
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-particle dispersion of carbon dots in the nano-hydroxyapatite lattice achieving solid-state green fluorescence†
Lunzhu
Wang‡
,
Xinru
Wang‡
,
Shuoshuo
Zhou
,
Jian
Ren
,
Liting
Liu
,
Cairong
Xiao
and
Chunlin
Deng
 *
*
School of Materials Science and Engineering, National Engineering Research Center for Tissue Restoration and Reconstruction, South China University of Technology, China. E-mail: chldeng@163.com
First published on 25th May 2023
Abstract
Carbon dots (CDs), as new carbon nanomaterials, have potential applications in multiple fields due to their superior optical properties, good biocompatibility, and easy preparation. However, CDs are typically an aggregation-caused quenching (ACQ) material, which has a huge limitation on the practical application of CDs. To solve this problem, in this paper, CDs were prepared by the solvothermal method using citric acid and o-phenylenediamine as precursors and dimethylformamide as solvent. Then using CDs as nucleating agents, solid-state green fluorescent CDs were synthesized by in situ growth of nano-hydroxyapatite (HA) crystals on the surface of CDs. The results show that CDs are stably dispersed single-particlely in the form of bulk defects in the nano-HA lattice matrices with a dispersion concentration of 3.10%, and solid-state green fluorescence of CDs is achieved with a stable emission wavelength peak position near 503 nm, which provides a new solution to the ACQ problem. CDs-HA nanopowders were further used as LED phosphors to obtain bright green LEDs. In addition, CDs-HA nanopowders showed excellent performance in cell imaging (mBMSCs and 143B) applications, which provides a new scheme for further applications of CDs in the field of cell imaging and even in vivo imaging.
Introduction
Carbon dots (CDs) are novel carbon nanomaterials, typically smaller than 10 nm in size, mainly composed of a sphere-like carbon nucleus formed from sp2/sp3 hybridized carbon and abundant surface functional groups.1,2 CDs have excellent fluorescence properties. Corresponding to the compositional structure, the fluorescence mechanism mainly considers the quantum confinement effect (QCE) of the conjugated structure of the carbon nucleus and the defect energy levels introduced by the surface structure.3,4 Therefore, the fluorescence of CDs can be regulated by controlling the size of the conjugated structure and the surface defects of CDs, and the fluorescence emission in the UV-Vis-NIR can be realized, and the far infrared region is also getting more and more attention from researchers.5–9 Compared with traditional fluorescent materials, CDs have the advantages of low production cost, simple preparation, environmental friendliness, tunable fluorescence, high quantum yield and good biocompatibility.10 The characteristics of CDs make them widely used in lighting display,11,12 biological imaging,13,14 photocatalysis,15,16 ion detection17,18 and so on.However, when CDs are in the aggregated state, the π–π superposition of the conjugated structure leads to the quenching (ACQ) phenomenon of CDs, resulting in strong luminescence loss or even quenching of CDs,19,20 which greatly limits the wide application of CD-based materials. To overcome the ACQ phenomenon of solid-state CDs, various methods have been explored. One is the surface ligand modification method, which grafts long organic or polymer chains on the surface of CDs to prevent direct interaction between CDs and can realize solid-state fluorescence of CDs from blue to red.21–24 Another is the matrix-assisted dispersion method, where CDs are dispersed in the polymer matrix network, such as PMMA, HEMA, PVP, etc., also achieving blue to red solid-state fluorescence.25–28 However, the above methods all require organic solvents to provide the compounding environment for CDs and polymers, which inevitably results in solvent residues, causing harm to humans and the environment. In addition, due to the low melting temperature of the polymer matrix, it easily denatures and quenches at higher operating temperatures, resulting in device failure.
Therefore, inorganic crystals can serve as ideal solid-state dispersion matrices of CDs.29–31 On the one hand, inorganic crystals have stable lattice matrices, which can disperse CDs stably in a fixed position, and no movement between CDs leads to ACQ, thus achieving solid-state fluorescence of CDs. On the other hand, when CDs are dispersed in inorganic crystals in the form of bulk defects, the functional groups on the surface of CDs will chemically bond with the surrounding anions and cations, which can introduce new surface energy levels for CDs and thus regulate the emission wavelength of CDs to achieve red-shift or blue-shift. At present, a small number of studies have dispersed CDs in BaSO4, CaCO3, etc., and realized solid-state fluorescence of CDs.32,33 However, there are few studies on nano-hydroxyapatite (HA) as the dispersion matrix of CDs. In addition, combined with the biological characteristics and applications of HA,34–36 solid-state fluorescence can be realized, which is of great significance to the research and development in the biomedical field.
In this work, as shown in Fig. 1, we designed a simple and effective synthesis method based on the structural characteristics of CDs, based on the dissolution–precipitation mechanism and crystal heterogeneous nucleation theory, to make CDs enter the nano-HA lattice single-particlely. CDs have good dispersion, small-size nanostructures and abundant functional groups on their surfaces, allowing them to serve as heterogeneous nucleation sites for nano-HA crystals. In a high-temperature and high-pressure hydrothermal environment with CDs as nucleating agents, single-particle dispersed CDs adsorb Ca2+ and PO43− in solution and grow in situ into nano-HA after heterogeneous nucleation. At this point, CDs successfully entered the nano-HA lattice as heterogeneous nuclei, and finally solid-state green fluorescence of CDs was achieved. It not only solves the problem that most CDs can only disperse luminescence in polar organic solvents (such as EtOH, DMF, DMSO, etc.), but also achieves the transition from liquid-phase dispersion to single-particle dispersion in the solid-state lattice for hydrophilic CDs. CDs-HA can be used as a nano-phosphor for lighting display due to its advantages of solid-state fluorescence, environmental friendliness and low-cost. In addition, CDs-HA has shown good performance in cell imaging.
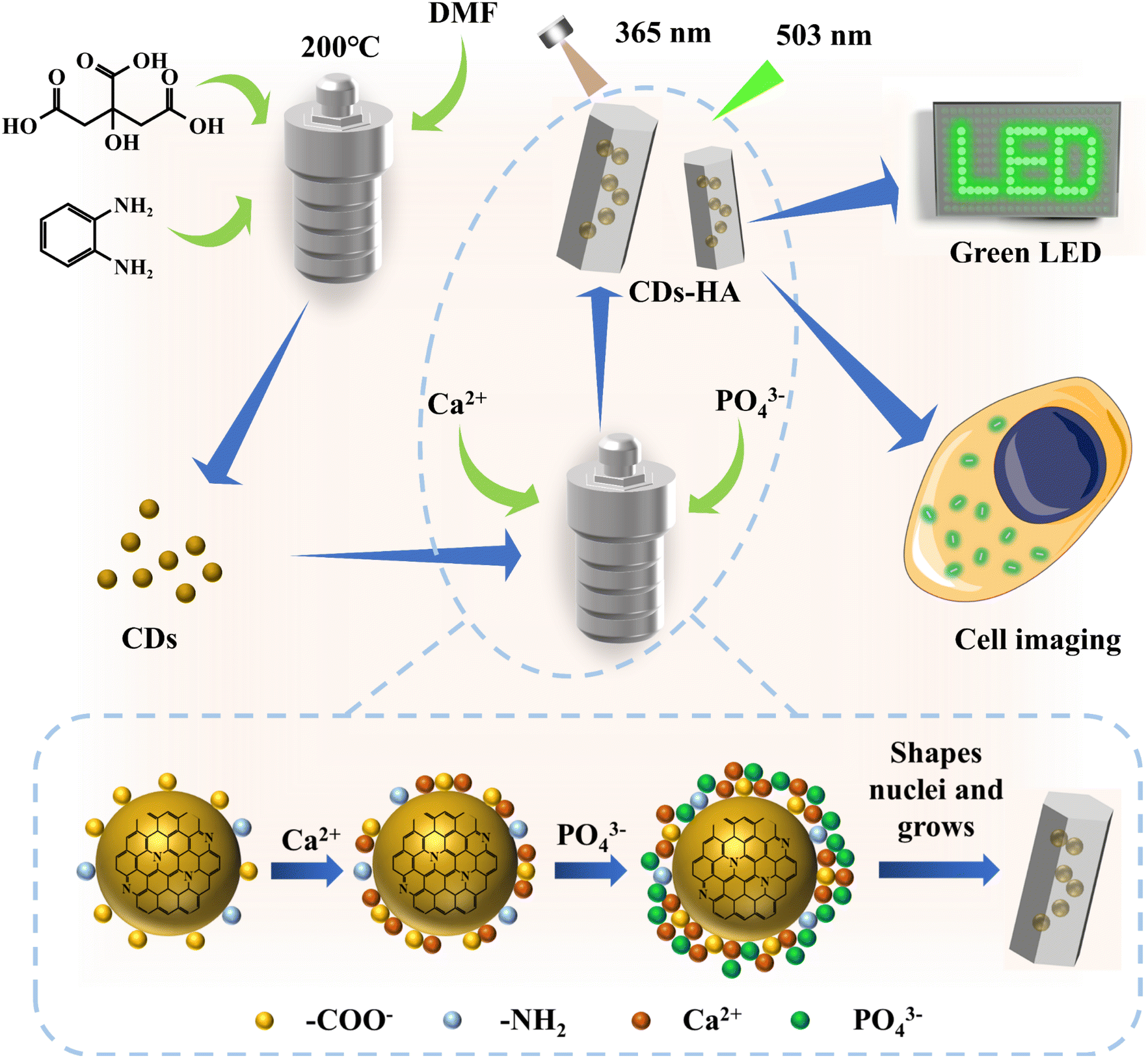 | ||
| Fig. 1 Schematic diagram of the preparation and application of CDs and CDs-HA. | ||
Experimental
Materials
The reagents required for the experiments included CA, o-phenylenediamine (o-PD), DMF, NaOH, HCl, Ca(NO3)2·4H2O, Na3PO4·10H2O and EtOH, all of which were analytically pure reagents purchased from Aladdin and could be used directly without further purification. The aqueous solutions were prepared with deionized water, and the interception molecular weight of the dialysis bag was 1000 Da.Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and phosphate buffer solution (PBS) were purchased from Gibco. Trypsin and penicillin-streptomycin solutions containing EDTA were purchased from Biosharp. The Actin-Tracker Red-Rhodamine, Calcein-AM/PI Cell Viability/Cytotoxicity Assay Kit, and Cell Counting Kit-8 (CCK-8) were purchased from Beyotime Biotechnology.
