
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of porous transition metal hydroxides from a poly(4-vinyl pyridine) film by controlling pH†
Gyeongwon
Ha‡
a,
Jaeyong
Lee‡
a,
Keon-Woo
Kim
a,
Chungryong
Choi
b and
Jin Kon
Kim
 *a
*a
aDepartment of Chemical Engineering, National Creative Research Initiative Center for Hybrid Nano Materials By High-level Architectural Design of Block Copolymer, Pohang University of Science and Technology, Pohang, Republic of Korea. E-mail: jkkim@postech.ac.kr
bDepartment of Polymer Science and Engineering, Kumoh National Institute of Technology, 61 Daehak-ro, Gumi, Gyeongbuk 39177, Republic of Korea
First published on 4th April 2023
Abstract
Non-noble transition metal hydroxides have been widely used in electrochemical devices because of low cost and multiple redox states. In particular, self-supported porous transition metal hydroxides are used to improve the electrical conductivity, as well as achieving fast electron and mass transfer and a large effective surface area. Herein, we introduce facile synthesis of self-supported porous transition metal hydroxides using a poly(4-vinyl pyridine) (P4VP) film. We used metal cyanide as a transition metal precursor capable of forming metal hydroxide anions in aqueous solution, which is the seed for transition metal hydroxides. To increase the coordination between P4VP and the transition metal cyanide precursors, we dissolved the precursors in buffer solutions with various pH. When the P4VP film was immersed in the precursor solution with lower pH, the metal cyanide precursors were sufficiently coordinated with the protonated nitrogen in P4VP. When reactive ion etching was performed on the precursor-containing P4VP film, the P4VP region without coordination was etched out and became pores. Then, the coordinated precursors were aggregated as metal hydroxide seeds and became the metal hydroxide backbone, resulting in the formation of porous transition metal hydroxide structures. We successfully fabricated various self-supported porous transition metal hydroxides (Ni(OH)2, Co(OH)2, and FeOOH). Finally, we prepared a pseudo-capacitor based on self-supported porous Ni(OH)2, which showed a good specific capacitance (780 F g−1 at 5 A g−1).
Introduction
Transition metal-based materials (oxides and hydroxides) have attracted significant attention in various electrochemical applications due to their low cost, high stability, and versatility in composition and morphology. In particular, transition metal hydroxides with various structures, such as layers,1,2 particles,3,4 sheets,5,6 wires,7–9 and porous structures,10–13 have been synthesized and widely used for water splitting,6,14,15 supercapacitors,5,16–18 energy storage materials,7,19–21 catalysts,22,23 and adsorbing contaminants,24 owing to their multiple redox states.Conventionally, transition metal hydroxide-containing electrodes are prepared from a mixture of active transition metal hydroxides and carbon binders onto current collectors.25–28 However, due to the peeling off of the mixture from the current collector and a serious interfacial resistance between transition metal hydroxides and a collector, the stability of the device is poor. In addition, the carbon binder reduces the active surface area and mass transfer of active ions and electrons. To overcome these disadvantages, transition metal hydroxides are constructed directly on an electrode without using the carbon binder, which is referred to as a self-supported electrode. Self-supported electrodes have several benefits compared with commercial electrodes made of powders,29,30 for instance, a simple process to make an electrode, fast electron and mass transfer, and a large effective surface area.
Especially, self-supported porous transition metal hydroxides are desirable, because they provide more active sites for the charge transfer reaction and ion adsorption.31 In addition, they can effectively relax the built-in stress of the active material during the repeated electrochemical reaction. Self-supported porous transition metal hydroxides have been synthesized by various methods: template-free synthesis (hydro-thermal32,33 and solvo-thermal34,35) and template-mediated synthesis, where an anodic aluminum oxide (AAO)36,37 membrane has been widely used as a template, and synthesis from a self-assembled block copolymer (BCP).38
For template-free synthesis, though hydro- (or solvo-) thermal methods provide a uniform product on a substrate directly, high temperature and pressure conditions as well as long reaction time (several hours) are needed. Also, the use of pre-seeds is required to grow self-supported transition metal hydroxides on a substrate surface. For template-mediated synthesis, AAO provides a high-purity and well-ordered structure, but needs one more step for removing the template after growing metal hydroxide structures. Self-assembled BCPs including functional groups (oxygen or nitrogen) capable of coordinating with metal precursors have been used to synthesize porous transition metal hydroxides. This method provides uniform pore size and can be applied to most substrates because of the relatively simple solution process. Takahashi and co-workers successfully synthesized various mesoporous transition metal hydroxide (Cr, Mn, Fe, Co, Ni, Cu, etc.) films on different substrates (silicon, glass, and ITO) by utilizing evaporation induced self-assembly of polyethylene oxide-block-polypropylene oxide-block-poly(ethylene oxide) copolymer (F127: BASF) and the precursors. However, the synthesis of a well-defined block copolymer with a controlled volume fraction of each block needs an additional synthetic step compared with a homopolymer, and self-assembled structures of thin films are achieved by thermal (or solvent) annealing. Thus, to synthesize porous metal hydroxides directly on a substrate, a homopolymer instead of a block copolymer is more desirable.
Here, we demonstrate the facile synthesis of self-supported porous transition metal hydroxides based on homopolymer templates which do not require high temperature and pressure, or preparation and removal of the well-ordered template. For this purpose, we chose metal cyanide precursors capable of forming metal hydroxide anions in aqueous solution and poly(4-vinyl pyridine) (P4VP) as a homopolymer template. P4VP was selected because the nitrogen atom in P4VP becomes easily protonated and coordinated with metal precursors.39–41 However, cyanide-containing metal precursors are very difficult in coordinating with P4VP due to poor protonation of the N atom in P4VP. This is because of basic solution when the precursors are dissolved in aqueous solution. In this study, we solved this problem by increasing the degree of the protonation of the N atom through the adjusting of pH in the precursor solution.
We successfully fabricated various self-supported porous transition metal hydroxides (Ni(OH)2, Co(OH)2, and FeOOH) after coordinating transition metal cyanide with the N atom in the P4VP polymer film in controlled pH buffer solutions followed by the removal of P4VP by reactive ion etching (RIE). Pores were formed after removing uncoordinated P4VP homopolymer by RIE. On the other hand, the coordinated precursors were aggregated as metal hydroxide seeds and then became the metal hydroxide backbone, resulting in the formation of self-supported porous transition metal hydroxide structures. We found that self-supported porous transition metal hydroxides were fabricated only under weakly acidic conditions (pH 7–5). Among many applications based on porous metal hydroxides, we prepared a supercapacitor made of self-supported porous Ni(OH)2, which showed a good specific capacitance (780 F g−1 at 5 A g−1) even though the device was not optimized for electrochemical performance.
Experimental section
P4VP synthesis and molecular characterization
All chemicals were purchased from Sigma Aldrich. P4VP was synthesized by reversible addition fragmentation chain transfer polymerization, and the detailed method is described in the ESI.† The molecular characteristics of P4VP were measured by size exclusion chromatography (SEC: 410 refractive index detector, Waters) using poly(2-vinyl pyridine) standards in dimethylformamide (DMF). The number-average molecular weight (Mn) and dispersity (Đ) of P4VP was 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and 1.2, respectively. The chemical structure was investigated by using a 1H nuclear magnetic resonance spectrum (1H NMR: AVANCE III 400 MHz, Bruker) (Fig. S1a†).
000 g mol−1 and 1.2, respectively. The chemical structure was investigated by using a 1H nuclear magnetic resonance spectrum (1H NMR: AVANCE III 400 MHz, Bruker) (Fig. S1a†).
pH of precursor solutions
Three precursors [nickel (K2Ni(CN)4), cobalt (K3Co(CN)6), and iron (K4Fe(CN)6)] were purchased from Sigma Aldrich. When they were dissolved in DI water alone, pH values measured with a pH meter (PH-8100PLUS, NAVMRO) of 0.1 M K2Ni(CN)4, K3Co(CN)6 and K4Fe(CN)6 were 9.1, 9.0, and 8.4, respectively. Because the protonation of the N atom in P4VP is difficult in the basic solution, we controlled the pH of the precursor solution by using buffer solution (Samchun Chemicals) with various pH ranging from 4 to 7. The concentration of all precursors in the buffer solution was fixed at 0.1 M.Fabrication of porous metal hydroxide structures and characterization
P4VP dissolved in DMF at a concentration of 10 wt% was spin-coated on ITO glass. The spin-coated P4VP film on the ITO glass was immersed in each precursor solution for 20 min. The uncoordinated precursors were removed by rinsing with DI water. Then, O2 RIE (VITA, Femto Science) was performed on the precursor-containing P4VP film to fabricate self-supported porous metal hydroxide structures (250 W and 100 sccm for 5–10 min depending on film thickness).Fourier transform infrared (FTIR: Vertex 70v, Bruker) was used to check the coordination of the precursors and P4VP. The fabricated metal hydroxide structures were observed by using a field emission scanning electron microscope (FESEM: S4800, Hitachi). The chemical composition and oxidation states of the structure were analyzed through X-ray photoelectron spectroscopy (XPS: Vg Escalab 250, Thermo Fisher Scientific). The C 1s binding energy (284.8 eV) was used to normalize all of XPS spectra.
Electrochemical measurement
Three electrode configurations with 1 M KOH solution were used. The fabricated porous metal hydroxide structures (1 cm2) on ITO, a Hg/HgO electrode, and a platinum electrode were used as working, reference, and counter electrodes, respectively. Cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) measurements were carried out with a potentiostat (Wave Driver 10, Pine Instrument).The specific capacitance (Csp) of the working electrode was calculated from GCD curves as follows:
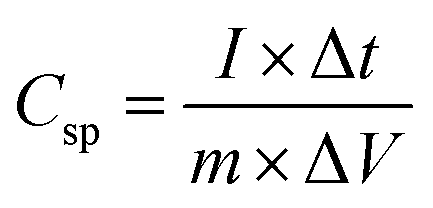
Results and discussion
Fig. 1a shows a schematic of the fabrication of self-supported porous transition metal hydroxides. When the P4VP film was immersed in a transition metal cyanide precursor solution, it was swollen by the solution and became easy to coordinate with the precursor. After the uncoordinated precursors were rinsed followed by drying, the thickness of the film increased to 380 nm from 260 nm (Fig. S2†). On the other hand, when the P4VP film was simply immersed into precursor-free solution followed by drying, the final film thickness did not change. Thus, the increased thickness originated from the coordinated precursors, because there was no void in the coordinated film (Fig. 1b). Based on this argument, 31 volume% of the film was coordinated with the precursors (see ESI S2†). Then, O2 RIE was performed on the P4VP film coordinated with the precursor to obtain a self-supported porous metal hydroxide structure. When O2 RIE was performed on the precursor-containing P4VP film, the uncoordinated P4VP film was etched out. On the other hand, coordinated precursors were aggregated as metal hydroxide seeds and then became the metal hydroxide backbone, which resulted in a gradual decrease in the total film thickness with increased RIE time (Fig. S3 in the ESI†). Because the etching of the P4VP film started from the top film surface, porous metal hydroxide structures were first formed near the top surface and finally became self-supported porous metal hydroxides throughout the entire film thickness at sufficiently long RIE time (5 min) (Fig. 1c–e). However, when we conducted dip-coating followed by heat treatment alone at 350 °C without O2 RIE to remove P4VP, particular Ni(OH)2 structures were observed; thus, self-supported porous structures were not formed, as shown in Fig. S4.†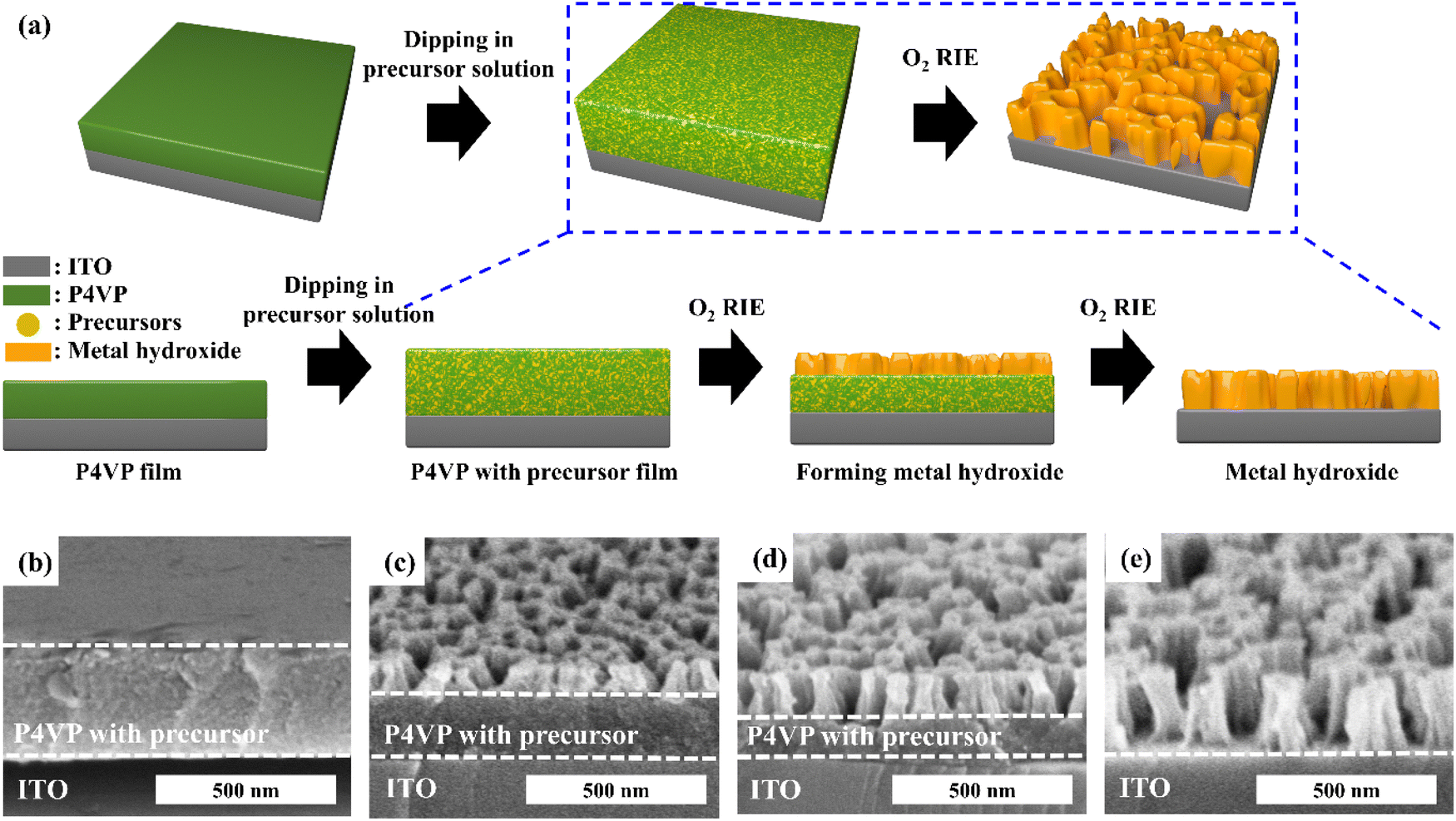 | ||
| Fig. 1 (a) Schematic of the fabrication of self-supported porous transition metal hydroxides. 60° tilt views of the FESEM image of porous nickel hydroxide prepared from K2Ni(CN)4 solution with pH = 5 depending on O2 RIE time: (b) before RIE, (c) 1 min, (d) 2 min, and (e) 5 min. The original film thickness is 260 nm. | ||
We studied the effect of pH of the precursor solution on the self-supported porous transition metal structures. Fig. 2 shows the Ni(OH)2 structure depending on the pH of the precursor solution. When DI water was used, the pH of the solution became 9.1 due to the CN group in the nickel precursor. In this situation, the Ni(OH)2 structure was not formed, because almost all N atoms in P4VP were not protonated. Thus, because almost all nickel precursors were not coordinated with N atoms, the whole film was etched out (Fig. 2a). At a pH of the precursor solution = 7, Ni(OH)2 started to form a discontinuous structure (Fig. 2b). When pH became lower than 6, porous Ni(OH)2 structures were formed because of increased amounts of coordinated precursors with N atoms in the P4VP film (Fig. 2c and d). On the other hand, when the pH became too low (say 4), some of the dissociated precursors were precipitated as solid states such as Ni(CN)2,42 which prevented the precursors from coordination with the protonated N atoms in P4VP. Thus, porous Ni(OH)2 was not obtained (Fig. S5†). Fig. 2e shows the area occupied by the nickel hydroxide on the surface and the height of the nickel hydroxide structure depending on the pH of the precursor solution. As the pH decreased, the nickel hydroxide had a larger surface area and higher thickness because a larger amount of the nickel precursor was coordinated.
