
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron oxide nanoparticles trigger endoplasmic reticulum damage in steatotic hepatic cells†
Mariia
Uzhytchak‡
a,
Mariia
Lunova‡
ab,
Barbora
Smolková
a,
Milan
Jirsa
b,
Alexandr
Dejneka
*a and
Oleg
Lunov
 *a
*a
aDepartment of Optical and Biophysical Systems, Institute of Physics of the Czech Academy of Sciences, Prague, 18221, Czech Republic. E-mail: dejneka@fzu.cz; lunov@fzu.cz
bInstitute for Clinical & Experimental Medicine (IKEM), Prague, 14021, Czech Republic
First published on 18th July 2023
Abstract
Iron oxide nanoparticles (IONPs) are being actively researched in various biomedical applications, particularly as magnetic resonance imaging (MRI) contrast agents for diagnosing various liver pathologies like nonalcoholic fatty liver diseases, nonalcoholic steatohepatitis, and cirrhosis. Emerging evidence suggests that IONPs may exacerbate hepatic steatosis and liver injury in susceptible livers such as those with nonalcoholic fatty liver disease. However, our understanding of how IONPs may affect steatotic cells at the sub-cellular level is still fragmented. Generally, there is a lack of studies identifying the molecular mechanisms of potential toxic and/or adverse effects of IONPs on “non-heathy” in vitro models. In this study, we demonstrate that IONPs, at a dose that does not cause general toxicity in hepatic cells (Alexander and HepG2), induce significant toxicity in steatotic cells (cells loaded with non-toxic doses of palmitic acid). Mechanistically, co-treatment with PA and IONPs resulted in endoplasmic reticulum (ER) stress, accompanied by the release of cathepsin B from lysosomes to the cytosol. The release of cathepsin B, along with ER stress, led to the activation of apoptotic cell death. Our results suggest that it is necessary to consider the interaction between IONPs and the liver, especially in susceptible livers. This study provides important basic knowledge for the future optimization of IONPs as MRI contrast agents for various biomedical applications.
Introduction
Iron oxide nanoparticles (IONPs) are among the first nanoparticle formulations to gain approval for clinical use.1–8 Despite decades of research, IONPs still remain an attractive nanomaterial composite for many biomedical applications, exhibiting antibacterial effects,9 contributing to delivery of therapeutics to disease sites,10 and inhibiting tumor growth.11 The unique magnetic properties of IONPs make them very promising nanoparticle formulations for advanced clinical diagnostic and theranostic applications.8,12–14 Initially, a number of IONP-based imaging contrast agents received approval from the US Food and Drug Administration (FDA) for magnetic resonance imaging (MRI),3,4 particularly for the detection of liver lesions.15 However, later research revealed the poor clinical performance and underestimated toxic effects of IONP-based MRI contrast agents, leading to their withdrawal from clinical use.3,4,16–18 Currently, only one formulation of IONPs, Feraheme or ferumoxytol, is in clinical application for the treatment of adult iron deficiency anemia.18–22 In fact, the FDA issued a black box warning for Feraheme due to the detection of fatal allergic (anaphylactic) reactions in patients.23,24 Some research suggests that ferumoxytol-based compounds might be potentially used as MRI contrast agents.25–28 Furthermore, there is intensive research on using IONP-based contrast agents to enhance MRI diagnostics of chronic liver diseases including non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), and cirrhosis (for a review see ref. 29 and references therein). Indeed, liver iron overload poses serious health concerns for patients with these pathologies.30–32The challenge associated with iron overload caused by IONPs requires development of improved and safer IONP-based MRI contrast agents. Additionally, we need a comprehensive understanding of toxicological mechanisms triggered by IONPs at cellular and sub-cellular levels. The liver serves as the central hub responsible for detoxifying and metabolizing drugs, as well as eliminating exogenous substances and waste products from the body.33–36 A majority of administered targeted nanoparticles (30 to 99% of injected dose) are sequestered and metabolized by the liver.18,37,38 Indeed, adverse effects associated with drug-induced liver injury (DILI) remain a major challenge in drug development.39,40 However, long-term safety studies and studies specifically monitoring potential drug-induced liver injury caused by IONP-based MRI contrast agents are underrepresented.18,41,42 Moreover, the sub-cellular targets and molecular foundations of IONP-driven adverse effects at the cellular level are still not fully understood.18,43–49 Studies that reveal the adverse effects of IONPs primarily rely on “healthy” models,18,43–49 which do not accurately represent the cellular responses triggered by IONPs under pathological conditions. Additionally, the use of IONPs as MRI contrast agents poses a risk of iron overload, especially at high doses, particularly in patients with chronic liver diseases.29 Consequently, the potential adverse effects of IONPs under NAFLD and NASH conditions represent a great safety concern. In fact, a recent study demonstrated that IONPs exacerbate hepatic steatosis and liver injury in NAFLD.50 However, sub-cellular mechanisms and targets of such effects at the cellular level remain not fully understood.
Taking into consideration the aforementioned points, in this study we investigated the in vitro effects of IONPs on a hepatic cell model under steatotic conditions. A handful of data is available regarding how hepatic cells respond to IONPs under steatotic conditions.50 The specific sub-cellular targets of these interactions have not been extensively investigated. Hence, this study aims to explore the factors that contribute to lipid accumulation in hepatic cells and the potential adverse effects induced by IONPs.
Experimental
Chemicals and antibodies
We have complied a detailed summary of the chemicals, fluorescent probes, assays, and antibodies utilized in this study. The information, including manufacturers, catalogue numbers, and dilutions, can be found in Tables S1–S3 of the ESI.†Cell line culturing
We used the human hepatoblastoma HepG2 cell line (American Type Culture Collection, ATCC, Manassas, VA, USA) and the human hepatocellular carcinoma cell lines Alexander (PLC/PRF/5, ATCC, Manassas, VA, USA) in this study. The cell cultures were cultivated in Minimum Essential Medium Eagle (BioConcept Ltd., Switzerland), supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific, US), 1% L-glutamine 100×, 200 mM (Serana Europe GmbH, Germany) and 1% penicillin/streptomycin (Thermo Fisher Scientific, US). The cell cultures were kept in a humidified 5% CO2 atmosphere at 37 °C. The culture medium (EMEM) was changed once per week. Cells were regularly checked for common culture contamination, such as Mycoplasma, using MycoAlert Detection Assay (Lonza, Switzerland). The cell lines were authenticated by short tandem repeat (STR) DNA profiling (ATCC, Manassas, VA, USA).Nanomaterial
In this study, we utilized, as a relevant model of MRI contrast agents, core–shell IONPs (fluidMAG-CMX) purchased from Chemicell (Chemicell GmbH, Berlin, Germany). These nanoparticles are composed of a magnetite core coated with a carboxymethyldextran shell. The hydrodynamic diameter of the nanoparticles falls within the range of 200 ± 20 nm (mean ± SD). We and others have thoroughly characterized those particles previously.51–55 In the current study, we used these nanoparticles as a model for an IONP-based MRI contrast agent to investigate their interactions with hepatic cells.Cell viability analysis
The viability of the cells was assessed using alamarBlue viability assay (Thermo Fisher Scientific, Waltham, MA, USA). The principle of the alamarBlue viability assay is based on enzymatic cleavage of the resazurin compound to resorufin by viable cells, which leads to an increase in fluorescence intensity. This fluorescence can be measured and quantified, reflecting the percentage of metabolically active cells in the culture. We performed the alamarBlue viability assay according to the manufacturer's guidelines and our verified treatment protocol.5,55 In short, the cells were grown in 96-well plates at a density of 5000 cells per well and then stimulated with different concentrations of oleic or palmitic acid for 24 or 48 hours. After the treatment, the alamarBlue reagent was added to each sample and incubated for 2 h at 37 °C. The fluorescence intensity was measured using a TECAN microplate reader SpectraFluor Plus (TECAN, Mannedorf, Switzerland) with excitation between 530 and 560 nm and emission at 590 nm. Readings were taken in quadruplicate, and three independent experiments were performed for each measurement. For co-stimulation with either oleic or palmitic acid and IONPs, the scheme presented in Fig. 1b was used. Cells were first treated with either oleic or palmitic acid and then, 24 h after, IONPs were added for an additional 24 h. After the treatment, the alamarBlue assay was performed as described above.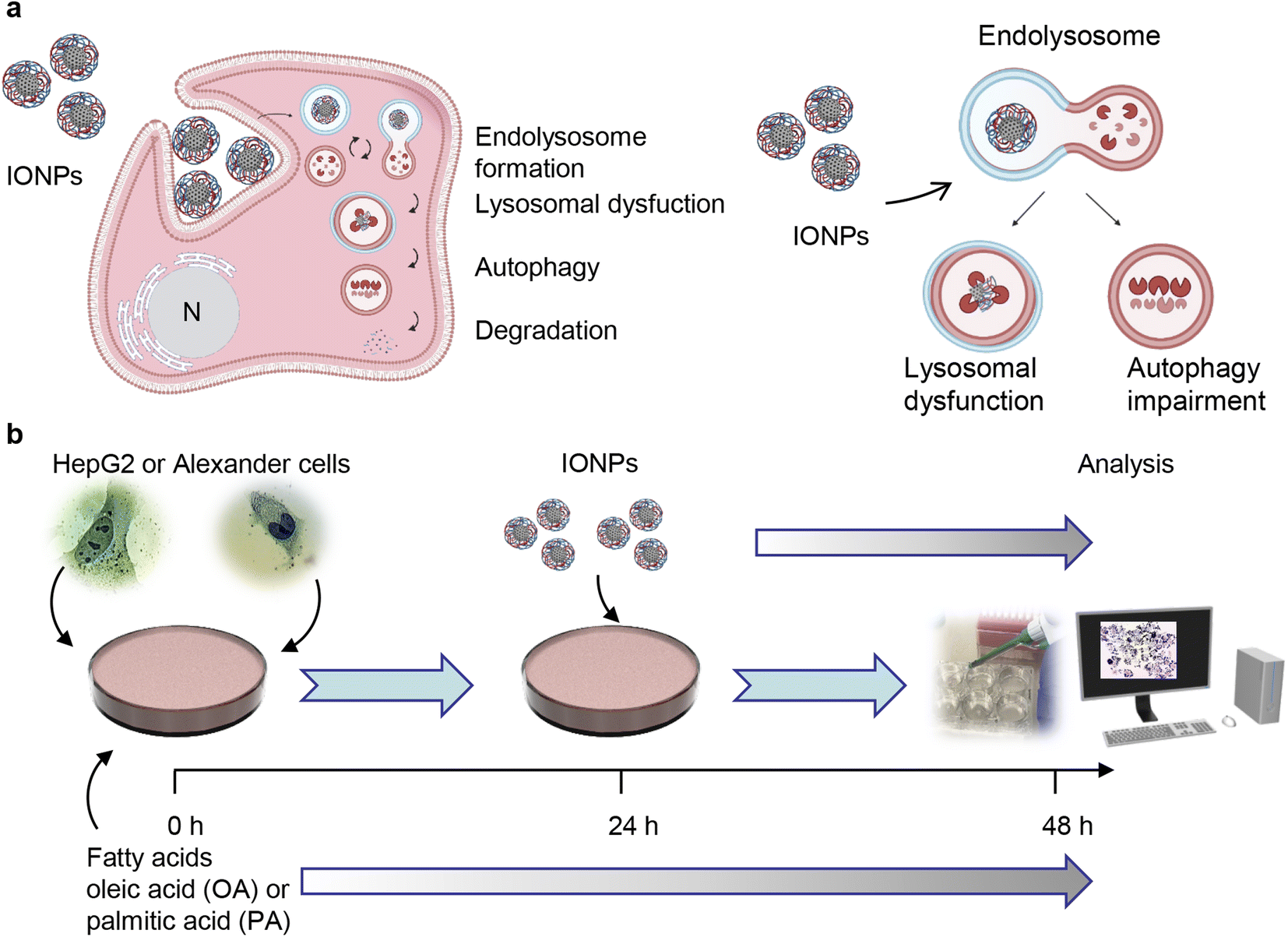 | ||
| Fig. 1 Design of the experimental setup. (a) Scheme of lysosomal dysfunction induced by iron oxide nanoparticles (IONPs). We showed previously that sub-lethal doses of IONPs accumulate in lysosomes of the hepatic cells.55 Such accumulation results in progressive impairment of lysosomal function. Created with https://BioRender.com. (b) Schematics of the synergetic treatment of Alexander and HepG2 cells with IONPs. | ||
Analysis of lipid accumulation in cell line culturing
Cells were treated with different concentrations of oleic or palmitic acid for 48 h. After the treatment the cells were washed with PBS and fixed with 4% paraformaldehyde in PBS pH 7.4, at room temperature for 10 min. The cells were labelled with Oil Red O to assess the total amount of neutral lipid accumulation. The Oil Red O reagent was added with agitation, followed by washing in 85% propylene glycol. After, the cells were washed with distilled water and counterstained with hematoxylin. The stained cells were imaged with a light microscope IM-2FL (Optika Microscopes, Ponteranica, Italy). ImageJ software (NIH, Bethesda, MD, USA) was used for image processing.Additionally, lipid accumulation was assessed using a specific fluorescent probe, LipidSpot™ lipid droplet stain (Biotium, Fremont, CA, USA). Cells were treated with oleic or palmitic acid for 48 h and then stained with the LipidSpot™ fluorescent probe according to the manufacturer's guidelines. Nuclei were counterstained with Hoechst 33342 (Thermo Fisher Scientific, Waltham, MA, USA). The stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan).
Detection of intracellular ROS
For the detection of reactive oxygen species (ROS) levels, we used a Cellular ROS/Superoxide Detection Assay Kit (Abcam, Cambridge, United Kingdom). Briefly, cells were seeded in 6-channel μ-Slides (Ibidi, Gräfelfing, Germany) at a density of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well and co-treated with either oleic or palmitic acid and IONPs according to the scheme presented in Fig. 1b. Afterwards, the cells were stained with the Oxidative Stress Detection Reagent (green) for ROS detection and the Superoxide Detection Reagent (orange) according to the manufacturer's instructions (Abcam, Cambridge, United Kingdom). The stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). As a positive control treatment with 1 mM H2O2 for 30 min was used. For quantitative analysis, the corrected total cell fluorescence (CTCF) of the full area of interest (ROI), i.e., selected region of a single cell, was calculated. The CTFC intensity of the single cell for separate “green” and “red” fluorescence channels was calculated for each image utilizing a previously described method.56 CTCF = integrated density − (area of selected cell × mean fluorescence of background readings). For background readings, we utilized a region placed in an area without fluorescent objects. Image quantification was performed using ImageJ software (NIH, Bethesda, MD, USA).
