
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluating the effect of two-dimensional molecular layout on DNA origami-based transporters†
Kodai
Fukumoto
ab,
Yuya
Miyazono
c,
Takuya
Ueda
cd,
Yoshie
Harada
*aef and
Hisashi
Tadakuma
 *acgh
*acgh
aInstitute for Protein Research, Osaka University, Osaka 565-0871, Japan. E-mail: tadakumahisashi@shanghaitech.edu.cn; yharada@protein.osaka-u.ac.jp
bDepartment of Biological Sciences, Graduate School of Science, Osaka University, Osaka 560-0043, Japan
cGraduate School of Frontier Science, The University of Tokyo, Chiba 277-8562, Japan
dGraduate School of Science and Engineering, Waseda University, Tokyo 162-8480, Japan
eCenter for Quantum Information and Quantum Biology, Osaka University, Osaka 560-0043, Japan
fPremium Research Institute for Human Metaverse Medicine (WPI-PRIMe), Osaka University, Osaka 565-0871, Japan
gSchool of Life Science and Technology, ShanghaiTech University, Shanghai 201210, People's Republic of China
hGene Editing Center, School of Life Science and Technology, ShanghaiTech University, Shanghai 201210, People's Republic of China
First published on 19th April 2023
Abstract
Cellular transport systems are sophisticated and efficient. Hence, one of the ultimate goals of nanotechnology is to design artificial transport systems rationally. However, the design principle has been elusive, because how motor layout affects motile activity has not been established, partially owing to the difficulty in achieving a precise layout of the motile elements. Here, we employed a DNA origami platform to evaluate the two-dimensional (2D) layout effect of kinesin motor proteins on transporter motility. We succeeded in accelerating the integration speed of the protein of interest (POI) to the DNA origami transporter by up to 700 times by introducing a positively charged poly-lysine tag (Lys-tag) into the POI (kinesin motor protein). This Lys-tag approach allowed us to construct and purify a transporter with high motor density, allowing a precise evaluation on the 2D layout effect. Our single-molecule imaging showed that the densely packed layout of kinesin decreased the run length of the transporter, although its velocity was moderately affected. These results indicate that steric hindrance is a critical parameter to be considered in the design of transport systems.
1. Introduction
In natural motile systems, motor proteins are aligned in a specific layout and exhibit power-efficient and reliable movements.1–7 Mimicking these natural systems to create efficient artificial motile systems is one of the ultimate goals of nanotechnology. The fundamental question of these motile system constructions is how collective motility (team activity) differs from the motile activity of a single molecule and how the individual motor contributes to overall collective movement.3,8–10 One approach to understanding collective movement is elucidating the effects of key parameters (e.g., motor number and layout) on motile activity (such as velocity and run length).Kinesin is a processive motor protein that walks along the microtubules and has been used as a model protein for collective movement. In previous studies, the effect of motor number has been examined; however, the effects of density and layout remain elusive.11–16 This is because of the (1) heterogeneity of the samples and (2) difficulty in controlling the number and density/layout separately. These drawbacks can be attributed to conventional assays (such as beads and gliding assays), where the motors are generally randomly adsorbed to the transporter, and the distributions of motor number and intermolecular distance are broad. To overcome these limitations, DNA-based assays that allow researchers to design and construct transporters with a defined number and layout of motor molecules have been developed.17–19 Although these studies have successfully evaluated the effect of one-dimensional (1D) intermolecular distance of motor proteins on motility, the effect of intermolecular distance remains controversial.20 Therefore, a more systematic examination of intermolecular distances is crucial.
DNA origami is a versatile method used to construct custom two- and three-dimensional structures and control precise molecular layouts.21–28 The molecular layout capability of DNA origami at nanometer resolution could provide a solid platform to elucidate the effect of intermolecular distance precisely. However, the advantages of DNA origami are yet to be completely exploited, partly because of the technical limitations of transporter construction. Previous studies used a hybridization method to integrate motor proteins,19,29–31 where single-stranded DNA (ssDNA)-bound motor proteins were hybridized to ssDNA handles protruding from the DNA nanostructure. However, a short ssDNA handle, reduces the yield of protein integration. In contrast, long double-stranded DNA (dsDNA) handle makes it difficult to evaluate the intermolecular distance precisely, especially for short-distance ranges. Subsequently, direct and covalent attachment of motor proteins have been reported,20,32,33 where protein-tag technology, such as SNAP-tag protein, was used to ensure the stable binding of the protein to the DNA nanostructure with a smaller handle. The central problems associated with these covalent binding tags are slow assembly speed and low integration yield. This reduction in integration may be partly owing to the charge repulsion between the protein and DNA nanostructure, where both are negatively charged in neutral buffer conditions [the isoelectric points (pIs) of two major tag proteins, SNAP-tag and HaloTag, are 6.0 and 4.9, respectively]. Therefore, a high molar ratio of the motor protein to the DNA nanostructure is required to compensate for the low integration yield, which makes the removal of the excess motor proteins during the purification step challenging. These contaminating free motor proteins impede the evaluation of transporter activity even when using single-molecule imaging. Moreover, in some cases, a long incubation time with a high protein concentration induces the aggregation of proteins, DNA nanostructures, or protein–DNA nanostructure complexes.
Here, we partially solved the problem of a low integration yield by introducing a positively charged peptide tag (poly-lysine tag composed of 5–10 lysine amino acids, Lys-tag) with an up to 700 times higher association rate (kon) of SNAPf-tag-fused kinesin. This Lys-tag allowed us to decrease the molar ratio of kinesin to the DNA nanostructure while maintaining high integration efficiency (>86%, where more than half of the transporters have the correct number in the case of the 4-kinesin integrated transporter). We successfully constructed and purified transporters with a high motor density (intermolecular distance of 7 nm, which matched the unit lattice size of the microtubule of 8 nm). Based on this system, we evaluated the effect of the 2D layout of kinesin molecules on artificial transporters. Our results showed that the densely packed layout of kinesin decreased the run length of the transporters, although the speed was similar. These results indicate that steric hindrance is a critical parameter to consider while designing artificial transporters.
2. Results
Effect of poly-lysine tag (Lys-tag) on SNAPf-kinesin motility
The positively charged poly-lysine peptide (Lys-tag) was fused to overcome the drawbacks (low assembly speed and low yield) of protein tags such as SNAP-tag, as previous literature suggested that the yield might depend on the pI of the protein.26 We introduced 5 (5 K) or 10 (10 K) lysine residues (Lys-tag) into the SNAPf-fused dimeric kinesin (Fig. 1A). Fusion proteins were expressed and purified (Fig. 1B). We first evaluated the effect of Lys-tag on the activities of SNAPf and kinesin. Lys-tag had a minimal effect o SNAPf substrate (O6-benzylguanine, BG) binding (ESI Fig. 1†). We also examined the motility of fluorescently-labeled kinesins (A647-kinesins) using total internal reflection fluorescence (TIRF) microscopy. We observed that the Lys-tagged kinesin-SNAPfs moved along axonemes at velocities similar to those without a Lys-tag (Fig. 1C and D, ±9% change compared to the 0 K construct), showing that the Lys-tag had a minimal effect on the motor activity of kinesin. These results are similar to those obtained for stalk (neck coiled-coil) mutants with a positive charge.34 By contrast, the Lys-tag lengthened the run lengths by increasing the number of lysine residues in the Lys-tag, probably because of the interactions between lysine and the tubulin C-terminal.34 The fluorescence intensities of the moving kinesin molecules also confirmed the minor effect of the Lys-tag (ESI Fig. 2,† −22% to +37% change compared to the 0 K construct). These results showed that the Lys-tag had minimal effects on SNAPf ligand-binding ability and kinesin motility.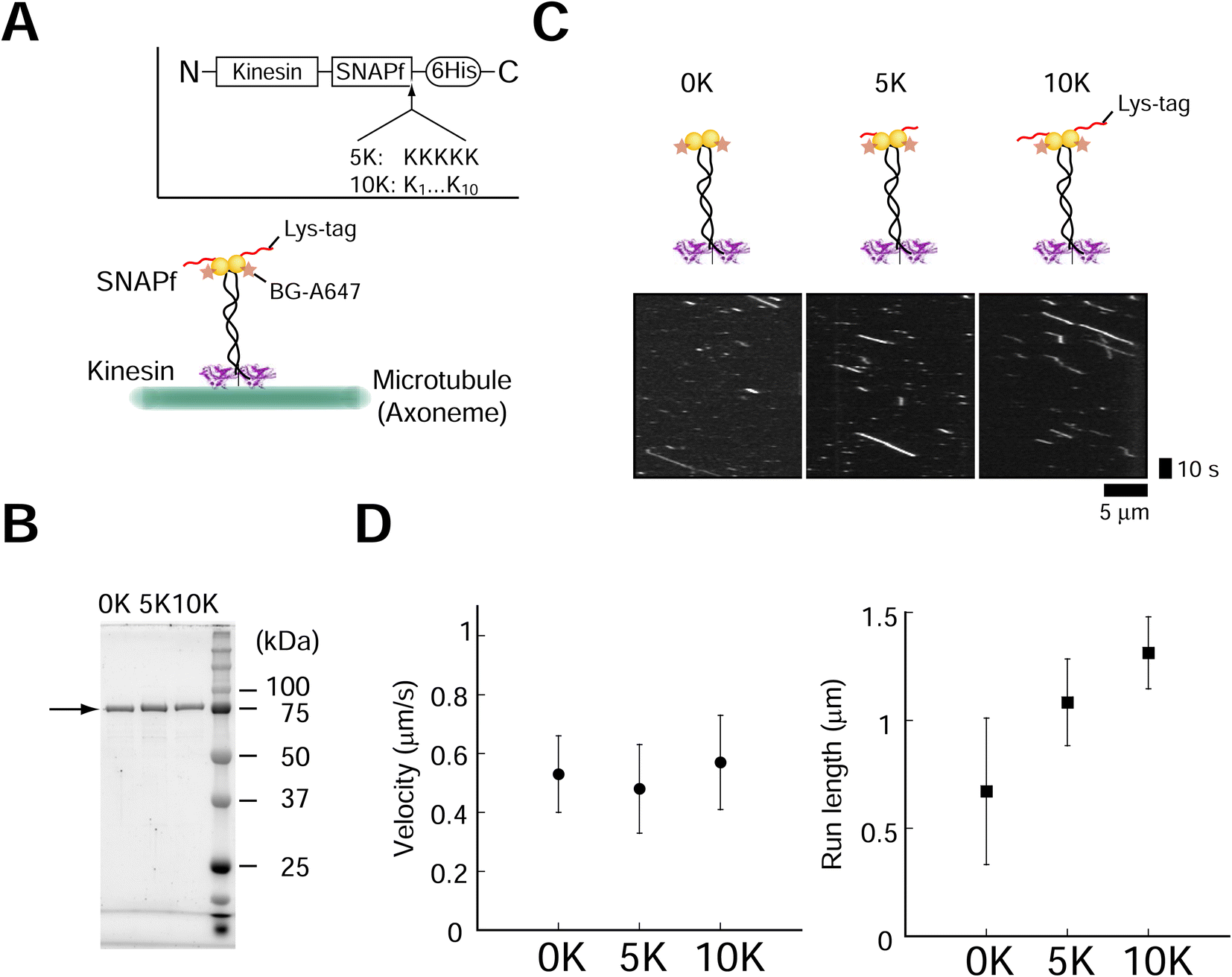 | ||
| Fig. 1 Characterization of the poly-lysine tag (Lys-tag). (A) Schematic of the dimeric kinesin-SNAPf construct with Lys-tag. (B) Results of 12% poly-acrylamide gel electrophoresis (SDS-PAGE) of kinesin-SNAPf-Lys-tag. The arrow indicates the position of kinesin. (C) Kymograph obtained using red laser (633 nm) excitation shows the processive movement of kinesin-SNAPf-Lys-tag. SNAPf proteins were labeled with BG-Alexa Fluor 647(BG-A647, BG: O6-benzylguanine) at a BG/SNAPf molar ratio of 2. Scale bar for the vertical axis = 10 s, horizontal axis = 5 μm. (D) Lys-tag had a modest effect on velocity (left, ±9% compared with the 0 K construct. P < 0.001 [one-way ANOVA]. 0–5 K, P ≥ 0.05; 0–10 K, P < 0.05 [Dunnett's test]), but slightly increased run length (right, P ≥ 0.05 [Kruskal–Wallis test]). Error bars indicate S.D. of total measurements (velocity) or S.E.M. determined by bootstrapping (run length). More than 61 motile particles were analyzed for each construct. | ||
Lys-tag accelerated the kinesin assembly speed with the DNA rod
We next investigated whether the Lys-tag improved the speed of kinesin assembly on DNA origami. A rod-shaped DNA origami (DNA rod) with a BG-handle was used (Fig. 2A, ESI Fig. 3;† DNA origami was also labeled with a Cy5 fluorophore). After incubation with kinesins, a clear band shift was observed for the DNA rod with a BG-handle but not for the BG-handle-less DNA rod (Fig. 2B, ESI Fig. 4†), indicating the specific attachment of the SNAPf-fused kinesin and BG-handle on the DNA rod. We calculated the second-order rate constants of the reactions (Fig. 2C), where the kinesin assembly speed increased by a factor of 3 with 5 K (2 × 104 M s−1) and by a factor of 700 with 10 K (4 × 106 M−1 s−1). Importantly, we found that the DNA rod, even without a BG-handle, tended to aggregate at high kinesin concentrations (>50 nM; ESI Fig. 5†), making it difficult to obtain a high yield of the kinesin–DNA rod complex. In stark contrast, the high-speed Lys-tag-fused kinesin assembly allowed us to mix the kinesin and DNA rod at a low concentration with a short incubation time. For example, 10 K-kinesin required 2 and 10 nM kinesin for 10 and 2 min, respectively, whereas 0 K-kinesin required 200 min even for 100 nM kinesin and showed extensively aggregated complexes (ESI Fig. 5†). Lys-tag acceleration of the assembly was further confirmed by performing different approaches to kinesin–DNA rod assembly experiments using fluorescently labeled kinesin (Cy3-kinesin), which can directly detect both specific and nonspecific interactions under various assay conditions. To avoid the overlap of free kinesin bands and kinesin–DNA rods, we supplemented the agarose gel with sodium dodecyl sulfate (SDS) (ref. 35; Fig. 2D and ESI Fig. 6†). Using this system, we observed the binding of fluorescently labeled kinesin molecules on the DNA rod, confirming a similar binding rate (2 × 106 M s−1; Fig. 2E and ESI Fig. 7†).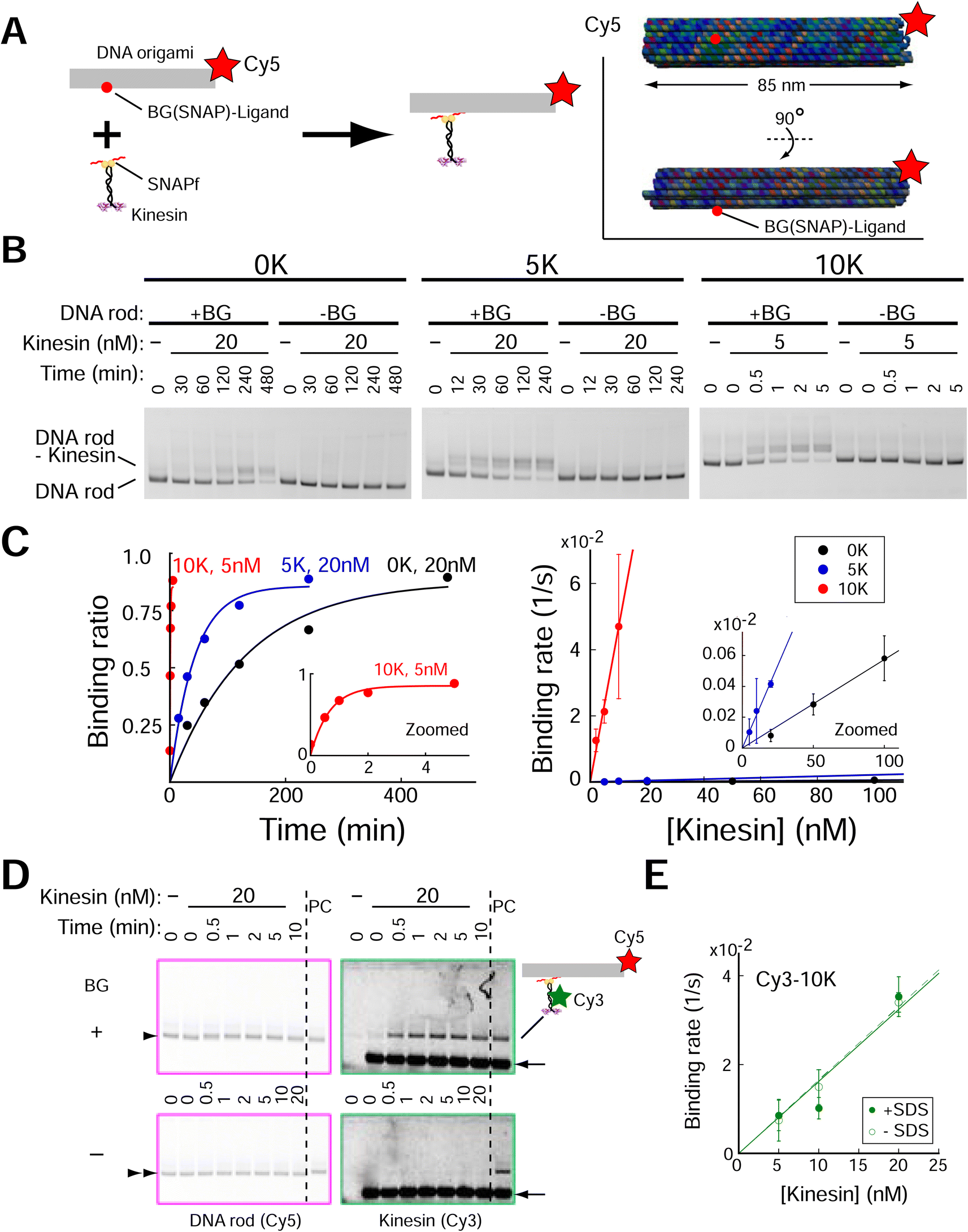 | ||
| Fig. 2 Lys-tag accelerated the speed of kinesin assembly on the DNA rod. (A) Schematic of the experiment. (Inset) DNA rod structure used in figure. Red circle indicates the position of the BG (SNAP)-ligand. (B) Results of 0.7% agarose gel electrophoresis using dimeric kinesin-SNAPf. Electrophoretic mobility shift assays show acceleration of assembly speed with Lys-tag. DNA rods (2 nM) were visualized through Cy5 fluorescence. +BG, DNA rod with the single-stranded DNA (ssDNA) handle attached with the SNAPf ligand (O6-benzylguanine, BG); -BG, DNA rod without BG-handle. (C) Left, quantification of agarose gel data in (B). Right, results of 3–5 independent experiments show that assembly speed (Kon of kinesins on DNA rods) accelerated with the increase in the number of Lys (6 × 103, 2 × 104, and 4 × 106 M−1 s−1 for 0 K, 5 K, and 10 K, respectively). (D) Images of 0.7% agarose gel supplemented with 0.1% SDS. Cy3-kinesin coincides with the Cy5-DNA rod with BG-handle (upper, arrowhead), but not with the DNA rod without BG-handle (lower, double arrowhead), confirming kinesin assembly on the DNA rod (5 nM). PC, DNA rod with BG-handle were incubated for 20 min with kinesin-SNAPf-10 K (20 nM). Note that signals of free kinesins (arrow) are saturated. (E) Similar kon values of kinesin-SNAPf-10 K were obtained by two different assays (−SDS, gel shift assay of the DNA rod similar to (B) and (C); +SDS, fluorescence intensity measurement of assembled Cy3-kinesin shown in (D); see ESI Fig. 6 and 7† for detail). Error bars indicate S.D. of three independent experiments. | ||
After we observed the acceleration of SNAPf-fused kinesin assembly on the DNA rod, we reasoned that the positive charge of the Lys-tag might accelerate the assembly of other proteins. We first confirmed whether a single peptide of Lys-tag was sufficient to accelerate assembly, as the kinesin used was a dimer with two Lys-tag peptides. We evaluated monomeric kinesin-SNAPf lacking the coiled-coil domain that mediates dimerization and found that the Lys-tag also accelerated the assembly speed of monomeric kinesin on the DNA rod. Interestingly, the acceleration factor of the 10 K Lys-tag was 300 (ESI Fig. 8†), which was much faster than that of dimeric kinesin with the same lysine number (5 K Lys-tag, acceleration factor of 3). We next evaluated HaloTag-fused dimeric kinesin and found that the Lys-tag also accelerated the HaloTag-fused kinesin 10 times (ESI Fig. 9†), with the acceleration factor appearing to depend on the net charge of the tag protein fragments, suggesting that the charge apparently affected the acceleration factor (ESI Fig. 10†).
Single-molecule motile properties of DNA rod–kinesin complexes
To explore the design principle of the artificial transporter, we first evaluated the effect of kinesin number on transporter motility using DNA origami with multiple kinesin anchoring sites (ESI Fig. 11;† DNA origami was also labeled with a Cy3 fluorophore). We used the advantage of Lys-tag and succeeded in reducing the kinesin concentration for transporter construction (ESI Fig. 12†). The low concentration of kinesin molecules also improved experimental quality, as it has been reported that a residual excess of unbound motor proteins impedes the sliding movement of the transporter.33 After construction and purification, we evaluated the number of kinesin molecules on the axoneme-bound transporter using a photobleaching assay (using the ATP analog adenylyl-imidodiphosphate (AMP-PNP); ESI Fig. 13†). As expected, we found that the number of photobleaching steps of the kinesin channel (Alexa Fluor 647, A647) increased with the number of BG-handles, while the number of photobleaching steps of the DNA rod channel (Cy3) remained constant. By fitting the histogram of the number of photobleaching steps with a binomial distribution, we estimated that the occupancy fractions of kinesin per handle for the two- and four-handle transporter were 0.94 and 0.86, respectively, which was further supported by the gel shift assay (ESI Fig. 14†). The reason behind that the occupancy fractions were still not 1 (100% construction yield) might be attributed to the defect in staple strand incorporation into DNA origami.36 After confirming the high yield of transporter construction, we observed transporter movement in the presence of ATP (Fig. 3A–C). We found that the fluorescence intensity of the kinesin channel of the moving spots increased with the number of BG-handles, as expected (ESI Fig. 15†). In contrast, no processive movement was observed with the DNA rod without a BG-handle (ESI Fig. 16†), showing that the observed movement was kinesin-dependent. The motile event of the transporter to the axoneme also increased with the number of kinesins attached to the transporter, although the frequencies of motile events were lower than those of free kinesin (ESI Fig. 17†). This reinforces the importance of removing excess free kinesin to minimize motile background molecules other than the transporter. In addition, it emphasizes the merit of using a Lys-tag to reduce the concentration of kinesin molecules at the transporter construction and ensures minimal contamination of excess free kinesin at observation. Notably, the run length of a single motor on the scaffold was greater than that of a single motor without the scaffold, possibly because of the higher rebinding probability of kinesin, as the DNA origami scaffold was expected to slow down motor diffusion after unbinding. This may have increased the run length of the DNA origami transporter. Therefore, the ionic (NaCl) concentration varied in different experiments based on the aim of each experiment (see Materials and Methods for details). The velocity remained moderately constant (<−5% change compared to free kinesin), whereas the run length increased with the number of BG-handles (Fig. 3D and E and ESI Fig. 18†), confirming the results of previous studies with other motor proteins (e.g., dynein19 and NCD20). These results confirm the formation of complexes and collective transport by kinesin teams.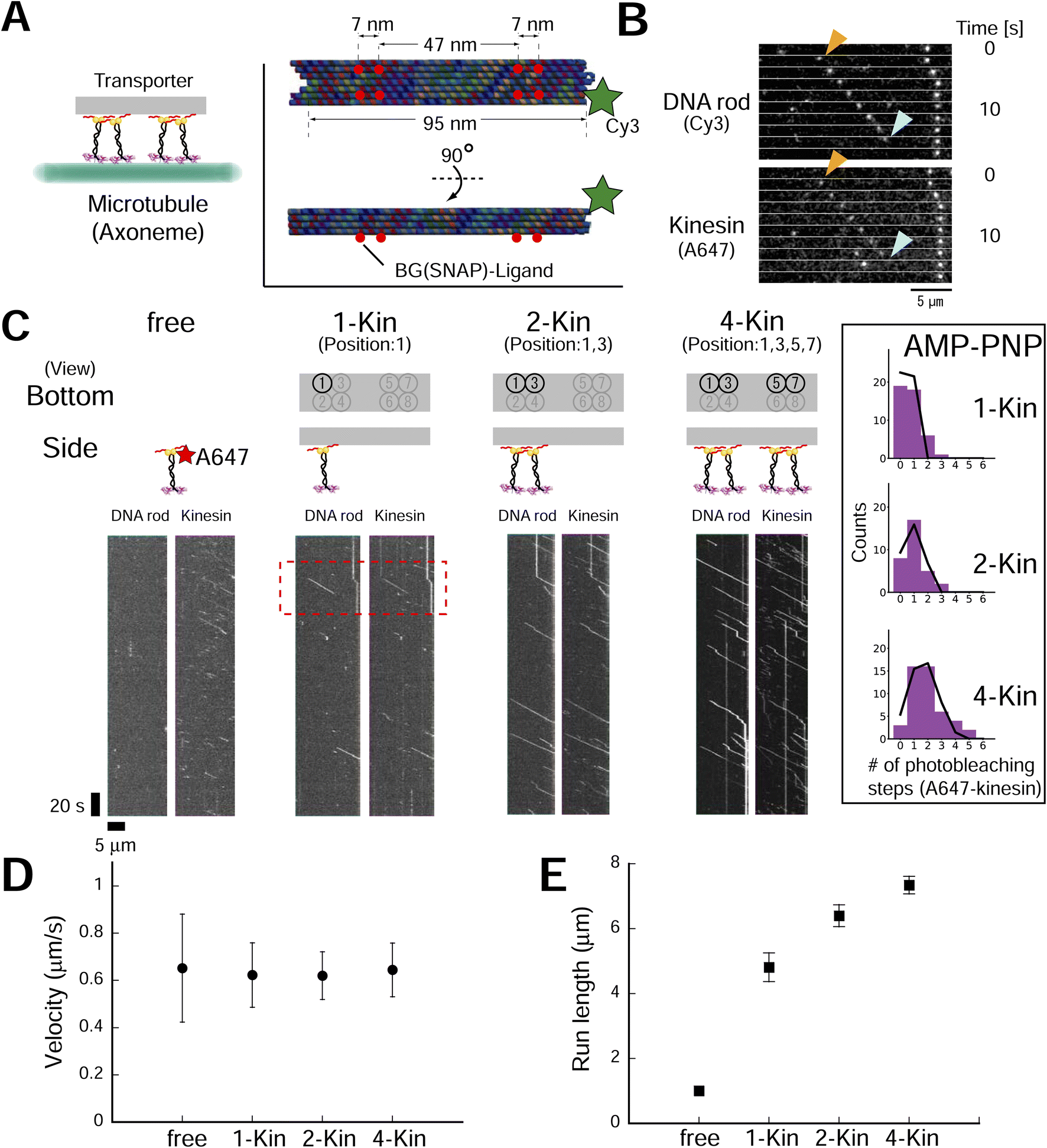 | ||
| Fig. 3 Kinesin number affects the single-molecule motile properties of rod–kinesin complexes. (A) Schematic of the experiment. (Inset) DNA rod structure used in figures, Fig. 4 and related ESI figures.† Red circles indicate the location of the BG (SNAP)–ligand. A part of SNAPf-tag in a dimer kinesin was labeled with A647 and remaining unoccupied SNAPf-tag is used to integrate kinesin onto the DNA rod. (B) Raw images that show the movement of transporters (A647-labeled dimeric kinesin and Cy3-labeled rods). (C) Kymographs of A647-labeled dimeric kinesin alone and Cy3-labeled rods with 1, 2, or 4 kinesins (A647-labeled). Scale bar for vertical axis = 20 s, horizontal axis = 5 μm. Red region of interest (ROI) indicates the images shown in (B). (Inset) Histograms of photobleaching step number of A647 (kinesin) in AMP-PNP condition. After correction or the label ratio (0.49), the occupancy fractions of kinesin per handle for the two- and four-handle transporter were estimated as 0.94 and 0.86, respectively. (D and E) Average velocities (D) of rod–kinesin were moderately constant (among rod–kinesin, P ≥ 0.05 [one-way ANOVA]), whereas run lengths (E) increased with increasing kinesin number (“1kin–2kin,” P < 0.01; “2kin–4kin,” P < 0.001 [Conover–Iman test]). The measurements were performed in 150 mM NaCl conditions (see methods for detail). Error bars indicate S.D. of total measurements (velocity) or S.E.M. determined by bootstrapping (run length) from two independent experiments (total of eight chambers). More than 93 motile particles were analyzed for each type. | ||
Effects of motor layouts on collective movement
We next explored the effect of kinesin molecular layout on transporter movement. Four kinesin molecules with three different layouts were used (Fig. 4). We used a higher ionic (NaCl) concentration to suppress the long run and accurately evaluate the run length. The fluorescence intensities of DNA rod (Cy3) and kinesin (A647) were similar for all three layouts (ESI Fig. 19†). This result supports our hypothesis that a high-yield Lys-tag approach allows for the construction of different layouts of similar quality. We found that the layout of the kinesin had a littlee effect on the velocity but significantly influenced on the run length (Fig. 4B and C, ESI Fig. 20,†p = 0.53 and p < 0.001 for velocity and run length, respectively). The transporter in the most densely packed layout demonstrated the shortest run length. Furthermore, to explore the minimum number of kinesins required in each layout to affect movement, we examined transporters containing two kinesins, which also showed that a shorter intermolecular length decreased the run length while maintaining a similar velocity (ESI Fig. 21 and 22†). These findings show that the spatial proximity of dimeric kinesins decreases the processivities of the transporters but has a moderate effect on the velocities. To further explore the motility characteristics of the transporter, we used monomeric kinesin, which cannot move processively at the single-molecule level. The collective movement of a group of kinesin monomers harnesses the motility of kinesin for the processive movement of the transporter. We found that the layout of two- and four-monomer kinesins had a more significant effect on run length than on velocity (ESI Fig. 23–26†).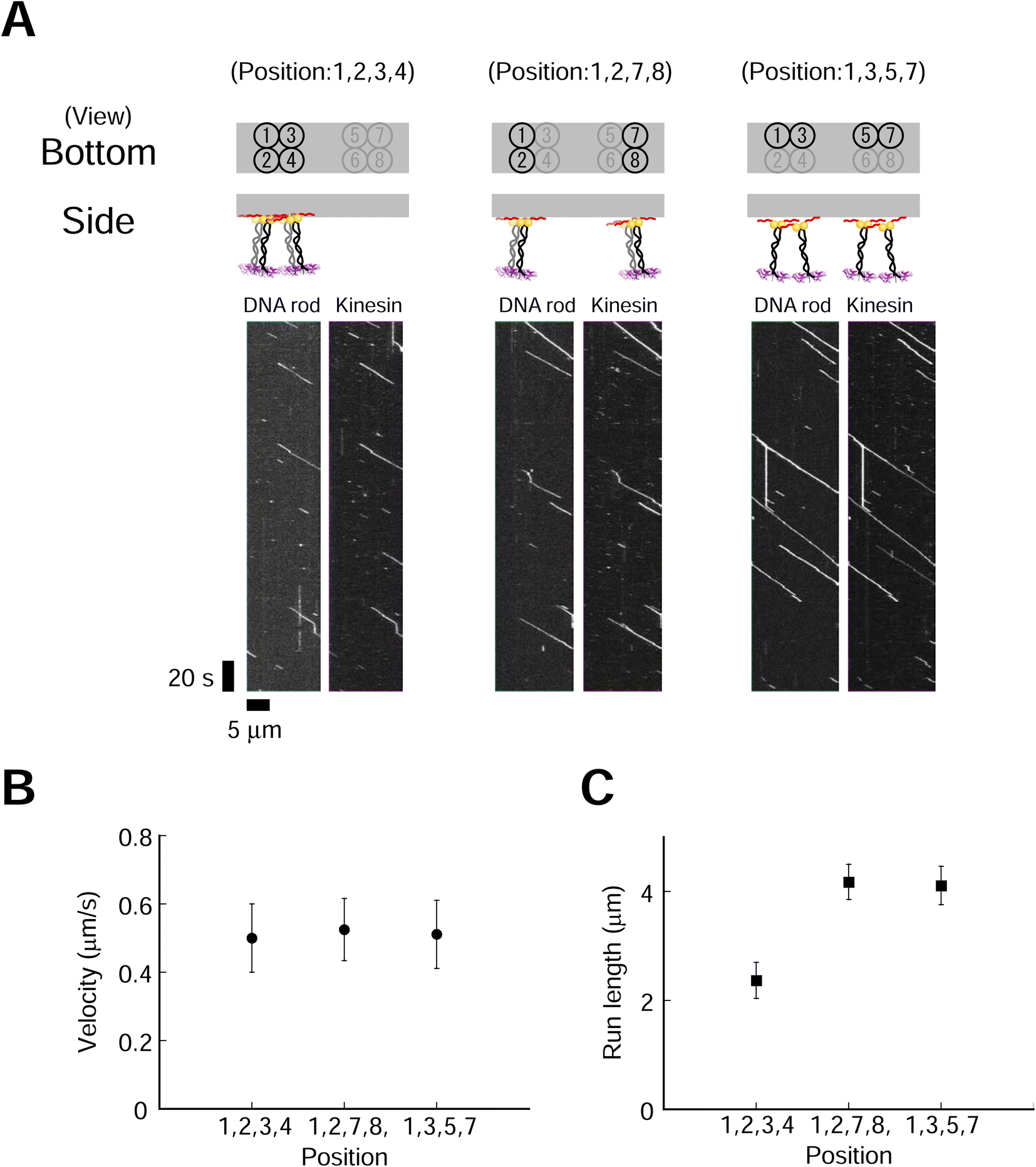 | ||
| Fig. 4 Kinesin layout affects the single-molecule motile properties of rod–kinesin complexes. (A) Kymographs of Cy3-labeled rods with three different layouts of four dimeric kinesins (kinesins were labeled with A647). Scale bar for vertical axis = 20 s, horizontal axis = 5 μm. (B and C) Average velocities (B) of rod–kinesin remained constant (P ≥ 0.05 [one-way ANOVA]), whereas run lengths (C) differed with different kinesin layouts (P < 0.001 [Kruskal–Wallis test]. “1,2,3,4-1,2,7,8,” P < 0.001; “1,2,3,4-1,3,5,7,” P < 0.001; “1,2,7,8-1,3,5,7,” P ≥ 0.05 [Conover–Iman test]). The measurements were performed in 250 mM NaCl condition (see methods for detail). Error bars indicate S.D. of total measurements (velocity) or S.E.M. determined by bootstrapping (run length) from 2–3 independent experiments (total 8–11 chambers). More than 179 motile particles were analyzed for each layout. | ||
3. Discussion
The key to building a sophisticated and efficient motile system is establishing a design principle. It is essential to evaluate the layout effect, because the architecture of motor proteins is integral to natural motile systems, such as muscles, cilia/flagella, and vesicle transporters. In this context, researchers have exploited the advantages of DNA nanostructure platforms to control the number and layout of molecules. However, understanding of the effect of the molecular layout (e.g., intermolecular distance) on the motility of transport systems is limited, partly owing to practical obstacles to transporter construction. Here, we introduced a Lys-tag to improve the construction process and successfully evaluated the effect of the 2D molecular layout. We found that the density of the kinesin motor protein affected the run length but only moderately affected the velocity. Both processive (dimer) and non-processive (monomer: catalytic domain only) kinesins support this conclusion, suggesting that steric hindrance control is the key to transporter design.In transporter construction, there are two major steps: (1) integration of motor proteins and (2) removal of excess unbound motor proteins from the transporter complex (purification of the transporter complex). The slow assembly of the protein on the DNA nanostructure hinders the first integration step. The necessity of a high concentration of molecules to drive an efficient reaction and achieve a high yield causes side effects, such as the aggregation of DNA nanostructures (ESI Fig. 5†). The second purification step is critical because of the intrinsic characteristics of the DNA nanostructure. The negative charge of the DNA nanostructure decreases the binding rate of the transporter (DNA nanostructure–protein complex) to the rail protein (e.g., microtubule) compared to that of the unbound free protein (ESI Fig. 17†). Thus, the complete removal of excess unbound motor proteins is critical for the assay. The trade-off between the requirement of a high concentration in the construction step and the desirability of a low concentration in the purification process makes transporter construction difficult. The Lys-tag partially resolved this trade-off and allowed us to decrease kinesin concentration during the integration process while maintaining a high final transporter yield. The lysine number had a strong effect on the acceleration factor of kon: 3 and 700 times for 5 K and 10 K of SNAPf, respectively (Fig. 2). Moreover, the acceleration factor of the Lys-tag differed from those of the SNAPf-tag and HaloTag (700 vs. 10 with 10 K Lys-tag). These results might be attributable to the difference in the estimated charge on the tag protein (−18.8, −9, −5.3, and +4.5 at pH 7.4, for Halo-0 K, Halo-10 K, SNAPf-0 K, and SNAPf-10 K, respectively), as the apparent association rate (kon) of the tag protein to the DNA nanostructure apparently depended on the charge (ESI Fig. 10†). This hypothesis is supported by the HaloTag mutant results, where replacing surface amino acids with positively charged amino acids improved the kon of the HaloTag protein to the DNA nanostructure.37 Further optimization of the peptide sequence and/or fusion to the DNA-binding motif should improve the assembly of many proteins on DNA nanostructures,38,39 where it might also be possible to design the integration process precisely using the difference in kon.
We evaluated the 2D layout effect on two key parameters of the transporter (velocity and run length) using a DNA origami platform with a defined number and layout of motor molecules. We found that the layout moderately affected the velocity, whereas the densely packed layout decreased the run length (when comparing transporters with the same motor number). Regarding velocity, the number of kinesins involved in the motile movement of a transporter should range from one to a fully defined number (e.g., 4 for a 4-Kin transporter) in our system. Using a gliding assay, it has been reported that the velocity of kinesin's collective movement is independent of motor density.11 Our results reflected this characteristic. The similar velocities of the transporter with different defined motor numbers support this view (Fig. 3). Another interpretation is that the number involving motile movement is less than the threshold number required to demonstrate the effect of counterforce produced by the microtubule-bound motors that do not produce the effective movement of the transporter while imposing negative interference.12,40 Future transporter studies with a larger number of motors are required to provide more precise information. For this line of experiments, improvement of the construction yield is the key,36 including improving the origami design, such as using rationally designed scaffold–staple pairs.41,42
In the present study, in contrast to velocity, molecular layout affected run length under these conditions. We observed short run lengths with densely packed layouts (when only comparing transporters with the same motor number). The run length was also affected by ionic strength (NaCl concentration; Fig. 3 and ESI Fig. 20†), suggesting that electrostatic interactions are essential for transporter activity43). Regarding the motile mechanism, there are two key parameters (on- and off-rates of the motor15,20). Currently, the reason for this remains unknown; however, we speculate that steric hindrance might affect both the on- and off-rates (see ESI† Discussion).
The effect of two-dimensional (2D) motor array, i.e., side-by-side arrangement versus along the axis of the microtubule rail, is an interesting issue regarding the movement of the transporter. Our results suggest that the effect was not strong under our experimental conditions (see ESI Fig. 21† and 4 for the two- and four-motor systems, respectively). However, our current data do not reveal the exact orientation of the DNA origami transporter relative to that of the microtubule rail. In future studies, further confirmation with qualitative analysis is required with a similar approach to the literature (14,15, i.e., attaching a marker to observe the individual motor and transporter movement). Currently, this is technically challenging owing to the large size of probes such as quantum dots (QD, 25 nm,14) and gold nanoparticles (40 nm,44) compared to the intermolecular distance between two kinesin molecules (such as a minimum of 7 nm in our study). Future advances in new probes with smaller sizes while providing sufficient photon numbers (such as nano-diamonds45) might overcome these technical issues.
4. Conclusion
The finding that a densely-packed layout decreased the run length reinforces the importance of the spatial layout in designing motile systems. The power of the DNA origami approach should reveal the similarities and differences between different motor proteins (e.g., kinesin, myosin, and dynein), explaining the differences in periodicity (intermolecular distance) in natural systems, such as muscles (for myosin) and cilia/flagella (for dynein).Moreover, combining theoretical model analysis29,31,46,47 with our evaluation of 2D layout effects should pave the way for constructing functional artificial transport/motile systems in natural and artificial cells, allowing the investigation of how other factors influence motility.48
5. Materials and Methods
DNA cloning
We used the cysteine-light human ubiquitous kinesin-1 493-residue dimer or 351-residue monomer fused to C-terminal SNAPf-tag or HaloTag followed by a six-histidine tag as previously described.17 Lys-tag (5 or 10 tandem repeats of AAG) was introduced between SNAPf/Halo and histidine tag by PCR. All constructs were verified by DNA sequencing.Protein purification
The proteins were expressed at 18 °C and purified by Ni-NTA chromatography (Qiagen) and ion exchange chromatography (Cytiva) as previously described,17 with modifications in ion exchange chromatography. Briefly, the dimeric proteins were purified using HiTrap-Q with buffer (200) (200 mM NaCl with 25 mM PIPES, pH 6.8, 2 mM MgCl2, 1 mM EGTA, 100 μM ATP, 500 μM DTT; the numbers in parentheses show the NaCl concentration) for diluting and washing and eluted at ∼0.45 M NaCl. Monomeric kinesin-SNAPf-0 K was purified using HiTrap-SP with buffer (100) for diluting and washing and eluted at ∼0.25 M NaCl. Monomeric kinesin-SNAPf-10 K was purified using HiTrap-SP with buffer (150) for diluting and buffer (200) for washing and eluted at ∼0.5 M NaCl. After purification, proteins were dialyzed against buffer A (25 mM PIPES, pH 7.0, 250 mM NaCl, 1 mM EGTA, 2 mM MgCl2, and 50 μM ATP).For binding assays, the dialyzed proteins were fluorescently labeled for 60 min at 4 °C with Cy3-NHS (Cytiva) at a kinesin subunit/dye molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. For transport assays, dialyzed kinesin-SNAPf-10 K dimer was reacted for 160 min at 4 °C with SNAP-Surface Alexa Fluor 647 (NEB) at a molar ratio of 4:3, or kinesin-SNAPf-10 K monomer was reacted for 60 min at 4 °C with Cy5-NHS (Cytiva) at a molar ratio of 1:8. The unreacted dyes were removed by His SpinTrap (Cytiva) and the eluted proteins were dialyzed against buffer A. The protein concentration was determined by Bradford assays or sodium dodecyl sulfate (SDS)-PAGE using BSA as a standard. Cy3 concentration was determined by measuring absorbance using the molar extinction coefficient of Cy3 at 550 nm (150000 M−1 cm−1). After the addition of 10% sucrose, the dialyzed proteins were frozen in liquid nitrogen and stored at −80 °C.
:10. For transport assays, dialyzed kinesin-SNAPf-10 K dimer was reacted for 160 min at 4 °C with SNAP-Surface Alexa Fluor 647 (NEB) at a molar ratio of 4:3, or kinesin-SNAPf-10 K monomer was reacted for 60 min at 4 °C with Cy5-NHS (Cytiva) at a molar ratio of 1:8. The unreacted dyes were removed by His SpinTrap (Cytiva) and the eluted proteins were dialyzed against buffer A. The protein concentration was determined by Bradford assays or sodium dodecyl sulfate (SDS)-PAGE using BSA as a standard. Cy3 concentration was determined by measuring absorbance using the molar extinction coefficient of Cy3 at 550 nm (150000 M−1 cm−1). After the addition of 10% sucrose, the dialyzed proteins were frozen in liquid nitrogen and stored at −80 °C.
For single-molecule imaging, we used the high-salt wash method to purify axonemes from sea urchin sperm flagella.17
SNAPf folding analysis
SNAP ligand (BG) was covalently attached to the amino-modified oligo DNA as previously described.19,28,32 The unreacted BG was removed using the QIAquick nucleotide removal kit (QIAGEN) and MicroSpin S-200 HR column (Cytiva). Later, dimeric kinesin-SNAPfs were reacted for 1.5 h at room temperature with the purified BG-conjugated oligo DNA at a SNAPf/BG-oligo molar ratio of 1:4 and run on SDS-PAGE, followed by staining with TaKaRa CBB Protein Safe Stain. The peptides were detected on the Typhoon FLA 9500 scanner (Cytiva), and the gel images were quantified using ImageJ.
Binding assay
The honeycomb-type DNA origami rod was designed using caDNAno (Maya plug-in; ESI Fig. 3, ESI Table 1†) and folded in 1× Barrel buffer (5 mM Tris-borate, pH 7.6, 20 mM Mg(OAc)2, 5 mM NaOAc, and 1 mM EDTA). Typically, 40 nM single-stranded M13mp18 DNA (tilibit) and 260 nM of each staple strand (6.5-fold excess) were mixed in 1× Barrel buffer and heated at 85 °C for 5 min, followed by annealing at 47.5 °C for 4 h. The folded DNA origami was purified by gel electrophoresis22 and concentrated by polyethylene-glycol (PEG) precipitation.49 The DNA origami concentration was determined by NANODROP 1000 (Thermo Scientific). The purified DNA rod was frozen in liquid nitrogen and stored at −80 °C. Before use, the DNA rod was mixed with kinesin in 5 mM Tris-borate (pH 7.6), 4 mM PIPES (pH 7.0), 1–4 mg mL−1 casein, 80 mM KCl, 20 mM Mg(OAc)2, 1 mM EDTA, 50 μM ATP, and 1 mM DTT and incubated at room temperature. After incubation, the reaction was stopped by immediately performing electrophoresis or by adding 20 μM BG-oligonucleotide. The concentrations of kinesin were optimized. For example, we did not perform the 10 K 20 nM experiments shown in Fig. 2, and instead estimated the 10 K's kon using 2–10 nM kinesin, because a short reaction time was expected (in the order of seconds, for which it is difficult to obtain reliable data).After incubating the DNA rod with kinesin, gel assays were performed in 0.5× TBE buffer (Medicago or Nacalai Tesque) supplemented with 12 mM MgCl2 and with or without 0.1% SDS and run for 1 h at 100 V. Dye-labeled DNA and kinesin were detected on a Typhoon FLA 9500 scanner (Cytiva) and analyzed with ImageJ. In the case of mobility shift assays, the binding ratio R was calculated by eqn (1):
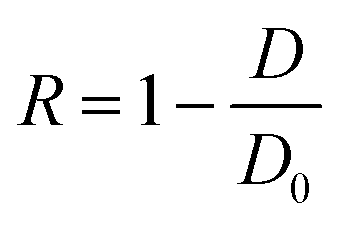 | (1) |
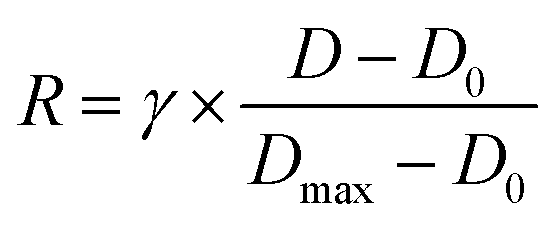 | (2) |
| R = β × (1 − e−αt[kinesin]) | (3) |
To determine the kinesin assembly speed (on-rate: kon), the binding rate was plotted against kinesin concentration ([kinesin]), and fitted with a linear least-squares function (α = kon [kinesin]).
Transport complex assembly for single-molecule imaging
The square lattice-type DNA origami rods were designed using caDNAno (ESI Fig. 11, ESI Tables 2 and 3†) and folded in 1× Barrel buffer (5 mM Tris-borate, pH 7.6, 20 mM Mg(OAc)2, 5 mM NaOAc, and 1 mM EDTA). Typically, 40 nM single-stranded P8064 DNA (tilibit) and 275 nM of each staple strand (7-fold excess) were mixed in 1× Barrel buffer and heated at 85 °C for 5 min, followed by annealing at 52.5 °C for 4 h. The folded DNA rod was purified as described above. Then, dimeric kinesin-SNAPf-10 K partially labeled with BG-A647 was mixed with the DNA rod at a BG/SNAPf molar ratio of 1:10 in 22 mM PIPES, pH 7.0, 0.8 mg mL−1 casein, 220 mM NaCl, 12 mM MgCl2, 0.9 mM EGTA, 43 μM ATP, and 1 mM DTT. The mixed sample was incubated for 5 h at room temperature and purified using MicroSpin S-300 or S-400 HR columns (Cytiva) equilibrated with 25 mM PIPES, pH 7.0, 0.8 mg mL−1 casein, 250 mM NaCl, 12 mM MgCl2, 1 mM EGTA, 50 μM ATP, and 1 mM DTT. The eluates were frozen in liquid nitrogen and stored at −80 °C after the addition of 10% sucrose.
Single-molecule imaging
Single-molecule images were visualized by a total internal reflection fluorescence (TIRF) microscope equipped on an inverted-type microscope (IX70; Olympus), as previously described.17,50 To prevent the nonspecific adsorption of the Lys-tag-fused proteins, we used a PEG-coated glass surface as previously described51 with the following modifications.A flow cell was constructed between a KOH-cleaned quartz glass slide and a coverslip by placing a polyethylene spacer of 40 μm thickness in between. The flow cell was coated with 1 mg mL−1 methoxy-poly(ethylene-glycol)-block-poly(L-lysine hydrochloride) (mPEG5K-bPLKF10; Alamanda Polymers) in BRB12 buffer for 0.5–6 h. After axoneme (purified from sea urchin sperm flagella) binding, the flow cell was washed and equilibrated with BRB12(X) buffer (BRB12 buffer supplemented with sodium chloride at the concentration described in each figure). The concentration of sodium chloride was optimized based on the aim of figure (see below). This is because we found that the experimental efficiency was traded off for the estimate accuracy, i.e., when using low ionic (NaCl) concentrations, we obtained more data on particle movements but many of these particles reached the end of an axoneme or field of view, rendering run length estimation difficult. Therefore, we chose different ionic (NaCl) concentrations based on the aim of each experiment. For example, for position (1,3) transporter of 2-kinesin, 150 and 210 mM of sodium chloride was used for Fig. 3 and ESI Fig. 21,† respectively. Fluorescence imaging was initiated after infusing fluorescently labeled kinesins (12–250 pM) or DNA rod–kinesin complexes (30–200 pM) in motility buffer (1 mg mL−1 casein, 4.5 mg mL−1 glucose, 50 U/mL glucose oxidase, 50 U/mL catalase, 2 mM Trolox, 0.5% 2-mercaptoethanol, and 1 mM ATP in BRB12(X) buffer) at 23 ± 1 °C.
The fluorescent dyes were illuminated with a He–Ne laser (633 nm for Cy5 and Alexa Fluor 647; 10 mW; 05-LHP-991; Melles Griot) and diode laser (532 nm for Cy3; 100 mM; Sapphire SF 532-100CW CDRH; Coherent Inc.). Fluorescence images were separated using Dual-View (Teledyne Photometrics) and then projected side-by-side onto an electron-multiplying charge-coupled device camera (iXon+ DU897C-CS0-#BV, Andor).
Single-molecule photobleaching assay
The assays were performed as described in the section “Single-molecule imaging,” except for the following modifications. Fluorescence imaging was initiated after infusing A647-labeled dimeric kinesin-SNAPf-10 K or DNA rod–kinesin complexes (2.5 pM) in motility buffer supplemented with 2 mM AMP-PNP instead of ATP. The time traces of A647 and/or Cy3 intensities were analyzed using custom Fiji macros and Python scripts, where each photobleaching point was finally determined by eye. The A647-labeled fractions were calculated from the histograms of step numbers with least-squares fitting to the binomial distribution.Data analysis for single-molecule imaging
Run length, velocity, and fluorescence intensities were analyzed using custom Fiji macros and Python scripts using kymographs as described previously,19 except for the following modifications. Kymographs were generated from axonemes with an average length of 18 μm. For run length, the variation (σ) of a single fluorophore was determined by Gaussian fitting (0.17 μm), and the particles with run lengths greater than 0.5 μm (∼3σ) were analyzed and defined as a motile event. The particles that bound to axonemes within 3.5 μm (spatial axis) or 10 s (time axis) at the edges of the kymographs in the direction of movement were excluded to reduce the influence of the particles that reached the edges, for which accurate measurement of the run lengths was not possible. In contrast, the particles that did not meet the criteria but reached the edges of the kymographs were treated as detached and included in the analysis. The average velocities were calculated by fitting the data with a Gaussian function. For statistical analyses, we first considered whether the data were parametric (i.e., assuming normal distribution of the data, e.g., velocity) or non-parametric (i.e., not assuming normal distribution of the data, e.g., run length). Then, we considered whether the data were composed of more than three groups. If more then three groups, we first checked whether there were significant changes in all groups using analysis of variance (ANOVA, for velocity) and Kruskal–Wallis tests (for run length). If we found any significant differences, we then considered whether the data had a control group. For many cases, the data had no control group, so we used a Tukey's test (velocity, parametric) or Conover–Iman test (run length, non-parametric). On the other hand, the data from free kinesins (Fig. 1) had a control group (0 K), so the velocities were analyzed using s Dunnett's test. We note that we did not further analyze the run length of free kinesin because of the lack of statistical difference.Author contributions
H. T. conceived, designed and supervised the study. K. F. performed the biochemical and single-molecule experiments and analyzed the data. H. T. designed and prepared the DNA rod. Y. M. contributed the kinesin attachment to the DNA nanostructure and the development of biochemical assay system. Y. H. supervised H. T. and K. F. T. U. supervised H. T. and Y. M. All authors discussed the results. H. T. and K. F. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank members of the Harada and Ueda Laboratories for providing comments on this manuscript; C. Shingyoji and I. Nakano for the axoneme; this work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas (‘Molecular Robotics’) (to H. T. 15H00798), Grant-in-Aid for Scientific Research (B) and a Grant-in-Aid for challenging Exploratory Research (to H. T. No. 16KT0068, 19H03197, 25650045; to Y. H. No. 18H01838, 22H02583), Grant-in-Aid for Transformative Research Areas (B) (to Y. H. No. 20H05785), Research Fellow (to K. F. No. 20J23072) from the Japan Society for the Promotion of Science (JSPS), MEXT – Quantum Leap Flagship (MEXT Q-LEAP) Program (to Y. H.) from the Japan Science and Technology Agency; National Natural Science Foundation of China (to H. T. No. 32171425, 32150710528); and by startup fund and Double First-class Initiative Fund from ShanghaiTech University (to H. T.).References
- I. R. Gibbons, Studies on the Protein Components Of Cilia from Tetrahymena Pyriformis, Proc. Natl. Acad. Sci. U. S. A., 1963, 50, 1002–1010, DOI:10.1073/pnas.50.5.1002
.
- H. E. Huxley, The mechanism of muscular contraction, Science, 1969, 164, 1356–1365, DOI:10.1126/science.164.3886.1356
- T. Yanagida, T. Arata and F. Oosawa, Sliding distance of actin filament induced by a myosin crossbridge during one ATP hydrolysis cycle, Nature, 1985, 316, 366–369, DOI:10.1038/316366a0
- R. D. Vale and R. A. Milligan, The Way Things Move: Looking Under the Hood of Molecular Motor Proteins, Science, 2000, 288, 88–95, DOI:10.1126/science.288.5463.88
- S. J. Kron and J. A. Spudich, Fluorescent actin filaments move on myosin fixed to a glass surface, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 6272–6276, DOI:10.1073/pnas.83.17.6272
- T. Funatsu, H. Higuchi and S. Ishiwata, Elastic filaments in skeletal muscle revealed by selective removal of thin filaments with plasma gelsolin, J. Cell Biol., 1990, 110, 53–62, DOI:10.1083/jcb.110.1.53
- S. E. Encalada, L. Szpankowski, C. H. Xia and L. S. Goldstein, Stable kinesin and dynein assemblies drive the axonal transport of mammalian prion protein vesicles, Cell, 2011, 144, 551–565, DOI:10.1016/j.cell.2011.01.021
- Y. Harada, A. Noguchi, A. Kishino and T. Yanagida, Sliding movement of single actin filaments on one-headed myosin filaments, Nature, 1987, 326, 805–808, DOI:10.1038/326805a0
- Y. Harada, K. Sakurada, T. Aoki, D. D. Thomas and T. Yanagida, Mechanochemical coupling in actomyosin energy transduction studied by in vitro movement assay, J. Mol. Biol., 1990, 216, 49–68, DOI:10.1016/S0022-2836(05)80060-9
- Y. Y. Toyoshima, S. J. Kron, E. M. McNally, K. R. Niebling, C. Toyoshima and J. A. Spudich, Myosin subfragment-1 is sufficient to move actin filaments in vitro, Nature, 1987, 328, 536–539, DOI:10.1038/328536a0
- J. Howard, A. J. Hudspeth and R. D. Vale, Movement of microtubules by single kinesin molecules, Nature, 1989, 342, 154–158, DOI:10.1038/342154a0
- P. Bieling, I. A. Telley, J. Piehler and T. Surrey, Processive kinesins require loose mechanical coupling for efficient collective motility, EMBO Rep., 2008, 9, 1121–1127, DOI:10.1038/embor.2008.169
- S. M. Block, L. S. Goldstein and B. J. Schnapp, Bead movement by single kinesin molecules studied with optical tweezers, Nature, 1990, 348, 348–352, DOI:10.1038/348348a0
- A. Seitz and T. Surrey, Processive movement of single kinesins on crowded microtubules visualized using quantum dots, EMBO J., 2006, 25, 267–277, DOI:10.1038/sj.emboj.7600937
- Q. Feng, K. J. Mickolajczyk, G.-Y. Chen and W. O. Hancock, Motor Reattachment Kinetics Play a Dominant Role in Multimotor-Driven Cargo Transport, Biophys. J., 2018, 114, 400–409, DOI:10.1016/j.bpj.2017.11.016
- R. Schneider, T. Korten, W. J. Walter and S. Diez, Kinesin-1 motors can circumvent permanent roadblocks by side-shifting to neighboring protofilaments, Biophys. J., 2015, 108, 2249–2257, DOI:10.1016/j.bpj.2015.03.048
- Y. Miyazono, M. Hayashi, P. Karagiannis, Y. Harada and H. Tadakuma, Strain through the neck linker ensures processive runs: a DNA-kinesin hybrid nanomachine study, EMBO J., 2010, 29, 93–106 CrossRef CAS PubMed
- D. K. Jamison, J. W. Driver, A. R. Rogers, P. E. Constantinou and M. R. Diehl, Two kinesins transport cargo primarily via the action of one motor: implications for intracellular transport, Biophys. J., 2010, 99, 2967–2977, DOI:10.1016/j.bpj.2010.08.025
- N. D. Derr,
et al., Tug-of-war in motor protein ensembles revealed with a programmable DNA origami scaffold, Science, 2012, 338, 662–665, DOI:10.1126/science.1226734
- K. Furuta,
et al., Measuring collective transport by defined numbers of processive and nonprocessive kinesin motors, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 501–506, DOI:10.1073/pnas.1201390110
- P. W. K. Rothemund, Folding DNA to create nanoscale shapes and patterns, Nature, 2006, 440, 297–302 CrossRef CAS PubMed
- S. M. Douglas,
et al., Self-assembly of DNA into nanoscale three-dimensional shapes, Nature, 2009, 459, 414–418 CrossRef CAS PubMed
- R. Iinuma,
et al., Polyhedra self-assembled from DNA tripods and characterized with 3D DNA-PAINT, Science, 2014, 344, 65–69 CrossRef CAS PubMed
- H. Yan, S. H. Park, G. Finkelstein, J. H. Reif and T. H. LaBean, DNA-templated self-assembly of protein arrays and highly conductive nanowires, Science, 2003, 301, 1882–1884, DOI:10.1126/science.1089389
- O. I. Wilner,
et al., Enzyme cascades activated on topologically programmed DNA scaffolds, Nat. Nanotechnol., 2009, 4, 249–254 CrossRef CAS PubMed
- B. Sacca,
et al., Orthogonal protein decoration of DNA origami, Angew. Chem., Int. Ed., 2010, 49, 9378–9383, DOI:10.1002/anie.201005931
- J. Fu, M. Liu, Y. Liu, N. W. Woodbury and H. Yan, Interenzyme substrate diffusion for an enzyme cascade organized on spatially addressable DNA nanostructures, J. Am. Chem. Soc., 2012, 134, 5516–5519 CrossRef CAS PubMed
- T. Masubuchi,
et al., Construction of integrated gene logic-chip, Nat. Nanotechnol., 2018, 13, 933–940, DOI:10.1038/s41565-018-0202-3
- R. F. Hariadi,
et al., Mechanical coordination in motor ensembles revealed using engineered artificial myosin filaments, Nat. Nanotechnol., 2015, 10, 696–700, DOI:10.1038/nnano.2015.132
- R. F. Hariadi, A. J. Appukutty and S. Sivaramakrishnan, Engineering Circular Gliding of Actin Filaments Along Myosin-Patterned DNA Nanotube Rings To Study Long-Term Actin-Myosin Behaviors, ACS Nano, 2016, 10, 8281–8288, DOI:10.1021/acsnano.6b01294
- K. Fujita, M. Ohmachi, K. Ikezaki, T. Yanagida and M. Iwaki, Direct visualization of human myosin II force generation using DNA origami-based thick filaments, Commun. Biol., 2019, 2, 437, DOI:10.1038/s42003-019-0683-0
- T. Torisawa,
et al., Autoinhibition and cooperative activation mechanisms of cytoplasmic dynein, Nat. Cell Biol., 2014, 16, 1118–1124, DOI:10.1038/ncb3048
- R. Ibusuki,
et al., Collective motility of dynein linear arrays built on DNA nanotubes, Biochem. Biophys. Res. Commun., 2020, 523, 1014–1019, DOI:10.1016/j.bbrc.2019.12.125
- K. S. Thorn, J. A. Ubersax and R. D. Vale, Engineering the processive run length of the kinesin motor, J. Cell Biol., 2000, 151, 1093–1100, DOI:10.1083/jcb.151.5.1093
- R. F. Sommese,
et al., Patterning protein complexes on DNA nanostructures using a GFP nanobody, Protein Sci., 2016, 25, 2089–2094, DOI:10.1002/pro.3020
- M. T. Strauss, F. Schueder, D. Haas, P. C. Nickels and R. Jungmann, Quantifying absolute addressability in DNA origami with molecular resolution, Nat. Commun., 2018, 9, 1600, DOI:10.1038/s41467-018-04031-z
- K. J. Koßmann,
et al., A Rationally Designed Connector for Assembly of Protein-Functionalized DNA Nanostructures, Chembiochem, 2016, 17, 1102–1106, DOI:10.1002/cbic.201600039
- E. Nakata, H. Dinh, T. A. Ngo, M. Saimura and T. Morii, A modular zinc finger adaptor accelerates the covalent linkage of proteins at specific locations on DNA nanoscaffolds, Chem. Commun., 2015, 51, 1016–1019, 10.1039/c4cc08167f
- T. M. Nguyen, E. Nakata, M. Saimura, H. Dinh and T. Morii, Design of Modular Protein Tags for Orthogonal Covalent Bond Formation at Specific DNA Sequences, J. Am. Chem. Soc., 2017, 139, 8487–8496, DOI:10.1021/jacs.7b01640
- H. Khataee and J. Howard, Force Generated by Two Kinesin Motors Depends on the Load Direction and Intermolecular Coupling, Phys. Rev. Lett., 2019, 122, 188101, DOI:10.1103/PhysRevLett.122.188101
- F. A. S. Engelhardt,
et al., Custom-Size, Functional, and Durable DNA Origami with Design-Specific Scaffolds, ACS Nano, 2019, 13, 5015–5027, DOI:10.1021/acsnano.9b01025
- R. Veneziano,
et al., In vitro synthesis of gene-length single-stranded DNA, Sci. Rep., 2018, 8, 6548, DOI:10.1038/s41598-018-24677-5
- R D Vale, T Funatsu, D W Pierce, L Romberg, Y Harada and T Yanagida, Direct observation of single kinesin molecules moving along microtubules., Nature, 1996, 380(6573), 451–453, DOI:10.1038/380451a0
- H. Isojima, R. Iino, Y. Niitani, H. Noji and M. Tomishige, Direct observation of intermediate states during the stepping motion of kinesin-1, Nat. Chem. Biol., 2016, 12, 290–297, DOI:10.1038/nchembio.2028
- R. Igarashi,
et al., Tracking the 3D Rotational Dynamics in Nanoscopic Biological Systems, J. Am. Chem. Soc., 2020, 142, 7542–7554, DOI:10.1021/jacs.0c01191
- A. Kunwar, M. Vershinin, J. Xu and S. P. Gross, Stepping, strain gating, and an unexpected force-velocity curve for multiple-motor-based transport, Curr. Biol., 2008, 18, 1173–1183, DOI:10.1016/j.cub.2008.07.027
- M. C. Ucar and R. Lipowsky, Collective force generation by molecular motors is determined by strain-induced unbinding, Nano Lett., 2020, 20, 669–676, DOI:10.1021/acs.nanolett.9b04445
- S. A. Abdellatef,
et al., Oscillatory movement of a dynein-microtubule complex crosslinked with DNA origami, Elife, 2022, 11, e76357, DOI:10.7554/eLife.76357
- E. Stahl, T. G. Martin, F. Praetorius and H. Dietz, Facile and Scalable Preparation of Pure and Dense DNA Origami Solutions, Angew. Chem., Int. Ed., 2014, 53, 12735–12740, DOI:10.1002/anie.201405991
- T. Funatsu, Y. Harada, M. Tokunaga, K. Saito and T. Yanagida, Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution, Nature, 1995, 374, 555–559, DOI:10.1038/374555a0
- T. Aoki, M. Tomishige and T. Ariga, Single molecule FRET observation of kinesin-1's head-tail interaction on microtubule, Biophysics, 2013, 9, 149–159, DOI:10.2142/biophysics.9.149
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00088e |
| This journal is © The Royal Society of Chemistry 2023 |