
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSize-controlled liquid phase synthesis of colloidally stable Co3O4 nanoparticles†
Johannes
Kießling
*a,
Sabine
Rosenfeldt
bc and
Anna S.
Schenk
 *ac
*ac
aPhysical Chemistry IV, University of Bayreuth, Universitaetsstrasse 30, 95447 Bayreuth, Germany. E-mail: Johannes.Kiessling@uni-bayreuth.de; Anna.Schenk@uni-bayreuth.de
bPhysical Chemistry I, University of Bayreuth, Universitaetsstrasse 30, 95447 Bayreuth, Germany
cBavarian Polymer Institute (BPI), University of Bayreuth, Universitaetsstrasse 30, 95447 Bayreuth, Germany
First published on 4th July 2023
Abstract
Spinel cobalt(II,III) oxide (Co3O4) represents a p-type semiconductor exhibiting promising functional properties in view of applications in a broad range of technological fields including magnetic materials and gas sensors as well as sustainable energy conversion systems based on photo- and electrocatalytic water splitting. Due to their high specific surface area, nanoparticle-based structures appear particularly promising for such applications. However, precise control over the diameter and the particle size distribution is required to achieve reproducible size-dependent properties. We herein introduce a synthetic strategy based on the decomposition of hydroxide precursors for the size-controlled preparation of purified Co3O4 nanoparticles with narrow size distributions adjustable in the range between 3–13 nm. The particles exhibit excellent colloidal stability. Their dispersibility in diverse organic solvents further facilitates processing (i.e. ligand exchange) and opens exciting perspectives for controlled self-assembly of the largely isometric primary particles into mesoscale structures. In view of potential applications, functional properties including absorption characteristics and electrocatalytic activity were probed by UV-Vis spectroscopy and cyclic voltammetry, respectively. In these experiments, low amounts of dispersed Co3O4 particles demonstrate strong light absorbance across the entire visible range and immobilized nanoparticles exhibit a comparably low overpotential towards the oxygen evolution reaction in electrocatalytic water splitting.
Introduction
In recent decades, the scientific community has upheld an intense interest in the development of nanostructured materials with adjustable size and morphology as well as the study of associated structure–property relationships. Due to their versatile, size-dependent functional properties, nanomaterials are appealing for many important technological applications including drug delivery, tissue engineering, sustainable energy storage and conversion systems, (opto-) electronics, and catalysis.1–3 In the field of heterogeneous catalysis, transition metal oxides based on earth-abundant elements, have received increasing interest in recent years. Cobalt(II,III) oxide (Co3O4) and related compounds, in particular, have been identified as robust and efficient catalyst materials promoting various environmentally relevant reactions.4–6 Aside from applications in Fischer–Tropsch synthesis and the oxidation of carbon monoxide, Co3O4 has shown excellent performance in the catalysis of both, the oxygen evolution reaction (OER) and the oxygen reduction reaction (ORR) in electrocatalytic water splitting and as a co-catalyst in photocatalysis.7–10 The latter represent highly promising reaction systems for clean energy conversion and benefit from the remarkable stability of spinel-type cobalt oxide in an aqueous medium even under highly acidic or alkaline conditions.The majority of synthetic approaches for the preparation of Co3O4 nanomaterials involve solvo- or hydrothermal sol–gel methods, followed by calcination at temperatures between 300–450 °C.11,12 These methods are mainly designed to generate larger, non-re-dispersible particles without capping ligands. While the omission of ligands can be advantageous for applications based on surface reactions requiring accessible facets of the nanoparticle (NP) units, e.g. in catalysis or charge/discharge reactions in energy storage systems,13 dispersed NPs offer desirable properties for large-scale processing, surface functionalization, and structuring as well as controlled assembly.
A number of methods for the synthesis of re-dispersible Co3O4 NPs have been reported in the literature. Agiral et al. developed a solvothermal synthetic protocol to fabricate re-dispersible 4 nm Co3O4 nanocubes.14 Another solvothermal method for the synthesis of re-dispersible Co3O4 NPs by oriented attachment was reported by Tsukiyama et al.15 Liu et al. recently described a hydrothermal method to prepare 8–9 nm sized spherical and cubic Co3O4 NPs.16 To probe the catalytic activity towards CO oxidation as a function of catalyst particle size, Iablokov et al. synthesized very small, spherical, and re-dispersible Co-NPs in the size range between 3–10 nm,17,18 which were then oxidized into Co3O4. Similarly, Saddeler et al. prepared spherical CoO NPs with diameters between 8–15 nm and subsequently oxidized into Co3O4,19 where the conversion is accompanied by ligand decomposition and the loss of dispersibility though. Recently, Haase et al. reported a method to prepare CoOx(OH) particles ranging from 1–9 nm.20
NP dispersions with tunable size and narrow size distribution are in great demand, as such systems may facilitate dedicated studies on size-dependent properties (e.g. catalysis, magnetism) and enable ordered self-assembly into customized meso- and microscale superstructures from solution.21 Therefore, our study aims to develop a convenient and reproducible liquid-phase strategy for the fabrication of primary, re-dispersible Co3O4 NPs with adjustable sizes over a comparably large range of diameters in just one precipitation system.
Typical liquid-phase nanoparticle syntheses involve the thermal decomposition of molecular compounds at high temperatures (250–320 °C) often accompanied by long reaction times (>10 h).22 In the case of cobalt oxides, such reactions mainly lead to the formation of CoO, instead of the oftentimes catalytically more active Co3O4 phase.23–25 During the electrocatalytic OER, for example, CoO is rapidly oxidized to Co3O4, towards which all further oxidation products revert in the catalytic cycle.26 For this reason, it can be desirable to directly introduce Co3O4 into the electrocatalytic reaction to avoid an uncontrolled structural transformation which would potentially impact the overall catalyst structure and possibly its stability.
The most commonly used direct reaction pathway for the synthesis of spinel-type Co3O4 is the initial precipitation of Co(OH)2 using alkali hydroxides, ammonia, or urea as reagents with various cobalt salts.27,28 Cobaltous hydroxides crystallize as layered double hydroxides (LDH) in the α-Co(OH)2 (hydrotalcite) and β-Co(OH)2 (brucite) form.29,30 LDH are capable of intercalating a large variety of anions within the interlayer space potentially altering the resulting morphology,31 which is then pseudomorphically translated into the oxide phase upon thermal conversion.32 As different crystal shapes are associated with a characteristic set of surface-exposed lattice planes, functional properties of Co3O4, such as catalytic activity and magnetism, are strongly affected by particle morphology.4,33,34 Another merit of utilizing Co(OH)2 as a precursor is the relatively mild decomposition temperature of α-Co(OH)2 at 165 °C resulting in the transformation to Co3O4,27 thus significantly reducing the energy required to generate spinel cobalt oxide NPs.
In view of functional properties, Iablokov et al. report an enhanced catalytic activity for Co3O4 NPs with diameters in the range between 5–8 nm towards the oxidation of CO, whereas the conversion rate decreases rapidly for larger-sized particles.18 The relationship between catalytic activity and NP size makes it reasonable to assume that similar size-dependent efficiency maxima exist for other catalytic processes occurring at the surfaces of spinel cobalt oxide NPs as well. However, the determination of size-dependent properties relies critically on the comparability of the investigated NPs with respect to their chemical nature and environment (e.g. composition, functionalization). Therefore, it is highly desirable to prepare an entire series of differently sized NPs based on the same inherent reaction conditions and educts. To the best of the authors' knowledge, no such protocol is currently available for the synthesis of Co3O4 NPs with systematically tunable particle size in the low nanometer regime. Moreover, the re-dispersibility of the particles is a desirable property for post-synthesis purification and storage.
To address the challenge posed by these requirements, we here establish a reaction system based on the precipitation of hydroxide precursor particles at mild temperatures (<100 °C) and subsequent in situ thermal conversion into spinel cobalt oxide. In contrast to other methods for the precipitation of cobalt hydroxide, an organic solvent (oleylamine) is used instead of an aqueous system. Oleylamine (OLA) simultaneously acts as the solvent and stabilizing ligand and limits monomer diffusion to achieve a stable growth regime. OLA further represents a bio-renewable chemical and may be recovered after the reaction if so desired.35 Similarly, the chosen stabilizer oleic acid (OA) is bio-renewable, as well as non-toxic, inexpensive, and well-researched while providing reliable stabilization for NPs in non-polar solvents.36,37 The injection of NaOH in an aqueous solution allows for facile, reliable size control as well as the colloidal stabilization of Co3O4 NPs in the low nano-regime by a reverse-micelle process.
Hence, a synthetic protocol relying only on commercially available reagents was designed to provide a facile, versatile, inexpensive, and potentially scalable method to produce Co3O4 NPs with controlled size and shape.
Experimental section
Chemicals and materials
Cobalt(II) nitrate hexahydrate (99%, for analysis), oleic acid (OA, synthesis grade), oleylamine (OLA, 98%), tetrabutylammonium hydroxide (1.0 M in MeOH), Co3O4 nanopowder (<50 nm, 99.5%), Nafion 117 solution (5% in lower aliphatic alcohols and water) and glassy carbon (spherical powder, 2–12 μm, 99.95%) were purchased from Sigma Aldrich. Ethanol (EtOH) and heptane were obtained from VWR. NaOH solution (0.1 M in distilled water) was purchased from Grüssing GmbH. Polytetrafluoroethylene (PTFE, 0.2 μm) syringe filters were obtained from Carl Roth GmbH. All chemicals and materials were used as received from the suppliers without further purification.Synthesis of Co3O4 nanoparticles
As a general procedure, Co3O4 nanoparticles were obtained via precipitation of a hydroxide-based precursor followed by thermal decomposition. For that purpose, Co(NO3)2·6H2O (2 mmol, 0.582 g) was dissolved in 20 mL oleylamine (OLA, >98% primary amine) in a sealed three-necked round-bottomed flask together with 2 mL of EtOH as phase mediator. The reactant solution was equilibrated at the desired reaction temperature (cf. Chapter 1 in the ESI†) to fully dissolve the salt before cobalt hydroxide precursor particles were precipitated with either 4 mL tetrabutylammonium hydroxide (TBAH) solution (1.0 M in MeOH, 4 mmol) or 2 mL NaOH solution (4 mmol in distilled H2O). After stirring the reagent mixture for 1 h, low-boiling solvents were removed in vacuo. Subsequently, the precursor particles were thermally converted into Co3O4 by heating to 180 °C for 1 h. The resulting nanoparticle dispersion was then cooled down to room temperature with an ice bath, followed by the addition of 2 mL oleic acid (OA), as well as 10 mL heptane before stirring the dispersion overnight. Afterwards, the nanoparticles were purified in several consecutive centrifugation and re-dispersion steps with EtOH and heptane to remove any traces of OLA and residual salts. The particles are stable during at least three precipitation and re-dispersion steps. After drying, the obtained cobalt oxide particles were re-dispersed in heptane and filtered through a 0.2 μm PTFE syringe filter.Specific reaction parameters and variations in the synthetic procedure implemented to obtain Co3O4 nanoparticles with different distinct sizes are detailed in the ESI† (cf. Chapter 1 in the ESI†). Major parameters influencing the particle size are the reaction temperature, the mode of base injection (instant injection vs. drop-wise addition), and the availability of oxygen as well as subsequent purification steps by precipitation and re-dispersion.
Characterization of composition, structure, and properties
Image analysis was performed using Gatan Digital Micrograph and Fiji software.38,39 The particle dimensions were obtained as Feret diameters. Average particle diameters were determined by fitting a log-normal distribution (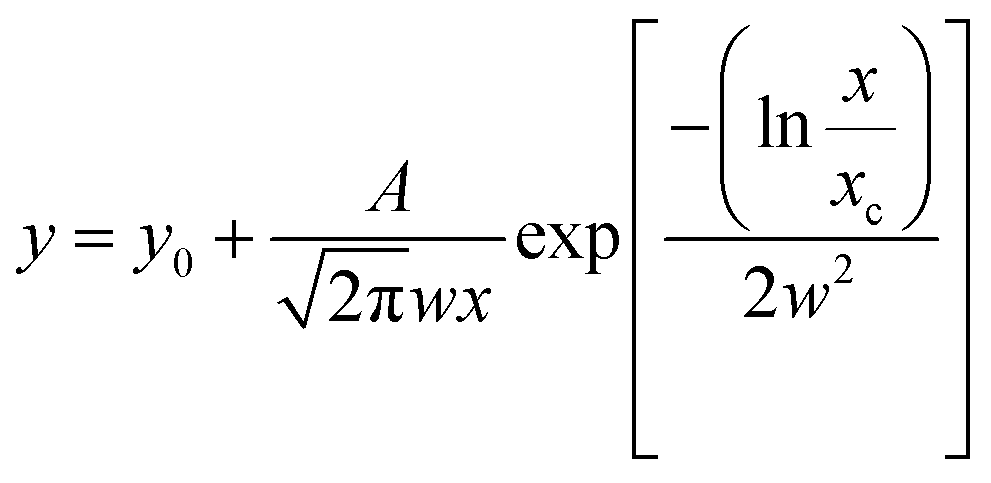 ; A = area, x = diameter, xc = center of the distribution, w = log standard deviation) to the count-size histograms obtained from Fiji. Particle size distributions were calculated from the full width at half maximum (FWHM) of the log-normal fit functions.
; A = area, x = diameter, xc = center of the distribution, w = log standard deviation) to the count-size histograms obtained from Fiji. Particle size distributions were calculated from the full width at half maximum (FWHM) of the log-normal fit functions.
SAED patterns were evaluated by using the DiffTools application for Gatan Digital Micrograph as well as Crystbox 1.10.40,41 Reference diffractograms were calculated from crystallographic information files (.cif) obtained from the Crystallographic Open Database (COD card: 9005896) with the free VESTA (visualization for electronic and structural analysis) software package.42 The reference diffractogram was recalculated from the conventional 2θ representation to match the scattering vectors obtained from SAED.
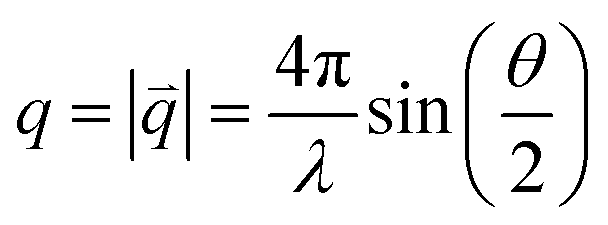 | (1) |
Modelling of the data was performed with the Scatter software (version 2.5) using the form factor models for polydisperse homogeneous spheres with a Gaussian size distribution.43 The obtained results were cross-compared to results obtained with the SasFit application.44
Electrochemical measurements were conducted in a potential window of −0.5 V to 1 V at a scan rate of 50 mV s−1 with an RDE rotation speed of 2000 rpm to cover the RedOx-active region of Co3O4 and to initiate the anodic oxygen evolution reaction (OER). The data were converted to the potential scale of the reversible hydrogen electrode (RHE) and corrected for the iR-drop according to the following equation
| ERHE = EAg/AgCl + 0.059 pH + E0Ag/AgCl (3 M KCl) − iRu | (2) |
Results and discussion
General synthetic procedure
Co3O4 NPs with tunable size in the low nanometer regime were prepared by a facile and versatile liquid phase synthesis based on the thermal decomposition of hydroxide precursors.The experimental system developed in this work consists of a complex solvent mixture of OLA, EtOH, and H2O. While a small amount of EtOH was added intentionally as a phase mediator to increase the solubility of the cobalt salt, water is contributed by the Co(NO3)2·6H2O reagent in the form of crystal water. Such minute quantities of H2O, however, did not affect the progress or products of the reaction as demonstrated in control experiments with an anhydrous Co(II) source obtained by in vacuo removal of crystal water from the Co(NO3)2·6H2O salt prior to the reaction. According to a study by Folkman et al. on a similar system, EtOH does not contribute a ligand effect during the reaction.45
As schematically illustrated in Fig. 1, the reaction protocol is based on the basic precipitation of green α-Co(OH)2 from the Co(II) solution. Precursor particles nucleating from the reaction mixture at elevated temperatures are in situ stabilized by OLA and subsequently thermally decomposed into Co3O4 at 180 °C. After thermal conversion into the oxide phase, the resulting NPs are additionally stabilized with OA. Characteristic colour changes in the reaction mixture are observed along the reaction path as documented in Fig. S1.† After injection of the base, the colour of the initially red Co(II) solution depends on the atmospheric conditions. Under N2, green hydrotalcite-type α-Co(OH)2 with a layered crystallographic structure and a strong capacity for ion intercalation is obtained.29 Without continuous nitrogen purging, the dispersion turns dark green and eventually brown, while under atmospheric conditions, a fast change towards a brown colour is observed due to the formation of a complex mixture of α-Co(OH)2 and its oxidation product CoO(OH). Above temperatures of 50 °C, α-Co(OH)2 is rapidly converted into light pink β-Co(OH)2 in N2 atmosphere.46 However, irrespective of the precursor composition, black Co3O4 NPs are obtained after in situ decomposition. The observed stages of the reaction were isolated as powders and analyzed by PXRD. The results are summarized in Fig. S2.† Adjustments of the specific reaction conditions in the precipitation system enable the isolation of NPs with sizes between 3 and 13 nm with narrow size distribution (Fig. S3†). Aliquots removed from the reaction mixture prior to thermal conversion were investigated by TEM revealing the preservation of NP size and shape, which points to a pseudomorphic transformation pathway (Fig. S4†). The assumed reaction scheme is presented in eqn (3).
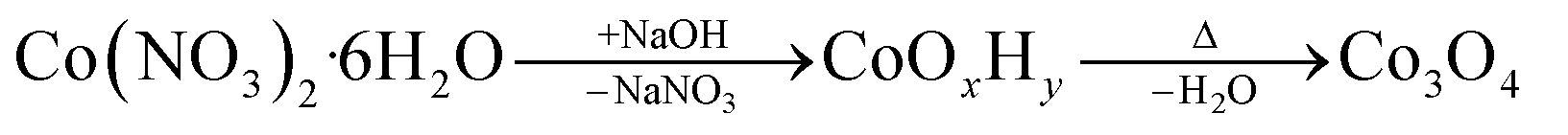 | (3) |
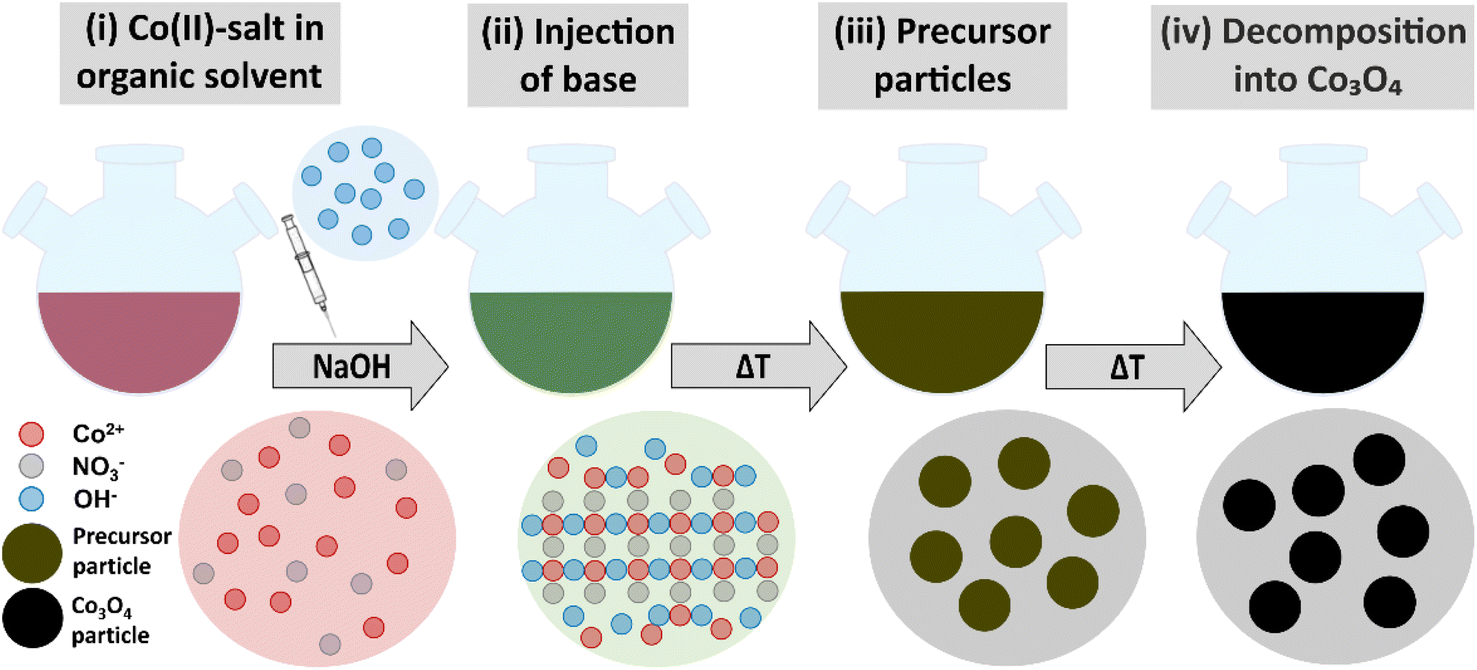 | ||
| Fig. 1 Schematic illustration of the reaction scheme. (i) A solution of red Co(NO3)2·6H2O in OLA is heated in a round-bottomed flask. (ii) Injection of a base (typically aqueous NaOH) induces the formation of green α-Co(OH)2. (iii) Further heating leads to the precipitation of hydroxide precursor NPs. In contact with air, the mixture adopts a brown-green color presumably due to the presence of an oxyhydroxide oxidation product. (iv) The precursor particles are pseudomorphically converted into black Co3O4 NPs. | ||
Remarkably, NPs created according to this method have been colloidally stable in dispersion (i.e. no agglomeration and sedimentation is observed) for more than 10 months, thus exhibiting excellent shelf-life for storage (see Fig. S5†).47 Aside from the stability of the resulting dispersions, the necessary reaction temperatures of 180 °C and below are significantly lower than in many reported liquid phase NP syntheses. The reaction time (∼1 h for nucleation and growth of precursor NPs and ∼1 h for conversion into Co3O4) is short compared to similar reaction systems in solvothermal synthesis.19,48 Hence, the here presented methodology indeed provides a platform for the controlled, resource-efficient preparation of colloidally stable Co3O4 NPs with tunable size.
Temperature dependence of the Co3O4 nanoparticle crystallization
When considering important reaction parameters critically affecting the solubility of reagents, and thus the nucleation and growth of NPs, temperature, reagent concentration, and solvent have to be optimized and controlled. In the here presented precipitation system, OLA is chosen as a medium, in which the solubility of ionic salts is limited and thus supersaturation is readily achieved. Nucleation of the hydroxide-based precursor is routinely induced by stoichiometric addition of base (here: NaOH). We assume that the phase transition occurs via homogenous nucleation as evidenced by control experiments performed with a tripled NaOH concentration (Fig. S6†). The increase in precipitation agent forcefully increases the supersaturation and thus the nucleation rate. The resulting particles are smaller with a significantly broader size distribution since the depletion of monomer occurs in a shorter time period.49To investigate the effects of the precipitation temperature (T) on the progress and products of the reaction, a series of syntheses were performed in the temperature range between 50–100 °C in steps of 10 °C while keeping the salt concentration (nCo(NO3)2·6H2O = 2 mmol) and all other reaction parameters constant to ensure comparability. The experiments are conducted under an air atmosphere. After stirring for 30 min at the desired temperature, the solutions were destabilized by injection of NaOH to induce nucleation. The results of these T-dependent experiments are summarized in Fig. 2.
 | ||
| Fig. 2 Temperature-dependence of NP size and shape. TEM micrographs of Co3O4 nanoparticles synthesized with a precipitation step performed at temperatures of (a) 50 °C, (b) 60 °C, (c) 70 °C, (d) 80 °C, (e) 90 °C and (f) 100 °C (same scale bar). Trends in the temperature-dependent modulation of particle size and shape are indicated by grey arrows. | ||
When analyzed by TEM, the particles precipitated at T = 50 °C exhibit a size of ca. 6.5 ± 2.7 nm (Fig. 2a). Their fractured appearance and irregular shapes suggest that at very low precipitation temperatures, the Co(NO3)2·6H2O salt may have been only partially dissolved in the medium. We speculate, that under these conditions, the solution is kept at saturation which leads to secondary nucleation. In this event, smaller, newly nucleated particles may readily attach to the surfaces of larger NPs to reduce the surface energy. Such an effect may explain the observed overall rough, irregular surface topography and larger “effective size” (i.e. core and surface-attached particles).50 This line of thought is supported by experiments performed at higher temperatures. When precipitation is carried out at T = 60 °C, the particle shape appears more homogeneous due to the increased solubility of the educts (Fig. 2b). Concomitantly, the average particle diameter is significantly reduced to 5.4 ± 1.9 nm due to the presumed absence of secondary nucleation and accretion of smaller particles onto the surfaces. However, even at 60 °C, the solubility of the reagent salt appears to be limited. When raising the precipitation temperature to T = 70 °C, we observed a substantial increase in yield in combination with a larger average particle size of 12.9 ± 4.6 nm and a centrosymmetric, more clearly defined particle shape (Fig. 2c). No indication of surface-attached smaller particles is seen in TEM micrographs of NPs deposited at 70 °C. At equilibration temperatures of 80 °C and 90 °C, similar results are obtained with average NP sizes of 12.5 ± 3.1 nm and 11.9 ± 3.3 nm, respectively (Fig. 2d and e). At T = 100 °C, however, a significant decrease in size to 8.3 ± 1.9 nm, is observed (Fig. 2f), thus indicating a drastic significant change in the conditions within the reaction system. Most importantly, the boiling point of water is reached at 100 °C, water and EtOH are boiling simultaneously. While it is unclear in which exact state of coordination water coexists with cobalt ions and OLA at elevated temperatures, we can assuredly infer that its mobility increases significantly at the boiling point.51,52 This may lower the solubility of OLA-capped NPs while having no effect on the cobalt salt. In support of this hypothesis, control experiments were performed. Without the addition of EtOH as a phase mediator or by replacing it with water, the NPs decrease further in size accompanied by a significantly lower yield. Thus, a negative influence of excess water on particle growth may be inferred.
Interestingly, the T-dependent changes in particle size are accompanied by a modulation in nanocrystal shape. Specifically, the smaller particles precipitated at T < 70 °C exhibit a generally spherical morphology, while a gradual transition toward cuboidal shapes is observed in NP samples obtained at temperatures of T ≥ 70 °C. Influences on particle size and morphology are further considered in the discussion section, vide infra.
In summary, these experiments demonstrate that in the here presented liquid-phase reaction system, adjustment of the average NP size is possible by systematic variation of the temperature at the precipitation step.
Size-controlled synthesis of Co3O4 nanoparticles
Building on these important observations, we implemented further adjustments to the process to improve particle size distributions and to achieve a series of NPs with even smaller dimensions. The smallest possible NP size achieved by pure thermal control is 5.4 ± 1.9 nm at 60 °C. Fig. 3 summarizes the results of these optimizations. Reactions were performed at 50 °C, 80 and 100 °C to assure reasonably spaced temperature intervals (here: a minimum of 20 °C) to facilitate the identification of effects of the varied parameters on particle size and shape. A substantial reduction in particle size (at a constant temperature) is observed when the reaction is performed under extended nitrogen purging (Fig. 3a–c). Specifically, NPs precipitated at T = 50 °C by an instantaneous injection of stoichiometric NaOH solution in the absence of oxygen exhibited an average size of 4.7 ± 1.0 nm (sample S5, Fig. 3c), which corresponds to a decrease by ∼2 nm as compared to the analogous reaction under air atmosphere (compare Fig. 2a; 6.5 ± 2.7 nm). Simultaneously, oxygen-free conditions appear to promote more clearly defined spherical particle morphologies.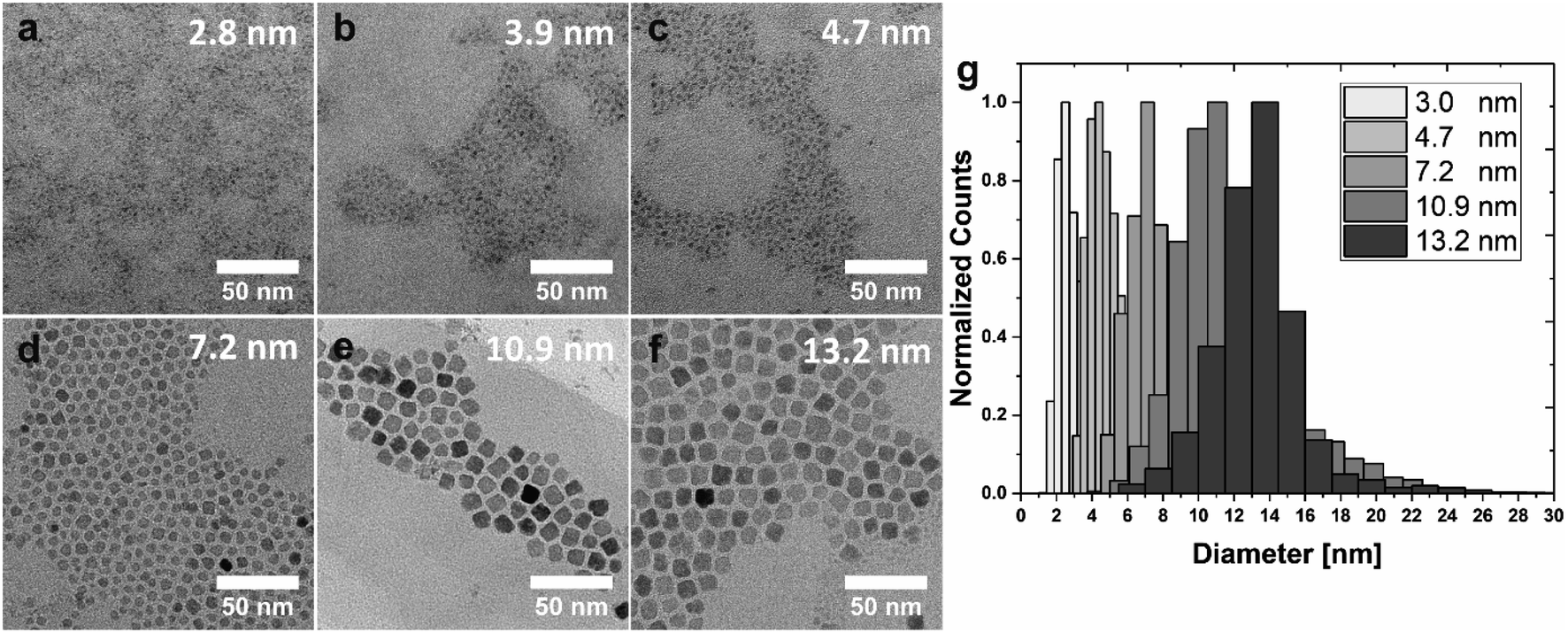 | ||
| Fig. 3 Size and shape of NPs in the low nanometer regime. (a–f) TEM micrographs of NPs in the size range from 2.8 nm to 13.2 nm, ordered according to an increase in particle size. From a–f, samples S3 (50 °C), S4 (50 °C), S5 (50 °C), Cb8 (100 °C), Cb11 (80 °C), and Cb13 (80 °C) are displayed. (g) Combined, normalized size-distribution histograms obtained by TEM image analysis. The particles of each sample focus on a narrow size margin, despite a relatively broad basis. Due to the strong overlap between 3–5 nm, the count-size histogram of S4 was omitted. | ||
For the synthesis of even smaller Co3O4 NPs, the mode of base addition needs to be modified, such that the precipitation agent is slowly dripped into the Co(NO3)2·6H2O solution over 15 min. Due to the limited amount of hydroxide available for particle formation, nucleation may only occur locally for each droplet of NaOH(aq). For this reason, only very small particles with an average size of 3.9 ± 1.2 nm (sample S4, Fig. 3b) can be formed under these conditions. By exchanging the aqueous NaOH solution with a 1.0 M solution of tetrabutylammonium hydroxide (TBAH) in MeOH a further reduction in particle diameter to an average size of 2.8 ± 1.0 nm can be achieved (sample S3, Fig. 3a). Since 4 mL of solution needs to be added for a stoichiometric amount of base, the application of EtOH as a phase mediator was omitted in this case (to keep the final volume constant). With an increasing amount of MeOH (during dropwise base addition), the solubility of the cobalt salt increases while the solubility of the resulting particles decreases, thus inhibiting further growth to yield extremely small-scale particles. Please note, that the overlap of size distributions cannot be fully prevented in the case of very small particles (<5 nm) close to the size of the critical nucleus. Particles of type S3 may also be synthesized with similar size in an air atmosphere (cf. Fig. S7†), but a nitrogen-derived sample is shown in Fig. 3 for consistency with samples S4 and S5 (both obtained under nitrogen atmosphere).
The aforementioned approach of decreasing particle size by nitrogen purging, however, is only reliably applicable at low reaction temperatures, since hydrotalcite α-Co(OH)2 is converted to brucite β-Co(OH)2 in the presence of nitrogen at elevated temperatures.46,53 While this conversion is slow, and can be neglected at T = 50° (as only very small amounts of brucite are detected in the X-ray diffractogram of sample S5), repeating the experiments at T = 80 °C leads to the precipitation of an insoluble pink solid from the dark green dispersion, thereby preventing the formation of colloidally stable Co3O4 NPs (cf. Fig. S2†). However, to overcome this problem, a blanket of nitrogen generated by a single surge of the inert gas was found to improve both, particle shape and size distribution at temperatures of 80 °C and above.
NPs in the size range between 7.2–13.2 nm are precipitated with NaOH as a base under a blanket of nitrogen and depicted in Fig. 3d–f. Well-defined cuboidal particles with an average size of 13.2 ± 2.1 nm are obtained by instantaneous injection of NaOH solution at T = 80 °C (sample Cb13, Fig. 3f). Slightly smaller particles with an average size of 10.9 ± 2.4 nm and facetted, cuboidal morphologies were synthesized by decreasing the routinely applied equilibration time at 80 °C from 30 min to 10 min (sample Cb11, Fig. 3e; refer to the discussion for more information, vide infra). Instantaneous injection of NaOH solution at T = 100 °C under a blanket of nitrogen leads to cuboidal NPs with an average size of 7.2 ± 1.6 nm (sample Cb8, Fig. 3d). A direct comparison with NPs depicted in Fig. 2f, which were also precipitated at 100 °C, but under exposure to air, demonstrates that the nitrogen blanket reduces particle size and slightly improves size distribution.
Since NP dispersions generally possess a certain size distribution, care is necessary when evaluating size-dependent properties due to potentially overlapping particle size distributions. The comparison of size distribution histograms obtained for the here described NPs demonstrates that the developed synthetic strategies lead to particles with focused average sizes (Fig. 3g, individual histograms cf. Fig. S3†). For better visibility, the distribution of the particles with an average size of 3.9 nm (sample S4, Fig. 3b) is omitted due to the significant overlap with samples S3 and S5 (exhibiting average sizes of 2.8 and 4.7 nm, respectively). The particles of 4.7, 7.2, and 10.9 nm average size (S5, Cb8, Cb11) possess comparable size distributions of 21–22%. Broader distributions of 31–35% are observed for the smallest particles (S3 and S4), while a narrower distribution of 15% was obtained for the largest sample (Cb13; see Fig. S3†). The histograms further reveal that small amounts of very large and very small particles are formed as a by-product of the synthesis (Fig. 3g) since nucleation and growth are not decoupled in this system. However, those by-products can be readily removed by size-selective precipitation during the purification process, if so desired. This is demonstrated in Fig. S8.† In this context, we would like to emphasize that the bulk of the distributions presented in this work was not artificially narrowed to illustrate the intrinsic potential of the developed synthetic strategies. The amount of OA ligands in the different samples was estimated based on thermogravimetric analysis displayed in Fig. S9.†
The reported average sizes and size distributions were obtained by TEM image analysis, which represents a semi-quantitative method. In order to complement these data, small-angle X-ray scattering (SAXS) was performed directly on NP dispersions as a volume-averaging technique omitting the need for further sample preparation and providing statistically accurate quantitative information on the size and size distribution of scattering objects in the nanometer regime (Fig. S10†).
A summary of the average dimensions of NPs extracted from TEM and SAXS data is provided in Table 1, demonstrating that particle sizes determined by SAXS are in good agreement with results from TEM image analysis.
| Sample | SAXS size [nm] | MinFeret [nm] | Feret [nm] |
|---|---|---|---|
| S3 | 2.0 ± 0.8 | 1.7 ± 0.7 | 2.8 ± 1.0 |
| S4 | 2.4 ± 1.0 | 2.3 ± 0.5 | 3.9 ± 1.2 |
| S5 | 3.2 ± 1.6 | 3.2 ± 0.6 | 4.7 ± 1.0 |
| Cb8 | 6.2 ± 1.1 | 5.7 ± 1.4 | 7.2 ± 1.6 |
| Cb11 | 9.0 ± 2.0 | 8.8 ± 1.9 | 10.9 ± 2.4 |
| Cb13 | 11.0 ± 1.7 | 10.7 ± 1.6 | 13.2 ± 2.1 |
Fig. S11† compares the scattering signals of dispersed Co3O4 NPs with SAXS measurements of the same particles (Cb13) deposited on Kapton foil as a dried film. A pronounced Debye–Scherrer ring observed for the immobilized NPs in the solid state indicates particle interaction and a very well-defined interparticle distance, and thus highlights the potential of the herein-described cuboidal NPs with narrow size distributions for controlled self-assembly into ordered superstructures.
Optical properties of colloidally stabilized Co3O4 nanoparticles
Absorption spectra were recorded in the ultraviolet-visible (UV-vis) and near-infrared (NIR) regions (Fig. 4) to study the composition and electronic structure of the synthesized NPs. Characteristic signals attributable to Co3O4 and OA ligands were identified for cuboidal particles with an average size of 13 nm (Cb13). Specifically, two strong bands centred at 390 nm (3.18 eV) and 677 nm (1.83 eV) represent charge transfer signals of Co3O4 are seen in Fig. 4a (black line).54 The remaining bands detected in the NP dispersion are in good agreement with reference spectra of OA and heptane and thus may be assigned to the ligand and solvent molecules. A more detailed analysis of the ligand signals may be found in the ESI† (Fig. S12 and S13†). Fig. 4b displays absorbance spectra obtained from NPs with diameters between 2.8–10.9 nm (samples S3, S4, S5, C8, and Cb11) at a mass concentration of 0.1 mg mL−1. At very small dimensions, a trend of systematically increasing absorption intensities with particle size is observed reaching a plateau for diameters of 7.2 nm and beyond.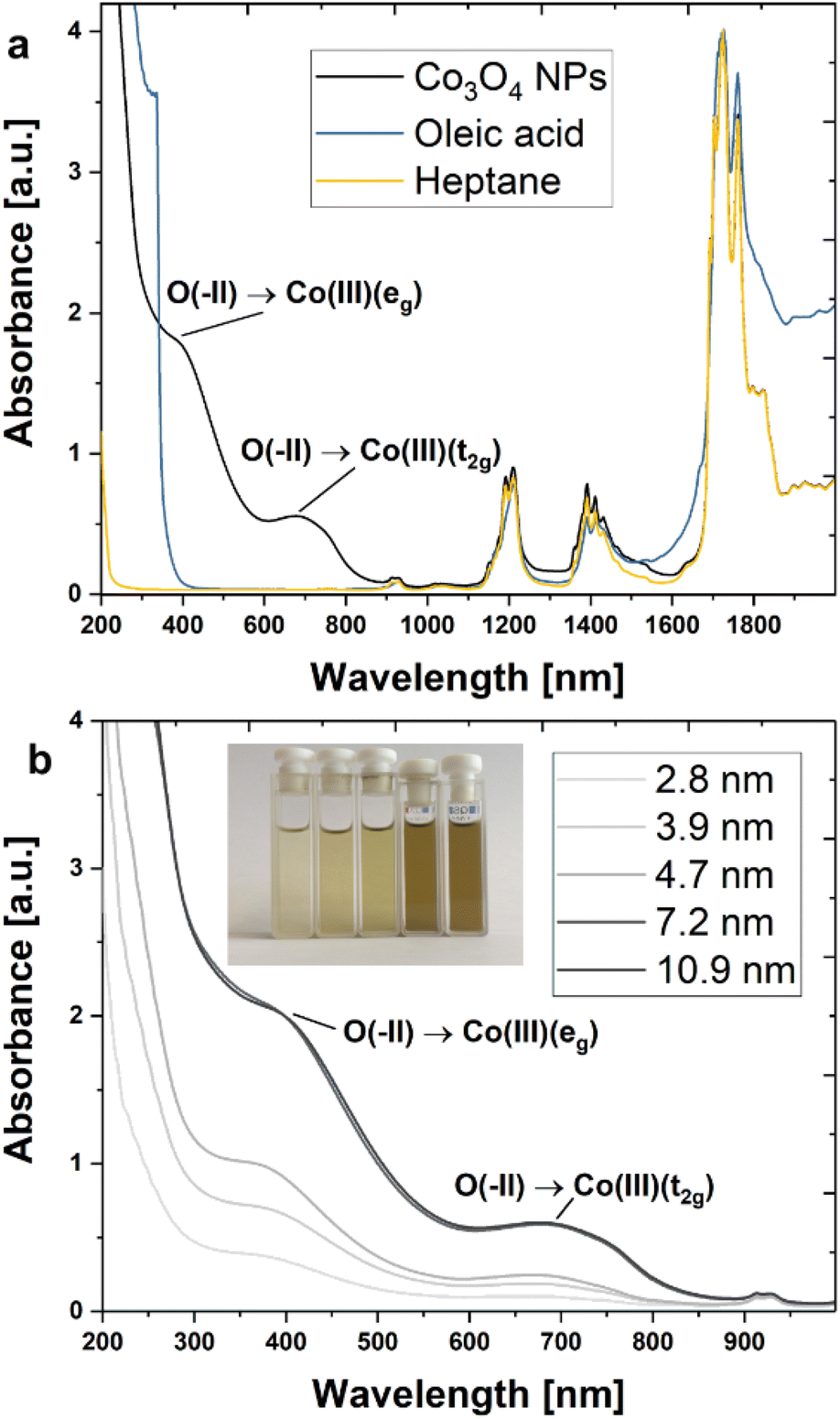 | ||
| Fig. 4 UV-vis/NIR spectra of Co3O4 NPs dispersed in heptane. (a) Absorption spectrum of OA-stabilized cuboidal Co3O4 NPs dispersed in heptane (Cb13) in the spectral range from 200–2000 nm (black line). For comparison, reference spectra of pure OA (blue line), as well as heptane (yellow line), are shown. (b) Series of absorption spectra for NPs with diameters between 2.8 and 10.9 nm showing size-dependent absorbance. Inset: photograph of cuvettes containing NP dispersions (left to right: increasing NP size from 2.8–10.9 nm). Intensification of colouration is observed with increasing NP size. | ||
From an optical point of view, Co3O4 is a complex material. The different oxidation states of Co2+/3+ and the two different coordination environments of the spinel structure (tetrahedral and octahedral) allow for six possible optical transitions in the UV-vis/NIR spectral range including two ligand-to-metal charge transfers (LMCT), two metal-to-metal charge transfers (MMCT), and two crystal field transitions for the tetrahedral and octahedral environments.55 In a semi-empirical approach, four transitions were attributed by Miedzinska et al. to the O2−(2p) → Co2+(t2g) (2.1 eV/590 nm) LMCT, the Co3+(t2g) → Co2+(t2g) (1.3 eV/954 nm) MMCT, the Co2+(t2g) → Co3+(eg) (1.0 eV/1240 nm) MMCT and the Co2+(eg) → Co2+(t2g) (0.8 eV/1550 nm) tetrahedral crystal field transition.54 More recent works suggest that the transition at 1.3 eV should be assigned to the O2−(2p) → Co3+(t2g) LMCT.56–58 Density functional theory (DFT) calculations by Xu et al. which are supported by experiments assign two transitions at 2.9 eV (440 nm) and 3.3 eV (376 nm) to the O2−(2p) → Co3+(eg) LMCT and the Co3+(t2g) → Co2+(t2g) MMCT, respectively.59 This attribution was in part confirmed by Jiang et al. who argue convincingly that the O2−(2p) → Co3+(eg) LMCT (440 nm) is excited by light irradiation at 400 nm. The same report also presents a typical absorbance spectrum for thin films of Co3O4.55
A recent summary of relevant literature and further studies on the optical properties of Co3O4 may be found with Turan et al.60 Nkeng et al. argue that the relative intensities of the peaks at 1333 nm (0.93eV) and 1512 nm (0.82 eV) in spinel-type cobalt oxide thin films are dependent on the layer thickness, reporting a significant decrease in signal intensity when reducing the thickness from 2000 nm to 300 nm.61 This observation may explain the absence of these absorption bands in the spectra of isolated Co3O4 NPs in suspension. Moreover, the authors discuss a size-related modulation of the spectrum in the spectral range between 500–1000 nm as compared to the bulk situation. Hence, we assign the prominent absorption bands detected at 390 and 677 nm to the O2−(2p) → Co3+(eg) LMCT and the O2−(2p) → Co3+(t2g) LMCT, respectively.
With decreasing particle size, the absorption bands undergo a conspicuous blue shift. This effect is confirmed by comparison with relevant literature-reported systems as summarized in Table 2 (for a detailed discussion of the size-dependent blue shift cf. Section 2.12, ESI†).
| Size [nm] | O2−(2p) → Co3+(eg) [nm] | O2−(2p) → Co3+(t2g) [nm] | Morphology | Source |
|---|---|---|---|---|
| 31.3 | ∼500 | ∼775 | Irregular | Makhlouf et al.58 |
| 22 | 441 | 756 | Thin film | Jiang et al.55 |
| 15.8 | ∼425 | ∼725 | Irregular | Makhlouf et al.58 |
| 10.9 | 390 | 677 | Cuboid NPs | This work |
| 4.7 | 376 | 669 | Spherical NPs | This work |
| 2.8 | 369 | 664 | Spherical NPs | This work |
Phase identification and crystallographic structure of NPs and precursors
The synthetic strategy presented in this work utilizes a co-precipitation approach via hydroxide-based precursor NPs, which in terms of composition comprise a mixture of α-Co(OH)2 and the oxidation product CoO(OH) as evidenced by selected-area electron diffraction (SAED) (Fig. S4a†). Pseudomorphic transformation of these precursor NPs into Co3O4 (Fig. 5 and S4b†) is readily achieved at elevated temperatures, such that particle size and shape are largely preserved (Fig. S4c–f†).After transformation, cuboidal NPs of sample Cb8 show two distinct sets of lattice fringes exhibiting a spacing of 2.82 Å (q = 3.54 1/nm) and 2.04 Å (q = 4.90 1/nm) (Fig. 5a), corresponding to characteristic distances between {022}- and {004}-type lattice planes of the spinel cobalt(II,III) oxide structure (Fig. 5b, data assigned according to COD reference 9005896). The observed well-defined lattice fringes in combination with the enclosed angle of 45° are consistent with a single crystalline Co3O4-cube. Fast Fourier Transformation (FFT) further confirms a highly periodic, well-ordered cubic lattice (Fig. 5c). SAED diffractograms (Fig. 5b) and corresponding 2D diffraction patterns (Fig. 5d–f) reveal a general agreement of the crystallographic structure of NPs in the size range between 4.7 and 13.2 nm with the spinel-Co3O4 lattice. However, the intensity of the signal decreases substantially with decreasing particle size, leading to weak and broad diffraction peaks for the very small NPs (samples S5 (green line) and Cb8 (yellow line)). Only the diffractogram of the largest cuboidal NPs (Cb13, blue line) can be reliably indexed. However, based on the general superposition of the peak maxima in combination with the results of UV-vis characterization (Fig. 4b) and XRD (Fig. S2†), it can be assumed that both the small spherical as well as the larger cuboidal NPs synthesized in this work can be identified as spinel-type Co3O4. An EELS measurement of sample Cb13 further confirms the formation of spinel Co3O4 (see Fig. S14†).
 | ||
| Fig. 5 Crystallographic characterization of Co3O4 NPs. (a) HR-TEM micrograph of an individual 7.2 nm-sized Co3O4 nanocube (Cb8). Two sets of lattice fringes with distances attributable to {022} and {004} lattice planes in the spinel-Co3O4 structure are highlighted in orange and yellow, respectively. (b) Electron diffractograms of samples S5 (green trace), Cb8 (yellow trace), and Cb13 (blue trace) obtained from SAED measurements. The peak profile of bulk Co3O4 is shown for comparison (red line). (c) Fast Fourier transformation corresponding to (a). (d–f) SAED patterns of NPs with 4.7 nm (S5), 7.2 nm (Cb8), and 13.2 nm (Cb13) size, respectively. The displayed data correspond to NPs in Fig. 3. | ||
Electrochemical characterization of Co3O4 nanoparticles and their properties in electrocatalytic water-splitting
In view of potential applications in clean energy conversion and storage systems, the electrocatalytic performance of nanoparticulate Co3O4 materials towards the OER (4 OH− → 2 H2O + O2 + 4 e−) during water electrolysis was investigated by cyclic voltammetry (CV). However, as the catalytic efficiency of NPs surface-passivated by OA ligands is limited, thus complicating electrochemical analysis, non-stabilized Co3O4 is chosen as a probe for initial experiments addressing the inherent properties of isolated, well-defined NPs (Fig. S15†) in comparison to a commercial powder of randomly agglomerated Co3O4 grains with irregular size and shape (Sigma Aldrich, Fig. S16†). For that purpose, a sample of cuboidal NPs (Cb13-2) was synthesized according to an analogous protocol as described for Cb13, but instead of stabilization with OA, the particles were first washed with OLA and EtOH to remove impurities post-synthetically followed by further washing in OLA. After several sonication and centrifugation cycles in pure EtOH, the particles were calcined at 300 °C for 2 h prior to immobilization on a rotating disc electrode (RDE) for electrochemical characterization. While the synthetic ligand-free NPs appear uniform in TEM analysis with average diameters of 11.3 ± 2.3 nm (Fig. S15†), the commercial nanopowder exhibits primary grains in the size range between 16–105 nm, partially agglomerated into micron-scale aggregates (Fig. S16†). The average size of Cb13-2 is slightly lower than that of Cb13. Possible reasons for this are the altered solubility due to the absence of OA as well as the purification steps intended to remove OLA residues from the particle surface.Representative cyclic voltammograms are presented in Fig. 6. Both, the synthetic and the commercial sample were cycled 10 times at a scan rate of 50 mV s−1 and showed characteristic redox couples around 1.2 and 1.55 V corresponding to electrochemically stimulated Co2+ → Co3+ and Co3+ → Co4+ transitions within the Co(II,III)-oxide catalyst material prior to the onset of the OER (E0 = 1.229 V vs. the Reversible Hydrogen Electrode, RHE).62
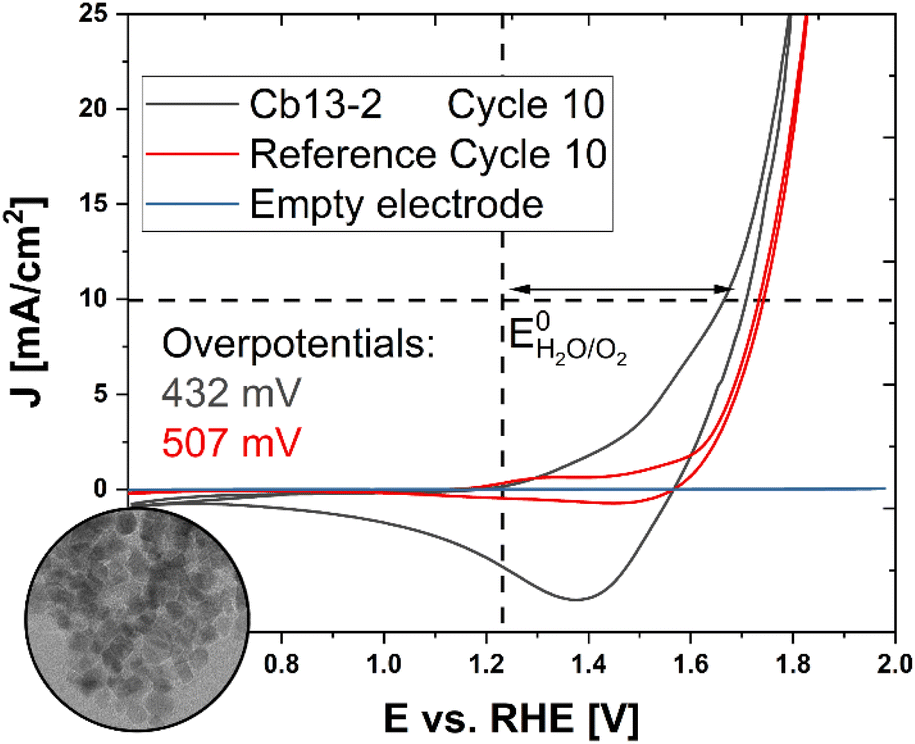 | ||
| Fig. 6 Electrochemical characterization and OER performance of ligand-free cuboidal NPs. Inset: TEM micrograph of non-stabilized NPs (Cb13-2). The complete TEM micrograph is displayed in Fig. S15.† Cyclic voltammograms of synthetic NPs (black curve) in comparison to a commercial Co3O4 nanopowder (red curve). Both samples were cycled 10 times at a scan rate of 50 mV s−1. The blue trace shows the electrochemical response of a bare glassy carbon RDE. The vertical dashed line indicates the position of EH2O/O20 and the horizontal dashed line marks the current density of J = 10 mA cm−2 at which the overpotentials ηi = 10 mA cm−2 were determined. | ||
The overpotential ηi = 10 mA cm−2 required for the OER, which represents a kinetic barrier towards the reaction and is widely regarded as a figure of merit for the activity of an electrocatalyst, was then determined for both specimens.63
While the reference exhibits an overpotential of ηi = 10 mA cm−2 = 507 mV, the ligand-free synthetic Cb13-2 NPs adopt a lower value of ηi = 10 mA cm−2 = 432 mV (cf. Fig. S17†) corresponding to an improved performance comparable with porous microscale tubular and spherulitic Co3O4 materials with a nanoparticle-based substructure.32,64
As the overpotential of an electrocatalyst depends on mass transport limitations and the binding affinity of reaction intermediates to the surface, structural parameters such as the availability and accessibility of active sites for chemisorption play a crucial role in the performance of the material, thus highlighting the importance of size- and shape-control in catalyst design.65,66 Accordingly, Esswein et al. have demonstrated that the catalytic efficiency of Co3O4 particles on Ni-foam increased slightly at smaller sizes (5.9 nm-534 mV, 21.1 nm-569 mV).67
As Cb13-2 exhibits a significantly lower overpotential than the reference under consistent conditions, we conclude that our well-defined NPs with tunable size demonstrate considerable potential for industrial application as a water oxidation catalyst. Therefore, the optimization of thorough post-synthetic removal of OA from the particle surface under preservation of the nanocrystal morphology and the systematic study of size-dependent electrochemical properties in the low nanometer regime are subject to further research.
Discussion of the influence of the reaction temperature on NP size and morphology
As elaborated in previous sections of the manuscript, our synthetic protocol enables us to systematically vary the size and associated structural or functional properties of Co3O4 NPs within one reaction system.Considering nanoparticle size as a function of reaction temperature, two key dependencies have to be monitored: firstly, salt solubility increases with temperature, thus providing a constant and stable source of solubilized reagents promoting nucleation and growth. Secondly, the solubility Cr of the nanoparticles themselves decreases with increasing temperature T as described by the Gibbs–Thomson equation (eqn (4)), which expresses the solubility of NPs (Cr) as a function of the bulk concentration of the solution (Cb), the surface energy (γ), the molar volume of the bulk crystal (ν), the particle radius (r) and the Boltzmann constant (kB).49
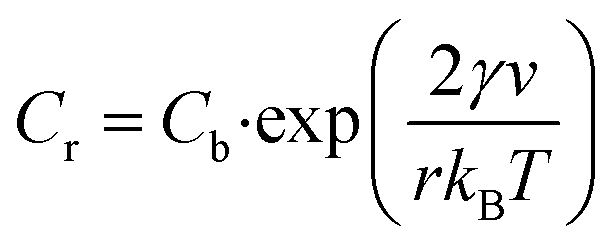 | (4) |
It is readily apparent from eqn (4) that the exponential term and thus the particle solubility decreases with increasing temperature and particle radius. For this reason, a relative decrease in the particle size is expected at increasing temperatures when the bulk concentration is kept constant due to inhibition of the growth reaction. However, the experimental observation of such a relationship may be restricted to a limited range of conditions, as the interplay between temperature and particle diameter is in general more complex than stated in eqn (4).
In our precipitation system, a trend of systematically decreasing NP diameters, which can be described by the Gibbs–Thomson equation, is observed in the temperature range between 70–90 °C (Fig. 2). Specifically, in this regime, the size decreases by 4% on average for every increase in temperature by 10 °C, while a rough estimation based on the Gibbs–Thomson equation yields that the particle solubility should decrease by approximately 3% in the same temperature interval. Even though solubility does not directly translate into size, the values are sufficiently close to concluding that the behaviour of the system can be at least partially explained by the Gibbs–Thomson effect.
This observation regarding the interplay between precursor solubility and particle size has inspired a modification to the reaction conditions of sample Cb13 (with the largest cuboidal nanocrystals), such that the stirring/dissolution time before the injection of NaOH was reduced from 30 min to 10 min. Indeed, the particle size in the resulting sample Cb11 reproducibly (based on 5 repeat experiments) decreases moderately from ca. 13 nm to ca. 11 nm presumably due to slower nucleation and growth kinetics at the lower effective precursor concentration. This observation again highlights the precursor solubility as an important parameter influencing the particle size. We speculate that the thorough dissolution and coordination of the Co(NO3)2·6 H2O in OLA requires a certain amount of time. From the decrease in size, we can infer that this process takes between 15–30 min at 80 °C, while a longer equilibration time does not influence the result.
Considering nanocrystal shape, two competing contributions are observed, kinetic and thermodynamic. The phase stability of NPs may be described in terms of the Gibbs free energy G, where the total Gibbs free energy G0total of a NP comprises contributions of bulk, surface, edges, and corners of the crystal (eqn (5)).68
| G0total = G0bulk + G0surface + G0edge + G0corner | (5) |
The surface energy is largely independent of particle size, but for extremely small particles of around 5 nm it is noticeably lowered compared to the bulk situation.69 Concomitantly, the contributions of edges and corners to the total free energy rapidly decrease as the particles increase in size,70 thus multi-faceted structures with shallower angles between the facets may be favoured in case of exceedingly small particles to reduce internal strain. Consequently, particles tend to form truncated shapes at very small sizes of ca. 5 nm which may converge towards an overall spherical appearance. For increasing particle sizes (>5 nm), internal strain accumulates and makes particles with fewer surface facets more favourable. As a result, the particle morphology develops towards the equilibrium shape dictated by the crystallographic unit cell,71–73i.e. a cube in the case of spinel type Co3O4. These considerations may explain the size-dependent transition from spherical to cuboidal/cubic appearance observed in this work (Fig. 3 and S10†).
Conclusions
In summary, we have introduced a facile, robust, and inexpensive liquid-phase synthetic protocol yielding spherical and cuboidal spinel-type Co3O4 NPs with narrow size distributions. In this approach, very small metal oxide particles in the size range between ca. 3–13 nm are formed by the in situ thermal decomposition of a hydroxide-based precursor precipitated under comparably mild reaction conditions, thus omitting the need for solvothermal procedures or environments with precisely controlled oxygen pressures. Remarkably, the decomposition temperature of 180 °C lies significantly lower than reported for many standard liquid phase nanoparticle syntheses (T > 300 °C).74,75The well-stabilized Co3O4 particles have demonstrated prolonged shelf-life in suspension such that agglomeration was prevented for more than 10 months, thus facilitating storage and transport under ambient conditions. Particle size and morphology can almost effortlessly be adjusted by thermal control between 50–100 °C in air. The NPs can be further customized by changing the employed base, the mode of base addition, the equilibration time, and the gas atmosphere (N2 in this work). All of the mentioned parameters affect the size and shape of the deposited NPs without a need to modify the concentration or solvent ratios. Specifically, very small homogeneous Co3O4 particles with spherical shapes and diameters between ca. 3–5 nm can be obtained at a reaction temperature of 50–60 °C. Higher temperatures (T = 70–100 °C), in contrast, promote the formation of cuboidal particles in the size range between ca. 7–13 nm.
In view of industrial applications, the obtained nanoparticles exhibit a variety of promising characteristics and properties. A comparably strong absorption signal spanning the near UV and the entire visible range is detectable even at low concentrations (ca. 0.1 mg mL−1), which renders the Co3O4 NPs an auspicious candidate for applications as a light-absorber material, e.g. in photocatalysis.76,77 Specifically, in the low nanometer regime, the absorbance appears to be centred around two strong charge-transfer bands, which are attributed to the O2−(2p) → Co3+(eg) LMCT and the O2−(2p) → Co3+(t2g) LMCT. With decreasing particle diameters, the intensities of these two signals are reduced. This effect is accompanied by a small, but systematic blue shift in the low nanometer regime (3–11 nm), which may enable the custom-designed adjustment of the absorption properties. Initial cyclic voltammetry experiments addressing the electrocatalytic performance of immobilized nanoparticles of 11 nm diameter revealed a reduction of the overpotential towards the OER reaction by 75 mV (432 mV) in comparison to a commercial Co3O4 nanopowder with irregularly shaped and partially agglomerated grains (507 mV). Electrochemical characterization thus highlights the potential of the herein-developed particles as an active catalyst for water electrolysis.
Considering their particularly small diameters and narrow size distribution, the here-described Co3O4 NPs build an excellent platform for dedicated future studies on size-dependent properties in the regime of dimensions <10 nm (e.g. with respect to electrocatalytic performance). The preparation of uniform metal oxide NPs further opens exciting perspectives for the controlled self-assembly of colloidal superstructures, potentially exhibiting emerging functional properties originating from cooperative effects between the individual units.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) via Collaborative Research Centre SFB 840 (A.S.S. TP B09). A.S.S. further thanks the Bavarian Academy of Sciences and Humanities (BAdW) for financial support through a Young Academy Fellowship. We acknowledge the Bavarian Polymer Institute (BPI) for providing access to small-angle X-ray scattering and electron microscopy facilities within the Keylabs “Mesoscale Characterization: Scattering Techniques” and “Electron and Optical Microscopy”, respectively. The authors are grateful to Markus Drechsler (University of Bayreuth) for assistance in TEM imaging and Benedikt Wirth for TGA sample preparation. This work benefited from the use of the Sasfit application (version 0.94.1).References
- N. D. Donahue, H. Acar and S. Wilhelm, Adv. Drug Deliv. Rev., 2019, 143, 68–96 CrossRef CAS PubMed.
- Y. Zhao, X. Li, B. Yan, D. Xiong, D. Li, S. Lawes and X. Sun, Adv. Energy Mater., 2016, 6, 1502175 CrossRef.
- Y. Wang, H. Arandiyan, J. Scott, A. Bagheri, H. Dai and R. Amal, J. Mater. Chem. A, 2017, 5, 8825–8846 RSC.
- X. Xie and W. Shen, Nanoscale, 2009, 1, 50 RSC.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- Y. Zhou, S. Sun, C. Wei, Y. Sun, P. Xi, Z. Feng and Z. J. Xu, Adv. Mater., 2019, 31, 1902509 CrossRef CAS PubMed.
- F. Fischer and H. Tropsch, Berichte der Dtsch. Chem. Gesellschaft A B Ser., 1926, 59, 830–831 Search PubMed.
- N. E. Tsakoumis, E. Patanou, S. Lögdberg, R. E. Johnsen, R. Myrstad, W. van Beek, E. Rytter and E. A. Blekkan, ACS Catal, 2019, 9, 511–520 CrossRef CAS.
- R. Jain, E. S. Gnanakumar and C. S. Gopinath, ACS Omega, 2017, 2, 828–834 CrossRef CAS PubMed.
- M. Grzelczak, J. Zhang, J. Pfrommer, J. Hartmann, M. Driess, M. Antonietti and X. Wang, ACS Catal, 2013, 3, 383–388 CrossRef CAS.
- Y. Wang, H. Xia, L. Lu and J. Lin, ACS Nano, 2010, 4, 1425–1432 CrossRef CAS PubMed.
- L. Tian, H. Zou, J. Fu, X. Yang, Y. Wang, H. Guo, X. Fu, C. Liang, M. Wu, P. K. Shen and Q. Gao, Adv. Funct. Mater., 2010, 20, 617–623 CrossRef CAS.
- J. Wang, R. Gao, D. Zhou, Z. Chen, Z. Wu, G. Schumacher, Z. Hu and X. Liu, ACS Catal, 2017, 7, 6533–6541 CrossRef CAS.
- A. Agiral, H. Sen Soo and H. Frei, Chem. Mater., 2013, 25, 2264–2273 CrossRef CAS.
- K. Tsukiyama, M. Takasaki, Y. Oaki and H. Imai, Langmuir, 2019, 35, 8025–8030 CrossRef CAS PubMed.
- Z. Liu, H. M. A. Amin, Y. Peng, M. Corva, R. Pentcheva and K. Tschulik, Adv. Funct. Mater., 2023, 33, 2210945 CrossRef CAS.
- V. Iablokov, S. K. Beaumont, S. Alayoglu, V. V. Pushkarev, C. Specht, J. Gao, A. P. Alivisatos, N. Kruse and G. A. Somorjai, Nano Lett, 2012, 12, 3091–3096 CrossRef CAS PubMed.
- V. Iablokov, R. Barbosa, G. Pollefeyt, I. Van Driessche, S. Chenakin and N. Kruse, ACS Catal, 2015, 5, 5714–5718 CrossRef CAS.
- S. Saddeler, U. Hagemann and S. Schulz, Inorg. Chem., 2020, 59, 10013–10024 CrossRef CAS PubMed.
- F. T. Haase, A. Bergmann, T. E. Jones, J. Timoshenko, A. Herzog, H. S. Jeon, C. Rettenmaier and B. R. Cuenya, Nat. Energy, 2022, 78(7), 765–773 CrossRef.
- M. A. Boles, M. Engel and D. V Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed.
- K. Sawano, K. Tsukiyama, M. Shimizu, M. Takasaki, Y. Oaki, T. Yamamoto, Y. Einaga, C. Jenewein, H. Cölfen, H. Kaiju, T. Sato and H. Imai, Nanoscale, 2020, 12, 7792–7796 RSC.
- M. Ghosh, E. V. Sampathkumaran and C. N. R. Rao, Chem. Mater., 2005, 17, 2348–2352 CrossRef CAS.
- K. An, N. Lee, J. Park, S. C. Kim, Y. Hwang, J.-G. Park, J.-Y. Kim, J.-H. Park, M. J. Han, J. Yu and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 9753–9760 CrossRef CAS PubMed.
- M. Sarif, J. Hilgert, I. Khan, R. A. Harris, S. Plana-Ruiz, M. Ashraf, E. Pütz, J. Schemberg, M. Panthöfer, U. Kolb, M. Nawaz Tahir and W. Tremel, Langmuir, 2020, 36, 13804–13816 CrossRef CAS PubMed.
- N. H. Chou, P. N. Ross, A. T. Bell and T. D. Tilley, ChemSusChem, 2011, 4, 1566–1569 CrossRef CAS PubMed.
- Z. P. Xu and H. C. Zeng, J. Mater. Chem., 1998, 8, 2499–2506 RSC.
- C. Nethravathi, S. Sen, N. Ravishankar, M. Rajamathi, C. Pietzonka and B. Harbrecht, J. Phys. Chem. B, 2005, 109, 11468–11472 CrossRef CAS PubMed.
- Z. Liu, R. Ma, M. Osada, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2005, 127, 13869–13874 CrossRef CAS PubMed.
- R. Ma, Z. Liu, K. Takada, K. Fukuda, Y. Ebina, Y. Bando and T. Sasaki, Inorg. Chem., 2006, 45, 3964–3969 CrossRef CAS PubMed.
- L. Hu, Q. Peng and Y. Li, J. Am. Chem. Soc., 2008, 130, 16136–16137 CrossRef CAS PubMed.
- A. S. Schenk, M. Goll, L. Reith, M. Roussel, B. Blaschkowski, S. Rosenfeldt, X. Yin, W. W. Schmahl and S. Ludwigs, Cryst. Growth Des., 2020, 20, 6407–6420 CrossRef CAS.
- L. Liu, Z. Jiang, L. Fang, H. Xu, H. Zhang, X. Gu and Y. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 27736–27744 CrossRef CAS PubMed.
- W. Li, Y. Wang, X. Y. Cui, S. Yu, Y. Li, Y. Hu, M. Zhu, R. Zheng and S. P. Ringer, ACS Appl. Mater. Interfaces, 2018, 10, 19235–19247 CrossRef CAS PubMed.
- S. Mourdikoudis and L. M. Liz-Marzán, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS.
- J.-W. Song, E.-Y. Jeon, D.-H. Song, H.-Y. Jang, U. T. Bornscheuer, D.-K. Oh and J.-B. Park, Angew. Chemie Int. Ed., 2013, 52, 2534–2537 CrossRef CAS PubMed.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141–153 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- C. T. Rueden, J. Schindelin, M. C. Hiner, B. E. DeZonia, A. E. Walter, E. T. Arena and K. W. Eliceiri, BMC Bioinformatics, 2017, 18, 529 CrossRef PubMed.
- D. R. G. Mitchell, Microsc. Res. Tech., 2008, 71, 588–593 CrossRef CAS PubMed.
- M. Klinger, J. Appl. Crystallogr., 2017, 50, 1226–1234 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- S. Förster, S. Fischer, K. Zielske, C. Schellbach, M. Sztucki, P. Lindner and J. Perlich, Adv. Colloid Interface Sci., 2011, 163, 53–83 CrossRef PubMed.
- J. Kohlbrecher and A. Studer, J. Appl. Crystallogr., 2017, 50, 1395–1403 CrossRef CAS.
- S. J. Folkman, M. Zhou, M. Nicki and R. G. Finke, Inorg. Chem., 2018, 57, 1517–1526 CrossRef CAS PubMed.
- Z. P. Xu and H. C. Zeng, Chem. Mater., 1999, 11, 67–74 CrossRef CAS.
- H. T. Phan and A. J. Haes, J. Phys. Chem. C, 2019, 123, 16495–16507 CrossRef CAS PubMed.
- M. R. Buck, A. J. Biacchi and R. E. Schaak, Chem. Mater., 2014, 26, 1492–1499 CrossRef CAS.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- W. Lv, W. He, X. Wang, Y. Niu, H. Cao, J. H. Dickerson and Z. Wang, Nanoscale, 2014, 6, 2531–2547 RSC.
- P. Batys, Y. Zhang, J. L. Lutkenhaus and M. Sammalkorpi, Macromolecules, 2018, 51, 8268–8277 CrossRef CAS PubMed.
- K. Karalis, D. Zahn, N. I. Prasianakis, B. Niceno and S. V. Churakov, Sci. Rep., 2021, 11, 19858 CrossRef CAS PubMed.
- Y. Du, K. M. Ok and D. O'Hare, J. Mater. Chem., 2008, 18, 4450 RSC.
- K. M. E. Miedzinska, B. R. Hollebone and J. G. Cook, J. Phys. Chem. Solids, 1987, 48, 649–656 CrossRef CAS.
- C.-M. Jiang, L. R. Baker, J. M. Lucas, J. Vura-Weis, A. P. Alivisatos and S. R. Leone, J. Phys. Chem. C, 2014, 118, 22774–22784 CrossRef CAS.
- C. Cheng, M. Serizawa, H. Sakata and T. Hirayama, Mater. Chem. Phys., 1998, 53, 225–230 CrossRef CAS.
- D. Barreca, C. Massignan, S. Daolio, M. Fabrizio, C. Piccirillo, L. Armelao and E. Tondello, Chem. Mater., 2001, 13, 588–593 CrossRef CAS.
- S. A. Makhlouf, Z. H. Bakr, K. I. Aly and M. S. Moustafa, Superlattices Microstruct, 2013, 64, 107–117 CrossRef CAS.
- X.-L. Xu, Z.-H. Chen, Y. Li, W.-K. Chen and J.-Q. Li, Surf. Sci., 2009, 603, 653–658 CrossRef CAS.
- E. Turan, E. Zeybekoğlu and M. Kul, Thin Solid Films, 2019, 692, 137632 CrossRef CAS.
- P. Nkeng, J. Electrochem. Soc., 1995, 142, 1777 CrossRef CAS.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- A. S. Schenk, S. Eiben, M. Goll, L. Reith, A. N. Kulak, F. C. Meldrum, H. Jeske, C. Wege and S. Ludwigs, Nanoscale, 2017, 9, 6334–6345 RSC.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- I. C. Man, H. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- A. J. Esswein, M. J. McMurdo, P. N. Ross, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2009, 113, 15068–15072 CrossRef CAS.
- A. S. Barnard and P. Zapol, J. Chem. Phys., 2004, 121, 4276–4283 CrossRef CAS PubMed.
- D. Vollath, F. D. Fischer and D. Holec, Beilstein J. Nanotechnol., 2018, 9, 2265–2276 CrossRef CAS PubMed.
- N. Moll, M. Scheffler and E. Pehlke, Phys. Rev. B, 1998, 58, 4566–4571 CrossRef CAS.
- C. L. Cleveland and U. Landman, J. Chem. Phys., 1991, 94, 7376–7396 CrossRef CAS.
- M. J. Yacamán, J. A. Ascencio, H. B. Liu and J. Gardea-Torresdey, J. Vac. Sci. Technol. B Microelectron. Nanom. Struct., 2001, 19, 1091 CrossRef.
- J. M. Rahm and P. Erhart, Nano Lett, 2017, 17, 5775–5781 CrossRef CAS PubMed.
- S. Mehdizadeh Taheri, M. Michaelis, T. Friedrich, B. Förster, M. Drechsler, F. M. Römer, P. Bösecke, T. Narayanan, B. Weber, I. Rehberg, S. Rosenfeldt and S. Förster, Proc. Natl. Acad. Sci., 2015, 112, 14484–14489 CrossRef PubMed.
- C. De Mello Donegá, P. Liljeroth and D. Vanmaekelbergh, Small, 2005, 1, 1152–1162 CrossRef PubMed.
- S. Koroidov, M. F. Anderlund, S. Styring, A. Thapper and J. Messinger, Energy Environ. Sci., 2015, 8, 2492–2503 RSC.
- G. Dong, H. Hu, X. Huang, Y. Zhang and Y. Bi, J. Mater. Chem. A, 2018, 6, 21003–21009 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis protocols, images of the reaction, particle size histograms, pseudomorphic transformation of the precursor into Co3O4, thermogravimetric analysis, size-selective precipitation, small-angle X-ray scattering (dispersions and assembly), absorbance spectra and band attribution, size-dependent light absorbance, electron energy loss spectra, TEM micrographs of ligand-free particles and reference nanopowder. See DOI: https://doi.org/10.1039/d3na00032j |
| This journal is © The Royal Society of Chemistry 2023 |