000 cells per well and co-treated with either oleic or palmitic acid and IONPs according to the scheme presented in Fig. 1b. Afterwards, the cells were stained with the Oxidative Stress Detection Reagent (green) for ROS detection and the Superoxide Detection Reagent (orange) according to the manufacturer's instructions (Abcam, Cambridge, United Kingdom). The stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). As a positive control treatment with 1 mM H2O2 for 30 min was used. For quantitative analysis, the corrected total cell fluorescence (CTCF) of the full area of interest (ROI), i.e., selected region of a single cell, was calculated. The CTFC intensity of the single cell for separate “green” and “red” fluorescence channels was calculated for each image utilizing a previously described method.56 CTCF = integrated density − (area of selected cell × mean fluorescence of background readings). For background readings, we utilized a region placed in an area without fluorescent objects. Image quantification was performed using ImageJ software (NIH, Bethesda, MD, USA).
Detection of apoptosis
Signs of early apoptosis, namely, phosphatidylserine expression and membrane permeability, were assessed utilizing a Dead Cell Apoptosis Kit (Thermo Fisher Scientific, Waltham, MA, USA) as described previously.5,57,58 Cells were co-treated with either oleic or palmitic acid and IONPs according to the scheme presented in Fig. 1b. Afterwards, the cells were labelled with the Dead Cell Apoptosis Kit following the manufacturer's guidelines. The Dead Cell Apoptosis Kit consists of an Alexa Fluor 488 Annexin V, which detects phosphatidylserine expression and propidium iodide, which is used to assess membrane permeability enabling discretion of necrotic cells. Treatment with camptothecin (30 μM for 24 hours) served as a positive control. Following staining, the cells were analyzed utilizing a CytoFLEX flow cytometer B53013 (Beckman Coulter, Brea, CA, USA). Data were acquired and analyzed using CytExpert software (Beckman Coulter, Brea, CA, USA).Lysosomal stability analysis by acridine orange (AO) release
Cells were seeded onto 96-well clear bottom plates (BD Biosciences, Franklin Lakes, NJ, USA) at a density of 5000 cells per well. The cells were co-treated with either oleic or palmitic acid and IONPs according to the scheme presented in Fig. 1b. Afterwards, the cells were labeled with 5 μg ml−1 AO in the culture medium for 15 min at 37 °C. Following labeling, the fluorescence of AO was measured by simultaneous acquiring the green signal (excitation 485 nm; emission 535 nm) and red signal (excitation 485 nm; emission 635 nm) using a fluorescence microplate reader, SpectraFluor Plus (TECAN, Mannedorf, Switzerland). Then the ratio of red/green fluorescence was calculated and normalized to the ratio of red/green fluorescence of the negative control giving a value of “AO Red/Green ratio normalized to control”. The normalized fluorescence data are presented as mean ± SEM. Readings were done in quadruplicate.Lipid peroxidation assay
The detection of lipid peroxidation upon co-treatment with either oleic or palmitic acid and IONPs was done using a BODIPY™ 581/591 C11 lipid peroxidation sensor (Thermo Fisher Scientific, Waltham, MA, USA). The assay principle is based on the fact that the BODIPY™ 581/591 C11 probe exhibits a shift in the fluorescence emission peak from ∼590 nm to ∼510 nm during oxidation, resulting in a decrease in red fluorescence.59,60 Thus, the change in the ratio of red/green fluorescence intensities is utilized as a sign of lipid peroxidation under oxidative stress.61 We assessed lipid peroxidation in accordance with the manufacturer's instructions and previously published protocols.55,61 In brief, cells were seeded onto 96-well clean bottom plates at a density of 5000 cells per well and co-treated with either oleic or palmitic acid and IONPs according to the scheme presented in Fig. 1b. After the treatment, the cells were loaded with the BODIPY™ 581/591 C11 probe at a concentration of 1 μM for 30 min at 37 °C. Then, the fluorescence of the BODIPY™ 581/591 C11 probe was assessed by simultaneously acquiring the green signal (excitation 485 nm; emission 535 nm) and red signal (excitation 562 nm; emission 635 nm) using a fluorescence microplate reader, SpectraFluor Plus (TECAN, Mannedorf, Switzerland). Then the ratio of red/green fluorescence was calculated. Readings were done in quadruplicate. The data are presented as mean ± SEM.Thioflavin T assay
To analyze endoplasmic reticulum stress (ER stress), we assessed misfolded or unfolded protein formation by thioflavin T assay.62,63 Thioflavin T, a small molecule, has high binding affinity to protein aggregates and shows enhanced fluorescence upon binding. In fact, the fluorescence of thioflavin T directly correlates with established indicators of unfolded protein response activation, enabling the detection and quantification of endoplasmic reticulum stress in live cells.62 After co-treatment with either oleic or palmitic acid and IONPs, the cells were loaded with 5 μM thioflavin T for 30 min. The green fluorescence intensities of thioflavin T (excitation 488 nm; emission 525 nm) were assessed using a CytoFLEX flow cytometer B53013 (Beckman Coulter, Brea, CA, USA). Data were acquired and analyzed using CytExpert software (Beckman Coulter, Brea, CA, USA).Immunofluorescence
Immunofluorescence staining was performed to assess the colocalization of cathepsin B with the lysosomal marker LAMP1. Cells were seeded in 6-channel μ-Slides (Ibidi, Gräfelfing, Germany) at a density of 10000 cells per well and co-treated with either oleic or palmitic acid and IONPs according to the scheme presented in Fig. 1b. Subsequently, the cells were washed with PBS and fixed with 4% paraformaldehyde in PBS pH 7.4 at room temperature for 10 min. Then the cells were permeabilized with 0.5% Triton X-100. Immunofluorescence staining was performed on fixed cells using primary antibodies against cathepsin B and LAMP1 and AlexaFluor 568- or AlexaFlour 488-conjugated secondary antibodies. The dilutions and catalogue numbers of the primary and secondary antibodies used are provided in ESI Table S2.† The stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). ImageJ software (NIH, Bethesda, MD, USA) was used for image processing and quantification.
Assessment of mitochondrial membrane potential
Cells were seeded in 6-channel μ-Slides (Ibidi, Gräfelfing, Germany) at a density of 10000 cells per well and co-treated with either oleic or palmitic acid and IONPs according to the scheme presented in Fig. 1b. Subsequently, the cells were stained with 1 μM JC-1 probe and imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). A treatment with 20% ethanol for 30 minutes was used as a positive control. JC-1 is a highly selective and sensitive dye to assess mitochondria potential. It enters the mitochondria and undergoes a reversible color change from red to green as the membrane potential decreases.64 In cells with high mitochondrial membrane potential (ΔmΦ), JC-1 forms complex J-aggregates that possess intense red fluorescence. In cells with low ΔmΦ, JC-1 remains in the monomeric form, which displays mostly green fluorescence. Therefore, the ratio of green to red fluorescence is used as an indicator of ΔmΦ changes. This ratio is not affected by other factors such as mitochondrial size, shape, and density, which may influence single-component fluorescence signals.64
Spinning disk confocal microscopy
To obtain clear subcellular details of DN localization, we employed a high-resolution IXplore SpinSR Olympus imaging system (Olympus, Tokyo, Japan). Cells were seeded in 6-channel μ-Slides (Ibidi, Gräfelfing, Germany) and co-treated with either oleic or palmitic acid and IONPs according to the scheme presented in Fig. 1b. Subsequently, the cells were stained for mitochondria, lysosomes, and endoplasmic reticulum utilizing specific fluorescent probes listed in Table S2.† The imaging system was equipped with an inverted microscope (IX83; Olympus, Tokyo, Japan) and a spinning disc confocal unit (CSUW1-T2S SD; Yokogawa, Musashino, Japan). Fluorescence images were obtained through a 100× silicone immersion objective (UPLSAPO100XS NA 1.35 WD 0.2 silicone lens, Olympus, Tokyo, Japan). The fluorophores were excited at appropriate wavelengths: 405 nm laser diode (50 mW), 488 nm laser diode (100 mW), and 561 nm laser diode (100 mW). Confocal images were acquired at a resolution of 2048 × 2048 pixels. The images were passed through suitable emission filters (BA420-460; BA575IF; BA510-550; Olympus, Tokyo, Japan) and simultaneously captured by two digital CMOS cameras ORCA-Flash4.0 V3 (Hamamatsu, Hamamatsu City, Japan). The acquisition software cellSens (Olympus, Tokyo, Japan) was used for fluorescence confocal image acquisition. For quantitative image analysis, approximately 5–10 random visual fields were selected per sample, using the same setting parameters (i.e. spinning disk speed, laser power and offset gain). ImageJ software (NIH, Bethesda, MD, USA) was used for image processing and quantification.Spinning disk super-resolution microscopy
To analyze the interactions between lipid droplets and lysosomes in living cells, we utilized an IXplore SpinSR Olympus super-resolution imaging system (Olympus, Tokyo, Japan). Cells were seeded in 6-channel μ-Slides (Ibidi, Gräfelfing, Germany) and incubated with palmitic acid and IONPs according to the scheme presented in Fig. 1b. Then the cells were stained for lipid droplets (LipidSpot™ 488 Lipid Droplet Stain) and lysosomes (LysoTracker™ Red DND-99). The catalog numbers of the fluorescent probes used are provided in ESI Table S2.† Fluorescence images were acquired using cellSens acquisition software (Olympus, Tokyo, Japan). ImageJ software (NIH, Bethesda, MD, USA) was used for image processing and analysis of the gray value intensities of lysosomes and lipid droplets.Image quantification
To measure lysosomal size and mitochondrial circularity, cells were stained with LysoTracker™ Green DND-26 and MitoTracker™ Red CMXRos (both probes from Thermo Fisher Scientific, Waltham, MA, USA). Nuclei were counterstained with Hoechst 33342 (Thermo Fisher Scientific, Waltham, MA, USA). The stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). The average lysosomal size per cell was measured using ImageJ software (NIH, Bethesda, MD, USA), and mitochondrial circularity was assessed using the particle analyzer plugin in ImageJ software.To analyze cathepsin B release from lysosomal compartments, colocalization analysis was performed. After the treatment, cells were immunostained against cathepsin B and LAMP1. The stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). Fluorescence images were acquired with cellSens software (Olympus, Tokyo, Japan). For quantitative colocalization analysis, we calculated Pearson's correlation coefficient. Pearson's correlation coefficient is a good estimate of overall association between probes, as it measures pixel-by-pixel correlation, mean-normalized to values from −1 (anticorrelation) to 1 (correlation).65,66 Pearson's correlation coefficient was calculated using Coloc 2 tool available in ImageJ.67
Cell extracts and western blot analysis
Aliquots of whole cell lysates containing equal amounts of protein were obtained using RIPA lysis buffer (Millipore, Burlington, MA, USA) in accordance with the manufacturer's instructions and our verified protocol.55,68 For the isolation of nuclear extracts from the cells, we used a NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific, Waltham, MA, USA). Protein lysates were subjected to SDS-PAGE electrophoresis and transferred to PVDF membranes. The membranes were blocked with 5% (w/v) fat-free dried milk or alternatively with 5% (w/v) bovine serum albumin (BSA) for 1 h. Afterwards, the membranes were incubated with various specific primary antibodies listed in ESI Table S3† at 4 °C overnight. Chemiluminescence signals were detected using an imaging system GBOX CHEMI XRQ (Syngene, Synoptics group, Cambridge, UK) and the acquisition software GeneTools (Syngene, Synoptics group, Cambridge, UK).Statistical analysis
Quantitative results of viability, early apoptosis markers, thioflavin T-fluorescence, the AO fluorescence ratio, and the BODIPY™ 581/591 C11 fluorescence ratio are presented as mean ± SEM. The statistical significance of differences between the groups was determined using ANOVA with subsequent application of the Newman–Keuls test. MaxStat Pro 3.6 software (MaxStat Software, Cleverns, Germany) and SigmaPlot 13 (Systat Software, Palo Alto, CA, USA) were used to perform all statistical analyses. Differences were considered statistically significant at (*) P < 0.05.Quantitative microscopy analysis (namely analysis of lysosomal size, mitochondrial circularity, ΔmΦ, ROS levels, and Pearson's correlation coefficient) was conducted following fluorescence quantitative microscopy guidelines.69–71 In brief, images from three independent experiments were subjected to quantitative analysis. In each experiment, 10 randomly selected fields from each sample were imaged. To determine the sample size, we employed an accepted statistical methodology.72 We used SigmaPlot 13 software (Systat Software, Palo Alto, CA, USA) to calculate the sample size, considering a minimum confidence level of 95% and a statistical power of 0.9. Under these conditions, the sample size was calculated to be 30. Thus, at least 30 randomly selected cells were used in fluorescence microscopy quantification.
Results and discussion
IONP treatment trigger toxicity in steatotic HepG2 and Alexander cells
In this study, we utilized core–shell iron oxide nanoparticles coated with a carboxymethyldextran shell as a representative model of IONPs.51–53,55 The physicochemical properties of these particles have been extensively characterized by us and others in previous studies.51–55 These IONPs consist of a magnetite (Fe3O4) core coated with a carboxymethyldextran shell, resulting in a hydrodynamic diameter of ∼200 nm.51–55 In a previous study, we demonstrated that these IONPs interact with serum proteins, forming a protein corona after 2 hours of incubation in a medium containing 10% serum.55 Indeed, IONPs containing polydisperse cores coated with dextran derivatives and exhibiting a large hydrodynamic diameter within the range of approximately 16–200 nm are actively being investigated as MRI contrast agents.18,73,74 Relatively large IONPs, ranging from 100 to 200 nm, are utilized as T2 contrast agents especially for liver MRI, due to their rapid and passive accumulation in the liver.1,18,74,75 Therefore, we selected these IONPs as the nanomaterial model to investigate the interactions between IONPs and hepatic cells.Phagocytic Kupffer cells are primarily responsible for IONP uptake in the liver.18,38,76 However, it has been demonstrated that other cell types, including hepatocytes, actively accumulate IONPs and participate in nanoparticle hepatobiliary elimination.18,37,38,76 The presence of hepatic steatosis, as observed through imaging or histology, serves as a diagnostic marker of NAFLD.77 Moreover, a major prerequisite of the NAFLD progression is the excessive aggregation of fat in hepatocytes.78 Therefore, it is reasonable to study molecular mechanisms underlying the interactions between IONPs and hepatic cells under steatotic conditions, using hepatocyte models. Additionally, hepatocytes perform many metabolic processes, including the metabolization of drugs40 and nanoparticles.22 Therefore, it is important to examine whether IONPs can also induce adverse effects on hepatocytes under steatotic conditions. Despite originating from cancer, HepG2 cells are widely used as a model for normal hepatocytes.79 This cell line has been recognized as a useful model for basic preclinical research focused on studying the toxic effects of heavy metals, nanoparticles, and drugs.79,80 Another cell model frequently used as a surrogate for hepatocytes is Alexander cells or PLC/PRF/5.81 To ensure reproducibility and comparability with other research groups, we employed established models of liver hepatic cells, namely HepG2 and Alexander cells.