Preparation of CDs
4 g CA and 1 g o-PD in 40 mL DMF were dissolved and sonicated for 15 min to make the reaction precursors mix well and dissolve. The mixture was transferred to a Teflon-lined reactor and reacted at 200 °C for 5 h. After the reaction, it was allowed to cool naturally, and the pH was adjusted to be weakly alkaline. The product was dialyzed in a 1000 Da dialysis bag for 48 h, during which the water was changed several times. After dialysis, the retained solution was freeze-dried for 72 h to obtain CD powders.In situ growth of nano-HA on the surface of CDs
The experiments were performed by the hydrothermal method. The calcium and phosphorus sources used were Ca(NO3)2 solution (0.25 mol L−1) and Na3PO4 solution (0.05 mol L−1), respectively. The procedure was as follows: a certain mass of CDs was weighed in Ca(NO3)2 solution (12.5 mL) and sonicated for 30 min. After the two were uniformly mixed, Na3PO4 solution (37.5 mL) was added to the mixture through a peristaltic pump at a rate of 0.5 mL min−1 with constant stirring. After finishing the drop addition, the pH was adjusted to 12, and the reaction was carried out in a teflon-lined reactor at a certain temperature for a certain time. After the reaction, the product was cooled naturally, washed with deionized water and EtOH 3 times, and dried at 60 °C for 12 h to obtain CDs-HA nanopowders. The reaction conditions are detailed in Table 1.| Samples | CDs (g) | Reaction time (h) | Reaction temperature (°C) |
|---|---|---|---|
| HA | 0 | 12 | 150 |
| 0.01-CDs-HA | 0.01 | 12 | 150 |
| 0.03-CDs-HA | 0.03 | 12 | 150 |
| 0.05-CDs-HA | 0.05 | 12 | 150 |
| 0.07-CDs-HA | 0.07 | 12 | 150 |
| 0.05-CDs-HA120 | 0.05 | 12 | 120 |
| 0.05-CDs-HA180 | 0.05 | 12 | 180 |
| 0.05-CDs-HA6 | 0.05 | 6 | 150 |
| 0.05-CDs-HA18 | 0.05 | 18 | 150 |
Characterization
The phase compositions of both CDs and CDs-HA samples were recorded by X-ray diffraction (Empyrean, PANalytical, XRD) in the 2θ range of 10–70° with a step size of 0.026° and a scan rate of 4° min−1. The morphology and size of CDs and CDs-HA samples were observed by transmission electron microscopy (Talos F200x/Talos F200c, Thermo Fisher, HR-TEM/TEM). The elemental composition of the CDs and CDs-HA sample surfaces was recorded by X-ray photoelectron spectroscopy (Escalab Xi+, Thermo Scientific, XPS). A Fourier transform infrared spectrometer (iS50, Thermo Scientific, FT-IR) was used to analyze the functional group composition of CDs and CDs-HA samples in the wavenumber range of 500 cm−1 to 4000 cm−1. The UV-Vis absorption spectrum of CDs was detected by using a UV spectrophotometer (UV2600, Shimadzu). The liquid-phase fluorescence of CDs and the solid-state fluorescence spectra of HA were detected by using a fluorescence spectrometer (F-7000, Hitachi). The CDs and CDs-HA samples were analyzed by using a thermal analyzer (STA449F, NETZSCH, TG) with a temperature rise rate of 10 °C min−1, temperature range of 30–900 °C and air atmosphere.CDs-HA applied to green LEDs
The GaN LED chips without phosphor coating were purchased from New Star Photoelectric CO., Ltd. The emission wavelength of the GaN LED chip was concentrated at 365 nm and the working current was 300 mA. The specific operations were as follows: an appropriate amount of 0.05-CDs-HA powder was dispersed evenly on top of the GaN LED chip. The whole process did not involve organic solvents. The spectrum of the LED was measured by using a HAAS-2000 high precision fast spectroradiometer. The color of the light was determined by using a CIE (CIE1931xy.V.1.6.0.2) calorimeter system.CDs-HA applied in cell imaging
Firstly, the cytotoxicity of CDs, HA and 0.05-CDs-HA to normal somatic cells was detected. Mouse bone marrow stem cells (mBMSCs) were cultured with complete medium configured with 90% DMEM, 10% FBS, and 1% penicillin-streptomycin, respectively. Cell activity was assayed by using the Cell Counting Kit-8 (CCK-8). mBMSCs were inoculated in 96-well plates with a density of 5 × 103 cells per well and cultured for 24 h. Then CDs, HA, and 0.05-CDs-HA (100 μg mL−1) were added to each well for 24 and 48 h. CCK-8 solution was added to the well plates and incubated for 2 h. The optical density (OD) values were detected at 450 nm by using an enzyme marker (Cytation 5, Biotek). The complete medium without any material was used as the control group and the cell group without material was used as the blank background.At the same time, live/dead cells were stained with a Calcein-AM/PI cell viability/cytotoxicity assay kit to observe cell viability. The mBMSCs were inoculated in 48-well plates at a density of 1.5 × 104 cells per well and incubated for 24 h. CDs, HA, and 0.05-CDs-HA (100 μg mL−1) were then added to each well for 24 and 48 h. After Calcein-AM/PI was added and incubated for 20 min away from light, its fluorescence staining images were observed and obtained by using an inverted fluorescence microscope (Axio Observer-7). Live cells were observed at 494 nm and dead cells were observed at 535 nm.
Co-localization experiments were then performed on cells (mBMSCs and 143B) using CDs-HA to evaluate their cellular imaging capabilities. The cells were inoculated in a confocal laser dish with a density of 1.5 × 105 per well for 24 h and then cultured for 4 and 8 h with 0.05-CDs-HA (100 μg mL−1). Cellular microfilaments were stained using Actin-Tracker Red-Rhodamine, and the cell morphology and imaging of 0.05-CDs-HA were observed and photographed under a laser scanning confocal microscope (TCS SP8, Leica, CLSM). The excitation wavelength of the dye was 561 nm and the excitation wavelength of 0.05-CDs-HA was 405 nm.
Statistical analysis
All data were expressed as mean ± standard deviation (SD) and analyzed using the one-way analysis of variance (ANOVA) with statistical significance assigned at *P < 0.05 (significant), **P < 0.01 (highly significant), and ***P < 0.001 (most significant).Results and discussion
As shown in Fig. 1, in order to make CDs have a larger conjugated structure and abundant surface functional groups, organic amine o-PD and organic acid CA are used as reaction precursors. In order to mix the precursors homogeneously, CDs were prepared by the solvothermal method using DMF as the reaction solvent. It is generally accepted that CDs can be regarded as composed of 3 parts, namely, the non-conjugated structure (sp3 hybridized carbon), the conjugated structure (sp2 hybridized carbon) of the carbon nucleus, and the functional groups on the surface. The morphology of CDs was first observed, and TEM characterization was performed on them, as shown in Fig. S1.† CDs are a kind of spherical nanoparticles with good dispersion and do not easily agglomerate, and their size is concentrated below 4 nm with an average diameter of 2.31 nm. To further observe the structure of CDs, HR-TEM characterization was performed. As shown in Fig. 2a, CDs exhibit distinct lattice stripes, which indicates that CDs have a certain crystal structure. The lattice spacing is 0.21 nm, which corresponds to the graphite (100) crystal plane, which indicates the presence of sp2 hybridized carbon structures in CDs. The physical phases of the CDs were characterized as shown in Fig. 2b. A diffraction peak with a broader peak shape and weaker intensity appears at 2θ = 23°, which corresponds to the (002) crystal plane of the graphite structure, which indicates the presence of the sp2 hybridized carbon structure in CDs again.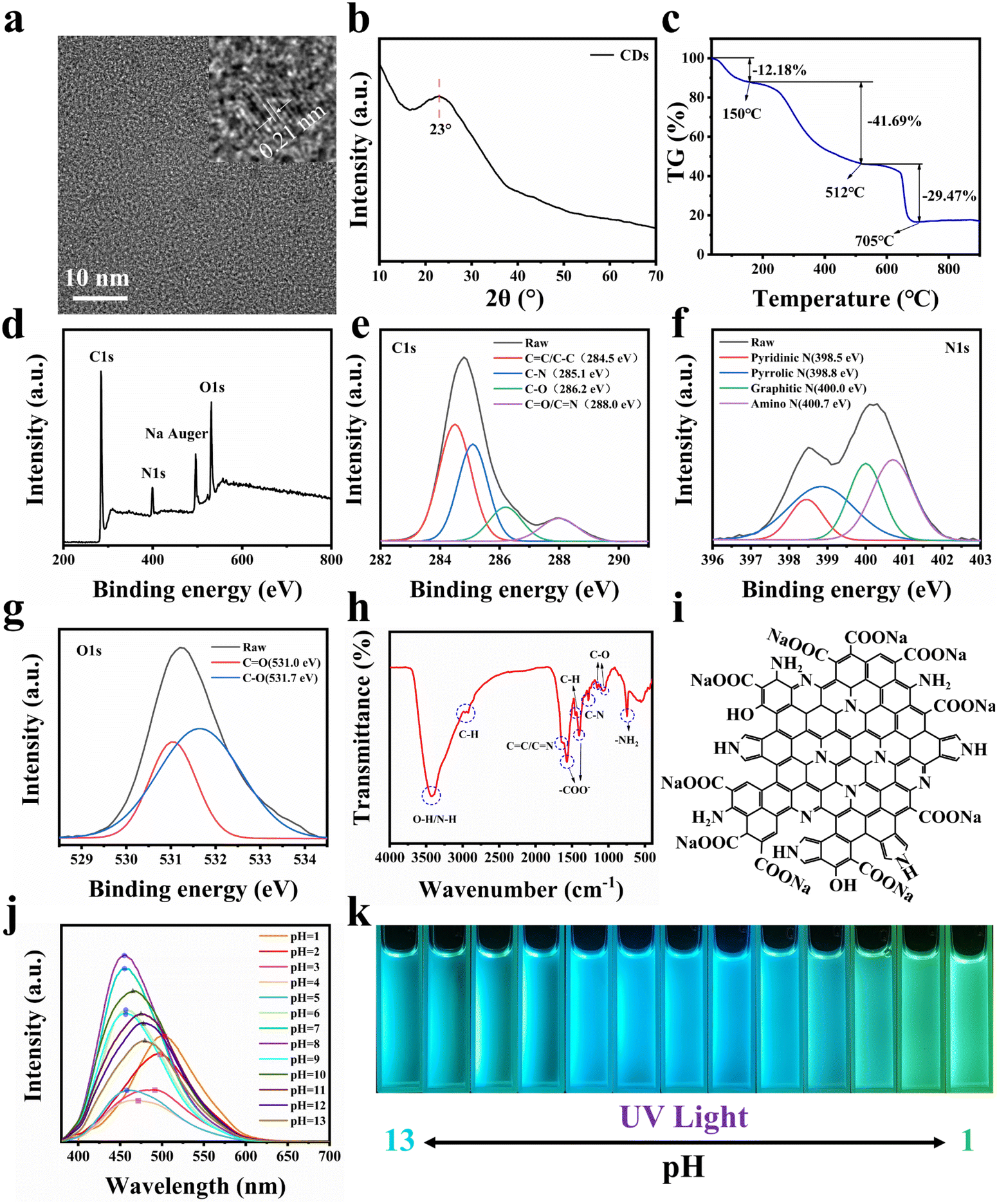 | ||
| Fig. 2 (a) HR-TEM and local magnification image of CDs. (b) XRD pattern of CDs. (c) Thermogravimetric curve of CDs. (d) XPS full spectrum of CDs. (e) C1s spectrum of CDs. (f) N1s spectrum of CDs. (g) O1s spectrum of CDs. (h) FT-IR spectrum of CDs. (i) Plane structure diagram of CDs. (j) Fluorescence spectrum of CD solution at pH = 1–13 and 365 nm excitation. (k) Fluorescence photos of CD solution at pH = 1–13 and 365 nm excitation. | ||
In addition, we attempted the structural analysis of CDs by thermogravimetry. As shown in Fig. 2c, CDs were tested at 0–900 °C under an air atmosphere, resulting in three stages of mass decline. Firstly, the mass of CDs decreases by 12.18% from 0–150 °C, which indicates that CDs have a large specific surface energy and a certain adsorption capacity for gases and water in the environment, and these adsorbed substances will be desorbed and discharged as the temperature rises. This is followed by a mass decrease of 41.69% from 150–512 °C. This is because the surface functional groups of CDs, such as –OH, –COO−, and –NH2, are further oxidized or dehydrated and escape from the surface as the temperature increases, and the surface structure of CDs is destroyed. The final mass drop of 29.47% from 512–705 °C implies that the sp3 hybridized carbon structure in the carbon nucleus reacts to generate gas and escapes. In addition, we found that there are still 16.66% mass residues, and these residues are stable sp2 hybridized carbon structures, which are conjugated structures in CDs, which supports the sp2 hybridized carbon structure of CDs again. According to the thermogravimetric curve, we can calculate the three structures of CDs sequentially, namely, the surface functional group, sp3 hybridized carbon and sp2 hybridized carbon with contents of 47.47%, 33.56 and 18.97%.
To analyze the chemical structure of CDs, we performed XPS tests on CDs, as shown in Fig. 2d. CDs are mainly composed of five elements, namely C, N, O, and H, plus deprotonated Na. For further high-resolution scanning of C1s, as shown in Fig. 2e, C1s can be fitted into 4 characteristic peaks with peak positions at 284.5, 285.1, 286.2, and 288.0 eV, representing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C, C–N, C–O, and CO/CN, respectively. Similarly, for the high-resolution scan of N1s, as shown in Fig. 2f, N1s can be fitted into 4 characteristic peaks with peak positions at 398.5, 398.8, 400.0, and 400.7 eV, corresponding to pyridine nitrogen, pyrrole nitrogen, graphite nitrogen, and amino nitrogen, respectively. In addition, for the high-resolution scan of O1s, as shown in Fig. 2g, O1s can be fitted into 2 characteristic peaks with peak positions at 531.0 and 531.7 eV, corresponding to CO and C–O, respectively.37–39 Combining the results of XPS, it can be tentatively concluded that CDs have a large number of CC and CN conjugated structures and abundant oxygen- and nitrogen-containing surface functional groups.
C/C–C, C–N, C–O, and CO/CN, respectively. Similarly, for the high-resolution scan of N1s, as shown in Fig. 2f, N1s can be fitted into 4 characteristic peaks with peak positions at 398.5, 398.8, 400.0, and 400.7 eV, corresponding to pyridine nitrogen, pyrrole nitrogen, graphite nitrogen, and amino nitrogen, respectively. In addition, for the high-resolution scan of O1s, as shown in Fig. 2g, O1s can be fitted into 2 characteristic peaks with peak positions at 531.0 and 531.7 eV, corresponding to CO and C–O, respectively.37–39 Combining the results of XPS, it can be tentatively concluded that CDs have a large number of CC and CN conjugated structures and abundant oxygen- and nitrogen-containing surface functional groups.
To further determine the chemical structure of CDs, we performed FT-IR characterization of CDs, as shown in Fig. 2h. The absorption at 3429 cm−1 corresponds to the stretching vibration of –OH and N–H. The absorption peaks at 2968 cm−1, 2926 cm−1 and 1460 cm−1 correspond to the C–H stretching and bending vibrations, respectively, and are attributed to the presence of aliphatic or aromatic C–H on the surface of CDs. The absorption peak at 1640 cm−1 represents the stretching vibration peak of CC/CN. The absorption peak at 1270 cm−1 represents the C–N stretching vibration peaks. The strong and sharp absorption peaks at 1570 cm−1 and 1400 cm−1 indicate the presence of –COO− in CDs. The absorption peaks at 1160 cm−1 and 1066 cm−1 represent C–O stretching vibration peaks and are attributed to the presence of C–OH and –COO− in CDs. The absorption peak at 750 cm−1 indicates that CDs have –NH2.40–42 Through the above analysis, we suggested that CDs have CC and CN conjugated structures and a large number of surface functional groups of –OH, –COO−, and –NH2. Through FT-IR characterization and analysis, we can qualitatively obtain a schematic diagram of the possible planar structure of CDs by combining the above conclusions of XPS characterization, as shown in Fig. 2i.
The optical properties of CDs were next characterized, and we investigated the effect of solution pH change on the fluorescence of CDs. H2O was used as the solvent, and NaOH and HCl were used to adjust the pH with an excitation wavelength of 365 nm, and the results are shown in Fig. 2j and k. When pH ≤ 4, with the decrease of pH, the emission wavelength of CDs gradually red-shifts and the emission intensity gradually increases, and at pH = 1, the emission wavelength reaches 507 nm, showing bright green light. When 5 ≤ pH ≤ 9, the emission wavelength of CDs remains at 455 nm, which does not change with pH, and the emission intensity reaches the maximum when pH = 8, showing bright blue light. When pH ≥ 10, with the increase of pH, the emission wavelength of CDs gradually red-shifts and the emission intensity gradually decreases, and at pH = 13, the emission wavelength reaches 480 nm, showing bright blue-green light. This phenomenon of pH response originates from the surface structure of CDs. The surface of CDs has pyridine nitrogen, pyrrole nitrogen, –NH2 and –COOH. Under acidic conditions, pyridine nitrogen, pyrrole nitrogen and –NH2 will be hydrolyzed and positively charged, and under basic conditions, –COOH will be ionized and negatively charged, and the increase in surface charge causes a change in the energy level of surface states and decreases the band gap of CDs, resulting in a red shift of the emission wavelength. This indicates that CDs in the solution state are susceptible to change under the influence of pH. Therefore, providing a stable chemical environment for CDs is significant for their further application.
To further understand the fluorescence mechanism of CDs, the green CD solution with pH = 1 was selected for the UV-Vis absorption spectroscopy test, as shown in Fig. S2.† There are two sharp absorption peaks at 271 and 277 nm, which are mainly caused by the π → π* leap of the CC bond. And as the wavelength increases, two weaker absorption peaks appear at 303 and 388 nm, which correspond to the n → π* leap of the CO and CN bonds, which are consistent with the structure of the above CDs.
Similarly, CD solution with pH = 1 was selected for an excitation wavelength-dependent study with a wavelength interval of 20 nm, and the results are shown in Fig. S3.† CDs can emit green fluorescence of 507 nm under excitation light ranging from 300 nm to 480 nm, and the optimal excitation wavelength is 420 nm. The emission wavelength of CDs does not change with the excitation wavelength, which, combined with Fig. 2j, suggests that the fluorescence mechanism of such CDs is influenced by the surface defect energy level.
In summary, CDs have small size nanostructures and abundant surface groups, including –OH, –COOH, pyridine nitrogen, pyrrole nitrogen, –NH2, etc. Therefore, CDs have great specific surface energy and a large amount of surface charge, which lead to the easy adsorption of Ca2+ and PO43− in solution, making it possible for them to be stably dispersed in the crystal as the nucleation sites for the in situ growth of nano-HA.
Therefore, after understanding the structure, composition, optical properties and fluorescence mechanism of CDs, we attempted to grow nano-HA in situ on the surface of CDs to disperse CDs single-particlely in the nano-HA lattice. After obtaining the samples, we performed HR-TEM characterization, and the results are shown in Fig. 3a–e. The nano-HA samples prepared by the hydrothermal method have high crystallinity, a small size and good dispersion, while CDs are dispersed single-particlely in the lattice of this nano-HA in the form of bulk defects, which are shown as small light-colored dots in the images due to the contrast degree. As can be seen from the images, with the addition of CDs increasing from 0 to 0.05 g, CDs in the nano-HA lattice gradually increase and do not adsorb on the crystal surface. Meanwhile with the addition increasing from 0.05 g to 0.07 g, it is found that CDs are more adsorbed on the surface of the crystal, showing as dark-colored dots on the surface of nano-HA. This indicates that the dispersion concentration of CDs reaches the maximum when the additive amount is 0.05 g. However, with the increase of the addition amount, CDs tend to be adsorbed on the crystal surface, and compared with the dispersion in the lattice, such surface adsorption is not strong and easy to fall off. In addition, in the powder state, CDs are more likely to aggregate and lead to fluorescence quenching.
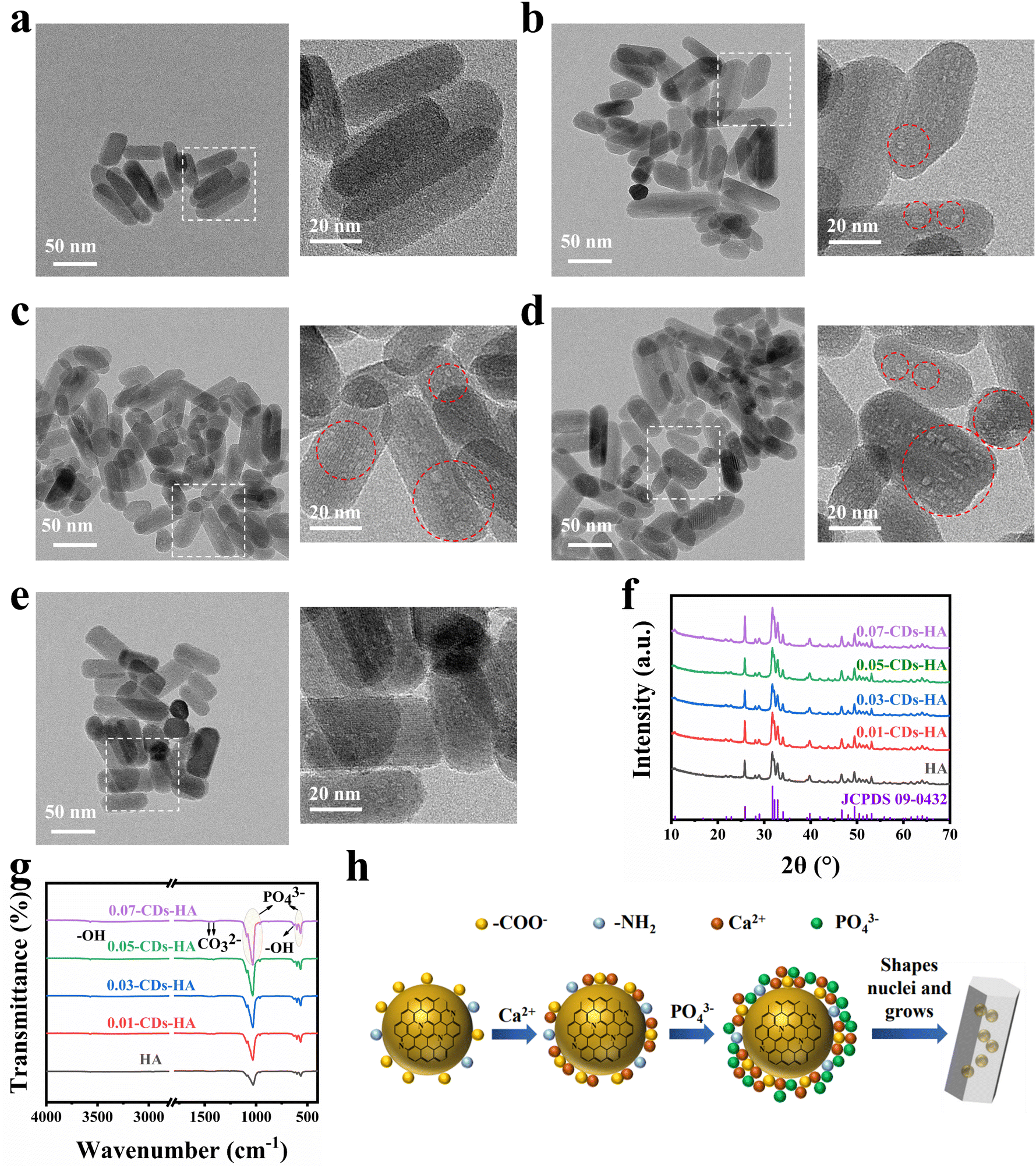 | ||
| Fig. 3 HR-TEM and local magnification image of (a) HA; (b) 0.01-CDs-HA; (c) 0.03-CDs-HA; (d) 0.05-CDs-HA; (e) 0.07-CDs-HA. (f) XRD patterns of HA and CDs-HA. (g) FT-IR spectra of HA and CDs-HA. (h) Schematic diagram of the process mechanism of in situ growth of nano-HA on the CD surface. | ||
XRD characterization was performed on 5 samples as shown in Fig. 3f. Using the standard card JCPDS: 09-0432 as a reference, the 5 samples correspond to the standard card one by one, indicating that the hydrothermal method and the participation of CDs will not produce other calcium and phosphorus phases, and the appropriate amount of CDs will not have a large effect on the crystallinity of nano-HA, and the physical phase is still the nano-HA phase. The XRD data were refined by Rietveld using GSAS software, and χ2 < 3 and Rwp < 0.05 indicate good quality and high confidence of the refined results, and the results are shown in Table 2 and Fig. S4.† Using the unit cell weight and unit cell density as references, we found that the cell mass and density of nano-HA increase abruptly when the addition of CDs increases from 0 to 0.01 g, indicating that the addition of CDs distorts the lattice of nano-HA. It implies that CDs are dispersed as a bulk defect inside the nano-HA lattice and that the excess ions adsorbed on the surface of CDs are distributed in the lattice interstices, leading to an increase in cell mass and density. Furthermore, the cell mass and density of nano-HA monotonically increase when the amount of CDs added is increased from 0.01 to 0.05 g, indicating that CDs are dispersed in the nano-HA lattice and have not yet reached saturation. And when the addition of CDs continues to increase to 0.07 g, we found that the cell mass and density begin to decrease, indicating that CDs have reached saturation at 0.05 g. And because the high addition of CDs leads to the aggregation of CDs in solution, they cannot enter the lattice and deposit on the surface of the nano-HA. This is consistent with the HR-TEM characterization results.
| Sample | χ 2 | R wp | Unit cell weight | Unit cell density (g cm−3) |
|---|---|---|---|---|
| HA | 1.541 | 0.0292 | 1003.693 | 3.150 |
| 0.01-CDs-HA | 1.711 | 0.0290 | 1015.055 | 3.181 |
| 0.03-CDs-HA | 1.508 | 0.0270 | 1018.965 | 3.196 |
| 0.05-CDs-HA | 1.605 | 0.0287 | 1023.483 | 3.209 |
| 0.07-CDs-HA | 1.711 | 0.0295 | 1019.817 | 3.200 |
Furthermore, FT-IR tests were performed on the 5 samples in ATR mode, as shown in Fig. 3g. The small and sharp peaks at 630 cm−1 and 3570 cm−1 correspond to the stretching and bending vibrations of –OH. 565 cm−1, 601 cm−1, 964 cm−1, 1033 cm−1 and 1093 cm−1 correspond to the characteristic absorption peaks of PO43−. 1414 cm−1 and 1456 cm−1 correspond to the characteristic absorption peaks of CO32−.43 All 5 samples have typical characteristic absorption peaks of nano-HA, and in a basic environment, CO32− substituted nano-HA is obtained. From the comparison of the 5 samples, the absorption peaks of –OH, CO32− and PO43− gradually increase with the increase of CDs from 0 g to 0.07 g. This indicates that the dispersion of CDs in the nano-HA lattice does not cause a change of the physical phase, but cause a change of the chemical composition. The increase of –OH originates from the CDs themselves, the increase of CO32− originates from the involvement of –COO− of CDs, and the increase of PO43− originates from the attraction of charged pyridine nitrogen, pyrrole nitrogen, and –NH2 on the surface of CDs to PO43− in solution.
Furthermore, the samples were analyzed for their constituent elements, and the blank group HA and the best added 0.05-CDs-HA samples were selected for XPS testing, as shown in Fig. S5.† The 0.05-CDs-HA sample contains the same elements as the blank group HA, which indicates that the addition of CDs does not hinder the generation of HA.
Through the above characterization and analysis, we derived the process mechanism of in situ growth of nano-HA on the surface of CDs, as shown in Fig. 3h. Firstly, CDs have large specific surface energy and abundant −COO− on the surface, which carries a large negative charge under deprotonation. In the high-temperature and high-pressure hydrothermal environment, CDs tend to adsorb Ca2+ from the solution, which enriches Ca2+ on the surface of CDs. Furthermore, the Ca2+ enriched on the surface of CDs attracts PO43− in solution, and then CDs act as heterophase nucleation sites for nano-HA. As the reaction proceeds, the solution pH decreases, and the pyridine nitrogen, pyrrole nitrogen and –NH2 on the surface of CDs will gradually hydrolyze and become positively charged, further attracting PO43− in solution to form heterogeneous nuclei and gradually grow into nano-HA crystals. As a result, CDs are naturally dispersed single-particlely in the nano-HA lattice, thus introducing more –OH, CO32− and PO43−, which increases the cell mass and density of nano-HA. This in situ growth mechanism is consistent with the above characterization results.
Next, the optical properties of the samples were characterized. Fig. 4a–d show the emission and absorption spectra of 0.01-CDs-HA, 0.03-CDs-HA, 0.05-CDs-HA and 0.07-CDs-HA powders at different excitation wavelengths. From the absorption spectra, the samples mainly have two absorption peaks at 365 and 460 nm, and the absorption peak at 365 nm is relatively enhanced with the increase of CD addition, while the absorption peak at 460 nm is gradually weakened. From the emission spectra, the emission peak does not change with the change of the excitation wavelength. All 4 samples emit bright green fluorescence of 503 nm at an excitation wavelength of 365 nm, which is similar to the emission wavelength of CDs in pH = 1 solution. It indicates that after the growth of nano-HA on the CD surface, the pyridine nitrogen, pyrrole nitrogen and –NH2 on the CD surface are similar to the solution at pH = 1, with a positive charge and electrostatic attraction with PO43−. Therefore, with the increase of CD addition, PO43− in the sample will also increase, which is consistent with the FT-IR result in Fig. 3g.
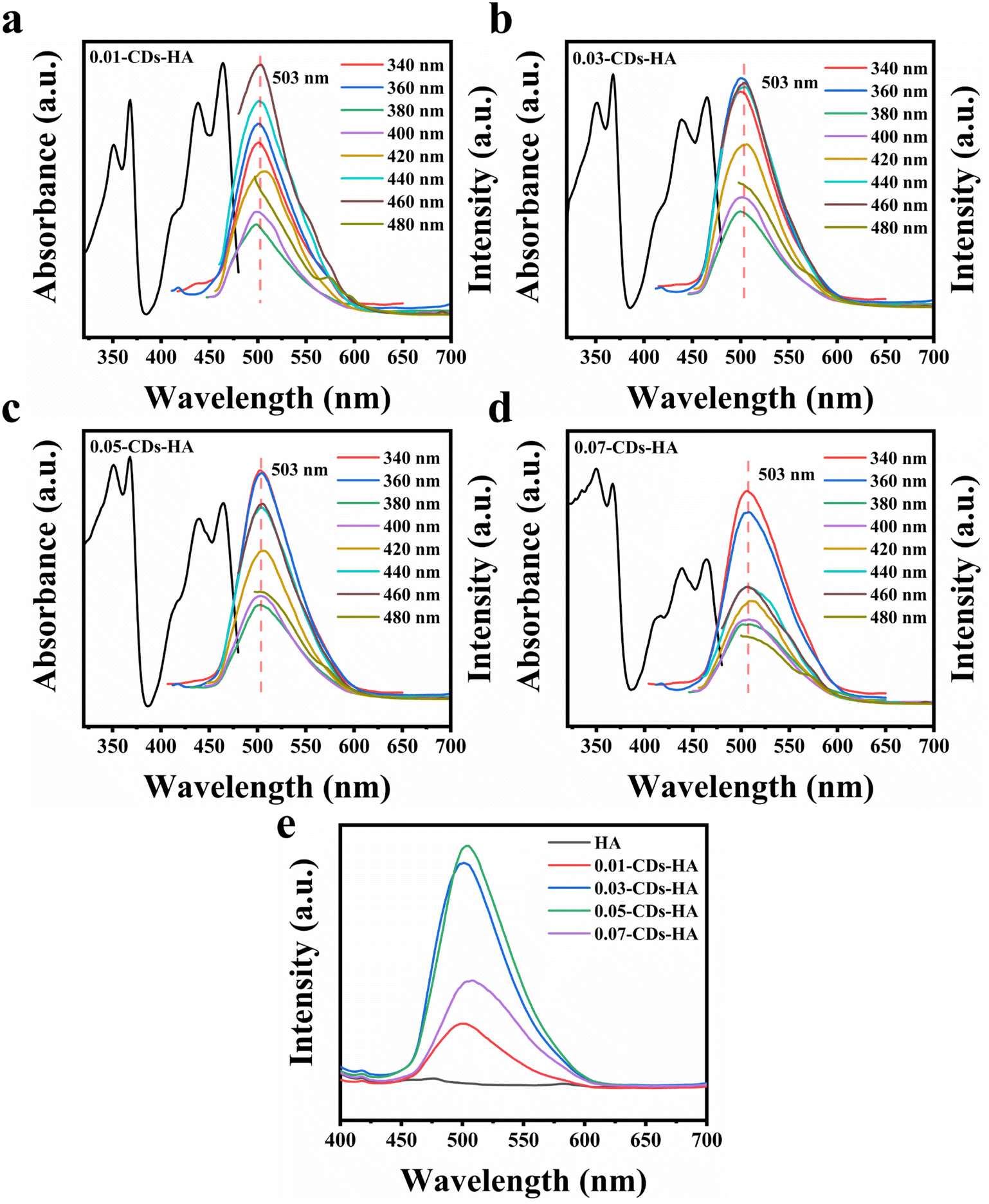 | ||
| Fig. 4 (a) Fluorescence spectrum of 0.01-CDs-HA. (b) Fluorescence spectrum of 0.03-CDs-HA. (c) Fluorescence spectrum of 0.05-CDs-HA. (d) Fluorescence spectrum of 0.07-CDs-HA. (e) Fluorescence spectra of HA and CDs-HA excited at 365 nm. | ||
In addition, the excitation wavelength of 365 nm was fixed to obtain the emission spectrum of CDs-HA, as shown in Fig. 4e. With the increase of CD addition from 0 to 0.05 g, the fluorescence intensity of CDs-HA powder gradually increases, and the highest fluorescence intensity is obtained at 0.05 g, namely 0.05-CDs-HA. And with the increase from 0.05 g to 0.07 g, the fluorescence intensity decreases instead. This indicates that the excess CDs do not act as nucleation growth sites, but are freely dispersed in the hydrothermal solution and will aggregate and settle, eventually manifesting as adsorption on the surface of the nano-HA crystals, leading to fluorescence quenching and intensity reduction. This is also consistent with the conclusion of Fig. 3a–e. Therefore, the optimal addition amount is 0.05 g, that is, the added concentration of CDs is 1 mg mL−1.
Furthermore, we calculated the dispersion concentration of CDs by thermogravimetric tests. Fig. S6† shows the thermogravimetric curves of blank HA and 0.05-CDs-HA samples. The 0–150 °C stage represents the volatilization of air and water adsorbed by the sample, and at this stage, HA and 0.05-CDs-HA have similar trends, which indicates that HA and 0.05-CDs-HA have similar surface structures and CDs are not adsorbed on the surface of nano-HA crystals. At the stage of 150–512 °C, –OH, –COO− and –NH2 successively react and dehydrate further inside the crystal and escape by diffusion, with a loss of 1.47%. Combined with Fig. 2c, the dispersion concentration of CDs can be calculated as 3.10%.
As the preparation method is hydrothermal, we investigated the influence of hydrothermal reaction conditions on the fluorescence properties of CDs-HA powder, including hydrothermal temperature and hydrothermal time. Fig. S7† shows the emission spectrum at the 365 nm excitation wavelength at hydrothermal temperatures of 120 °C, 150 and 180 °C, respectively. We found that the hydrothermal temperature has a small effect on the emission peak position and intensity. The higher the temperature, the higher the intensity of the emission wavelength, and the emission peak position is slightly red-shifted. This is because the rise of temperature intensifies the movement of the CDs and ions in the solution, making it easier for CDs to disperse in the HA lattice. And when the temperature is higher than 150 °C, the CDs will further start to dehydrate and carbonize, leading to a change in the emission peak position. Fig. S8† shows the emission spectrum at the 365 nm excitation wavelength for hydrothermal times of 6, 12 and 18 h. The hydrothermal time has no effect on the emission peak position, but has an enhanced effect on the emission intensity, that is, the longer the time, the higher the emission intensity. This is because with the increase of time, more CDs will participate in the reaction as nucleation growth sites and be dispersed in the nano-HA lattice, resulting in enhanced emission, but this enhancement is not unlimited.
After characterizing the sample's performance, we selected 0.05-CDs-HA as the phosphor to make green LEDs. As can be seen from Fig. 5a, 0.05-CDs-HA is white powder in daylight, which is the same as nano-HA without CDs added. This indicates that the binding mode of CDs and nano-HA is not surface adsorption, but dispersed in the lattice, and emits bright green fluorescence under the irradiation of 365 nm UV-light. Unlike the conventional method of dispersing CDs in organic polymer solutions, CDs-HA solves the ACQ problem of CDs, so that CDs-HA powder can be directly coated on top of GaN LED chips. Fig. 5b shows the photo of the LED working effect. The LED passes 300 mA current and emits bright green light after the CDs-HA layer conversion. Furthermore, we performed relevant tests on the LED, as shown in Fig. 5c and d. The LED has a wide emission spectrum of 460–700 nm with a CIE coordinate of (0.304, 0.4573) and a color temperature of 6253 K. In addition, the LED has a color rendering index of 69.8, a radiation flux of 3.288 lm, and a luminous efficiency of 2.57 lm W−1, which is comparable to LEDs made with CDs only and can basically meet the requirements of general lighting, which has an important prospect in the application of green lighting.
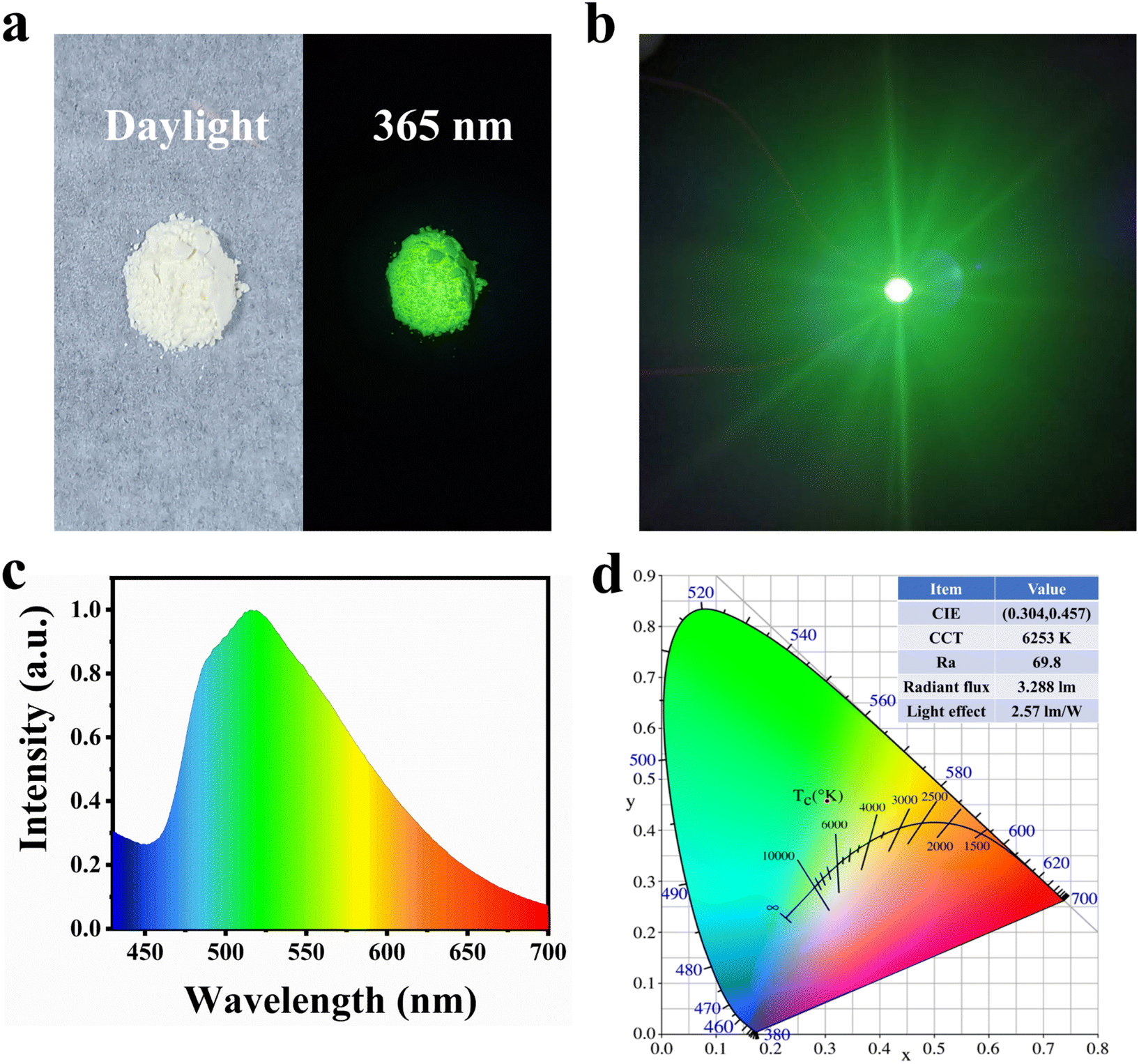 | ||
| Fig. 5 (a) Daylight (left) and 365 nm excitation (right) photographs of 0.05-CDs-HA. (b) Working photo of a green LED made from 0.05-CDs-HA. (c) The fluorescence spectrum of the green light LED. (d) Chromaticity diagram and data diagram of the green LED. | ||
In addition, combined with the biomedical applications of nano-HA, CDs-HA has potential applications in bioimaging applications. Fig. 6a shows the evaluation of the cell activity of CDs, HA, and 0.05-CDs-HA co-cultured with mBMSCs for 24 and 48 h. Compared with the control group, the CDs group shows a significant difference, with an inhibition rate of 80% on mBMSCs, showing low toxicity, which may be related to the small size structure of CDs. However, HA and 0.05-CDs-HA groups show no significant difference, and there is almost no toxicity to mBMSCs. Therefore, the dispersion of CDs in the HA lattice can further reduce the toxicity of CDs to mBMSCs on the basis of low toxicity, and this dispersion method broadens the application of CDs in biomedical fields. Further live-dead staining experiments were performed to observe cell viability, as shown in Fig. 6b. The cell densities at 48 h are all greater than those at 24 h, and the cell morphology is shuttle-shaped with a large number of tentacles, and almost no cell death occurs. These results indicate that mBMSCs have good cell activity and vigorous growth, and 0.05-CDs-HA is almost non-toxic to mBMSCs, which is the basis of the biological application of CDs-HA. On this basis, we used 0.05-CDs-HA for cell imaging of mBMSCs and 143B, as shown in Fig. 6c. Within 4 h, 0.05-CDs-HA can be smoothly taken up by cells and dispersed in the cytoplasm, and in the cellular environment, 0.05-CDs-HA fluorescence can remain stable without quenching. Under a laser scanning confocal microscope, we can clearly observe the location and distribution of CDs-HA in cells, and then we can trace the metabolic pathways of nano-HA in cells. On this basis, if CDs-HA can achieve a longer wavelength or even NIR emission, then in vivo imaging can be realized, which is undoubtedly very promising for biological imaging applications.
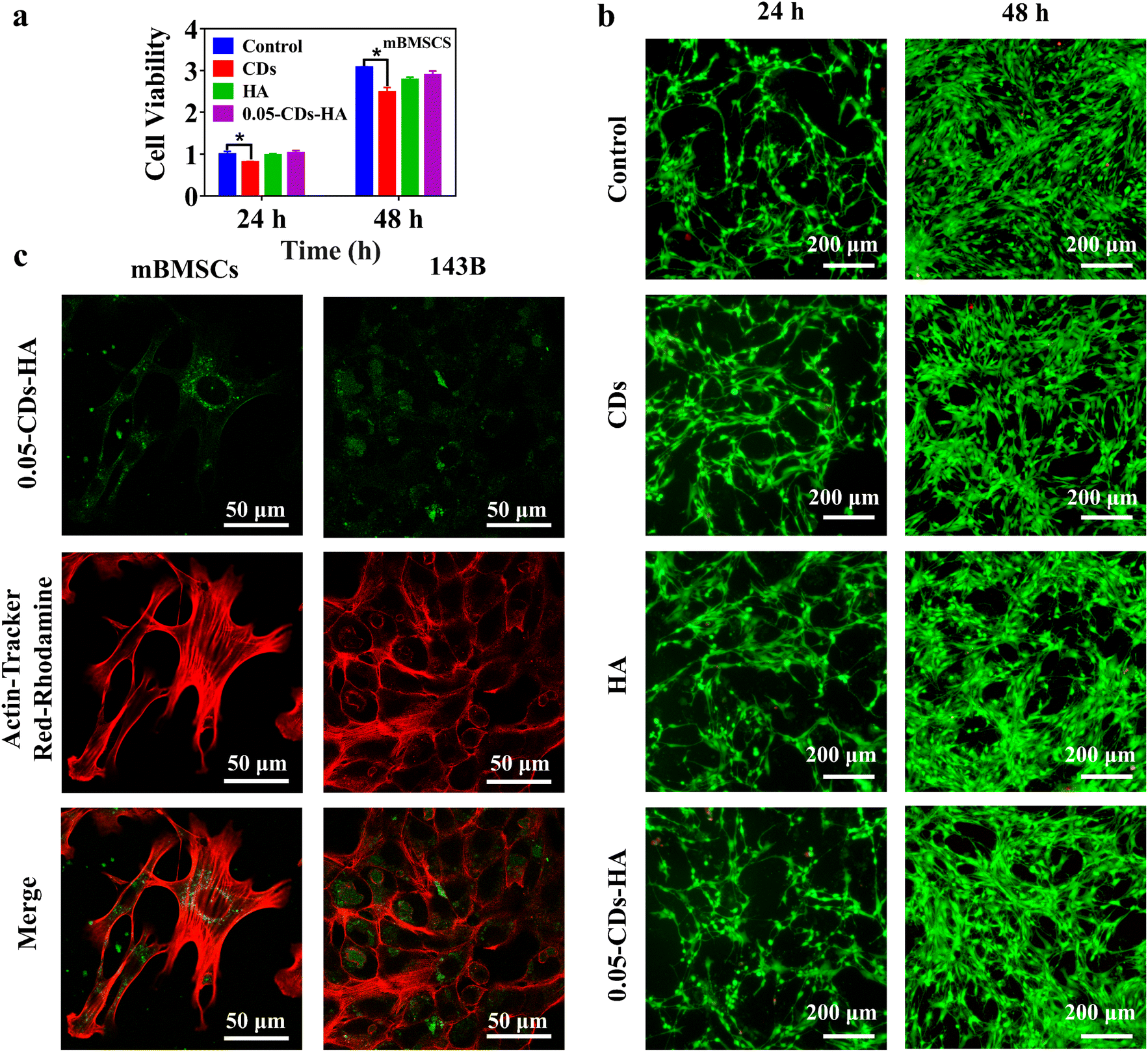 | ||
| Fig. 6 (a) CCK-8 results of the control group, CDs, HA and 0.05-CDs-HA co-cultured with mBMSCs for 24 and 48 h. (b) Live and dead staining images of the control group, CDs, HA and 0.05-CDs-HA co-cultured with mBMSCs for 24 and 48 h. (c) Co-localization images of 0.05-CDs-HA with cells (mBMSCs and143B). | ||
Conclusions
In summary, we dispersed CDs single-particlely in the nano-HA lattice by a hydrothermal method through in situ growth. CDs-HA nanopowders were prepared, and solid-state green luminescence of CDs was achieved with a stable emission wavelength peak near 503 nm and a dispersion concentration of 3.10% of CDs, and the effects of reaction parameters (CD addition, hydrothermal temperature, and hydrothermal time) on CDs-HA fluorescence were investigated. This method of growing inorganic nanocrystals on the surface of CDs based on the nano-size and surface structure characteristics of CDs effectively solves the ACQ problem faced by CDs in applications and provides a new solution for the wide application of CDs. Furthermore, CDs-HA nanopowders were coated on a LED chip as phosphors to obtain a green LED with a CIE coordinate of (0.304, 0.4573) and a color temperature of 6253 K. In addition, applying CDs-HA to cell imaging (mBMSCs and 143B), the location of CDs-HA within cells can be clearly observed with good cell imaging capability, which provides a new idea to realize in vivo imaging and shows great potential in biomedical applications.Author contributions
Lunzhu Wang and Xinru Wang contributed equally to this work. Lunzhu Wang conceived and designed the work. Lunzhu Wang and Xinru Wang synthesized and characterized the materials, performed the LED applications, analyzed and discussed the results, and wrote the paper. Shuoshuo Zhou and Jian Ren performed the cell imaging applications. Liting Liu and Cairong Xiao provided suggestions for the revision of the paper. Chunlin Deng provided funding, ideas and guidance for the whole work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant no 51972120 and no 51772105) and the Natural Science Foundation of Guangdong Province (No. 2023A1515012952).References
- L. Ðorđević, F. Arcudi, M. Cacioppo and M. Prato, Nat. Nanotechnol., 2022, 17, 112 CrossRef PubMed
.
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736 CrossRef CAS PubMed
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo, S. Prantl, R. Godin, J. R. Durrant and E. Reisner, Angew. Chem., Int. Ed., 2017, 56, 6459 CrossRef CAS PubMed
- H. Ding, S. Yu, J. Wei and H. Xiong, ACS Nano, 2016, 10, 484 CrossRef CAS PubMed
- S. Qu, D. Zhou, D. Li, W. Ji, P. Jing, D. Han, L. Liu, H. Zeng and D. Shen, Adv. Mater., 2016, 28, 3516 CrossRef CAS PubMed
- Y. Liu, W. Duan, W. Song, J. Liu, C. Ren, J. Wu, D. Liu and H. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 12663 CrossRef CAS PubMed
- Y. J. Chung, J. Kim and C. B. Park, ACS Nano, 2020, 14, 6470 CrossRef CAS PubMed
- X. Miao, D. Qu, D. Yang, B. Nie, Y. Zhao, H. Fan and Z. Sun, Adv. Mater., 2018, 30, 1704740 CrossRef PubMed
- D. Li, P. Jing, L. Sun, Y. An, X. Shan, X. Lu, D. Zhou, D. Han, D. Shen, Y. Zhai, S. Qu, R. Zbořil and A. L. Rogach, Adv. Mater., 2018, 30, 1705913 CrossRef PubMed
- J. Guo, Y. Lu, A. Xie, G. Li, Z. Liang, C. Wang, X. Yang and S. Chen, Adv. Funct. Mater., 2022, 32, 2110393 CrossRef CAS
- T. Hu, Z. Wen, C. Wang, T. Thomas, C. Wang, Q. Song and M. Yang, Nanoscale Adv., 2019, 1, 1413 RSC
- F. Yuan, T. Yuan, L. Sui, Z. Wang, Z. Xi, Y. Li, X. Li, L. Fan, Z. Tan, A. Chen, M. Jin and S. Yang, Nat. Commun., 2018, 9, 2249 CrossRef PubMed
- H. Li, X. Yan, D. Kong, R. Jin, C. Sun, D. Du, Y. Lin and G. Lu, Nanoscale Horiz., 2020, 5, 218 RSC
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2015, 54, 5360 CrossRef CAS PubMed
- J. Fang, T. Debnath, S. Bhattacharyya, M. Döblinger, J. Feldmann and J. K. Stolarczyk, Nat. Commun., 2020, 11, 5179 CrossRef CAS PubMed
- T. F. Yeh, C. Y. Teng, S. J. Chen and H. Teng, Adv. Mater., 2014, 26, 3297 CrossRef CAS PubMed
- X. Zhang, X. Tan and Y. Hu, J. Hazard Mater., 2021, 411, 125184 CrossRef CAS PubMed
- L. Pan, S. Sun, A. Zhang, K. Jiang, L. Zhang, C. Dong, Q. Huang, A. Wu and H. Lin, Adv. Mater., 2015, 27, 7782 CrossRef CAS PubMed
- Y. Deng, X. Chen, F. Wang, X. Zhang, D. Zhao and D. Shen, Nanoscale, 2014, 6, 10388 RSC
- Y. Chen, M. Zheng, Y. Xiao, H. Dong, H. Zhang, J. Zhuang, H. Hu, B. Lei and Y. Liu, Adv. Mater., 2016, 28, 312 CrossRef CAS PubMed
- L. Ai, Z. Song, M. Nie, J. Yu, F. Liu, H. Song, B. Zhang, G. I. N. Waterhouse and S. Lu, Angew. Chem., Int. Ed., 2022, e202217822 Search PubMed
- B. Xu, J. Li, J. Zhang, H. Ning, X. Fang, J. Shen, H. Zhou, T. Jiang, Z. Gao, X. Meng and Z. Wang, Adv. Sci., 2022, e2205788 Search PubMed
- J. Shao, S. Zhu, H. Liu, Y. Song, S. Tao and B. Yang, Adv. Sci., 2017, 4, 1700395 CrossRef PubMed
- Y. Chen, J. He, C. Hu, H. Zhang, B. Lei and Y. Liu, J. Mater. Chem. C, 2017, 5, 6243 RSC
- J. Li, H. Zhao, X. Zhao and X. Gong, Nanoscale, 2021, 13, 9561 RSC
- T. Zhang, F. Zhao, L. Li, B. Qi, D. Zhu, J. Lü and C. Lü, ACS Appl. Mater. Interfaces, 2018, 10, 19796 CrossRef CAS PubMed
- X. Yang, L. Sui, B. Wang, Y. Zhang, Z. Tang, B. Yang and S. Lu, Sci. China Chem., 2021, 64, 1547 CrossRef CAS
- Y. Han, X. Zhao, A. Vomiero, X. Gong and H. Zhao, J. Mater. Chem. C, 2021, 9, 12255 RSC
- Z. Qiao, Q. Huo, M. Chi, G. M. Veith, A. J. Binder and S. Dai, Adv. Mater., 2012, 24, 6017 CrossRef CAS PubMed
- W. Li, W. Zhou, Z. Zhou, H. Zhang, X. Zhang, J. Zhuang, Y. Liu, B. Lei and C. Hu, Angew. Chem., Int. Ed., 2019, 58, 7278 CrossRef CAS PubMed
- Q. Li, M. Zhou, Q. Yang, Q. Wu, J. Shi, A. Gong and M. Yang, Chem. Mater., 2016, 28, 8221 CrossRef CAS
- D. C. Green, M. A. Holden, M. A. Levenstein, S. Zhang, B. R. G. Johnson, J. Gala de Pablo, A. Ward, S. W. Botchway and F. C. Meldrum, Nat. Commun., 2019, 10, 206 CrossRef PubMed
- S. Guo, M. Yang, M. Chen, J. Zhang, K. Liu, L. Ye and W. Gu, Dalton Trans., 2015, 44, 8232 RSC
- F. Ye, H. Guo, H. Zhang and X. He, Acta Biomater., 2010, 6, 2212 CrossRef CAS PubMed
- M. Epple, Acta Biomater., 2018, 77, 1 CrossRef CAS PubMed
- H. Xiong, S. Du, J. Ni, J. Zhou and J. Yao, Biomaterials, 2016, 94, 70 CrossRef CAS PubMed
- J. Hu, Y. Sun, X. Geng, J. Wang, Y. Guo, L. Qu, K. Zhang and Z. Li, Light: Sci. Appl., 2022, 11, 185 CrossRef CAS PubMed
- M. Yu, X. Guo, H. Lu, P. Li, R. Huang, C. Xu, X. Gong, Y. Xiao and X. Xing, Carbon, 2022, 199, 395 CrossRef CAS
- J. Wan, S. Xu, J. Li, M. Yu, K. Zhang, G. Wei and Z. Su, Nanoscale, 2022, 14, 11359 RSC
- Y. Xu, B. Wang, M. Zhang, J. Zhang, Y. Li, P. Jia, H. Zhang, L. Duan, Y. Li, Y. Li, X. Qu, S. Wang, D. Liu, W. Zhou, H. Zhao, H. Zhang, L. Chen, X. An, S. Lu and S. Zhang, Adv. Mater., 2022, 34, 2200905 CrossRef CAS PubMed
- H. Liu, L. Mo, H. Chen, C. Chen, J. Wu, Z. Tang, Z. Guo, C. Hu and Z. Liu, Adv. Healthcare Mater., 2022, 11, 2101448 CrossRef CAS PubMed
- Z. Sun, F. Yan, J. Xu, H. Zhang and L. Chen, Nano Res., 2022, 15, 414 CrossRef CAS
- H. Yang, J. Hong, L. Wei and C. Deng, Ceram. Int., 2018, 44, 16844 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00106g |
| ‡ These authors contributed equally to this study and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2023 |