 catalyst for HCHO under the assistance of visible light irradiation. Additionally, in evaluating the thermal regeneration stability and reutilization of Cu2O@MnO2 in catalytic HCHO removal at room temperature (Fig. 3f), it was found that the catalytic activity of the sample did not change significantly after repeated thermal regeneration experiments for 6 cycles, meaning the sample showed good catalytic stability.
Furthermore, the differences in the light absorption properties between various samples were explored by UV-Vis-NIR diffuse-reflectance spectra, as shown in Fig. S8.† The absorption edge located in the visible region is observed in the absorption spectrum of Cu2O (2.18 eV), which means it can absorb visible light to excite the electron transfer between its valence band (VB) and conduction band (CB).43 The composite Cu2O@MnO2 shows higher absorbance than a pure Cu2O catalyst, which is attributed to the improved light absorption capacity of the material by the Mn d–d transition of manganese oxide.44 Therefore, Cu2O@MnO2 not only achieves light-assisted HCHO oxidation degradation compared with a pure MnO2 catalyst, but also obtains higher light utilization capacity compared with a pure photocatalyst Cu2O. In conclusion, the bimetallic oxide “60 min Cu2O@MnO2” catalyst with exposed phase interfaces achieves superior activity and stability for catalytic HCHO degradation with the assistance of visible light compared with a traditional MnO2 catalyst or other Cu2O@MnO2 composites, showing huge prospects for commercial application in the removal of indoor HCHO and other organic pollutant gases.
2.3 Bactericidal and bacteriostatic properties for E. coli and S. aureus
The bactericidal and bacteriostatic properties of bimetallic oxide Cu2O@MnO2 for Gram bacteria such as E. coli and S. aureus were evaluated with bacteriostatic zone, minimum inhibitory concentration (MIC) and bacterial growth curve tests.45,46 Photographs of the bacteriostatic zones of different samples cultured in agar seeded with 109 CFU mL−1E. coli and S. aureus for 24 h are shown in Fig. 4. It was found that all the Cu2O@MnO2 samples except pure MnO2 produced a blue bacteriostatic zone on the agar medium, indicating that the bimetallic oxides have certain bacteriostatic abilities compared with pure MnO2.47 Based on the diagram in Fig. 4m, the distance between the outer diameter of the bacteriostatic zone and the edge of the material was measured with a Vernier caliper and recorded as bacteriostatic zone width to quantify and compare the bacteriostatic abilities of different materials for various pathogenic bacteria. In Fig. 4n and o, the variation tendencies of the bacteriostatic zone width for E. coli and S. aureus with an increase in Cu2O deposition time in various Cu2O@MnO2 samples are the same. The antibacterial capacity of Cu2O@MnO2 gets stronger and stronger with the increase in Cu2O deposition time, and it reaches a peak in the “120 min Cu2O@MnO2” sample, and then decreases slightly. It can be inferred that, although Cu2O makes an obvious enhancement to the bactericidal ability of Cu2O@MnO2, excessive Cu2O basically covers the substrate MnO2 completely to inhibit the interfacial effect of the bimetallic oxide, thus reducing its bacteriostatic ability. Thus, bimetallic oxide Cu2O@MnO2 with exposed phase interfaces has a stronger bactericidal property for E. coli and S. aureus than a Cu-based bacteriostatic agent Cu2O alone.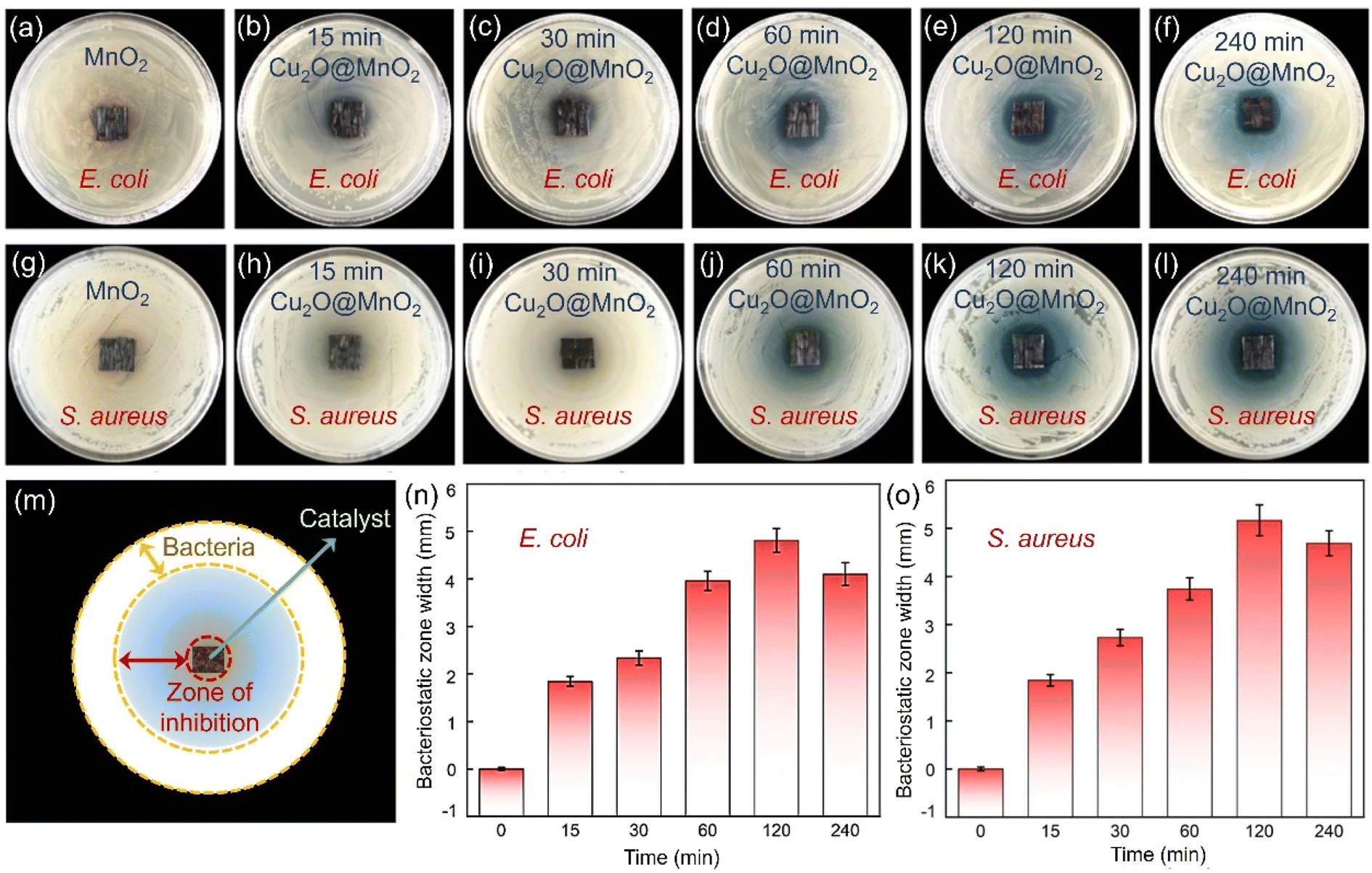 | ||