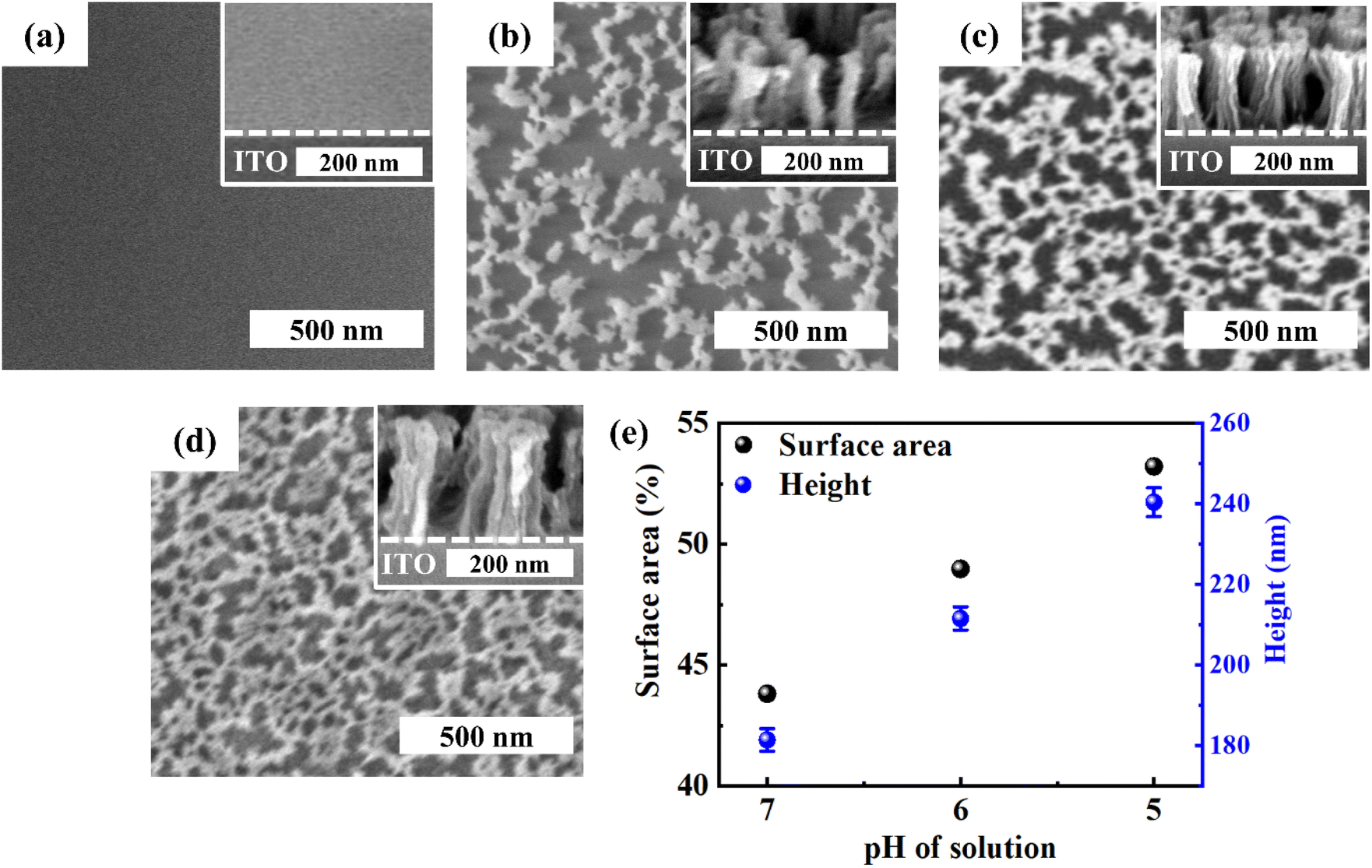 | ||
| Fig. 2 Top and 60° tilt (inset) views of FESEM images of Ni(OH)2 structures depending on the pH of the K2Ni(CN)4 solutions: (a) pH = 9.1, (b) pH = 7, (c) pH = 6, and (d) pH = 5. (e) The surface area occupied by Ni(OH)2 and the height of the structures. Occupied surface area is calculated by using Image J. | ||
These results could be explained by different degrees of coordination and dissociation of precursors depending on pH. Potassium metal cyanide precursors (Kx[M(CN)y]) (M = Ni, Fe, and Co) were dissociated in aqueous solution as follows:
| Kx[M(CN)y] → xK+ + [M(CN)y]x− | (1) |
The cyanide complex anion may undergo further dissociation and release cyanide ions (and finally hydroxy ions) as follows:
| [M(CN)y]x− → zCN− + Mn+ + [M(CN)m]x− | (2a) |
| CN− + H2O → HCN + OH− | (2b) |
Those hydroxy ions react with metal cyanide anions, producing metal hydroxide cyanide anions ([M(OH)l(CN)m−l]x−) (eqn (3a)). But, some metal ions could directly combine with cyanide ions to form metal cyanide (M(CN)n) precipitates (eqn (3b)).
| [M(CN)m]x− + lOH− → [M(OH)l(CN)m−l]x− + kCN− | (3a) |
| Mn+ + nCN− → M(CN)n(s) | (3b) |
We consider that the coordination between these metal hydroxide cyanide anions and protonated nitrogen (N+) in P4VP is the key factor for synthesizing porous transition metal hydroxides. All of the above reactions are known to increase as the pH decreases.43 Also, the amount of N+ in P4VP increases with decreasing pH. Thus, under basic conditions (pH > 7), the number of N+ and metal hydroxide cyanide anions is too small so that the degree of coordination between P4VP and metal hydroxide cyanide anions is insufficient to form metal hydroxides. With decreasing pH from 7 to 5, the number of both N+ and metal hydroxide cyanide anions increases, which results in increased coordination. However, under strongly acidic conditions (pH < 5), though the amount of N+ and metal hydroxide cyanide anions for the coordination is sufficient, the amount of precipitated metal cyanide is also increased,42–44 as is evident from Fig. S6.† Because this precipitate prevents the effective coordination between N+ and the metal cyanide precursor, porous metal hydroxide structures are not formed (Fig. S5†). Therefore, to obtain metal hydroxides from the precursor containing the cyanide group, a moderate pH is needed.
We also investigated the formation of Ni(OH)2 structures depending on the initial P4VP film thickness and dipping time with a buffer solution having pH = 5. We observed that porous Ni(OH)2 structures were formed regardless of the initial P4VP film thickness. Also, the height of porous Ni(OH)2 structures increased with increasing initial P4VP film thickness as the height of the initial P4VP increased (Fig. S7†). When the dipping time was short, the amount of coordination was small. Thus, Ni(OH)2 showed individual particles. However, as the dipping time increased, a sufficient amount of coordination occurred, resulting in the formation of porous Ni(OH)2 structures. The coordination became saturated within a short immersion time (10 min) (Fig. S8†).
The amount of coordinated metal hydroxide with N+ was checked by FTIR. Since the metal hydroxide anion contains a CN group, the strong intensity of the CN group peak indicates a large coordination. In fact, as shown in Fig. S9,† the peak intensity of the CN group increased with decreasing pH.
We also successfully synthesized self-supported porous cobalt and iron (oxy)hydroxides using the corresponding cyanide-containing precursors, as shown in Fig. 3. When we used a precursor solution in pure DI water where pH is ∼9, porous structures were not formed due to very small amount of N+, which resulted in very low coordination with the precursors. Similar to the nickel hydroxide FTIR results, the CN group peak indicating the amount of coordination increased with decreasing pH as shown in Fig. S10.† However, unlike nickel hydroxide structures, cobalt hydroxide and iron oxyhydroxide structures need more acidic conditions, because cobalt and iron cyanide precursors are more stable than nickel at fixed pH.45 Thus, both have a lower degree of dissociation than nickel in aqueous solution to form metal hydroxide cyanide anions.
 | ||
| Fig. 3 Top and 60° tilt (inset) views of FESEM images of Co(OH)2 (upper panel) and FeOOH (lower panel) depending on the pH of the corresponding precursor solutions. (a and e) DI water, (b and f) buffer solution with pH = 7, (c and g) buffer solution with pH = 6, and (d and h) buffer solution with pH = 5. | ||
Fig. 4 shows the chemical state identified by XPS. Clear Ni 2p, O 1s, and C 1s peaks indicate Ni(OH)2 (Fig. 4a–c). Peak fitting analysis of Ni 2p, Ni(OH)2 revealed one Ni2+ state for Ni at binding energies of 855.5 and 873.5 eV with a spin energy separation of 18 eV, each binding energy representing Ni 2p3/2 and Ni 2p1/2, respectively. The intense shake-up satellite peaks at 861.8 and 880.0 eV provided additional evidence for the presence of Ni2+. The O 1s spectra of Ni(OH)2 fit well with the binding energy of 530.9 eV, indicating that the oxygen is present in the form of a hydroxide (Fig. 4c).7Fig. 4d and g clearly show the peaks of Co 2p and Fe 2p, and O 1s and C 1s of Co(OH)2 and FeOOH. In the case of Co(OH)2, the two main peaks at 781.3 and 797.3 eV correspond Co 2p3/2 and 2p1/2, respectively, and two satellite peaks at 785.8 and 801.7 eV were observed (Fig. 4e). Also, the O 1s spectra peak of 531.4 eV indicates that the oxygen is present in the form of a hydroxide (Fig. 4f).46Fig. 4h–i show the detailed XPS peaks of FeOOH. Two main peaks located at 711.6 and 725.2 eV with two satellites peaks at 719.5 and 733.2 eV were observed (Fig. 4h). In addition, three deconvoluted peaks, Fe–O–Fe, Fe–O–H, and H–O–H, were observed at 529.8, 531.8 and 533.4 eV, respectively (Fig. 4i).47 These results show that FeOOH was successfully synthesized.
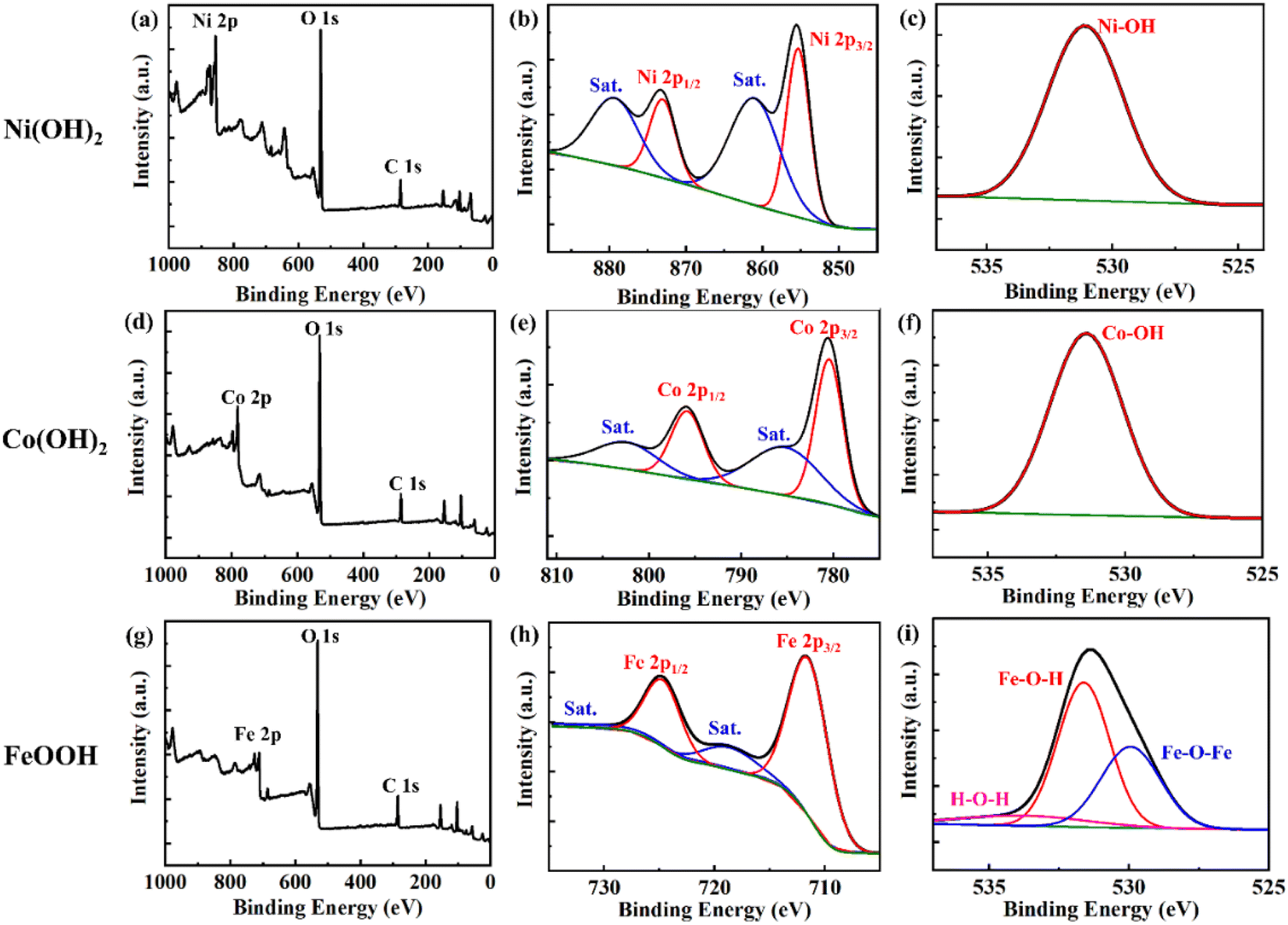 | ||
| Fig. 4 XPS spectra of the three metal hydroxides. (a) XPS survey spectra, (b) Ni 2p, and (c) O 1s spectra of Ni(OH)2. (d) XPS survey, (e) Co 2p, and (f) O 1s spectra of Co(OH)2. (g) XPS survey, (h) Fe 2p, and (i) O 1s spectra of FeOOH. | ||
Among the three self-supported porous metal hydroxides, Ni(OH)2 was selected for electrochemical experiments. We used a three-electrode configuration in 1.0 M KOH aqueous solution. We used three self-supported porous Ni(OH)2 structures prepared at different pHs of the precursor solution. It is known that the electrochemical performance of the metal (or metal-based) structure with a porous structure is better than that without a porous structure because of the large surface area.47Fig. 5a shows CV curves at a scan rate 10 mV s−1 of the porous Ni(OH)2 structure prepared at different pH of the precursor solution. Oxidation and reduction peaks were clearly observed according to the faradaic reaction of Ni(OH)2 as follows:
| Ni(OH)2 + OH− ↔ NiOOH + H2O + e− | (4) |
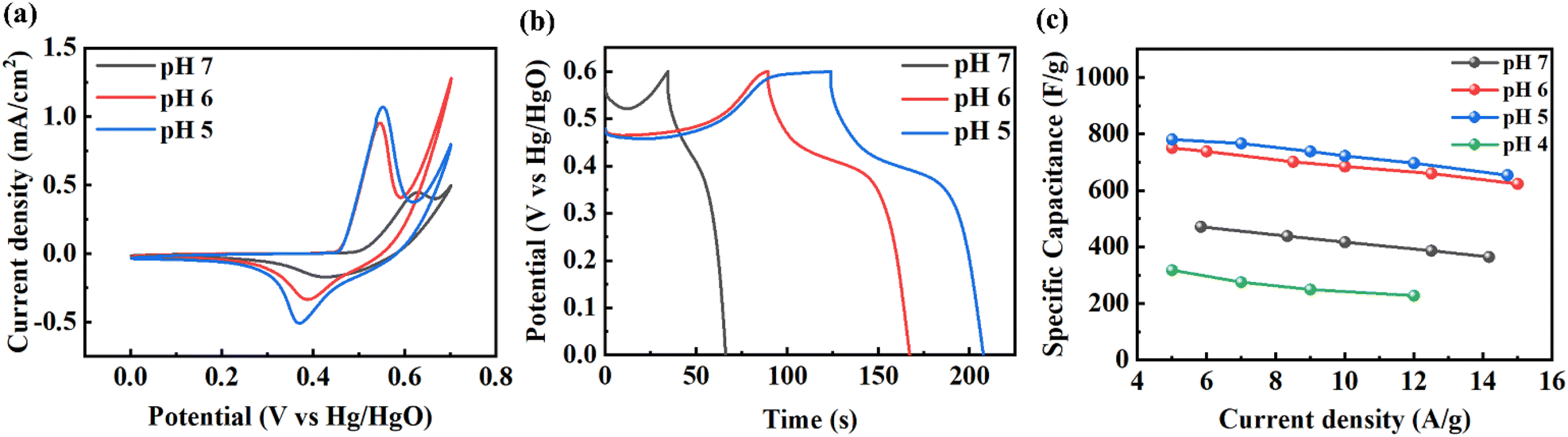 | ||
| Fig. 5 Electrochemical measurement of the porous Ni(OH)2 structure prepared at various pH of precursor solution. (a) Cyclic voltammetry (CV) curves at a scan rate of 10 mV s−1, (b) galvanostatic charge–discharge (GCD) curves at a current density of 100 μA cm−2, and (c) specific capacitance at various current densities. | ||
As the pH of the precursor solution decreased from 7 to 6, the position of the redox peaks slightly moved to the left side, because particle structures were changed to porous structures. However, as the pH of the precursor solution was further decreased to 5, the surface area of Ni(OH)2 increased and the porous structure was maintained. Thus, the position of the peak was almost constant and had a larger CV area. Fig. 5b shows the charge–discharge curves at a current density of 100 μA cm−2. The energy storage of Ni(OH)2 shows a pseudocapacitive characteristic dominated by the faradaic reaction, since all samples show a stable discharge peak at a voltage near 0.4 V related to the reduction process of Ni(OH)2. From GCD, we obtained specific capacitance, as shown in Fig. 5c. Similar to CV and GCD plots, this also shows better efficiency as the pH decreased, indicating that the porous structure improved the performance. In the case of pH = 4, though most of the structure was covered by Ni(CN)2, a small amount of Ni(OH)2 remained. But, the electrochemical performance was even poorer than that prepared at pH = 7, where the particulate structures were formed. As a result, we obtained a maximum specific capacitance of 780 F g−1 at 5 A g−1. This value is better than the reported values of supercapacitors prepared from self-supported Ni(OH)2 on various substrates (Table S1†).