We previously demonstrated that even non-toxic doses of IONPs have a significant impact on lysosomal activity, leading to lysosomal dysfunction and impaired autophagy (Fig. 1a).55 It is important to note that iron overload represents a serious safety concern in liver diseases, such as NAFLD, NASH, and cirrhosis.30–32 Furthermore, studies have indicated that IONPs may worsen NAFLD and NASH pathologies.29,50 However, the precise cellular targets responsible for these adverse effects of IONPs are not fully understood. Additionally, most studies in this area typically utilize only one cell line, limiting the direct comparison of observed effects on closely related cell lines. Considering these points, we investigated whether IONPs may possess unsuspected adverse effects on the steatotic in vitro model (Fig. 1b) by using two hepatic cell lines (HepG2 and Alexander cells). These two cell lines exhibit differences in overall gene expression profiles.81 For instance, we previously demonstrated that HepG2 express elevated levels of Bcl-2 protein (an important regulator of apoptosis).58,63 This difference in Bcl-2 protein levels resulted in different responses of HepG2 and Alexander cells to lysosomal disturbance and toxicity triggered by different IONPs.55,57 Thus, it is interesting to investigate and compare these cell lines in terms of potential toxic effects induced by IONPs in steatotic hepatic cells.
Generally, to induce excessive fat accumulation in hepatic cell treatment with oleic (OA) or palmitic (PA) acid is used.82–86 Accumulating evidence suggests that OA and PA bear substantial differences in cellular responses, particularly in the execution of lipotoxicity.82–86 Therefore, in this study, our intention is to compare the cellular effects of OA and PA in combination with IONPs in hepatic cells. Firstly, we needed to determine the optimal concentration range of fatty acids that induces steatosis without causing profound lipotoxicity. HepG2 cells are predominantly used in most in vitro steatotic hepatic models.82–86 However, there are limited data available on steatosis establishment in Alexander cells. Therefore, it is interesting to compare responses in these cell lines. In fact, HepG2 cells showed higher resistance to lipotoxicity compared to Alexander cells when exposed to either OA or PA (Fig. 2a). The appropriate concentrations of OA and PA for HepG2, showing no significant signs of lipotoxicity while inducing lipid droplet accumulation, ranged from 50 to 300 μM (Fig. 2b). For Alexander cells, the appropriate concentrations of OA and PA that exhibited low cytotoxicity after 48 hours (Fig. 2a) and significant lipid droplet accumulation (Fig. 2b) were in the range of 5 to 20 μM. Furthermore, high-resolution confocal imaging revealed a dramatic increase in the number and size of lipid droplets in the cytosol of Alexander and HepG2 cells incubated with either OA or PA compared to control cells (Fig. 2c). Notably, lipid droplets were found to be more massive with OA treatment in contrast to PA stimulation (Fig. 2c).
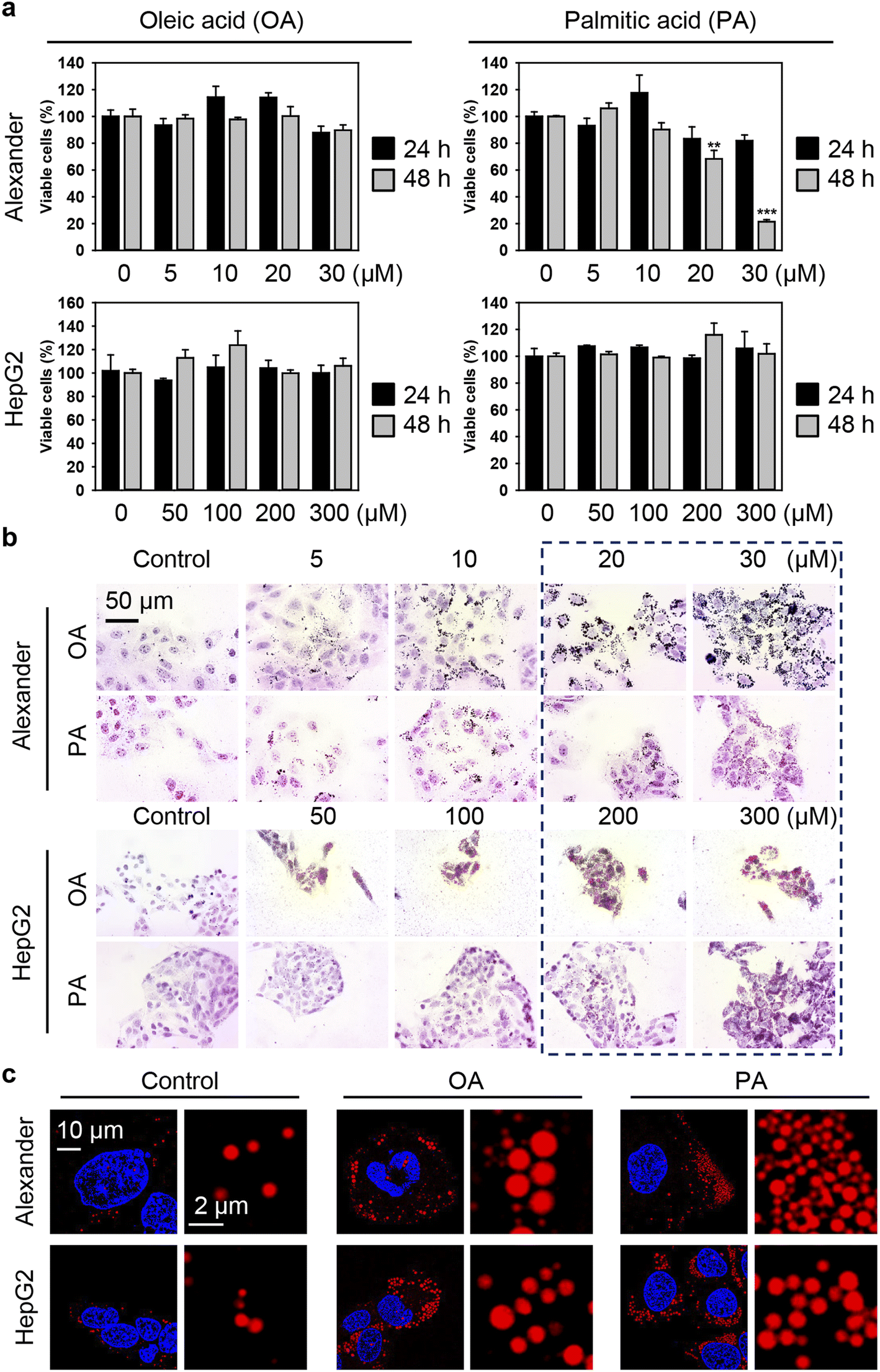 | ||
| Fig. 2 Establishing steatotic hepatic cells. (a) Cytotoxicity of oleic acid (OA) and palmitic acid (PA) in distinct cell lines: Alexander and HepG2. Cells were treated with indicated concentrations of either OA or PA for 24 h or 48 h. Cytotoxicity was assessed using alamarBlue assay. The data were normalized to control values (no fatty acid exposure), which were set as 100% cell viability. Control cells were untreated. Data are expressed as mean ± SEM (n = 3). (**) P < 0.01 and (***) P < 0.001 denote significant differences. (b) Oil Red O staining of Alexander and HepG2 cells treated with indicated concentrations of either OA or PA for 48 h. Hematoxylin was used as a counterstain. (c) Confocal imaging of lipid droplets in Alexander and HepG2 cells. Alexander cells treated with either OA or PA (both 20 μM) for 48 h. HepG2 cells treated with either OA or PA (both 200 μM) for 48 h. Lipid droplets were labeled with LipidSpot™ (Biotium, Inc.). Nuclei were counterstained with Hoechst 33342 (Thermo Fisher Scientific). Stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). Images were deconvolved using CellSens software (Olympus, Japan). | ||
There is a growing concern that the potential toxicity of different nanomaterials may be intensified in liver pathologies, such as NAFLD, NASH, and cirrhosis.29,50,87–90 Especially this is relevant for IONPs being investigated as a contrast agent for monitoring NAFLD, NASH, and cirrhosis.29,50 Thus, we assessed whether treatment with free fatty acids followed by IONP stimulation could lead to enhanced cytotoxicity (Fig. 1b). Co-treatment with OA and increasing concentrations of IONPs did not show any significant impairment in the viability of both Alexander and HepG2 cells (Fig. 3a and b). In contrast, co-treatment with PA and increasing concentrations of IONPs resulted in toxic responses in both cell lines (Fig. 3a and b). Our findings are consistent with previous reports demonstrating that PA is more lipotoxic than OA.82–86 Interestingly it was shown that OA is more steatogenic but less lipotoxic than PA in hepatic cell lines.82 Overall, monounsaturated fatty acids are known to be less toxic than saturated ones in hepatocytes.91 However, little is known about molecular determinants responsible for the differences in cytotoxicity progression between PA and OA.92 One hypothesis suggests that the low cytotoxicity of OA is associated with the rewiring of saturated fatty acid metabolism towards triglyceride synthesis and concomitant decrease in total cholesterol.84
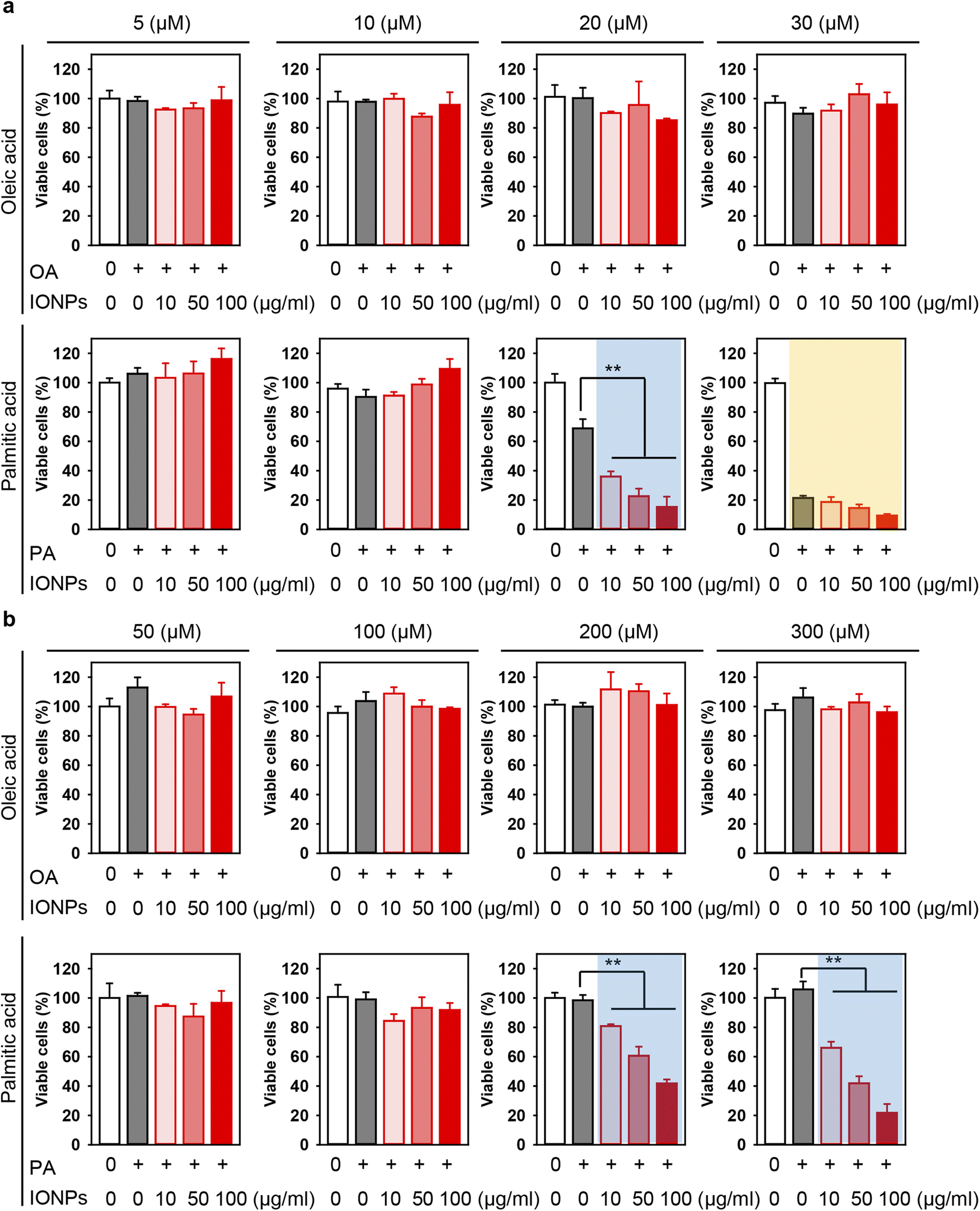 | ||
| Fig. 3 Effect of co-treatment with IONPs and fatty acids on Alexander and HepG2 cell viability. Alexander (a) or HepG2 (b) cells were co-treated with indicated concentrations of IONPs and either OA or PA in accordance with scheme Fig. 1b. Cytotoxicity was assessed using alamarBlue assay. The data were normalized to control values (no fatty acid and IONP exposure), which were set as 100% cell viability. Control cells were untreated. Data are expressed as mean ± SEM (n = 3). (**) P < 0.01 denote significant differences. | ||
In fact, co-treatment with 20 μM PA and increasing concentrations of IONPs resulted in IONP dose-dependent cytotoxicity in Alexander cells (Fig. 3a), demonstrating a synergetic effect of PA and IONP treatment. Therefore, for further mechanistic studies we selected 20 μM PA treatment for Alexander cells. In the case of HepG2 cells, co-treatment with either 200 or 300 μM PA and increasing concentrations of IONPs resulted in IONP dose-dependent cytotoxicity (Fig. 3b). For the subsequent mechanistic studies, we selected 200 μM PA treatment for HepG2 cells. It is evident that HepG2 exhibited significantly higher resistance to co-treatment with PA and IONPs compared to Alexander cells (Fig. 3). Our findings align with a previous study showing that HepG2 is more resistant to PA-induced toxicity compared to other hepatic cell lines.85 These discrepancies may be explained by the different levels of Bcl-2 protein expression in those cell lines.58,63 Bcl-2 protein is a well known regulator of apoptosis showing anti-apoptotic function.93 We have previously demonstrated that these differences in Bcl-2 expression levels result in different lysosomal disturbance and toxicity profiles triggered by IONPs in HepG2 and Alexander cells.55,57
Recently it has been shown that IONP doses up to 90 mg kg−1 induced mild toxic effects in vivo in rats.94 On the other hand, a dose of 100 mg kg−1 of ultrasmall IONPs (2–4 nm) was found to be lethal in mice after less than 10 h post-injection.95 The recommended daily dietary dose of iron is 8 mg for men and 18 mg for women according to the US Office of Dietary Supplements at National Institute Health.96 The highest supplemental iron intake should not exceed 45 mg for healthy adults, which can result in gastrointestinal adverse reactions.96 The recommended dietary iron doses would translate into 0.13 mg Fe per kg, which is at least four times lower than the dosage for IONP-based MRI contrast agents.29 Therefore, physiologically relevant IONP doses used in MRI may possess toxic effects.18
In our previous study, we demonstrated that the treatment of hepatic cells with IONPs at a concentration of 50 μg ml−1 led to impaired lysosomal function without concomitant cytotoxic responses.55 Physiologically relevant doses (up to 40 mmol Fe per kg) after intravenous injection of IONP contrast agents result in blood levels in the range of 50 μg ml−1, which are sequestered by the liver.97–100 We found that IONPs at a concentration of 50 μg ml−1 induce elevated toxicity in the presence of PA (Fig. 3). Taking into account physiologically relevant concentrations and the literature discussed above, we proceeded further with a concentration of 50 μg ml−1 of IONPs to investigate basic molecular determinants of cytotoxicity.
Total reactive oxygen species (ROS) level is elevated upon IONP treatment in steatotic HepG2 and Alexander cells
Uncontrolled accumulation of reactive oxygen species (ROS) has been identified as one of the main contributors to cytotoxicity induced by different metal nanoparticles, specifically IONPs.18,95,101,102 Additionally, PA overload may promote ROS accumulation and subsequent toxicity.103,104 Thus, we analyzed whether co-treatment with fatty acids and IONPs results in elevated ROS accumulation. We used a ROS/superoxide detection assay kit that contains two fluorescent probes, namely 2′,7′-dichlorofluorescein diacetate (DCFDA) and dihydroethidium (hydroethidine or DHE). DCFDA is a probe that measures the total ROS level (hydroxyl, peroxyl and other ROS activity). DHE is specific for superoxide levels. Consistent with cytotoxicity results, neither IONPs alone nor OA induced elevation of total ROS and superoxide in both cell lines (Fig. 4, S1 and S2†). Co-treatment with OA and IONPs did not affect ROS and superoxide production either (Fig. 4, S1 and S2†). Interestingly, treatment with sole PA resulted in significant elevation of both ROS and superoxide levels in both cell lines (Fig. 4). Furthermore, co-treatment with PA and IONPs resulted in an increase in superoxide accumulation. However, that increase was not significantly different from single PA treatment in both Alexander and HepG2 cells (Fig. 4). It is worth noting that co-treatment with PA and IONPs resulted in an increase of the total ROS level, which was significantly higher than that induced by PA alone (Fig. 4).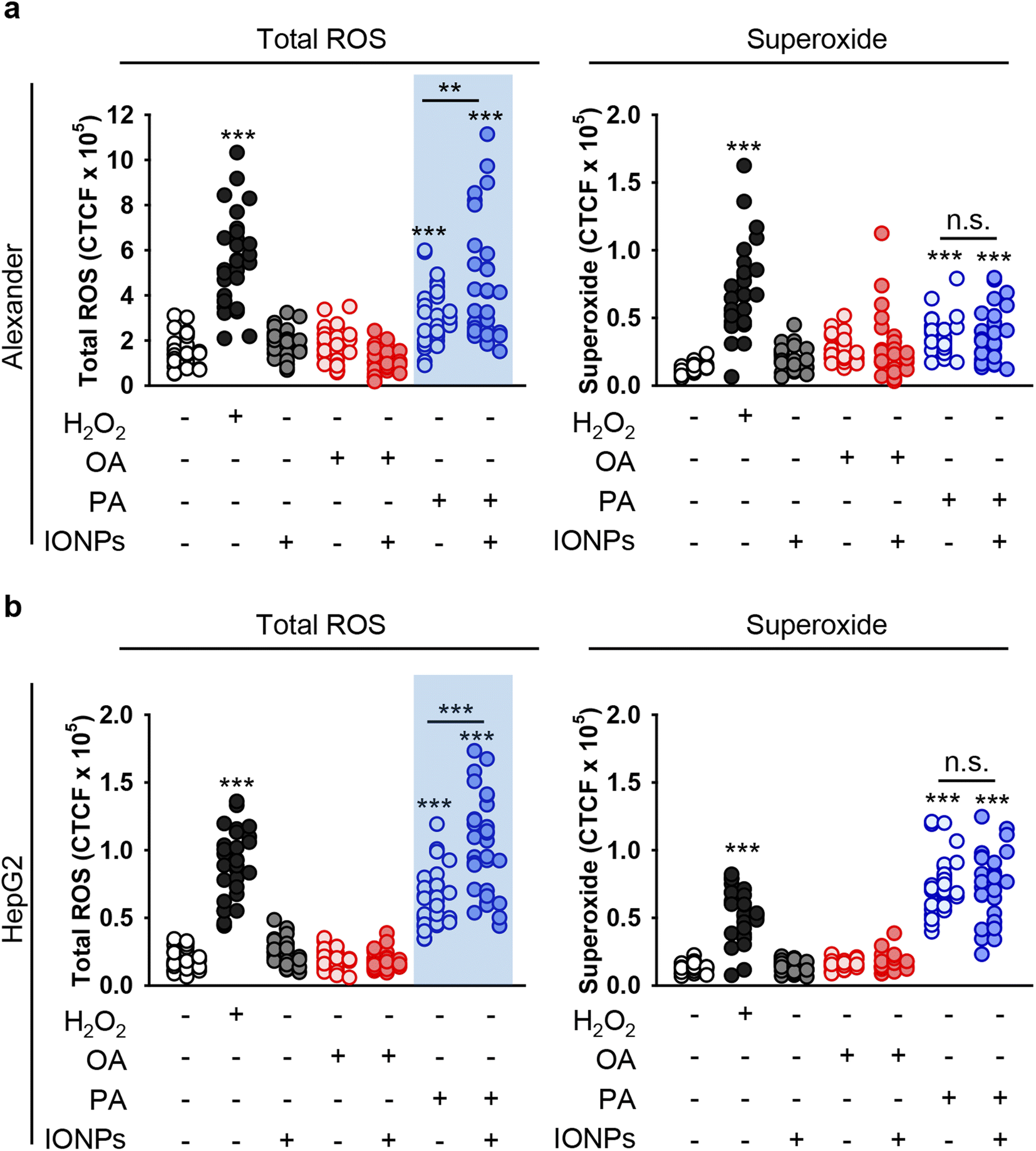 | ||
| Fig. 4 Analysis of ROS induction in hepatic cell lines upon co-treatment with IONPs and fatty acids. Alexander (a) or HepG2 (b) cells were co-treated with IONPs (50 μg ml−1) and either OA or PA (20 μM for Alexander and 200 μM for HepG2 cells) in accordance with scheme Fig. 1b. Treated cells were stained with a ROS/Superoxide Detection Assay Kit (Abcam, Cambridge, United Kingdom) and imaged by confocal microscopy. ImageJ software (NIH) was used for image processing and quantification. (**) P < 0.01 and (***) P < 0.001 denote significant differences. Positive control 100 μM H2O2 for 30 min was used. | ||
Our intention was to uncover a basic mechanistic explanation for the cytotoxicity induced by co-treatment with PA and IONPs. Therefore, we analyzed if co-treatment with fatty acids and IONPs leads to higher levels of lipid peroxidation. Single treatment with either IONPs or OA did not affect the level of lipid peroxidation in either cell line (Fig. S3a†). Co-treatment with OA and IONPs also did not elevate the level of lipid peroxidation (Fig. S3a†). On the other hand, co-treatment with PA and IONPs resulted in increased lipid peroxidation. However, this increase was similar to that observed with single PA treatment (Fig. S3a†). Thus, we concluded that lipid peroxidation is not a primary reason for the cytotoxicity triggered by the co-treatment of PA and IONPs.
Lysosomal destabilization, accompanied by profound lysosomal membrane permeabilization, largely contributes to excessive ROS accumulation and resultant cell death.105,106 Furthermore, we investigated the signs of lysosomal destabilization upon co-treatment with fatty acids and IONPs. The acridine orange assay for lysosomal integrity revealed that PA and IONP co-treatment resulted in lysosomal leakage similar to single PA stimulation (Fig. S3b†). Treatment with either OA or IONPs alone did not affect lysosomal integrity (Fig. S3b†). Co-treatment with OA and IONPs also did not lead to lysosomal leakage (Fig. S3b†). It is known that lysosomal leakage may destabilize mitochondria, leading to toxic response.105,106 Furthermore, we analyzed whether the morphology of lysosomes and mitochondria changed upon co-treatment with fatty acids and IONPs. In fact, all treatments led to an increase of the lysosomal size (Fig. 5a, b, S4 and S5†). However, there were no significant differences observed between the different treatments (Fig. 5a, b, S4 and S5†). Importantly, we found no differences between PA and IONP co-treatment and single PA treatment (Fig. 5a and b). Moreover, there were no noticeable changes in mitochondria circularity with any treatment in both cell lines (Fig. 5a, c, S4 and S5†). Interestingly, all treatments led to mitochondria membrane depolarization, as revealed by JC-1 staining in both cell lines (Fig. S6 and S7†). It is worth noting that no significant differences in mitochondria depolarization were observed between the different treatments (Fig. S6 and S7†).
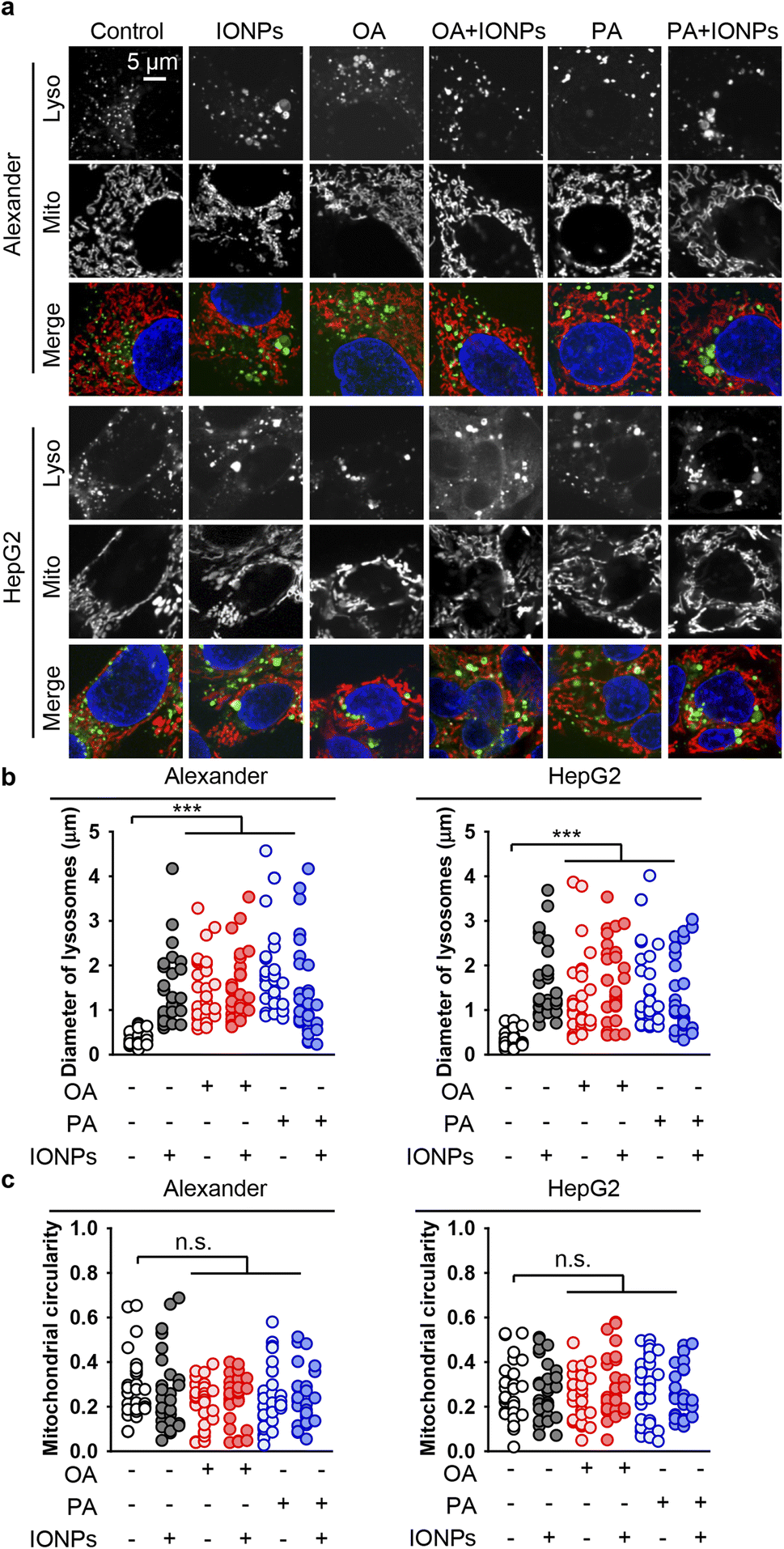 | ||
| Fig. 5 Effect of co-treatment with IONPs and fatty acids on the morphology of lysosomes and mitochondria in Alexander and HepG2 cells. (a) Alexander or HepG2 cells were co-treated with IONPs (50 μg ml−1) and either OA or PA (20 μM for Alexander and 200 μM for HepG2 cells) in accordance with scheme Fig. 1b. Cells were labeled with LysoTracker™ Green DND-26 (green) and MitoTracker™ Red CMXRos (red). Nuclei were stained with Hoechst 33342 nuclear stain (blue). (b) Measurements of the lysosomal diameter upon co-treatment with IONPs and fatty acids. Confocal images (a) were quantified using ImageJ software. Quantification results are presented as means of n = 30 cells. (***) P < 0.001 denotes significant differences with respect to the control (no treatment). (c) Measurements of the mitochondrial circularity upon co-treatment with IONPs and fatty acids. Confocal images (a) were quantified using ImageJ software. Quantification results are presented as means of n = 30 cells. | ||
Taken together, these data imply that lysosomal leakage and mitochondria membrane depolarization might be involved in cytotoxicity triggered by co-treatment with PA and IONPs. However, neither lysosomal leakage nor mitochondria membrane depolarization is the primary signal that mediates the cytotoxicity of IONPs in steatotic hepatic cells.
IONP treatment leads to endoplasmic reticulum stress in steatotic HepG2 and Alexander cells
Furthermore, to decipher mechanistic reasons for the toxicity triggered by the co-treatment of PA and IONPs, we assessed early signs of apoptosis. We performed annexin V and propidium iodide staining. Stimulation of Alexander and HepG2 cells with either IONPs or OA only did not induce phosphatidylserine translocation to the outer cell membrane leaflet, as revealed by annexin V staining (Fig. 6a and S8†). Additionally, we found no membrane permeability as confirmed by propidium iodide exclusion in either IONP or OA treatment (Fig. 6a and S8†). Co-treatment with OA and IONPs also did not result in an increase in annexin V or propidium iodide positive staining (Fig. 6a and S8†). Consistent with the cytotoxicity, we observed a significant increase in annexin V and propidium iodide positive staining for cells co-treated with PA and IONPs (Fig. 6a and S8†). This result prompted us to examine other apoptotic markers to verify if apoptosis signaling is involved in cytotoxicity triggered by the co-treatment with PA and IONPs.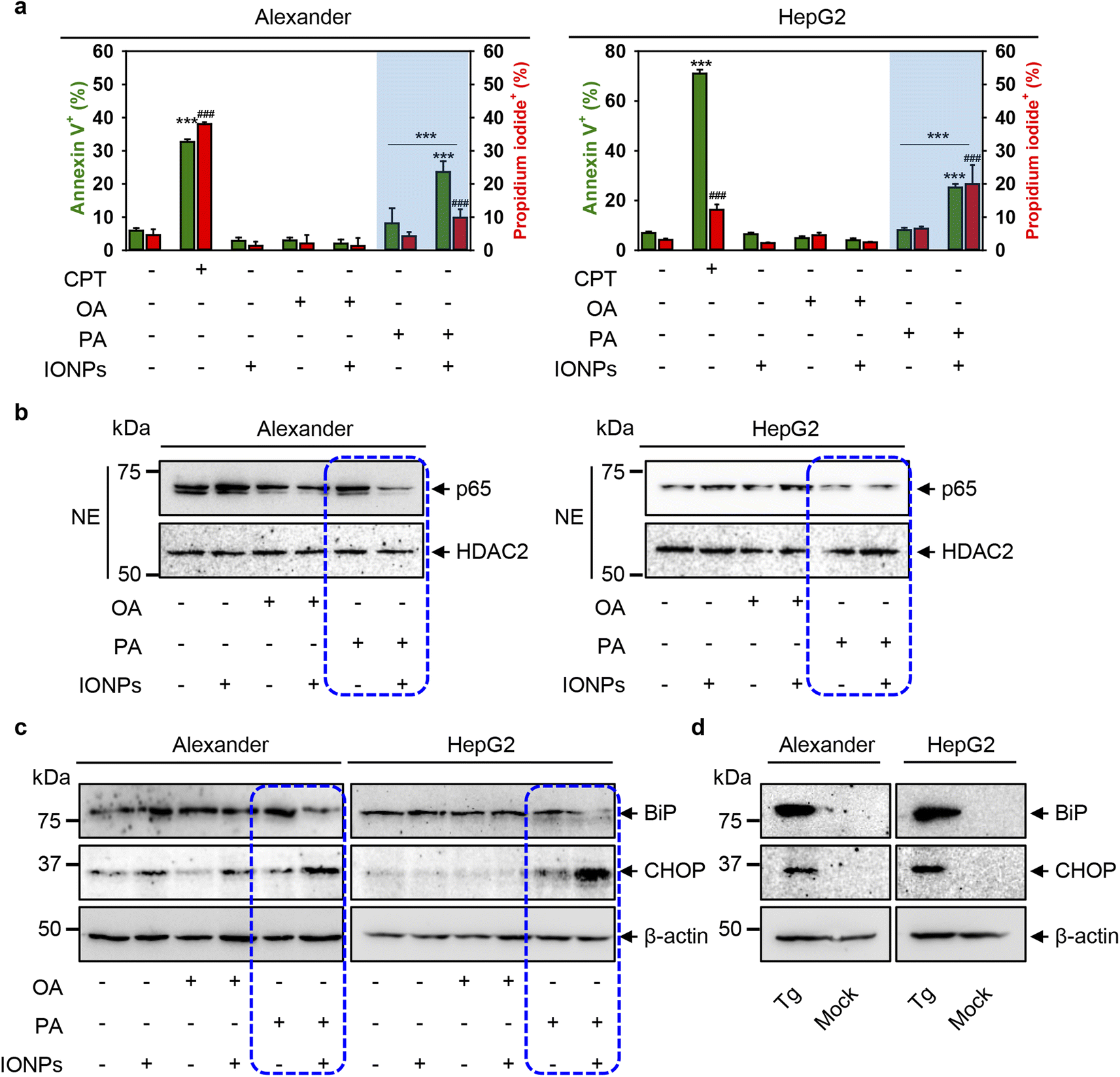 | ||
| Fig. 6 Cell death signaling pathway analysis upon co-treatment with IONPs and fatty acids of Alexander and HepG2 cells. Alexander or HepG2 cells were co-treated with IONPs (50 μg ml−1) and either OA or PA (20 μM for Alexander and 200 μM for HepG2 cells) in accordance with scheme Fig. 1b. (a) After treatment the cells were labelled with annexin V and propidium iodide (PI). Labelled cells were analyzed by flow cytometry. As a positive control, cells were treated with 30 μM camptothecin (CPT) for 24 h. Quantification results are presented as means of n = 5. (###) P < 0.001 and (***) P < 0.001 denote significant differences. (b) Nuclear extracts (NEs) from cells co-treated with IONPs and fatty acids. HDAC2 serves as a nuclear marker. Expressions of p65 was analyzed by immunoblotting. (c) Expressions of BiP and CHOP were analyzed by immunoblotting. Actin denotes the loading control. (d) Expressions of BiP and CHOP were analyzed by immunoblotting upon treatment with 1 μM thapsigargin (Tg) for 12 h (known positive control for ER stress). Actin denotes the loading control. | ||
Of note, the nuclear factor κB (NF-κB) is a well-known transcription factor involved in cell survival and apoptosis regulation in response to different stimuli, such as chemotherapeutic drugs, oxidative stress, UV light, and many others.107–109 NF-κB is known to promote the cell survival via upregulation of anti-apoptotic genes that inhibit the apoptotic machinery in various cell types.109,110 Specifically, inhibition of nuclear localization of one member of NF-κB, namely p65 (RelA), which harbors the transcription activation domain, potentiates apoptosis through oxidative damage.111,112 In fact, downregulation of p65 in HepG2 cells has been shown to promote apoptosis.113 We analyzed the nuclear levels of p65 upon co-treatment with fatty acids and IONPs. Immunoblot analysis revealed that the nuclear levels of p65 were downregulated in both cell lines upon co-treatment with PA and IONPs (Fig. 6b). Taken together, the increase in annexin V positive cells and the downregulation of nuclear p65 levels indicate that co-treatment with PA and IONPs leads to apoptosis execution.
However, our aim was to elucidate the molecular reasons for cell death triggered by the co-treatment of PA and IONPs. It is well known that excessive accumulation of lipids leads to lipotoxicity.114 Additionally, accumulating evidence highlights that chronic endoplasmic reticulum (ER) stress may lead to lipotoxicity in the liver.114 It was found that ER stress can trigger apoptosis.115 Specifically, excessive doses of PA have been shown to induce ER stress, leading to oxidative burst and subsequent apoptosis in hepatic cells.116–118 Here, we used PA and IONP concentrations that do not induce toxic responses. It is worth noting that single PA treatment resulted in the elevation of ROS (Fig. 4), increased lipid peroxidation (Fig. S3a†) and lysosomal dysfunction (Fig. S3b†). However, single PA stimulation did not result in toxicity in HepG2 and only slightly impaired the viability of Alexander cells (Fig. 3). These data imply that single PA treatment may increase the susceptibility of cells to agents affecting lysosomal activity. On the other hand, the combination of two stimuli (PA and IONPs) resulted in marked decrease in the viability of both Alexander and HepG2 cells (Fig. 2). Considering the abovementioned studies on ER stress and PA lipotoxicity, we hypothesized that co-treatment with PA and IONPs may lead to apoptosis via initiation of ER stress.
ER stress is an adaptive pro-survival mechanism activated in cells upon various stimuli. ER stress, mediated by the unfolded protein response (UPR), helps to restore ER homeostasis. Prolonged ER stress and excessive accumulation of UPR signaling can lead to cell death.114,119,120 To analyze whether co-treatment of PA and IONPs triggers ER stress in Alexander and HepG2 cells, we examined the protein expression levels of the main molecular indicators of the UPR and ER stress, specifically immunoglobulin heavy-chain binding protein (BiP) and C/EBP homologous protein (CHOP). Immunoblot analysis revealed that co-treatment with PA and IONPs resulted in a slight downregulation of BiP protein expression levels in both cell lines (Fig. 6c). However, CHOP was significantly upregulated in both cell lines upon co-treatment with PA and IONPs (Fig. 6c). Interestingly, the well-known inducer of ER stress thapsigargin led to upregulation of both BiP and CHOP protein levels (Fig. 6d). It has been shown that certain stimuli may lead to feedback-mediated suppression of the ER chaperones like BiP and GRP94.121 Specifically, in HepG2 cells, saturated fatty acids were found to activate ER stress, resulting in the upregulation of CHOP and downregulation of BiP.122 These data suggest that molecular foundations of ER stress triggered by the co-treatment of PA and IONPs are distinct from ER stress induced by thapsigargin.
To further validate ER stress and UPR activation induced by the co-treatment of PA and IONPs, we utilized thioflavin T assay to analyze the accumulation of misfolded or unfolded proteins under ER stress.62,63 Thioflavin T enhances its fluorescence upon binding to protein aggregates, serving as an indicator of UPR activation.62,63 Indeed, co-treatment with PA and IONPs resulted in a significant increase in thioflavin T fluorescence, indicating the accumulation of misfolded or unfolded proteins (Fig. 7a). Additionally, we investigated the impact of PA and IONP co-treatment on the ER structure. We performed high-resolution confocal imaging of ER in cells labeled with the fluorescent ER dye ER-Tracker™ Red. In the control cell ER appeared as elongated tubular structures (Fig. 7b). The full-size images can be found in ESI Fig. S9.† Treatment with thapsigargin resulted in ER fragmentation and the presence of vesicular and globular structures (Fig. 7b). Co-treatment with PA and IONPs led to marked ER damage, as evidenced by completely diffusive staining of the ER (Fig. 7b). Collectively, these observations indicate that the co-treatment of PA and IONPs drastically alters the configuration of the ER, leading to the accumulation of misfolded or unfolded proteins and subsequent induction of ER stress.
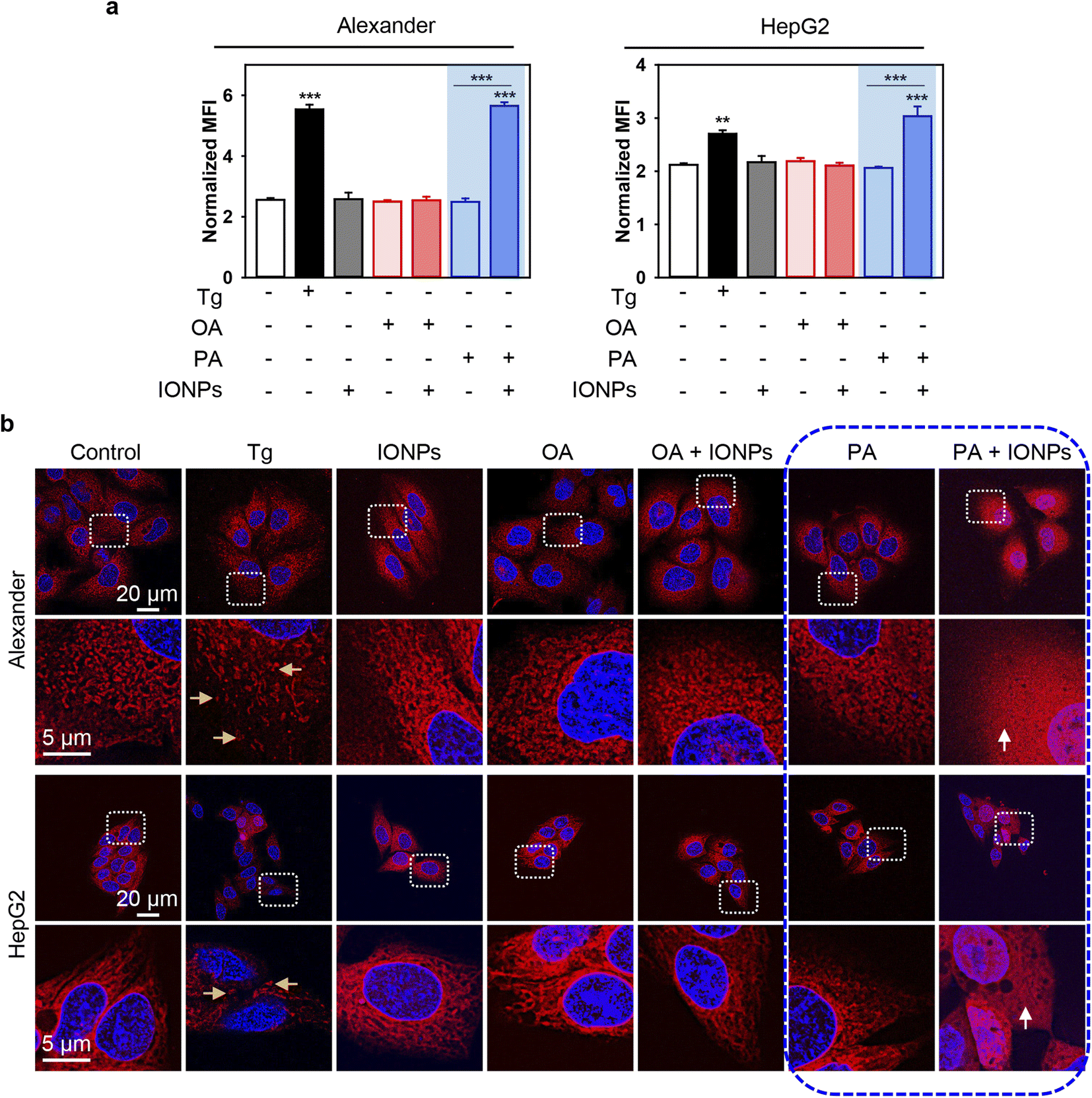 | ||
| Fig. 7 ER stress analysis upon co-treatment with IONPs and fatty acids of Alexander and HepG2 cells. Alexander or HepG2 cells were co-treated with IONPs (50 μg ml−1) and either OA or PA (20 μM for Alexander and 200 μM for HepG2 cells) in accordance with scheme Fig. 1b. (a) Cells were stained with 5 μM thioflavin T (green). Labelled cells were analyzed by flow cytometry. Positive control – treatment with 1 μM thapsigargin (Tg) for 12 h. Quantification results are presented as means of n = 3. (**) P < 0.01 and (***) P < 0.001 denote significant differences. (b) Cells were stained with ER-Tracker Red and visualized by confocal microscopy. Nuclei were stained with Hoechst 33342 nuclear stain (blue). Positive control – treatment with 1 μM thapsigargin (Tg) for 12 h. Representative confocal microscopic images show minor ER alterations after IONP, OA, PA, and OA + IONP treatments, transformation of tubule-lamellar into ring-link and/or fragmented ER structures after treatment with Tg (yellow arrows), and widespread ER diffusive staining after dual treatment PA + IONPs (white arrows). Stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). Images were deconvolved using CellSens software (Olympus, Japan). | ||
However, we still lacked a mechanistic link that could explain why the co-treatment of PA and IONPs induced ER stress and activated cell death. Previous studies have demonstrated that ER stress-induced apoptosis may independently of the mitochondrial damage result in lysosomal destabilization followed by the release of cathepsin B from lysosomes into the cytosol.123 This, in turn, triggers apoptosis execution.123,124 Therefore, we examined the sub-cellular localization of cathepsin B by immunofluorescence. Confocal imaging revealed that the co-treatment of PA and IONPs significantly decreased the colocalization of cathepsin B with the lysosomal marker LAMP1 (Fig. 8a, b, S10 and S11†). It is worth noting that lysosomal integrity assay indicated lysosomal leakage upon the co-treatment of PA and IONPs (Fig. S3b†). However, this leakage was similar to that observed with single PA treatment (Fig. S3b†). Taken together, these observations indicate that the co-treatment of PA and IONPs specifically leads to the release of cathepsin B into the cytoplasm. We were puzzled as to why the co-treatment of PA and IONPs, but not single PA stimulation, resulted in the relocation of cathepsin B from lysosomes to the cytosol.
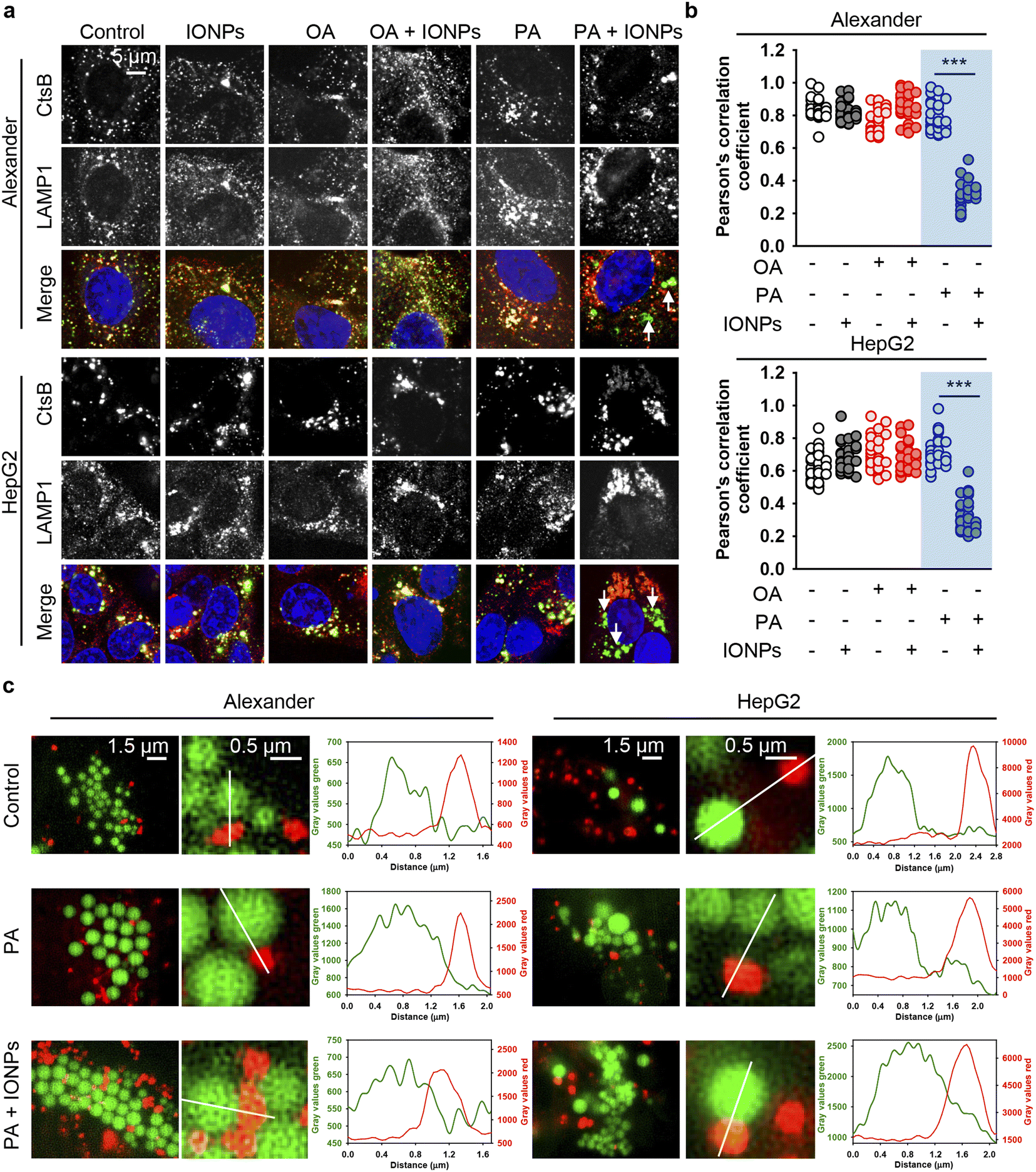 | ||
| Fig. 8 The release of cathepsin B upon co-treatment with IONPs and fatty acids of Alexander and HepG2 cells. (a) Alexander or HepG2 cells were co-treated with IONPs (50 μg ml−1) and either OA or PA (20 μM for Alexander and 200 μM for HepG2 cells) in accordance with scheme Fig. 1b. After cells were fixed, they were permeabilized and immunostained with anti-CtsB (green) and anti-LAMP1 (red) antibodies. Nuclei were stained with Hoechst 33342 nuclear stain (blue). Stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). (b) Analysis of CtsB/LAMP1 colocalization in Alexander or HepG2 cells. Pearson's correlation coefficient for the CtsB/LAMP1 pair was calculated using the Coloc 2 tool available in ImageJ software (NIH) and is presented as a mean of n = 30 cells. (***) P < 0.001 denotes significant differences. (c) Live-cell super-resolution microscopy of interactions between lysosomes and lipid droplets. Alexander or HepG2 cells were co-treated with IONPs (50 μg ml−1) and either OA or PA (20 μM for Alexander and 200 μM for HepG2 cells). Then the cells were labelled with LysoTracker™ Red DND-99 (red) and LipidSpot™ 488 Lipid Droplet Stain (green). Super-resolution imaging was done using an IXplore SpinSR Olympus super-resolution system. Graphs are line scans drawn through the lysosomes and lipid droplets and show the gray intensity values of lysosomes (red) and lipid droplets (green). | ||
It is known that fatty acids are transported into the ER, where they are metabolized into neutral lipids that accumulate and are stored as lipid droplets (LDs).125,126 Under normal conditions LDs are actively metabolized by lysosomes.125,126 Specifically, hepatocytes metabolize LDs utilizing the lysosome-directed process of autophagy.125,126 Furthermore, lysosomes and LDs can directly interact, facilitating the transfer of LD content into lysosomes.127 This process results into LD catabolism by lysosomes.127
On the one hand, excessive accumulation of lipids can disturb lysosomal function, leading to increased lysosomal membrane permeability subsequently provoking lipotoxicity.128–131 Dysregulation of LD catabolism and biogenesis result into uncontrolled lipid accumulation, ER stress and the progression of toxic responses.125,131,132 Therefore, we analyzed LD–lysosome interactions in Alexander and HepG2 cells. To visualize individual organelles in live cells, we employed super-resolution spinning disk microscopy. This super-resolution microscopy is alike structured illumination microscopy giving a spatial resolution of 120 nm with an ultra-fast speed.133 Ultra-fast imaging enables life cell super-resolution visualization with minimized phototoxicity.63,133 We labelled LDs and lysosomes with LipidSpot™ 488 and LysoTracker™ Red DND-99, respectively, and observed interactions between these two compartments in living cells under super-resolution. Indeed, co-treatment with PA and IONPs, but not PA treatment alone, resulted in increased LD–lysosome interactions (Fig. 8c). These increased interactions, along with the lysosomal dysfunction induced by IONPs,55 may explain why co-treatment with PA and IONPs leads to a cytotoxic response. The accumulation of PA and uptake of IONPs disturb lysosomal function (Fig. 9), resulting in impaired lipid metabolism and the activation of ER stress, which is accompanied by the release of cathepsin B from lysosomes into the cytosol (Fig. 9). Ultimately, the release of cathepsin B, along with the activation of ER stress, provokes apoptotic cell death.
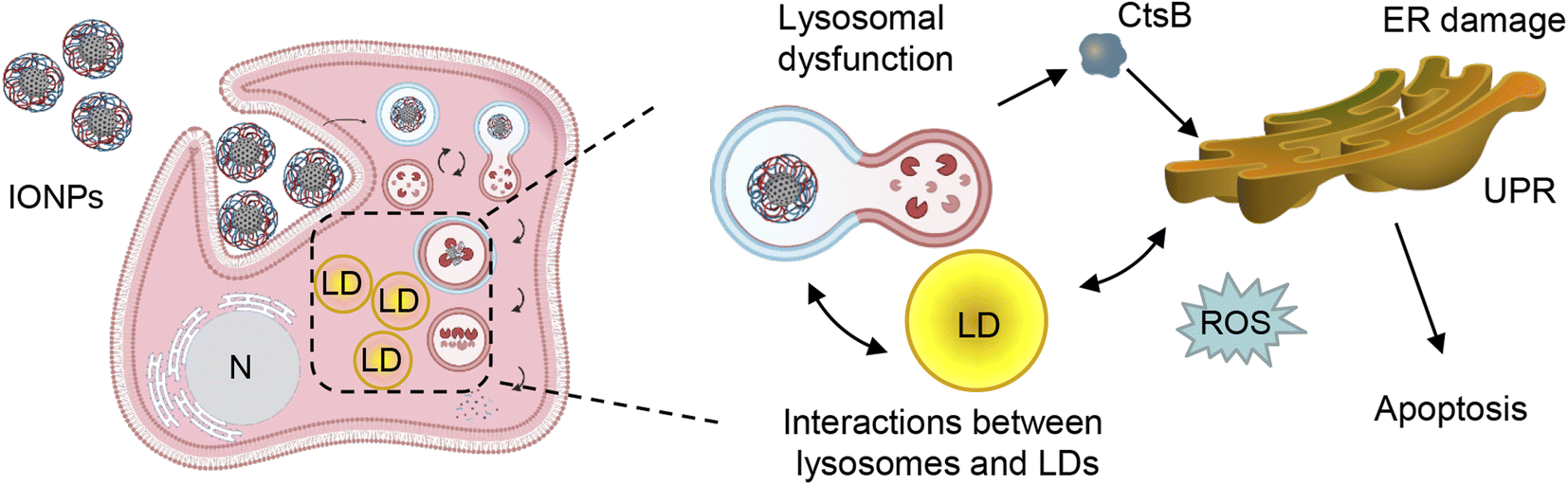 | ||
| Fig. 9 A scheme illustrating the proposed pathway activation after co-treatment with IONPs and fatty acids of Alexander and HepG2 cells. UPR – unfolded protein response; LD – lipid droplets; ER – endoplasmic reticulum; ROS – reactive oxygen species; N – nucleus; CtsB – cathepsin B. Created with https://BioRender.com. | ||
Conclusions
In summary, in this study we explored the potential impact of IONPs on steatotic hepatic (Alexander and HepG2) cells in vitro. Both cell lines treated with OA did not show a response to IONP stimulation. In contrast, co-treatment with PA and IONPs significantly exacerbated cell injury, leading to pronounced cell death. We found that the accumulation of PA and subsequent uptake of IONPs by both cell lines resulted in impaired lysosomal function. This impairment of lysosomal function, combined with lipid accumulation, triggered the execution of ER stress, as evidenced by significant upregulation of CHOP protein levels. The excessive ER stress was accompanied by the release of cathepsin B from lysosomes into the cytosol, which ultimately led to apoptotic cell death.We are fully aware of the limitations of our study. We understand that in vitro analysis bears substantial weaknesses in comparison with in vivo experiments as it fails to replicate the conditions found in a living organism. However, in vitro experiments provide tight control over the chemical and physical environment, reduce costs, offer higher throughput, and minimize animal use. Therefore, for the purpose of unraveling the initial basic molecular determinants of IONP toxicity in fatty acid-loaded hepatic cells, in vitro analysis is justified. Furthermore, in vivo models also have their own limitations when it comes to translating the results to humans.134,135 Specifically for liver injury, currently there is no model (in vitro or in vivo) approved by regulatory agencies in Europe and the US.40 Therefore, we utilized widely used surrogates of hepatocytes in the existing literature, namely the HepG2 and Alexander cell lines.79,80 Our findings indicate the need for further research that would involve in vivo models. For instance, it would be worth to study the reaction of other cell types with IONPs in an in vivo model of NAFLD. Additionally, investigating the interactions between different cell types and the inflammatory response upon IONP treatment in vivo would be of interest.
Indeed, the potential toxicity of IONPs and their adverse effects on human health have been extensively studied. However, we would like to emphasize the importance of conducting studies that analyze toxicity in models considering pathological conditions. While it has been suggested that IONPs may exacerbate liver injury and worsen hepatic steatosis in mice with NAFLD,29,50 the molecular mechanism and sub-cellular targets of such nanoparticle impacts remain poorly investigated. Our study provides a mechanistic explanation for the cytotoxicity triggered by IONPs in steatotic hepatic cells, laying the foundation for future optimization of IONPs in biomedical applications. Our findings provide a novel insight into the toxicological effects of IONPs and shed light on the sub-cellular targets underlying IONP-induced cell injury.
Author contributions
Mariia Uzhytchak: methodology, validation, formal analysis, and review & editing. Mariia Lunova: methodology, validation, formal analysis, and review & editing. Barbora Smolková: formal analysis, validation, and methodology. Milan Jirsa: supervision, formal analysis, validation, and methodology. Alexandr Dejneka: methodology, project administration, review & editing, funding acquisition, and supervision. Oleg Lunov: conceptualization, methodology, data curation, funding acquisition, supervision, and writing-original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Operational Programme Research, Development and Education financed by European Structural and Investment Funds and the Czech Ministry of Education, Youth and Sports (Project No. SOLID21 – CZ.02.1.01/0.0/0.0/16_019/0000760) and MH CZ – DRO Institute for Clinical and Experimental Medicine – IKEM, IN 00023001. Graphical abstract, Fig. 1 and 9 were created using https://Biorender.com.References
- W. Wu, C. Z. Jiang and V. A. Roy, Nanoscale, 2016, 8, 19421–19474 RSC.
- E. T. Ahrens and J. W. M. Bulte, Nat. Rev. Immunol., 2013, 13, 755–763 CrossRef CAS PubMed.
- Y. X. Wang, Quant. Imaging Med. Surg., 2011, 1, 35–40 Search PubMed.
- Y. X. Wang and J. M. Idee, Quant. Imaging Med. Surg., 2017, 7, 88–122 CrossRef PubMed.
- O. Lunov, M. Uzhytchak, B. Smolkova, M. Lunova, M. Jirsa, N. M. Dempsey, A. L. Dias, M. Bonfim, M. Hof, P. Jurkiewicz, Y. Petrenko, S. Kubinova and A. Dejneka, Cancers, 2019, 11, 1873 CrossRef CAS PubMed.
- S. Tong, H. B. Zhu and G. Bao, Mater. Today, 2019, 31, 86–99 CrossRef CAS PubMed.
- A. Farzin, S. A. Etesami, J. Quint, A. Memic and A. Tamayol, Adv. Healthcare Mater., 2020, 9, e1901058 CrossRef PubMed.
- W. Wu, Z. H. Wu, T. Yu, C. Z. Jiang and W. S. Kim, Sci. Technol. Adv. Mater., 2015, 16, 023501 CrossRef PubMed.
- Y. Liu, P. C. Naha, G. Hwang, D. Kim, Y. Huang, A. Simon-Soro, H. I. Jung, Z. Ren, Y. Li, S. Gubara, F. Alawi, D. Zero, A. T. Hara, D. P. Cormode and H. Koo, Nat. Commun., 2018, 9, 2920 CrossRef PubMed.
- C. Kaittanis, T. M. Shaffer, A. Ogirala, S. Santra, J. M. Perez, G. Chiosis, Y. M. Li, L. Josephson and J. Grimm, Nat. Commun., 2014, 5, 3384 CrossRef PubMed.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS PubMed.
- D. Ho, X. L. Sun and S. H. Sun, Acc. Chem. Res., 2011, 44, 875–882 CrossRef CAS PubMed.
- N. Ahmed, H. Fessi and A. Elaissari, Drug Discovery Today, 2012, 17, 928–934 CrossRef CAS PubMed.
- S. Gul, S. B. Khan, I. U. Rehman, M. A. Khan and M. I. Khan, Front. Mater., 2019, 6, 179 CrossRef.
- J. W. M. Bulte, Am. J. Roentgenol., 2009, 193, 314–325 CrossRef PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS PubMed.
- M. Kendall and I. Lynch, Nat. Nanotechnol., 2016, 11, 206–210 CrossRef CAS PubMed.
- A. Frtus, B. Smolkova, M. Uzhytchak, M. Lunova, M. Jirsa, S. Kubinova, A. Dejneka and O. Lunov, J. Controlled Release, 2020, 328, 59–77 CrossRef CAS PubMed.
- M. H. Schwenk, Pharmacotherapy, 2010, 30, 70–79 CrossRef CAS PubMed.
- V. S. Balakrishnan, M. Rao, A. T. Kausz, L. Brenner, B. J. G. Pereira, T. B. Frigo and J. M. Lewis, Eur. J. Clin. Invest., 2009, 39, 489–496 CrossRef CAS PubMed.
- https://www.amagpharma.com/news/amag-pharmaceuticals-announces-fda-approval-of-supplemental-new-drug-application-snda-for-feraheme-ferumoxytol-injection/ .
- M. Uzhytchak, B. Smolkova, M. Lunova, A. Frtus, M. Jirsa, A. Dejneka and O. Lunov, Adv. Drug Delivery Rev., 2023, 197, 114828 CrossRef CAS PubMed.
- R. Rubin, JAMA, J. Am. Med. Assoc., 2015, 313, 1704 Search PubMed.
- https://www.fda.gov/Drugs/Drug-Safety-and-Availability/Fda-Drug-Safety-Communication-Fda-Strengthens-Warnings-and-Changes-Prescribing-Instructions-Decrease .
- G. B. Toth, C. G. Varallyay, A. Horvath, M. R. Bashir, P. L. Choyke, H. E. Daldrup-Link, E. Dosa, J. P. Finn, S. Gahramanov, M. Harisinghani, I. Macdougall, A. Neuwelt, S. S. Vasanawala, P. Ambady, R. Barajas, J. S. Cetas, J. Ciporen, T. J. DeLoughery, N. D. Doolittle, R. W. Fu, J. Grinstead, A. R. Guimaraes, B. E. Hamilton, X. Li, H. L. McConnell, L. L. Muldoon, G. Nesbit, J. P. Netto, D. Petterson, W. D. Rooney, D. Schwartz, L. Szidonya and E. A. Neuwelt, Kidney Int., 2017, 92, 47–66 CrossRef CAS PubMed.
- S. A. Wells, T. Schubert, U. Motosugi, S. D. Sharma, C. A. Campo, S. Kinner, K. M. Woo, D. Hernando and S. B. Reeder, Radiology, 2020, 294, 108–116 CrossRef PubMed.
- B. J. McCullough, O. Kolokythas, J. H. Maki and D. E. Green, J. Magn. Reson. Imaging, 2013, 37, 1476–1479 CrossRef PubMed.
- M. R. Bashir, L. Bhatti, D. Marin and R. C. Nelson, J. Magn. Reson. Imaging, 2015, 41, 884–898 CrossRef PubMed.
- Q. B. Zhou and Y. S. Wei, Chem. Res. Toxicol., 2017, 30, 73–80 Search PubMed.
- U. Ahmed, P. S. Latham and P. S. Oates, World J. Gastroenterol., 2012, 18, 4651–4658 Search PubMed.
- M. Lunova, C. Goehring, D. Kuscuoglu, K. Mueller, Y. Chen, P. Walther, J. C. Deschemin, S. Vaulont, J. Haybaeck, C. Lackner, C. Trautwein and P. Strnad, J. Hepatol., 2014, 61, 633–641 CrossRef CAS PubMed.
- J. M. Fernandez-Real and M. Manco, Lancet Diabetes Endocrinol., 2014, 2, 513–526 CrossRef CAS PubMed.
- A. Mardinoglu, J. Boren, U. Smith, M. Uhlen and J. Nielsen, Nat. Rev. Gastroenterol. Hepatol., 2018, 15, 365–377 CrossRef CAS PubMed.
- G. K. Michalopoulos and B. Bhushan, Nat. Rev. Gastroenterol. Hepatol., 2021, 18, 40–55 CrossRef PubMed.
- E. S. Bjornsson and R. J. Andrade, J. Hepatol., 2022, 76, 435–445 CrossRef PubMed.
- M. I. Lucena, J. Sanabria, M. Garcia-Cortes, C. Stephens and R. J. Andrade, Lancet Gastroenterol. Hepatol., 2020, 5, 862–874 CrossRef PubMed.
- K. M. Tsoi, S. A. MacParland, X. Z. Ma, V. N. Spetzler, J. Echeverri, B. Ouyang, S. M. Fadel, E. A. Sykes, N. Goldaracena, J. M. Kaths, J. B. Conneely, B. A. Alman, M. Selzner, M. A. Ostrowski, O. A. Adeyi, A. Zilman, I. D. McGilvray and W. C. W. Chan, Nat. Mater., 2016, 15, 1212–1221 CrossRef CAS PubMed.
- Y. N. Zhang, W. Poon, A. J. Tavares, I. D. McGilvray and W. C. W. Chan, J. Controlled Release, 2016, 240, 332–348 CrossRef CAS PubMed.
- R. J. Andrade, N. Chalasani, E. S. Bjornsson, A. Suzuki, G. A. Kullak-Ublick, P. B. Watkins, H. Devarbhavi, M. Merz, M. I. Lucena, N. Kaplowitz and G. P. Aithal, Nat. Rev. Dis. Prim., 2019, 5, 58 CrossRef PubMed.
- J. C. Fernandez-Checa, P. Bagnaninchi, H. Ye, P. Sancho-Bru, J. M. Falcon-Perez, F. Royo, C. Garcia-Ruiz, O. Konu, J. Miranda, O. Lunov, A. Dejneka, A. Elfick, A. McDonald, G. J. Sullivan, G. P. Aithal, M. I. Lucena, R. J. Andrade, B. Fromenty, M. Kranendonk, F. J. Cubero and L. J. Nelson, J. Hepatol., 2021, 75, 935–959 CrossRef CAS PubMed.
- G. Rostoker, M. Griuncelli, C. Loridon, R. Couprie, A. Benmaadi, C. Bounhiol, M. Roy, G. Machado, P. Janklewicz, G. Drahi, H. Dahan and Y. Cohen, Am. J. Med., 2012, 125, 991–999 CrossRef PubMed.
- G. Rostoker and N. D. Vaziri, Heliyon, 2019, 5, e02045 CrossRef PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- K. T. Gause, A. K. Wheatley, J. Cui, Y. Yan, S. J. Kent and F. Caruso, ACS Nano, 2017, 11, 54–68 CrossRef CAS PubMed.
- T. J. Anchordoquy, Y. Barenholz, D. Boraschi, M. Chorny, P. Decuzzi, M. A. Dobrovolskaia, Z. S. Farhangrazi, D. Farrell, A. Gabizon, H. Ghandehari, B. Godin, N. M. La-Beck, J. Ljubimova, S. M. Moghimi, L. Pagliaro, J. H. Park, D. Peer, E. Ruoslahti, N. J. Serkova and D. Simberg, ACS Nano, 2017, 11, 12–18 CrossRef CAS PubMed.
- S. Y. Qin, A. Q. Zhang, S. X. Cheng, L. Rong and X. Z. Zhang, Biomaterials, 2017, 112, 234–247 CrossRef CAS PubMed.
- Y. H. Cheng, C. He, J. E. Riviere, N. A. Monteiro-Riviere and Z. Lin, ACS Nano, 2020, 14, 3075–3095 CrossRef CAS PubMed.
- S. Sindhwani, A. M. Syed, J. Ngai, B. R. Kingston, L. Maiorino, J. Rothschild, P. MacMillan, Y. W. Zhang, N. U. Rajesh, T. Hoang, J. L. Y. Wu, S. Wilhelm, A. Zilman, S. Gadde, A. Sulaiman, B. Ouyang, Z. Lin, L. S. Wang, M. Egeblad and W. C. W. Chan, Nat. Mater., 2020, 19, 566–575 CrossRef CAS PubMed.
- M. L. Zhu, H. Q. Chen, S. Zhou, L. N. Zheng, X. Li, R. X. Chu, W. Chen, B. Wang, M. Wang, Z. F. Chai and W. Y. Feng, Nanotoxicology, 2021, 15, 761–778 CrossRef CAS PubMed.
- M. Uzhytchak, A. Lynnyk, V. Zablotskii, N. M. Dempsey, A. L. Dias, M. Bonfim, M. Lunova, M. Jirsa, S. Kubinova, O. Lunov and A. Dejneka, Appl. Phys. Lett., 2017, 111, 243703 CrossRef.
- Y. C. Lu, P. C. Luo, C. W. Huang, Y. L. Leu, T. H. Wang, K. C. Wei, H. E. Wang and Y. H. Ma, Nanoscale, 2014, 6, 10297–10306 RSC.
- M. Marcus, M. Karni, K. Baranes, I. Levy, N. Alon, S. Margel and O. Shefi, J. Nanobiotechnol., 2016, 14, 37 CrossRef PubMed.
- J. Domey, C. Bergemann, S. Bremer-Streck, I. Krumbein, J. R. Reichenbach, U. Teichgraber and I. Hilger, Nanotoxicology, 2016, 10, 20–31 CAS.
- M. Uzhytchak, B. Smolkova, M. Lunova, M. Jirsa, A. Frtus, S. Kubinova, A. Dejneka and O. Lunov, Cells, 2020, 9, 1015 CrossRef CAS PubMed.
- O. Gavet and J. Pines, Dev. Cell, 2010, 18, 533–543 CrossRef CAS PubMed.
- K. Levada, S. Pshenichnikov, A. Omelyanchik, V. Rodionova, A. Nikitin, A. Savchenko, I. Schetinin, D. Zhukov, M. Abakumov, A. Majouga, M. Lunova, M. Jirsa, B. Smolkova, M. Uzhytchak, A. Dejneka and O. Lunov, Nano Convergence, 2020, 7, 17 CrossRef CAS PubMed.
- B. Smolkova, M. Lunova, A. Lynnyk, M. Uzhytchak, O. Churpita, M. Jirsa, S. Kubinova, O. Lunov and A. Dejneka, Cell. Physiol. Biochem., 2019, 52, 119–140 CrossRef CAS PubMed.
- Y. M. A. Naguib, Anal. Biochem., 1998, 265, 290–298 CrossRef CAS PubMed.
- G. P. C. Drummen, L. C. M. van Liebergen, J. A. F. Op den Kamp and J. A. Post, Free Radical Biol. Med., 2002, 33, 473–490 CrossRef CAS PubMed.
- E. H. W. Pap, G. P. C. Drummen, V. J. Winter, T. W. A. Kooij, P. Rijken, K. W. A. Wirtz, J. A. F. Op den Kamp, W. J. Hage and J. A. Post, FEBS Lett., 1999, 453, 278–282 CrossRef CAS PubMed.
- D. R. Beriault and G. H. Werstuck, Biochim. Biophys. Acta, Mol. Cell Res., 2013, 1833, 2293–2301 CrossRef CAS PubMed.
- M. Lunova, B. Smolkova, M. Uzhytchak, K. Z. Janouskova, M. Jirsa, D. Egorova, A. Kulikov, S. Kubinova, A. Dejneka and O. Lunov, Cell. Mol. Life Sci., 2020, 77, 2815–2838 CrossRef CAS PubMed.
- T. Zuliani, R. Duval, C. Jayat, S. Schnebert, P. Andre, M. Dumas and M. H. Ratinaud, Cytometry, Part A, 2003, 54a, 100–108 CrossRef PubMed.
- E. M. Manders, J. Stap, G. J. Brakenhoff, R. van Driel and J. A. Aten, J. Cell Sci., 1992, 103(pt. 3), 857–862 CrossRef CAS PubMed.
- E. T. Arena, C. T. Rueden, M. C. Hiner, S. Wang, M. Yuan and K. W. Eliceiri, Wiley Interdiscip. Rev.: Dev. Biol., 2017, 6, e260 Search PubMed.
- https://Imagej.Net/Coloc_2 .
- O. Lunov, V. Zablotskii, O. Churpita, M. Lunova, M. Jirsa, A. Dejneka and S. Kubinova, Sci. Rep., 2017, 7, 600 CrossRef PubMed.
- J. Jonkman, C. M. Brown, G. D. Wright, K. I. Anderson and A. J. North, Nat. Protoc., 2020, 15, 1585–1611 CrossRef CAS PubMed.
- J. Y. Lee and M. Kitaoka, Mol. Biol. Cell, 2018, 29, 1519–1525 CrossRef CAS PubMed.
- N. Hamilton, Traffic, 2009, 10, 951–961 CrossRef CAS PubMed.
- R. B. Dell, S. Holleran and R. Ramakrishnan, ILAR J., 2002, 43, 207–213 CrossRef CAS PubMed.
- J. Estelrich, M. J. Sanchez-Martin and M. A. Busquets, Int. J. Nanomed., 2015, 10, 1727–1741 CAS.
- H. Wei, O. T. Bruns, M. G. Kaul, E. C. Hansen, M. Barch, A. Wisniowska, O. Chen, Y. Chen, N. Li, S. Okada, J. M. Cordero, M. Heine, C. T. Farrar, D. M. Montana, G. Adam, H. Ittrich, A. Jasanoff, P. Nielsen and M. G. Bawendi, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2325–2330 CrossRef CAS PubMed.
- A. Eftekhari, A. Arjmand, A. Asheghvatan, H. Svajdlenkova, O. Sausa, H. Abiyev, E. Ahmadian, O. Smutok, R. Khalilov, T. Kavetskyy and M. Cucchiarini, Front. Chem., 2021, 9, 674786 CrossRef CAS PubMed.
- W. Ngo, S. Ahmed, C. Blackadar, B. Bussin, Q. Ji, S. M. Mladjenovic, Z. Sepahi and W. C. W. Chan, Adv. Drug Delivery Rev., 2022, 185, 114238 CrossRef CAS PubMed.
- N. Chalasani, Z. Younossi, J. E. Lavine, A. M. Diehl, E. M. Brunt, K. Cusi, M. Charlton and A. J. Sanyal, Gastroenterology, 2012, 142, 1592–1609 CrossRef PubMed.
- Q. R. Lu, X. Y. Tian, H. Wu, J. C. Huang, M. X. Li, Z. B. Mei, L. Zhou, H. Y. Xie and S. S. Zheng, Front. Physiol., 2021, 12, 710420 CrossRef PubMed.
- V. A. Arzumanian, O. I. Kiseleva and E. V. Poverennaya, Int. J. Mol. Sci., 2021, 22, 13135 CrossRef CAS PubMed.
- V. Mersch-Sundermann, S. Knasmuller, X. J. Wu, F. Darroudi and F. Kassie, Toxicology, 2004, 198, 329–340 CrossRef CAS PubMed.
- K. Fukuyama, M. Asagiri, M. Sugimoto, H. Tsushima, S. Seo, K. Taura, S. Uemoto and K. Iwaisako, PLoS One, 2021, 16, e0245939 CrossRef CAS PubMed.
- M. Ricchi, M. R. Odoardi, L. Carulli, C. Anzivino, S. Ballestri, A. Pinetti, L. I. Fantoni, F. Marra, M. Bertolotti, S. Banni, A. Lonardo, N. Carulli and P. Loria, J. Gastroenterol. Hepatol., 2009, 24, 830–840 CrossRef CAS PubMed.
- Y. Tian, H. H. Feng, L. Han, L. Wu, H. M. Lv, B. Y. Shen, Z. Li, Q. L. Zhang and G. W. Liu, Front. Immunol., 2018, 9, 147 CrossRef PubMed.
- X. M. Chen, L. Z. Li, X. H. Liu, R. X. Luo, G. N. Liao, L. Li, J. P. Liu, J. Q. Cheng, Y. R. Lu and Y. N. Chen, Life Sci., 2018, 203, 291–304 CrossRef CAS PubMed.
- A. U. Nissar, L. Sharma and S. A. Tasduq, Toxicol. Res., 2015, 4, 1344–1358 CrossRef CAS.
- O. Khalifa, N. S. AL-Akl, K. Errafii and A. Arredouani, Sci. Rep., 2022, 12, 2226 CrossRef CAS PubMed.
- Y. Cao, M. Roursgaard, A. Kermanizadeh, S. Loft and P. Moller, Int. J. Toxicol., 2015, 34, 67–76 CrossRef CAS PubMed.
- J. B. Jia, F. F. Li, H. Y. Zhou, Y. H. Bai, S. J. Liu, Y. G. Jiang, G. B. Jiang and B. Yan, Environ. Sci. Technol., 2017, 51, 9334–9343 CrossRef CAS PubMed.
- A. Khatchadourian and D. Maysinger, Mol. Pharm., 2009, 6, 1125–1137 CrossRef CAS PubMed.
- T. Sun, Y. Y. Kang, J. Liu, Y. L. Zhang, L. L. Ou, X. N. Liu, R. F. Lai and L. Q. Shao, J. Nanobiotechnol., 2021, 19, 108 CrossRef PubMed.
- H. Malhi, S. F. Bronk, N. W. Werneburg and G. J. Gores, J. Biol. Chem., 2006, 281, 12093–12101 CrossRef CAS PubMed.
- A. Eynaudi, F. Diaz-Castro, J. C. Borquez, R. Bravo-Sagua, V. Parra and R. Troncoso, Front. Nutr., 2021, 8, 775382 CrossRef PubMed.
- J. Kale, E. J. Osterlund and D. W. Andrews, Cell Death Differ., 2018, 25, 65–80 CrossRef CAS PubMed.
- J. Han, Y. Tian, M. Wang, Y. Li, J. Yin, W. Qu, C. Yan, R. Ding, Y. Guan and Q. Wang, Front. Pharmacol., 2022, 13, 1011065 CrossRef CAS PubMed.
- L. Wu, W. Wen, X. F. Wang, D. H. Huang, J. Cao, X. Y. Qi and S. Shen, Part. Fibre Toxicol., 2022, 19, 24 CrossRef CAS PubMed.
- https://ods.od.nih.gov/Factsheets/Iron-Healthprofessional/ .
- M. X. Yu and J. Zheng, ACS Nano, 2015, 9, 6655–6674 CrossRef CAS PubMed.
- Q. Y. Feng, Y. P. Liu, J. Huang, K. Chen, J. X. Huang and K. Xiao, Sci. Rep., 2018, 8, 2082 CrossRef PubMed.
- B. Hamm, T. Staks, M. Taupitz, R. Maibauer, A. Speidel, A. Huppertz, T. Frenzel, R. Lawaczeck, K. J. Wolf and L. Lange, J. Magn. Reson. Imaging, 1994, 4, 659–668 CrossRef CAS PubMed.
- O. Lunov, T. Syrovets, C. Rocker, K. Tron, G. U. Nienhaus, V. Rasche, V. Mailander, K. Landfester and T. Simmet, Biomaterials, 2010, 31, 9015–9022 CrossRef CAS PubMed.
- V. Mirshafiee, B. B. Sun, C. H. Chang, Y. P. Liao, W. Jiang, J. H. Jiang, X. S. Liu, X. Wang, T. Xia and A. E. Nel, ACS Nano, 2018, 12, 3836–3852 CrossRef CAS PubMed.
- Y. Yao, Y. T. Zang, J. Qu, M. Tang and T. Zhang, Int. J. Nanomed., 2019, 14, 8787–8804 CrossRef CAS PubMed.
- L. L. Yang, J. C. Wei, F. Y. Sheng and P. Li, Mol. Nutr. Food Res., 2019, 63, 1801432 CrossRef PubMed.
- R. A. Egnatchik, A. K. Leamy, Y. Noguchi, M. Shiota and J. D. Young, Metab., Clin. Exp., 2014, 63, 283–295 CrossRef CAS PubMed.
- F. J. Wang, R. Gomez-Sintes and P. Boya, Traffic, 2018, 19, 918–931 CrossRef CAS PubMed.
- P. Boya, K. Andreau, D. Poncet, N. Zamzami, J. L. Perfettini, D. Metivier, D. M. Ojcius, M. Jaattela and G. Kroemer, J. Exp. Med., 2003, 197, 1323–1334 CrossRef CAS PubMed.
- B. Büchele, W. Zugmaier, O. Lunov, T. Syrovets, I. Merfort and T. Simmet, Anal. Biochem., 2010, 401, 30–37 CrossRef PubMed.
- V. Baud and M. Karin, Nat. Rev. Drug Discovery, 2009, 8, 33–40 CrossRef CAS PubMed.
- D. Verzella, A. Pescatore, D. Capece, D. Vecchiotti, M. V. Ursini, G. Franzoso, E. Alesse and F. Zazzeroni, Cell Death Dis., 2020, 11, 210 CrossRef PubMed.
- J.-L. Luo, H. Kamata and M. Karin, J. Clin. Invest., 2005, 115, 2625–2632 CrossRef CAS PubMed.
- X. Pan, T. Arumugam, T. Yamamoto, P. A. Levin, V. Ramachandran, B. Ji, G. Lopez-Berestein, P. E. Vivas-Mejia, A. K. Sood, D. J. McConkey and C. D. Logsdon, Clin. Cancer Res., 2008, 14, 8143–8151 CrossRef CAS PubMed.
- Y. Dai, M. Rahmani, P. Dent and S. Grant, Mol. Cell. Biol., 2005, 25, 5429–5444 CrossRef CAS PubMed.
- Y. Shi, S. Y. Wang, M. Yao, W. L. Sai, W. Wu, J. L. Yang, Y. Cai, W. J. Zheng and D. F. Yao, World J. Gastroenterol., 2015, 21, 12814–12821 CrossRef CAS PubMed.
- J. Han and R. J. Kaufman, J. Lipid Res., 2016, 57, 1329–1338 CrossRef CAS PubMed.
- T. Yorimitsu and D. J. Klionsky, Autophagy, 2007, 3, 160–162 CrossRef CAS PubMed.
- Y. Zhang, R. L. Xue, Z. N. Zhang, X. Yang and H. Y. Shi, Lipids Health Dis., 2012, 11, 1 CrossRef PubMed.
- Y. R. Wei, D. Wang, F. Topczewski and M. J. Pagliassotti, Am. J. Physiol.: Endocrinol. Metab., 2006, 291, E275–E281 CrossRef CAS PubMed.
- S. Xu, S. M. Nam, J. H. Kim, R. Das, S. K. Choi, T. T. Nguyen, X. Quan, S. J. Choi, C. H. Chung, E. Y. Lee, I. K. Lee, A. Wiederkehr, C. B. Wollheim, S. K. Cha and K. S. Park, Cell Death Dis., 2015, 6, e1976 CrossRef CAS PubMed.
- C. Lebeaupin, D. Vallee, Y. Hazari, C. Hetz, E. Chevet and B. Bailly-Maitre, J. Hepatol., 2018, 69, 927–947 CrossRef CAS PubMed.
- D. Imrie and K. C. Sadler, J. Hepatol., 2012, 57, 1147–1151 CrossRef CAS PubMed.
- J. A. Gomez and D. T. Rutkowski, eLife, 2016, 5, e20390 CrossRef PubMed.
- X. Gu, K. Li, D. R. Laybutt, M.-l. He, H.-L. Zhao, J. C. N. Chan and G. Xu, Life Sci., 2010, 87, 724–732 CrossRef CAS PubMed.
- W.-C. Huang, Y.-S. Lin, C.-L. Chen, C.-Y. Wang, W.-H. Chiu and C.-F. Lin, J. Pharmacol. Exp. Ther., 2009, 329, 524–531 CrossRef CAS PubMed.
- A. C. Johansson, H. Appelqvist, C. Nilsson, K. Kagedal, K. Roberg and K. Ollinger, Apoptosis, 2010, 15, 527–540 CrossRef CAS PubMed.
- E. Scorletti and R. M. M. Carr, J. Hepatol., 2022, 76, 934–945 CrossRef CAS PubMed.
- J. A. Olzmann and P. Carvalho, Nat. Rev. Mol. Cell Biol., 2019, 20, 137–155 CrossRef CAS PubMed.
- R. J. Schulze, E. W. Krueger, S. G. Weller, K. M. Johnson, C. A. Casey, M. B. Schott and M. A. McNiven, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 32443–32452 CrossRef CAS PubMed.
- E. Rampanelli, P. Ochodnicky, P. C. Johannes, L. M. Butter, N. Claessen, A. Calcagni, L. Kors, L. A. Gethings, S. J. L. Bakker, M. H. de Borst, G. J. Navis, G. Liebisch, D. Speijer, M. A. V. Weerman, B. Jung, J. Aten, E. Steenbergen, G. Schmitz, A. Ballabio, S. Florquin, J. M. F. G. Aerts and J. C. Leemans, J. Pathol., 2018, 246, 470–484 CrossRef CAS PubMed.
- T. Yamamoto, Y. Takabatake, A. Takahashi, T. Kimura, T. Namba, J. Matsuda, S. Minami, J. Y. Kaimori, I. Matsui, T. Matsusaka, F. Niimura, M. Yanagita and Y. Isaka, J. Am. Soc. Nephrol., 2017, 28, 1534–1551 CrossRef CAS PubMed.
- S. Zhang, X. Q. Peng, S. Yang, X. Y. Li, M. Y. Huang, S. B. Wei, J. X. Liu, G. P. He, H. Y. Zheng, L. Yang, H. Y. Li and Q. Fan, Cell Death Dis., 2022, 13, 132 CrossRef CAS PubMed.
- Z. Q. Zhang, P. F. Yue, T. Q. Lu, Y. Wang, Y. Q. Wei and X. W. Wei, J. Hematol. Oncol., 2021, 14, 79 CrossRef PubMed.
- S. N. Fu, L. Yang, P. Li, O. Hofmann, L. Dicker, W. Hide, X. H. Lin, S. M. Watkins, A. R. Ivanov and G. S. Hotamisligil, Nature, 2011, 473, 528–531 CrossRef CAS PubMed.
- S. Hayashi and Y. Okada, Mol. Biol. Cell, 2015, 26, 1743–1751 CrossRef CAS PubMed.
- D. Reijnders, G. H. Goossens, G. D. A. Hermes, E. P. J. G. Neis, C. M. van der Beek, J. Most, J. J. Holst, K. Lenaerts, R. S. Kootte, M. Nieuwdorp, A. K. Groen, S. W. M. O. Damink, M. V. Boekschoten, H. Smidt, E. G. Zoetendal, C. H. C. Dejong and E. E. Blaak, Cell Metab., 2016, 24, 63–74 CrossRef CAS PubMed.
- H. S. Warren, R. G. Tompkins, L. L. Moldawer, J. Seok, W. Xu, M. N. Mindrinos, R. V. Maier, W. Xiao and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E345 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: List of chemical probes used in the study (Table S1). List of fluorescent probes used in the study (Table S2). List of antibodies used in the study (Table S3). Analysis of ROS induction in Alexander cells upon co-treatment with IONPs and fatty acids (Fig. S1). Analysis of ROS induction in HepG2 cells upon co-treatment with IONPs and fatty acids (Fig. S2). Analysis of lipid peroxidation and lysosomal integrity in hepatic cell lines upon co-treatment with IONPs and fatty acids (Fig. S3). Effect of co-treatment with IONPs and fatty acids on the morphology of lysosomes and mitochondria in Alexander cells (Fig. S4). Effect of co-treatment with IONPs and fatty acids on the morphology of lysosomes and mitochondria in HepG2 cells (Fig. S5). Assessment of mitochondrial membrane depolarization upon co-treatment with IONPs and fatty acids on the morphology of lysosomes and mitochondria in Alexander cells (Fig. S6). Assessment of mitochondrial membrane depolarization upon co-treatment with IONPs and fatty acids on the morphology of lysosomes and mitochondria in HepG2 cells (Fig. S7). Representative flow charts of annexin V and propidium iodide staining of Alexander and HepG2 cells (Fig. S8). Cathepsin B release analysis of Alexander cells co-treated with IONPs and fatty acids (Fig. S9). Cathepsin B release analysis of HepG2 cells co-treated with IONPs and fatty acids (Fig. S10). Uncropped immunoblot scans. See DOI: https://doi.org/10.1039/d3na00071k |
| ‡ These authors contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2023 |