| Fig. 4 Photographs of the bacteriostatic zone effect of Cu2O@MnO2 samples with different deposition times for (a–f) E. coli and (g–l) S. aureus. (m) Diagram of the effective area of the bacteriostatic zone. Bacteriostatic zone width of Cu2O@MnO2 samples with different deposition times for (n) E. coli and (o) S. aureus. | ||
The “120 min Cu2O@MnO2” sample with good bacteriostatic performance in the above bacteriostatic zone tests underwent further MIC tests, as shown in Fig. S9.† For the pathogenic bacterium E. coli (Fig. S9a†), the pure MnO2 material still does not show any antibacterial activity against the bacteria, while the bimetallic oxide Cu2O@MnO2 with different concentrations show different bactericidal abilities. The MIC of Cu2O@MnO2 was between 400 and 600 μg mL−1 when the bacterial concentration was 105 CFU mL−1. Thus, to determine the final MIC value of the bacteriostatic material, the bacterial concentration was further diluted to 104 CFU mL−1. It was found that only 5 E. coli colonies grew when the concentration of the “120 min Cu2O@MnO2” antibacterial material was 300 μg mL−1, while no colonies grew when the concentration was in the range of 400 μg mL−1 to 800 μg mL−1. Therefore, the MIC value of “120 min Cu2O@MnO2” was determined to be 400 μg mL−1 for E. coli with a bacterial concentration of 105 CFU mL−1.48 In a similar way, the MIC value of “120 min Cu2O@MnO2” for S. aureus with a bacterial concentration of 105 CFU mL−1 was calculated to be 10 μg mL−1 (Fig. S9b†). For bimetallic oxide Cu2O@MnO2, the MIC value for E. coli is 40 times greater than that for S. aureus, meaning that the composite has better ability to inactivate S. aureus. Moreover, the bacterial growth curve (Fig. S10a†) shows that the E. coli growth process under the action of “120 min Cu2O@MnO2” includes three typical phases: slow growth, logarithmic growth and steady phase. As the concentration of “120 min Cu2O@MnO2” gradually increased, the logarithmic phase of the E. coli growth curve tended to be smooth. In particular, the logarithmic growth phase could be stable for a long time when the bacterial inhibitor concentration reached 320 μg mL−1, and the antibacterial efficiency reached 57.64%. In addition, the long-term bacteriostatic effect on S. aureus went up to 92.4% when the concentration of Cu2O@MnO2 reached 80 μg mL−1, and there was no colony propagation within 24 h after the introduction of the antibacterial agent, showing the strong bactericidal ability of the antibacterial agent (Fig. S10b†). Therefore, it can be inferred that bimetallic oxide Cu2O@MnO2 has better inactivation ability on the pathogenic bacterium S. aureus than on E. coli, which shows bacteriostatic performance against E. coli and excellent bactericidal performance against S. aureus.
2.4 The interfacial effects of bimetallic oxide
In order to further explore the interface effects of the bimetallic oxide, models of Cu2O@MnO2 (Fig. 5a), Cu2O (Fig. S11†) and MnO2 (Fig. S12†) were established for theoretical calculation by VASP. According to the differential charge density distribution shown in Fig. 5b, the charge density increases at the interface between Cu2O and MnO2 of the bimetallic oxide, indicating that a strong electron migration process occurs here. In particular, a reconstruction of electron state and density was realized between the interfacial Olatt and its neighboring metal atoms by electron exchange between them (Fig. 5c), which could not only create a more secure binding between the two metal oxides, but could also increase the amount of delocalized electrons in the phase interface to obtain the electron-rich region.49 Furthermore, the total density of states (DOS) of Cu2O@MnO2, Cu2O and MnO2 were calculated, as shown in Fig. 5d. The bimetallic oxide shows much higher occupation than that of the single metal oxides near the Fermi level, meaning a promoted electron transfer of Cu2O@MnO2. In Fig. 5e, the projected density of states (PDOS) in the d-band from Mn and Cu atoms also show a similar tendency to the total DOS, which reveals that the increased DOS of Cu2O@MnO2 near the Fermi level is mainly derived from the contribution of metal atom d orbitals. Moreover, in Fig. 5f, the whole PDOS peak of the O atom at the interface of the bimetallic oxide right-shifted a little compared with the metal oxide of MnO2 and Cu2O. This indicated that the energy of O atoms is promoted to a higher energy level by forming a Cu2O/MnO2 interface, which causes the lattice oxygen atom to gain or lose electrons more easily.50 Moreover, by comparing O2 adsorption energy on the material surface (Fig. 5g), it can be inferred that the bimetallic oxide interface is more likely to capture O2 in the air and activate it into reactive oxygen species to participate in the subsequent oxidative degradation of HCHO and pathogenic bacteria.51 In conclusion, the deep coupling interface of the bimetallic oxide Cu2O@MnO2 plays a critical role in regulating the electron density of the two phases to obtain an electron-rich region, which improves the ability of lattice oxygen to gain and lose electrons in the catalyst and the adsorptive-active ability of O2 molecules under the action of the interface for the efficient degradation of organic pollutants and pathogens. | ||
| Fig. 5 Theoretical investigations. (a) Computational models and (b) localized electric field distribution of Cu2O@MnO2. (c) 2D isosurface map of the cross-section consisting of Cu, Mn, and O atoms in Cu2O@MnO2. Calculated PDOS in (d) total band, (e) d-band, (f) s-band and p-band of Cu2O@MnO2, Cu2O and MnO2. (g) O2 adsorption energy on Cu2O@MnO2, Cu2O and MnO2. | ||
In general, compared with the pure catalyst MnO2 and bacteriostatic agent Cu2O, bimetallic oxide Cu2O@MnO2 with exposed phase interfaces respectively shows superior HCHO removal efficiency and pathogen inactivation ability, which could be attributed to the following two factors. On the one hand, the modification of MnO2 by Cu2O particles induces the generation of reactive oxygen species via the separated electron–hole pairs under the assistance of visible light for the oxidative degradation of HCHO and inactivation of bacterial DNA double strands, achieving the double-effect purification of chemical and biological indoor pollutants by an Mn-based composite;52,53 on the other hand, the bimetallic oxide Cu2O@MnO2 with exposed phase interfaces improves the migration of lattice oxygen and the adsorption–activation of O2 on the surface of materials to generate more reactive oxygen species for the degradation of HCHO and pathogenic bacteria.54 In conclusion, this work effectively provides innovative ideas and exploration experience for the construction of composite materials with exposed phase interfaces and the design of multifunctional purification materials for indoor chemical and biological indoor environmental pollutants.
3. Conclusion
In this work, a bimetallic oxide Cu2O@MnO2 with exposed phase interfaces was prepared by a two-step electrochemical method with surfactant conditioning for the dual-effect purification of indoor HCHO and pathogenic bacteria, which shows the composite structure of non-continuously dispersed Cu2O particles anchored on flower-like MnO2. In an evaluation of material purification performance, Cu2O@MnO2 shows superior HCHO removal efficiency and pathogen inactivation ability compared with the pure catalyst MnO2 and bacteriostatic agent Cu2O, respectively. It shows dynamic HCHO removal efficiency of 97.2% with a WHSV of 120000 mL g−1 h−1 under the action of light at room temperature. For the pathogenic bacterium S. aureus, the minimum inhibitory concentration of Cu2O@MnO2 for 104 CFU mL−1S. aureus is 10 μg mL−1, and the long-term bacteriostatic effect was up to 92.4% when the concentration of Cu2O@MnO2 reached 80 μg mL−1. The different phase components in the bifunctional catalyst Cu2O@MnO2 show their own advantages for the removal of the two kinds of pollutant. Specifically, the MnO2 component is mainly responsible for the catalytic-oxidative decomposition of HCHO at room temperature by activating O2 to  and the Cu2O component is mainly responsible for inactivating pathogenic bacteria by the synergistic effect of Cu+ ions and photocatalyzed active hydroxyl groups. According to the material characterization and theoretical calculation, its excellent catalytic-oxidative activity can be attributed to the electron-rich region at the phase interfaces fully exposed to the reaction atmosphere, inducing the capture and activation of O2 on the material surface, and then promoting the generation of reactive oxygen species used for the oxidative-removal of HCHO and bacteria. Additionally, Cu2O as a photocatalytic semiconductor further enhances the catalytic ability of Cu2O@MnO2 under the assistance of visible light. In conclusion, this work will provide efficient theoretical guidance and a practical basis for the ingenious construction of multiphase coexisting composites in the field of multi-functional indoor pollutant purification strategies.
and the Cu2O component is mainly responsible for inactivating pathogenic bacteria by the synergistic effect of Cu+ ions and photocatalyzed active hydroxyl groups. According to the material characterization and theoretical calculation, its excellent catalytic-oxidative activity can be attributed to the electron-rich region at the phase interfaces fully exposed to the reaction atmosphere, inducing the capture and activation of O2 on the material surface, and then promoting the generation of reactive oxygen species used for the oxidative-removal of HCHO and bacteria. Additionally, Cu2O as a photocatalytic semiconductor further enhances the catalytic ability of Cu2O@MnO2 under the assistance of visible light. In conclusion, this work will provide efficient theoretical guidance and a practical basis for the ingenious construction of multiphase coexisting composites in the field of multi-functional indoor pollutant purification strategies.
4. Experimental section
4.1 Synthesis of MnO2–CC
A piece of carbon cloth (CC, 10 × 10 cm2, 1.8 g) was immersed in a mixed solution of concentrated sulfuric acid and nitric acid (volume ratio of 4:1) and kept warm at 60 °C for 30 minutes. The acid-treated CC was cleaned with deionized water several times and then placed in a vacuum drying furnace at 80 °C for 3 hours to complete the pre-treatment of the substrate material. Next, with a piece of CC as the anode and a platinum sheet as the cathode, electrochemical deposition was carried out in 0.05 M KMnO4 electrolyte solution with deposition voltage, time and temperature of 12 V, 60 min and 80 °C, respectively. Subsequently, the electrodeposited CC was fully washed with deionized water and annealed in a vacuum drying oven at 110 °C for 12 h to obtain the catalyst MnO2–CC. For more details of the above preparation process of the MnO2–CC catalyst, see our previous published work.29
4.2 Synthesis of bimetallic oxide Cu2O@MnO2–CC (abbreviated to Cu2O@MnO2)
Firstly, 9.987 g of copper sulfate pentahydrate (CuSO4·5H2O) and 10 g of PEG-6000 were dissolved in a beaker with 400 mL of deionized water, and stirred magnetically at room temperature for about 30 min until the drug was fully dissolved. Then, 26.3 mL of lactic acid liquid (C3H6O3) was added to the above solution, and the beaker was sealed with plastic wrap and placed in an ultrasonic cleaning machine for ultrasonic processing for 20 min. After the pH value of the above configured solution had been adjusted to 12.5 with 4 M NaOH solution, precursor electrolyte solution containing the complex of Cu[C3H6O3]2 was obtained. Then, with Pt sheet as the anode and MnO2–CC as the cathode, electrochemical deposition under a constant current of 15 mA was carried out in the above electrolyte at 60 °C. After the electrodeposition, the cathode material was taken out and washed with deionized water, and was dried at 60 °C for 12 hours to obtain bimetallic oxide Cu2O@MnO2. In particular, the loading amount and dispersion of Cu2O anchored on MnO2 are controlled by controlling the deposition time, and samples prepared with electrodeposition times of 15, 30, 60, 120 and 240 min were named “15 min Cu2O@MnO2”, “30 min Cu2O@MnO2”, “60 min Cu2O@MnO2”, “120 min Cu2O@MnO2” and “240 min Cu2O@MnO2”, respectively.4.3 Efficiency evaluation of catalytic HCHO oxidation removal
Firstly, the static HCHO removal efficiency of the prepared samples was evaluated in a home-made test chamber, and the relevant parameters were as follows: the test chamber volume was 48000 mL, the test environment was kept at 25 °C and 50% relative humidity (RH), and the amount of samples to be tested was consistent in each test (four pieces of sample containing a total of 1.0 g of Cu2O@MnO2). Before starting the static test, an appropriate amount of 38% HCHO solution was dripped into the closed test chamber and the HCHO concentration was stabilized at 7 ppm by motor-driven air circulation (this process could take 45 min). Subsequently, by operating the motor outside the test chamber, the sample to be tested was exposed to air containing 7 ppm HCHO, and the change in HCHO concentration in the test chamber over time was monitored with an HCHO tester (PPM-HTV, UK, the measuring range is 0–10 ppm, the test accuracy is 0.01 ppm) in real time under the test conditions of dark state and light state, respectively. In particular, the dark state condition was built by covering the outside of the closed test chamber with shading cloths, and the visible light conditions were simulated by external xenon lamps (PL-XQ, 500 W). In addition, single-pass dynamic HCHO removal efficiency tests were performed on the prepared samples over a long period to evaluate their catalytic stability. In the dynamic tests, two pieces of sample (two pieces of the sample containing a total of 0.5 g of Cu2O@MnO2) were placed into the transparent glass reaction tube, and the intake and outlet of the reaction tube were connected to standard HCHO gas (RH: 45%, O2: 21 wt%, HCHO initial concentration: ∼7 ppm, the rest is N2) and an HCHO concentration tester (PPM-HTV, UK, the measuring range is 0–10 ppm, the test accuracy is 0.01 ppm), respectively. In order to exclude experimental errors, the HCHO concentration value at the outlet was read after continuous ventilation with standard gas for 5 min. The continuous catalytic stability of the sample was evaluated by a long-term dynamic test (air flow: 1000 mL min−1, WHSV: 120000 mL g−1 h−1) under the test conditions of visible light simulated by external xenon lamps. The dynamic HCHO removal efficiency is calculated with the following formula:| HCHO removal efficiency = Cinlet − Coutlet/Cinlet × 100% | (1) |
| ln(Ct) = (−k0 + CADR/V)t + ln(C0) | (2) |
4.4 Bactericidal and antimicrobial capacity evaluation
In all the following tests, all laboratory equipment and samples were sterilized at 120 °C for 30 min to ensure the accuracy of the tests. In this work, E. coli and S. aureus were used as the target species of Gram-negative and Gram-positive bacteria, respectively; the bactericidal and bacteriostatic properties of the samples were comprehensively evaluated by antibacterial zone tests, growth curve tests and MIC tests. In the bacteriostatic zone test, 100 mL of bacterial solution (about 109 CFU mL−1, CFU: colony forming units) was evenly spread on the surface of a prepared semi-solid agar plate, at the center of which was placed a piece of tailored sample (15 mm × 15 mm). After 24 h of culture in a sterile chamber at 37 °C, the bacteriostatic ability of different samples was evaluated and compared by measuring the width of the bacteriostatic zone. In addition, in order to further explore the inhibition effect of the samples on bacteria, a growth curve test and MIC test also were carried out in this work, and Cu2O@MnO2 powders to be tested were obtained by separation–grinding–ultrasonic-drying before the tests. In the growth curve test, 1 mL of the target bacterial solution with a concentration of 109 CFU mL−1 was injected into 200 mL of agar medium, and all experiments were conducted in a sterile incubator at 37 °C with a rotation speed of 250 rpm. After samples of different concentrations were injected into the above bacterial solution, 3 mL was extracted every 1 hour and the absorbance of the solution at a wavelength of 600 nm was determined with an ultraviolet spectrophotometer. In particular, according to the bacteriostatic ability of the samples, the bacterial solutions of E. coli and S. aureus were collected continuously for 24 hours and 16 hours, respectively, to draw the growth curve. In the MIC test for E. coli, the target bacterial solution with a concentration of 109 CFU mL−1 was diluted into two groups of bacterial solutions with concentrations of 104 CFU mL−1 and 105 CFU mL−1. Next, bacteriostatic agar was prepared by adding different concentrations of Cu2O@MnO2 solutions (100 μg mL−1, 200 μg mL−1, 300 μg mL−1, 400 μg mL−1, 600 μg mL−1, 800 μg mL−1) into nutrient agar. Subsequently, the above two groups of bacterial solution were coated on the bacteriostatic agar, which was placed in an incubator at a constant temperature of 37 °C for 48 hours and then taken out to observe the bacteriostatic situation. For the MIC test for S. aureus, the concentration of Cu2O@MnO2 was simply changed in the bacteriostatic agar to 5, 10, 20, 40, 80 g mL−1 and the preceding steps were repeated.4.5 Characterizations
In this work, the surface morphology and microstructure of the materials were characterized with an SU 9000 high-resolution field-emission SEM produced by the Hitachi company. The composition and phase structure of the prepared samples were investigated with an XRD produced by Bruker AXS in Germany. In particular, Cu-Kα was taken as a ray source with a scanning range of 10–90° and operating voltage of 30 kV. The composition phase and element content of the material were determined with a Raman spectrometer (Raman, LabRAM HR800) with an excitation wavelength of 532 nm and inductively coupled plasma emission spectroscopy (ICP-OES, Thermo Fisher Scientific). TEM was used for further study of the structure of the prepared material, the model for which was a Tecnai G2 F20 S-TWIN with an acceleration voltage of 200 kV and point resolution of 0.24 nm produced by FEI in the United States. In addition, a UV-Vis spectrophotometer (UV-3600, Shimadzu, Japan) was used for determination of the absorbance of the bacterial solution at 600 nm wavelength. UV-Vis-NIR diffuse-reflectance spectra (DRS) were achieved on a Hitachi U-4100 spectrophotometer. EPR analyses were conducted on a Bruker EPR 300E spectrometer to explore the concentration of surface oxygen vacancies on different samples. Moreover, the parameters set for the theoretical calculations by VASP are described in detail in the ESI (I)† for this work.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors gratefully acknowledge the National Natural Science Foundation of China (NSFC, 52070006), the National Innovation Demonstration Zones and Collaborative Innovation Platform Project for Fuzhou–Xiamen–Quanzhou (Grant No. 2021FX05), and Beijing University Students Innovation and Entrepreneurship Training Inter school Cooperation Program (202298026).References
- C. He, C. Jie, Z. Xin, D. Mark, P. Samuel and H. Zhengping, Chem. Rev., 2019, 119, 4471–4568 CrossRef CAS PubMed.
- K. Huang, W. Sun, G. Feng, J. Wang and J. Song, Sustain. Cities Soc., 2020, 54, 101947–101957 CrossRef.
- D. Qin, B. Guo, J. Zhou, H. Cheng and X. Chen, Sci. Rep., 2020, 10, 332–340 CrossRef CAS PubMed.
- H. Han, J. Zhu, D. Q. Wu, F. X. Li, X. L. Wang, J. Y. Yu and X. H. Qin, Adv. Funct. Mater., 2019, 29, 1806594 CrossRef.
- Ministry of Health, Indoor Air Quality Standard, National Standard, China Environmental Science Press, GB/T 18883-2002 Search PubMed.
- Anonymous, J. Clin. Pathol., 2006, 6, 691 Search PubMed.
- L. Wang, X. Zhang, X. Yu, F. Gao, Z. Shen, X. Zhang, S. Ge, J. Liu, Z. Gu and C. Chen, Adv. Mater., 2019, 31, 1901965 CrossRef PubMed.
- Z.-m. Xiu, Q.-b. Zhang, H. L. Puppala, V. L. Colvin and P. J. J. Alvarez, Nano Lett., 2012, 12, 4271–4275 CrossRef CAS PubMed.
- J. Y. Zheng, K. L. Zhou, W. K. Zhao, Y. Wang, J. He, X. Wang, H. Wang, H. Yan and C. B. Han, J. Colloid Interface Sci., 2022, 628, 359–370 CrossRef PubMed.
- J. Y. Zheng, W. K. Zhao, L. Song, H. Wang, H. Yan, G. Chen, C. B. Han and J. Zhang, Green Energy Environ., 2022 DOI:10.1016/j.gee.2022.01.008.
- J. Wang, D. Li, P. Li, P. Zhang, Q. Xu and J. Yu, RSC Adv., 2015, 5, 100434–100442 RSC.
- S. Tu, Y. Chen, X. Zhang, J. Yao, Y. Wu, H. Wu, J. Zhang, J. Wang, B. Mu, Z. Li and Q. Xia, Appl. Catal., A, 2021, 611, 117975–117985 CrossRef CAS.
- C. Wang, Y. Li, L. Zheng, C. Zhang, Y. Wang, W. Shan, F. Liu and H. He, ACS Catal., 2021, 11, 456–465 CrossRef CAS.
- W. K. Zhao, J. Y. Zheng, C. B. Han, J. Ruan, Y. Lu, K. L. Zhou, T. R. Zhai, H. Wang and H. Yan, Chem. Eng. J., 2022, 440, 135877 CrossRef.
- Z. Wang, H. Yu, Y. Xiao, L. Zhang, L. Guo, L. Zhang and X. Dong, Chem. Eng. J., 2020, 394, 125014 CrossRef CAS.
- Z. Lin, W. Shen, J.-P. Corriou, X. Chen and H. Xi, J. Colloid Interface Sci., 2022, 608, 1769–1781 CrossRef CAS PubMed.
- H. Gao, J. Zhang, R. Wang and M. Wang, Appl. Catal., B, 2015, 172, 1–6 CrossRef.
- Y. Lu, X. Zhang, Y. Chu, H. Yu, M. Huo, J. Qu, J. C. Crittenden, H. Huo and X. Yuan, Appl. Catal., B, 2018, 224, 239–248 CrossRef CAS.
- J. Nie, X. Yu, Z. Liu, Y. Wei, J. Zhang, N. Zhao, Z. Yu and B. Yao, Appl. Surf. Sci., 2022, 576, 151842 CrossRef CAS.
- J. Zhou, Y. Wang, W. Pan, H. Xiang, P. Li, Z. Zhou and M. Zhu, J. Mater. Sci. Technol., 2021, 81, 58–66 CrossRef CAS.
- X. Shi, C. Xue, F. Fang, X. Song, F. Yu, M. Liu, Z. Wei, X. Fang, D. Zhao, H. Xin and X. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 8386–8392 CrossRef CAS PubMed.
- S. Rong, T. He and P. Zhang, Appl. Catal., B, 2020, 267, 118375 CrossRef CAS.
- J. Ye, M. Zhou, Y. Le, B. Cheng and J. Yu, Appl. Catal., B, 2020, 267, 118689–118698 CrossRef CAS.
- S. Zhang, H. Wang, H. Si, X. Jia, Z. Wang, Q. Li, J. Kong and J. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 40285–40295 CrossRef CAS PubMed.
- L. Miao, J. Wang and P. Zhang, Appl. Surf. Sci., 2019, 466, 441–453 CrossRef CAS.
- L. Du, P. Li, W. Gao, X. Ding, W. Jiao and Y. Liu, J. Taiwan Inst. Chem. Eng., 2021, 118, 29–37 CrossRef CAS.
- M. Cao, H. Wang, X. Wang, F. Chen, S. Ji, S. Pasupathi and R. Wang, J. Alloys Compd., 2019, 805, 977–983 CrossRef CAS.
- C. Chai, A. Liu, Y. Lv, J. Mu, X. Zhang and H. Che, Mater. Lett., 2017, 196, 308–311 CrossRef CAS.
- J. Y. Zheng, W. K. Zhao, X. Wang, Z. Zheng, Y. Zhang, H. Wang, H. Yan, X. Song and C. B. Han, Chem. Eng. J., 2020, 401, 125790–125812 CrossRef CAS.
- M. Li, D. Wang, J. Li, Z. Pan, H. Ma, Y. Jiang, Z. Tian and A. Lu, Chin. J. Catal., 2017, 38, 597–606 CrossRef CAS.
- T. Lan, C. Mundt, M. Tran and S. Padalkar, J. Mater. Res., 2017, 32, 1656–1664 CrossRef CAS.
- A. K. Mohapatra, S. Mohanty and S. K. Nayak, Polym. Compos., 2014, 35, 283–293 CrossRef CAS.
- W. Yang, J. Xu, L. Niu, J. Zhao, C. Wang and X. Liu, Part. Sci. Technol., 2017, 36, 844–849 CrossRef.
- Z. Wang, Z. Qu, X. Quan, Z. Li, H. Wang and R. Fan, Appl. Catal., B, 2013, 134–135, 153–166 CrossRef CAS.
- L. Dorner, P. Schmutz, P. Kagi, M. V. Kovalenko and L. P. H. Jeurgens, Langmuir, 2020, 36, 8075–8085 CrossRef CAS PubMed.
- R. Yang, Y. Fan, R. Ye, Y. Tang, X. Cao, Z. Yin and Z. Zeng, Adv. Mater., 2021, 33, e2004862 CrossRef PubMed.
- N. Zou, Q. Nie, X. Zhang, G. Zhang, J. Wang and P. Zhang, Chem. Eng. J., 2019, 357, 1–10 CrossRef CAS.
- Y. Huang, K. Ye, H. Li, W. Fan, F. Zhao, Y. Zhang and H. Ji, Nano Res., 2016, 9, 3881–3892 CrossRef CAS.
- A. Zhang, R. Gao, L. Hu, X. Zang, R. Yang, S. Wang, S. Yao, Z. Yang, H. Hao and Y.-M. Yan, Chem. Eng. J., 2021, 417, 129186 CrossRef CAS.
- W. Ahmad, E. Park, H. Lee, J. Y. Kim, B. C. Kim, J. Jurng and Y. Oh, Appl. Surf. Sci., 2021, 538, 147504 CrossRef CAS.
- Y. Huang, Y. Liu, W. Wang, M. Chen, H. Li, S.-c. Lee, W. Ho, T. Huang and J. Cao, Appl. Catal., B, 2020, 278, 119294 CrossRef CAS.
- S. Giraudet, B. Boulinguiez and P. Le Cloirec, Energy Fuels, 2014, 28, 3924–3932 CrossRef CAS.
- C. Yao, A. Yuan, H. Zhang, B. Li, J. Liu, F. Xi and X. Dong, J. Colloid Interface Sci., 2019, 533, 144–153 CrossRef CAS PubMed.
- J. Wang, G. Zhang and P. Zhang, Appl. Catal., B, 2018, 239, 77–85 CrossRef CAS.
- N. Jo, B. Kim, S.-M. Lee, J. Oh, I. H. Park, K. J. Lim, J.-S. Shin and K.-H. Yoo, Biosens. Bioelectron., 2018, 102, 164–170 CrossRef CAS PubMed.
- R. Diaz, V. Afreixo, E. Ramalheira, C. Rodrigues and B. Gago, Clin. Microbiol. Infect., 2018, 24, 97–104 CrossRef CAS PubMed.
- L. Feng, S. Zhang, B. Tao, B. Tan, B. Xiang, W. Tian and S. Chen, Colloids Surf., B, 2020, 190, 110898 CrossRef CAS PubMed.
- N. J. Hos, N. Jazmati, D. Stefanik, M. Hellmich, H. AlSael, W. V. Kern, S. Rieg, H. Wisplinghoff, H. Seifert and A. J. Kaasch, J. Infect., 2017, 74, 248–259 CrossRef PubMed.
- T. R. Kafle, B. Kattel, S. Wanigasekara, T. Wang and W. L. Chan, Adv. Energy Mater., 2020, 10, 1904013 CrossRef CAS.
- H. Wu, N. Zhang, H. Wang and S. Hong, Chem. Phys. Lett., 2013, 568–569, 84–89 CrossRef CAS.
- H. M. Dao, I. Husain, V. K. Shankar, S. I. Khan, S. N. Murthy and S. Jo, Chem. Commun., 2020, 56, 12017–12020 RSC.
- W. Liu, M. Shi, Y. Li, Z. Wu, L. Yang, S. Zhang, X. Xiao, C. Liu, W. Dai, C. Chen, X. Tu, J. Zou and X. Luo, Appl. Catal., B, 2022, 312, 121427 CrossRef CAS.
- S. Jang, S. Jung, S. Song, S. Lee, H. Lee, E. Cho, H. J. Lee, S. Park, B. Youn and K. H. Park, Environ. Sci.: Nano, 2021, 8, 1226–1235 RSC.
- S. Saidin, M. A. Jumat, N. A. A. Mohd Amin and A. S. Saleh Al-Hammadi, Mater. Sci. Eng., C, 2021, 118, 111382 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00922f |
| ‡ The authors contribute equally to the article. |
| This journal is © The Royal Society of Chemistry 2023 |