Conclusion
We introduced a facile synthesis of self-supported porous transition metal hydroxides with a large surface area using a P4VP film after dipping transition metal cyanide precursors in controlled pH buffer solution. Since the coordination between the transition metal precursors and the nitrogen atom in P4VP is affected by pH, self-supported porous metal hydroxides could be obtained only at controlled pH (pH 7–pH 5). By using different transition metal precursors, we synthesized diverse self-supported porous transition metal hydroxides (Ni(OH)2, Co(OH)2 and FeOOH). A pseudo-capacitor based on porous Ni(OH)2 showed a good specific capacitance (780 F g−1 at 5 A g−1) without device optimization.Author contributions
Gyeongwon Ha and Jaeyong Lee performed research and wrote the paper, Keon-woo Kim analysed the data, Chungryong Choi discussed the results, and Jin Kon Kim conceived the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Creative Research Initiative Program supported by the National Research Foundation of Korea (No. 2022R1A3A3002149).Notes and references
- G. R. Williams and D. O'Hare, J. Mater. Chem., 2006, 16, 3065–3074 RSC.
- M.-Q. Zhao, G.-L. Tian, Q. Zhang, J.-Q. Huang, J.-Q. Nie and F. Wei, Nanoscale, 2012, 4, 2470–2477 RSC.
- G. M. Koenig Jr, I. Belharouak, H. Deng, Y.-K. Sun and K. Amine, Chem. Mater., 2011, 23, 1954–1963 CrossRef.
- G. Lee, C. V. Varanasi and J. Liu, Nanoscale, 2015, 7, 3181–3188 RSC.
- Z. Pan, Y. Jiang, P. Yang, Z. Wu, W. Tian, L. Liu, Y. Song, Q. Gu, D. Sun and L. Hu, ACS Nano, 2018, 12, 2968–2979 CrossRef CAS PubMed.
- Y. Jia, L. Zhang, G. Gao, H. Chen, B. Wang, J. Zhou, M. T. Soo, M. Hong, X. Yan and G. Qian, Adv. Mater., 2017, 29, 1700017 CrossRef PubMed.
- T. Zhou, Z. Cao, P. Zhang, H. Ma, Z. Gao, H. Wang, Y. Lu, J. He and Y. Zhao, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
- F. Chen, H. Wang, S. Ji, V. Linkov and R. Wang, Chem. Eng. J., 2018, 345, 48–57 CrossRef CAS.
- W. Xu, R. Lan, D. Du, J. Humphreys, M. Walker, Z. Wu, H. Wang and S. Tao, Appl. Catal., B, 2017, 218, 470–479 CrossRef CAS.
- Y. Wang, B. Zhang, W. Pan, H. Ma and J. Zhang, ChemSusChem, 2017, 10, 4170–4177 CrossRef CAS PubMed.
- X. Xia, J. Tu, Y. Zhang, J. Chen, X. Wang, C. Gu, C. Guan, J. Luo and H. J. Fan, Chem. Mater., 2012, 24, 3793–3799 CrossRef CAS.
- J. Xiao and S. Yang, RSC Adv., 2011, 1, 588–595 RSC.
- D.-S. Kong, J.-M. Wang, H.-B. Shao, J.-Q. Zhang and C.-n. Cao, J. Alloys Compd., 2011, 509, 5611–5616 CrossRef CAS.
- P. Babar, A. Lokhande, V. Karade, B. Pawar, M. G. Gang, S. Pawar and J. H. Kim, ACS Sustain. Chem. Eng., 2019, 7, 10035–10043 CrossRef CAS.
- W. Ma, R. Ma, C. Wang, J. Liang, X. Liu, K. Zhou and T. Sasaki, ACS Nano, 2015, 9, 1977–1984 CrossRef CAS PubMed.
- R. R. Salunkhe, K. Jang, S.-w. Lee, S. Yu and H. Ahn, J. Mater. Chem., 2012, 22, 21630–21635 RSC.
- Y. Zhu, C. Cao, S. Tao, W. Chu, Z. Wu and Y. Li, Sci. Rep., 2014, 4, 1–7 Search PubMed.
- X. Wang, C. Yan, A. Sumboja, J. Yan and P. S. Lee, Adv. Energy Mater., 2014, 4, 1301240 CrossRef.
- R. Chen, J. Xue, X. Gao, C. Yu, Q. Chen, J. Zhou, G. Sun and W. Huang, Nanoscale, 2020, 12, 22075–22081 RSC.
- R. Patel, J. T. Park, M. Patel, J. K. Dash, E. B. Gowd, R. Karpoormath, A. Mishra, J. Kwak and J. H. Kim, J. Mater. Chem. A, 2018, 6, 12–29 RSC.
- D. P. Dubal, R. Holze and P. Gomez-Romero, Sci. Rep., 2014, 4, 1–10 Search PubMed.
- Y. Gao, X. Liu and G. Yang, Nanoscale, 2016, 8, 5015–5023 RSC.
- X. Chen, H. Wang, R. Meng, B. Xia and Z. Ma, ACS Appl. Energy Mater., 2020, 3, 1305–1310 CrossRef CAS.
- X. Guo, P. Yin, K. Kanamori, K. Nakanishi and H. Yang, J. Sol-Gel Sci. Technol., 2018, 88, 114–128 CrossRef CAS.
- T. Mukhiya, B. Dahal, G. P. Ojha, D. Kang, T. Kim, S.-H. Chae, A. Muthurasu and H. Y. Kim, Chem. Eng. J., 2019, 361, 1225–1234 CrossRef CAS.
- H. Wu, Y. Zhang, W. Yuan, Y. Zhao, S. Luo, X. Yuan, L. Zheng and L. Cheng, J. Mater. Chem. A, 2018, 6, 16617–16626 RSC.
- X.-q. Niu, X.-l. Wang, D.-h. Wang, Y. Li, Y.-j. Zhang, T. Yang, T. Yu and J.-p. Tu, J. Mater. Chem. A, 2015, 3, 17106–17112 RSC.
- M. Duraivel, S. Nagappan, K. H. Park and K. Prabakar, Electrochim. Acta, 2022, 411, 140071 CrossRef CAS.
- M. Guo, L. Zhou, Y. Li, Q. Zheng, F. Xie and D. Lin, J. Mater. Chem. A, 2019, 7, 13130–13141 RSC.
- J. Yang, C. Yu, X. Fan, C. Zhao and J. Qiu, Adv. Funct. Mater., 2015, 25, 2109–2116 CrossRef CAS.
- F. Shi, L. Li, X.-l. Wang, C.-d. Gu and J.-p. Tu, RSC Adv., 2014, 4, 41910–41921 RSC.
- S. Dutta, A. Indra, Y. Feng, T. Song and U. Paik, ACS Appl. Mater. Interfaces, 2017, 9, 33766–33774 CrossRef CAS PubMed.
- D. Wang, Q. Li, C. Han, Q. Lu, Z. Xing and X. Yang, Nat. Commun., 2019, 10, 1–12 CrossRef PubMed.
- F.-X. Ma, L. Yu, C.-Y. Xu and X. W. D. Lou, Energy Environ. Sci., 2016, 9, 862–866 RSC.
- W. Song, X. Teng, Y. Liu, J. Wang, Y. Niu, X. He, C. Zhang and Z. Chen, Nanoscale, 2019, 11, 6401–6409 RSC.
- L. Guo, Y. Ren, J. Liu, S. Y. Chiam and W. K. Chim, Small, 2014, 10, 2611–2617 CrossRef CAS PubMed.
- A. Yourdkhani and G. Caruntu, J. Mater. Chem., 2011, 21, 7145–7153 RSC.
- N. Tarutani, Y. Tokudome, M. Jobbágy, F. A. Viva, G. J. Soler-Illia and M. Takahashi, Chem. Mater., 2016, 28, 5606–5610 CrossRef CAS.
- D. O. Shin, D. H. Lee, H.-S. Moon, S.-J. Jeong, J. Y. Kim, J. H. Mun, H. Cho, S. Park and S. O. Kim, Adv. Funct. Mater., 2011, 21, 250–254 CrossRef CAS.
- J. H. Mun, Y. H. Chang, D. O. Shin, J. M. Yoon, D. S. Choi, K.-M. Lee, J. Y. Kim, S. K. Cha, J. Y. Lee, J.-R. Jeong, Y.-H. Kim and S. O. Kim, Nano Lett., 2013, 13, 5720–5726 CrossRef CAS PubMed.
- S. K. Cha, J. H. Mun, T. Chang, S. Y. Kim, J. Y. Kim, H. M. Jin, J. Y. Lee, J. Shin, K. H. Kim and S. O. Kim, ACS Nano, 2015, 9, 5536–5543 CrossRef CAS PubMed.
- D. Milne, Sewage Ind. Wastes, 1950, 1192–1199 CAS.
- E. Gail, S. Gos, R. Kulzer, J. Lorösch, A. Rubo, M. Sauer, R. Kellens, J. Reddy, N. Steier and W. Hasenpusch, Ullmann's Encycl. Ind. Chem., 2000, 10, 673–710 Search PubMed.
- P. Poskozim, R. Shute, R. Taylor and J. Wysocki, J. Inorg. Nucl. Chem., 1973, 35, 687–691 CrossRef CAS.
- E. Yilmaz, F. Ahlatcı, E. Y. Yazici, O. Celep and H. Deveci, Mugla J. Sci. Technol., 2017, 3, 171–177 Search PubMed.
- J. Wang, T. Xie, Q. Deng, Y. Wang, Q. Zhu and S. Liu, New J. Chem., 2019, 43, 3218–3225 RSC.
- J. Liu, M. Zheng, X. Shi, H. Zeng and H. Xia, Adv. Funct. Mater., 2016, 26, 919–930 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00076a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |