
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChallenges and opportunities for SERS in the infrared: materials and methods
Chiara
Deriu
 *a,
Shaila
Thakur
a,
Olimpia
Tammaro
a and
Laura
Fabris
ab
*a,
Shaila
Thakur
a,
Olimpia
Tammaro
a and
Laura
Fabris
ab
aDepartment of Applied Science and Technology, Politecnico di Torino, 10129 Turin, Italy. E-mail: chiara.deriu@polito.it
bDepartment of Materials Science and Engineering, Rutgers University, Piscataway, NJ 08854, USA
First published on 22nd March 2023
Abstract
In the wake of a global, heightened interest towards biomarker and disease detection prompted by the SARS-CoV-2 pandemic, surface enhanced Raman spectroscopy (SERS) positions itself again at the forefront of biosensing innovation. But is it ready to move from the laboratory to the clinic? This review presents the challenges associated with the application of SERS to the biomedical field, and thus, to the use of excitation sources in the near infrared, where biological windows allow for cell and through-tissue measurements. Two main tackling strategies will be discussed: (1) acting on the design of the enhancing substrate, which includes manipulation of nanoparticle shape, material, and supramolecular architecture, and (2) acting on the spectral collection set-up. A final perspective highlights the upcoming scientific and technological bets that need to be won in order for SERS to stably transition from benchtop to bedside.
 Chiara Deriu | Chiara Deriu obtained her PhD in Chemistry from Florida International University (FIU), Miami, USA, in 2020, where she worked on tailoring the stabilization of nanomaterials for the detection of drugs by SERS. Following, she received a postdoctoral appointment at FIU to work on the computational modeling of adsorbates on bimetallic clusters. She then joined Politecnico di Torino (Italy) in September 2021, where she is an ERC-funded postdoctoral researcher. Her scientific interests lie at the intersection of physical chemistry and the analytical sciences, with particular attention to nanoscale surface chemistry and its effects on both nanofabrication and SERS analytical performance. |
 Shaila Thakur | Shaila Thakur obtained her PhD in Chemical Engineering from Indian Institute of Technology (IIT) Kharagpur, India in 2017. She worked as a postdoctoral scholar at National Center for Biological Sciences (NCBS) Bangalore (India) from 2020–2021. She joined Politecnico di Torino (Italy) as a postdoctoral fellow in 2022. She currently works on an ERC-funded project based on the utilization of plasmonic nanostructures for probing prostate cancer biomarkers. Her research interests include nanomaterial synthesis, biosensing, and interaction of nanomaterials with cells, DNA/RNA and proteins. |
 Olimpia Tammaro | Olimpia Tammaro is a postdoctoral researcher at the Department of Applied Sciences and Technologies at the Politecnico di Torino (Italy). She received an MSc in Materials Engineering from University of Naples “Federico II” (Italy) in 2016. She obtained her PhD in the group of Prof. P. A. Netti at IIT – CABHC (Italy) in 2020. The topic of her doctoral thesis was the development through a microfluidic platform of multi-modal probes. She then carried out postdoctoral research at the Politecnico di Torino (Italy) in the field of chemistry for technologies, with a focus on the synthesis of functional materials. |
 Laura Fabris | Laura Fabris received her B.S./M.S. degree in Chemistry in 2001 from the University of Padova, where she was awarded a PhD in 2006. She was then a postdoc at the University of California Santa Barbara. In 2009 she moved to Rutgers University in the Department of Materials Science and Engineering where she remained first as Assistant then as Associate Professor. Upon receiving an ERC Consolidator Grant, she moved to Politecnico di Torino (Italy) in the Department of Applied Science and Technology where she is a Full Professor. Her research combines fundamental notions of nanomaterials chemistry and spectroscopy for the development of new technologies addressing important challenges in medicine, biology, and energy. |
1 Introduction
When setting up a new laboratory infrastructure for surface-enhanced Raman spectroscopy (SERS), scientists have nowadays the privilege to choose among the widest-to-date set of commercially available instrumentation: from tunable lasers to microscope-integrated spectrometers, from easily couplable instruments that allow for operando measurements, to portable and miniaturized technology enabling measurements in the field or in vivo. This was certainly not the case twenty years ago. Raman instruments were often built in-house, the knowledge on the manipulation of materials at the nanoscale, and with it, the whole world of plasmonics, was still very limited. Surely, applications of SERS to everyday analytical issues were pioneering, when not still out of reach.Fueled by the nanotechnology revolution that characterized the 2000s, SERS has managed to rapidly advance from niche, obscure analytical technique, to a thriving field at the forefront of analytical innovation. The technological advancements in bottom-up fabrication with increasing control over size and shape,1–5 the development of high precision top-down and soft patterning methods,6–8 the increasing understanding of nanoscale assembly and its integration across larger length scales,9 all greatly contributed to a transformative era for SERS.10 In a synergistic tie, nanotechnology and SERS prompted new fundamental research over the last twenty years, opening the doors to plasmonics,11,12 thus leading to a deeper understanding of the relationships between physical characteristics (material, size, shape, dielectric environment) of SERS-active nanomaterials and their optical properties.13–16
The 2000s are also to be remembered as the years that saw a sharp increase in the interest in the biological world: the sequencing of the whole human genome,17 the cloning of mammals,18–21 and the development of pharmacogenomics and molecular diagnostics that set the basis for what we now call personalized medicine.22–27 The SERS community was certainly not an outsider in this global trend; aided by its single molecule sensitivity, it promised access to the study of otherwise inaccessible low concentration biological species.
SERS has an advantage over traditional Raman spectroscopy to allow for trace detection, but it still faces some challenges similar to Raman spectroscopy when dealing with biological analytes like cells and tissues. This is because such challenges—self-absorption and autofluorescence—are inherent to the molecular structure and complexity of biological systems.28 With self-absorption we define the physical phenomenon by which sample-scattered photons are absorbed by the sample itself, before reaching the detector, while with autofluorescence we indicate the spontaneous fluorescence emission that a molecule, typically characterized by delocalized π electrons, undergoes upon laser irradiation. These affect the intensity and the signal to noise ratio (S/N) of the detected signal, respectively, and are especially prominent when excitation in the visible range is utilized.28–30 This is because common tissue constituents, such as hemoglobin, are chromophores that have both the highest absorption coefficients and fluorescence-related electronic transitions in the visible spectrum.28,29
While it is true that the plasmonic nanoparticles also act as fluorescence quenchers,31 a fluorescence background, also called SERS continuum,31 is often present in SERS spectra of biological systems.32,33 This may be due to the interplay between the analyte's distance from the SERS-active surface, the size and three-dimensional conformation of the analyte on the SERS-active surface, and the concomitant presence of other species (e.g., nanoparticle synthesis by-products, capping agents, residual biological matrix components). The most common strategy that is utilized in traditional Raman spectroscopy to mitigate autofluorescence is to decrease the energy provided to the system, by utilizing lasers emitting at longer wavelengths, for example in the near infrared (NIR).
Incidentally, the use of NIR light can also solve self-absorption phenomena. For instance, the main constituents of tissue, namely whole blood and water, show the lowest absorption coefficients in the regions from 700 to 1000 nm (first biological window, NIR I) and from 1000 nm to 1400 nm (second biological window, NIR II) (Fig. 1). By reducing absorption, the penetration of the radiation also increases, making imaging of deeper (>500 μm to cm)28 tissues possible. Furthermore, less energetic excitation sources make Raman spectroscopy amenable to in vivo measurements, as they are less likely to impart photochemical damage to samples. Since these principles of Raman spectroscopy also apply to SERS, the problem of autofluorescence and self-absorption phenomena in biological specimen can be solved or minimized by switching to NIR excitation wavelengths. This, however, does not come without its own set of challenges.
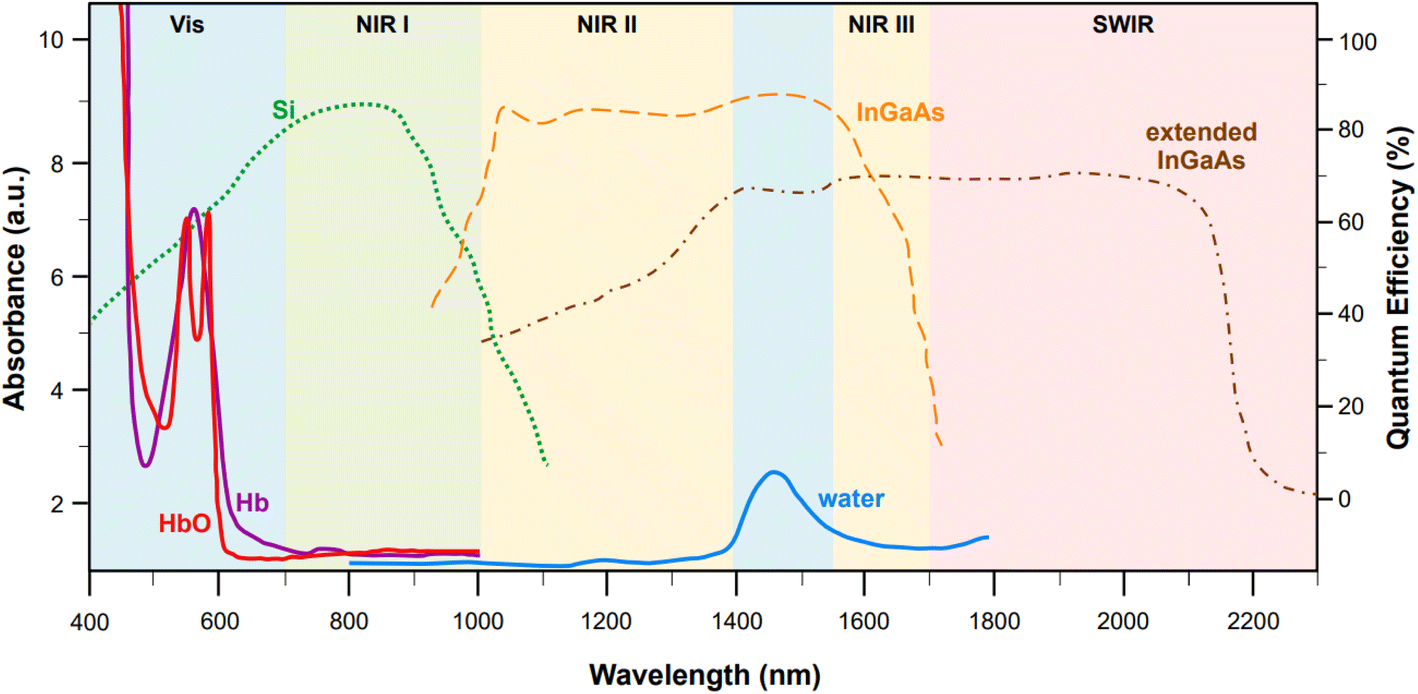 | ||
Fig. 1 Representative absorption spectra (y axis on the left, optical length of 1 mm) of oxygenated hemoglobin (HbO, red),38 deoxygenated hemoglobin (Hb, purple),38 and water (blue).38 The quantum efficiencies (y axis on the right) for the main NIR and SWIR detectors are also reported: silicon-based back-thinned deep depletion EMCCD (green, dotted),39 lattice-matched InGaAs (orange, dashed),39 and extended InGaAs (brown, dot-and-dash).40 The following spectral ranges are highlighted: NIR I (700–1000 nm, first biological window), NIR II (1000–1400 nm second biological window), NIR III (1550–1700 nm), and SWIR (>1700 nm). Figure inspired by Smith et al., 2009![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39 and Hong et al., 2017.38 39 and Hong et al., 2017.38 | ||
2 Overview of challenges
While moving to higher wavelengths can reduce or eliminate autofluorescence and self-absorption phenomena, it also causes a concurrent decrease in the intensity of the signal according to the fourth power relationship between the Raman cross section and the frequency of the excitation source (σ ∝ ω4), thus causing a loss of sensitivity.31 Unfortunately, sensitivity problems can also arise from limitations in the current technology for the fabrication of NIR detectors. Silicon-based electron multiplying charge-coupled devices (EMCCD) with back-thinned deep depletion design are characterized by a maximum quantum efficiency at 850 nm (Fig. 1, green dotted profile), which makes them the most popular and effective solution for systems fitted with multiple excitation lines, spanning from the visible (i.e., 514, 532, 633 nm) to 785 nm or 830 nm in the NIR I. However, because of silicon's band gap, the sensitivity of these detectors rapidly approaches zero at wavelengths above 1000 nm. Therefore, different detectors, such as those based on InGaAs, must be utilized in order to access higher wavelengths in the NIR.Standard InGaAs detectors (lattice-matched In0.53Ga0.47As on InP) have sensitivity that ranges from 1000 nm to 1700 nm (Fig. 1, dashed orange profile),34 allowing access to the second biological window. The sensitivity range can be further pushed to higher wavelengths via indium enrichment (extended InxGa1−xAs detectors, 0.53 < x < 1),35,36 enabling detection windows centered from 1637 to 1811 nm,36 or even higher up in the short wavelength (SW) IR (∼2200–3000 nm)34,35 (Fig. 1, brown dot-and-dash profile). Certainly, the tunability of the InGaAs detection window via indium enrichment is an interesting property with important technological outcomes; regrettably, indium enrichment also translates to an increase in the lattice mismatch between the InGaAs alloy and the InP substrate, on which the former is typically epitaxially grown or vapor deposited.34,36 This mismatch causes structural defects that ultimately reduce the optical performances of the material,36 leading to dark current-induced sensitivity loss,37 which can be only in part modulated via either thermoelectronic- or cryo-cooling. At present, research towards commercially mature, cost effective, low-noise NIR and SWIR detectors is still in progress.
The introduction of excitation wavelengths in the NIR also generates a need to intervene on the design of the enhancing substrate. To understand this necessity, we must recall that SERS is a plasmonic analytical technique, and thus, it relies on the excitation of the localized surface plasmon resonance (LSPR). The LSPR can only occur if the following equation is satisfied:41
| εnp(λ) = −χεenv | (1) |
Salt-induced aggregation can shift the LSPR of a SERS-active substrate to higher wavelengths.31,42 It is indeed very common to perform measurements with aggregated colloidal Au nanospheres when the excitation line is at 785 nm,43–46 and it has been recently discussed how planar substrates based on large (i.e., obtained by complete colloidal sol disruption) fractal aggregates of silver nanospheres can also be effectively utilized with excitation sources in the NIR I (680–920 nm).47 However, such planar substrates might not be the optimal analytical strategy for all SERS applications (e.g., ex vivo or in vivo analysis of tissues), and standard colloidal aggregated nanospheres (Lee, Meisel's,48 Turkevich's,49 Frens'50) would still keep longer excitation wavelengths in the NIR II and SWIR inaccessible. The necessity to manipulate the LSPR by means other than salt-induced aggregation is therefore apparent.
To this end, it must be recalled that the extinction behavior of a plasmonic nanoparticle as a function of wavelength, E(λ), can be described by:14,41
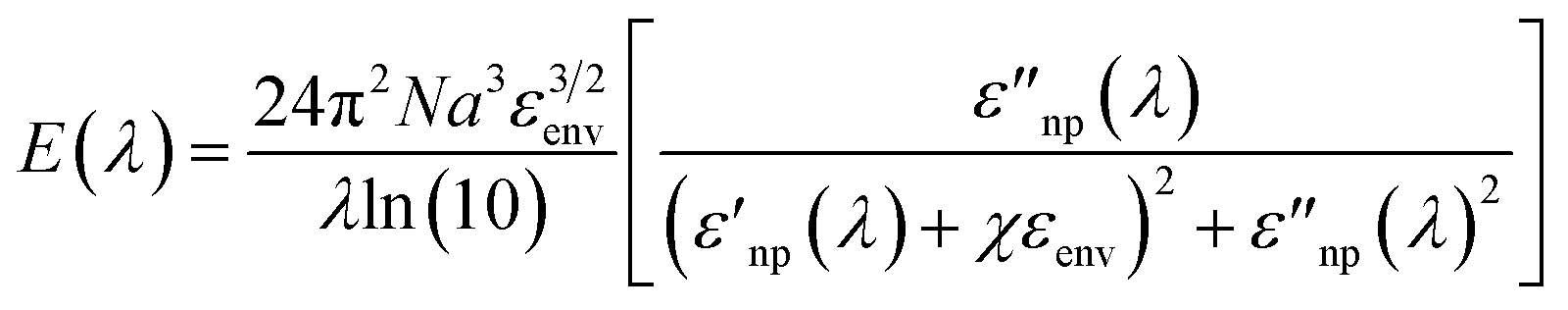 | (2) |
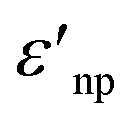 and
and 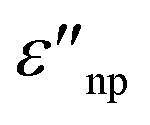 are the real and imaginary part of the relative permittivity of the metal, εnp, describing its absorption and scattering behavior, respectively, and χ is the shape factor, as seen in eqn (1). If resonance conditions are met (eqn (1)), and the metal's absorption
are the real and imaginary part of the relative permittivity of the metal, εnp, describing its absorption and scattering behavior, respectively, and χ is the shape factor, as seen in eqn (1). If resonance conditions are met (eqn (1)), and the metal's absorption 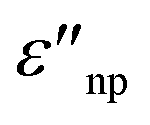 is small and positive, then E(λ) reaches its maximum (λLSPR). In practical terms, this means that the extinction, and thus, its maximum λLSPR, can be tuned by acting on three fundamental parameters: size (a), shape (χ), and chemical identity of the nanomaterial
is small and positive, then E(λ) reaches its maximum (λLSPR). In practical terms, this means that the extinction, and thus, its maximum λLSPR, can be tuned by acting on three fundamental parameters: size (a), shape (χ), and chemical identity of the nanomaterial 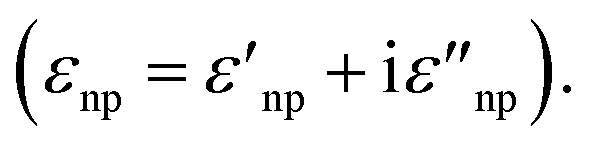 In particular, it can be observed that as the size and anisotropic character of nanoparticles is increased, their λLSPR is also increased (i.e., red shifted), while doping, defect engineering, and hybridization can be exploited to fabricate novel nanomaterials with native NIR and SWIR activity (Fig. 2).
In particular, it can be observed that as the size and anisotropic character of nanoparticles is increased, their λLSPR is also increased (i.e., red shifted), while doping, defect engineering, and hybridization can be exploited to fabricate novel nanomaterials with native NIR and SWIR activity (Fig. 2).
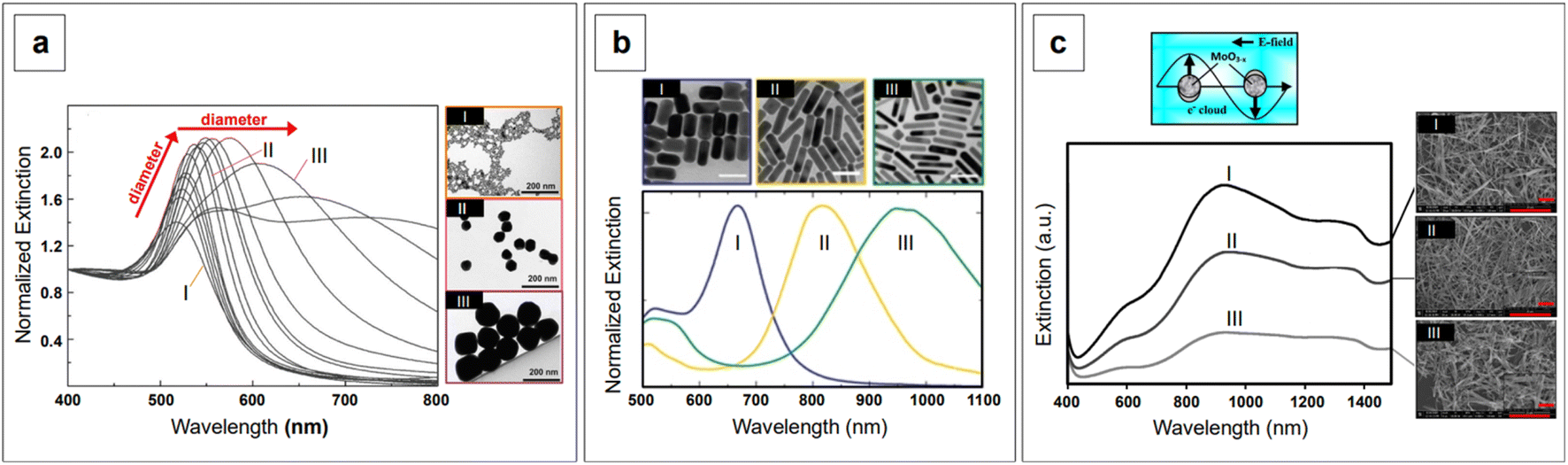 | ||
| Fig. 2 Effect of size, shape, and dielectric function on the plasmon band of nanomaterials. (a) The λLSPR of citrate-reduced gold nanospheres widens and shifts to the red and NIR as the size increases from 8.4 ± 1.0 nm (I), to 64.8 ± 3.4 nm (II), and 123.6 ± 10.6 nm (III). Adapted with permission from Bastús et al., Langmuir, 2011, 27, 11098–11105. Copyright 2011 American Chemical Society. (b) Nanorods show two plasmon bands, corresponding to the transverse (∼500 nm) and longitudinal components. The wavelength of the latter increases with increasing aspect ratio (AR): gold nanorods having AR of 2 ± 1 show a longitudinal λLSPR at about 660 nm (I, blue), those with AR of 4.2 ± 1 show a longitudinal λLSPR at about 810 nm (II, yellow), and those with AR of 6 ± 2 show a longitudinal λLSPR at about 960 nm (III, green). Scale bars in the TEM micrographs are 50 nm. Adapted with permission from Springer Nature: Zijlstra et al., Nature, 2009, 459, 410–413. Copyright 2009. (c) Extinction spectra of three NIR-active MoO3−x nanomaterials with different oxygen vacancies and corresponding Field Emission SEM (FE-SEM) micrographs. Adapted with permission from Patil et al., J. Phys. Chem. C, 2020, 124, 21082–21093. Copyright 2020 American Chemical Society. | ||
Although the relationships between the above listed parameters and the optical properties of nanoparticles have been extensively investigated in the past twenty years, and a lot of energy has been put into improving size and shape control at the nanofabrication level, there are still some criticalities that make substrate tuning a non-trivial task. For example, reaching morphological uniformity and high morphological yield of anisotropic substrates, especially in surfactant-free systems, still represents a challenge in current practice. Surfactant-free systems are particularly interesting for direct detection applications; while nanoparticle capping is unavoidable in any colloidal system,51 the presence of strongly interacting shape-directing surfactants (e.g., cetyltrimethylammonium bromide, CTAB) at the solid/liquid interface of an enhancing substrate can interfere with measurements by increasing the distance at which the analyte can interact with the SERS-active surface.52,53
SERS is indeed a distance-dependent phenomenon that relies on the adsorption of analytes on or in close proximity to plasmon-sustaining nanostructures (within 1 nm for maximum enhancement, within 3 nm for lower, longer-range enhancement)54 to achieve their detection.55,56 In thermodynamic terms, adsorption will happen if the association constant, Kad, between the analyte and the nanostructured surface is larger than all of the other constants at play, namely, Kad between substrate and stabilizers, substrate and nanofabrication by-products, and between individual analyte molecules.53 When the system does not allow for adequate adsorption of an analyte for its direct detection, or when a very complex matrix has to be dealt with (e.g., biological fluids, cells, tissues) and high selectivity and specificity are required, indirect detection strategies must be utilized instead.
Indirect detection by SERS is effected by means of functionalized nanoparticles, called SERS nanotags, that allow for the switching on and off of a signal given by a reporter molecule, upon folding, binding, or other recognition event. Differently engineered nanotags in the same colloidal sol or assay57–59 can then be exploited for multiplexing, allowing for the concomitant indirect detection or imaging of different analytical targets during the same measurement. For this approach to be successful, the reporter molecule must be carefully selected, such that it can be easily conjugated to the metal core of the nanotag, and it can give off a bright signal, even at trace concentrations. This implies that a good reporter for SERS nanotags must have a large SERS cross section. While in the visible and NIR I range a wide array of large cross section reporter molecules is available (crystal violet, rhodamine 6G, and 4-mercaptobenzoic acid among others), bright NIR II and SWIR reporters for SERS are scarce and research on their design and synthesis is still ongoing and niche.
Lastly, we recall that by avoiding self-absorbance, NIR light can penetrate tissues, reaching about 1 cm of depth when 785 nm lasers are utilized.60 Increasing wavelengths into the NIR II and SWIR further improves transparency at such depths by concomitantly reducing scattering.61,62 However, such an extent of penetration depth might not be sufficient for applications that require non-destructive access to deeper targets within the human body, such as abdominal organs or intracranial regions. This issue clearly limits the range of application of NIR-SERS to the medical and intraoperative fields. Traditional Raman spectroscopy can benefit of alternative spectral collection arrangements, such that the radiation can reach deeper targets. This variation of the technique is called Spatially Offset Raman Spectroscopy (SORS), and its Surface Enhanced (SE) counterpart, SESORS, can be similarly implemented, affording depths of penetration from 4 to 5 cm.63 As a consequence, this spectral collection configuration brings about new and complex challenges in that, for a SERS signal to be generated by a deep target, a suitable enhancing substrate must be delivered in its vicinity. Therefore, nanoparticle dose, delivery method, biocompatibility, toxicity, and clearance are all aspects that must be carefully evaluated when implementing a realistic SESORS analytical protocol.
In the following sections, we will explore how the challenges herein outlined have been tackled by the SERS community, providing examples of relevant literature from the past six years (2017–2022). While there is a broad range of recent reviews on SERS,64,65 especially on substrates66–70 and biomedical applications,71–78 we intend to offer a different take on the matter and focus on aspects that are seldom discussed, although within popular topics such as anisotropic growth. An example of these are surface chemistry and its relationship with crystal growth, monodispersity, and final SERS performance, as well as the insights coming from computational chemistry, which aid in the elucidation of mechanisms that are still experimentally elusive. We hope to guide the reader in a journey that illustrates the reasons behind certain practical choices presented by NIR-focused literature cases, and stimulate the debate around the criticalities that still need to be addressed to finally push (NIR-)SERS into the real world.
2.1 Excitation wavelength and nanomaterial design
As previously discussed, the use of NIR and SWIR excitation wavelengths necessarily implies some degree of intervention on the nanomaterial design, with respect to the most commonly utilized substrates for SERS in the visible range. Three main intervention strategies can be identified:(1) acting on the anisotropy of the nanoparticle morphology to shift the LSPR to the NIR and SWIR regions;
(2) acting on the chemical composition (εnp) and architecture of the nanomaterials (e.g., core–shell, multimaterial hybridization, doping, etc.) to obtain novel SERS-active substrates in the NIR and SWIR;
(3) using NIR and SWIR reporters to obtain NIR and SWIR nanotags for indirect SERS detection.
This effect entails the accumulation of dipolar fields at the tips of prolate structures, such as the tips of nanorods or nanostar branches, resulting in electric fields that are much more intense than those at flattened regions.31 Since plasmonic nanostructures behave like nano-antennas, in that the radiative process from the nanostructure (i.e., near-field) to the far-field is what ultimately enables SERS (electromagnetic mechanism),56 it follows that a radiative process that is generated by stronger electric fields will generate inherently larger surface-enhanced Raman signals. In other words, the tips of rods or branches are intrinsic hotspots.31,79–82 Additionally, the more a prolate geometry is needle-like, the higher the concentration of dipolar fields, and thus, the larger the resulting signal enhancement.80 This can be easily observed in Fig. 3a, which shows the NIR-SERS spectra (λexc 785 nm) of standard probe crystal violet, obtained utilizing AuAg nanostars with varying degrees of branch sharpness, as well as AuAg nanospheres.83 All nanostars give raise to a larger amplification of the signal compared to nanospheres, and among the nanostars, the intensity of the spectra is positively correlated to the sharpness of the branches.
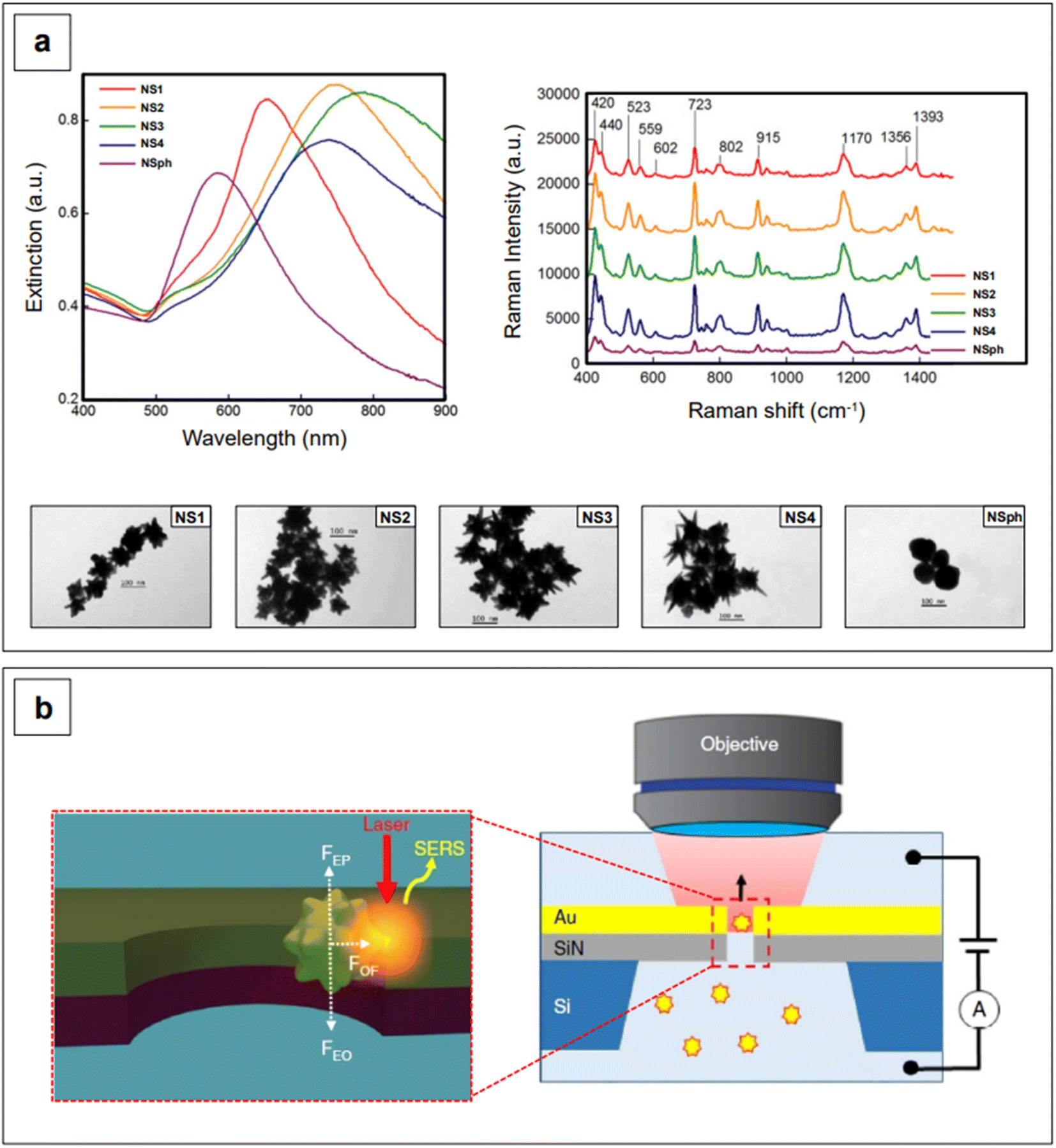 | ||
| Fig. 3 (a) Extinction spectra (left) and TEM micrographs (bottom) of AuAg nanostars with varying degrees of branch length and sharpness (NS from 1 to 4), and of AuAg nanospheres (NSph). The capping agent is CTAB for all colloidal formulations. These nanomaterials were utilized for the NIR-SERS detection of model analyte crystal violet at a final concentration of 1 μM (right). It can be easily observed that all branched morphologies give rise to intense SERS spectra compared to the nanospheres, and that the sharpest morphologies yield strongest SERS signals among the branched morphologies (lightning rod effect). Adapted with permission from He et al., J. Opt., 2015, 17, 114013–114026. Copyright IOP Publishing. All rights reserved. (b) Schematic of the flow-through detection platform developed by Huang et al. A nanostar is trapped in a nanohole via the application of a voltage and laser illumination at 785 nm. The trapping is the result of the combination of electrophoretic (FEP), electroosmotic (FEO), and optical (FOF) forces and allows for the formation of on-demand, controllable tip-wall hotspots. Adapted from Huang et al., Nat. Commun., 2019, 10, 5321; figure licensed under CC BY 4.0, https://creativecommons.org/licenses/by/4.0/. | ||
The intrinsic hotspots of rod-like or rod-containing (i.e., branched) colloidal nanoparticles are such that, under suitable excitation conditions, they may be used for SERS measurements without need for prior aggregation.84 This constitutes an advantage in terms of signal reproducibility, in that the more common extrinsic (i.e., salt-induced) hotspots are inherently stochastic, as stochastic is also the probability of a molecule adsorbing on a hotspot.85 An interesting way of generating extrinsic hotspots in a more controlled way was recently implemented by Huang et al.,86 who coupled label-free gold nanostars with gold nanohole arrays for the single molecule NIR-SERS detection of both individual DNA bases and single nucleobases within DNA oligonucleotides. Gold nanoholes were size-tuned and suitably spaced to exhibit their LSPR in the NIR I, and then embedded as a partitioning wall in a PDMS microfluidic chamber. Gold nanostars were then incubated with either the individual DNA bases or the DNA oligonucleotides and let diffuse from one compartment to the other inside the microfluidic chamber. By concomitant application of a 785 nm laser light and an electrical potential, a plasmonic gradient inside the nanohole and two equal and opposing electrokinetic and electroosmotic forces are generated, respectively (Fig. 3b). The plasmonic gradient gives rise to an optical force that brings the nanostar-analyte complex towards the nanohole sidewalls, creating on-demand, controllable tip-wall hotspots in which the analyte can reside. On the other hand, the combined effect of the two equal and opposing electrokinetic and electroosmotic forces causes the electrophoretic motion and entropic flow of the nanostar through the nanohole to be abated. This electro-opto-kinetic trapping and on-demand hotspot manipulation can be sustained up to a few minutes, allowing for sufficient time to detect rare single molecule events (e.g., the detection of A as a single nucleobase in 5′-CCCCCCCCCA-3′) across multiple acquisitions.
Oblique Angle Deposition (OAD) can also be utilized to fabricate planar substrates based on anisotropic elements and with controlled spatial distribution of hotspots. Li et al.87 developed an OAD-based gold nanorod planar substrate with nanorod spacing optimized to be active in the NIR I, and applied it to the fast (8 minutes), label-free, and direct detection of the protective antigen (PA) produced by Bacillus anthracis upon anthrax infection.88 Their method was demonstrated to have a sensitivity of 100 pg mL−1 both in standard solutions and in human serum albumin (HSA), and discrimination of HSA from HSA/PA mixtures was aided by principal component analysis (PCA).88 Although real serum samples contain species other than HSA, the determined limit of detection is four orders of magnitude lower than the typical levels of protective antigen found in infected human serum, and, thus, reasonably fit-to-purpose.88
The anisotropic arrays described by Li et al.87,88 are not the most common type of SERS-ctive substrate; in fact, colloidal suspensions have a much wider prevalence in analytical work, due to their low cost and versatility. Colloidal nanoparticles can be delivered in situ,89 incubated into cells or injected through tissues,90,91 utilized for measurements in transmission configuration,92 and immobilized on planar supports to produce arrays.93–95 However, the elucidation of the mechanisms that enable anisotropic growth in colloidal sols is still subject of animated debate and research, in that several factors limit the realistic formulation of a punctual and unambiguous unified theory. While the growing accessibility of computational methods such as molecular dynamics simulations has recently aided in the exploration of otherwise inaccessible nanoscale phenomena, the real-time atomic-scale imaging and chemical mapping instrumentation that would be requisite to observe such processes directly and in their native environment is not widely accessible yet (e.g., cryogenic transmission electron microscopy, cryo-TEM), or existent. Furthermore, the enormous number of different synthetic protocols that have been implemented in the last two decades poses in and of itself a limitation to the extent of the reaction space parameters that can be realistically explored and compared experimentally. However, from a strictly practical standpoint, it is well known that anisotropic gold and silver nanoparticles can be achieved via the introduction of shape-directing agents, and their rational modulation allows fine plasmonic tuning for tailored NIR-active substrates. Shape-directing agents can be categorized into two groups—surfactants and small ions (i.e., silver and halides). In the case of gold nanorods, both agents are utilized for the control of the aspect ratio for plasmon tuning, as well as for morphological yield and crystallinity control.
CTAB is probably the most famous among the shape-directing surfactants and it is essential in the fabrication of nanorods. The general mechanism by which it assists in the formation of these nanoparticles is understood to be the result of the different affinity the surfactant has towards different Au facets, such that growth on the longitudinal axis is faster than on the transverse axis, leading to rod-shaped nanoparticles.96,97 As previously mentioned, advances in the field of computational chemistry have recently allowed for the elucidation of mechanistic aspects that are otherwise experimentally elusive. Of particular importance is the modeling of surfactants as micelles and the study of their behavior at the interface with nanoscale plasmonic surfaces in solution. For example, molecular dynamics simulations by Meena and Sulpizi98 and Da Silva and Meneghetti99 have shown that the adsorption of CTAB on the seed and growing nanorod likely occurs as individual micelles. In analogy to what postulated in the early developments of nanorods synthesis,96,97 CTAB micelles adsorb with different packing density depending on the crystal facet and surface curvature.98–100 This creates facet- and curvature-specific intramicellar channels of varying width, where the solution environment can easily reach the surface of the seed or growing rod.98–100 This affects the growth kinetics and ultimately results in a faster deposition of reduced gold on the facets that are less densely packed by the CTAB micelles (i.e., curved, (111) facets).98–100
The event that breaks the symmetry of the initial isotropic gold seed is not generally attributed to CTAB.94 For penta-twinned nanorods, this event is identified with the stochastic formation of twinning planes in the growing seed,101 while for single-crystal nanorods, which are obtained in the presence of silver, the general belief is that silver itself is responsible for symmetry breaking.102–105 Because symmetry breaking is an inherently stochastic process, more families of nanorods inevitably coexist in the same colloidal sol (“popcorn” mechanism),106 thus affecting its homogeneity. Colloidal polydispersity is reflected by the width of the longitudinal LSPR band in extinction spectroscopy, and it is generally associated to a non-optimal batch to batch reproducibility of the SERS enhancement. This issue has been addressed by Liz-Marzán's group,107 who introduced the use of small AR nanorods as seeds. This variation in the original procedure causes an effective temporal and spatial decoupling of symmetry breaking and growth events, thus leading to an improved morphological homogeneity and reproducibility of the SERS signal.107 This is an elegant example of how the understanding of fundamental nanoscale processes can lead to very practical outcomes at the analytical level. The necessary high yield and uniformity of the rod-shaped seeds was achieved by the introduction of n-decanol as a co-surfactant;107 by finely tuning the n-decanol:CTAB ratio, growth of low-dispersion, higher aspect ratio nanorods was achieved (Fig. 4a).107 It must be recalled that the possibility of utilizing co-surfactants to obtain nanorods with higher AR, and thus NIR II activity, was found also early in the nanofabrication development of this morphology,96 although with lower shape uniformity, which is reflected by the wide longitudinal LSPR band (Fig. 4b).
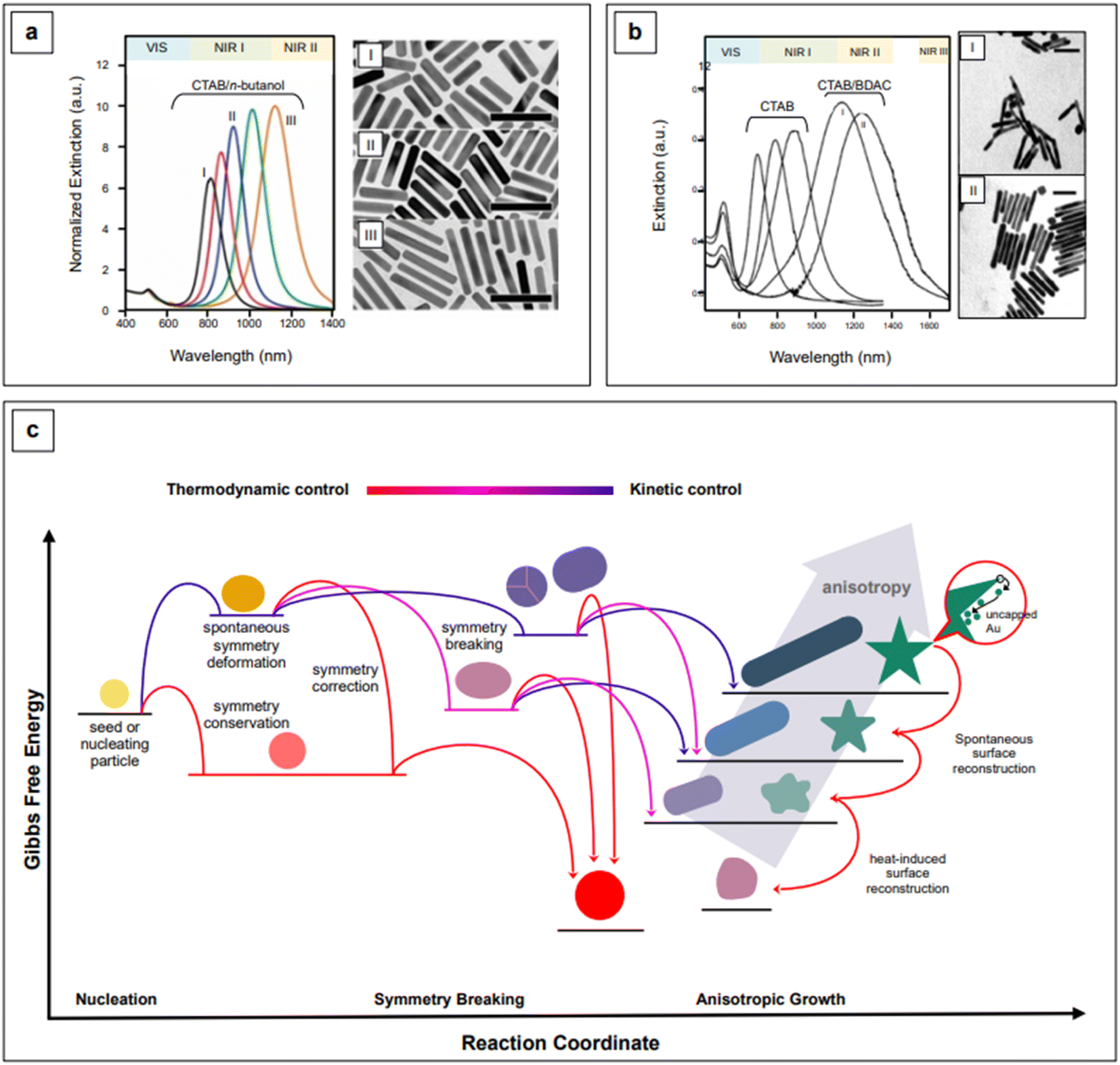 | ||
| Fig. 4 Comparison between high aspect ratio nanorods synthesized in co-surfactant mixtures with (a) small anisotropic seeds and (b) traditional, spherical seeds. Panel a adapted with permission from González-Rubio et al., ACS Nano, 2019, 13, 4424–4435. Copyright 2019 American Chemical Society; panel b adapted with permission from Nikoobakht et al., Chem. Mater., 2003, 15, 1957–1962. Copyright 2003 American Chemical Society. (c) Kinetic vs. thermodynamic control in nanoparticle growth. Figure inspired by González-Rubio et al., 2020.110 | ||
A practical advantage of the nanorods developed by Liz-Marzán's group is that they are characterized by narrow longitudinal LSPR bands that can be straightforwardly tuned from the NIR I (760 nm) to the NIR II (1125 nm), by simple manipulation of the pH via HCl:HAuCl4 ratio, from 40 to 160, and control of the reaction temperature at 16 °C.107 The combination of a lower pH, which decreases the redox potential of ascorbic acid, and a lower temperature (T < room T) slows the kinetics of the growth reaction, thus achieving a more controlled manipulation of anisotropy.107,108 Anisotropic morphologies are indeed kinetic products (Fig. 4c),109,110 and reaction kinetics strongly influences the morphology and morphological yield of the final product. As previously noted, differences in thermodynamic parameters, such as the affinity of a particular capping agent for a specific crystal facet, do have kinetic outcomes—a more strongly bound or more tightly organized capping agent can slow down the deposition rate of the precursor noble metal.99,100,107 This has been recently demonstrated by Chen et al.111 for the formation of silver nanocubes with (poly)vinylpyrrolidone (PVP) as the shape director. For pentatwinned gold nanorods, Sánchez-Iglesias et al.112 have further explored the topic by achieving aspect ratio tuning in the NIR I and II via the sole temperature control, in the presence of CTAC as CTAB's co-surfactant.
If the elucidation of anisotropic growth in nanorods still proves to be challenging, the elucidation and fine control of the development of branched morphologies are even more elusive. Branched nanoparticles, often called nanostars (but also nanourchins113–115 or nanoflowers116–118), are typically synthesized via seed-mediated processes, in the presence of silver ions from silver nitrate, as well as a surfactant or a polymer (e.g., CTAB,119,120 Triton-X,121–123 anionic dioctyl sodium sulfosuccinate, AOT, and AOT/dimethyldodecylbenzylammonium bromide,124 PVP,125,126 chitosan,127,128etc.). While the role of silver is understood to be—at a minimum—that of a symmetry breaking agent like with nanorods, the role of the various surfactants and polymers has not been fully elucidated yet. This is to be attributed not only to the low accessibility and affordability of in situ atomic-scale organic phase-compatible characterization techniques, but also to the high degrees of freedom that this morphological class inherently bears. Compared to nanorods, where the morphological parameters are essentially circumscribed to length and width, nanostar subclasses can be identified based on a wider range of parameters, such as the length and thickness of branches, their sharpness, their number, the presence or absence of tip decorations. This readily translates to an enormous number of variations along the same fundamental shape (Fig. 5), and thus, an accompanying enormous number of synthetic protocols that can be hardly rationalized for mechanistic purposes. However, by analogy with what has been illustrated for nanorods and for anisotropic growth in general, it can be said that a combination of selective adsorption,121 configuration at the solid/liquid interface,129 and modulation of redox potentials and reaction kinetics53,109,123 is likely to compound in the generation of a specific branched nanoparticle.
 | ||
| Fig. 5 Examples of branched nanoparticles. (a) Highly symmetrical and homogeneous Ag-coated gold tetrapods. Reprinted from Spectrochim. Acta Part A, 211, Zhu et al., 154–165, Copyright 2019, with permission from Elsevier. (b) Highly symmetrical and homogeneous six-branches AuAg nanostars. Used with permission of Royal Society of Chemistry, from Atta et al., Nanoscale, 11, 2019; permission conveyed through Copyright Clearance Center, Inc. (c) Gold-copper starfish-like nanostars. Adapted from Sanchez et al., RSC Adv., 2021, 11, 25788–25794; figure licensed under CC BY-NC 3.0, https://creativecommons.org/licenses/by-nc/3.0/. (d) Citrate-capped one-pot multibranched AuAg nanostars. Adapted with permission from Deriu et al., J. Phys. Chem. C, 2022, 126, 2023–2040. Copyright 2022 American Chemical Society. (e) Predominantly hexapod silver nanostars. Reprinted from Garcia-Leis et al., Colloids Surf., A, 535, 49–60, Copyright 2017, with permission from Elsevier. (f) Branches-on-branches gold nanostars. Adapted with permission from Atta et al., J. Phys. Chem. C, 2016, 120, 20749–20758. Copyright 2016 American Chemical Society. | ||
An important difference compared to nanorods is that surfactant-free procedures for the fabrication of colloidal gold nanostars can also be implemented,83,113,130–133 indicating that surfactants are not as necessary as in the fabrication of nanorods. In fact, while surfactant-free nanorods can be obtained only in the presence of a template,134,135 this is not necessary for the surfactant-free synthesis of gold nanostars; rather, a quick look at the literature might suggest that the only indispensable reagent is silver. It has indeed been amply shown that, regardless of the synthetic strategy (i.e., surfactant-mediated or surfactant-free, seed-mediated or one pot), the removal of silver from branched gold nanoparticle syntheses can lead to the formation of spheroids.83,123,131,136 This has been reconducted to the role of silver in inducing anisotropy-favoring twinning defects on the nucleating crystals or growing seeds.53,123
However, a more accurate look at the literature would show that synthetic protocols for the fabrication of branched gold nanoparticles can also be achieved without silver, often in association with reducing agents different than ascorbic acid.113,130,132,137 This demonstrates the previously mentioned complexity of this type of growth, and the difficulty in finding a unifying mechanistic theory. However, when silver is utilized, it has been frequently observed to influence the branches extent, length, and sharpness83,123,131,136,138,139—an effect that is important for NIR and hotspot tuning.83 Moreover, as observed in the stabilization of symmetry-breaking outcomes in the evolving seed in nanorod synthesis,103,104 silver has been associated to the stabilization of shape via underpotential deposition.123
As previously discussed, anisotropic nanoparticles are kinetic products,109,110 and they have a higher density of high energy facets (i.e., {110} and {100}) compared to isotropic shapes such as spheroids, as well as a larger number of low coordination surface atoms.140 Therefore, their formation and the retention of their shape and crystallinity is not thermodynamically favored, thus leading to surface reconstruction (Fig. 4c).123,140 This phenomenon is particularly evident in gold anisotropic nanoparticles, due to the known high mobility of this element.123,141 Atta et al.123 studied the fate of silver in the evolution of highly uniform, seed-mediated, six-branches gold nanostars in Triton X-100, and found that their morphology, and thus the LSPR, was stable when an optimal concentration of 100 μM AgNO3 was utilized for their nanofabrication. In all other instances, a decrease in the length of the spikes and rounding of the tips was observed. Line-scanned energy-dispersive X-ray spectroscopy (EDS) data of the nanostars obtained with varying Ag+ concentration showed that silver is uniformly present throughout the nanoparticle, but only forms a thicker layer on the side walls of the branches when 100 μM is used; this is believed to prevent the thermodynamically favored diffusion of gold atoms from the tips towards the core, thus allowing shape retention.
Nanoparticle restructuring towards more thermodynamically favored shapes was also noticed in the surfactant-free gold nanostars optimized by Xie et al.,137 in which silver is absent, and in those optimized by He et al.,83 in which silver is present as a trace constituent (Au:Ag of 18:1). In these cases, boiling and aging, respectively, blueshift the plasmon, indicating the collective regression of the nanoparticle population to spheroids or a less anisotropic shape.83,137 Thermally induced restructuring of individual plasmonic nanostars was recently observed in situ, thanks to the advances in electron microscopy cells with both heating and tilting capability.142 These were utilized in the development of a scanning TEM (STEM) acquisition method based on the collection of high angle annular dark field (HAADF) 2D projections of individual nanostars and consequent 3D shape reconstruction (HAADF-STEM tomography). The novelty of this method is that it can be performed at a rate that is 10 times faster than conventional electron tomography approaches, thus allowing the direct observation of individual nanostars during temperature-induced reshaping.142 During these in situ heating experiments, it was directly observed for the first time that gold atoms migrate from the tip of the branches towards the core of the nanostar as a function of increasing temperature, and that the majority of the reshaping occurs within the first minute of heating, regardless of the temperature.142
Because of the tight relationship between morphological and optical properties in plasmonic nanoparticles, and because laser illumination during a SERS experiment might cause plasmon-induced heating, heat-mediated shape restructuring of nanoparticles should be taken into serious consideration. This is particularly true when implementing NIR-SERS analytical protocols, as they are likely to make use of highly anisotropic morphologies in conjunction with high energy and long acquisition times (i.e., to counterbalance the energy dependence of the signal intensity), which tend to generate heat within the sample.
Regardless of applied heat, surface restructuring of highly anisotropic nanoparticles is thermodynamically favored, and a common strategy to hinder and slow down the process is the use of surface ligands. Besides modulating colloidal stability, surface ligands can indeed minimize surface energy. Shape conversion of gold nanostars can thus be prevented by capping them with strongly interacting surfactants, such as CTAB,83 weakly chemisorbed small molecules such as citrate,53,143 or other ionic species such as HEPES129 and carbonate.53 It must be recalled that the choice of nanoparticle capping agents is not only critical for the retention of shape and colloidal stability,53,140 but also for optimal signal enhancement53 and biocompatibility.144,145 The relevance of the latter aspect will be apparent in Sections 2.1.3 and 2.2 of this review.
The nanoparticles developed by Atta et al.123 are of particular interest in the NIR as they have very thin and long branches, which shifts the plasmon from the usual NIR I to the NIR II, offering the possibility to explore the second biological window. The second biological window and the SWIR region are seldom explored compared to the NIR I, not only because of the technological limitations that we have previously discussed (i.e., performance of NIR I and SWIR detectors), but also because highly homogeneous and controlled thin branching are difficult to obtain, and there is at present little to no knowledge on how to tailor this shape such that no core ripening or thickening of the branches occurs during growth (besides impeding restructuring). Atta et al.123 also observed how, beyond the optimal silver concentration they found for their synthetic protocol, silver deposited at the core, enlarging it. Moreover, it has been observed that the seed size also influences the branching extent and individual branch growth.146,147
In gradient array growth experiments on planar substrates that were intended to unveil the morphological transition of poly(N-isopropylacrylamide)-encapsulated gold seeds into silver-free nanostars, Kuttner et al.147 observed that new branches are formed at the expense of branch growth with increasing seed diameter, which was attributed to the progressively larger surface that was available for autocatalytic reduction, and thus, for the nucleation of new branches. The same authors also studied the interplay of seed size with the concentration of gold precursor, noticing that for bigger (>12 nm) seeds to grow into longer-branched nanostars and counteract an overall isotropic growth in volume, a gold precursor supply higher than the usual 0.25 mM concentration was necessary.147 After prior screening of SERS efficiency using model analyte nitrothiophenol, and identification of a nanostar morphology along the gradient array that was best suited for NIR I 785 nm irradiation, Kuttner et al.147 were able to exploit the encapsulated nanoparticles as hydrophobic platforms for the label-free surface enhanced resonance Raman (SERRS) detection of the elusive Pseudomonas aeruginosa infection biomarker pyocyanin. This was achieved via a tempering-promoted entrapment of the analyte at the nanoparticle-polymer interface, which allowed poly(N-isopropylacrylamide) to undergo volume-phase transition without altering the optical properties of the encapsulated nanostars, thus enabling pyocyanin detection at clinically relevant concentrations (dynamic range 10−5 to 10−7 M) by bringing it closer to the plasmonic surface.147
Label-free SERS, that is, SERS in the absence of nanotags, can be challenging for the reasons illustrated in the overview section of this review; in fact, the majority of recent works on NIR-SERS, and in particular those targeted at the detection of biomolecules in complex biological media, are characterized by the use of functionalized anisotropic nanoparticles. A selection of these anisotropic nanotag-based studies will be discussed in the dedicated section. It must also be mentioned that, although the present section is focused on the examination of nanorods and nanostars, these are not the only morphologies available to the SERS scientist. Other anisotropic materials with fit-to-purpose SERS activity in the NIR can also be fabricated. For example, interested readers can refer to the works on NIR-active silver nanowires by Becucci et al.148 and on gold nanoplates by Luo et al.,149 or the synthetic study on the role of iodide ions in the transition of copper nanowires to nanoplates by Kumar's group.150
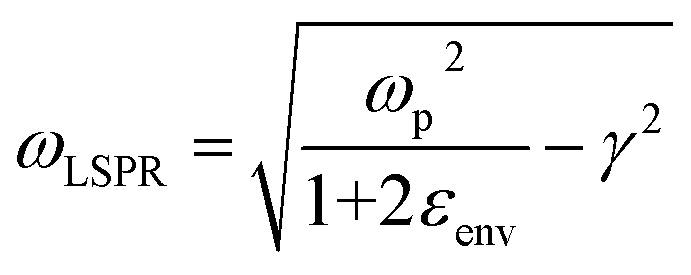 | (3) |
However, what constitutes the main advantage of using semiconductors as SERS substrates is also their main challenge. The LSPR bands generated by these materials are not as intense as those observed for noble metals, as semiconductors intrinsically have scarce electrons in the conduction band.157 Consequently, they have a very low enhancement factor (10 to 102) compared to the commonly used noble metals (106 to 1010).158 It must be recalled that the overall SERS enhancement is understood to be the product of two different phenomena, the electromagnetic effect, cited in the preceding section, and the chemical effect, of which charge transfer is the main enabling mechanism. Charge transfer is an analyte-specific mechanism that entails a chemical interaction between the analyte and the SERS substrate, in which an electron can be transferred through vibronic coupling from the Fermi level of the substrate (i.e., metal) to a frontier orbital of the analyte molecule, or vice versa.159 While this phenomenon does not account for the majority of the total enhancements observed in SERS using traditional plasmonic nanomaterials, it represents instead the main mechanism by which semiconductor-based SERS is enabled. Because the comparatively weaker signal obtained by semiconductors is due to their low electron density, and thus, low amount of charge transfer from the valence to the conduction band, the biggest challenge for the direct implementation of semiconductors in SERS is to engineer them such to improve and promote the chemical enhancement.
To achieve this materials engineering objective, the possible ways in which charge transfer can occur must be taken into consideration. These can be broadly classified into three types: (1) movement of electrons within the atoms of semiconductor, (2) electronic excitations in the target molecule, and (3) mutual transfer of electrons between the semiconductor and target molecule by photo-induced charge transfer (PICT).157 While the exploitation of the latter mechanism is self-explanatory, the first two need some clarification. The transfer of electrons within the semiconductor depends on their band gap, density of electrons in the valence and conduction bands, and on the wavelength of incident light. Similarly, the transfer of electrons in the target molecule depends on the highest unoccupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Upon excitation, electrons from the ground state (HOMO) transit to a higher energy level (LUMO); these electrons, being thermodynamically unstable, move back to the relaxed state in HOMO. The energy released in this process contributes to the Raman signal.160 Based on this principle, the positions of the conduction and valence bands of a semiconductor can be adjusted to come close to the HOMO–LUMO of the target analyte, thus facilitating the charge transfer process, and with it, the SERS chemical enhancement.
Tailoring of the band gap of a semiconductor can be achieved in three main ways: by fabricating heterojunctions, through doping, and via defect engineering (Fig. 6). With heterojunction, we define the interface resulting from the coupling of two different semiconductors; this physical coupling not only tailors the energy level positions of the resulting material, but also facilitates SERS measurement by the formation of hotspots at the junctions. Doping is defined as the deliberate introduction of impurities into an intrinsic (i.e., pure) semiconductor, thus modifying its chemical structure. This introduces additional energy levels in the resulting material, the extrinsic semiconductor, which can be exploited to facilitate SERS-enabling electronic transitions. Finally, defect engineering intervenes on the nanocrystal growth to introduce defects such as oxygen incorporation and abstraction. For applications in the NIR, the two most common tailoring strategies for semiconductor-based SERS are doping and defect engineering. Heterojunctions have been utilized in SERS applications, but their application is still primarily confined to the visible spectrum.161–163
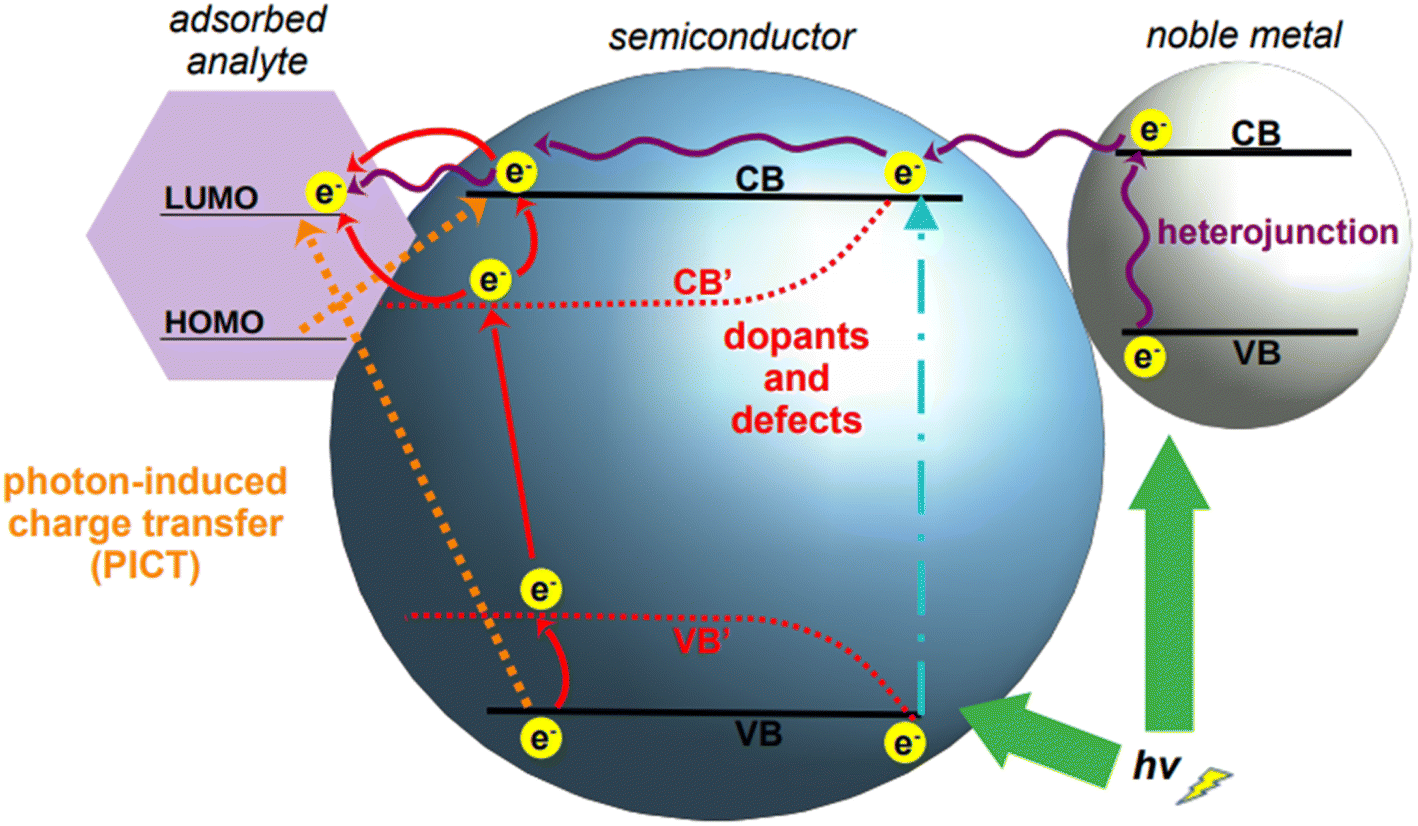 | ||
| Fig. 6 Mechanisms of charge transfer increase in semiconductors. Upon suitable laser illumination, electrons in the native valence band, VB, of a semiconductor are transferred to the conduction band, CB (cyan dot-and-dash line); electrons in the conduction band can be increased by formation of a heterojunction with a noble metal (right), which feeds its electrons into the conduction band of the semiconductor. Charge transfer can then occur between the enriched conduction band of the semiconductor and an adsorbed analyte (purple wavy arrows). The introduction of doping agents and defects in the crystal structure of a semiconductor modifies the band structure of the resulting material, typically lowering the band gap (ΔCB′–VB′ < ΔCB–VB). Upon appropriate laser illumination, electrons are transferred from VB to the new energy levels VB′ and CB′ and to the original CB. Depending on the relative position of the energy levels of the engineered semiconductor and those of the adsorbed analyte, charge transfer can occur from either CB or CB′ to the analyte (red arrows). A third mechanism can occur when electrons are transferred by photon-induction from the valence band of the semiconductor to the LUMO of the adsorbed analyte, or from the HOMO of the adsorbed analyte to the conduction band of the semiconductor (dotted orange arrows). | ||
Semiconductor nanoparticles can be obtained by a variety of bottom-up fabrication techniques,164 such as precipitation,165 template assisted,166 hydrothermal,160 and sol–gel167 syntheses. The same techniques can be utilized also for the doping of semiconductors by incorporation of dopant ions into the crystal lattice during nanoparticle growth;168 alternatively, doping can also be achieved by post-synthetic methods such as diffusion or impregnation.169,170 As depicted in Fig. 6, the introduction of dopants in the crystal lattice of semiconductors creates new energy levels, called sub-energy levels, thereby lowering the band gap. The specific position of the new energy levels and their proximity to the conduction or valence band depends on the charge and number of free electrons in the outermost orbital of the dopant ion. New sub-energy levels can also be achieved by doping-induced formation of defects in the crystal lattice, such as free electrons or electron holes.160 In the case of free electrons, upon excitation they are trapped by the newly created intermediate bands, which prevent electron–hole recombination, thereby increasing the charge transfer.
To date, numerous studies have focused on the synthesis of doped semiconductor nanoparticles of controlled morphology with different concentration of dopant ions.171 However, there is a huge gap between the high volume of literature on the synthesis of these materials, and the application of doped semiconductor nanomaterials for SERS detection. A recent effort to synthesize doped semiconductor nanomaterials for SERS detection was made by Wang and co-workers.172 They doped ZnO oblong nanoparticles (d = 21–87 nm) with varying gallium content using a sol–gel method, and tested their SERS activity using 4-mercaptopyridine as the model analyte. As-synthesized ZnO nanoparticles exhibit a typical UV absorption band at 356 nm, which is attributed to band edge adsorption. By doping these nanoparticles with different concentrations of gallium, the authors were able to generate LSPR bands and tune them in the NIR, without intervening on the morphology, as it is generally done with standard plasmonic materials such as gold and silver. A doping concentration of 5% provided the maximum intensity of the SERS signal. The enhancement was mainly due to the charge transfer mechanism arising from the substitutional incorporation of the trivalent gallium in place of the divalent zinc, which allowed one electron from gallium to behave as a free charge carrier in the crystal lattice, thus producing the LSPR. These free electrons generate LSPRs up to 5% of gallium doping, after which the electron concentration saturates and the LSPR decreases. The enhancement factor (EF) for the detection of 4-mercaptopyridine at a doping ratio of 5% was 6.66 × 104.
Another study employed indium doping of CdO nanoparticles and tested their SERS activity with 4-mercaptopyridine molecules.173 Indium-doped CdO nanoparticles were synthesized hydrothermally with varying amount of indium doping (0.025–0.200 atomic percentage) and it was observed that the charge transfer, as well as the morphologies of doped particles, is dependent on the doping concentration. With an In3+ doping content below 0.05, the nanoparticles formed polygonal shapes, which became more spherical with the increase of doping in the limit of 0.05. When the atomic percentage of doping was further increased (between 0.05 and 0.20), the nanoparticle morphology changed to a heterogeneous structure consisting of nanofibers embedded with 50–100 nm nanoparticles (Fig. 7a). High resolution XPS measurements of O 1s showed that doped and undoped CdO nanoparticles are characterized by a different distribution of oxygen species (oxygen vacancies, ionic oxygen, and surface oxygen, Fig. 7b). This is expected, as doping in metal oxides changes the electronic state of oxygen. Because of the absence of free electrons in the conduction band, bare CdO nanoparticles do not show any SERS signal. However, the additional energy levels generated via doping allowed for the production of a SERS signal (Fig. 7c). The observed enhancements were in the order of 103 (λexc 785 nm), with a degree of charge transfer that was maximized at the lower doping ratios that were tested (0.025–0.075, spectra from II to IV, Fig. 7c). The charge transfer mechanism was rationalized to arise from enriched electrons in the conduction band of the nanoparticle to the LUMO of the analyte, mercaptopyridine.
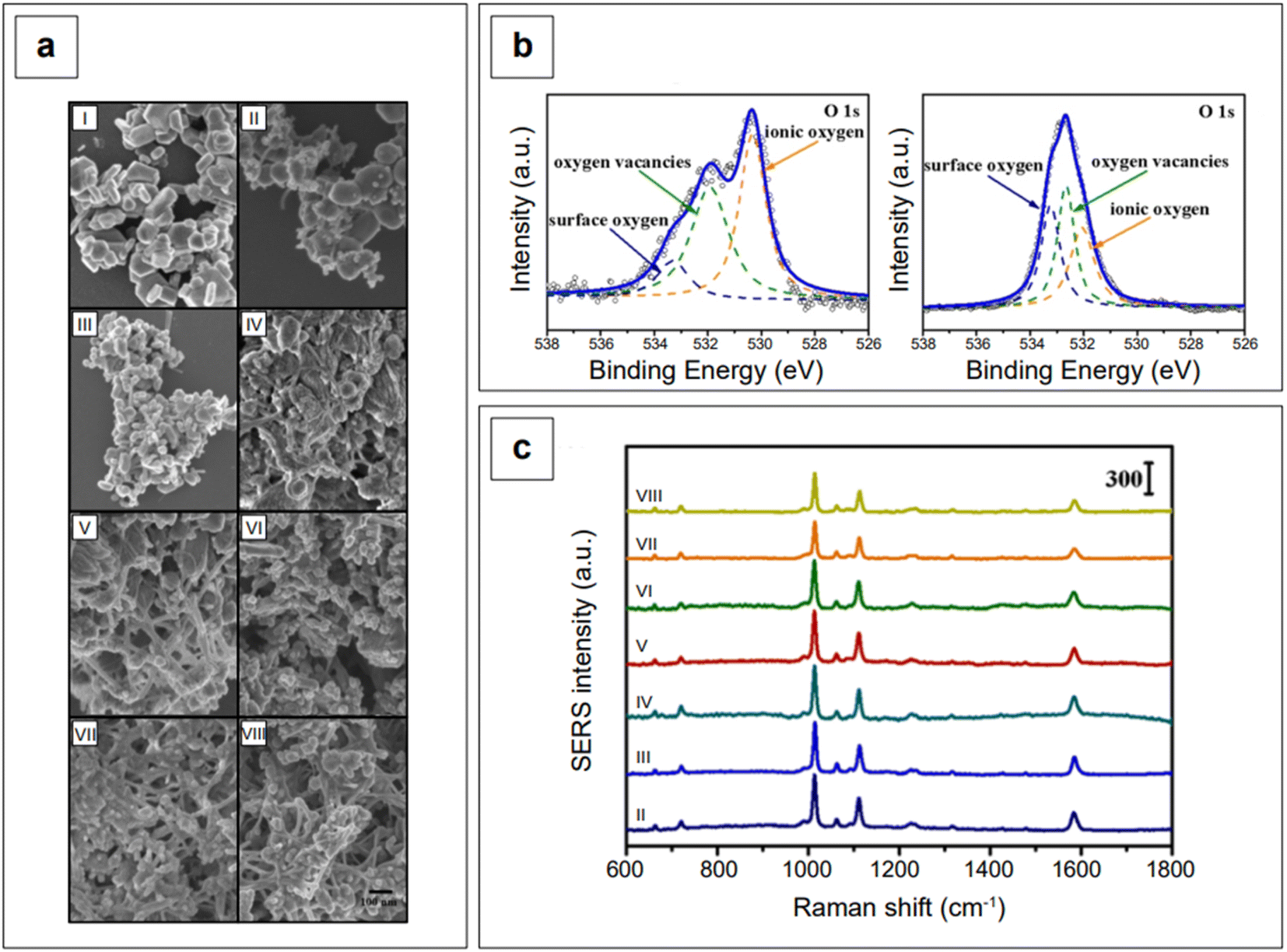 | ||
| Fig. 7 (a) SEM micrographs of indium-doped CdO with varying In3+ atomic percentages: (I) x = 0, (II) x = 0.025, (III) x = 0.050, (IV) x = 0.075, (V) x = 0.100, (VI) x = 0.125, (VII) x = 0.150, and (VIII) x = 0.200. (b) High resolution O 1s XPS spectra of bare CdO (left) and indium-doped CdO (x = 0.075, IV sample) nanoparticles. (c) SERS spectra of mercaptopyridine obtained using the various iterations of indium-doped CdO substrates depicted in panel a. Mercaptopyridine (1 mM) was first allowed to adsorb on the substrates, then the mixture was centrifuged, rinsed, and dried prior to analysis. The highest enhancements are observed at the lowest doping loadings (II, III, and IV spectra). Adapted with permission from Zhang et al., J. Phys. Chem. C, 2021, 125, 17125–17132. Copyright 2021 American Chemical Society. | ||
Doping for SERS purposes is often carried out to include plasmonic metals, such as silver or copper. The introduction of these metals causes an increase in the metallic character of the semiconductor, thus improving the ability of the final nanomaterial to elicit the SERS effect by concomitant electromagnetic and charge transfer mechanisms. An example of this type of doped semiconductors utilized for SERS detection in the NIR is the Ag-doped TiO2 implemented by Zhou et al.174 This doped material was then decorated with Ag nanospheres synthesized by sol-hydrothermal method to further enhance the SERS signal via their ability to yield an electromagnetic enhancement, and the formation of hotspots at the junctions between the main surface and the decorations. Interestingly, UV/Vis diffuse reflectance spectroscopy indicated that the reduction in the band gap of TiO2 after Ag-doping was not sufficient to promote analyte-substrate charge transfer on its own. In fact, the 785 nm laser utilized for the measurements was found to cause charge transfer from the HOMO of the analyte to the lower energy level of the Ag/Ag-doped TiO2 system, then to the conduction band of TiO2, and finally to the LUMO of analyte (PICT mechanism).
It might be apparent now that metal oxides are among the most popular semiconductors. The engineering of their band gap is most frequently carried out by engineering defects such as oxygen vacancies. Haldavnekar et al.175 utilized this strategy for the fabrication of undoped quantum sized ZnO with SERS capabilities. Quantum-sized (1.7–18.99 nm) ZnO nanoparticles were synthesized by a multi-photon ionization technique in which ZnO seeds were first created from bulk zinc by pulsed-laser ablation in the presence of atmospheric oxygen, and let self-assemble by melting, collision, and coalescence to form nanorods. The resulting nanomaterial was a hybrid system consisting of nanorods arranged as a ZnO nanodendritic platform, decorated with quantum sized ZnO nanoparticles on the top (Fig. 8a). The authors postulated that the high energy generated in the pulsed laser ablation technique introduced crystal defects in the ZnO structure, as well as additional electrons in the conduction band. The collective oscillation of the resulting increased electron density gave rise to an LSPR in the NIR range, enabling overall SERS enhancements with standard probes crystal violet, rhodamine 6G, 4-aminothiophenol, and 4-mercaptobenzoic acid around the order of 106. These overall enhancements were also the result of charge transfer, as previously described for all semiconductor systems. The charge transfer in this case was attributed to the presence of defects (oxygen vacancies, defects in crystal geometries at the corners and edges, stacking faults, etc.) and identified to originate from electronic transfer from the HOMO of the analytes to the conduction band of the semiconductor, and from the valence band of the semiconductor to the LUMO of the analytes.
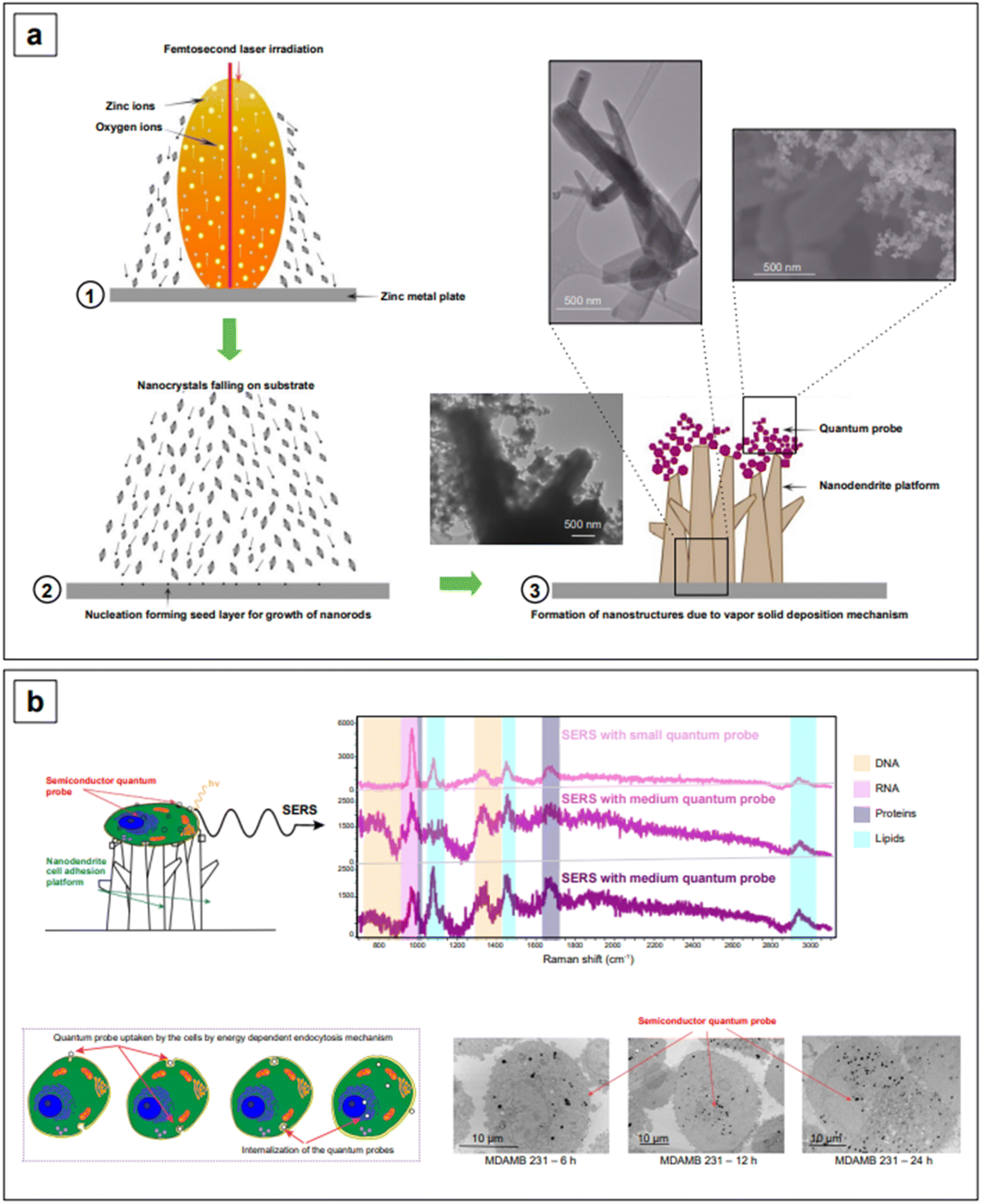 | ||
| Fig. 8 (a) Fabrication and mechanism of formation of the ZnO nanodendrite-quantum probe assembly. (b) Single-cell detection by SERS. The nanodendrite platform has cell-adhering properties; the quantum probes are internalized by cells, increasingly with time, in an ATP-dependent process. The dimension of the quantum probe also plays a role in the extent of internalization. SERS spectra of adhered cells demonstrate multiplex and label-free detection of cell components: DNA (beige), RNA (pink), proteins (purple), and lipids (cyan). Adapted from Haldavnekar et al., Nat. Commun., 2018, 9, 3065. Figures licensed under Creative Commons CC BY license, https://creativecommons.org/licenses/by/4.0/. | ||
The same group utilized the described ZnO assemblies for differentiating cancer cells from non-cancerous cells via the SERS signatures of their DNA, RNA, protein, and lipid constituents. For example, based on the signal intensities of lipids versus proteins, they categorized cancer cells as having low lipid:protein ratio compared to healthy cells. The cellular uptake of nanoparticles was also investigated to characterize the limits of detection of the individual biomolecule classes inside the cells. They postulated that the signal intensity of lipids was the highest and the DNA/RNA was the lowest because a smaller number of nanostructures reaches the nucleus, and it took more time for the nanoparticles to reach the nucleus compared to the cytoplasm. This work provides strong evidence that the size and shape of nanoparticles decide the cellular uptake and thusly affect the signal intensity of individual biomolecules inside the cells. Further research into the development of nanostructures with a better cellular uptake can extend the limits of simultaneous detection of biomolecules inside cells and tissues.
Band gap engineering is not the only strategy that can be adopted to improve the SERS performance of semiconductors. Another common strategy is to fabricate composite materials. With composite materials, we here identify those substrates resulting from the coupling of two different materials in which only one of them is a semiconductor, such as gold on graphene. Contrary to what has been said about heterojunctions and doping, coupling of semiconductors to SERS-active non-semiconductors (i.e., plasmonic metals) as composites does not yield any modification and tailoring of the band gap of the resulting composite material. However, the combined chemical enhancement of the semiconductor (e.g., TiO2, graphene oxide, …) with the intense LSPR of the plasmonic metal (e.g., gold) gives rise to an improved system for SERS detection. An example of this approach is given by Lee and Kim,176 who reported an easy procedure to obtain gold nanoparticles of about 7 nm in size, dispersed on a graphene oxide surface, using a photoreduction method to promote the formation of Au0 (Fig. 9a). The obtained solution was subsequently drop-casted on a hydrophobic paper to fabricate a paper-based SERS sensor for the detection of fungicide thiram at the micromolar level, under 785 nm excitation.176
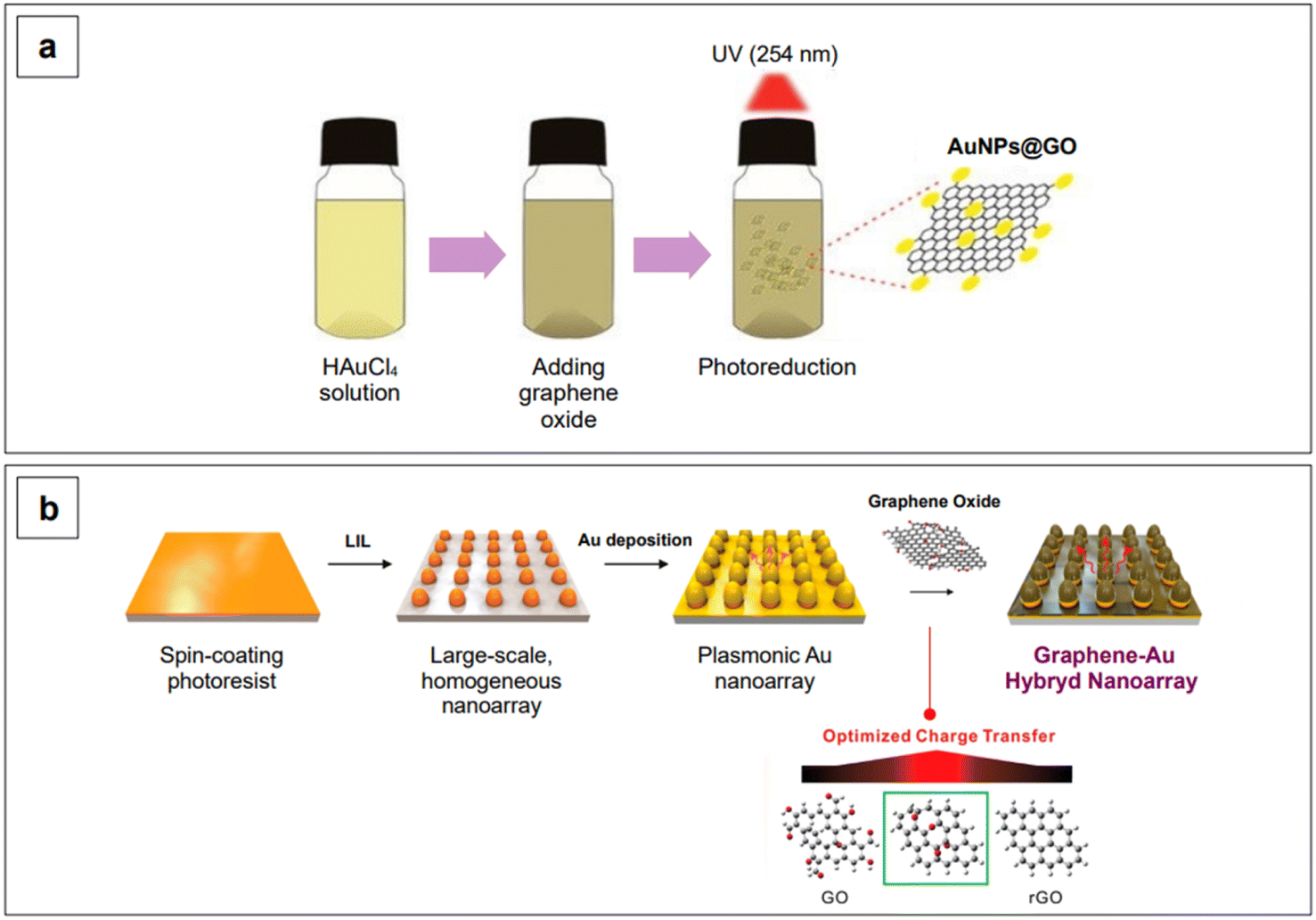 | ||
| Fig. 9 Examples of synthetic strategies for gold–graphene oxide composite production. (a) Photoreduction method using UV light. Adapted from Lee, D.-J. and Kim, D. Y., Sensors, 2019, 19, 5471. Figure licensed under Creative Commons CC BY license, https://creativecommons.org/licenses/by/4.0/. (b) Homogeneous plasmonic nanoarray generated by laser interference lithography (LIL) on a polymeric substrate, followed by gold coating by physical vapor deposition. An additional graphene oxide layer was established by electrostatic interactions after prior functionalization of gold with cysteamine. The degree of reduction of graphene oxide was tuned based on the maximization of the SERS performance of the substrate. Adapted with permission from Yang et al., Nano Lett., 2019, 19, 8138–8148. Copyright 2019 American Chemical Society. | ||
In a recent work, Liu et al.177 fabricated a gold–graphene oxide composite, Au@GO, incorporated with Fe3O4 nanoparticles and poly(dopamine), and utilized it for the SERS detection of phenanthrene in standard solution (LOD 10−2 ng mL−1). The magnetic character of Fe3O4 allows for the reuse of the SERS-active substrate, while the presence of poly(dopamine) prevents the aggregation of the gold nanoparticle component. When a suitably structured analyte is to be detected, such as phenanthrene, the peculiar π–π stacking of graphene or graphene oxide sheets favors π–π interactions with the adsorbed analyte. This promotes a maximization of the electromagnetic enhancement component in graphene-based plasmonic composites by shortening the distance between the analyte and the plasmon-sustaining surface.152,178,179 π–π interactions are also leveraged in graphene-only substrates (i.e., Graphene-Enhanced Raman Scattering, GERS180), optimizing the analyte-substrate contact and favoring signal enhancement via charge transfer mechanism. However, GERS is essentially confined to applications in the visible range, and for more information on this topic, the reader is referred to specialized literature.181–184
Yang et al.185 reported a graphene–Au hybrid SERS nanoarray for the characterization of neuronal differentiation, where the Raman signal enhancement is obtained by combining the electromagnetic mechanism of the plasmonic Au cones nanoarray with the chemical mechanism of graphene oxide. The degree of subsequent reduction of the latter was tuned to ensure better interaction with the target molecule.185 The array was obtained by laser-interference lithography (LIL) followed by gold deposition and graphene oxide functionalization via prior cysteamine linking. The homogeneous pattern created by LIL ensures a consistent enhancement without high point-to-point signal variations (Fig. 9b). The resulting graphene oxide/reduced graphene–Au nanoarray exhibits SERS activity in both the visible (λexc 514 and 633 nm) and NIR range (λexc 785 nm), although the highest local electric field responses were obtained when exciting at 633 nm, due to the specific morphological design of the plasmonic component of the nanoarray.185 Further tuning in the NIR could be achieved by intervening on the morphological aspects of the nanoarray, as seen for colloidal nanoparticles in Section 2.1.1.
Another interesting example of NIR-SERS composite plasmonic-semiconductor substrates has been described by Yang et al.,186 who have also incorporated up-conversion nanoparticles for use in both luminescence resonance energy transfer (LRET) and NIR-SERS experiments (λexc 980 nm). These substrates consisted of heterojunctions arising from the combination of Ag nanorices with W18O49 nanowire films coated with up-conversion nanoparticles (NaYF4:Yb3+, Tm3+). The plasmons generated in the Ag/W18O49 nanowires not only contributed to the SERS signal but also improved the up-conversion photoluminescence efficiency of NaYF4:Yb3+, Tm3+ nanoparticles. Three-dimensional finite element simulations demonstrated that the maximum SERS enhancement is reached when the heterojunction interface is aligned in such a way that the tip of a silver nanorice is in vertical contact with the sides of a W18O49 nanowire. In the size optimization of the up-conversion component, it was found that 10 nm NaYF4:Yb3+, Tm3+ nanoparticles showed maximum LSPR response in the NIR, while a significant decrease was observed upon further increasing of the size.
The combination of plasmonic and other materials can also be achieved via the fabrication of core–shell structures. Plasmonic metals such as Au, Ag, and Cu are typically used as the core, while other materials, such as other transition metals, semiconductors, or SiO2, are generally selected as the shell phase. This nanostructure architecture tunes the electronic structure and the optical properties of the final material by exploiting the phenomenon of borrowing SERS activity. When a target molecule is adsorbed on the shell surface of a plasmonic–nonplasmonic core–shell nanostructure, there is no direct contact with the plasmonic metal; however, the shell architecture allows the nonplasmonic material to borrow SERS activity from the plasmonic core, amplifying the Raman signal of the analyte by the electromagnetic mechanism. Because the SERS enhancement is strongly related to the distance between the analyte and the plasmon-sustaining material,152 the nonplasmonic shell is required to be very thin. As previously described for traditional spherical nanoparticles (Fig. 2a), the position of the λLSPR of core–shell nanoparticles is influenced by the size of the plasmonic component; for example, the larger the core size, the higher the λLSPR. It is therefore apparent that large cores must be utilized to push the SERS activity of these nanostructures in the NIR. Unfortunately, ultrathin shells without defects (pinhole-free shells) are difficult to prepare, and thus, for applications in the NIR, the research has moved towards inverse architectures, in which the plasmonic metal is utilized to fabricate a thick shell, while another material (another transition metal, a semiconductor, SiO2, …) is utilized as the core.
Interestingly, it is possible to create a complex core–shell structure using SiO2 as the shell also on an anisotropic core, as reported by Atta et al.187 In their work, they demonstrated that it is possible to obtain both anisotropic SiO2 shells that follow the morphology of gold nanostars, and isotropically etched core-covering shells that leave the nanostar branches exposed, without altering their length or sharpness. The first type of core–shell structure is obtained during the initial phases of the silica shell growth, as this initially follows the anisotropic morphology of the nanostar. The second type of core–shell structure, on the other hand, is obtained after prior growth of a full, isotropic silica shell, with an optimized protocol that ensures the morphological preservation of the nanostar core. After the formation of a thick silica shell around the nanostars, a silica etching step is performed using a mild etching reagent, NaBH4, instead of the more common and stronger reagent NaOH. This demonstrated to be the crucial step to chemoselectively etch silica, leaving the branches of the gold nanostar exposed and unaltered.
Rare-earth semiconductors can also be utilized to form shells on plasmonic cores, the most common of which is CeO2. Bao et al.188 proposed a method that is easily adaptable to several combinations of metal@mesoporous oxide architectures, by simple variation of the pre-formed metal core or shell precursors. The same protocol also allows a facile modification of the shell thickness from 4 to 30 nm, by varying the Ce3+ added to the colloidal gold sol. The plasmonic behavior of the gold core (55 nm) was tuned to the NIR region owing to the confinement effect of the CeO2 porous shell. The structure based on gold core and CeO2 shell was tested to detect toluene vapor at concentrations of 10 ppm (λexc 785 nm), and the observed SERS enhancement is attributed to the high number of micropores in the shell, that can trap target molecules, bringing them closer to the plasmonic core where the SERS effect is higher.
Among novel structures, shell components can also be made with Metal–Organic Frameworks (MOFs).189 MOFs are highly ordered porous materials synthesized by combining metal ions or clusters with an organic ligand, following the commonly adopted supramolecular approach. Due to their rational design and functionalization, this class of materials is widely applied in multiple fields such as catalysis,190 gas storage,191 drug delivery,192 and sensing,193 including SERS. For instance, Zhang and co-workers194 presented a one-pot synthesis for a series of SERS substrates composed of a Mg-based MOF, MOF-74, as the shell of varying thickness, and a gold core. While dimethylformamide (DMF) is the most commonly used solvent in MOF syntheses, these Au-MOF core–shell nanoparticles were synthesized in DMF–ethanol. The resulting nanoparticles were utilized for the detection of model analyte 4-nitrothiophenol by NIR-SERS (λexc 785 nm) with a LOD of 69 nM, as well as for the in situ monitoring of plasmon-assisted reactions. MOFs have also been utilized in composites, such as in the very recent work by Zhao et al.,195 in which a Fe-MOF was utilized as a substrate to anchor gold nanorods, and implemented for the catalytic degradation and SERS detection of methylene blue, with a LOD of 9.3 × 10−12 M.195
Exploiting the concept seen for thicker plasmonic shell-nonplasmonic core nanoparticles, NIR-active substrates can also be fabricated by creating assemblies via plasmonic decorations on non-plasmonic large nanoparticles. In a very recent work, Bock et al.196 prepared silica nanoparticles decorated with Au nanoparticles and demonstrated SERS activity and fit-to-purpose homogeneity for potential applications for in vivo imaging. In brief, after the separate synthesis of SiO2 (∼190 nm) and Au (∼3 nm) nanoparticles, the latter were introduced into the SiO2 sol to form SiO2@Au seeds. The final product was then obtained by a seed-mediated method in which the Au growth was controlled by varying the concentration of the Au3+ precursor. By increasing the Au precursor concentration, the λLSPR also increases. This behavior was explained by the simultaneous presence of multiple factors: a higher number of Au nanoparticles, a narrow nanogap among the Au nanoparticles (1 nm for the highest Au precursor concentration), and thus, a stronger plasmonic coupling of the Au nanoparticles on the SiO2 surface. The highest enhancements upon 785 nm illumination were therefore achieved at those Au concentrations that reduced the gap between adjacent Au nanoparticles on the surface of silica nanoparticles, generating hotspots. The Au-decorated silica nanoparticles were then labeled with more than ten Raman-active compounds and tested as potential SERS tags during in vivo NIR-SERS measurements.
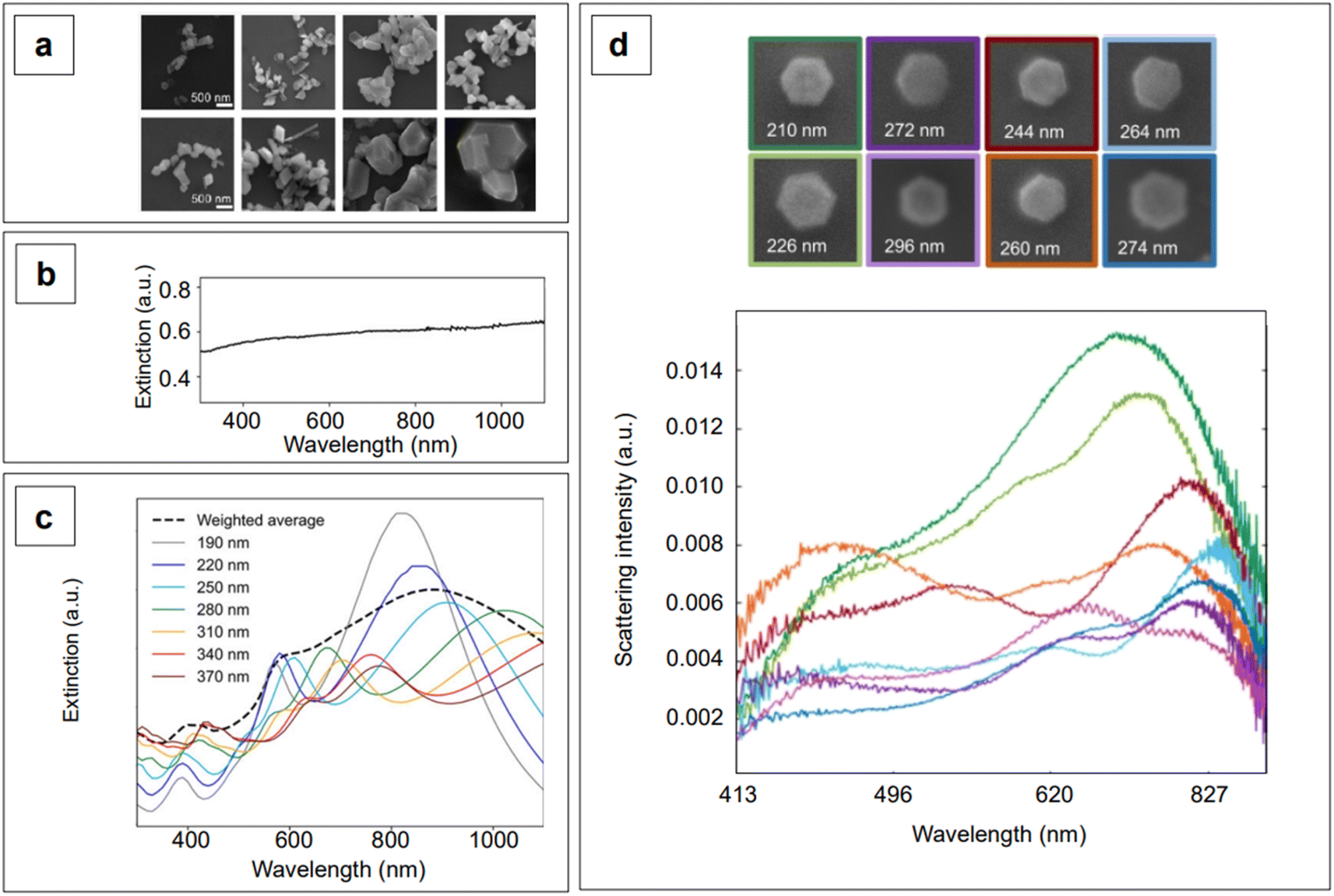
Although the majority of alternative materials that we have listed so far are metal oxides, some alternative metals,197–199 as well as transition metal nitrides (e.g., TiN dimers and ZrN)200 and chalcogenides (e.g., CuTe nanocubes,201 AuCu nanostars/MoS2202), also show potential for use as SERS-active plasmonic substrates in NIR applications. Transition metal nitrides TiN and ZrN, for example, exhibit high electron conductivity200 compared to metal oxides, have optical properties in the NIR thats are comparable to gold,200,203,204 and their electronic structure can be easily tailored via nitrogen vacancies engineering,197 as previously illustrated for oxygen vacancies in metal oxides. Among the alternative metallic nanomaterials beyond gold and silver, magnesium nanoparticles have garnered interest in the last few years, demonstrating interesting optical properties, including near-field scattering that can be exploited in enhanced spectroscopies applications such as SERS.205 Hopper et al.199 elucidated and optimized the colloidal synthesis of Mg nanoparticles with varying size and broad LSPR in the visible and NIR ranges (Fig. 10a and b). The broad plasmonic response was attributed to the polydispersity of the sample, as well as to nanoparticle aggregation caused by the absence of stabilizing agents. However, numerical simulations by Discrete Dipole Approximation (DDA, Fig. 10c) show a correlation with the extinction spectra obtained by dark field scattering of individual nanoparticles (Fig. 10d), indicating promising avenues for less polydisperse versions of these materials. It must be recalled that magnesium, as any other non-noble metal, has high susceptibility for oxidation and chemical reactivity in general; this disadvantage, however, can be exploited to generate SERS-active composite nanomaterials, such as Au- and Ag-decorated Mg nanoparticles obtained by galvanic replacement.206 A graphical summary of the alternative materials discussed in this section is reported in Fig. 11, along with a list of the diverse synthetic strategies that have been adopted for their fabrication.
 | ||
| Fig. 10 (a) Magnesium nanoparticles as prepared by Hopper et al.,199 obtained with overall increasing reactant concentrations (from left to right) and decreasing reaction temperature (top to bottom). (b) Experimental extinction spectrum of colloidal magnesium nanoparticles dispersed in isopropyl alcohol and having an average size of 300 ± 60 nm. (c) DDA-simulated extinction spectra of magnesium nanoparticles having varying size, hexagonal shape as found in real samples, and thickness ∼0.1 times their tip-to-tip length. The weighted average obtained on the basis of the experimental size distribution is reposted as a black dashed profile. (d) Color-matched SEM micrographs (top) and scattering spectra (bottom) of Mg hexagonal platelets of varying tip-to-tip size. Adapted from Hopper et al., J. Phys. Chem. C, 2022, 126, 563–577; figures licensed under CC BY 4.0, https://creativecommons.org/licenses/by/4.0/. | ||
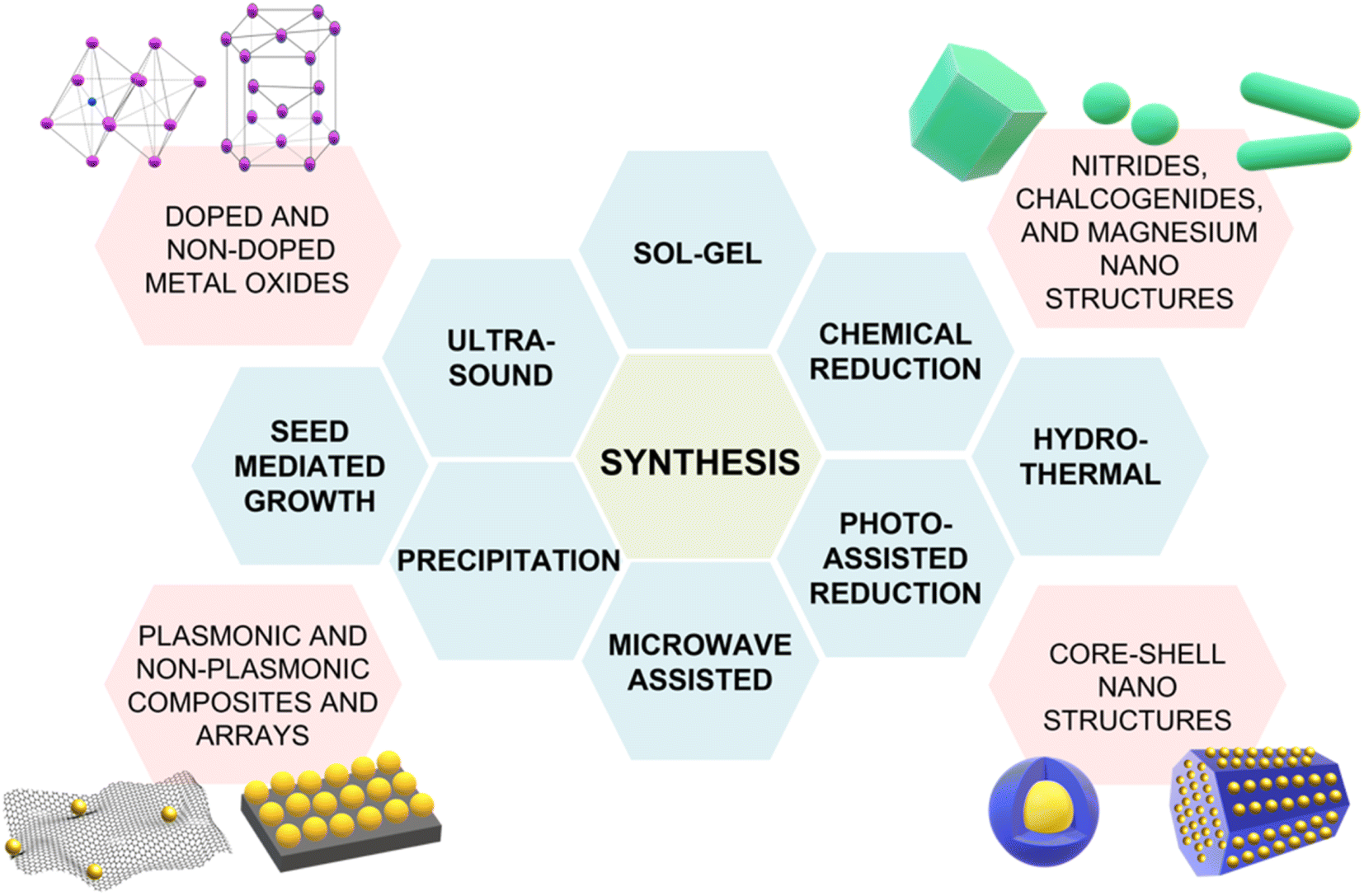 | ||
| Fig. 11 Summary of the alternative NIR-active SERS substrates discussed in this section and their varying synthetic strategies: sol–gel,172 chemical reduction,199 hydrothermal synthesis,173 photo-assisted reduction,176 microwave-assisted synthesis,207 precipitation,188 seed-mediated growth,196 ultrasound.208 | ||
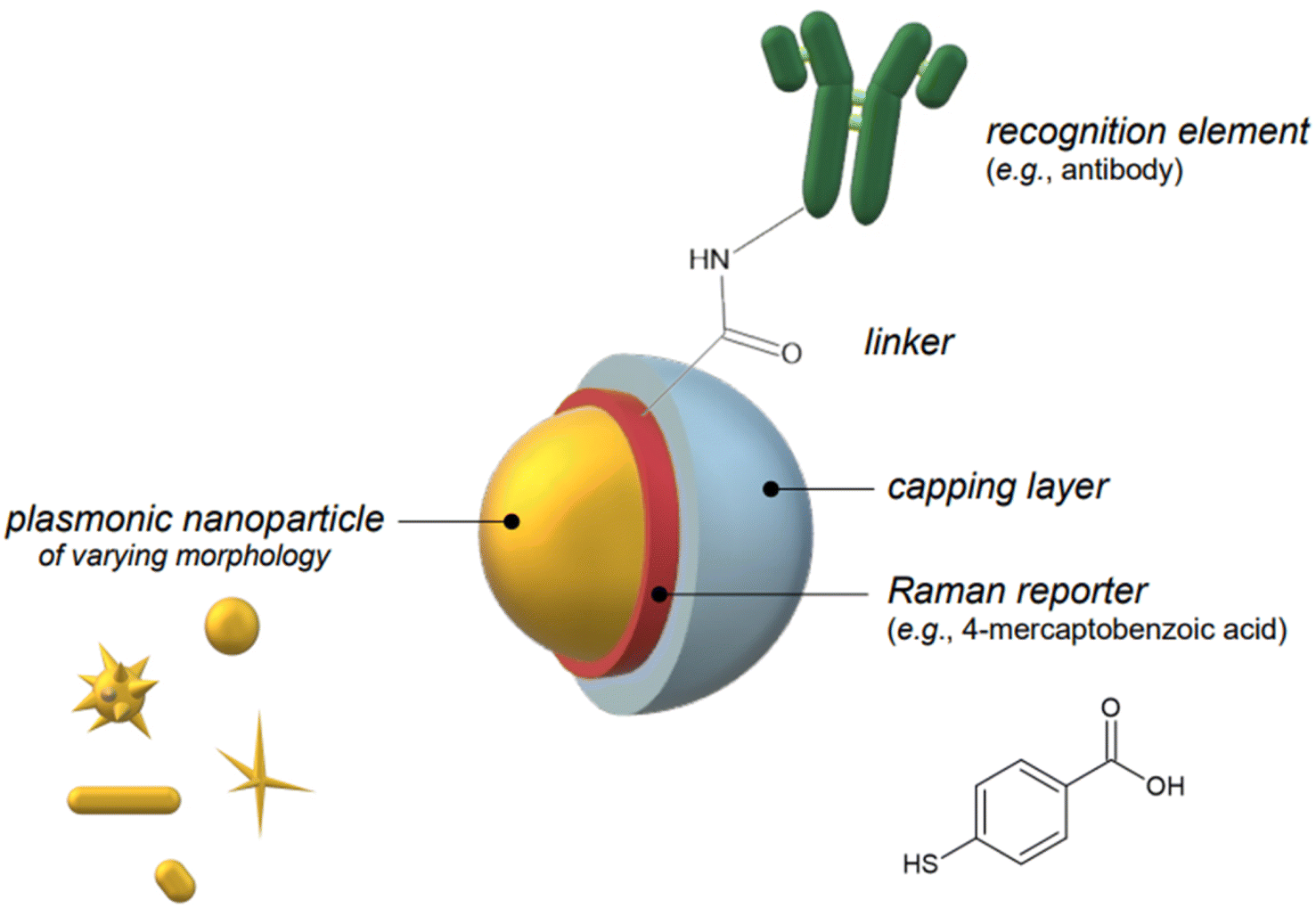 | ||
| Fig. 12 Schematic representation of the components of a SERS nanotag: a plasmonic nanoparticle of varying morphology (yellow), an embedded or chemisorbed Raman reporter (red), a biocompatible capping layer (e.g., silica, a polymer such as polyethylene glycol or bovine serum albumin, etc.; pale blue), and a recognition element such as an antibody (green), covalently conjugated to the Raman reporter via the use of a linker molecule. The SERS nanotag components are not in scale. | ||
Because SERS nanotags are especially utilized in complex biological samples such as cell cultures and tissues, biocompatibility and stability in physiological conditions are of paramount importance. For these reasons, biocompatible capping systems, typically polymeric in nature such as bovine serum albumin (BSA) or varyingly functionalized polyethylene glycol (PEG: PEG-SH, NH2-PEG-SH, etc.), are utilized to prevent opsonization.209 The latter consists in the formation of a multi-layer protein corona around the nanoparticle, which causes passivation of the surface from analyte adsorption and, ultimately, irreversible colloid aggregation.209,210 The growth of silica shells around plasmonic nanoparticles, which was described in the previous section of this review, can also be implemented as an alternative to the use of natural or synthetic polymers, as they not only offer a platform to tune the plasmonic response of the nanoparticle system, but also achieve biocompatibility and stability in physiological media. Examples of this strategy can be seen in the works by Kircher's group,90,211–214 who utilizes SERS nanotags for in vivo SERRS and SESORRS (see Section 2.2) imaging and thus, have stringent biocompatibility needs. An additional requirement that a nanotag capping system might have is the selectivity towards a particular biological target. For example, Dugandžić et al.215 capped their silica-encapsulated nanotags with mannose for fast uptake by mature macrophages, which are known to express a mannose-specific C-type lectin receptor.
The recognition element can be a peptide,214,216 an antibody,90,115 or an aptamer,213,217 and it is typically covalently bound to the nanoparticle via the Raman reporter, taking advantage of thiol chemistry or via the use of coupling agents such as heterobifunctional cross-linkers.218,219 For example, Song et al.216 utilized a commonly adopted bioconjugation strategy known as EDC-NHS chemistry218–220 (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, N-hydroxysuccinimide) to anchor arginylglycylaspartic acid (RGD), a peptide recognition element for adenocarcinoma biomarker αvβ3 integrin, to gold nanostars. This strategy is utilized when the bioconjugation reaction involves an amine and a carboxylate; in this case, the amine comes from the RGD peptide, while the carboxylate comes from the Raman reporter 4-mercaptobenzoic acid (4-MBA). As shown in Scheme 1, EDC activates the carboxylate by forming a reactive O-acylisourea ester conjugate, making the charged form of NHS, SuO−, able to yield an NHS-ester by transesterification.219–221 The formed NHS-ester is an amine-reactive species that reacts with the primary amine in the arginine's residue of RGD, yielding the final reporter-recognition element adduct, RGD/4-MBA.218–220 Such a bioconjugate was obtained after prior incubation of 4-MBA with gold nanostars, during which the Raman reporter chemisorbs to the gold substrate (Fig. 13a). Another example of bioconjugation reaction for the fabrication of SERS nanotags is illustrated by Bhamidipati et al.,217 who utilized a thiolated aptamer as the recognition element for epithelial cell adhesion molecule (EpCAM), a protein that is utilized as a metastasis biomarker of different oncological diseases. The bioconjugation reaction was undergone step-wise, utilizing a heterobifunctional cross-linker, sulfo-succinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate (sulfo-SMCC, Fig. 13b). 4-Aminothiophenol was pre-adsorbed onto gold nanostars via its sulfhydryl group, leaving its primary amine able to react with the NHS-ester terminus of sulfo-SMCC, yielding an amide bond.218,222 The introduction of the thiolated aptamer at this point initiates a reaction with the maleimide terminus of sulfo-SMCC, producing a stable thioether bond between the aptamer and the linker/reporter system.218,222 In those cases in which a silica shell is chosen as the capping material, prior functionalization of silica is necessary to perform any subsequent bioconjugation reaction. For example, in a work by Kircher's group on the ex vivo and in vivo SERRS imaging of breast cancer lung metastases,90 SERRS nanotags for transmembrane glycoprotein Tissue Factor (TF) were obtained by first growing a silica shell around gold nanostars in the presence of resonant Raman reporter IR780. Subsequently, the silica surface was functionalized with (3-mercaptopropyl)trimethoxy-silane (MPTMS), providing sulfhydryl groups for further grafting of the recognition element, an antibody targeted against glycoprotein TF. Anti-TF conjugation was then achieved by utilizing the heterobifunctional linker poly(ethylene glycol) (N-hydroxy-succinimide 5-pentanoate) ether N′-(3-maleimidopropionyl)amino ethane (NHS-PEG4k-Mal), which reacts in a similar way as illustrated for sulfo-SMCC (Fig. 13c).
 | ||
| Scheme 1 EDC-NHS chemistry exemplified by the arginylglycylaspartic acid peptide/4-mercaptobenzoic acid (RGD/4-MBA) conjugate utilized in the fabrication of the SERS nanotags developed by Song et al.216 Abbreviations: EDC, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide; NHS, N-hydroxysuccinimide; SuO-, anionic N-hydroxysuccinimide. | ||
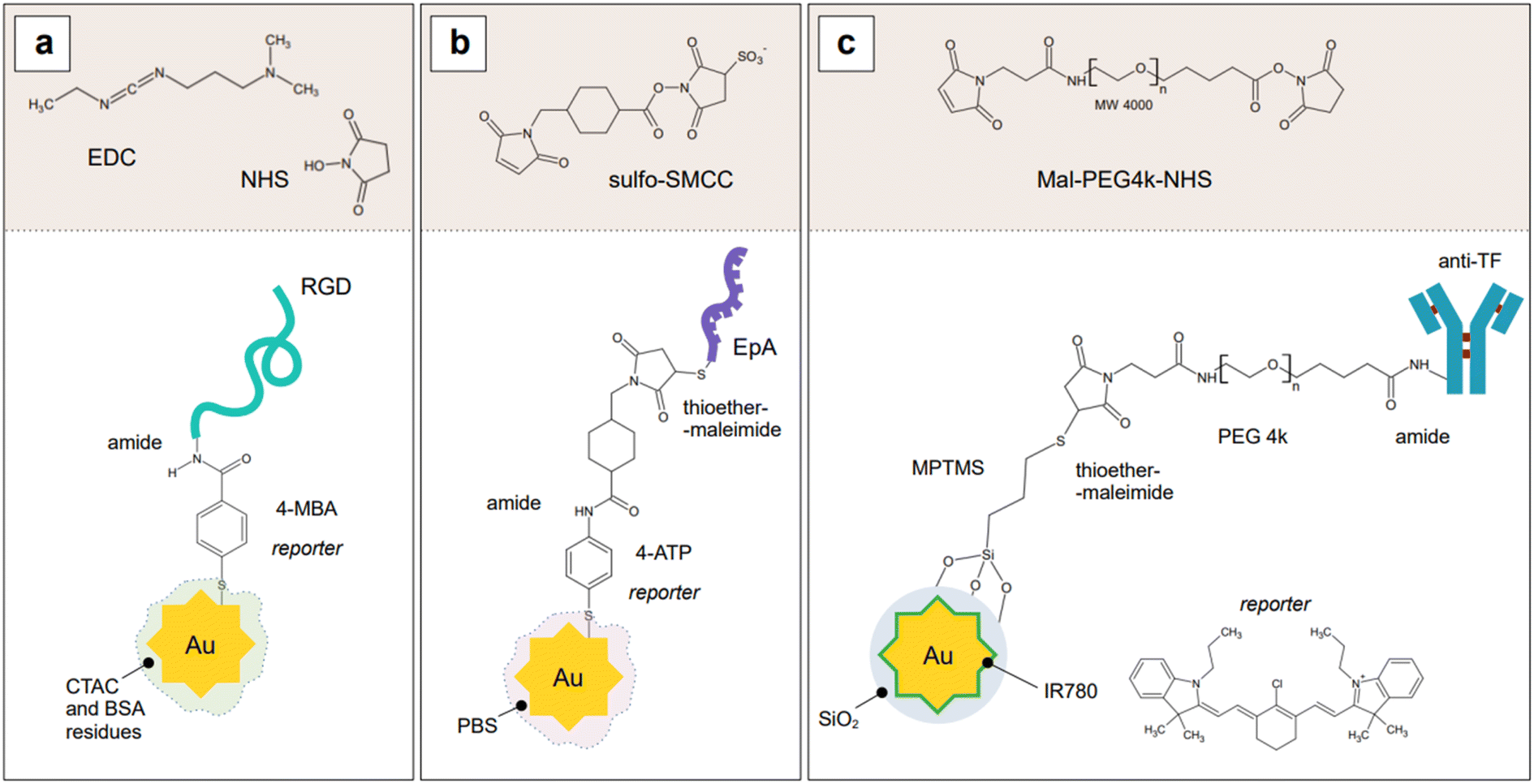 | ||
| Fig. 13 Architecture of SERS nanotags and crosslinking chemistry. (a) Schematic representation of the SERS nanotags developed by Song et al.,216 obtained utilizing EDC-NHS chemistry. Gold nanostars were incubated with 4-mercaptobenzoic acid (4-MBA) and conjugated via the formation of an amide bond to the arginylglycylaspartic acid peptide (RGD). The capping layer is likely constituted by remaining residues of cetyltrimethylammonium chloride (CTAC) and bovine serum albumin (BSA) originating from synthesis and SERS nanotag fabrication, respectively. (b) Schematic representation of the SERS nanotags developed by Bhamidipati et al.,217 obtained utilizing sulfo-SMCC as the cross-linking agent. Surfactant-free gold nanostars were incubated with 4-aminothiophenol (4-ATP), which was then allowed to react with the NHS-ester terminus of the cross-linking agent. The maleimide terminus was subsequently allowed to react with the thiolated aptamer, EpA. The capping layer is likely formed by phosphate ions coming from the PBS buffer utilized to redisperse the SERS nanotags after fabrication and purification. (c) Schematic representation of the SERS nanotags developed by Nayak et al.,90 obtained utilizing Mal-PEG4k-NHS as the cross-linking agent. Surfactant-free gold nanostars were encapsulated in silica shells, obtained in the presence of Raman reporter IR780, which remains embedded in the silica shell. The shell was then functionalized with mercaptopropyltrimethoxysilane, MPTMS, providing it with sulfhydryl groups to react with the maleimide group of the cross-linking agent. The antibody anti-TF was then conjugated via the NHS functionality of the cross-linking agent. Drawings are not in scale. | ||
It must be noted that case-specific experimental parameters like pH, temperature, pre-treatments, incubation environment and time, all compound into an efficient functionalization of nanoparticles into SERS nanotags. For example, EDC chemistry is most efficient at a pH of 4.5, but it can be also successfully carried out at higher pH values, close to neutral, if required by the system (e.g., use of PBS buffer).218 As another example, reducing conditions (e.g., incubation in tris(2-carboxyethyl)phosphine, TCEP223) are used as a pre-treatment to the adsorption of thiols, to cleave any disulfide bonds resulting from either self-reaction between probes or the presence of protecting capping groups. As for thiol-mediated functionalization reactions of gold substrates, temperature has proven to be a crucial parameter. Liu et al.224 demonstrated that functionalization of gold nanoparticles at freezing conditions (−20 °C) leads to an increase in the loadings of thiolated oligonucleotides, compared to traditional room temperature and reagent-based (i.e., salting out, low pH) procedures. This is the result of the formation of micropockets of segregated reactants during the freezing process. This segregation causes an increase in the local concentration of the thiolated reactants, thus increasing their rate of adsorption and, with it, their resulting packing density on the surface.224 While this procedure was originally established for the fabrication of nanoflares,224,225 it has been seamlessly transferred to analogous systems such as SERS nanotags.226,227
The systems described so far all make use of nanostars as the plasmonic component of the nanotag. This is because control of the hotspots formation via the use of intrinsic features such as nanostars branches is the preferred way of creating them in these analytical systems. Indeed, they allow for a reproducible, constant enhancement compared to the formation of hotspots by salt-induced aggregation, thus allowing fully quantitative studies. An alternative, although more complex, way of generating hotspots is the assembly of dimers or sandwich superstructures among SERS nanotags, such as in the case of the previously mentioned work by Bhamidipati et al.217 The SERS nanotags described in Fig. 13b were utilized as the components of a planar array-based sandwich assay for the detection of EpCAM, both as a soluble protein in standard solution (binding buffer: 1× PBS, pH 7.4, 4.5 g L−1 glucose, 5 mM MgCl2) and as a transmembrane protein in cell culture, under a 785 nm illumination. EpCAM-deficient HeLa cell lines were utilized as negative controls and thus, as substrates for assay selectivity studies. The planar array was prepared by first adhering surfactant-free gold nanostars onto a silanized and BSA-coated glass substrate, and by subsequently functionalizing the nanostars with the same thiolated aptamer utilized in the fabrication of the SERS tags. To aid in the minimization of non-specific target binding, a 6-mercaptohexanol backfilling was established on the substrate-adhered nanostars by simple incubation. EpCAM protein was then incubated on such prepared substrates, after which washing and subsequent incubation with the SERS nanotags were performed. Captured EpCAM targets are in this way immobilized between the two plasmonic elements of the sandwich, thus benefiting from an increased enhancement given by the formation of hotspots between the SERS nanotags and the immobilized nanostars. This method allowed for a LOD of 10 pM for the soluble protein and 10 cells for the transmembrane protein when a truncated aptamer, EpA, was utilized as the recognition element.
When sandwich configurations are implemented such that transmission measurements are required, for example in solution, considerations on the λmax of the plasmon band of the final dimer or aggregate must be taken into account. As seen in Section 2, the formation of dimers, trimers, and multiple aggregates shifts the plasmon band to higher wavelengths. This can be an advantage, in that it can be used to tune the enhancing substrate such that the LSPR can be excited by higher wavelength excitation sources than those that would excite its native, well-disperse colloidal form. However, the extent of this shift for measurements in transmission must never exceed the amount given by the difference between the native λLSPR and the wavelength of the excitation source. In other words, when working in transmission mode, pre-resonance conditions between the enhancing substrate and the laser source must be maintained for ideal LSPR excitation, and thus, ultimately, for best analytical performance. Readers interested in the physics behind this best practice point can refer to Sivapalan et al.228 The practical effect of this can be observed in Tatar et al.,115 where two versions of the same solution-based sandwich assay were implemented utilizing nanostars–nanostars and nanostars–nanosphere SERS tags dimers. The dimers were obtained by the bridging upon analyte capture of a 4-ATP/antibody SERS nanotag based on nanostars with “SERS amplifiers”, PEG-SH/antibody-functionalized gold nanospheres or nanostars. The plasmon band of both dimer types falls past the utilized 785 nm excitation: this resulted in lower SERS signal than expected for a sandwich configuration. However, the nanostar–nanostar dimer system is able to compensate for this drawback because of the lightning rod effect given by the branched morphology. The nanostar–nanostar configuration of the assay was thus able to achieve an LOD in the range of 1 to 3 ng mL−1 for the detection of biomarker carcinoembryonic antigen-related cell adhesion molecule 5 (CEA-CAM5).
Because the Raman reporter is the actual element that produces the SERS signal, its quality must be such to guarantee low limits of detection and low fluorescence background. This can be achieved if the chosen reporter has the general characteristics of having a clean, defined spectrum, high photostability, and a large SERS cross section with low fluorescence quantum yield under the chosen illumination conditions. This implies that molecules that behave as effective Raman reporters under visible excitation might not behave equally well when illuminated in the NIR II, III, or SWIR. While common visible range Raman reporters such as 4-mercaptobenzoic acid,57,216,229 4-aminothiophenol,115,217 methylene blue,230 1,2-bis(4-pyridyl)ethylene (BPE),57 or rhodamine 6G,230 can still be successfully utilized in the NIR I, such as under 785 and 1064 nm illuminations, moving towards higher wavelengths brings about the need of engineering this aspect, too, in analogy to what was illustrated for the nanoparticle morphology (Section 2.1.1). Moreover, exploitation of resonance effects to further increase the signal by working under NIR-SERRS conditions also requires engineering of the reporter molecule such that it possesses resonances under the given illumination.
Although this line of research has not seen many additions in the most recent years, groups such as Kircher's and Graham's and Faulds' produced notable work in the years between 2015 and 2017, deserving a mention here. Harmsen et al.231 developed for the first time five 785 nm-resonant chalcogenpyrylium reporters for improved SERRS tags with attomolar sensitivity. These reporters were designed to exhibit low fluorescence due to increased intersystem crossing given by the presence of chalcogens, an absorption maximum that was tunable with chalcogen atom substitution (i.e., S vs. Se), and high affinity for gold via the introduction of coplanar 2-thienyl groups that allow for direct adsorption. Synthesis is achieved by coupling reaction products 5 and 8 in Scheme 2a, after prior methylation and dehydration of known chalchogenpyranones 4 and 6 with methylmagnesium bromide, respectively, and condensation of 7 with N,N-dimethylthioformamide and hydrolysis of the resulting iminium salt intermediate.231 SERRS nanotags prepared with thiopyrylium Reporter 3 produced the highest SERRS signal, and this was attributed to the better adsorption ability of 2-thienyl groups to the gold surface as compared to phenyl groups. This was hypothesized in the rational design of the reporters,231 as 2-thyenyl groups are known to be more efficient at charge delocalization compared to phenyl substituents,232 which could aid in π-metal interactions. Moreover, 2-thienyl groups are known for shifting the absorption maximum of a compound,232 thus resulting in Reporter 3 having the most red-shifted λmax. As a consequence, SERRS intensity could be ranked (P < 0.05) in terms of the number of 2-thyenyl groups, as follows: Reporters 1a/1b (no 2-thienyl group) < Reporters 2a/2b (two 2-thienyl groups) < Reporter 3 (four 2-thienyl groups).231 Nanotags assembled with Reporter 3 showed attomolar sensitivity and were utilized for in vivo tumor margin delineation with an antibody targeting the epidermal growth factor receptor, EGFR.231
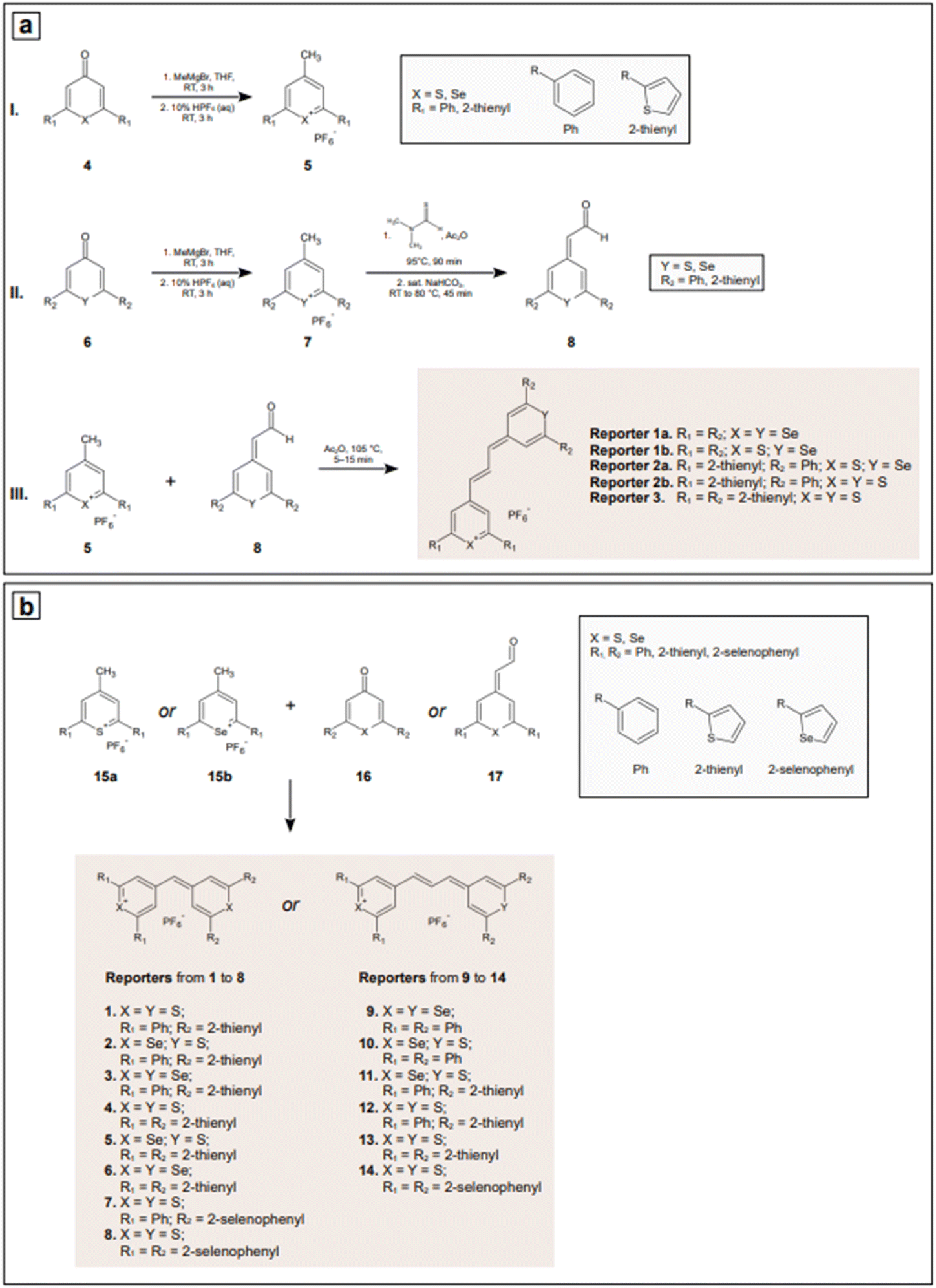 | ||
| Scheme 2 (a) Reaction scheme for the synthesis of 785 nm-resonant chalcogenpyrylium reporters as prepared by Harmsen et al.231 Abbreviations: THF, tetrahydrofuran; RT, room temperature; Ph, phenyl; Ac2O, acetic anhydride; sat., saturated. (b) Precursors and final chalcogenpyrylium reporters active under 1280 nm excitation, as prepared by Bedics et al.233 | ||
The synthesis presented by Harmsen et al.231 was then expanded by Bedics et al.233 to fabricate a library of 14 non-resonant chalcogenpyrylium reporters with activity in the NIR II (λexc 1280 nm) and picomolar sensitivity. In this work, the relationship between the SERS performance of the nanotag and the structural elements of the reporter, such as the number and type of chalcogen atoms and the number of methine groups, was further studied. In addition to the previously identified 2-thienyl groups,231 it was found that 2-selenophenyl substituents also make optimal moieties for the adsorption of the reporters onto gold nanoparticles.233,234 In fact, when comparing the SERS signal obtained from Reporter 13 (four 2-thienyl substituents) with that obtained from Reporter 14 (four 2-selenophenyl substituents) after prior adsorption onto hollow gold nanospheres, a significant difference in intensity can be observed, where the 2-selenophenyl-substituted reporter shows the highest values. This is in line with prior reports that indicate a greater affinity for gold of selenolates than thiolates.235,236 The library of reporters was in this case obtained by synthesizing starting materials 4-methylthiopyrylium or 4-methylselenopyrylium anions 15a and 15b and either chalcogenopyranone 16 or (4-chalcogeno-pyranylidene)acetaldehyde derivative 17, which were then condensed in a similar manner as seen in Harmsen et al.,231 to yield a series of monomethine (reporters from 1 to 8) or polymethine (9 to 14) reporters (Scheme 2b).
Bedics et al.233 observed that polymethine reporters exhibit higher SERS intensity under 1280 nm excitation compared to monomethine reporters, and attributed this behavior to the number of sp2 carbons in the chalcogenpyrylium backbone. For this reason, Kearns et al.237 prepared two chalcogenpyrylium pentamethine reporters having four 2-thienyl and four 2-selenophenyl substituents, respectively, and tested them after assembly as gold nanosphere-based SERS nanotags, verifying the trends highlighted by Bedics et al.233—2-selenophenyl substituents and longer polymethine bridges are the optimizing structural parameters for brighter, NIR-active chalcogenpyrylium SERS reporters.233,237 The prepared pentamethine chalcogenpyrylium reporters have absorption maxima at 959 nm (2-selenophenyl-substituted thiopyrylium) and 986 nm (2-thienyl-substituted selenopyrylium), and allowed for limits of detection of 51 ± 5 pM and 63 ± 5 pM when utilized in 100 nm hollow gold nanosphere-based nanotags under non-resonant 1550 NIR III illumination. These LODs are one order of magnitude higher than those achievable under the same experimental conditions utilizing commercially available reporters in the SERS nanotag assembly, as BPE. Given the absorption maxima of these pentamethine compounds, they were subsequently successfully implemented as resonant reporters in the fabrication of femtomolar-sensitive SERS nanotags for 1064 nm illumination.238
The library of chalcogenpyrylium reporters obtained by Bedics et al.,233 although very structurally similar to each other, shows non-overlapping SERS spectra. This is an intrinsic characteristic of Raman and SERS spectra, which combine the molecular fingerprint capability to narrow bands, thus making Raman reporters amenable to multiplexing. As opposed to the common singleplex strategy, multiplexing allows for the concomitant detection of multiple targets in a single measurement. Multiplexing is typically implemented by making use of different nanotags at the same time; heterogeneously functionalized (i.e., mixed-ligand) nanotags can also be in principle implemented, but their fabrication poses challenges in terms of differential enrichment given by the different reporters, which may negatively impact sensitivity and quantitation. Although more control and knowledge about mixed-ligand nanoparticles fabrication processes are progressively achieved,239 the use of a series of mono-reporter SERS nanotags in place of single, mixed-reporter SERS nanotags is the current preferred approach in SERS multiplexing.
An example of multiplexed detection using NIR-active SERS nanotags can be seen in the work by Zhang et al.,230 who developed a lateral flow SERS immunoassay for the multiplexed detection, on a single test line, of three myocardial infarction biomarkers, creatine kinase-MB isoenzymes (CK-MB), cardiac troponin I (cTnI), and myoglobin (Myo) (Fig. 14). Three SERS nanotags active under 785 nm excitation and targeted each at one of the three biomarkers were prepared utilizing silver-core gold-shell nanoparticles as the plasmonic element, antibodies targeted at CK-MB, cTnI, and Myo as the recognition element, and methylene blue, Nile blue A, and rhodamine 6G as the Raman reporters. The latter were incorporated by direct adsorption to silver, and subsequently embedded in the gold shell of the resulting bimetallic nanosphere; optimal enhancement is therefore obtained by exploiting the core–shell interface as a hotspot. The tags were further functionalized with SH-PEG-COOH as the stabilizing coating, which was utilized for the bioconjugation reaction with the antibodies using EDC-NHS chemistry. Indirect, multiplexed detection of the cardiac biomarkers on the lateral flow assay strip was implemented using a sandwich configuration, obtained by immobilizing antibodies targeted at the biomarkers on the same test line, which hybridize with the SERS nanotag/biomarker complexes formed in the conjugation pad area of the strip (left panel, Fig. 14).
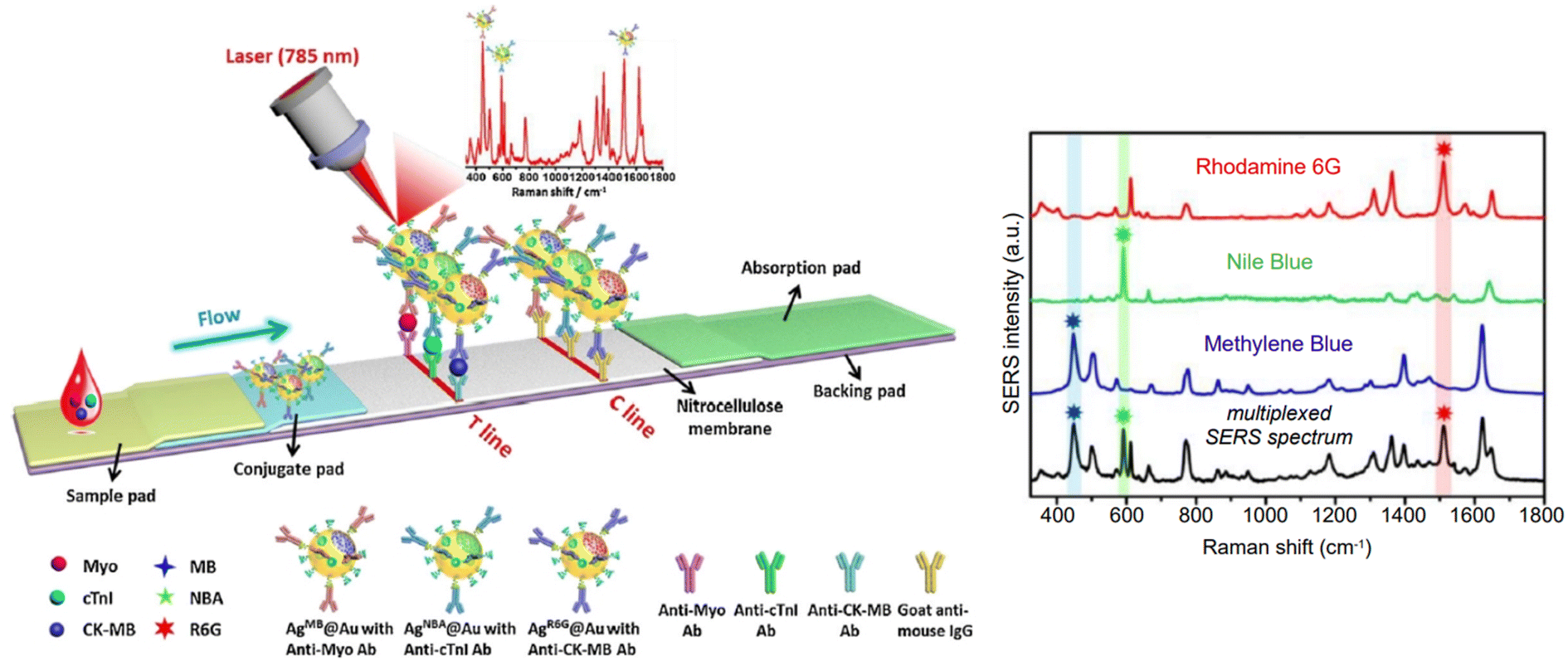 | ||
| Fig. 14 Schematic representation of the multiplexed NIR-SERS single test line lateral flow assay developed by Zhang et al.230 A mock-up or clinical blood sample is deposited on the sample pad. By capillary action, the analytes (Myo, myoglobin, purple sphere; cTnI, cardiac troponin I, cyan sphere; CK-MB, creatine kinase-MB isoenzymes, blue sphere) travel towards the conjugate pad, where they react with targeted SERS nanotags, pre-deposited on a single test line (T). A positive control line (C) is also implemented. The test line is imaged by SERS mapping, giving rise to a multiplexed SERS spectrum (red on the schematic, black on the right panel) when a positive sample is analyzed. The right panel illustrates the interplay between the three spectra of the nanotag reporters and the bands that are used for calibration and quantitation purposes. Drawings are not in scale. Reprinted with minor adaptations from Zhang et al., Sens. Actuators B Chem., 2018, 277, 502–509, Copyright (2018), with permission from Elsevier. | ||
The immobilization of all three nanotag/biomarker complexes on the same test line constitutes an improvement of a previous lateral flow design from the same research group, which had three separate test lines (i.e., sequential singleplex design), and thus, required longer times for the readout SERS signal to be acquired. The new design allowed for the total detection time to be reduced to one third, amounting to 17 minutes, while maintaining the limit of detection for each biomarker essentially unaltered, in the clinically relevant low to sub-pg mL−1 range. This is an example of how multiplexed detection can be especially of aid in time-sensitive applications, such as in the development of diagnostic tools for emergency room situations: myocardial infarction, as in this case, as well as potentially other acute cardiovascular diseases, or poisoning and overdosing events.
2.2 Collection set-up
In the introductory section we discussed how infrared radiation can penetrate tissues more in depth compared to visible light, reaching about 1 cm below the surface if a 785 nm source is used.60 What if we need to illuminate a target that is located at a larger depth? As previously mentioned, the concept of Spatially Offset Raman Spectroscopy can be exploited in SERS or SERRS terms too, setting up SESORS or SESORRS experiments. These deep Raman techniques require an intervention on the design of the instrumentation, in that the collection probe has to be placed at an offset position compared to the laser source,240 as depicted in Fig. 15a. The offset is necessary as it is the element that enables larger depth sampling; photons migrating laterally into a sample statistically tend to populate deeper volumes, because those that approach the air-sample interface tend to escape the sample instead, resulting undetected by an offset collection probe.241,242 Readers interested in the basics of SORS as an introduction to SESORS can refer to a recent tutorial review by Mosca et al.240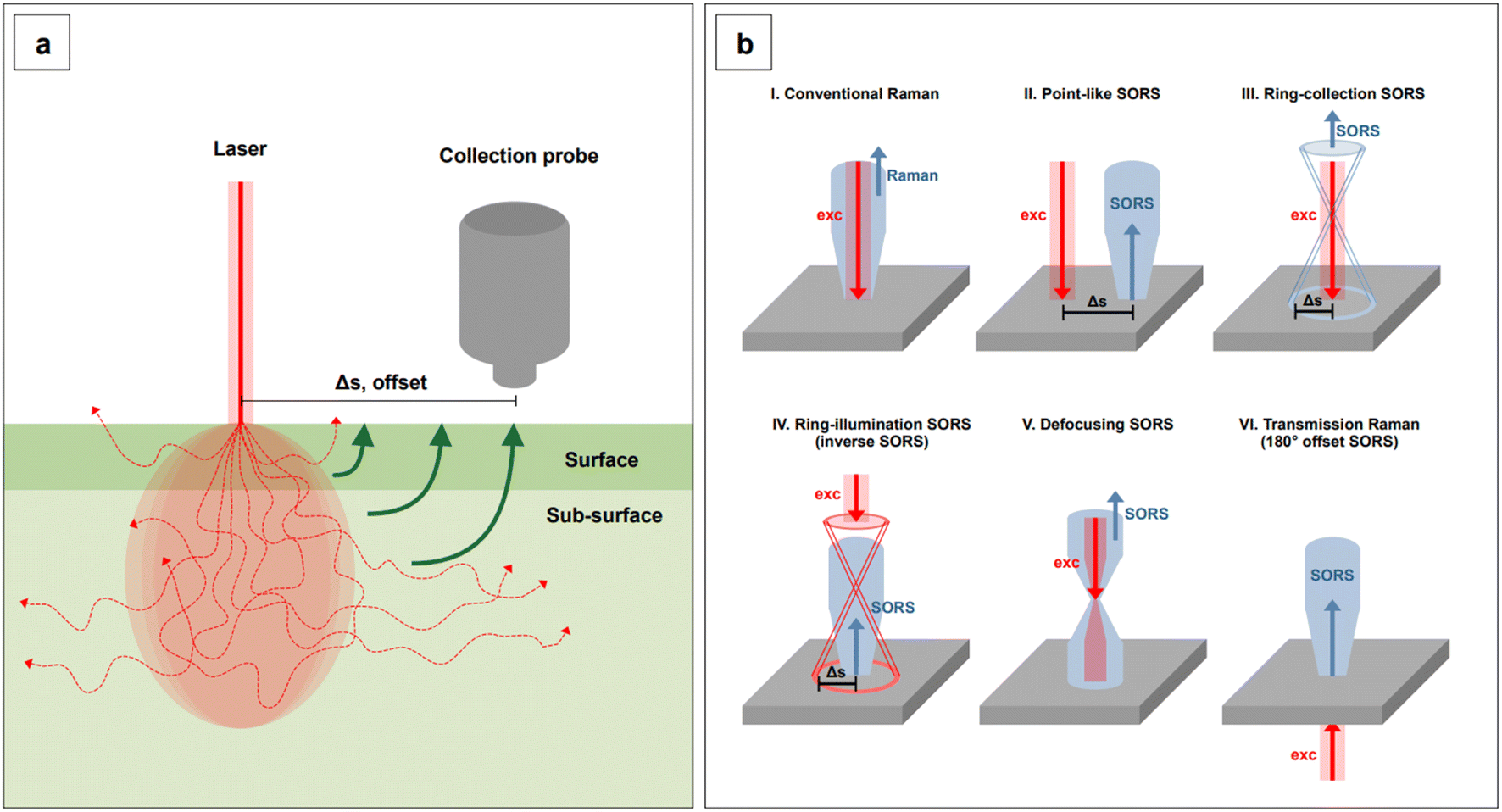 | ||
| Fig. 15 (a) Schematic representation of the principles of Spatially Offset Raman Spectroscopy (SORS). Raman photons diffuse from the sample to the surface: photons originated from deeper layers tend to first spread sideways, thus allowing a selective detection of them when the collection probe is placed at an offset (Δs) from the illumination point. (b) Five possible source-collection probe configurations that allow for SORS and SESORS measurements, as compared to conventional Raman (I): (II) point-like configuration (backscattering); (III) ring-collection; (IV) ring-illumination; (V) defocusing; (VI) transmission configuration. Figures inspired by Mosca, S., et al., 2021.240 | ||
The first account of SESO(R)RS was published by Stone et al. in 2010,243 and demonstrated the applicability of surface-enhancement in a SORS context for the detection of lesions located 1.5 to 2.5 cm below the surface of a porcine tissue sample. The measurements were performed in transmission mode (Fig. 15b, VI) and were enabled by the injection of resonant (λexc 830 nm) SERS tags based on citrate-reduced silver nanospheres into the tissue sample. By the sole examination of this pioneering experiment, it is easy to notice the many factors that must be considered in a SESORS or SESORRS experiment: the delivery of SERS-active substrates inside the sample at the depth of interest, the SERS-substrate formulation, and thus, its biocompatibility and stability in biological media, as well as its concentration or number of administered nanoparticles per target cell (i.e., dose). The latter was determined by Stone et al.243 by taking into account the thickness of the sample, the collection geometry (i.e., transmission), the resulting sampled volume and number of cells in that volume, and the possible lesion size. For a 10 mm lesion, less resonant SERS tags per cell are necessary (estimated 3.5 SERS tags per cell), while the number increases to about 3500 per cell for a 1 mm lesion.243 In another early work by Sharma et al.,244 who demonstrated the feasibility of SESORS for the detection of species through bone and in backscattering geometry, the necessary dose of SERS tags to generate a SESORS signal was determined in absolute terms, and it was 2 × 1012 particles total. Because nanoparticles in tissue medium are not static, and they naturally tend to undergo diffusion along with other biophysical processes, the actual number of illuminated nanoparticles is plausibly less than the estimated, indicating that lower nanoparticle doses are likely possible.244
Despite nanoparticle dose is an essential element for the translation of SESO(R)RS to real life clinical applications, few focused studies have been carried out on the subject. It must also be noted that, necessarily, these studies are nanoparticle- or nanotag-specific, as well as wavelength-, geometry-, and offset-specific. For example, Nicolson et al.245 have studied the dose of resonant and non-resonant tags for SESORS using a handheld instrument fitted with a 830 nm excitation line at its optimal246 8 mm offset and in backscattering geometry (Fig. 15b, II). To accurately control the concentration of the SERS tags and obtain limits of detection that were independent from diffusion into tissue, Nicolson et al.245 performed their measurements in cuvettes, which were shielded by tissue samples of increasing thickness, up to 5 mm. This sampling depth is clinically relevant for through-skin breast cancer applications.245,247
In the same paper, Nicolson et al.245 also demonstrated the benefits of utilizing resonant tags to achieve higher signal-to-noise ratio, and with it, the possibility of administering lower nanoparticle doses. The selected 830 nm-resonant chalcogenpyrylium231 reporter, dye 823, was able to yield SESORRS signals that were eleven times higher than those obtained with the standard, commercially available non-resonant reporter, BPE. The same dye was also utilized to fabricate nanotags for the SESORRS detection of breast cancer 3D models based on multicellular tumor spheroids (MTS) through 15 mm porcine tissue,248 as well as to perform multiplex imaging on analogous MTS models, in conjunction with other two 830 nm-resonant chalcogenpyrylium reporters.249 Due to their brightness, resonant SERS tags also allow for larger sampling depths compared to non-resonant tags.248,249 This is particularly important for measurements performed in backscattering geometry, which is known to access shallower depths compared to transmission measurements.250,251
The spatial component inherent to SESO(R)RS can be exploited to retrieve information about the depth of a target inclusion; this is particularly relevant for diagnostic or intra-operative applications, where the localization of a lesion could be required, either in vivo or ex vivo. Methods for this type of determination have been recently developed for both transmission and backscattering geometries by two independent groups. Mosca et al.250 proposed an approach based on a totally non-invasive acquisition of calibration data, that is, without any need to obtain information from internal components of the sample. Consequently, this method can be utilized in transmission configuration. The reported root-mean-square error of prediction is 6.7%. Berry et al.,251 on the other hand, developed a ratiometric approach to be utilized in backscattering geometry, based on the use of non-resonant nanotags and Principal Component Analysis (PCA). PCA was primarily utilized to determine the bands of the nanotags and tissue that contributed to the largest variance in the system. These were then utilized to calculate the natural logarithm of the nanotag to tissue intensity ratios at different known depths and build a linear model for each nanotag system. Two different non-resonant nanotags were studied to investigate the importance of the identity of the reporter, showing the slope of the intensity ratio as a function of inclusion depth exclusively depends on the optical properties of the tissue. In prediction studies, this method showed a root-mean-square error between 5.015 and 4.727%, depending on the utilized nanotag reporter.
Although SERS and SERRS nanotags populate the SESO(R)RS literature, less common, direct SESORS can also be implemented. Moody et al.252 developed a direct SESORS method to detect neurotransmitters through bone (0.5–1.25 mm), both in mock brain samples and in a euthanized rat subject, under 785 nm irradiation. Mock samples were obtained by the superimposition of a rat skull onto agarose gel slabs that were previously incubated with standard solutions of neurotransmitters serotonin, melatonin, epinephrine, norepinephrine, and dopamine at physiologically relevant concentrations (low hundreds of nM (ref. 253)), both individually and in mixtures. To compensate for post-mortem decomposition processes, the brain of the euthanized rat subject was enriched with an injection of 200 μL of 100 μM serotonin, which was allowed to diffuse prior to analysis. Citrate-reduced gold nanospheres were chosen as the enhancing substrates, brought to pH 2 to invert the surface charge from negative to positive for neurotransmitter-substrate affinity reasons, and let incubate and diffuse into the gel slabs. The nanoparticle delivery into the skull of the euthanized rat subject was performed via through-skull injection instead. The measurements on mock samples demonstrated that it is possible to obtain direct SESORS spectra of neurotransmitters at concentrations that are relevant for human applications, with LODs that do not differ much from those calculated for the same species in standard solutions and synthetic biological fluids in regular SERS experiments.252,253 While the thickness of the euthanized rat's skull allowed for through-bone SERS detection of serotonin even in the absence of an offset, maximum signal was obtained with a 1 mm offset in backscattering geometry, thus showing a potential of the SESORS technique to allow for the study neurotransmitters in vivo, in a minimally invasive way.252
For the detection of targets that are more complex than small molecules, and especially in those cases where imaging has to be performed, elaborate nanotags must be utilized. A paradigmatic example of this case is the first in vivo SESORRS study performed by Kircher's group.214 Nanotags based on cyclic-RGDyK peptide-functionalized silica-encapsulated resonant-dye labeled gold nanostars were fabricated to target integrin, a protein that is overexpressed in glioblastoma multiforme (GBM) tumors. SESORRS imaging of GBM masses was performed in vivo, on glioblastoma-bearing mice under general anesthesia, after prior (18–24 hours) injection of a 100 μL 8 nM dose of nanotags via the tail vein (Fig. 16a). Measurements were collected both with and without offset, demonstrating the higher accuracy of SESORRS over conventional, non-offset imaging collection set-ups (Fig. 16b), as well as comparable results to MRI (Fig. 16c).
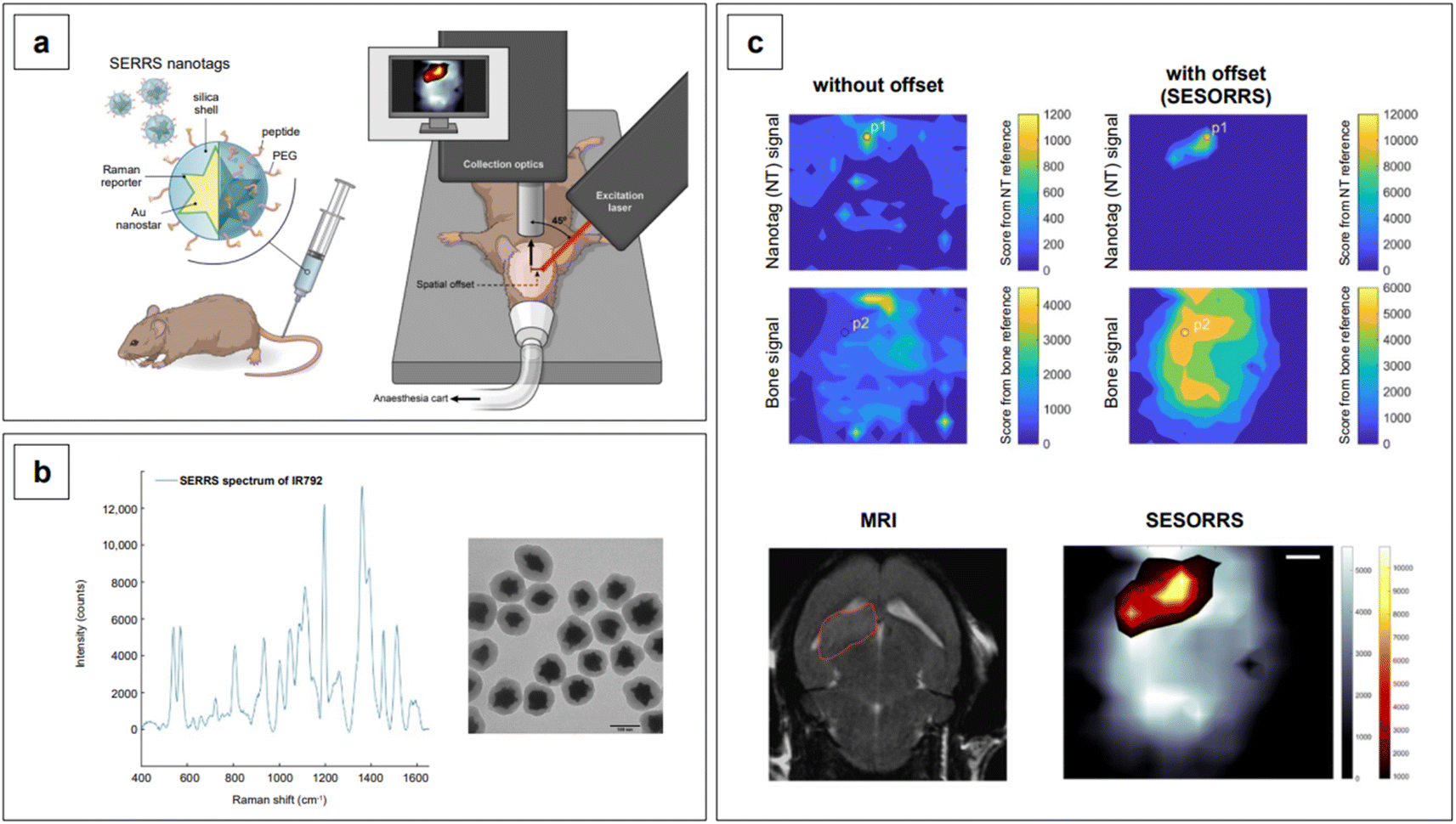 | ||
| Fig. 16 (a) Schematic representation of the SERRS nanotag architecture and clinical setup for the in vivo SESORRS study. (b) Representative SERRS spectrum of the utilized Raman reporter, dye IR792, and TEM micrograph of the SERRS nanotags. (c) (top) Heatmaps of the nanotag and bone signals acquired without any offset (left) and with an offset of 2.5 mm (right). (bottom, left) 2D axial T2-weighted MRI with an horizontally inverted orientation shows the presence of a left frontal tumor, circled in red. (bottom, right) Comparatively analogous tumor margins are shown by the SESORRS heatmap, obtained by the merging of the individual bone and nanotag heatmaps. Adapted from Nicolson et al., Theranostics, 2019, 9, 5899–5913; figures licensed under CC BY 4.0, https://creativecommons.org/licenses/by/4.0/. | ||
It might be now apparent to the reader that because SESO(R)RS is a new technique, it is still being explored in its more fundamental aspects by relatively few groups. This notwithstanding, the two recent studies by Kircher's and Sharma's groups demonstrate a clear potential for clinical translation. To fully enable this process, in-depth studies on toxicity and biocompatibility of the enhancing substrates, as well as on nanoparticle administration, distribution, bioaccumulation, and kinetics of elimination must be undertaken. The interested reader can refer to recent reviews on these topics and learn about the state of the art in nanotoxicology.254–257
3 Perspective and conclusions
Throughout this review we have illustrated how the community has been addressing the challenges that arise when targeting or manipulating biological samples in sensing applications using NIR excitation sources. However, many more still remain open. Before concluding, we want therefore to give an overview of what we believe are the important problems to tackle moving forward. This list is based on our perspective, hence it is not exhaustive, but we hope it will provide the reader with some useful reflection points.SERS measurements at any wavelength can certainly benefit from a homogeneous enhancing substrate, as this is directly responsible of the level of uniformity of the signal enhancement that can be produced. Substrate homogeneity is however fundamental in biological applications because of the intrinsic inhomogeneity and variability of the biological targets which, inherently, produce an intrinsically variable SERS response. Therefore, to ensure that the signal variation is due to the target and not to the substrate, one should aim to minimize substrate heterogeneity, especially when the analyte is present at low concentration. This heterogeneity is in general particularly prominent when colloidally synthesized nanoparticles are drop-casted or chemisorbed onto a substrate to form planar arrays. The heterogeneity arises not only from aggregation upon solvent evaporation (e.g., coffee rings), but also from the often-limited morphological uniformity of the nanoparticles themselves, caused by a lack of understanding and control of the synthetic protocols. This is especially true in the case of one-pot syntheses with fast reaction kinetics. One-pot protocols inherently offer less reaction control because they lack separation between synthesis steps such as nucleation, seed formation, and growth—a level of control that can instead be achieved by adopting seed-mediated approaches. The latter, in contrast, lead to improved morphological modulation and hence to a better tuning of the plasmonic properties; a better control of the reaction kinetics in the fabrication of the enhancing substrates ultimately achieves a more homogeneous and reproducible SERS response.
The SERS response would also benefit from the identification of protocols that eliminate the need for surfactants in seed-mediated anisotropic growth. In fact, while being ideal for morphological manipulation, surfactants can be difficult to remove and can thus hinder performance by increasing the separation between the nanoparticle surface and the target. Lastly, the properties of the nanoparticles would benefit from a more in-depth exploration of the role of stoichiometry, as recent literature highlighted its potentially pivoting role in morphological control. For example, in a surfactant-free, one pot synthesis of nanostars, different ratios of ascorbic acid to gold and silver were utilized to yield a series of nanostars with increased branch length (modulated via silver).83 In the reaction coordinates that have been explored, one particular combination did not yield branched structures, but nanospheroids. This result has been later associated to the stoichiometry of the reaction:53 when L-ascorbic acid is in excess, branched nanoparticles can be obtained, while, when it is the limiting agent, nanospheroids are synthesized instead. This could be due to the manipulation of nucleation vs. growth and deposition vs. diffusion rates via stoichiometry, and/or to the onset of surface-promoted disproportionation reactions upon excess of metal precursors, with a concomitant drift towards low-energy structures. Stoichiometry in the synthesis of nanoparticles is a factor that is seldom investigated or mentioned at all,53,131,258 while it could be a tool in uncovering the rules that predict the shape of a nanoparticle, in the same way we can predict reaction products in organic syntheses.
The importance of morphological control cannot be discounted even when employing nanoparticles to build SERS tags. In this instance, ensuring (hypothetically) that the nanoparticles synthesized are always identical would lead to obtaining tags with identical SERS response, guaranteeing that, similar to what mentioned before, the measured SERS variability can only be associated to concentration changes in the target. Importantly however, considering that SERS tags are suspended in water or buffer and not anchored on a surface such as in the case of SERS arrays, nanoparticle reshaping can often occur, primarily due to defective or inadequate surface functionalization. One, for instance, should think about the fate of SERS tags in DNA extraction mixtures. In these solutions the presence of dithiotreitol (DTT) leads to the cleavage of the Au–S bond and the deprotection of the Au atoms on the nanoparticle's surface, which increases their mobility, finally leading to reshaping, consequent loss of SERS activity, and even colloidal destabilization and precipitation. Au ad-atom mobility, especially important at high energy locations on the nanoparticles, can be reduced by the presence of atomic Ag, which has been shown by Atta et al.123 to deposit as a surface monolayer or bilayer on 6-branched gold nanostars. The presence of atomic Ag on the surface of the nanoparticle drastically reduces spike reshaping by limiting atomic mobility in surface Au, similar to what a well-bound surface ligand would do. Thus, both surface chemistry and surface functionalization of nanoparticles employed in SERS sensing, and especially as SERS tags, should be well understood and rationalized if we want to introduce them in the practical world.53,123,259 This is particularly important in critical environments and bodily fluids, where, for instance, the presence of enzymes or the formation of a protein corona can drastically alter the properties of the nanoparticles as they had been designed or synthesized. Taken all together, these factors are key in determining the quality and repeatability of the SERS measurement, which are imperative to a successful translation of the technique to the clinical practice.
Thinking about clinical application leads us to consider the importance of collaborating with (bio)statisticians to bring power analysis, the inverse problem approach, and, in general, a rigorous (vs. black box) statistical treatment of data to the SERS community. While mandatory when dealing with clinical specimens, improving the way we handle SERS data, especially with the advent of instrumentation with ultrafast collection capability, would allow us to reap further benefits, such as an improved interpretation of full SERS spectra, as compared to the approach that only uses individual peak positions. This approach is fundamental in biological applications of direct SERS, where the spectra are complex and often not well characterized (i.e., the signal is not generated by a known reporter but by the actual target, whose Raman fingerprint may not have been fully assigned yet). This improved analysis can provide richer information on the biological analyte and increase discrimination capability. All this, along with the richer information that we can extract by developing ad hoc statistical and deep learning tools, could improve the robustness and reliability of SERS and render it an important analytical and diagnostic tool in the biological and medical fields.
On the other side of the collaborative spectrum, one should remember the importance of establishing and sustaining collaborations with industry to push for the development of instruments with fast (and stable) acquisition rates. It must be recalled that adsorption is a dynamic process, and the association events between the components of a SERS sample (analyte, plasmonic metal, capping agents, synthetic by-products, etc.) are not static in nature, but represent instead instantaneous pictures of a dynamic equilibrium, i.e., what might be happening on the surface of the nanomaterial during a given time interval. Molecules on a surface adsorb, desorb, diffuse,260 and this dynamicity has a significant impact on the measurement of analytes, especially at ultratrace concentrations (<1 nM, single molecule), where it is often said that SERS spectra are observed as “blinking” events.261 This intermittence in the observation of SERS spectra in the ultratrace regime is caused not only by the stochastic probability of a molecule adsorbing on a hotspot, the stochastic nature of nanoparticle-extrinsic (i.e., aggregation-induced) hotspot formation, but also by the dynamic nature of the equilibria established during adsorption and the kinetics of the adsorption. The latter can be particularly problematic in that adsorption of certain analytes can occur at time scales that are faster than the sampling speed of the Raman instrument utilized for SERS measurements, effectively impeding single molecule detection.262,263 This is especially true in the case of flow-through detection set-ups (e.g., microfluidics, lab-on-a-chip, nanopore sequencing264), which are particularly relevant to clinical diagnostics applications. They are also important when analyzing large and complex bioanalytes, such as proteins, which, during interaction with the SERS substrate may be deformed by the highly curved surface and thus provide a fingerprint pattern that is not representative of the molecule in its functional state. Finally, it will be important to leverage the collaboration with industry to develop equipment of low cost and high portability, which will be key to enable the use of the technique in laboratories where dangerous (or poorly portable) targets are studied, such as viruses.
Collaboration with industry brings up the necessity to make SERS substrates fabrication a more commercially appealing endeavor, which translates into achieving high yield, scaled-up synthetic processes. Unfortunately, nanomaterial fabrication is extremely sensitive to mass transfer parameters, and scale-up approaches via simple proportional increase of reagents and batch vessels volumes (i.e., scale-out approach) are unfeasible, because they do not preserve these parameters.265 Micromixer-based flow reactor processes, on the other hand, allow for the preservation of the mass transfer characteristics of an original batch process by increasing the number of parallel reactors as opposed to their individual volume (i.e., numbering-up approach).265 However, the lack of fundamental knowledge of the complex relationships between all mass transfer parameters, especially for anisotropic growth syntheses, still constitutes an obstacle in the straightforward application of these scale-up approaches, and thus, in the large-scale fabrication of SERS substrates. As mentioned for other aspects of SERS research, computational simulations of the hydrodynamic flows,266 as well as the application of industrial processes-specific machine learning algorithms265,267–269 can aid in the advancement of this field, minimizing the bottleneck that currently halts the commercial potential of these materials, and thus, the establishment of SERS as a routine analytical platform.
In consideration of our focus on applications in the infrared, it will be fundamental to exploit the increasing accessibility and cost-effectiveness of SWIR detectors (e.g., extended InGaAs detectors),34 that can overcome the instrumental issues that have so far drastically limited SERS analysis beyond 1000 nm. In turn, technological progress in this area could also benefit the telecommunications industry when coupled with the development of highly anisotropic plasmonic materials with resonances in the SWIR.270,271 Along with the design of SWIR-resonant plasmonic materials, we must not forget the development of new resonant and non-resonant SERS reporters for NIR and SWIR applications, which has seen a stall in the recent years. However, developments in the design of NIR fluorophores, which are typically also Raman active, can be leveraged to extract useful tips for future revival of this branch of research. Research on fluorophores is more active, although the synthesis of red-shifted dyes remains challenging. Theoretical information for rational design of these molecules can be compiled and leveraged as it concerns the reduction of the fluorescence quantum yield (QY), as it would reduce the fluorescence background that is known to affect the SERS response. In a recent review on NIR-II fluorescent dyes by Li et al.,60 the following points were listed as key guidelines in the design of these types of reporters: (1) increasing the conjugation chain length, and varying the heteroatom from oxygen to other chalcogens, results in redshifted spectra (as seen in the works of Harmsen,231 Bedics,233 and Kearns238); (2) a rigid aromatic structure bearing halogen atoms like Cl/Br/I is unfavorable to the fluorescence process and can decrease the QY via increased intersystem crossing caused by the heavy atom effect; (3) functional groups like carboxyl, cyano, and carbonyl can lower the HOMO and increase the energy gap between HOMO and LUMO, hence leading to a certain loss of QY. Taking into account all these guidelines would enable the synthesis of NIR-II fluorophores that can be applied also as Raman reporters, similar to what is commonly done with rhodamine 6G, crystal violet, and others.
Finally, and most importantly from the materials design standpoint, going forward it will be necessary to leverage the use of doped metal oxide nanoparticles (such as ZnO) as infrared SERS substrates. Fundamental studies on these materials, focusing on composition, morphology, electronic energy levels, and their electromagnetic and chemical enhancement mechanisms will be needed to fill the gaps that are still significant in their application for the detection of bioanalytes, such as disease biomarkers. While some of the research that has already been carried out to study the SERS properties of plasmonic materials active in the visible will have to be repeated for infrared-active materials, much of the knowledge we have put together over the past 25 years will result tremendously useful: it will guide us in this understanding and discovery process that is now in its infancy, but promises to bring significant scientific and technological breakthroughs in the years to come.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (grant agreement No. 865819).References
- C. J. Murphy, T. K. Sau, A. M. Gole, C. J. Orendorff, J. Gao, L. Gou, S. E. Hunyadi and T. Li, J. Phys. Chem. B, 2005, 109, 13857–13870 CrossRef CAS PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef CAS.
- S. J. Hurst, E. K. Payne, L. Qin and C. A. Mirkin, Angew. Chem., Int. Ed., 2006, 45, 2672–2692 CrossRef CAS PubMed.
- C. G. Khoury and T. Vo-Dinh, J. Phys. Chem. C, 2008, 112, 18849–18859 CrossRef CAS PubMed.
- S. Barbosa, A. Agrawal, L. Rodríguez-Lorenzo, I. Pastoriza-Santos, R. A. Alvarez-Puebla, A. Kornowski, H. Weller and L. M. Liz-Marzán, Langmuir, 2010, 26, 14943–14950 CrossRef CAS PubMed.
- L. Gunnarsson, E. J. Bjerneld, H. Xu, S. Petronis, B. Kasemo and M. Käll, Appl. Phys. Lett., 2001, 78, 802–804 CrossRef CAS.
- N. Félidj, S. L. Truong, J. Aubard, G. Lévi, J. R. Krenn, A. Hohenau, A. Leitner and F. R. Aussenegg, J. Chem. Phys., 2004, 120, 7141–7146 CrossRef PubMed.
- L. A. Dick, A. D. McFarland, C. L. Haynes and R. P. van Duyne, J. Phys. Chem. B, 2002, 106, 853–860 CrossRef CAS.
- L. Wang, L. Xu, H. Kuang, C. Xu and N. A. Kotov, Acc. Chem. Res., 2012, 45, 1916–1926 CrossRef CAS PubMed.
- J. P. Camden, J. A. Dieringer, J. Zhao and R. P. van Duyne, Acc. Chem. Res., 2008, 41, 1653–1661 CrossRef CAS PubMed.
- S. A. Maier and H. A. Atwater, J. Appl. Phys., 2005, 98, 011101 CrossRef.
- W. A. Murray and W. L. Barnes, Adv. Mater., 2007, 19, 3771–3782 CrossRef CAS.
- M. M. Miller and A. A. Lazarides, J. Phys. Chem. B, 2005, 109, 21556–21565 CrossRef CAS PubMed.
- C. L. Haynes, A. J. Haes, A. D. McFarland and R. P. Van Duyne, in Topics in fluorescence spectroscopy, ed. C. D. Gedde and J. R. Lakowicz, Springer Science + Business Media Inc, New York, 2005, pp. 47–99 Search PubMed.
- K.-S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 19220–19225 CrossRef CAS PubMed.
- J. E. Millstone, S. J. Hurst, G. S. Métraux, J. I. Cutler and C. A. Mirkin, Small, 2009, 5, 646–664 CrossRef CAS PubMed.
- International Human Genome Sequencing Consortium, Nature, 2004, 431, 931–945 CrossRef PubMed.
- J. Grisham, Nat. Biotechnol., 2000, 18, 365 CrossRef CAS PubMed.
- T. Shin, D. Kraemer, J. Pryor, L. Liu, J. Rugila, L. Howe, S. Buck, K. Murphy, L. Lyons and M. Westhusin, Nature, 2002, 415, 859 CrossRef CAS PubMed.
- C. Galli, I. Lagutina, G. Crotti, S. Colleoni, P. Turini, N. Ponderato, R. Duchi and G. Lazzari, Nature, 2003, 424, 635 CrossRef CAS PubMed.
- Q. Zhou, J.-P. Renard, G. le Friec, V. Brochard, N. Beaujean, Y. Cherifi, A. Fraichard and J. Cozzi, Science, 2003, 302, 1179 CrossRef CAS PubMed.
- L. Mancinelli, M. Cronin and W. Sadée, AAPS PharmSci, 2000, 2, 29–41 CrossRef PubMed.
- J. de Leon, Expert Rev. Mol. Diagn., 2006, 6, 277–286 CrossRef CAS PubMed.
- K. Jain, Expert Rev. Mol. Diagn., 2002, 2, 299–301 CrossRef CAS PubMed.
- R. Sachidanandam, D. Weissman, S. C. Schmidt, J. M. Kakol, L. D. Stein, G. Marth, S. Sherry, J. C. Mullikin, B. J. Mortimore, D. L. Willey, S. E. Hunt, C. G. Cole, P. C. Coggill, C. M. Rice, Z. Ning, J. Rogers, D. R. Bentley, P.-Y. Kwok, E. R. Mardis, R. T. Yeh, B. Schultz, L. Cook, R. Davenport, M. Dante, L. Fulton, L. Hillier, R. H. Waterston, J. D. McPherson, B. Gilman, S. Schaffner, W. J. van Etten, D. Reich, J. Higgins, M. J. Daly, B. Blumenstiel, J. Baldwin, N. Stange-Thomann, M. C. Zody, L. Linton, E. S. Lander and D. Altshuler, Nature, 2001, 409, 928–933 CrossRef CAS PubMed.
- F. S. Collins and V. A. McKusick, JAMA, 2001, 285, 540 CrossRef CAS PubMed.
- K. K. Jain, Technol. Cancer Res. Treat., 2005, 4, 645–650 CrossRef CAS PubMed.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed.
- T. Hirschfeld and B. Chase, Appl. Spectrosc., 1986, 40, 133–137 CrossRef CAS.
- H. J. Butler, L. Ashton, B. Bird, G. Cinque, K. Curtis, J. Dorney, K. Esmonde-White, N. J. Fullwood, B. Gardner, P. L. Martin-Hirsch, M. J. Walsh, M. R. McAinsh, N. Stone and F. L. Martin, Nat. Protoc., 2016, 11, 664–687 CrossRef CAS PubMed.
- E. C. LeRu and P. Etchegoin, Principles of Surface-Enhanced Raman Spectroscopy, Elsevier, Amsterdam, 2009 Search PubMed.
- E. C. le Ru, P. G. Etchegoin, J. Grand, N. Félidj, J. Aubard and G. Lévi, J. Phys. Chem. C, 2007, 111, 16076–16079 CrossRef CAS.
- F. Shan, X. Y. Zhang, X. C. Fu, L. J. Zhang, D. Su, S. J. Wang, J. Y. Wu and T. Zhang, Sci. Rep., 2017, 7, 6813 CrossRef PubMed.
- X. Chen, Y. Gu and Y. Zhang, in Epitaxy, ed. M. Zhong, InTech Open, London, 2018, pp. 203–234 Search PubMed.
- Q. L. Kleipool, R. T. Jongma, A. M. S. Gloudemans, H. Schrijver, G. F. Lichtenberg, R. M. van Hees, A. N. Maurellis and R. W. M. Hoogeveen, Infrared Phys. Technol., 2007, 50, 30–37 CrossRef CAS.
- B. Smiri, M. ben Arbia, D. Ilkay, F. Saidi, Z. Othmen, B. Dkhil, A. Ismail, E. Sezai, F. Hassen and H. Maaref, Mater. Sci. Eng., B, 2020, 262, 114769 CrossRef CAS.
- X. Ji, B. Liu, H. Tang, X. Yang, X. Li, H. Gong, B. Shen, P. Han and F. Yan, AIP Adv., 2014, 4, 087135 CrossRef.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010 CrossRef CAS.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- Horiba, Synapse InGaAs/Symphony II InGaAs Deep Cooled NIR Scientific Cameras, https://www.horiba.com/int/products/detail/action/show/Product/synapse-ingaassymphony-ii-ingaas-1483/, accessed 10 June 2022 Search PubMed.
- K. M. Kosuda, J. M. Bingham, K. L. Wustholz and R. P. van Duyne, in Comprehensive Nanoscience and Technology, ed. D. L. Andrews, G. D. Scholes and G. P. Wiederrecht, Elsevier, Amsterdam, 2001, pp. 263–301 Search PubMed.
- L. Litti and M. Meneghetti, Phys. Chem. Chem. Phys., 2019, 21, 15515–15522 RSC.
- C. Deriu, I. Conticello, A. M. Mebel and B. McCord, Anal. Chem., 2019, 91, 4780–4789 CrossRef CAS PubMed.
- E. Akanny, A. Bonhommé, C. Commun, A. Doleans-Jordheim, F. Bessueille, S. Bourgeois and C. Bordes, Anal. Bioanal. Chem., 2019, 411, 5563–5576 CrossRef CAS PubMed.
- H. T. Phan, T. S. Heiderscheit and A. J. Haes, J. Phys. Chem. C, 2020, 124, 14287–14296 CrossRef CAS PubMed.
- D. Yılmaz and M. Culha, Langmuir, 2022, 38, 6281–6294 CrossRef PubMed.
- R. Pilot and M. Massari, Chem. Phys., 2021, 2, 100014 Search PubMed.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- G. Frens, Nature, 1973, 241, 20–22 CAS.
- C. J. Murphy and J. M. Buriak, Chem. Mater., 2015, 27, 4911–4913 CrossRef CAS.
- R. A. Alvarez-Puebla and L. M. Liz-Marzán, Chem. Soc. Rev., 2012, 41, 43–51 RSC.
- C. Deriu, A. Bracho and B. McCord, J. Phys. Chem. C, 2022, 126, 2023–2040 CrossRef CAS.
- S. S. Masango, R. A. Hackler, N. Large, A.-I. Henry, M. O. McAnally, G. C. Schatz, P. C. Stair and R. P. van Duyne, Nano Lett., 2016, 16, 4251–4259 CrossRef CAS PubMed.
- M. Moskovits, J. Raman Spectrosc., 2005, 36, 485–496 CrossRef CAS.
- M. Moskovits, Phys. Chem. Chem. Phys., 2013, 15, 5301–5311 RSC.
- M. Sánchez-Purrà, B. Roig-Solvas, A. Versiani, C. Rodriguez-Quijada, H. de Puig, I. Bosch, L. Gehrke and K. Hamad-Schifferli, Mol. Syst. Des. Eng., 2017, 2, 401–409 RSC.
- D. Zhang, L. Huang, B. Liu, E. Su, H. Y. Chen, Z. Gu and X. Zhao, Sens. Actuators, B, 2018, 277, 502–509 CrossRef CAS.
- R. Chen, B. Liu, H. Ni, N. Chang, C. Luan, Q. Ge, J. Dong and X. Zhao, Analyst, 2019, 144, 4051–4059 RSC.
- L. Li, X. Dong, J. Li and J. Wei, Dyes Pigm., 2020, 183, 108756 CrossRef CAS.
- M. Balu, T. Baldacchini, J. Carter, T. B. Krasieva, R. Zadoyan and B. J. Tromberg, J. Biomed. Opt., 2009, 14, 010508 CrossRef PubMed.
- G. Hong, S. Diao, A. L. Antaris and H. Dai, Chem. Rev., 2015, 115, 10816–10906 CrossRef CAS PubMed.
- N. Stone, M. Kerssens, G. R. Lloyd, K. Faulds, D. Graham and P. Matousek, Chem. Sci., 2011, 2, 776–780 RSC.
- J. Langer, D. J. de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. Javier García de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. le Ru, H. K. Lee, J. F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J. M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L. L. Tay, K. George Thomas, Z. Q. Tian, R. P. van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- D. Li, D. Yao, C. Li, Y. Luo, A. Liang, G. Wen and Z. Jiang, TrAC, Trends Anal. Chem., 2020, 127, 115885 CrossRef CAS.
- G. Barbillon, Nanomaterials, 2020, 10, 1–17 Search PubMed.
- M. P. Oyarzún, A. Tapia-Arellano, P. Cabrera, P. Jara-Guajardo and M. J. Kogan, Sensors, 2021, 21, 1–30 CrossRef PubMed.
- M. Blanco-Formoso, N. Pazos-Perez and R. A. Alvarez-Puebla, J. Raman Spectrosc., 2021, 52, 554–562 CrossRef CAS.
- B. Andreiuk, F. Nicolson, L. M. Clark, S. R. Panikkanvalappil, Kenry, M. Rashidian, S. Harmsen and M. F. Kircher, Nanotheranostics, 2022, 6, 10–30 CrossRef PubMed.
- I. B. Becerril-Castro, I. Calderon, N. Pazos-Perez, L. Guerrini, F. Schulz, N. Feliu, I. Chakraborty, V. Giannini, W. J. Parak and R. A. Alvarez-Puebla, Analysis Sensing, 2022, 2, e20220000 Search PubMed.
- T. D. Payne, A. S. Moody, A. L. Wood, P. A. Pimiento, J. C. Elliott and B. Sharma, Analyst, 2020, 145, 3461–3480 RSC.
- G. Q. Wallace and J. F. Masson, Analyst, 2020, 145, 7162–7185 RSC.
- S. S. Panikar, D. Cialla-May, E. de la Rosa, P. Salas and J. Popp, TrAC, Trends Anal. Chem., 2021, 134, 116122 CrossRef CAS.
- V. Moisoiu, S. D. Iancu, A. Stefancu, T. Moisoiu, B. Pardini, M. P. Dragomir, N. Crisan, L. Avram, D. Crisan, I. Andras, D. Fodor, L. F. Leopold, C. Socaciu, Z. Bálint, C. Tomuleasa, F. Elec and N. Leopold, Colloids Surf., B, 2021, 208, 112064 CrossRef CAS PubMed.
- V. Cupil-Garcia, P. Strobbia, B. M. Crawford, H. N. Wang, H. Ngo, Y. Liu and T. Vo-Dinh, J. Raman Spectrosc., 2021, 52, 541–553 CrossRef CAS.
- M. E. Berry, H. Kearns, D. Graham and K. Faulds, Analyst, 2021, 146, 6084–6101 RSC.
- W. A. Hassanain, C. L. Johnson, K. Faulds, D. Graham and N. Keegan, Analyst, 2022, 147, 4674–4700 RSC.
- H. Chen, Z. Cheng, X. Zhou, R. Wang and F. Yu, Anal. Chem., 2022, 94, 143–164 CrossRef CAS PubMed.
- A. Guerrero-Martínez, S. Barbosa, I. Pastoriza-Santos and L. M. Liz-Marzán, Curr. Opin. Colloid Interface Sci., 2011, 16, 118–127 CrossRef.
- P. F. Liao and A. Wokaun, J. Chem. Phys., 1982, 76, 751–752 CrossRef CAS.
- L. Rodríguez-Lorenzo, R. A. Álvarez-Puebla, I. Pastoriza-Santos, S. Mazzucco, O. Stéphan, M. Kociak, L. M. Liz-Marzán and F. J. García de Abajo, J. Am. Chem. Soc., 2009, 131, 4616–4618 CrossRef PubMed.
- T. Chung, S.-Y. Lee, E. Y. Song, H. Chun and B. Lee, Sensors, 2011, 11, 10907–10929 CrossRef CAS PubMed.
- S. He, M. W. C. Kang, F. J. Khan, E. K. M. Tan, M. A. Reyes and J. C. Y. Kah, J. Opt., 2015, 17, 114013–114026 CrossRef.
- A. Garcia-Leis, A. Torreggiani, J. V. Garcia-Ramos and S. Sanchez-Cortes, Nanoscale, 2015, 7, 13629–13637 RSC.
- C. D. L. de Albuquerque, R. G. Sobral-Filho, R. J. Poppi and A. G. Brolo, Anal. Chem., 2018, 90, 1248–1254 CrossRef CAS PubMed.
- J. A. Huang, M. Z. Mousavi, Y. Zhao, A. Hubarevich, F. Omeis, G. Giovannini, M. Schütte, D. Garoli and F. de Angelis, Nat. Commun., 2019, 10, 5321 CrossRef PubMed.
- B. Li, T. Wang, Q. Su, X. Wu and P. Dong, Sensors, 2019, 19, 3742 CrossRef CAS PubMed.
- B. Li, T. Wang, W. Bai, Q. Su, X. Wu and P. Dong, IEEE Sens. J., 2021, 21, 18425–18434 CAS.
- L. Minati, D. Maniglio, F. Benetti, A. Chiappini and G. Speranza, Materials, 2021, 14, 7272 CrossRef CAS PubMed.
- T. R. Nayak, C. Andreou, A. Oseledchyk, W. D. Marcus, H. C. Wong, J. Massagué and M. F. Kircher, Nanoscale, 2017, 9, 1110–1119 RSC.
- K. Dardir, H. Wang, B. E. Martin, M. Atzampou, C. B. Brooke and L. Fabris, J. Phys. Chem. C, 2020, 124, 3211–3217 CrossRef CAS.
- S. He, Y. M. E. Kyaw, E. K. M. Tan, L. Bekale, M. W. C. Kang, S. S. Y. Kim, I. Tan, K. P. Lam and J. C. Y. Kah, Anal. Chem., 2018, 90, 6071–6080 CrossRef CAS PubMed.
- J. Dong, X. Zhao, W. Gao, Q. Han, J. Qi, Y. Wang, S. Guo and M. Sun, Nanoscale Res. Lett., 2019, 14, 118 CrossRef PubMed.
- Y. Sun, S. Ge, J. Xue, X. Zhou, W. Lu, G. Li and X. Cao, Biomed. Opt. Express, 2020, 11, 7062 CrossRef CAS PubMed.
- N. Albarghouthi, P. MacMillan and C. L. Brosseau, Analyst, 2021, 146, 2037–2047 RSC.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- P. L. Gai and M. A. Harmer, Nano Lett., 2002, 2, 771–774 CrossRef CAS.
- S. K. Meena and M. Sulpizi, Angew. Chem., Int. Ed., 2016, 55, 11960–11964 CrossRef CAS PubMed.
- J. A. da Silva and M. R. Meneghetti, Langmuir, 2018, 34, 366–375 CrossRef CAS PubMed.
- J. A. da Silva, P. A. Netz and M. R. Meneghetti, Langmuir, 2020, 36, 257–263 CrossRef CAS PubMed.
- C. J. Johnson, E. Dujardin, S. A. Davis, C. J. Murphy and S. Mann, J. Mater. Chem., 2002, 12, 1765–1770 RSC.
- M. J. Walsh, S. J. Barrow, W. Tong, A. M. Funston and J. Etheridge, ACS Nano, 2015, 9, 715–724 CrossRef CAS PubMed.
- M. J. Walsh, W. Tong, H. Katz-Boon, P. Mulvaney, J. Etheridge and A. M. Funston, Acc. Chem. Res., 2017, 50, 2925–2935 CrossRef CAS PubMed.
- W. Tong, M. J. Walsh, P. Mulvaney, J. Etheridge and A. M. Funston, J. Phys. Chem. C, 2017, 121, 3549–3559 CrossRef CAS.
- W. Tong, H. Katz-Boon, M. J. Walsh, M. Weyland, J. Etheridge and A. M. Funston, Chem. Commun., 2018, 54, 3022–3025 RSC.
- J. A. Edgar, A. M. McDonagh and M. B. Cortie, ACS Nano, 2012, 6, 1116–1125 CrossRef CAS PubMed.
- G. González-Rubio, V. Kumar, P. Llombart, P. Díaz-Núñez, E. Bladt, T. Altantzis, S. Bals, O. Peña-Rodríguez, E. G. Noya, L. G. Macdowell, A. Guerrero-Martínez and L. M. Liz-Marzán, ACS Nano, 2019, 13, 4424–4435 CrossRef PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- Y. Xia, X. Xia and H.-C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- G. González-Rubio, L. Scarabelli, A. Guerrero-Martínez and L. M. Liz-Marzán, ChemNanoMat, 2020, 6, 698–707 CrossRef.
- Z. Chen, J. W. Chang, C. Balasanthiran, S. T. Milner and R. M. Rioux, J. Am. Chem. Soc., 2019, 141, 4328–4337 CrossRef CAS PubMed.
- A. Sánchez-Iglesias, K. Jenkinson, S. Bals and L. M. Liz-Marzán, J. Phys. Chem. C, 2021, 125, 23937–23944 CrossRef PubMed.
- A. Waragai, T. Nakagawa, W. L. Hinze and Y. Takagai, Langmuir, 2021, 37, 10702–10707 CrossRef CAS PubMed.
- X. Lin, C. Li, C. He, Y. Zhou, Z. Wang, N. Duan and S. Wu, ACS Appl. Nano Mater., 2021, 4, 8231–8240 CrossRef CAS.
- A. S. Tatar, C. Farcău, A. Vulpoi, S. Boca and S. Astilean, Spectrochim. Acta, Part A, 2022, 273, 121069 CrossRef CAS PubMed.
- F. Liebig, R. Henning, R. M. Sarhan, C. Prietzel, M. Bargheer and J. Koetz, Nanotechnology, 2018, 29, 185603 CrossRef PubMed.
- A. Jakhmola, R. Vecchione, F. Gentile, M. Profeta, A. C. Manikas, E. Battista, M. Celentano, V. Onesto and P. A. Netti, Mater. Today Chem., 2019, 14, 100203 CrossRef CAS.
- L. A. Osminkina, O. Žukovskaja, S. N. Agafilushkina, E. Kaniukov, O. Stranik, K. A. Gonchar, D. Yakimchuk, V. Bundyukova, D. A. Chermoshentsev, S. A. Dyakov, N. A. Gippius, K. Weber, J. Popp, D. Cialla-May and V. Sivakov, Appl. Surf. Sci., 2020, 507, 144989 CrossRef CAS.
- T. K. Sau, A. L. Rogach, M. Döblinger and J. Feldmann, Small, 2011, 7, 2188–2194 CrossRef CAS PubMed.
- H. I. Khan, M. U. Khalid, A. Abdullah, A. Ali, A. S. Bhatti, S. U. Khan and W. Ahmed, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2018, 36, 03E101 Search PubMed.
- P. Pallavicini, A. Donà, A. Casu, G. Chirico, M. Collini, G. Dacarro, A. Falqui, C. Milanese, L. Sironi and A. Taglietti, Chem. Commun., 2013, 49, 6265–6267 RSC.
- A. J. Blanch, M. Döblinger and J. Rodríguez-Fernández, Small, 2015, 11, 4550–4559 CrossRef CAS PubMed.
- S. Atta, M. Beetz and L. Fabris, Nanoscale, 2019, 11, 2946–2958 RSC.
- F. Liebig, R. Henning, R. M. Sarhan, C. Prietzel, C. N. Z. Schmitt, M. Bargheer and J. Koetz, RSC Adv., 2019, 9, 23633–23641 RSC.
- P. Senthil Kumar, I. Pastoriza-Santos, B. Rodríguez-González, F. Javier García De Abajo and L. M. Liz-Marzán, Nanotechnology, 2008, 19, 015606 CrossRef PubMed.
- Y. Pu, Y. Zhao, P. Zheng and M. Li, Inorg. Chem., 2018, 57, 8599–8607 CrossRef CAS PubMed.
- T. T. V. Phan, V. T. Nguyen, S.-H. Ahn and J. Oh, Eur. Polym. J., 2019, 118, 492–501 CrossRef CAS.
- P. T. Huynh, G. D. Nguyen, K. T. le Tran, T. M. Ho, B. T. Duong, V. Q. Lam and T. V. K. Ngo, J. Nanomater., 2021, 2021, 1–10 CrossRef.
- W. Xi and A. J. Haes, J. Am. Chem. Soc., 2019, 141, 4034–4042 CrossRef CAS PubMed.
- O. M. Bakr, B. H. Wunsch and F. Stellacci, Chem. Mater., 2006, 18, 3297–3301 CrossRef CAS.
- H. Yuan, C. G. Khoury, H. Hwang, C. M. Wilson, G. A. Grant and T. Vo-Dinh, Nanotechnology, 2012, 23, 075102 CrossRef PubMed.
- L. Minati, F. Benetti, A. Chiappini and G. Speranza, Colloids Surf., A, 2014, 441, 623–628 CrossRef CAS.
- K. Chandra, K. S. B. Culver, S. E. Werner, R. C. Lee and T. W. Odom, Chem. Mater., 2016, 28, 6763–6769 CrossRef CAS.
- C. A. Foss, G. L. Hornyak, J. A. Stockert and C. R. Martin, J. Phys. Chem., 1994, 98, 2963–2971 CrossRef CAS.
- N. Kameta and H. Shiroishi, Chem. Commun., 2018, 54, 4665–4668 RSC.
- H. Yuan, M. Wanhong, C. Chen, J. Zhao, J. Liu, H. Zhu and X. Gao, Chem. Mater., 2007, 19, 1592–1600 CrossRef CAS.
- J. Xie, J. Y. Lee and D. I. C. Wang, Chem. Mater., 2007, 19, 2823–2830 CrossRef CAS.
- A. M. Fales, H. Yuan and T. Vo-Dinh, J. Phys. Chem. C, 2014, 118, 3708–3715 CrossRef CAS PubMed.
- A. S. de Silva Indrasekara, S. F. Johnson, R. A. Odion and T. Vo-Dinh, ACS Omega, 2018, 3, 2202–2210 CrossRef CAS PubMed.
- H. T. Phan and A. J. Haes, J. Phys. Chem. C, 2019, 123, 16495–16507 CrossRef CAS PubMed.
- E. Tosatti and F. Ercolessi, Mod. Phys. Lett. B, 1991, 5, 413–425 CrossRef CAS.
- H. Vanrompay, E. Bladt, W. Albrecht, A. Béché, M. Zakhozheva, A. Sánchez-Iglesias, L. M. Liz-Marzán and S. Bals, Nanoscale, 2018, 10, 22792–22801 RSC.
- C. Deriu, A. N. Morozov and A. M. Mebel, J. Phys. Chem. A, 2022, 126, 5236–5251 CrossRef CAS PubMed.
- A. Borowik, K. Butowska, K. Konkel, R. Banasiuk, N. Derewonko, D. Wyrzykowski, M. Davydenko, V. Cherepanov, V. Styopkin, Y. Prylutskyy, P. Pohl, A. Krolicka and J. Piosik, Nanomaterials, 2019, 9, 973 CrossRef CAS PubMed.
- X. Shi, H. L. Perry and J. D. E. T. Wilton-Ely, Nanotheranostics, 2021, 5, 155–165 CrossRef PubMed.
- H.-M. Song, Q. Wei, Q. K. Ong and A. Wei, ACS Nano, 2010, 4, 5163–5173 CrossRef CAS PubMed.
- C. Kuttner, V. Piotto and L. M. Liz-Marzán, Chem. Mater., 2021, 33, 8904–8914 CrossRef CAS.
- M. Becucci, M. Bracciali, G. Ghini, C. Lofrumento, G. Pietraperzia, M. Ricci, L. Tognaccini, S. Trigari, C. Gellini and A. Feis, Nanoscale, 2018, 10, 9329–9337 RSC.
- X. Luo, L. Qiao, Z. Xia, J. Yu, X. Wang, J. Huang, C. Shu, C. Wu and Y. He, Langmuir, 2022, 38, 6454–6463 CrossRef CAS PubMed.
- D. C, P. Sahu and D. V. R. Kumar, J. Chem. Sci., 2022, 134, 64 CrossRef CAS.
- P. Drude, Ann. Phys., 1900, 306, 566–613 CrossRef.
- E. C. le Ru and P. G. Etchegoin, Principles of Surface-enhanced Raman Spectroscopy and related plasmonic effects, Elsevier, Amsterdam, 2009 Search PubMed.
- X. Liu, Front. Mater., 2018, 5, 59 CrossRef.
- B. Yang, S. Jin, S. Guo, Y. Park, L. Chen, B. Zhao and Y. M. Jung, ACS Omega, 2019, 4, 20101–20108 CrossRef CAS PubMed.
- X. Wang, E. Zhang, H. Shi, Y. Tao and X. Ren, Analyst, 2022, 147, 1257–1272 RSC.
- G. Song, S. Cong and Z. Zhao, Chem. Sci., 2022, 13, 1210–1224 RSC.
- X. Wang and L. Guo, Angew. Chem., Int. Ed., 2020, 132, 4259–4267 CrossRef.
- S. Cong, Y. Yuan, Z. Chen, J. Hou, M. Yang, Y. Su, Y. Zhang, L. Li, Q. Li, F. Geng and Z. Zhao, Nat. Commun., 2015, 6, 7800 CrossRef CAS PubMed.
- J. R. Lombardi, R. L. Birke, T. Lu and J. Xu, J. Chem. Phys., 1986, 84, 4174–4180 CrossRef CAS.
- Samriti, V. Rajput, R. K. Gupta and J. Prakash, J. Mater. Chem. C, 2022, 10, 73–95 RSC.
- M. Li, X. Fan, Y. Gao and T. Qiu, J. Phys. Chem. Lett., 2019, 10, 4038–4044 CrossRef CAS PubMed.
- X. Jiang, L. Xu, W. Ji, W. Wang, J. Du, L. Yang, W. Song, X. Han and B. Zhao, Appl. Surf. Sci., 2022, 584, 152609 CrossRef CAS.
- S. Xie, K. Lai, C. Gu, T. Jiang, L. Zhou, X. Zheng, X. Shen, J. Han and J. Zhou, Mater. Today Nano, 2022, 18, 100179 CrossRef CAS.
- A. D. Terna, E. E. Elemike, J. I. Mbonu, O. E. Osafile and R. O. Ezeani, Mater. Sci. Eng., B, 2021, 272, 115363 CrossRef CAS.
- E. W. Seelig, B. Tang, A. Yamilov, H. Cao and R. P. H. Chang, Mater. Chem. Phys., 2003, 80, 257–263 CrossRef CAS.
- R. Spectroscopy, X. Wang, W. Shi, Z. Jin, W. Huang, J. Lin, G. Ma, S. Li, L. Guo, X. Wang, Z. Jin, J. Lin, G. Ma, L. Guo, D. Shi, S. Li and D. Huang, Angew. Chem., Int. Ed., 2017, 56, 9851–9855 CrossRef PubMed.
- T. Taib, M. R. Johan and W. J. Basirun, Phys. B, 2021, 606, 412638 CrossRef CAS.
- B. Bonelli, O. Tammaro, F. Martinovic, R. Nasi, G. Dell'Agli, P. Rivolo, F. Giorgis, N. Ditaranto, F. A. Deorsola and S. Esposito, ACS Omega, 2021, 6, 24562–24574 CrossRef CAS PubMed.
- X. Chen, N. Deng, X. Zhang, Y. Yang, J. Li, B. Hong, J. Fang, J. Xu, D. Jin, X. Peng, X. Wang and H. Jin, Chem. Phys. Lett., 2019, 722, 96–103 CrossRef CAS.
- L. Ma, Q. Zhang, J. Li, X. Lu, C. Gao, P. Song and L. Xia, Materials, 2021, 14, 922 CrossRef CAS PubMed.
- S. Thakur and S. Neogi, Appl. Nanosci., 2021, 11, 159–171 CrossRef CAS.
- Y. Wang, M. Zhang, H. Ma, H. Su, A. Li, W. Ruan and B. Zhao, ACS Appl. Mater. Interfaces, 2021, 13, 35038–35045 CrossRef CAS PubMed.
- M. Zhang, Y. Wang, X. Wang, B. Zhao and W. Ruan, J. Phys. Chem. C, 2021, 125, 17125–17132 CrossRef CAS.
- L. Zhou, J. Zhou, W. Lai, X. Yang, J. Meng, L. Su, C. Gu, T. Jiang, E. Y. B. Pun, L. Shao, L. Petti, X. W. Sun, Z. Jia, Q. Li, J. Han and P. Mormile, Nat. Commun., 2020, 11, 1785 CrossRef CAS PubMed.
- R. Haldavnekar, K. Venkatakrishnan and B. Tan, Nat. Commun., 2018, 9, 3065 CrossRef PubMed.
- D.-J. Lee and D. Y. Kim, Sensors, 2019, 19, 5471 CrossRef CAS PubMed.
- J. Liu, Y. Liu, Y. Cao, S. Sang, L. Guan, Y. Wang and J. Wang, Micromachines, 2022, 13, 128 CrossRef PubMed.
- X. Ling, L. G. Moura, M. A. Pimenta and J. Zhang, J. Phys. Chem. C, 2012, 116, 25112–25118 CrossRef CAS.
- W. Xu, N. Mao and J. Zhang, Small, 2013, 9, 1206–1224 CrossRef CAS PubMed.
- X. Ling, S. Huang, S. Deng, N. Mao, J. Kong, M. S. Dresselhaus and J. Zhang, Acc. Chem. Res., 2015, 48, 1862–1870 CrossRef CAS PubMed.
- Z. Wang, S. Wu, L. Colombi Ciacchi and G. Wei, Analyst, 2018, 143, 5074–5089 RSC.
- G. Darabdhara, M. R. Das, S. P. Singh, A. K. Rengan, S. Szunerits and R. Boukherroub, Adv. Colloid Interface Sci., 2019, 271, 101991 CrossRef CAS PubMed.
- H. Lai, F. Xu, Y. Zhang and L. Wang, J. Mater. Chem. B, 2018, 6, 4008–4028 RSC.
- X. Ling, S. Huang, S. Deng, N. Mao, J. Kong, M. S. Dresselhaus and J. Zhang, Acc. Chem. Res., 2015, 48, 1862–1870 CrossRef CAS PubMed.
- L. Yang, J.-H. Lee, C. Rathnam, Y. Hou, J.-W. Choi and K.-B. Lee, Nano Lett., 2019, 19, 8138–8148 CrossRef CAS PubMed.
- Y. Yang, Y. Cong, J. Shang, Y. Liu, G. Fang, J. Zhang and B. Dong, Sens. Actuators, B, 2021, 330, 129199 CrossRef CAS.
- S. Atta, S. Rangan and L. Fabris, ChemNanoMat, 2020, 6, 53–57 CrossRef CAS.
- H. Bao, Y. Guo, T. Zhang, H. Fu, S. Zhu, L. Zhou, H. Zhang, Y. Li, W. Cai, H. Bao, Y. Guo, T. Zhang, H. Fu, S. Zhu, L. Zhou, H. Zhang, Y. Li and W. Cai, Adv. Mater. Interfaces, 2022, 9, 2102107 CrossRef CAS.
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Introduction to Reticular Chemistry: Metal-Organic Frameworks and Covalent Organic Frameworks, Wiley-VCH, Weinheim, 2019 Search PubMed.
- N. Martín and F. G. Cirujano, Org. Biomol. Chem., 2020, 18, 8058–8073 RSC.
- S. Denning, A. A. A. Majid, J. M. Lucero, J. M. Crawford, M. A. Carreon and C. A. Koh, ACS Appl. Mater. Interfaces, 2020, 12, 53510–53518 CrossRef CAS PubMed.
- J. Cao, X. Li and H. Tian, Curr. Med. Chem., 2019, 27, 5949–5969 CrossRef PubMed.
- E. A. Dolgopolova, A. M. Rice, C. R. Martin and N. B. Shustova, Chem. Soc. Rev., 2018, 47, 4710–4728 RSC.
- Y. Zhang, Y. Hu, G. Li and R. Zhang, Microchim. Acta, 2019, 186, 1–10 CrossRef PubMed.
- X. Zhao, T. Yang, D. Wang, N. Zhang, H. Yang, X. Jing, R. Niu, Z. Yang, Y. Xie and L. Meng, Anal. Chem., 2022, 94, 4484–4494 CrossRef CAS PubMed.
- S. Bock, Y. S. Choi, M. Kim, Y. Yun, X. H. Pham, J. Kim, B. Seong, W. Kim, A. Jo, K. M. Ham, S. G. Lee, S. H. Lee, H. Kang, H. S. Choi, D. H. Jeong, H. Chang, D. E. Kim and B. H. Jun, J. Nanobiotechnol., 2022, 20, 130 CrossRef CAS PubMed.
- Y. Gutiérrez, A. S. Brown, F. Moreno and M. Losurdo, J. Appl. Phys., 2020, 128, 080901 CrossRef.
- J. Asselin, C. Boukouvala, E. R. Hopper, Q. M. Ramasse, J. S. Biggins and E. Ringe, ACS Nano, 2020, 14, 5968–5980 CrossRef CAS PubMed.
- E. R. Hopper, T. M. R. Wayman, J. Asselin, B. Pinho, C. Boukouvala, L. Torrente-Murciano and E. Ringe, J. Phys. Chem. C, 2022, 2022, 39 Search PubMed.
- S. Juneja and M. S. Shishodia, Opt. Commun., 2019, 433, 89–96 CrossRef CAS.
- W. Li, R. Zamani, P. Rivera Gil, B. Pelaz, M. Ibáñez, D. Cadavid, A. Shavel, R. A. Alvarez-Puebla, W. J. Parak, J. Arbiol and A. Cabot, J. Am. Chem. Soc., 2013, 135, 7098–7101 CrossRef CAS PubMed.
- J. E. Sanchez, S. A. Jaramillo, E. Settles, J. J. Velazquez Salazar, A. Lehr, J. Gonzalez, C. Rodríguez Aranda, H. R. Navarro-Contreras, M. O. Raniere, M. Harvey, D. M. Wagner, A. Koppisch, R. Kellar, P. Keim and M. Jose Yacaman, RSC Adv., 2021, 11, 25788–25794 RSC.
- U. Guler, G. v. Naik, A. Boltasseva, V. M. Shalaev and A. v. Kildishev, Appl. Phys. B, 2012, 107, 285–291 CrossRef CAS.
- U. Guler, A. v. Kildishev, A. Boltasseva and V. M. Shalaev, Faraday Discuss., 2015, 178, 71–86 RSC.
- J. Asselin, C. Boukouvala, E. R. Hopper, Q. M. Ramasse, J. S. Biggins and E. Ringe, ACS Nano, 2020, 14, 5968–5980 CrossRef CAS PubMed.
- J. Asselin, C. Boukouvala, Y. Wu, E. R. Hopper, S. M. Collins, J. S. Biggins and E. Ringe, J. Chem. Phys., 2019, 151, 244708 CrossRef PubMed.
- E. A. Jenrette, M. J. Farrell, J. A. Flowers and A. K. Pradhan, Nanomanufacturing, 2021, 1, 3–13 CrossRef.
- T. Daeneke, B. J. Carey, A. F. Chrimes, J. Z. Ou, D. W. M. Lau, B. C. Gibson, M. Bhaskaran and K. Kalantar-zadeh, J. Mater. Chem. C, 2015, 3, 4771–4778 RSC.
- L. Fabris, J. Opt., 2015, 17, 114002 CrossRef.
- H. Wang, K. Dardir, K.-B. Lee and L. Fabris, Bioconjugate Chem., 2019, 30, 2555–2562 CrossRef CAS PubMed.
- C. Andreou, S. A. Kishore and M. F. Kircher, J. Nucl. Med., 2015, 56, 1295–1299 CrossRef CAS PubMed.
- C. Andreou, V. Neuschmelting, D.-F. Tschaharganeh, C.-H. Huang, A. Oseledchyk, P. Iacono, H. Karabeber, R. R. Colen, L. Mannelli, S. W. Lowe and M. F. Kircher, ACS Nano, 2016, 10, 5015–5026 CrossRef CAS PubMed.
- S. Pal, S. Harmsen, A. Oseledchyk, H.-T. Hsu and M. F. Kircher, Adv. Funct. Mater., 2017, 27, 1606632 CrossRef PubMed.
- F. Nicolson, B. Andreiuk, C. Andreou, H. T. Hsu, S. Rudder and M. F. Kircher, Theranostics, 2019, 9, 5899–5913 CrossRef CAS PubMed.
- V. Dugandžić, D. Drikermann, O. Ryabchykov, A. Undisz, I. Vilotijević, S. Lorkowski, T. W. Bocklitz, C. Matthäus, K. Weber, D. Cialla-May and J. Popp, J. Biophotonics, 2018, 11, e201800013 CrossRef PubMed.
- C. Song, F. Li, X. Guo, W. Chen, C. Dong, J. Zhang, J. Zhang and L. Wang, J. Mater. Chem. B, 2019, 7, 2001–2008 RSC.
- M. Bhamidipati, H. Y. Cho, K. B. Lee and L. Fabris, Bioconjugate Chem., 2018, 29, 2970–2981 CrossRef CAS PubMed.
- Thermo Scientific, Bioconjugation and crosslinking technical handbook, https://assets.thermofisher.com/TFS-Assets/BID/Handbooks/bioconjugation-technical-handbook.pdf, accessed 24 October 2022 Search PubMed.
- G. T. Hermanson, in Bioconjugate Techniques, Elsevier, Amsterdam, 2013, pp. 229–258 Search PubMed.
- G. T. Hermanson, in Bioconjugate Techniques, Elsevier, Amsterdam, 2013, pp. 259–273 Search PubMed.
- W. H. Brown, C. S. Foote, B. L. Iverson and E. V. Ansly, Organic Chemistry, Brooks/Cole, Belmont, 6th edn, 2010 Search PubMed.
- G. T. Hermanson, in Bioconjugate Techniques, Elsevier, Amsterdam, 2013, pp. 299–339 Search PubMed.
- J. A. Burns, J. C. Butler, J. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648–2650 CrossRef CAS.
- B. Liu and J. Liu, J. Am. Chem. Soc., 2017, 139, 9471–9474 CrossRef CAS PubMed.
- D. S. Seferos, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477–15479 CrossRef CAS PubMed.
- P.-C. Guan, H. Zhang, Z.-Y. Li, S.-S. Xu, M. Sun, X.-M. Tian, Z. Ma, J.-S. Lin, M.-M. Gu, H. Wen, F.-L. Zhang, Y.-J. Zhang, G.-J. Yu, C. Yang, Z.-X. Wang, Y. Song and J.-F. Li, Anal. Chem., 2022, 94, 17795–17802 CrossRef CAS PubMed.
- W. Teng, J. Zhao, Q. Li, P. Shi, J. Zhang, M. Yan and S. Zhang, Sens. Diagn., 2023, 2, 132–139 RSC.
- S. T. Sivapalan, B. M. DeVetter, T. K. Yang, T. van Dijk, M. V. Schulmerich, P. S. Carney, R. Bhargava and C. J. Murphy, ACS Nano, 2013, 7, 2099–2105 CrossRef CAS PubMed.
- D. Li, L. Jiang, J. A. Piper, I. S. Maksymov, A. D. Greentree, E. Wang and Y. Wang, ACS Sens., 2019, 4, 2507–2514 CrossRef CAS PubMed.
- D. Zhang, L. Huang, B. Liu, E. Su, H. Y. Chen, Z. Gu and X. Zhao, Sens. Actuators, B, 2018, 277, 502–509 CrossRef CAS.
- S. Harmsen, M. A. Bedics, M. A. Wall, R. Huang, M. R. Detty and M. F. Kircher, Nat. Commun., 2015, 6, 6570 CrossRef CAS PubMed.
- S. Rajappa, in Comprehensive Heterocyclic Chemistry, Elsevier, Amsterdam, 1984, pp. 741–861 Search PubMed.
- M. A. Bedics, H. Kearns, J. M. Cox, S. Mabbott, F. Ali, N. C. Shand, K. Faulds, J. B. Benedict, D. Graham and M. R. Detty, Chem. Sci., 2015, 6, 2302–2306 RSC.
- T. Nakamura, R. Kimura, F. Matsui, H. Kondoh, T. Ohta, H. Sakai, M. Abe and M. Matsumoto, Langmuir, 2000, 16, 4213–4216 CrossRef CAS.
- F. K. Huang, R. C. Horton, D. C. Myles and R. L. Garrell, Langmuir, 1998, 14, 4802–4808 CrossRef CAS.
- S. Y. Lee, E. Ito, H. Kang, M. Hara, H. Lee and J. Noh, J. Phys. Chem. C, 2014, 118, 8322–8330 CrossRef CAS.
- H. Kearns, M. A. Bedics, N. C. Shand, K. Faulds, M. R. Detty and D. Graham, Analyst, 2016, 141, 5062–5065 RSC.
- H. Kearns, F. Ali, M. A. Bedics, N. C. Shand, K. Faulds, M. R. Detty and D. Graham, R. Soc. Open Sci., 2017, 4, 170422 CrossRef PubMed.
- O. Zeiri, ACS Sens., 2020, 5, 3806–3820 CrossRef CAS PubMed.
- S. Mosca, C. Conti, N. Stone and P. Matousek, Nat. Rev. Methods Primers, 2021, 1, 21 CrossRef CAS.
- N. A. Macleod and P. Matousek, Appl. Spectrosc., 2008, 62, 291A–304A CrossRef CAS PubMed.
- F. Martelli, S. del Bianco, A. Ismaelli and G. Zaccanti, Light Propagation Through Biological Tissue and Other Diffusive Media: Theory, Solutions, and Software (SPIE Press Monograph vol. PM193), 2010 Search PubMed.
- N. Stone, K. Faulds, D. Graham and P. Matousek, Anal. Chem., 2010, 82, 3969–3973 CrossRef CAS PubMed.
- B. Sharma, K. Ma, M. R. Glucksberg and R. P. van Duyne, J. Am. Chem. Soc., 2013, 135, 17290–17293 CrossRef CAS PubMed.
- F. Nicolson, L. E. Jamieson, S. Mabbott, K. Plakas, N. C. Shand, M. R. Detty, D. Graham and K. Faulds, Analyst, 2018, 143, 5358–5363 RSC.
- F. Nicolson, L. E. Jamieson, S. Mabbott, N. C. Shand, D. Graham and K. Faulds, J. Raman Spectrosc., 2017, 48, 1828–1838 CrossRef CAS.
- P. R. Bakic, C. Zhang and A. D. A. Maidment, Med. Phys., 2011, 38, 3165–3176 CrossRef PubMed.
- F. Nicolson, L. E. Jamieson, S. Mabbott, K. Plakas, N. C. Shand, M. R. Detty, D. Graham and K. Faulds, Chem. Sci., 2018, 9, 3788–3792 RSC.
- F. Nicolson, L. E. Jamieson, S. Mabbott, K. Plakas, N. C. Shand, M. R. Detty, D. Graham and K. Faulds, Chem. Commun., 2018, 54, 8530–8533 RSC.
- S. Mosca, P. Dey, T. A. Tabish, F. Palombo, N. Stone and P. Matousek, J. Biophotonics, 2020, 13, e201960092 CrossRef PubMed.
- M. E. Berry, S. M. Mccabe, N. C. Shand, D. Graham and K. Faulds, Chem. Commun., 2022, 58, 1756 RSC.
- A. S. Moody, T. D. Payne, B. A. Barth and B. Sharma, Analyst, 2020, 145, 1885–1893 RSC.
- A. S. Moody and B. Sharma, ACS Chem. Neurosci., 2018, 9, 1380–1387 CrossRef CAS PubMed.
- T. A. Qiu, P. L. Clement and C. L. Haynes, Chem. Commun., 2018, 54, 12787–12803 RSC.
- W. Najahi-Missaoui, R. D. Arnold and B. S. Cummings, Int. J. Mol. Sci., 2020, 22, 385 CrossRef PubMed.
- C. Egbuna, V. K. Parmar, J. Jeevanandam, S. M. Ezzat, K. C. Patrick-Iwuanyanwu, C. O. Adetunji, J. Khan, E. N. Onyeike, C. Z. Uche, M. Akram, M. S. Ibrahim, N. M. el Mahdy, C. G. Awuchi, K. Saravanan, H. Tijjani, U. E. Odoh, M. Messaoudi, J. C. Ifemeje, M. C. Olisah, N. J. Ezeofor, C. J. Chikwendu and C. G. Ibeabuchi, J. Toxicol., 2021, 2021, 1–21 CrossRef PubMed.
- R. Singh, A. Sharma, J. Saji, A. Umapathi, S. Kumar and H. K. Daima, Nano Convergence, 2022, 9, 21 CrossRef CAS PubMed.
- S. Kumar, K. S. Gandhi and R. Kumar, Ind. Eng. Chem. Res., 2007, 46, 3128–3136 CrossRef CAS.
- S. R. Jackson, J. R. McBride, S. J. Rosenthal and D. W. Wright, J. Am. Chem. Soc., 2014, 136, 5261–5263 CrossRef CAS PubMed.
- K. Christmann, Introduction to Surface Physical Chemistry, in Topics in Physical Chemistry, Springer-Verlag, Berlin/Heidelberg, 1991, vol. 1 Search PubMed.
- D. P. dos Santos, M. L. A. Temperini and A. G. Brolo, Acc. Chem. Res., 2019, 52, 456–464 CrossRef CAS PubMed.
- M. Belkin, S.-H. Chao, M. P. Jonsson, C. Dekker and A. Aksimentiev, ACS Nano, 2015, 9, 10598–10611 CrossRef CAS PubMed.
- C. Chen, Y. Li, S. Kerman, P. Neutens, K. Willems, S. Cornelissen, L. Lagae, T. Stakenborg and P. van Dorpe, Nat. Commun., 2018, 9, 1733 CrossRef PubMed.
- D. Deamer, M. Akeson and D. Branton, Nat. Biotechnol., 2016, 34, 518–524 CrossRef CAS PubMed.
- N. A. Jose, M. Kovalev, E. Bradford, A. M. Schweidtmann, H. Chun Zeng and A. A. Lapkin, Chem. Eng. J., 2021, 426, 131345 CrossRef CAS.
- K.-J. Wu, G. M. de Varine Bohan and L. Torrente-Murciano, React. Chem. Eng., 2017, 2, 116–128 RSC.
- E. Bradford, A. M. Schweidtmann and A. Lapkin, J. Glob. Optim., 2018, 71, 407–438 CrossRef.
- A. M. Schweidtmann, A. D. Clayton, N. Holmes, E. Bradford, R. A. Bourne and A. A. Lapkin, Chem. Eng. J., 2018, 352, 277–282 CrossRef CAS.
- A. D. Clayton, A. M. Schweidtmann, G. Clemens, J. A. Manson, C. J. Taylor, C. G. Niño, T. W. Chamberlain, N. Kapur, A. J. Blacker, A. A. Lapkin and R. A. Bourne, Chem. Eng. J., 2020, 384, 123340 CrossRef CAS.
- B. Smiri, F. Saidi, A. Mlayah and H. Maaref, J. Mater. Sci.: Mater. Electron., 2020, 31, 10750–10759 CrossRef CAS.
- T. Qi, Y. Yu, Y. Hu, K. Li, N. Guo and Y. Jia, Nanomaterials, 2022, 12, 1258 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |